

Abstract
Genome-wide association studies (GWAS) have mapped over 90% of disease- and quantitative-trait-associated variants within the non-coding genome. Non-coding regulatory DNA (e.g., promoters and enhancers) and RNA (e.g., 5′ and 3′ UTRs and splice sites) are essential in regulating temporal and tissue-specific gene expressions. Non-coding variants can potentially impact the phenotype of an organism by altering the molecular recognition of the cis-regulatory elements, leading to gene dysregulation. However, determining causality between non-coding variants, gene regulation, and human disease has remained challenging. Experimental and computational methods have been developed to understand the molecular mechanism involved in non-coding variant interference at the transcriptional and post-transcriptional levels. This review discusses recent approaches to evaluating disease-associated single-nucleotide variants (SNVs) and determines their impact on transcription factor (TF) binding, gene expression, chromatin conformation, post-transcriptional regulation, and translation.
Keywords: non-coding variants, gene regulation, transcription factors, massively parallel reporter assay, RNA processing
1. Non-coding Genetic Variants in Human Diseases
The haploid human genome is ~3.2 billion base pairs, with about 98% comprising non-protein-coding DNA [1–4]. Genome-wide association studies (GWAS) have revealed that over 90% of disease- and trait-associated variants have been mapped within the non-coding genome [5–9]. This raises the question: How do single-nucleotide mutations outside the protein-coding genome impact cellular and organismal phenotype? A possible reason is that the non-coding genome potentially regulates gene expression [9,10]. Cis-regulatory elements (CREs) are non-coding DNA sequences that regulate gene expression, including promoters, enhancers, insulators, and silencers. Promoters are near the transcription start site (TSS), where the transcriptional machinery is recruited to form the pre-initiation complex [11–13]. Enhancers are one of the most abundant CREs responsible for enhancing transcription and regulating the spatial and temporal expression of genes in a tissue-specific manner [14]. They can be located as far as megabases upstream or downstream from the target gene and have been shown to physically interact with the promoters of the target genes through protein-mediated DNA looping [12,15].
An early example of a non-coding single-nucleotide variant/polymorphism (SNV/SNP) associated with a human disease was reported in 1982 in the β-globin gene (HBB) promoter and was linked to β-thalassemia [16]. In 2005, it was reported that this non-coding mutation resulted in the loss of a binding site for GATA1, which interacts with other transcription factors (TFs), such as CCAAT-enhancer-binding proteins (C/EBPs) and Krueppel-like factor 1 (KLF1), to modulate HBB expression [7,17,18]. Advances in DNA sequencing and functional genomics assays have propelled studies on the role of non-coding variants in regulatory regions of the genome to understand human pathophysiology, genetic diagnosis, and treatments. Non-coding variants can impact cellular and organismal phenotypes by altering the molecular recognition of CREs and disrupting transcriptional and post-transcriptional regulation of gene expression [19]. This review discusses advances in identifying functional non-coding SNVs and quantifying their impact on gene regulation. We mostly focus on research in GWAS SNVs but will also highlight examples of work on non-GWAS variants and their role in human diseases.
2. Non-coding Variants in Transcription Factor-DNA Binding
SNVs can modulate genomic binding by regulatory proteins, such as transcription factors (TFs), which are sequence-specific DNA-binding proteins that bind to CREs (e.g., promoters and enhancers) and recruit the transcriptional machinery needed to regulate gene expression (Fig. 1A) [20–23]. TFs target their specific binding sites through their DNA binding domains (DBDs), which in eukaryotes recognize short sequences of 6–12 bp [24–26]. Non-coding SNVs have been shown to alter TF-DNA recognition, leading to gene dysregulation (Fig. 1B) [6,27,28]. These variants can increase or decrease the affinity of TFs for a specific DNA sequence through the creation or disruption of TF-binding motifs [29–31].
Fig. 1. Non-coding variants can alter transcription factor (TF)–DNA binding activity, transcriptional machinery recruitment, and gene expression.
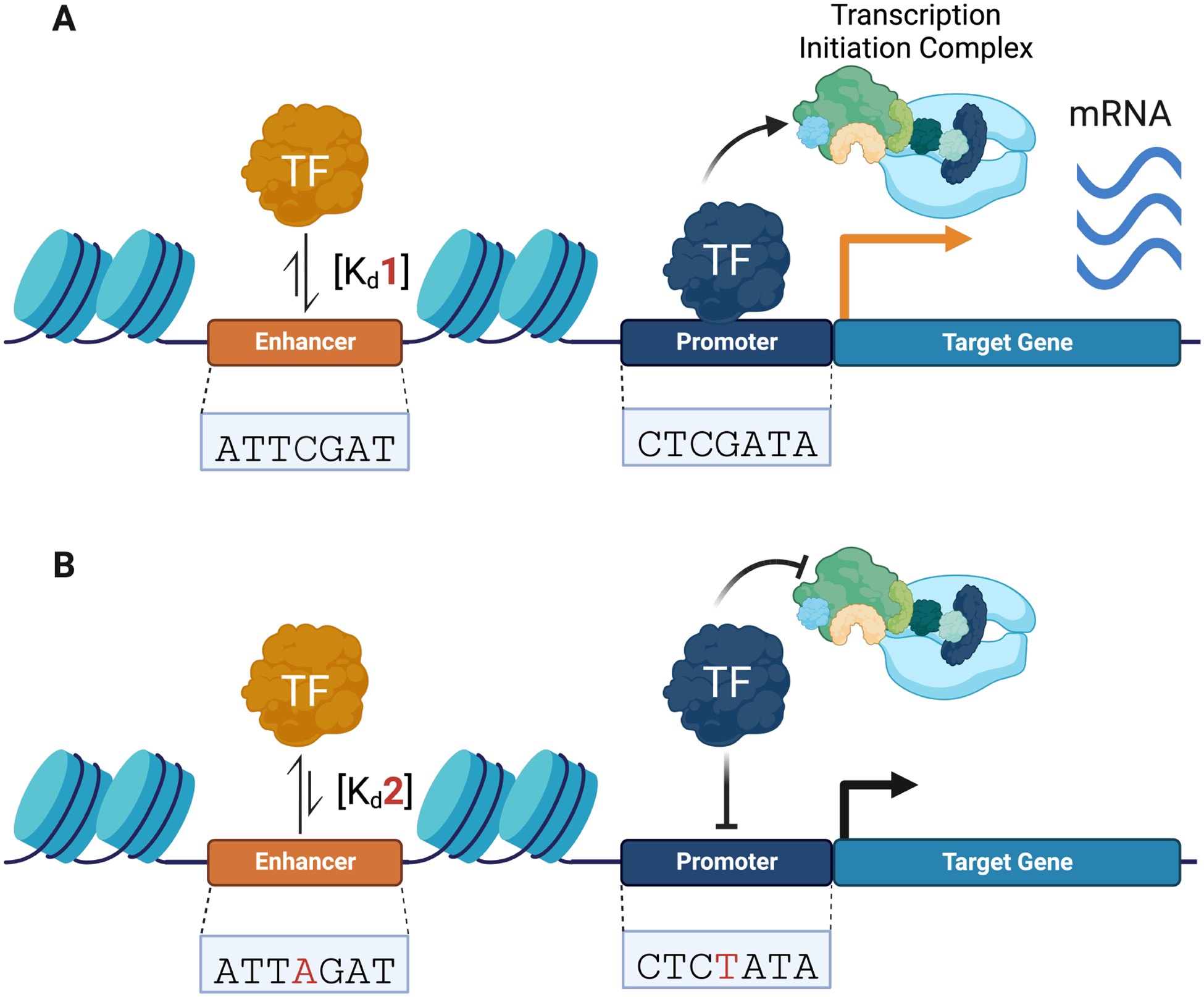
(A) TFs bind to regulatory DNA (e.g., promoters and enhancers) and recruit transcriptional machinery to initiate gene expression. (B) Non-coding variants can change TF–DNA binding affinities, altering transcriptional complex recruitment and gene expression. Changes in TF–DNA binding affinities are represented by equilibrium arrows. Changes in gene expression are represented by a black (decrease) and orange (increase) arrow.
Previous studies have determined changes in TF affinity through its binding site with in vitro assays, such as electrophoretic mobility shift assays (EMSA) [32]. Recently, Peña-Martínez et al. [33] identified five cardiovascular disease/trait-associated SNPs (rs7350789, rs61216514, rs7719885, rs747334, and rs3892630) predicted to alter the cardiac TF NKX2–5 DNA binding affinity and validated these predictions through EMSA. Although EMSA can be implemented to evaluate how non-coding SNPs can impact the formation of the TF-DNA complex and quantify changes in dissociation constant (Kd), it is a low throughput method [34,35]. High-throughput methods to determine TF-DNA binding preferences [36], such as protein binding microarrays (PBMs) [37], mechanically induced trapping of molecular interactions (MITOMI) [38], systematic evolution of ligands by exponential enrichment followed by sequencing (SELEX-seq) [39,40], and bacterial and yeast one-hybrid (B1H) [41,42], have contributed a wealth of information on the intrinsic TF DNA-binding specificity.
The Fordyce lab developed microfluidic-based high-throughput approaches to determine differences in TF affinities through Binding Energy Topography by sequencing (BET-seq) [43] and simultaneous transcription factor affinity measurements via microfluidic protein arrays (STAMMP) [44]. BET-seq can estimate Gibbs free energy of binding (ΔG) for over one million DNA sequences in parallel at high energetic resolution by determining the DNA sequencing count at a TF concentration. Using BET-seq, they measured changes in binding energy for all possible combinations of 10 nucleotide flanking regions (NNNNNCACGTGNNNNN) in the yeast TFs Pho4 and Cbf1 [43]. They were able to quantify changes in binding energies as small as ~0.5 kcal/mol between flanking regions, equivalent to mutating the core motif of Pho4 and Cbf1. Using STAMMP, they can express and purify over 1500 TFs while measuring affinities in parallel by determining the occupancy of fluorescently labeled DNA (Alexa-647) and TF (GFP). Through this approach, they expressed ~210 Pho4 missense mutants and measured binding affinities for DNA sequences with substitutions along the core binding motif and the 5′/3′ flanking regions, resulting in >1800 Kd measurements in a single experiment [44].
Jung et al. [45] developed high-performance fluorescence anisotropy (HiP-FA), a microscopy-based fluorescence polarization method using fluorophore-labeled DNA. TF–DNA complexes have a larger molecular weight than the unbound DNA, resulting in a decreased rotational speed and increased FA. Using HiP-FA, Jung et al. [45] determined the DNA-binding specificity for 26 purified TF DBDs from Drosophila and changes in affinity for all 33 possible 1-mismatch variants in the homeobox protein Bicoid (Bcd) 11-mer consensus sequence. Bray et al. [46] developed the Customizable Approach to Survey Complex Assembly at DNA Elements (CASCADE), a PBM-based method to profile cofactor recruitment by TFs through antibody labeling. They used CASCADE to profile cofactor recruitment at 1712 SNPs associated with eQTLs and chromatin accessibility (caQTLs) changes that altered binding motifs for multiple ETS–family TF–cofactor complexes in myeloid cells. Through this approach, Bray et al. [46] found that non-coding variants also impact cofactor recruitment, which is essential in regulating gene expression. Yan et al. [47] developed SNP-SELEX, a high-throughput multiplexed TF–DNA binding assay, and evaluated the differential binding of 270 human TFs on 95,886 type-2 diabetes-associated SNPs (permutated to all four bases and included SNPs in linkage disequilibrium). An oligo pool was synthesized with 40 bp genomic DNA centered on the SNP and flanking regions for polymerase chain reaction (PCR) amplification and barcoding for sequencing. Using full-length TFs and DBDs, they performed six rounds of enrichment and measured 828 million TF–DNA interactions [47].
Despite the advancements in high-throughput assays to measure changes in binding affinity, the number of TF (>1600 in humans) and GWAS SNP (>500,000) combinations greatly exceeds the capacity of these techniques [8,48,49]. Many computational approaches have implemented position weight matrices (PWMs) and position frequency matrices (PFMs), which describe TF binding preferences, to identify SNVs that alter TF binding motifs. PWMs and PFMs are typically generated from in vitro experimental data, such as mechanically induced trapping of molecular interactions (MITOMI) [50], PBMs [37], SELEX-seq [39], and B1H [41] and from chromatin immunoprecipitation followed by sequencing (ChIP-seq) [51–53]. The development of these in vitro methods has led to the development of motif-based predictive models, such as SNP2TFBS [54] and atSNP [55], which use PWMs from the JASPAR [56] database to predict the impact of non-coding variants in TF binding. These predictive models can integrate variants from databases, such as the 1000 Genomes Project [57] and dbSNP [48], to make in silico calculations that determine the disruption or formation of a TF binding site (TFBS) compared to a reference genome [54,55]. Examples of other bioinformatics resources that aid in identifying SNPs altering TFBS are sTRAP [58], motifbreakR [59], Raven [60], rSNP-MAPPER [61], OncoCis [62], and HaploReg [63]. However, models that rely solely on PWMs may not be sufficient to predict changes in affinity accurately.
Predictions using PWMs assume nucleotides contribute to binding in an additive and independent manner but ignore sequence features such as dinucleotides, DNA shape, and complex intracellular patterns [64–66]. Nishizaki et al. [67] developed an SNP effect matrix pipeline (SEMpl), a computational approach that considers data of TF endogenous binding (ChIP-seq), chromatin accessibility (DNase-seq), and TF-binding patterns (PWMs) to predict intracellular-binding patterns more accurately. SEMpl significantly outperforms the traditional PWM models at predicting changes in affinity by non-coding SNPs using in vitro validation through EMSA [67]. However, the previously mentioned techniques are less effective at predicting tissue-specific binding events altered by non-coding variants. Boytsov et al. [68] recently developed ANANASTRA, an upgraded version of ADASTRA [69], a web server that can accurately predict allele-specific binding events of TFs in different cell types [68]. This program requires inputs from four databases: allele-specific binding events from GTRD (ChIP-seq data) [70], binding patterns from HOCOMOCO (TF motif predictions) [71], a list of variants from dbSNP (rs-IDs) [48], and tissue-specific context from the GTEx project (eQTL) [72].
Machine learning models, such as support vector machine (SVM) and deep learning-based convolutional neural networks (CNN), have been widely used to predict changes in TF binding due to SVMs [73–75]. VandenBosch et al. [76] used ATAC-seq data to train a gapped k-mer SVM (gkm-SVM) model to predict changes in TF binding to all possible SNPs on 3773 human retinal CREs. Alternatively, CNNs, such as DeepFun [77] and AgentBind [78], are deep learning-based frameworks trained with ChIP-seq and DNase-seq to accurately predict tissue and cell type-specific TF differential binding because of non-coding variants. To further predict the functionality of non-coding SNPs, Wang et al. [79] developed DeFine, a CNN that also implements Hi-C data to map genes affected by risk variants while quantifying real-valued TF binding intensities.
3. Non-coding Variants in Gene Expression
Non-coding variants can impact cellular/organismal phenotypes as a downstream effect of altering TF–DNA binding by changing gene expression and the dysregulation of gene regulatory networks (GRNs) (Fig. 1B). Gene reporter assays are a popular method for quantifying the impact of regulatory variants by measuring the promoter and enhancer activity on a reporter gene [80,81]. Jiang et al. [82] identified three novel regulatory SNVs from 195 conotruncal heart defect patients that impaired GATA6 binding at the promoter of TBX1, resulting in decreased expression as determined by a dual-luciferase reporter assay. Many of the traditional enzyme-mediated gene reporter assays, such as luciferase [83] and β-galactosidase [84], are effective at evaluating changes in expression caused by non-coding variants but with a low-to-medium throughput.
Massively parallel reporter assays (MPRA) are an emerging high-throughput technique that substitutes standard enzyme assays with mRNA expression detection [85]. A library of thousands of regulatory elements or genomic-variant candidates is cloned into an expression vector with unique barcodes that can be quantified through DNA and RNA sequencing to determine the gene expression fold change or through flow cytometry in the case of fluorescent proteins. Lu et al. [86] used MPRA to evaluate 3073 GWAS systemic lupus erythematosus (SLE)-risk variants and observed allele-dependent enhancer activity in 16% of the risk variants. Through this approach, they nominated 51 causal variants in 27 SLE-risk loci with allelic impact on gene regulation. Another high-throughput assay to measure regulatory element activity is self-transcribing active regulatory region sequencing (STARR-seq). In STARR-seq, candidate CREs are cloned downstream of a minimal promoter and an open reading frame, removing the need to use barcodes by directly sequencing the transcribed element [87]. Toropainen et al. [88] used a multiplex STARR-seq assay to evaluate the enhancer activity of 34,344 vascular disease trait GWAS variants and observed allele-specific enhancer activity for 5711 SNPs. For example, rs17293632:C>T was nominated as a causal variant in smooth muscle cells by creating an AP-1 motif and reducing the expression of SMAD3, a TF that has been extensively characterized in smooth muscle cells of the vascular wall [88]. Going a step further to evaluate regulatory SNVs in a developing animal has occurred through the development of a high-throughput enhancer-insertion mouse reporter assay named enSERT, which uses CRISPR/Cas9-directed mutagenesis to quantify the enhancer activity of multiple variants in developing mouse embryos through β-galactosidase staining. Kvon et al. [89] developed this method and evaluated mutations on all nucleotides of ZRS (789 bp), a limp-specific enhancer. They observed abnormal enhancer activity from 71% of previously reported polydactyly-causal variants, providing further insight into causality and molecular mechanisms [89].
Experimental MPRA datasets have been implemented to train predictive models to enhance the prediction of functional non-coding variants. Yang et al. [90] developed presence-only with an elastic net penalty (PO-EN), a semi-supervised model that integrates MPRA data with epigenetic features (chromatin accessibility, methylation, histone modifications, etc.) to predict the regulatory effects of genetic variants. The developers of PO-EN reported greater accuracy at identifying GWAS SNPs with differential enhancer activity in a tissue- and cell-specific manner than other deep-learning models. Dong et al. [91] developed Score of Unified Regulatory Feature (SURF), a computational model that incorporates MPRA data to Regulome DB [92] functional genomics features (e.g., chromatin accessibility, histone variants, and TFBS) to predict the effect of variants on gene expression. SURF was tested in the Fifth Critical Assessment of Genome Interpretation (CAGI5) regulation saturation challenge. SURF outperformed other models in predicting the effect of 17,500 SNPs in disease-associated promoters and enhancers [91]. Movva et al. [93] developed a CNN-based method that utilizes MPRA data to predict and interpret the transcriptional regulatory activity of non-coding variants, Deep RegulAtory GenOmic Neural Network (MPRA-DragoNN). MPRA-DragoNN successfully predicted patterns in TF activity and gene expression events affected by reduced LDL cholesterol level-associated variants from GWAS [93].
4. Non-coding Variants in CRE Interactions
For over 30 years, DNA looping has been used to model how distal regulatory elements, such as enhancers, are brought near promoters to regulate gene expression (Fig. 2A) [94]. Advances in chromosome conformation capture (3C) technologies, such as circular 3C (4C) and 3C carbon copy (5C), have led to a better understanding of genome conformation, dynamics, and physical proximity between genomic elements [95–97]. These methods rely on restriction enzyme digestion of crosslinked chromatin and ligation of proximal elements to determine spatial proximity between genomic regions [98]. Coupled with massively parallel DNA sequencing, 3C assays have fueled widespread adoption and increased understanding of the genome structure on varying scales [97]. The human genome is organized in topologically associating domains (TADs), which provide an additional level of gene regulation by allowing distal CREs to interact with target promoters [99]. Understanding long-range genomic interactions is necessary to understand the potentially disruptive role of CRE variants in human diseases (Fig. 2B). High-throughput chromosome conformation capture (Hi-C) methods have proven more effective at identifying functional variants than mapping the nearest gene of GWAS single nucleotide polymorphisms (SNPs) [100]. CREs are capable of long-range interactions over one megabase (Mb) through DNA looping, skipping several genes [15,101].
Fig. 2. Non-coding variants can alter Cis-regulatory element (CRE) interactome.
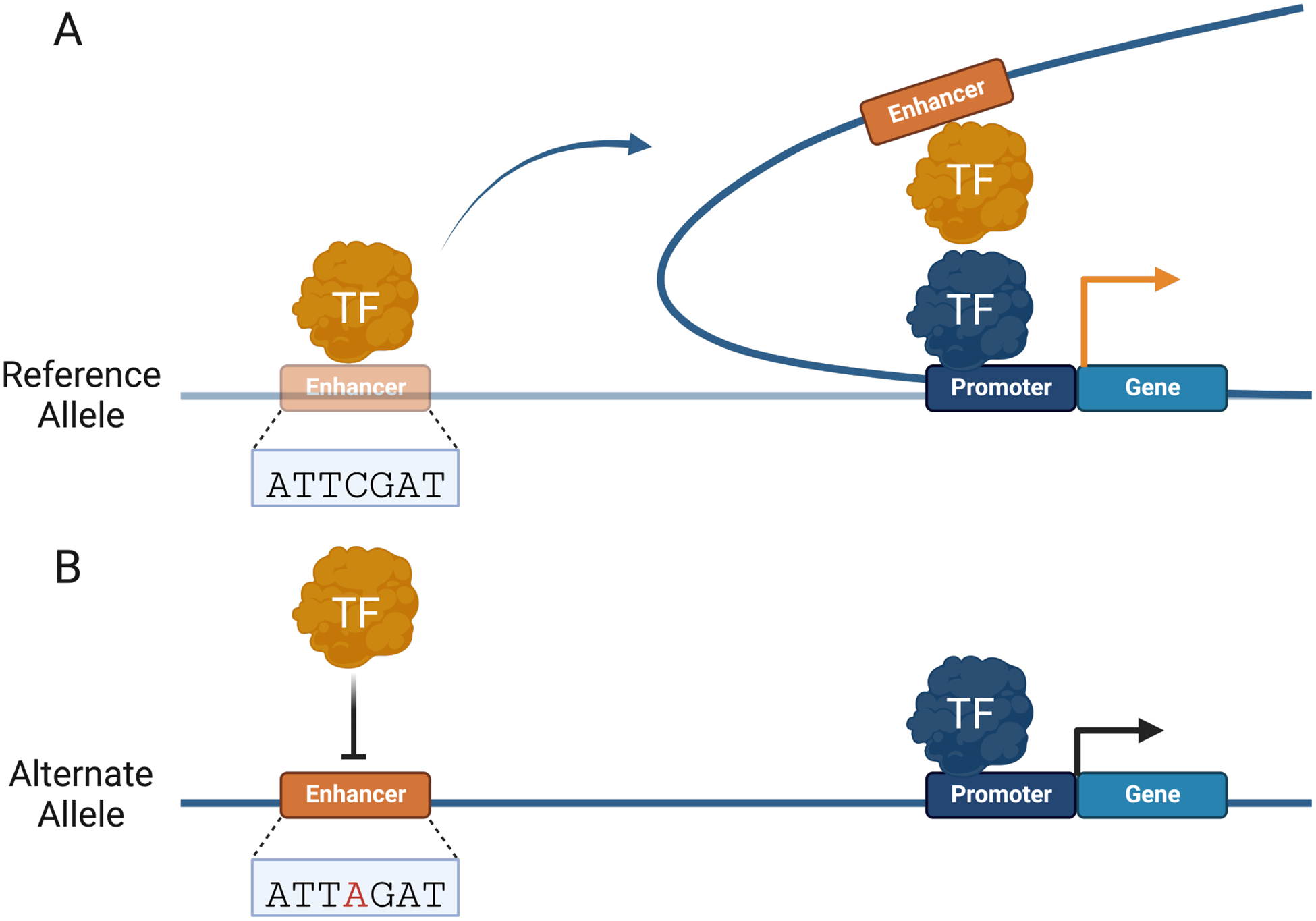
(A) TFs facilitate promoter–enhancer interactions by forming topologically associating domains (TADs) to regulate gene expression. (B) Non-coding variants can alter TAD boundaries and CRE interactions that regulate gene expression. Changes in gene expression are represented by an orange (increase) and black (decrease) arrow.
Promoter-capture Hi-C (PCHi-C) measures the frequency of genome-wide promoter interactions [102]. Orlando et al. [103] screened 19,023 promoter fragments to identify non-coding driver SNVs that alter the colorectal cancer (CRC) cell regulatory landscape. They identified a recurrently mutated CRE that resulted in increased interactions with the ETV1 promoter and a significant upregulation of ETV1, commonly overexpressed in CRC. Selvarajan et al. [104] used PCHi-C to determine the effect of genome-wide coronary artery disease (CAD)-associated non-coding SNPs within liver-specific enhancers. They identified 1277 potential CAD-causal SNPs with allele-specific regulatory activity and 621 target genes that may contribute to CAD phenotypes (compared to only 138 with eQTL analysis). They found PCHi-C to be a powerful technique for identifying target genes affected by non-coding variants, outperforming previous methods such as expression quantitative trait loci (eQTL) analysis.
Contrary to promoters, some enhancers have been shown to regulate the expression of multiple genes [105]. As such, PCHi-C has been adapted to understand how the enhancer-to-enhancer interactome is affected by genomic variations. Madsen et al. [106] used an enhancer-capture Hi-C (ECHi-C) capture array (library of 76,846 121nt RNA probes) to study the effects of genomic variants on human mesenchymal stem cells (hMSC) differentiation to adipocytes. Through this approach, they captured 17,235 putative active enhancers at 0, 1, and 10 days of adipocyte differentiation and observed that most eQTL variants increase enhancer interactomes. They found that the variant rs41281051: T>C is associated with increased interactions with the LAMB1 locus and decreased LAMB1 expression in subcutaneous adipose tissue [106]. Hi-C library preparation followed by chromatin immunoprecipitation (HiChIP) provides an additional layer of regulatory information than PCHi-C by effectively mapping tissue-specific promoter–enhancer interactions in different cell types [107]. Chandra et al. [101] used H3K27ac (marks active enhancers) HiChIP to evaluate cell-specific and genotype-dependent effects of SNPs on various immune cell types. Most of the variants had a tissue-specific impact on the promoter–enhancer interactions, such as CD4+ T cells (rs8087912) and natural killer cells (rs13379920), which exhibited a significant decrease when compared to monocytes, resulting in a decreased expression of EPB41L3 and TM6SF1, respectively.
There have been significant advances in experimental approaches to understanding non-coding variant effects on phenotypes. However, due to the overwhelming number of identified GWAS SNPs in the human genome (>500,000), prioritizing the variants to evaluate remains a challenge [48]. Computational approaches, such as predictive models and machine learning, can address this challenge and prioritize functional non-coding variants for validation. Meng et al. [108] used Hi-C data from human embryonic stem cells (hESC) to develop a deep learning model (DeepHiC) to predict the impact of SNPs on long-range chromatin interactions. Using ~8 million non-coding SNPs from the 1000 Genomes Project [57], they were able to successfully identify five osteoporosis-associated functional variants (rs9533090, rs9594738, rs8001611, rs9533094, and rs9533095) in an eQTL of TNSFS11 [108]. Computational approaches have also been developed to identify cell-specific functions of non-coding variants. Yu et al. [109] developed a Single-Nucleus Analysis Pipeline for Hi-C (SnapHiC) to analyze 3471 neuropsychiatric disorder-associated SNPs. They observed different interactions for the same variants in different prefrontal cortical cells. For example, two enhancers containing Alzheimer’s-associated SNPs (rs112481437 and rs138137383) resulted in astrocyte-specific loops to the APOE gene TSS [109]. Other computational approaches have constructed gene regulatory networks (GRNs) of GWAS SNPs from 3C techniques (i.e., Hi-C and ChIA-PET) to predict causal risk variants [110]. Gao et al. [111] developed the Annotation of Regulatory Variants using Integrated Networks (ARVIN) and identified over 1000 risk variants for seven autoimmune diseases using disease-relevant GRNs for known causal SNPs. Using ARVIN, they successfully predicted an average of 160 risk SNPs with a significant overlap of the eQTL analysis [111].
5. Non-coding Variants in Post-transcriptional Regulation
Non-coding variants can occur within the 5′ and 3′ untranslated regions (UTRs) and introns, impeding potentially altering mRNA processing (e.g., splicing, polyadenylation and cleavage, and ribosome binding and assembly) (Fig. 3A,B). Non-coding SNVs can change the binding affinity between RNA-binding proteins (RBPs) and pre-mRNA, impacting on phenotypes through post-transcriptional dysregulation [112]. Krooss et al. [113] described the pathomechanism of a non-GWAS SNP found in four families with moderate to severe hemophilia B. The variant created a U1snRNP binding site in the 3′ UTR region of the coagulation factor 9 (F9) mRNA (c.2545A>G). The binding of U1snRNP inhibited polyadenylation and proper 3′-end processing, which resulted in mRNA degradation and reduced expression of F9 [113]. Bauwens et al. [114] identified eight non-GWAS variants in a group of German and Belgian patients diagnosed with ABCA4-associated diseases. The variants that occurred within ABCA4 introns 2, 7, 21, 30, and 36 resulted in eight pathogenic splice variants determined by minigene splicing assays, a method that clones variant sequences into expression vectors and identifies them through reverse transcription polymerase chain reaction (RT-PCR) [114]. However, both gene expression and splicing present tissue-and cell-specific patterns, making it challenging to detect functional variants. Bronstein et al. [115] implemented whole-genome sequencing (WGS) and RNA-seq alongside patient-induced pluripotent stem cell (iPSC) transcriptome analysis to detect tissue-specific splicing patterns caused by non-coding variants. They cultured iPSC-derived retinal organoids from a family with inherited retinal degenerations and used RNA-seq to identify a novel pathogenic splice variant (chr8:g.87618576G>A) in the CNGB3 gene caused by an intronic SNV [115]. WGS and iPSC from pedigrees provided an innovative alternative for the functional analysis of genomic variants where no prior knowledge or association had been established.
Fig. 3. Non-coding variants can disrupt mRNA processing and translation initiation.
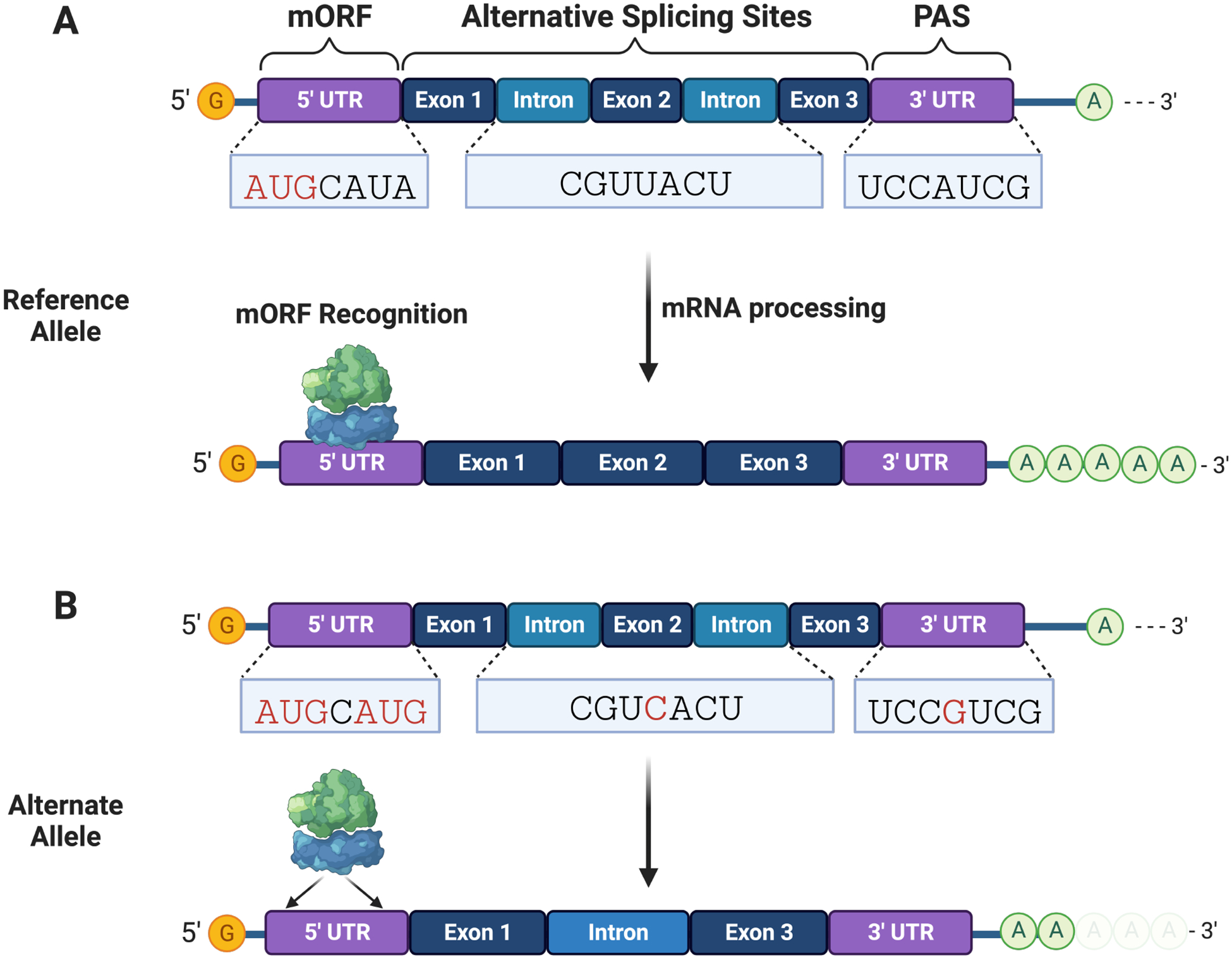
(A) mRNA interactions with RNA-binding proteins and ribosomes are needed for processing (e.g., splicing and adenylation) and translation initiation, respectively. (B) Non-coding variants can alter splice and polyadenylation sites needed for stable mRNA processing and expression of functional protein isoforms. mRNA variants can create translation sites that compete with the main open reading frame (mORF). PAS, polyadenylation sites.
Variants within the 5′ UTR of a gene can affect protein translation by interfering with ribosome scanning and assembly. Zhou et al. [116] screened 14 genetically undiagnosed Saethre–Chotzen syndrome (SCS) patients and identified the first (non-GWAS) SCS-associated non-coding SNV (c.−263C>A and c.−255G>A) within TWIST1. These variants created translation start sites within the 5′ UTR of the TWIST1 mRNA, which decreased translation of the main open reading frame (mORF), causing a more than 75% reduction in TWIST1, as determined by gene reporter assays [116]. Lim et al. [117] developed Pooled full-length UTR Multiplex Assay on Gene Expression (PLUMAGE), a high-throughput method that clones a luciferase gene and barcode downstream of the 5′ UTR variant to quantify mRNA transcription and translation efficiency in parallel. Using PLUMAGE on tissues from prostate cancer patients, they identified 326 mutations within the 5′ UTRs, of which 35% (114/326) was associated with altered transcription and translation [117]. Griesemer et al. [118] developed a Massively Parallel Reporter Assay for the 3′ UTR (MPRAu), a high throughput approach to quantify allelic expression imbalances in 3′ UTR variants in a cell-specific manner [118]. Through this approach, they tested 12,173 3′ UTR variants and identified 2368 variants that altered transcription levels across six cell types (HEK293, HEPG2, HMEC, K562, GM12878, and SK-N-SH).
With the overwhelming number of non-coding variants, computational approaches have been developed to identify and prioritize functional variants that occur in mRNA untranslated regions. Chen et al. [119] developed a computational pipeline coupled with experimental validation to identify functional variants within polyadenylation sites (PAS). By implementing four resources of human polyadenylation maps and two disease-associated databases, they identified 68 pathogenic variants within PAS that were validated using a modified luciferase reporter vector (mpCHECK2) designed to evaluate polyadenylation in gene expression [119]. Paggi et al. [120] developed a deep learning-based computational method to predict mRNA splicing points known as the Long Short-term memory network Branchpoint Retriever (LaBranchoR). LaBranchoR predictions identified 106 pathogenic variants affecting mRNA splicing, showing a substantial overlap of pathogenic variants from ClinVar and the Human Gene Mutation Database (HGMD) [120]. In contrast, Sample et al. [121] developed Optimus 5-Prime, a CNN trained on data from polysome profiling and RNA-seq, to predict the effect of 5′ UTR variants on ribosomal loading. They were able to predict ribosome loading for over 40,000 variants and were able to identify 45 functional disease-associated SNPs in the 5′ UTR [121].
6. Future Directions and Author Recommendations
Technological advances and reduced costs in DNA sequencing have resulted in an ever-increasing number of disease/trait-associated variants. This has resulted in a need to develop innovative computational and experimental strategies to determine the role and causal mechanisms of non-coding variants in human diseases and quantitative traits. The first challenge is to select or prioritize from the existing GWAS variants (>500,000). Our group and others have implemented computational approaches to prioritize variants based on a particular disease, gene target, or protein of interest (TFs or RBPs) [33,47,86,103]. We recommend incorporating multi-omics and functional genomics datasets (genomic, transcriptomic, epigenomic, etc.), which can improve the predictive power of the computational models to identify variants with a temporal- or tissue-specific impact [68,91,111,121,122]. In our previous work on cardiac TFs, we implemented predictive models (PWM- and SVM-based) to prioritize cardiovascular disease (CVD)-associated SNVs from the GWAS catalog [33,75]. Since our work has focused on CVD-associated SNVs, we have trained our predictive models with cardiac TF ChIP-seq data from human-induced cardiomyocytes (hiPSC-CM). We have also prioritized genomic variants mapped in regions active in cardiac tissue or during heart development by incorporating ChIP-seq and DNase I hypersensitivity genomic footprints (DGF) from cardiac tissue. Our recommendation and most strategies reviewed here rely on mining public databases or previous knowledge. When these options are unavailable, pedigree WGS combined with patient-derived iPSCs and transcriptomics of differentiated cells provides an alternative to identify de novo variants in specific cases [113–115,123–126].
This manuscript aimed to discuss the vast advancements in functional assays to identify causal variants for multiple human diseases and propel collaborations to describe their complete genetic mechanisms. In the future, we believe that these computational and experimental methods will be combined to achieve a genome-wide understanding of the role of SNV in human diseases. For instance, 97% of congenital heart disease (CHD)-associated variants have been mapped within the non-coding genome, including intronic, intergenic, UTRs, and regulatory regions [127–132]. Elucidating the genome-wide impact of these non-coding variants in complex biological systems, from human cardiomyocytes to CHD patients, will require a combination of methods to assay all levels of genetic regulation. Thus, a combined analysis of high-throughput technology is required to understand the impact of CHD-associated SNVs on chromatin structure (e.g., HiChIP [101]), TF–DNA and TF–cofactor interactions (e.g., CASCADE [46] and SNP-SELEX [47]), gene expression (e.g., MPRA [86] and STARR-seq [88]), RNA processing (e.g., MPRAu [118]), and translation (e.g., PLUMAGE [117]). The findings generated by such an integrative approach can produce crucial data needed to train effective models, which prioritize the functional impact of genomic variants that can be scaled to multiple diseases. Going further, knowing the causal mechanism of pathogenic SNVs is crucial for treating or even curing diseases through gene editing by CRISPR-based methods [133–135].
7. Concluding Remarks
Recent advancements have allowed us to understand and identify functional non-coding variants that can play a role in human diseases. Although these mutations occur outside the protein-coding genome, they can impact on phenotype by altering how regulatory proteins, such as TFs and RBP, interact with CREs and dysregulate gene expression. Non-coding variants can impact different stages of gene regulation by affecting (i) chromatin interactions (promoter and enhancer interactomes), (ii) TF affinity for their binding sites, (iii) transcriptional activity of target genes, (iv) post-transcriptional regulation (mRNA stability and splicing), and (v) translation initiation (ribosome recognition).
New methods have been developed to perform high-throughput functional evaluations of variants to determine causal mechanisms linked to human diseases (Table 1, Ref. [43–47,82,86,88,89,101,103,104,106,113–118]). Changes in chromatin interaction maps, TF–DNA binding affinity, gene expression, and translation efficiency provide evidence to support the role of many disease-associated variants. However, with the overwhelming and increasing number of variants in the non-coding genome, identifying functional variants remains challenging. Experimental data has been implemented to design computational approaches to predict and identify functional pathogenic variants. Computational pipelines and machine learning tools (SVMs and CNNs) can decipher tissue- and cell-specific patterns to predict variants with functional activity and prioritize in vitro validation (Table 2, Ref. [55,67,68,74,77–79,90,91,93,108,109,111,119–121]).
Table 1.
Summary of experimental methods to identify non-coding functional variants.
| Method | Throughput | Detection | Cell- and tissue-specific | Experiment | Ref | |
|---|---|---|---|---|---|---|
| CRE-interactome | PCHi-C | High | Promoter-CRE interactome, target gene | Yes | In vivo (cell line) | [103,104] |
| ECHi-C | High | Enhancer-CRE interactome | Yes | In vivo (cell line) | [106] | |
| HiChIP | High | Cell-type CRE interactome | Yes | In vivo (cell line) | [101] | |
| TF–DNA binding | BET-seq | High | Binding free energy | No | In vitro | [43] |
| STAMMP | High | Binding affinity | No | In vitro | [44] | |
| HiP-FA | High | Binding affinity and specificity | No | In vitro | [45] | |
| CASCADE | High | Cofactor recruitment by TFs | Yes | In vivo (cell line) | [46] | |
| SNP-SELEX | High | Binding affinity | No | In vitro | [47] | |
| Gene expression | Luciferase reporter assay | Low | Bioluminescence | Yes | In vivo (cell line) | [82] |
| MPRA | High | RNA-seq/flow cytometry | Yes | In vivo (cell line) | [86] | |
| STARR-seq | High | RNA-seq | Yes | In vitro (cell) | [88] | |
| enSERT | High | lacZ staining | Yes | In vivo | [89] | |
| Post-transcriptional regulation | Luciferase reporter assay | Low | Bioluminescence | No | In vivo | [116] |
| Low | Bioluminescence | No | In vitro | [113] | ||
| Minigene splicing assays | Low | RNA-seq | No | In vitro (from patients) | [114] | |
| Patient iPSC WGS | High | RNA-seq | Yes | In vivo | [115] | |
| MPRAu | High | RNA-seq | Yes | In vitro (cells) | [118] | |
| Plumage | High | RNA-seq and bioluminescence | Yes | In vitro | [117] |
PCHi-C, promoter-capture Hi-C; ECHi-C, enhancer-capture Hi-C; HiChIP, Hi-C library preparation followed by chromatin immunoprecipitation; BET-seq, Binding Energy Topography by sequencing; STAMMP, simultaneous transcription factor affinity measurements via microfluidic protein arrays; HiP-FA, high-performance fluorescence anisotropy; CASCADE, Customizable Approach to Survey Complex Assembly at DNA Elements; MPRA, massively parallel reporter assays; STARR-seq, self-transcribing active regulatory region sequencing; iPSC, induced pluripotent stem cell; WGS, whole-genome sequencing; MPRAu, Massively Parallel Reporter Assay for 3′ UTR.
Table 2.
Summary of computational methods to predict non-coding functional variants.
| Program | Type | Training data | Prediction | Cell- and tissue-specific | Ref | |
|---|---|---|---|---|---|---|
| CRE interactions | DeepHiC | Deep learning | Hi-C | Long-range chromatin interactions | Yes | [108] |
| SnapHiC | Computational pipeline | Hi-C | CRE interactions | Yes | [109] | |
| Arvin | Network-based predictive model | Hi-C, ChIA-PET | GRNs | Yes | [111] | |
| TF–DNA binding | atSNP | Motif-based predictive model | PWMs | TF binding | No | [55] |
| SEMpl | Computational pipeline | ChIP-seq, DNase-seq, PWMs | TF binding | No | [67] | |
| ANANASTRA | Computational pipeline | ChIP-seq, PWMs, rs-IDs, eQTL | TF binding | Yes | [68] | |
| deltaSVM | SVM | ATAC-seq | TF binding | Yes | [74] | |
| DeepFun/AgentBind | Deep neural networks | ChIP-seq, DNase-seq | TF binding | Yes | [77,78] | |
| DeFine | CNN | ChIP-seq, Hi-C | TF binding, mapped gene | Yes | [79] | |
| Gene expression | PO-EN | Semi-supervised model | MPRA | Enhancer activity | Yes | [90] |
| SURF | Deep learning | DNase-seq, ChIP-seq, MPRA | Gene expression, TF binding | Yes | [91] | |
| MPRA-DragoNN | CNN | MPRA | Gene expression | Yes | [93] | |
| Post-transcriptional regulation | Variant PAS Pipeline | Computational pipeline | Polyadenylation maps | PAS variants | No | [119] |
| LaBranchoR | Deep learning | Splicing branchpoints | mRNA splicing points | No | [120] | |
| Optimus 5-prime | CNN | Polysome profiling, RNA-seq | Ribosome loading, gene expression | No | [121] |
SnapHiC, Single-Nucleus Analysis Pipeline for Hi-C; SEMpl, SNP effect matrix pipeline; PO-EN, presence-only with elastic net penalty; SURF, Score of Unified Regulatory Feature; PAS, polyadenylation sites; SVM, support vector machine; CNN, convolutional neural networks; PWMs, position weight matrices.
Despite all the progress in understanding the role of disease-associated variants within the non-coding regulatory genome, determining causality remains challenging. We hypothesize that the number of regulatory variants will continue to increase significantly while the molecular mechanisms of most reported variants remain unknown. The increased throughput and ability to functionally validate disease-associated non-coding variants will contribute to the rapid development of diagnostic methods and treatments for these diseases.
Acknowledgment
We would like to give special thanks to Yamil Miranda-Negron for his support during the preparation of the manuscript and revisions. We also thank Diego Pomales-Matos, Leandro Sanabria-Alberto, Alejandro Rivera-Madera, Jean L. Messon-Bird, Adriana C. Barreiro-Rosario, and Jeancarlos Rivera-Del Valle for their support during the preparation of the manuscript.
Funding
This project was supported by NIH-SC1GM127231. EGPM was funded by the NIH RISE Fellowship (5R25GM061151-20) and the NSF BioXFEL Fellowship (STC-1231306).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Saenko VA, Rogounovitch TI. Genetic Polymorphism Predisposing to Differentiated Thyroid Cancer: A Review of Major Findings of the Genome-Wide Association Studies. Endocrinology and Metabolism (Seoul, Korea). 2018; 33: 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Taft RJ, Pheasant M, Mattick JS. The relationship between non-protein-coding DNA and eukaryotic complexity. BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology. 2007; 29: 288–299. [DOI] [PubMed] [Google Scholar]
- [3].Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome International Human Genome Sequencing Consortium* The Sanger Centre: Beijing Genomics Institute/Human Genome Center. Nature. 2001; 409, 860–921. [DOI] [PubMed] [Google Scholar]
- [4].Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, et al. The complete sequence of a human genome. Science (New York, N.Y.). 2022; 376: 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lee PH, Lee C, Li X, Wee B, Dwivedi T, Daly M. Principles and methods of in-silico prioritization of non-coding regulatory variants. Human Genetics. 2018; 137: 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhang F, Lupski JR. Non-coding genetic variants in human disease. Human Molecular Genetics. 2015; 24: R102–R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Deplancke B, Alpern D, Gardeux V. The Genetics of Transcription Factor DNA Binding Variation. Cell, 2016; 166: 538–554. [DOI] [PubMed] [Google Scholar]
- [8].Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Research. 2019; 47: D1005–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science (New York, N.Y.). 2012; 337: 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Vierstra J, Lazar J, Sandstrom R, Halow J, Lee K, Bates D, et al. Global reference mapping of human transcription factor footprints. Nature. 2020; 583: 729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Elkon R, Agami R. Characterization of noncoding regulatory DNA in the human genome. Nature Biotechnology. 2017; 35: 732–746. [DOI] [PubMed] [Google Scholar]
- [12].Cremer M, Cremer T. Nuclear compartmentalization, dynamics, and function of regulatory DNA sequences. Genes, Chromosomes & Cancer. 2019; 58: 427–436. [DOI] [PubMed] [Google Scholar]
- [13].Haberle V, Stark A. Eukaryotic core promoters and the functional basis of transcription initiation. Nature Reviews. Molecular Cell Biology 2018; 19: 621–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jindal GA, Farley EK. Enhancer grammar in development, evolution, and disease: dependencies and interplay. Developmental Cell. 2021; 56: 575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Meddens CA, van der List ACJ, Nieuwenhuis EES, Mokry M. Non-coding DNA in IBD: from sequence variation in DNA regulatory elements to novel therapeutic potential. Gut. 2019; 68: 928–941. [DOI] [PubMed] [Google Scholar]
- [16].Orkin SH, Kazazian HH Jr, Antonarakis SE, Goff SC, Boehm CD, Sexton JP, et al. Linkage of beta-thalassaemia mutations and beta-globin gene polymorphisms with DNA polymorphisms in human beta-globin gene cluster. Nature. 1982; 296: 627–631. [DOI] [PubMed] [Google Scholar]
- [17].Al Zadjali S, Wali Y, Al Lawatiya F, Gravell D, Alkindi S, Al Falahi K, et al. The β-globin promoter −71 C>T mutation is a β+ thalassemic allele. European Journal of Haematology. 2011; 87: 457–460. [DOI] [PubMed] [Google Scholar]
- [18].Gordon CT, Fox VJ, Najdovska S, Perkins AC. C/EBPdelta and C/EBPgamma bind the CCAAT-box in the human beta-globin promoter and modulate the activity of the CACC-box binding protein, EKLF. Biochimica et Biophysica Acta. 2005; 1729: 74–80. [DOI] [PubMed] [Google Scholar]
- [19].van der Lee R, Correard S, Wasserman WW. Deregulated Regulators: Disease-Causing cis Variants in Transcription Factor Genes. Trends in Genetics: TIG. 2020; 36: 523–539. [DOI] [PubMed] [Google Scholar]
- [20].Inukai S, Kock KH, Bulyk ML. Transcription factor-DNA binding: beyond binding site motifs. Current Opinion in Genetics & Development. 2017; 43: 110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Song W, Kir S, Hong S, Hu Y, Wang X, Binari R, et al. Tumor-Derived Ligands Trigger Tumor Growth and Host Wasting via Differential MEK Activation. Developmental Cell. 2019; 48: 277–286.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lee D, Kapoor A, Safi A, Song L, Halushka MK, Crawford GE, et al. Human cardiac cis-regulatory elements, their cognate transcription factors, and regulatory DNA sequence variants. Genome Research. 2018; 28: 1577–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rodríguez-Martínez JA, Reinke AW, Bhimsaria D, Keating AE, Ansari AZ. Combinatorial bZIP dimers display complex DNA-binding specificity landscapes. eLife. 2017; 6: e19272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Geertz M, Maerkl SJ. Experimental strategies for studying transcription factor-DNA binding specificities. Briefings in Functional Genomics. 2010; 9: 362–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wang Z, He W, Tang J, Guo F. Identification of Highest-Affinity Binding Sites of Yeast Transcription Factor Families. Journal of Chemical Information and Modeling. 2020; 60: 1876–1883. [DOI] [PubMed] [Google Scholar]
- [26].Martha L. Bulyk AJ. Marian Walhout, Chapter 4 - Gene Regulatory Networks. In: Marian Walhout AJ, Marc Vidal, Job Dekker, eds. Handbook of Systems Biology (pp. 65–88). Academic Press: Cambridge, MA, USA. 2013. [Google Scholar]
- [27].Zhao J, Li D, Seo J, Allen AS, Gordân R. Quantifying the Impact of Non-coding Variants on Transcription Factor-DNA Binding. Research in Computational Molecular Biology. 2017; 10229: 336–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shrestha S, Sewell JA, Santoso CS, Forchielli E, Carrasco Pro S, Martinez M, et al. Discovering human transcription factor physical interactions with genetic variants, novel DNA motifs, and repetitive elements using enhanced yeast one-hybrid assays. Genome Research. 2019; 29: 1533–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014; 158: 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Khurana E, Fu Y, Chakravarty D, Demichelis F, Rubin MA, Gerstein M. Role of non-coding sequence variants in cancer. Nature Reviews. Genetics 2016; 17: 93–108. [DOI] [PubMed] [Google Scholar]
- [31].Le ATH, Krylova SM, Krylov SN. Determination of the Equilibrium Constant and Rate Constant of Protein-Oligonucleotide Complex Dissociation under the Conditions of Ideal-Filter Capillary Electrophoresis. Analytical Chemistry. 2019; 91: 8532–8539. [DOI] [PubMed] [Google Scholar]
- [32].Hellman LM, Fried MG. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nature Protocols. 2007; 2: 1849–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Peña-Martínez EG, Rivera-Madera A, Pomales-Matos DA, Sanabria-Alberto L, Rosario-Cañuelas BM, Rodríguez-Ríos JM, et al. Disease-associated non-coding variants alter NKX2–5 DNA-binding affinity. Biochimica et Biophysica Acta. Gene Regulatory Mechanisms 2023; 1866: 194906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hou G, Harley ITW, Lu X, Zhou T, Xu N, Yao C, et al. SLE non-coding genetic risk variant determines the epigenetic dysfunction of an immune cell specific enhancer that controls disease-critical microRNA expression. Nature Communications. 2021; 12: 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Christensen AH, Andersen CB, Wassilew K, Svendsen JH, Bundgaard H, Brand SM, et al. Rare non-coding Desmoglein-2 variant contributes to Arrhythmogenic right ventricular cardiomyopathy. Journal of Molecular and Cellular Cardiology. 2019; 131: 164–170. [DOI] [PubMed] [Google Scholar]
- [36].Stormo GD, Zhao Y. Determining the specificity of protein-DNA interactions. Nature Reviews. Genetics 2010; 11: 751–760. [DOI] [PubMed] [Google Scholar]
- [37].Berger MF, Philippakis AA, Qureshi AM, He FS, Estep PW 3rd, Bulyk ML. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nature Biotechnology. 2006; 24: 1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fordyce PM, Gerber D, Tran D, Zheng J, Li H, DeRisi JL, et al. De novo identification and biophysical characterization of transcription-factor binding sites with microfluidic affinity analysis. Nature Biotechnology. 2010; 28: 970–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Slattery M, Riley T, Liu P, Abe N, Gomez-Alcala P, Dror I, et al. Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins. Cell. 2011; 147: 1270–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, Rastas P, et al. DNA-binding specificities of human transcription factors. Cell. 2013; 152: 327–339. [DOI] [PubMed] [Google Scholar]
- [41].Noyes MB, Meng X, Wakabayashi A, Sinha S, Brodsky MH, Wolfe SA. A systematic characterization of factors that regulate Drosophila segmentation via a bacterial one-hybrid system. Nucleic Acids Research. 2008; 36: 2547–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Berenson A, Fuxman Bass JI. Enhanced Yeast One-Hybrid Assays to Study Protein-DNA Interactions. Methods in Molecular Biology (Clifton, N.J.). 2023; 2599: 11–20. [DOI] [PubMed] [Google Scholar]
- [43].Le DD, Shimko TC, Aditham AK, Keys AM, Longwell SA, Orenstein Y, et al. Comprehensive, high-resolution binding energy landscapes reveal context dependencies of transcription factor binding. Proceedings of the National Academy of Sciences of the United States of America. 2018; 115: E3702–E3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Aditham AK, Markin CJ, Mokhtari DA, DelRosso N, Fordyce PM. High-Throughput Affinity Measurements of Transcription Factor and DNA Mutations Reveal Affinity and Specificity Determinants. Cell Systems. 2021; 12: 112–127.e11. [DOI] [PubMed] [Google Scholar]
- [45].Jung C, Bandilla P, von Reutern M, Schnepf M, Rieder S, Unnerstall U, et al. True equilibrium measurement of transcription factor-DNA binding affinities using automated polarization microscopy. Nature Communications. 2018; 9: 1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bray D, Hook H, Zhao R, Keenan JL, Penvose A, Osayame Y, et al. CASCADE: high-throughput characterization of regulatory complex binding altered by non-coding variants. Cell Genomics. 2022; 2: 100098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yan J, Qiu Y, Ribeiro Dos Santos AM, Yin Y, Li YE, Vinckier N, et al. Systematic analysis of binding of transcription factors to noncoding variants. Nature. 2021; 591: 147–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Research. 2001; 29: 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The Human Transcription Factors. Cell. 2018; 172: 650–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Maerkl SJ, Quake SR. A systems approach to measuring the binding energy landscapes of transcription factors. Science (New York, N.Y.). 2007; 315: 233–237. [DOI] [PubMed] [Google Scholar]
- [51].Ambrosini G, Groux R, Bucher P. PWMScan: a fast tool for scanning entire genomes with a position-specific weight matrix. Bioinformatics (Oxford, England). 2018; 34: 2483–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Stormo GD. Modeling the specificity of protein-DNA interactions. Quantitative Biology. 2013; 1: 115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Orenstein Y, Shamir R. A comparative analysis of transcription factor binding models learned from PBM, HT-SELEX and ChIP data. Nucleic Acids Research. 2014; 42: e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kumar S, Ambrosini G, Bucher P. SNP2TFBS - a database of regulatory SNPs affecting predicted transcription factor binding site affinity. Nucleic Acids Research. 2017; 45: D139–D144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Shin S, Hudson R, Harrison C, Craven M, Keleş S. atSNP Search: a web resource for statistically evaluating influence of human genetic variation on transcription factor binding. Bioinformatics (Oxford, England). 2019; 35: 2657–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Research. 2020; 48: D87–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Devuyst O. The 1000 Genomes Project: Welcome to a New World. Peritoneal Dialysis International: Journal of the International Society for Peritoneal Dialysis. 2015; 35: 676–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Thomas-Chollier M, Hufton A, Heinig M, O’Keeffe S, Masri NE, Roider HG, et al. Transcription factor binding predictions using TRAP for the analysis of ChIP-seq data and regulatory SNPs. Nature Protocols. 2011; 6: 1860–1869. [DOI] [PubMed] [Google Scholar]
- [59].Coetzee SG, Coetzee GA, Hazelett DJ. motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics (Oxford, England). 2015; 31: 3847–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Andersen MC, Engström PG, Lithwick S, Arenillas D, Eriksson P, Lenhard B, et al. In silico detection of sequence variations modifying transcriptional regulation. PLoS Computational Biology. 2008; 4: e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Riva A. Large-scale computational identification of regulatory SNPs with rSNP-MAPPER. BMC Genomics. 2012; 13: S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Perera D, Chacon D, Thoms JAI, Poulos RC, Shlien A, Beck D, et al. OncoCis: annotation of cis-regulatory mutations in cancer. Genome Biology. 2014; 15: 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ward LD, Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Research. 2016; 44: D877–D881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Siddharthan R. Dinucleotide weight matrices for predicting transcription factor binding sites: generalizing the position weight matrix. PLoS ONE. 2010; 5: e9722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Tomovic A, Oakeley EJ. Position dependencies in transcription factor binding sites. Bioinformatics (Oxford, England). 2007; 23: 933–941. [DOI] [PubMed] [Google Scholar]
- [66].Bulyk ML, Johnson PLF, Church GM. Nucleotides of transcription factor binding sites exert interdependent effects on the binding affinities of transcription factors. Nucleic Acids Research. 2002; 30: 1255–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Nishizaki SS, Ng N, Dong S, Porter RS, Morterud C, Williams C, et al. Predicting the effects of SNPs on transcription factor binding affinity. Bioinformatics (Oxford, England). 2020; 36: 364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Boytsov A, Abramov S, Aiusheeva AZ, Kasianova AM, Baulin E, Kuznetsov IA, et al. ANANASTRA: annotation and enrichment analysis of allele-specific transcription factor binding at SNPs. Nucleic Acids Research. 2022; 50: W51–W56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Abramov S, Boytsov A, Bykova D, Penzar DD, Yevshin I, Kolmykov SK, et al. Landscape of allele-specific transcription factor binding in the human genome. Nature Communications. 2021; 12: 2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kolmykov S, Yevshin I, Kulyashov M, Sharipov R, Kondrakhin Y, Makeev VJ, et al. GTRD: an integrated view of transcription regulation. Nucleic Acids Research. 2021; 49: D104–D111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kulakovskiy IV, Vorontsov IE, Yevshin IS, Sharipov RN, Fedorova AD, Rumynskiy EI, et al. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Research. 2018; 46: D252–D259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nature Genetics. 2013; 45: 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Quan L, Mei J, He R, Sun X, Nie L, Li K, et al. Quantifying Intensities of Transcription Factor-DNA Binding by Learning From an Ensemble of Protein Binding Microarrays. IEEE Journal of Biomedical and Health Informatics. 2021; 25: 2811–2819. [DOI] [PubMed] [Google Scholar]
- [74].Lee D, Gorkin DU, Baker M, Strober BJ, Asoni AL, McCallion AS, et al. A method to predict the impact of regulatory variants from DNA sequence. Nature Genetics. 2015; 47: 955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Peña-Martínez EG, Pomales-Matos DA, Rivera-Madera A, Messon-Bird JL, Medina-Feliciano JG, Sanabria-Alberto L, et al. Prioritizing cardiovascular disease-associated variants altering NKX2–5 and TBX5 binding through an integrative computational approach. The Journal of Biological Chemistry. 2023; 299: 105423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].VandenBosch LS, Luu K, Timms AE, Challam S, Wu Y, Lee AY, et al. Machine Learning Prediction of Non-Coding Variant Impact in Human Retinal cis-Regulatory Elements. Translational Vision Science & Technology. 2022; 11: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Pei G, Hu R, Jia P, Zhao Z. DeepFun: a deep learning sequence-based model to decipher non-coding variant effect in a tissue-and cell type-specific manner. Nucleic Acids Research. 2021; 49: W131–W139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Zheng A, Lamkin M, Zhao H, Wu C, Su H, Gymrek M. Deep neural networks identify sequence context features predictive of transcription factor binding. Nature Machine Intelligence. 2021; 3: 172–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Wang M, Tai C, E W, Wei L. DeFine: deep convolutional neural networks accurately quantify intensities of transcription factor-DNA binding and facilitate evaluation of functional non-coding variants. Nucleic Acids Research. 2018; 46: e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Lenhard B, Sandelin A, Carninci P. Metazoan promoters: emerging characteristics and insights into transcriptional regulation. Nature Reviews. Genetics 2012; 13: 233–245. [DOI] [PubMed] [Google Scholar]
- [81].Gasperini M, Tome JM, Shendure J. Towards a comprehensive catalogue of validated and target-linked human enhancers. Nature Reviews. Genetics 2020; 21: 292–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Jiang X, Li T, Liu S, Fu Q, Li F, Chen S, et al. Variants in a cis-regulatory element of TBX1 in conotruncal heart defect patients impair GATA6-mediated transactivation. Orphanet Journal of Rare Diseases. 2021; 16: 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Smale ST. Luciferase assay. Cold Spring Harbor Protocols. 2010; 2010: pdb.prot5421. [DOI] [PubMed] [Google Scholar]
- [84].Smale ST. Beta-galactosidase assay. Cold Spring Harbor Protocols. 2010; 2010: pdb.prot5423. [DOI] [PubMed] [Google Scholar]
- [85].Melnikov A, Murugan A, Zhang X, Tesileanu T, Wang L, Rogov P, et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nature Biotechnology. 2012; 30: 271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Lu X, Chen X, Forney C, Donmez O, Miller D, Parameswaran S, et al. Global discovery of lupus genetic risk variant allelic enhancer activity. Nature Communications. 2021; 12: 1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Lee D, Shi M, Moran J, Wall M, Zhang J, Liu J, et al. STAR-RPeaker: uniform processing and accurate identification of STARR-seq active regions. Genome Biology. 2020; 21: 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Toropainen A, Stolze LK, Örd T, Whalen MB, Torrell PM, Link VM, et al. Functional noncoding SNPs in human endothelial cells fine-map vascular trait associations. Genome Research. 2022; 32: 409–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Kvon EZ, Zhu Y, Kelman G, Novak CS, Plajzer-Frick I, Kato M, et al. Comprehensive In Vivo Interrogation Reveals Phenotypic Impact of Human Enhancer Variants. Cell. 2020; 180: 1262–1271.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Yang Z, Wang C, Erjavec S, Petukhova L, Christiano A, Ionita-Laza I. A semi-supervised model to predict regulatory effects of genetic variants at single nucleotide resolution using massively parallel reporter assays. Bioinformatics (Oxford, England). 2021; 37: 1953–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Dong S, Boyle AP. Predicting functional variants in enhancer and promoter elements using RegulomeDB. Human Mutation. 2019; 40: 1292–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Research. 2012; 22: 1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Movva R, Greenside P, Marinov GK, Nair S, Shrikumar A, Kundaje A. Deciphering regulatory DNA sequences and noncoding genetic variants using neural network models of massively parallel reporter assays. PLoS ONE. 2019; 14: e0218073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Mossing MC, Record MT Jr. Upstream operators enhance repression of the lac promoter. Science. 1986; 233: 889–892. [DOI] [PubMed] [Google Scholar]
- [95].Zhao Z, Tavoosidana G, Sjölinder M, Göndör A, Mariano P, Wang S, et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra-and interchromosomal interactions. Nature Genetics. 2006; 38: 1341–1347. [DOI] [PubMed] [Google Scholar]
- [96].Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science (New York, N.Y.). 2002; 295: 1306–1311. [DOI] [PubMed] [Google Scholar]
- [97].Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Research. 2006; 16: 1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].McCord RP, Kaplan N, Giorgetti L. Chromosome Conformation Capture and Beyond: Toward an Integrative View of Chromosome Structure and Function. Molecular Cell. 2020; 77: 688–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Tena JJ, Santos-Pereira JM. Topologically Associating Domains and Regulatory Landscapes in Development, Evolution and Disease. Frontiers in Cell and Developmental Biology. 2021; 9: 702787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Tak YG, Farnham PJ. Making sense of GWAS: using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome. Epigenetics & Chromatin. 2015; 8: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Chandra V, Bhattacharyya S, Schmiedel BJ, Madrigal A, Gonzalez-Colin C, Fotsing S, et al. Promoter-interacting expression quantitative trait loci are enriched for functional genetic variants. Nature Genetics. 2021; 53: 110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Schoenfelder S, Javierre BM, Furlan-Magaril M, Wingett SW, Fraser P. Promoter Capture Hi-C: High-resolution, Genome-wide Profiling of Promoter Interactions. Journal of Visualized Experiments: JoVE. 2018; 57320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Orlando G, Law PJ, Cornish AJ, Dobbins SE, Chubb D, Broderick P, et al. Promoter capture Hi-C-based identification of recurrent noncoding mutations in colorectal cancer. Nature Genetics. 2018; 50: 1375–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Selvarajan I, Toropainen A, Garske KM, López Rodríguez M, Ko A, Miao Z, et al. Integrative analysis of liver-specific non-coding regulatory SNPs associated with the risk of coronary artery disease. American Journal of Human Genetics. 2021; 108: 411–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Karnuta JM, Scacheri PC. Enhancers: bridging the gap between gene control and human disease. Human Molecular Genetics. 2018; 27: R219–R227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Madsen JGS, Madsen MS, Rauch A, Traynor S, Van Hauwaert EL, Haakonsson AK, et al. Highly interconnected enhancer communities control lineage-determining genes in human mesenchymal stem cells. Nature Genetics. 2020; 52: 1227–1238. [DOI] [PubMed] [Google Scholar]
- [107].Shi C, Rattray M, Orozco G. HiChIP-Peaks: a HiChIP peak calling algorithm. Bioinformatics (Oxford, England). 2020; 36: 3625–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Meng XH, Xiao HM, Deng HW. Combining artificial intelligence: deep learning with Hi-C data to predict the functional effects of non-coding variants. Bioinformatics (Oxford, England). 2021; 37: 1339–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Yu M, Abnousi A, Zhang Y, Li G, Lee L, Chen Z, et al. SnapHiC: a computational pipeline to identify chromatin loops from single-cell Hi-C data. Nature Methods. 2021; 18: 1056–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].He B, Chen C, Teng L, Tan K. Global view of enhancer-promoter interactome in human cells. Proceedings of the National Academy of Sciences of the United States of America. 2014; 111: E2191–E2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Gao L, Uzun Y, Gao P, He B, Ma X, Wang J, et al. Identifying noncoding risk variants using disease-relevant gene regulatory networks. Nature Communications. 2018; 9: 702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Cohen OS, Weickert TW, Hess JL, Paish LM, McCoy SY, Rothmond DA, et al. A splicing-regulatory polymorphism in DRD2 disrupts ZRANB2 binding, impairs cognitive functioning and increases risk for schizophrenia in six Han Chinese samples. Molecular Psychiatry. 2016; 21: 975–982. [DOI] [PubMed] [Google Scholar]
- [113].Krooss S, Werwitzke S, Kopp J, Rovai A, Varnholt D, Wachs AS, et al. Pathological mechanism and antisense oligonucleotide-mediated rescue of a non-coding variant suppressing factor 9 RNA biogenesis leading to hemophilia B. PLoS Genetics. 2020; 16: e1008690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Bauwens M, Garanto A, Sangermano R, Naessens S, Weisschuh N, De Zaeytijd J, et al. ABCA4-associated disease as a model for missing heritability in autosomal recessive disorders: novel noncoding splice, cis-regulatory, structural, and recurrent hypomorphic variants. Genetics in Medicine: Official Journal of the American College of Medical Genetics. 2019; 21: 1761–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Bronstein R, Capowski EE, Mehrotra S, Jansen AD, Navarro-Gomez D, Maher M, et al. A combined RNA-seq and whole genome sequencing approach for identification of non-coding pathogenic variants in single families. Human Molecular Genetics. 2020; 29: 967–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Zhou Y, Koelling N, Fenwick AL, McGowan SJ, Calpena E, Wall SA, et al. Disruption of TWIST1 translation by 5’ UTR variants in Saethre-Chotzen syndrome. Human Mutation. 2018; 39: 1360–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Lim Y, Arora S, Schuster SL, Corey L, Fitzgibbon M, Wladyka CL, et al. Multiplexed functional genomic analysis of 5’ untranslated region mutations across the spectrum of prostate cancer. Nature Communications. 2021; 12: 4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Griesemer D, Xue JR, Reilly SK, Ulirsch JC, Kukreja K, Davis JR, et al. Genome-wide functional screen of 3’UTR variants uncovers causal variants for human disease and evolution. Cell. 2021; 184: 5247–5260.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Chen M, Wei R, Wei G, Xu M, Su Z, Zhao C, et al. Systematic evaluation of the effect of polyadenylation signal variants on the expression of disease-associated genes. Genome Research. 2021; 31: 890–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Paggi JM, Bejerano G. A sequence-based, deep learning model accurately predicts RNA splicing branchpoints. RNA (New York, N.Y.). 2018; 24: 1647–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Sample PJ, Wang B, Reid DW, Presnyak V, McFadyen IJ, Morris DR, et al. Human 5’ UTR design and variant effect prediction from a massively parallel translation assay. Nature Biotechnology. 2019; 37: 803–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Benaglio P, D’Antonio-Chronowska A, Ma W, Yang F, Young Greenwald WW, Donovan MKR, et al. Allele-specific NKX2–5 binding underlies multiple genetic associations with human electrocardiographic traits. Nature Genetics. 2019; 51: 1506–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Kashima Y, Sakamoto Y, Kaneko K, Seki M, Suzuki Y, Suzuki A. Single-cell sequencing techniques from individual to multi-omics analyses. Experimental & Molecular Medicine. 2020; 52: 1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Nawy T. Single-cell sequencing. Nature Methods. 2014; 11: 18. [DOI] [PubMed] [Google Scholar]
- [125].Park ST, Kim J. Trends in Next-Generation Sequencing and a New Era for Whole Genome Sequencing. International Neurourology Journal. 2016; 20: S76–S83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].van El CG, Cornel MC, Borry P, Hastings RJ, Fellmann F, Hodgson SV, et al. Whole-genome sequencing in health care: recommendations of the European Society of Human Genetics. European Journal of Human Genetics: EJHG. 2013; 21: 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Kathiresan S, Srivastava D. Genetics of human cardiovascular disease. Cell. 2012; 148: 1242–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Lusis AJ. Genetic factors in cardiovascular disease. 10 questions. Trends in Cardiovascular Medicine. 2003; 13: 309–316. [DOI] [PubMed] [Google Scholar]
- [129].Heshmatzad K, Naderi N, Maleki M, Abbasi S, Ghasemi S, Ashrafi N, et al. Role of non-coding variants in cardiovascular disease. Journal of Cellular and Molecular Medicine. 2023; 27: 1621–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Villar D, Frost S, Deloukas P, Tinker A. The contribution of non-coding regulatory elements to cardiovascular disease. Open Biology. 2020; 10: 200088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Dallapiccola B, Mingarelli R, Digilio MC, Marino B, Novelli G. Genetics of congenital heart diseases. Giornale Italiano Di Cardiologia. 1994; 24: 155–166. [PubMed] [Google Scholar]
- [132].Morton SU, Quiat D, Seidman JG, Seidman CE. Genomic frontiers in congenital heart disease. Nature Reviews. Cardiology 2022; 19: 26–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Liao J, Chen S, Hsiao S, Jiang Y, Yang Y, Zhang Y, et al. Therapeutic adenine base editing of human hematopoietic stem cells. Nature Communications. 2023; 14: 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Behan FM, Iorio F, Picco G, Gonçalves E, Beaver CM, Migliardi G, et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 2019; 568: 511–516. [DOI] [PubMed] [Google Scholar]
- [135].Han R, Li L, Ugalde AP, Tal A, Manber Z, Barbera EP, et al. Functional CRISPR screen identifies AP1-associated enhancer regulating FOXF1 to modulate oncogene-induced senescence. Genome Biology. 2018; 19: 118. [DOI] [PMC free article] [PubMed] [Google Scholar]