

Abstract
The development of degradable polymers has commanded significant attention over the past half century. Approaches have predominantly relied on ring-opening polymerization of cyclic esters (e.g., lactones, lactides) and N-carboxyanhydrides, as well as radical ring-opening polymerizations of cyclic ketene acetals. In recent years, there has been a significant effort applied to expand the family of degradable polymers accessible via olefin metathesis polymerization. Given the excellent functional group tolerance of olefin metathesis polymerization reactions generally, a broad range of conceivable degradable moieties can be incorporated into appropriate monomers and thus into polymer backbones. This approach has proven particularly versatile in synthesizing a broad spectrum of degradable polymers including poly(ester), poly(amino acid), poly(acetal), poly(carbonate), poly(phosphoester), poly(phosphoramidate), poly(enol ether), poly(azobenzene), poly(disulfide), poly(sulfonate ester), poly(silyl ether), and poly(oxazinone) among others. In this review, we will highlight the main olefin metathesis polymerization strategies that have been used to access degradable polymers, including (i) acyclic diene metathesis polymerization, (ii) entropy-driven and (iii) enthalpy-driven ring-opening metathesis polymerization, as well as (iv) cascade enyne metathesis polymerization. In addition, the livingness or control of polymerization reactions via different strategies are highlighted and compared. Potential applications, challenges and future perspectives of this new library of degradable polyolefins are discussed. It is clear from recent and accelerating developments in this field that olefin metathesis polymerization represents a powerful synthetic tool towards degradable polymers with novel structures and properties inaccessible by other polymerization approaches.
Keywords: Degradable polymers, Ring-Opening metathesis polymerization, Cascade enyne metathesis polymerization, Acyclic diene metathesis polymerization
1. Introduction
Since Hermann Staudinger proposed his macromolecular hypothesis, the field of polymer science has exploded [1]. Countless synthetic polymers with outstanding thermal, optical, mechanical, and chemical properties have been developed and commercialized over the past 100 years [2]. Synthetic polymers are now ubiquitous in modern society, approaching a staggering production level of more than 350 million tons worldwide as of 2018 [3]. Indeed, almost every facet of our society benefits from the numerous applications of synthetic polymers, including industrial applications (e.g., packaging, coating, and building materials) as well as higher-value-added uses such as medical devices, energy harvesting, and microelectronics [4–6]. Unlike naturally occurring biopolymers (e.g., proteins, nucleic acids, and cellulose) which can be rapidly degraded via hydrolysis and enzymatic digestion [7, 8], most synthetic polymers are highly durable, resisting degradation in biological environments [9]. While this exceptional longevity of synthetic polymers is desirable for maintaining a material’s strength and properties during its usage, the majority of polymeric or plastic waste eventually ends up in landfills or in incinerators, resulting in serious air and water pollution [10]. Moreover, the non-degradability of synthetic polymers may cause unwanted in vivo bioaccumulation, immune responses, and toxicity, eventually limiting their translation from proof-of-concept biomedical research to clinical applications [11].
Degradable synthetic polymers containing weak covalent bonds in their backbone— such as esters, acetals, and carbonate groups have promise, or have been directly applied in drug delivery, tissue engineering, as sacrificial materials for semiconductor fabrication, and as recyclable materials for environmental remediation [9, 12–16]. In these applications, polymers can be programmed to degrade or depolymerize at specific stages following different pathways including but not limited to hydrolysis, proteolysis, reduction in the presence of biomolecules (e.g., cysteine and glutathione), and irradiation-mediated degradation (e.g., light). For instance, biodegradable polymers used for constructing drug delivery systems and tissue-engineering scaffolds should maintain their structural integrity, and eventually degrade into smaller fragments that can be excreted from the body [17, 18]. This requires careful design of degradable polymers, allowing for proper degradation kinetics in vivo. Historically, synthetic approaches to preparing degradable polymers have included (i) step-growth polymerization (e.g., condensation polymerization of lactic acid) [19], and (ii) chain-growth polymerization methods, particularly ring-opening polymerization (ROP). [20–23] According to the ring-opening mechanisms and monomer scope, three classes of ROP have been adopted to generate degradable polymers (Fig. 1).
Fig. 1.

Synthetic degradable polymers through ring-opening polymerization via various chain-growth mechanisms. (a) Radical ROP of cyclic ketene acetals and thionolactone. (b) Anionic/cationic/metal/organo-catalyzed ROP of lactones, lactides, cyclic carbonate, and N-carboxyanhydrides, among others; (c) ROMP of cyclic olefin monomers bearing cleavable linkages.
Among them, radical ring-opening polymerization (rROP) is useful for polymerizing cyclic ketene acetals and more recently, thionolactones into degradable vinyl polymers [9, 21]. Anionic/cationic/coordination/organo-catalyzed ring-opening polymerizations of lactides, lactones, and N-carboxyanhydrides lead to polyesters and polypeptides [24–27]. Ring-opening metathesis polymerization (ROMP) of cyclic olefin monomers bearing cleavable moieties can give rise to polyolefins with degradable polymer backbones [28–31]. Critically, the recent convergence of controlled/living polymerization concepts and ROP has enabled the synthesis of well-defined degradable polymers with precise control of molecular weights, low molecular mass dispersity, complex architectures (e.g., block copolymer), and loading of side-chain functional groups [28, 32, 33]. For example, when rROP is used in conjunction with reversible deactivation radical polymerization (RDRP), various functional polymers with substantial amount of backbone esters and thioesters can be produced [34–37].
Olefin metathesis has been widely exploited in the synthesis of small molecules and novel lipids, including at industrial scales [38]. Regarding polymer fabrication, its real strength lies in the inherent functional group tolerance and resistance towards air and moisture, thanks to the development of well-defined metathesis catalysts [39]. Living and controlled polymerizations of chemically and structurally diverse monomers have been extensively reported through olefin metathesis in the literature [40–48]. In the past decades, polymer chemists have begun to exploit olefin metathesis to incorporate degradable bonds into polymer backbones, giving rise to a new library of degradable polymers inaccessible by other polymerization methods (Fig. 2) [12, 22, 28–31, 49–61]. These new monomers and polymers display a high degree of modularity in the design of degradable polymers. In acknowledgement of this field’s burgeoning prevalence, we aim to highlight efforts to prepare degradable and depolymerizable polyolefins, organized according to their polymerization mechanisms, including (i) acyclic diene metathesis polymerization (ADMET); (ii) entropy-driven ring-opening metathesis polymerization (entropy-driven ROMP); (iii) strain-induced or enthalpy-driven ring-opening metathesis polymerization (enthalpy-driven ROMP); and (iv) cascade enyne metathesis polymerization (CEMP). The degree of control obtained by each approach will be systematically evaluated, discussed and put into perspective. Moreover, we will elucidate structure-property relationships of degradable polyolefins by highlighting the diversity of degradable functionalities that have been incorporated into polymer backbones, including ester, acetal, phosphoester, phosphoramidate, enol ether, carbonate, silyl ether, azobenzene, and disulfide, among others. Finally, current challenges, potential applications, opportunities, and future directions (such as metal-free and catalytic living ROMP protocols) will be critically assessed.
Fig. 2.
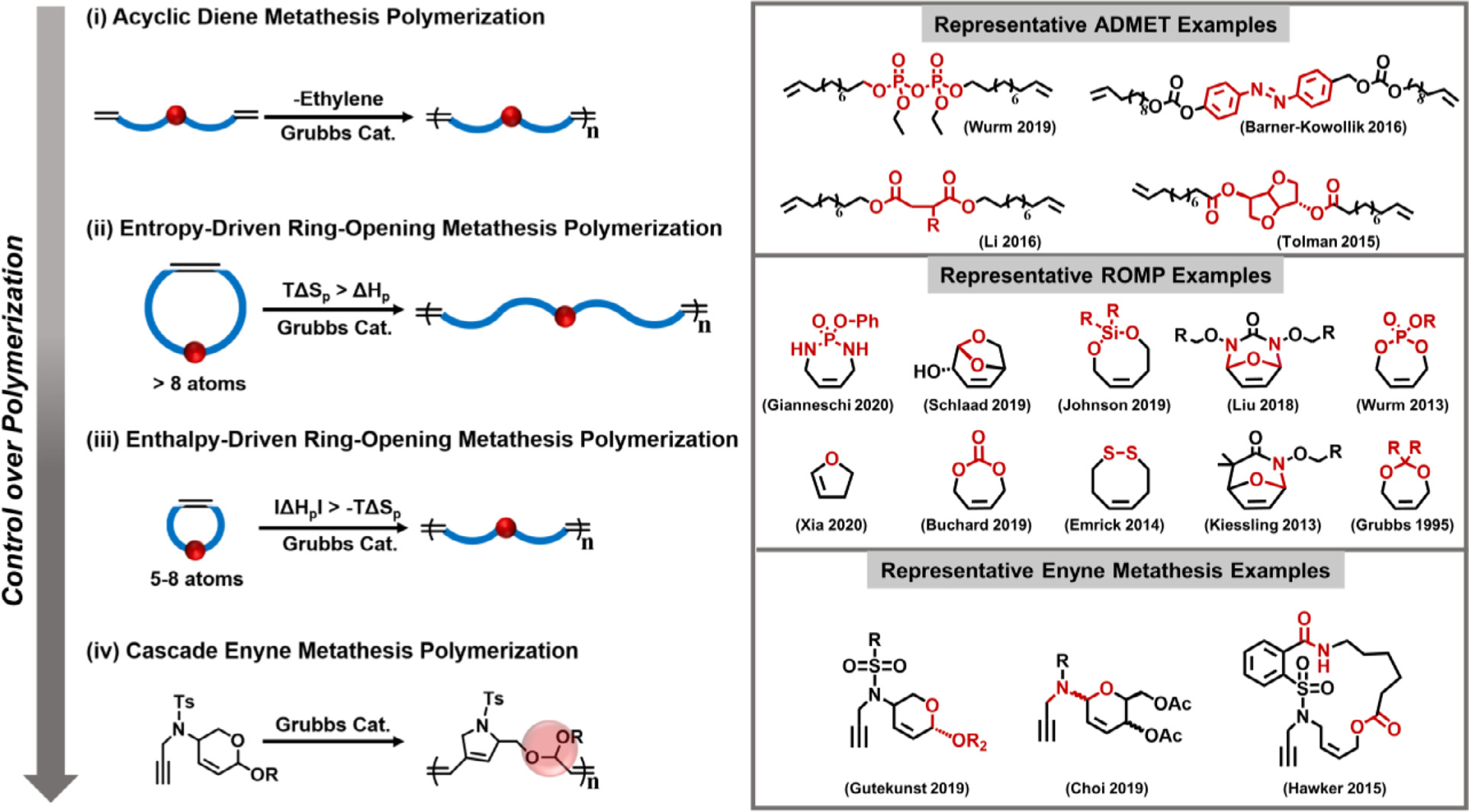
Synthetic approaches to degradable polymers via olefin metathesis polymerization. Four different approaches have been developed, including (i) acyclic diene metathesis polymerization (ADMET); (ii) entropy-driven ring-opening metathesis polymerization (entropy-driven ROMP); (iii) strain-induced or enthalpy-driven ring-opening metathesis polymerization (enthalpy-driven ROMP); and (iv) cascade enyne metathesis polymerization (CEMP).
2. Synthetic strategies of degradable polymers via olefin metathesis polymerizations
As is well known, the blossoming of metathesis chemistry is closely related to the development of transition metal-based metathesis catalysts. In 1986, Schrock and coworkers developed the first well-defined metathesis catalysts based on tungsten and molybdenum, making it possible to synthesize hydrocarbon polyolefins with high molecular weights and low dispersity [62–64]. The functional group tolerance of these metathesis polymerizations was expanded with the creation of late transition metal catalysts containing ruthenium by Grubbs and coworkers [44]. The rapid advent of these ruthenium complexes, now known as Grubbs catalysts, has led to a considerable expansion of olefin metathesis chemistry, enabling the incorporation of various functionalities into polymer backbones and side chains [38]. Through further optimization of ancillary ligands, a series of Grubbs catalysts with enhanced activity and air stability have been created, granting the fabrication of well-defined and functionalized polyolefins with predictable molecular weights and complex architectures (Fig. 3). In this section, we will review different olefin metathesis polymerization strategies used for the preparation of degradable polymers, along with a discussion of the degree of control over polymer molecular weight, dispersity, and architecture via each polymerization approach.
Fig. 3.
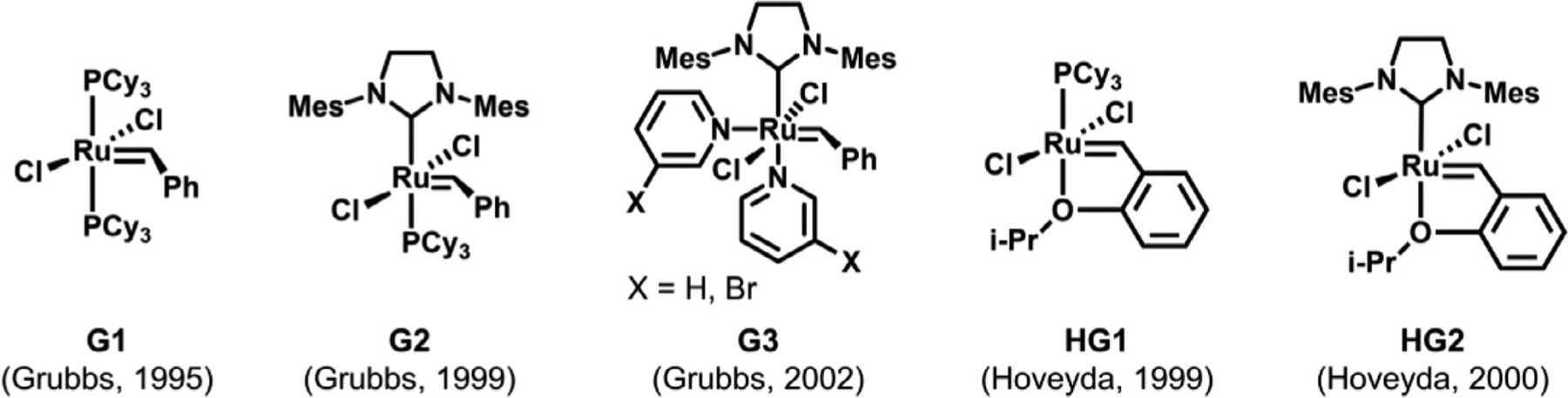
Structures of representative ruthenium-based olefin metathesis catalysts [65–70].
2.1. Acyclic diene metathesis polymerization
Acyclic diene metathesis (ADMET) polymerization is a step-growth polymerization of α, ω-dienes which liberates ethylene as a byproduct [71]. The elimination of ethylene under vacuum serves as the driving force of the polymerization. Because of the step-growth nature of ADMET, highly pure monomer and quantitative conversion are typically required for performing the polymerization reactions. Early work on ADMET polymerization mainly focused on the polymerization of all aliphatic symmetric α, ω-dienes, leading to alkylene copolymers with unprecedented control of pendant functional group spacing [72]. The precise placement of side-chain functionalities along the polymer backbone facilitated structure-property investigations of ADMET polymers. Recently, the scope of ADMET polymerization has been expanded beyond polymers with aliphatic hydrocarbon backbones, as evidenced by the development of novel α, ω-diene monomers containing heteroatom linkages which can undergo ADMET to form a diverse array of degradable polymers (Table 1).
Table 1.
Summary of representative degradable polymers via ADMET.
| Polymer type | Polymerization approaches | Degradation stimulus | Ref. |
|---|---|---|---|
| Polyacetal | ADMET | N/A | Wagener (1998) [49] |
| Poly(amino acid) | ADMET | N/A | Wagener (2001) [73] |
| Polyester | ADMET | pH | Meier (2011) [74] |
| Poly(sulfonate ester) | ADMET | Thermal | Simon (2014) [75] |
| Polyphosphonate | ADMET | pH | Wurm (2014) [76] |
| Polyester | ADMET | pH | Tolman (2015) [51] |
| Polyester | ADMET | Thermal | Li (2016) [53] |
| Polyazobenzene | ADMET | Enzyme | Barner-Kowollik (2016) [54] |
| Polypyrophosphate | ADMET | pH | Wurm (2019) [52] |
Aliphatic polyesters are perhaps the most frequently studied group of biodegradable polymers thanks to their excellent biodegradability and biocompatibility. [77] A set of aliphatic polyesters including polylactide, polyhydroxybutyrate, polycapro-lactone, and poly(lactide-co-glycolide) have been commercialized for more than half a century now, eliciting widespread applications in the biomedical field [78]. They are typically produced via anionic/metal catalyzed ROP of lactides and lactones, as well as radical ROP of cyclic ketene acetals [9, 78]. The recent development of ADMET has significantly increased the structural diversity of aliphatic polyesters with new physiochemical properties [51, 53, 74]. Meier et al. leveraged ADMET to prepare a set of biodegradable polymers including polyesters, polyanhydrides, and a random copolymer poly(ester-co-anhydride) from undec-10-enoic acid [74]. The solution stability of these polymers was assessed under acidic (i.e., H2SO4 in THF) and enzymatic conditions (i.e., Novozyme 435 in THF), respectively. According to degradation kinetics, the polyanhydride was much less stable than the polyester in both acidic and enzymatic digestions. Interestingly, the degradation rate of poly(ester-co-anhydride) was between the two homopolymers, suggesting the potential to achieve tunable degradability by adjusting the comonomer ratio of ester and anhydride.
Tolman and coworkers achieved the synthesis of sustainable polyesters using derivatives of glucose and castor oil (Fig. 4) [51]. Two diene monomers including glucarodilactone undercenoate (GDLU) and isosorbide undercenoate (IU) were synthesized and polymerized via ADMET polymerization to yield two homopolymers, PGDLU and PIU, as well as a copolymer, P(GDLU-co-IU) (Fig. 4 a). To elucidate the relationship between chemical structure and degradability, these polymers were subjected to acidic, basic, and neutral conditions, respectively. Degradation kinetics revealed that GDLU containing polymers, i.e., PGDLU and P(GDLU-co-IU) exhibited faster hydrolytic degradation rates than that of PIU. This can be attributed to the low hydrolytic stability of the lactone structure in the GDLU motif (Fig. 4 b).
Fig. 4.

pH-responsive polyesters via ADMET polymerization. (a) Synthesis of sustainable polyesters by ADMET polymerization of biomass-derived monomers IU and GDLU. (b) Degradation studies of PIU, PGDLU, P(GDLU-co-IU) under acidic, basic, and neutral conditions over a six-day period. [51], Copyright 2015. Reproduced with permission from the American Chemical Society.
Li and coworkers have developed thermally-degradable polyesters with tunable degradation temperatures (Fig. 5) [53]. First, a diene monomer, di(10-undecynyl) maleate was polymerized via ADMET, leading to the parent polyester containing maleate vinyl groups. Post-polymerization modification of the parent polyester through thiol-Michael additions afforded three polyesters with different pendant amino groups (Fig. 5 a). Because of the nucleophilicity of the side-chain amines, thermally induced degradation of the polyesters occurred by intramolecular cyclization and the concomitant formation of six-membered lactam derivatives (Fig. 5 b). Critically, degradation kinetics revealed that an increase of the side-chain steric bulk and a decrease of the nucleophilicity of the amines could enhance the stability the polyesters by increasing the degradation temperatures from 0 °C to 50 °C.
Fig. 5.

Thermally labile polyesters via ADMET polymerization. (a) Synthesis of polyesters by ADMET polymerization of di(10-undecynyl) maleate and subsequent thiol-Michael addition. (b) Degradation mechanisms of polyesters through heat-induced intramolecular cyclization. The thermal stability of polyesters is tunable by modifying the side-chain chemistry. [53], Copyright 2016. Reproduced with permission from the American Chemical Society.
Beyond classical carboxylate esters, polymers containing sulfonate esters in the backbone can also be achieved by ADMET polymerization. Simon and coworkers designed a series of sulfonate ester containing α, ω-diene monomers with varying numbers of methylene spacers [75]. ADMET polymerization of those monomers and subsequent catalytic hydrogeneration gave rise to saturated poly(sulfonate ester)s, which are structurally similar to high-density polyethylene (HDPE). The effect of sulfonate ester moieties on polymer crystallization was further elucidated by differential scanning calorimetry (DSC), showing that the melting temperatures and crystallinity of poly(sulfonate ester)s were markedly smaller than those of HDPE. More importantly, thermo-gravimetric analysis (TGA) of poly(sulfonate ester)s displayed an onset of thermal decomposition at 247 °C, significantly lower than that of HDPE (> 450 °C). This study suggests that poly(sulfonate ester)s could be useful as degradable polyethylene-like materials.
Synthetic polypeptides or poly(amino acids), non-sequence controlled analogues of natural-occurring proteins, are considered an ideal class of biocompatible and biodegradable polymers [79]. Akin to proteins, synthetic polypeptides are expected to undergo enzymatic digestion in vivo via proteolytic degradation pathways [80, 81]. Various efforts have been made to synthesize these types of polymers which contain amino acids in polymer backbones [73, 82, 83]. Wagener and coworkers demonstrated the first ADMET polymerization of α, ω-dienes bearing amino acid moieties [73]. In their study, a series of diene monomers with different lengths of methylene spacers between the alkene and the amino acid functionality were synthesized and polymerized in the presence of second-generation Grubbs catalyst G2. The polymerization kinetic study of these monomers revealed that the length of methylene spacers plays a pivotal role in mitigating the intramolecular complexation of the ruthenium catalyst. In the case of monomers with two and three methylene spacers, only oligomers or low molecular weight polymers of 4, 700 g/mol were generated. As the spacer length increased to eight, the molecular weights of polymers significantly increased to 33,000 g/mol. While those polyolefins possessing amino acids in the backbone units are expected to be biodegradable, their degradation behavior was not evaluated.
Polyacetals and polyketals hold considerable promise as biodegradable drug delivery systems for the treatment of chronic inflammatory diseases [84]. In comparison with esters and hydrazones, acetals and ketals are more sensitive to the acidic environments of tumors while exhibiting enhanced stability in blood at physiological pH [85]. Moreover, their degradation products including alcohol and aldehydes/ketones are non-acidic, thus alleviating potential inflammatory issues. Wagener et al. reported the first example of polyacetals via ADMET polymerization [49]. A benzaldehyde acetal moiety was inserted in the α, ω-diene monomer which was then polymerized with first-generation Grubbs catalyst. The resulting polymer was amorphous (Tg = −46 °C) and exhibited a high molecular weight of 23, 000 g/mol. = Murphy and coworkers further demonstrated the generality of this ADMET approach by synthesizing polyacetals and polyketals with various aldehyde precursors [86]. Most of these polymers were liquid due to their low glass transition temperatures. However, a polyacetal based on anthracene aldehyde was solid and mechanically strong enough for formulation as microparticles. The hydrolysis study of this anthracene-containing polyacetal was carried out in a mixed solvent of 90% dioxane and 10% aqueous buffer at 37 °C, revealing a half-life of 3 days at pH 4.5 and 20 days at pH 7.4. These results confirmed the degradability of polyacetals in acidic environment, as well as their relatively high stability under neutral conditions.
In nature, phosphate esters form the covalent linkages in the backbone of nucleotides in nucleic acids [87]. In addition, they are critical in the energy-transfer process of adeno-sine triphosphate. The unique and interesting properties of phosphate bonds have inspired polymer chemists to develop synthetic poly(phosphoester)s [88–90]. Several illustrative examples of phosphorus-based polymers involving ADMET chemistry have been reported by the Wurm group [52, 76]. In their initial work, a phosphonate monomer, di(undec-10-en-1-yl) methyl-phosphonate was designed and synthesized [76]. ADMET polymerization of the phosphonate monomer led to unsaturated aliphatic polyphosphonates with molecular weights up to 23,000 g/mol. Saturated polyphosphonates were further obtained upon hydrogenation in the presence of Palladium on carbon. Very recently, the same group used this ADMET technique coupled with post-polymerization hydrogeneration to access the first example of polypyrophosphates (Fig. 6) [52]. Despite the hydrophobicity of this polymer, the pyrophosphate group in the polymer backbone exhibited fast hydrolysis, prompting polymer degradation in aqueous environments. Additionally, the hydrolyzed fragments can be further digested by microorganisms, highlighting the potential of polypyrophosphates in biodegradable drug delivery systems. Notably, the analogous polyphosphates showed negligible biodegradation (< 3% after 30 days) under the same conditions.
Fig. 6.

pH-Sensitive polypyrophosphates via ADMET polymerization. (a) Synthesis of saturated polypyrophosphates (P1-H) by ADMET polymerization and post-polymerization hydrogeneration. (b) Weight loss of P1-H films under acidic, basic, and neutral conditions (black) and 31P NMR intensity of pyrophosphate in P1-H films under acidic and neutral conditions (blue). (c) P1-H film before and after incubation in aqueous solutions at different pH values. [52], Copyright 2019. Reproduced with permission from the American Chemical Society.
Enzymatically degradable polymers via ADMET polymerization were demonstrated by Barner-Kowollik (Fig. 7) [54]. In this research, an azobenzene-containing α, ω-diene monomer was polymerized and terminated with a monoacrylate functionalized polyethylene glycol, giving rise to an amphiphilic triblock copolymer containing cleavable azobenzene groups in the middle block. The degradation of azobenzene moieties relies on a two-electron reduction mechanism in the presence of azobenzene reductase, which is also known as azoreductase. This results in the formation of a self-immolative aniline derivative which can fully degrade to 4-aminobenzyl alcohol: a low toxicity product.
Fig. 7.

Enzyme-responsive and amphiphilic triblock PEG-b-poly(azobenzene)-b-PEG via ADMET polymerization. The central block contains azobenzene moieties which are cleavable in the presence of azoreductase. [54], Copyright 2016. Reproduced with permission from the Royal Society of Chemistry.
2.2. Entropy-driven ring-opening metathesis polymerization
To avoid low, poorly controlled and broad molecular weights inherent to step-growth polymerization reactions, [71] polymer chemists have sought to access degradable polymers via ROMP, which adopts a chain-growth mechanism [44]. On the basis of the type of thermodynamic driving force, ROMP can be categorized into two classes: (i) entropy-driven ROMP and (ii) enthalpy-driven ROMP [91]. Entropy-driven ROMP involves high-molecularmass, low-strain monomers or macrocycles, of which the polymerization would increase the conformational entropy as well as the entropy of mixing [92]. The latter relies on the ring-opening and concomitant strain release of low-molecular-mass, highly strained cyclic alkene monomers, resulting in a significant decrease in the enthalpy of the polymerization. Critically, entropy-driven ROMP is usually carried out under concentrated monomer conditions and even in bulk, with the aim of decreasing the loss of translational entropy in the polymerization. In principle, a very broad range of conceivable chemical functionalities could be incorporated into a macrocycle via state-of-the-art ring-closing olefin metathesis (RCM) to generate viable monomers [93]. Several examples of cyclic alkene macrocyclic monomers bearing cleavable linkages have been demonstrated for entropy-driven ROMP (Table 2).
Table 2.
Summary of representative degradable polymers via entropy- and enthalpy-driven ROMP.
| Polymer type | Polymerization approaches | Degradation stimulus | Ref. |
|---|---|---|---|
| Poly(glycolipid) | Entropy-driven ROMP | N/A | Gross (2007) [94] |
| Polyester | Entropy-driven ROMP | N/A | Meyer (2015) [95,96] |
| Polydisulfide | Entropy-driven ROMP | Reducing agents | Schlaad (2017) [97,98] |
| Polyester | Entropy-driven ROMP | Mechano + pH | Craig (2020) [99,100] |
| Polyacetal | Enthalpy-driven ROMP | pH | Grubbs (1995) [55] |
| Polyphosphoester | Enthalpy-driven ROMP | pH | Wurm (2013) [31] |
| Polydisulfide | Enthalpy-driven ROMP | Reducing agent | Emrick (2014) [56] |
| Polyoxazinone | Enthalpy-driven ROMP | pH | Kiessling (2013) [50,101] |
| Polyoxadiazinone | Enthalpy-driven ROMP | pH | Liu (2018) [57] |
| Polyacetal | Enthalpy-driven ROMP | pH | Schlaad (2019) [102] |
| Polycarbonate | Enthalpy-driven ROMP | N/A | Buchard (2019) [103] |
| Poly(silyl ether) | Enthalpy-driven ROMP | pH | Johnson (2019) [12,28] |
| Polyacetal | Enthalpy-driven ROMP | pH | Xia (2020) [29] |
| Poly(enol ether) | Enthalpy-driven ROMP | pH | Xia (2020) [30] |
| Polyester | Enthalpy-driven ROMP | Mechano + pH | Wang (2020) [104] |
| Polyester + Polyacetal | Enthalpy-driven ROMP | pH | Sampson (2020) [105] |
| Polyphosphoramidate | Enthalpy-driven ROMP | pH | Gianneschi (2020) [22] |
Schlaad et al. leveraged RCM to obtain macrocyclic monomers based on L-cystine (Fig. 8) [97]. The monomer contains a series of cleavable bonds including disulfide, amide, and ester, rendering it and the resulting polymer degradable (Fig. 8 a). Entropy-driven ROMP led to a polymer which exhibited an apparent molecular weight of up to 80 KDa. Moreover, a moderate control over the molecular weight was achieved, as indicated by a linear growth in molecular weight as a function of monomer conversion and polymerization time (Fig. 8 b). However, the dispersity was approximately 2 (Fig. 8 c). This is in agreement with the nature of equilibrium polymerizations which are characterized by a large extent of competing reactions (secondary metathesis) such as backbiting and cyclo-depolymerization. Finally, the degradability of the polymer was demonstrated by reductive cleavage of the main-chain disul-fides (Fig. 8 d).
Fig. 8.

Synthesis and degradation of redox-responsive poly(disulfide)s derived from L-Cystine. (a) Entropy-driven ROMP of macrocyclic disulfide monomers which were produced by RCM. (b) SEC traces of purified polymer 3a obtained at different reaction times. (c) Evolution of apparent weight-average molecular weight of polymer 3a as a function of monomer conversion (values in parentheses are dispersity indexes from SEC). (d) SEC traces of the polymer 3a and the degradation products obtained by reduction of main-chain disulfide after different degradation times. [97], Copyright 2017. Adapted with permission from the Royal Society of Chemistry.
Meyer and coworkers reported the synthesis of sequenced-controlled polyesters by entropy-driven ROMP (Fig. 9 a). [95] In this work, a library of macrocyclic monomers consisting of lactic acid, glycolic acid, and Ɛ-caprolactone-derived units were prepared via multi-step synthesis including RCM as the last step. Entropy-driven ROMP of the resulting monomers using the second-generation Grubbs catalyst G2, gave rise to sequence-defined polyesters, whose molecular weights were tunable by varying the monomer-to-catalyst ratio (Fig. 9 b). Impressively, polymers were generated with dispersities below 1.3 (Fig. 9 c). These results indicated that good control was achieved over the polymerizations.
Fig. 9.

Sequence-defined polyesters via entropy-driven ROMP. (a) Synthesis of polyesters by entropy-driven ROMP of macrocyclic monomers. (b) Evolution of molecular weights of polyesters with varying [M]/[cat] ratios. The molecular weights determined by SEC are in red, whereas the dotted black line represents a theoretical plot of living polymerization. (c) Evolution of polyester molecular weight versus time (black), and dispersity index versus time (red). [95], Copyright 2015. Adapted with permission from the American Chemical Society.
Recently, the same group described the entropy-driven ROMP of strainless macrocyclic monomers comprised of lactic, glycolic, 6-hydroxy hexanoic, and syringic acids [96]. Both cis- and trans-olefin isomers were prepared and compared for their polymerization kinetics, which indicated that cis-olefins are markedly more reactive than their trans analogues. The exploitation of the cis-olefin monomers led to an enhanced livingness for the polymerizations, improved molecular weight control, decreased dispersity, and the ability to perform chain extension with a second monomer. While a degradation study of the resulting sequence-controlled polyesters was not performed, it is expected that the approach should enable a tunable hydrolysis rate by varying the sequence of the ester units [106].
Besides RCM-centered monomer synthesis, macrocyclic olefin-based monomers can also be directly obtained from nature. Lac-tonic sophorolipid (LSL) is a 26-membered macrocyclic glycolipid lactone which can be biosynthesized through fermentation from the yeast candida bombicola. [94] Gross and coworkers pioneered the synthesis of poly(LSL) by harnessing entropy-driven ROMP. [94] In the optimized polymerization conditions (i.e., high monomer concentration, high temperature, and Grubbs second-generation catalyst G2), it was found that the obtained polymeric molecular weights were close to the theoretical values based on a living polymerization model. Nevertheless, the relatively broad molecular weight distributions (Ð = 1.6–1.8) suggested that pronounced secondary metathesis was present in the entropy-driven ROMP of LSL.
2.3. Enthalpy-driven ring-opening metathesis polymerization
Over the past decades, a variety of functional monomers containing degradable moieties have been designed to polymerize through enthalpy-driven ROMP to introduce backbone degradability [22]. Unlike the highly strained norbornenes (27.2 kcal/mol) [107], most of these monomers are low or moderately strained five- to eight-membered cyclic olefins [22, 28, 30]. Enthalpy-driven ROMP of these small cyclic alkene monomers with low strain typically leads to an equilibrium, in which the propagation rate is equal to the depolymerization rate [108, 109]. Therefore, an equilibrium monomer concentration exists in these systems, and optimizing reaction condition is crucial to drive the equilibrium towards polymer formation [110]. In this section, we will provide a comprehensive review of the use of these low-strain cyclic olefins (LSCOs) in preparing main-chain degradable polymers (Table 2). We will highlight different polymerization strategies, including the use of high monomer concentrations, low reaction temperatures, and copolymerization with highly strained monomers, to promote living and controlled ROMP.
2.3.1. Dioxepin based monomers
Cis −4,7-dihydro-1,3-dioxepin (Dxp) and its derivatives are the most explored LSCOs for enthalpy-driven ROMP because of the facile monomer syntheses and the incorporation of acid labile acetal/ketal linkages, giving rise to full or partial backbone degradable polymers (Fig. 10). In 1995, Grubbs and coworkers reported the first attempt to polymerize Dxp through enthalpy-driven ROMP [55]. Using first-generation Grubbs catalyst G1, ROMP of Dxp 1a yielded a polyacetal with a broad molecular weight (MW) distribution (M n = 17.2 kDa, Đ > 1.5), while homopolymerization of phenyl Dxp 1b was not achieved. The researchers then performed a copolymerization of Dxp with cyclooctadiene (COD). Despite the formation of high molecular weight copolymers, more than 40% Dxp was unreacted, leading to a low incorporation of the acetal moieties, ranging from 4% to 30% depending on the feed ratio. Moreover, the broad dispersity of Dxp-COD copolymers (Đ = 1.51~3.46) indicated poor control over the polymerization and existence of secondary metathesis during ROMP. Following acid-triggered hydrolysis in organic solvent, the copolymers degraded into 1,4-hydroxytelechelic polybutadienes, which can be further hydrogenated to obtain polyethylene.
Fig. 10.
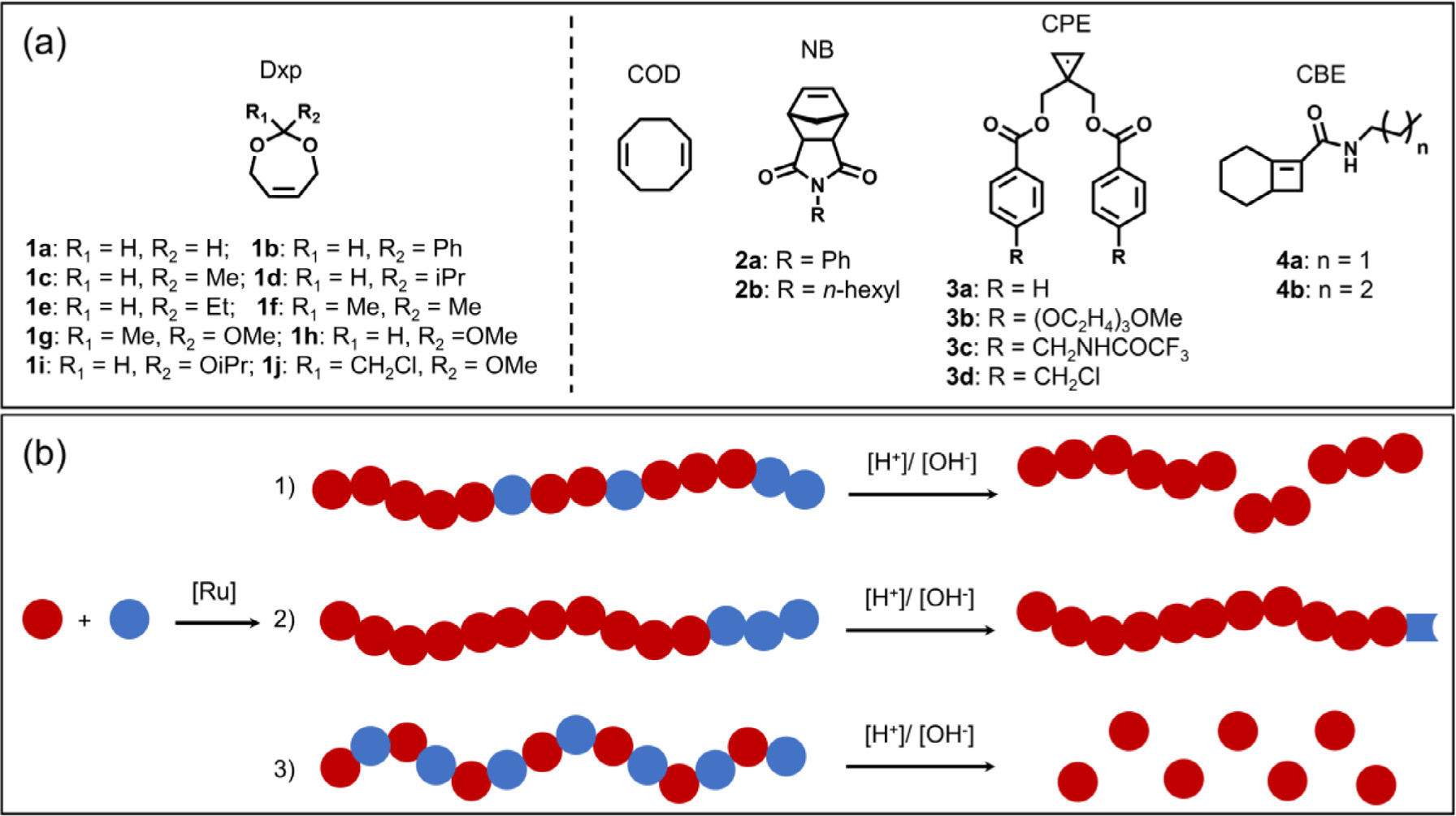
Degradable polyacetals/ketals via ring-opening metathesis copolymerization of dioxepins and highly strained monomers. (a) Structures of dioxepins and highly strained comonomers. (b) Different degradation profiles of copolymers prepared from Dxp (blue) and comonomers (red). (1) Random copolymers that degrade into fragments of various sizes; (2) Block copolymers where dioxepin is incorporated as a sacrificial monomer for end-functionalization; (3) Alternating copolymers that completely disintegrate into small fragments.
In follow-up work, Kilbinger confirmed that the pendant R groups have a strong impact on the reactivity of Dxp towards polymerization, probably through the Thorpe-Ingold (gemdisubstituent) effect [111], and also influence the backbone degradation kinetics. The sterically bulky Dxps 1b and 1d only oligomerized in the presence of Grubbs catalyst, and thus have been used as sacrificial monomers to achieve end-functionalization of polynorbornenes [112, 113]. In addition, Dxp has been explored as a linker between two polynorbornene blocks. However, due to the slow initiation of the Dxp carbene, the second block was polymerized in an uncontrolled manner [43]. In 2019, Wurm and coworkers demonstrated enthalpy-driven ROMP of orthoester versions of Dxp (1g-1j) with COD using first-generation Hoveyda-Grubbs catalyst HG1 [114]. Similar to Grubbs’ early example, the resulting copolymers suffered from broad MW distributions (Đ > 2) and poor orthoester incorporation (between 10 to 30%), giving rise to high MW degradation products upon hydrolysis.
Recently, Xia and coworkers reported the first controlled alternating ROMP (AROMP) of Dxps and 1,1-disubstituted cyclo-propenes (CPEs) in the presence of third-generation Grubbs catalyst G3 [29, 115]. The highly strained CPE provides enough thermodynamic driving force to promote the ring opening of Dxps. The bulky substituents on the CPE monomer suppressed its homopolymerization and, to some extent, inhibited the secondary metathesis of the alternating backbone. Under optimal AROMP conditions, the researchers obtained polyacetals/ketals with controlled molecular weights and narrow dispersity (Đ < 1.25) in 8 to 48 h. 1H NMR analysis confirmed the nearly equal composition of Dxp and CPE in the isolated polymer. HPLC-MS further verified the major degradation product (70~85%) to be the diallyl alcohol, which was generated from the CPE-Dxp alternating dyad. The degradation kinetics of these copolymers was found to be dependent on the substituents on Dxp, with poly(3a-alt-1f) showing the fastest hydrolysis (completion in 1 h) in THF solution containing 1 vol% TFA. By incorporating functional handles on the CPE monomers (3c with protected amine and 3d with benzyl chloride), the researchers noted the potential for these copolymers to afford further diversification through post-polymerization modifications. Moreover, the same group demonstrated the synthesis of block copolymers bearing Dxp and cyclohexene as LSCOs in the first and second block through AROMP, indicating that the metallocarbene remained active post incorporation of LSCOs.
Contemporaneously, the Sampson group described AROMP of Dxps and bicyclo[4.2.0]oct-1(8)-ene-8-carboxamides (CBE), affording alternating copolymers with moderate MW distributions (Đ = 1.2~1.6) [105]. Degradation was achieved in aqueous TFA solution, showing similar substituent-dependent hydrolysis kinetics observed in Xia’s work. It’s worth noting that in the same paper, the researchers demonstrated AROMP between CBE and lactone based 9, 10-membered cyclic olefins, yielding ester-containing polymers.
Cyclic carbonate, 4,7-dihydro-1,3-dioxepin-2-one, is so far the only dioxepin derivative capable of homopolymerization via enthalpy-driven ROMP, affording high molar mass polycarbonates [103]. Quantitative monomer conversion of this moderately-strained cyclic monomer (12.6 kcal/mol) was rapidly achieved within 50 min in the presence of G2 at 22 °C in DCM. The relative high MW distribution (Ð = 1.58–2.69) might be attributed to the carbonate coordination to the proceeding metallocarbene, which can facilitate the chain transfer. Additionally, the chosen Grubbs catalyst is known for the production of polymers with uncontrolled MW because of slow initiation and rapid propagation rates [116]. Interestingly, the researchers observed that more than 80% trans-olefin was produced in the polymer backbone through ROMP, leading to an amorphous material with glass transition temperature (Tg) at −22 °C. Conversely, exclusive cis-olefin formation was realized via ring-opening transesterification polymerization (ROTEP), which gave rise to semicrystalline polymer materials with a melting temperature (Tm) of 115 °C. The ability of the cyclic carbonate to undergo stereoselective polymerization enables the formation of homopolymers with distinctive physiochemical properties, and ABA triblock polymers that can potentially serve as a thermoplastic elastomer. Nevertheless, the impact of backbone stereochemistry on degradation kinetics remains unexplored.
2.3.2. Bicyclic monomers
In 2013, Kiessling and coworkers developed a new class of monomer that leads to the preparation of fully degradable polyoxazinones through enthalpy-driven ROMP (Fig. 11) [50]. The bicyclic oxazinone based monomers were prepared through aza-[4+3] cycloaddition, and have a moderate ring strain (13.4 kcal/mol) that is comparable to that of trans-cyclooctene, rendering them polymerizable under ROMP conditions (Fig. 11 a). To mitigate backbiting, the reaction was conducted in THF, a catalyst-coordinating solvent, at room temperature in the presence of third-generation Grubbs catalyst G3. Under the investigated conditions, the researchers observed 73~87% monomer conversion within 1 h and obtained well defined polymers (Mn = 6.1~41 kDa,) with low dispersity (Đ = 1.4~1.5). More importantly, the molecular weight of the polymer increased proportionally as the monomer-to-catalyst ratio increased, demonstrating the character of living polymerization. The resulting polyoxazinone (5e, Mn = 18.5 kDa) was stable for a prolonged time (18 h) in a mixed solvent of THF and MeOH at neutral pH. Rapid degradation was observed under either low or high pH (pH < 1 or pH > 13.1) (Fig. 11 b and c). Notably, polymerization of bicyclic oxazinones with different hydroxamic ester substituents (5a-5f) was explored, verifying the generality of enthalpy-driven ROMP for a wide range of functional monomers. Post-polymerization modifications of the polyoxazinones for spectral and biological activities were explored in subsequent work [101] and will be discussed in the application section.
Fig. 11.

Acid/base labile polyoxazinones via enthalpy-driven ROMP of bicyclic oxazinones. (a) Schematic overview of polymerization and degradation. (b) Degradation profile of polymer 5e under acidic and basic conditions. (c) Degradation of polymer 5f in 3:1 THF/MeOH (pH = 0.25) monitored by comparing the ratio of polymeric pyrene emission (λmax = 480 nm) and monomeric pyrene emission (λmax = 370 nm). Empty diamonds represent low emission owing to poor initial polymer solubility. [50], Copyright 2013. Adapted with permission from John Wiley and Sons Inc.
Besides bicyclic oxazinone, enthalpy-driven ROMP of a structurally similar monomer, oxadiazabicyclooctenone (bicyclic dioxazinone) was later demonstrated by Liu and coworkers to construct pH-sensitive polymers [57]. However, the incorporation of an extra oxygen in the monomer structure can reinforce the coordination with the metallocarbene, and concomitantly facilitate secondary metathesis. As a result, the polymerization of bicyclic dioxazinones (Đ = 1.6~1.9) was less controlled compared to the bicyclic oxazinones in the presence of G3 and G2. The resulting poly(oxadiazinone)s showed rapid degradation under acidic or basic conditions, agreeing well with the results in Kiessling’s study. The researchers also performed random copolymerization of bicyclic oxadiazinones and norbornenes. While both monomers achieved high conversions, the resulting copolymers showed varying degrees of dioxazinone incorporation, which is highly dependent on the substituents on the norbornenes.
Another interesting bicyclic LSCO is the biomass-derived levoglucosenol (Fig. 12) [102]. The monomer was readily obtained by reduction of levoglucosenone, a commercial product obtained via pyrolysis of cellulose (Fig. 12 a). Successful polymerization of levoglucosenol with approximately 60% monomer conversion was achieved in 24 h at room temperature using the mono-ortho-substituted NHC derivative of second-generation Grubbs catalysts, yielding high molar mass polyacetals (Mn up to 100 kDa) (Fig. 12 a and b). The researchers observed a non-living polymerization pattern, as demonstrated by the rapid formation of high molar-mass polymers at the beginning of the reaction, followed by backbiting and chain end deactivation at prolonged polymerization times (Fig. 12 c). When high initial concentrations of levoglucosenol (2~4 M) and low temperature (0~5 °C) were employed, monomer conversion was modestly increased to approximately 70%. However, the dispersity still remained quite high even under these optimized conditions (Đ ≥ 2). Moreover, the resulting polyacetals completely degraded in acidic 1,4-dioxane in 40 days at room temperature (Fig. 12 d). This study highlights the potential to acquire novel and sustainable ROMP monomer from renewable feedstock, leading to environmentally friendly thermoplastics.
Fig. 12.
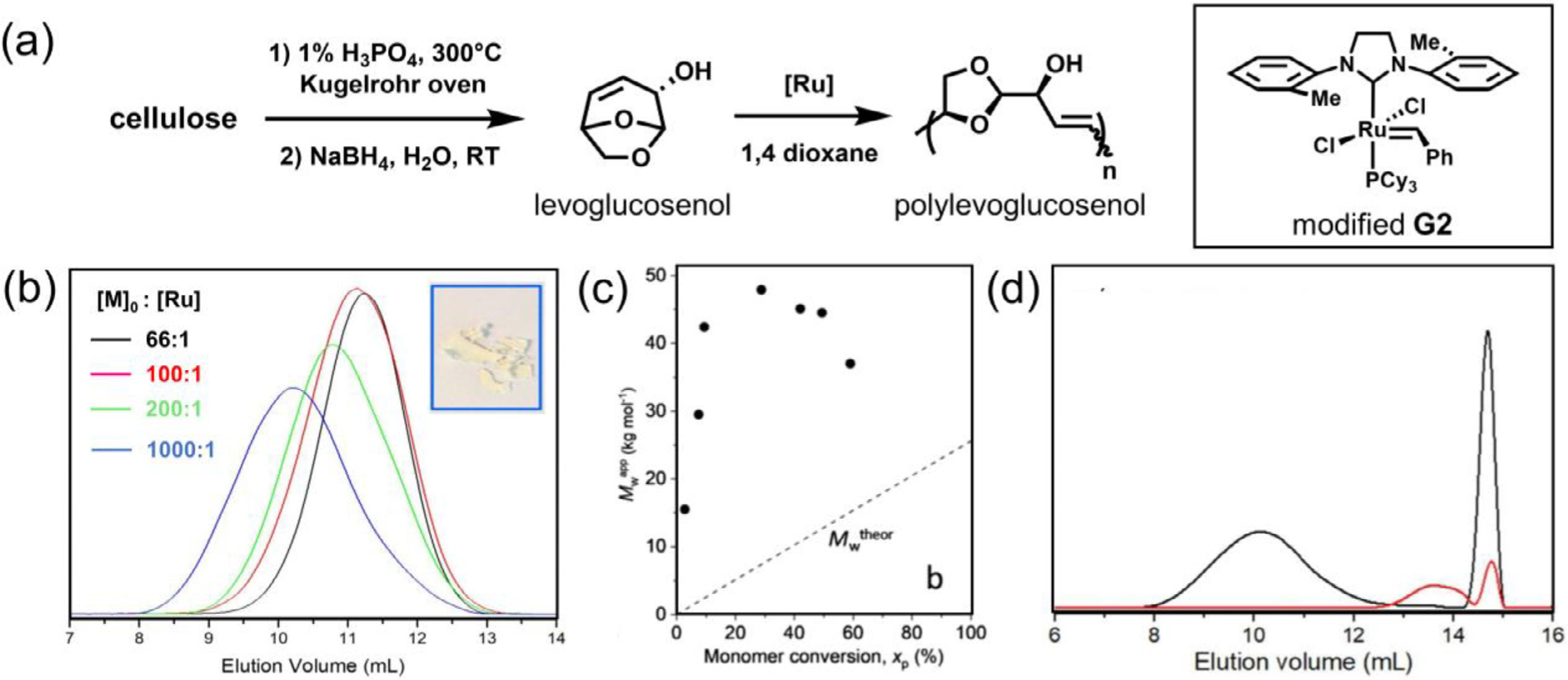
Enthalpy-driven ROMP of biomass-derived levoglucosenol to yield acid labile polyacetals. (a) Synthesis of levoglucosenol from cellulose and subsequent polymerization by ROMP. (b) SEC traces of polylevoglucosenol obtained from different catalyst-to-monomer ratio. Isolated polylevoglucosenol appeared as a white solid at room temperature. (c) Evolution of weight-average molar mass (Mwapp) with monomer conversion (xp) is significantly deviated from the theoretical values, indicative of non-controlled polymerization and pronounced chain transfer. (d) SEC traces of polylevoglucosenol before (black) and after (red) degradation. The peak at the elution volume of ~14.5 mL refers to the internal reference (toluene). Condition: p-toluenesulfonic acid/water for 40 days. [102], Copyright 2019. Adapted with permission from John Wiley and Sons Inc.
2.3.3. Other heteroatom monomers
LSCOs bearing other heteroatom moieties including phosphoester, phosphoramidate, disulfide and silyl ether, have also been developed for enthalpy-driven ROMP, leading to novel main-chain degradable polymers with versatile functionalities and possibly orthogonal degradation. However, the incorporation of these heteroatoms poses significant challenges in achieving controlled ROMP due to the tendency of these groups to interact with the metal catalysts, slowing down propagation and facilitating secondary metathesis.
The first attempt to polymerize 7-membered cyclic phosphoester monomers through enthalpy-driven ROMP was reported by Wurm and coworkers in 2013 [31]. The homopolymerization of cyclic phosphoester monomers yielded polyphosphoesters with molecular weights up to 5 kDa with Đ = 2 regardless of the monomer-to-catalyst ratio, suggesting significant backbiting and potential catalyst deactivation/decomposition. Copolymerization of the cyclic phosphoesters with another low strain monomer cis-cyclooctene (7.4 kcal/mol) was further performed. Despite high monomer conversion, the resulting copolymers had broad MW distribution (Đ > 1.72) and low incorporation of the phosphoester moiety (~30%), which led to high molecular weight degradation products post acidolysis. Similarly, a disulfide-containing cyclic olefin, (Z)-3,4,7,8-tetragtdro-1,2-dithiocine, failed to homopolymerize through ROMP. Random copolymerization of the cyclic disulfide monomer with various cis-cyclooctenes led to copolymers that showed rapid degradation under the treatment with tri-n-butylphosphine, photogenerated radicals and thiol-disulfide exchange [56].
In 2020, Gianneschi and coworkers demonstrated the first example of a degradable polyphosphoramidate accessed via enthalpy-driven ROMP at low temperatures (Fig. 13) [22]. The researchers found that while the cyclic phosphoramidate monomer, 2-phenoxy-1,3,4,7-tetrahydro-1,3,2-diazaphosphepine 2-oxide (PTDO), has a low strain energy (10.86 kcal/mol) and only oligomerized at room temperature, well-defined homopolymers with molar masses up to 44.3 kDa and low dispersity (Đ can ≤ 1.2) be achieved at low temperature (2 °C) in the presence of G3 (Fig. 13 a and b). This result agrees with the thermodynamic principle for enthalpy-driven ROMP of low strain cyclic monomers (e.g., cyclopentene), in which lower reaction temperatures favor the shift of equilibrium towards polymerization [110, 117]. Moreover, secondary metathesis such as backbiting would be mitigated at low temperatures. Before equilibrating at 55% monomer conversion between 3 to 5 h, the reaction followed pseudo first order kinetics and showed good agreement between theoretical and targeted molecular weight. However, broadening of the SEC signal was observed at prolonged reaction times, indicative of the events of secondary metathesis after reaching equilibrium. The resulting polyphosphoramidate completely degraded into small molecules phosphoric acid and 1,4-diamino-2-butene in acidic DMSO within 240 h (Fig. 13 c).
Fig. 13.

Degradable polyphosphoramidate via low temperature ROMP. (a) Schematic of PTDO polymerization and degradation. (b) SEC traces of PPTDO of different degrees of polymerization (DP = 55, 94, 216). (c) Degradation of PPTDO and production of phosphoric acid monitored by 31P NMR. Condition: 0.25 M HCl in DMSO-d6 at room temperature. (d) SEC traces of NBOEG homopolymer and NBOEG-PTDO random copolymer before and after acid treatment. (e) Transmission electron microscopy (TEM) image of micellar nanoparticles assembled from NBOEG-PTDO random copolymer. Scale bar: 100 nm. [22], Copyright 2020. Adapted with permission from the American Chemical Society.
Furthermore, the researchers demonstrated nearly 50% PTDO incorporation into polynorbornenes through a copolymerization strategy, which led to either complete (Fig. 13 d) or partial backbone degradation. When norbornene with pendent oligoethylene glycol (NBOEG) was used for copolymerization with PTDO, the resulting NBOEG-PTDO copolymers were amphiphilic and successfully self-assembled into micellar nanoparticles (Fig. 13 e). No discernable cytotoxicity of the NBOEG-PTDO micelles was observed in HeLa cells, highlighting the potential of these novel polymers in biomedical applications.
Silyl ether based LSCOs represent another class of monomer that has been reported for controlled ring opening metathesis copolymerization to afford degradable polynorbornene-based copolymers (Fig. 14) [28]. Based on computational calculations, these monomers have a strained energy less than 8 kcal/mol, making them unlikely to homopolymerize. Nevertheless, Johnson and coworkers showed that these cyclic silyl ether monomers were capable of efficiently copolymerizing with norbornenes to generate well-defined copolymers when the molar ratio of norbornene to cyclic silyl ether was 1:1 (Fig. 14 a). As the molar ratio of cyclic silyl ether to norbornene further increased, an increase in MW distribution as well as a decrease in monomer conversion were observed, which is similar to the AROMP example mentioned above [29]. Through a screening of monomers with different ring size, the researchers demonstrated that the 8-membered cyclic silyl ether possessed the optimal balance between statistical copolymerization efficiency and synthetic accessibility as compared to the 7- and 9-membered analogues. The resulting silyl ether-norbornene copolymer completely degraded into low molar mass species via acid-catalyzed hydrolysis in a dioxane/water mixture (Fig. 14 b). By tuning the substituents on the silyl ether, variations in degradation kinetics were achieved (Fig. 14 c). A variety of norbornenes, including macromonomers were subjected to copolymerization, leading to degradable polynorbornenes with a variety of architectures including linear, bottlebrush, and brush-arm star polymers (BASPs) (Fig. 14 d). Through in vitro and in vivo assessment, the PEG-based bottlebrush copolymers showed minimal cytotoxicity and enhanced long-term tissue clearance, which will be discussed in the application section.
Fig. 14.
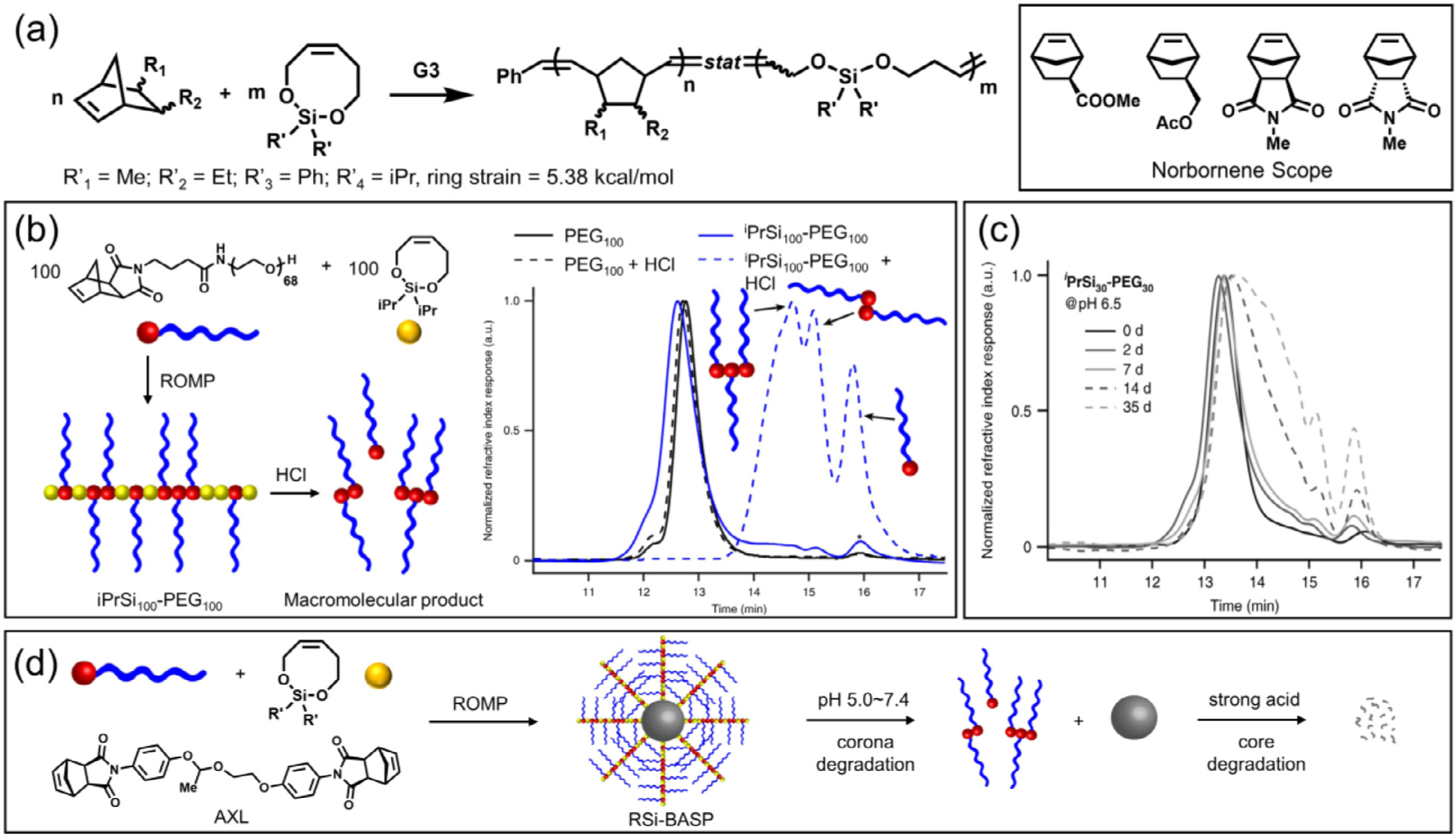
Copolymerization of cyclic silyl ether olefin with a variety of norbornenes to afford degradable polynorbornene-based linear, bottlebrush, and star arm copolymers. (a) Synthetic scheme of copolymerization by ROMP. (b) Synthesis and degradation of iPrSi100-PEG100 bottlebrush polymer. As indicated by the SEC traces, only the bottlebrush polymer containing silyl ether functionality degraded into oligomeric PEG fragments post acid treatment. (c) SEC traces of iPrSi30-PEG30 degradation in 35 days. Condition: pH = 6.5 in phosphate-citrate buffer at 37 °C. (d) Synthesis and degradation of BASPs. By varying the strength of applied acid, selective and sequential degradation of bottlebrush arm and acetal core can be achieved. [28], Copyright 2019. Adapted with permission from Springer Nature.
Vinyl ethers have long been conceived as terminating agents for ROMP due to the formation of a thermodynamically stable Fischer carbene. Nevertheless, Xia and coworkers recently illustrated the successful enthalpy-driven ROMP of a commercially available cyclic enol ether, 2,3-dihydrofuran (DHF), which led to poly(enol ether) with molar mass ranging from 6.0 to 127.7 kDa (Fig. 15 a and b) [30]. The polymerization was promoted by an extremely high initial DHF concentration as the reaction was performed in neat monomer. Approximately 80% monomer conversion was reached as the polymerization equilibrated after 4 h at room temperature. Through the Van’t Hoff equation, the researchers extrapolated the thermodynamic parameters for the ROMP of DHF, revealing that ΔH = −5.0 kcal/mol and ΔS = −19.4 cal/mol•K, which closely resemble the values reported for ROMP of cyclopentene [118]. Despite a broad dispersity (Đ = 1.35~2.14), the polymerization was highly efficient even at 0.01 mol% catalyst loading, giving rise to PDHF with a molar mass greater than 100 kDa. Upon treatment with acidic THF, the PDHF hydrolyzed into small molecular 4-hydroxybutanal and its furan isomer (Fig. 15 c). Interestingly, the researchers demonstrated that PDHF can be depolymerized back to the cyclic enol ether through in situ ring-closing metathesis (Fig. 15 d). The depolymerization behavior of PDHF is similar to the recently reported depolymerizable polypentenamers [119]. To date, this work is the only LSCO example that affords both a degradable and depolymerizable polymeric backbone, demonstrating the potential of this platform in chemically recyclable material.
Fig. 15.
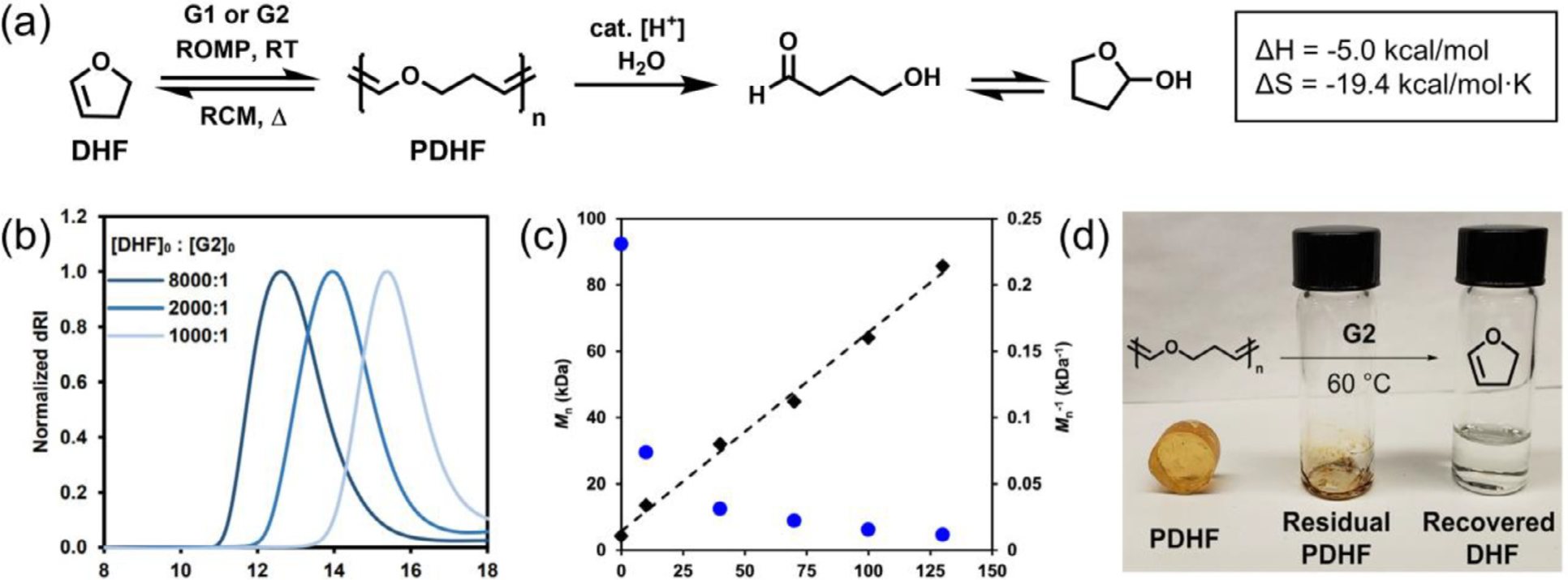
Enthalpy-driven ROMP of cyclic enol ether to afford degradable and depolymerizable poly(enol ether). (a) Scheme for DHF polymerization, depolymerization and degradation. (b) SEC traces of PDHF obtained using different catalyst-to-monomer ratio. (c) Molecular weight (blue dot) and inverse molecular weight (black square) of PDHF during TFA triggered hydrolysis. (d) PDHF depolymerization and monomer recovered via ring-closing metathesis at 60 °C. [30], Copyright 2019. Adapted with permission from the American Chemical Society.
Recently, a follow up work by Gutekunst demonstrated the alternating polymerization behavior of a cyclic enol ether with enyne monomers via cascade enyne metathesis polymerization. The details will be discussed in the next section [61].
2.4. Cascade enyne metathesis polymerization
Entropy-driven and enthalpy-driven ROMP strategies not only can produce degradable polymers with high molecular weights, but also can provide modest control over the molecular weights of degradable polymers (vide supra). However, the major examples of cyclic olefin monomers bearing degradable moieties have low ring strain or are even strainless (in the case of macrocycles). In view of this, a thermodynamic equilibrium is typically found in ROMP of such monomers, which are accompanied by noticeable secondary metathesis events, compromising the livingness of the polymerization [108]. In the quest for robust synthetic tools that can render the polymerization of low-strain monomers with enhanced living characteristics, polymer chemists have recently developed a new approach which is referred to as cascade enyne metathesis polymerization (CEMP) (Table 3) [58–61]. The original concept of CEMP was established in 2012 by Choi and coworkers who reported an extremely fast tandem ring-opening/ring-closing metathesis (RO/RCM) polymerization of a monomer containing two rather unreactive functional groups: cyclohexene and a terminal alkyne [120]. Since then, various eligible monomers are produced by modifying the ring size of the cycloalkene moieties, the length of the alkynes, and linker units [121, 122]. In a typical CEMP process, a NHC-ligated Grubbs catalyst first reacts with the terminal alkyne on the monomer to form a vinyl ruthenium carbene, which further undergoes a thermodynamically driven intramolecular cyclization and concomitant ring-opening (RO/RCM reaction) with the proximal alkene to produce a stable diene and a low strain pyrrolidine moiety, leading to the fast polymerization.
Table 3.
Summary of representative degradable polymers via CEMP.
The first example of degradable polymers via CEMP was illustrated by Gutekunst and Hawker in 2015 (Fig. 16) [58]. In their work, a series of unstrained macrocyclic monomers containing an alkyne-based “polymerization trigger” and various functionalities (e.g., ester, amide, sulfonamide) were prepared (Fig. 16 a). CEMP of these monomers exhibited ultra-fast polymerization kinetics, achieving high monomer conversions of up to 92% within 10 min. Moreover, SEC analysis indicated the resulting polymers had molecular weights similar to theoretical values, and with low dispersity (Ð = 1.07–1.39). Since the polymers contain esters in the main chain, the researchers further demonstrated the degradability of polymers by incubating them in 9:1 dichloromethane/methanol in the presence of 3 mol % triazabicyclodecene (TBD). Under this digestion condition, alcoholysis of the polymers via TBD-catalyzed transesterification occurred in a fast fashion, leading to a dramatic reduction of polymer molecular weight from 32.6 kDa to 1.8 kDa within just 30 seconds (Fig. 16 b).
Fig. 16.

Sequence-defined polyesters via cascade enyne metathesis polymerization. (a) Synthesis of macrocyclic enyne monomer and subsequent CEMP. (b) TBD-catalyzed degradation of polyester in a mixed solvent of 9:1 DCM/MeOH. SEC analysis of degraded products at different reaction times. [58], Copyright 2015. Reproduced with permission from the American Chemical Society.
The complete degradation was observed after 2 h, as evidenced by the SEC signal of low molecular weight products (Mn = 200). While the CEMP strategy was quite effective in the production of well-defined polymers, the synthesis of macrocycles required lengthy reaction sequences and suffered from chain transfer at high conversions, limiting their potential in degradable materials.
Later, Gutekunst et al. expanded the monomer scope of the CEMP strategy to a class of enyne acetal monomers which contains an alkyne handle and a six-membered, unstrained cyclic acetal alkene (Fig. 17) [59]. By changing substituents, the researchers were able to access a diverse set of enyne acetal monomers M1–M6 at gram scale (Fig. 17 a). The homopolymerization of M1–M5 gave rise to monomer conversions of more than 90% within 6 to 60 min in the presence of third-generation Grubbs catalyst G3 at room temperature. In addition, a first order kinetic profile, along with a linear increase in molecular weight with monomer conversion was observed in the CEMP of M1 (Fig. 17 b). The molecular weights of resulting polyacetals can be easily tuned in the range of 26–171 kDa by varying the monomer-to-catalyst ratio. Further, chain extension of poly(M1) with M5 was successfully performed, leading to a block copolymer poly(M1)-b-poly(M5), as evidenced by a significant and clean shift in the SEC chromatography to a shorter elution time (Fig. 17 c). These results unequivocally verified the livingness of CEMP enyne acetal monomers. Finally, the degradation rate of these polyacetals was investigated in THF/water (20/1 v/v) mixtures at various pH (Fig. 17 d). Poly(M1) showed gradual hydrolysis under neutral conditions, reaching 33% and 63% reduction in molecular weight at 24 and 48 h, respectively. When triethylamine (TEA) was added, the molecular weights remained unchanged over the course of the experiments, indicative of the structural integrity of the polymer at basic pH. On the other hand, the addition of acetic acid or trifluoroacetic acid accelerated the hydrolysis, as confirmed by faster degradation kinetics than that corresponding to neutral conditions.
Fig. 17.

pH-Responsive polyacetals via CEMP. (a) Modular synthesis of polyacetals by CEMP of enyne acetal monomers. (b) SEC traces of poly(M1) at different targeted degrees of polymerization (DP). (c) SEC chromatogram of the poly(M1) macroinitiator and diblock copolymer upon the chain-extension with M5. (d) Degradation of poly(M1) in neutral, basic, and acidic aqueous media. [59], Copyright 2019. Reproduced with permission from John Wiley and Sons Inc.
Very recently, the same group developed CEMP of enyne monomers and cyclic enol ether, yielding degradable alternating copolymers with > 90% alternating dyads [61]. While ruthenium Fischer carbenes are typically considered inert in metathesis processes, the researchers verified that the facile addition of alkoxy-substituted ruthenium alkylidenes to the highly reactive terminal alkynes enabled the rapid copolymerization of cyclic enol ether and enyne monomers in a controlled chain-growth polymerization profile. The molecular weights of the resulting poly(vinyl ether) increased linearly as a function of monomer-to-catalyst ratio. Additionally, block copolymer synthesis was achieved, demonstrating the high fidelity of the ruthenium carbene chain end, and the livingness of the polymerization. Rapid degradation of the copolymer was observed in dichloromethane/water/TFA (1/1/1, V/V/V) mixtures, revealing that complete hydrolysis was reached within 30 min. Nevertheless, the alternating copolymer showed good stability in the solid state or in neutral water without significant change in molecular weight over the course of the experiments (up to 1 week).
Given the depletion of fossil fuels and environmental issues arising from non-degradable petroleum-based plastics, biomass-derived polymers, which are biocompatible and biodegradable, hold tremendous promise for next-generation plastics [123]. In 2019, Choi and coworkers demonstrated the first example in CEMP of sugar-derived enyne monomers in which the six-membered sugar ring can be opened and incorporated into the polymer backbone [60]. Polymerizations of these sugar-derived monomers were well-controlled, as proven by the high molecular weights of the obtained polymers (up to 135 kDa) with generally narrow molecular weight distributions (1.04−1.44). Polymerization kinetics stud ies suggested that both the bulky 2,4,6-triisopropylbenzenesulfonyl substituent and monomer stereochemistry played critical roles in the polymerization. The living nature of the polymerization was further corroborated by the successful production of a block copolymer from two different sugar-derived monomers. Moreover, degradation experiments of the resulting polymers were conducted in acetone-d6/D2O (9/1 v/v) mixtures with varying HCl concentrations, showing a pH-dependent hydrolysis into small molecule pyrroles.
3. Applications
The most significant advantage of olefin metathesis polymerization over other polymerization approaches is the facile functionalization and diversification of the polymeric backbone. The mild reaction conditions and excellent functional group tolerance enable direct graft-through polymerization of monomers bearing various functional groups (e.g. peptides, oligonucleotides, saccharides) [124]. A number of non-degradable, norbornene-based functional polymers have been fabricated in this way for biomedicine, lithography and electronic purposes [42, 125–127]. In the case of degradable polyolefins, most of the research effort remains illustrative, focusing on optimizing polymerization condition for more controlled and living-like polymerization. Nevertheless, recent studies on degradable polyolefins have begun to enter a new phase in proof-of-concept applications, emphasizing the potential of these materials as biodegradable drug delivery vehicles and recyclable thermoplastics [12, 28, 101].
In Kiessling’s original work, a water soluble glycopolymer was achieved through ROMP of a protected mannose loaded oxazinone followed by hydrolysis (Fig. 18 a). In the follow-up work, Kiessling reported further backbone diversification of their polyoxazinones via post polymerization modifications (PPM) and provided primary results on their biological activities. Through the use of a ketone bearing polyoxazinone, the researchers successfully appended different functional moieties including fluorophores, reporter groups and bioactive epitopes to the polymer backbone via oxime conjugation (Fig. 18 b) [101]. By altering the feed ratio of applied alkoxy-lamines, a series of copolymers with different degrees of ligand content (immunogenic epitope 2,4-dinitrophenol (DNP, R’ 1), OEG (R’ 2), water-soluble fluorophore AlexaFluor488 (AF488, R’ 3) and mannose) were prepared. By treating A20HL B cells with these copolymers that expressed a DNP-binding receptor (BCR), the researchers confirmed that the polyoxazinone backbone itself cannot engage in biological interactions, but can be employed as an antigen to activate immune signaling when DNP is attached (Fig. 18 c and d). Similarly, copolymers that contained the oligoguanidinium (R’ 4) artificial transduction domain (ATD) were capable of being internalized by two types of B cells (A20 and A20HL) through active cell penetration (Fig. 18 e). This preliminary study demonstrated the potential of polyoxazinone as a degradable and cell permeable biological probe. It’s worthwhile to note that the researchers also attempted PPM using copper mediated azide-alkyne cycloaddition (CuAAC) and strain promoted cycloaddition. However, due to the high density of heteroatom that can potentially chelate to the metallocenter and suppress the click reaction, the researchers observed the formation of insoluble gel during CuAAC reaction and broadening of MW distribution in the strain promoted click reaction. Attempts to construct the oxazinone bearing succinimidyl ester also failed due to monomer decomposition, revealing the nonnegligible challenges to diversifying the degradable polymers while keeping the backbone structure intact.
Fig. 18.
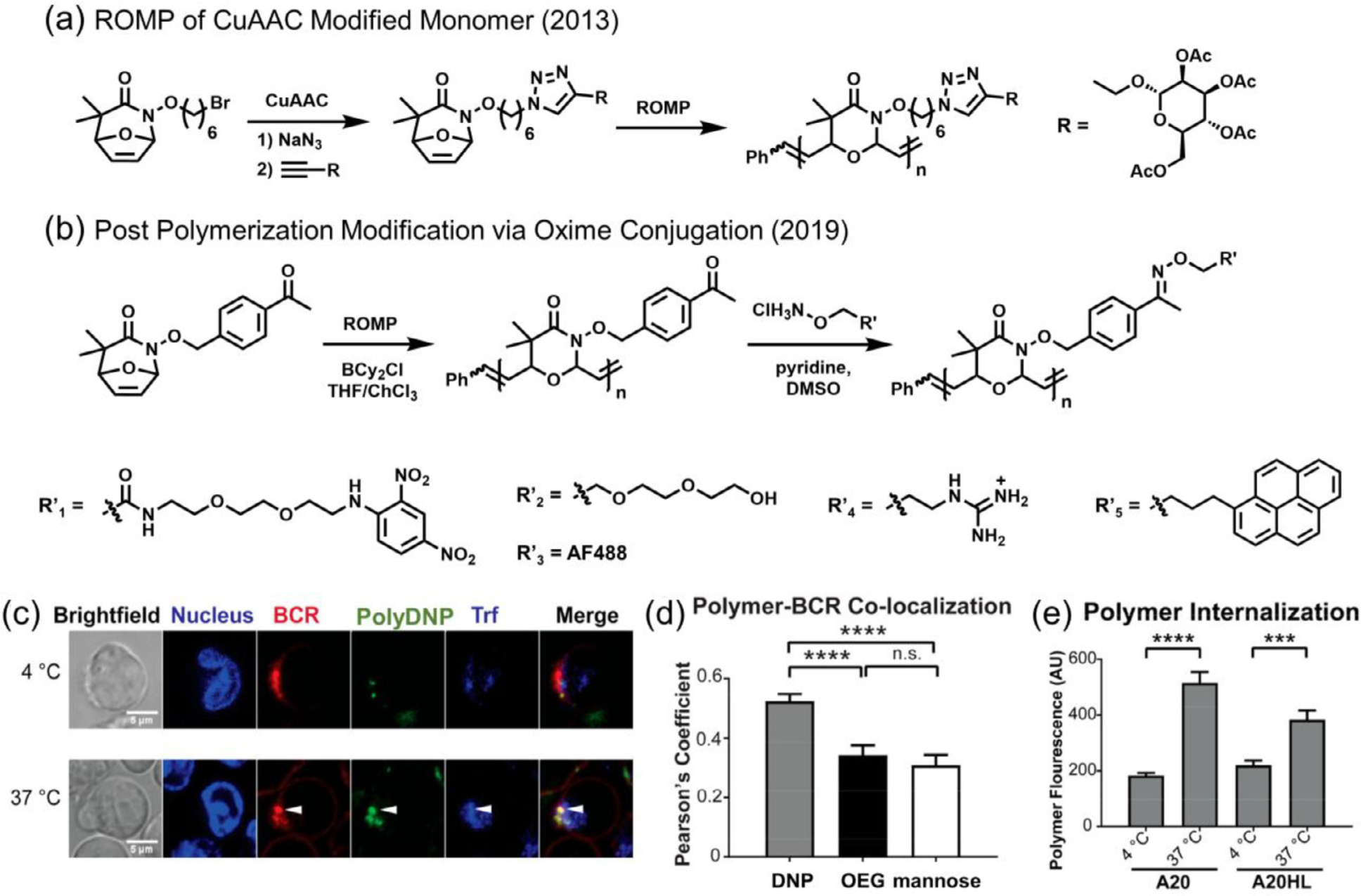
Functionalization of polyoxazinones and cellular take up studies. (a) Synthesis of protected glycopolymer. (b) Synthesis of polyoxazinone followed by functionalization via oxime conjugation. (c) Fluorescence microscopy images of DNP containing polyoxazinone (polyDNP) trafficking at 4 and 37 °C, confirming the BCR-mediated endocytosis. Markers include Hoechst (nuclear staining), anti-BCR antibody (red) and transferrin (blue) as an early endosomal marker. Arrow indicates colocalization. (d) Quantification of polymer colocalization with BCR at 37 °C. (e) Internalization of ATD containing polyoxazinone by two types of B cells at 4 and 37 °C, confirming active cell penetration. [101], Copyright 2019. Adapted with permission from the American Chemical Society.
Johnson and coworkers also evaluated the biological performance of their silyl ether-based bottlebrush copolymers both in vitro and in vivo (Fig. 19 a) [28]. In the cell studies, all copolymers showed minimal cytotoxicity at 0.75 mg/ml concentration. In the animal study, 5 mg/ml polymer in 5% aqueous glucose was administered to female BALB/c mice through tail vein injection. Cy5.5 fluorescence in the blood and organs was evaluated at different time points to determine the pharmacokinetics of the injected polymers. Although no significant difference was observed for the short-term pharmacokinetics, the installation of the degradable silyl ether facilitated the long-term clearance (6 to 10 weeks) of the copolymer in the blood, liver and spleen as compared to the non-degradable control (Fig. 19 b and c). Since bioaccumulation/ deposition can lead to immune response and toxicity, the silyl ether system can be potentially used as a safer drug delivery platform. Depending on the nature of the drug applied, it can be either encapsulated or introduced as a norbornene comonomer.
Fig. 19.

Animal studies on fluorescently labelled bottlebrush (co)polymer. (a) Synthesis of fluorescently labeled block (co)polymers. (b) Pharmacokinetics of the polymers in blood samples after three and six weeks determined by fluorescence analysis. (c) Biodistribution of polymers in the organ. [28], Copyright 2019. Adapted with permission from Springer Nature.
Not limited to biomedical applications, the researchers further demonstrated that the silyl ether monomer can be employed as an additive to fabricate reprocessible thermosets (Fig. 20) [12]. The researchers have shown that the placement of silyl ether linkages (7.5~15%) within the strands of polydicyclopentadienes (pDCPD), a thermoset that has been mainly used for the construction of automotive components, did not harm the mechanical properties of the material, but allowed it to degrade into soluble product under tetrabutylammonium fluoride (TBAF) or hydrofluoric acid (HF) treatment (Fig. 20 a–c). The degradation product of 10% iPrSi-doped material can be repolymerized with DCPD in the presence of Grubbs catalyst G2 to afford a recycled pDCPD with comparable stress–strain behavior and elastic moduli to the native material (Fig. 20 d).
Fig. 20.

Fabrication, degradation and reprocessing of silyl ether doped polydicyclopentadiene (pDCPD). (a) Schematic of the synthesis of main chain degradable silyl ether doped pDCPD. (b) iPrSi-doped pDCPD samples showed iPrSi volume-fraction-dependent dissolution in a THF solution of TBAF. (c) Young’s modulus of iPrSi-doped pDCPD samples. X axis represents the percent iPrSi in the pDCPD strand. (d) Dried pDCPD fragments from degradation of 10% iPrSi-doped material. The fragments contained cyclopentene functional groups and exchangeable alkene-containing crosslinks available for further crosslinking. [12], Copyright 2020. Reproduced with permission from Springer Nature.
This proof-of-concept work opens up new possibilities to achieve degradable and recyclable materials and hold the potential for industrial scale manufacturing. We can expect that through the use of different comonomers and altering the substitutes on the silyl ether, materials with tunable mechanical properties and degradation kinetics can be achieved.
In addition to these systems, several other platforms that have been highlighted in the above sections hold the potential for applied functions. For example, polyphosphoramidates are a potentially biocompatible material as they completely degrade into small molecule phenyl phosphoric acid and 1,4-diamino-2-butene, which is an unsaturated analog of biogenic amines involved in cell growth and differentiation [128]. Its copolymer with OEG bearing norbornene self-assembled into sub-150 nm micellar nanoparticles and did not show discernable cytotoxicity at high concentrations (150 μg/ml), enabling it to be employed as a relatively safe drug delivery vehicle [22]. DHF can be efficiently polymerized into high molecular weight and thermally stable (up to 320 °C) PDHF, which can be chemically recycled back to pure monomer, implying its potential as a reprocessible material [30]. Polylevoglucosenol, which is fabricated from a renewable feedstock, can be potentially utilized as a thermoplastic due to the rearrangement of hydrogen bonding at high temperatures (Tg ~ 100 °C, thermal table up to 220 °C) [102].
4. Conclusion and future perspectives
This review focuses on the scope of olefin metathesis strategies available for the preparation of backbone-degradable polymers. These methods have proven particularly versatile in synthesizing degradable polymers because of the excellent tolerance of functional groups in the olefin metathesis process. A library of degradable functionalities has been incorporated into the polymer backbone, including but not limited to pH-sensitive linkages (e.g, ester, acetal, oxazinone, silyl ether, enol ether, phosphoester, phosphoramidate), redox-responsive moieties (i.e., disulfide), and enzymatically cleavage bonds (e.g., azobenzene, amide). The diverse structures obtained by olefin metathesis polymerization methodology are unprecedented, and are inaccessible by other polymerization methods, opening up the possibilities for various applications.
Nevertheless, several challenges still remain in the synthesis and application of these materials. ADMET represents a type of step-growth polymerization which provides limited control over the molecular weights of polymer products. Consequently, degradable polymers produced by ADMET are typically characterized by relatively low molecular weights and broad molecular weight distributions. Improved control has been achieved in entropy- and enthalpy-driven ROMP systems, giving rise to degradable polymers with high molecular weights which are tunable by varying the monomer-to-catalyst ratio. However, most examples of degradable polymers made by ROMP rely on low and moderate strain cyclic alkene monomers (for enthalpy-driven ROMP), and unstrained macrocycles (for entropy-driven ROMP). This results in a thermodynamic equilibrium, where the polymerization rate equals the depolymerization rate, and secondary metathesis pre-dominates. Therefore, the molecular weight dispersity of degradable polymers remains high in ROMP of most monomers except for bicyclic oxazinone and PTDO. To mitigate the low-strain issue of cyclic alkene monomers, enthalpy-driven ROMP can be carried out at high monomer concentration, low temperature, and in the presence of highly strained comonomers such as norbornene. Most recently, the livingness of CEMP has been shown to outperform the other three olefin metathesis polymerization strategies, as evidenced by the tunable molecular weights, low dispersities, as well as the ability to conduct chain extension of the resulting degradable polymers. However, the monomer scope for CEMP is significantly narrower than that for other olefin metathesis polymerizations.
In addition, it is also noteworthy that all of the examples of degradable polymers outlined in this review utilized ruthenium-based metal catalyst, which bestow a ruthenium carbene on each polymer chain during the polymerization. Multiple purification approaches including extraction, absorption, and converting the catalyst into water-soluble species have been developed to remove the ruthenium. In some cases, these efforts can lead to trace amounts of metal residue (less than 0.1 μg/5 mg crude in olefin metathesis reaction) [129–132]. However, complete removal of these heavy metal initiators could prove difficult leading to residual heavy metals in the final polymer products, potentially hindering biological applications and clinical translation of these materials. There is a significant need to develop new olefin metathesis methods to prepare degradable polymers in the absence of metal initiators. Recently, Boydston et al. demonstrated a breakthrough in metal-free ROMP of 5-norbornene and endo-dicyclopentadiene using organophotoredox catalysts [133]. Moreover, Kilbinger and coworkers developed catalytic living ROMP of norbornene monomers, requiring the use of only catalytic amounts of Grubbs catalysts (< 9 ppm) [134]. While the polymer scope of these new ROMP methods is limited to non-degradable polymers, we expect that continuing efforts to develop and optimize protocols for metal-free ROMP and catalytic living ROMP will eventually facilitate the synthesis of degradable polymers.
Furthermore, the stability or shelf-life of degradable polyolefins poses another concern in regard to real world applications. With the backbone rich in degradable linkages, the polymers face risks for unintended degradation under ambient conditions, such as exposure to heat and water, during handling, storage and use. Recently, both Craig and Wang reported the use of mechanically gated degradable polymers to address this stability issue.[99, 104] In their work, a cyclobutane mechanophore is installed as a fused ring in the polymerizable macrocycle containing the degradable linkage. The mechanophore “locked” the polymer degradability and only allowed acid triggered hydrolysis when it is mechanically “unlocked” via sonication. If scalable, this strategy which involves multiple triggers may offer a new approach to fabricate degradable polymers with improved stability.
Given the strong motivation for more, and diversified degradable polymers in the field of biomaterials, environmental protection, and recyclable materials, we believe that olefin metathesis polymerization will expand the scope and open the door to new classes of degradable and recyclable polymeric materials with unique properties and new applications.
Acknowledgement
The authors are grateful for the support of the NIH through the NHLBI (R01HL139001), and the U.S. Army Research Office through the MURI program (W911NF-15-1-0568).
Abbreviations:
- ADMET
acyclic diene metathesis polymerization
- AF488
AlexaFluor488
- AROMP
alternating ring-opening metathesis polymerization
- AROP
anionic ring-opening polymerization
- ATD
artificial transduction domain
- AXL
acetal based cross linker
- BASP
brush-arm star polymer
- BCR
B cell receptor
- CBE
bicyclo[4.2.0]oct-1(8)-ene-8-carboxamide
- CEMP
cascade enyne metathesis polymerization
- COD
cyclooctadiene
- CPE
cyclopropene
- CuAAC
copper mediated azide-alkyne cycloaddition
- Cy5.5
cyanine 5.5
- Cy5.5-MM
macromonomer carrying cyanine 5.5
- Ð
dispersity
- DCM
dichloromethane
- DCPD
dicyclopentadiene
- DHF
2,3-dihydrofuran
- DMSO
dimethyl sulfoxide.
Footnotes
Declaration of Competing Interest
The authors declare there is no conflict of interest regarding publication of this review article.
CRediT authorship contribution statement
Hao Sun: Conceptualization, Formal analysis, Methodology, Writing – original draft, Writing – review & editing. Yifei Liang: Formal analysis, Methodology, Writing – original draft, Writing – review & editing. Matthew P. Thompson: Writing – review & editing. Nathan C. Gianneschi: Conceptualization, Supervision, Writing – review & editing.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi: 10.1016/j.progpolymsci.2021.101427.
References
- [1].Sun H, Kabb CP, Sims MB, Sumerlin BS. Architecture-transformable polymers: reshaping the future of stimuli-responsive polymers. Prog Polym Sci 2019;89:61–75. [Google Scholar]
- [2].Abd-El-Aziz AS, Antonietti M, Barner-Kowollik C, Binder WH, Boker A, Boyer C, et al. The next 100 years of polymer science. Macromol Chem Phys 2020;221:2000216. [Google Scholar]
- [3].Corbari L, Maltese A, Capodici F, Mangano MC, Sara G, Ciraolo G. Indoor spectroradiometric characterization of plastic litters commonly polluting the Mediterranean Sea: toward the application of multispectral imagery. Sci Rep 2020;10:19850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Stuart MAC, Huck WTS, Genzer J, Muller M, Ober C, Stamm M, et al. Emerging applications of stimuli-responsive polymer materials. Nat Mater 2010;9:101–13. [DOI] [PubMed] [Google Scholar]
- [5].Zhu JD, Zhu P, Yan CY, Dong X, Zhang XW. Recent progress in polymer materials for advanced lithium-sulfur batteries. Prog Polym Sci 2019;90:118–63. [Google Scholar]
- [6].Bian LY, Zhu EW, Tang J, Tang WH, Zhang FJ. Recent progress in the design of narrow bandgap conjugated polymers for high-efficiency organic solar cells. Prog Polym Sci 2012;37:1292–331. [Google Scholar]
- [7].Sun H, Cao W, Zang N, Clemons TD, Scheutz GM, Hu Z, et al. Proapoptotic peptide brush polymer nanoparticles via photoinitiated polymerization-induced self-assembly. Angew Chem Int Ed 2020;59:19136–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sun H, Yang L, Thompson MP, Schara S, Cao W, Choi W, et al. Recent advances in amphiphilic polymer-oligonucleotide nanomaterials via living/controlled polymerization technologies. Bioconjug Chem 2019;30:1889–904. [DOI] [PubMed] [Google Scholar]
- [9].Delplace V, Nicolas J. Degradable vinyl polymers for biomedical applications. Nat Chem 2015;7:771–84. [DOI] [PubMed] [Google Scholar]
- [10].Hong M, Chen EYX. Chemically recyclable polymers: a circular economy approach to sustainability. Green Chem 2017;19:3692–706. [Google Scholar]
- [11].Fairbanks BD, Gunatillake PA, Meagher L. Biomedical applications of polymers derived by reversible addition - fragmentation chain-transfer (RAFT). Adv Drug Deliver Rev 2015;91:141–52. [DOI] [PubMed] [Google Scholar]
- [12].Shieh P, Zhang W, Husted KEL, Kristufek SL, Xiong B, Lundberg DJ, et al. Cleavable comonomers enable degradable, recyclable thermoset plastics. Nature 2020;583:542–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Snyder RL, Fortman DJ, De Hoe GX, Hillmyer MA, Dichtel WR. Reprocessable acid-degradable polycarbonate vitrimers. Macromolecules 2018;51:389–97. [Google Scholar]
- [14].Tran H, Feig VR, Liu K, Wu HC, Chen R, Xu J, et al. Stretchable and fully degradable semiconductors for transient electronics. ACS Cent Sci 2019;5:1884–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gross RA, Kalra B. Biodegradable polymers for the environment. Science 2002;297:803–7. [DOI] [PubMed] [Google Scholar]
- [16].Lober A, Verch A, Schlemmer B, Hofer S, Frerich B, Buchmeiser MR. Monolithic polymers for cell cultivation, differentiation, and tissue engineering. Angew Chem Int Ed 2008;47:9138–41. [DOI] [PubMed] [Google Scholar]
- [17].Buchmeiser MR. Monolithic biocompatible and biodegradable scaffolds for tissue engineering. J Polym Sci Polym Chem 2009;47:2219–27. [Google Scholar]
- [18].Farah S, Anderson DG, Langer R. Physical and mechanical properties of PLA, and their functions in widespread applications - a comprehensive review. Adv Drug Deliver Rev 2016;107:367–92. [DOI] [PubMed] [Google Scholar]
- [19].Dove AP, Meier MAR. Step-growth polymerization in the 21st century. Macromol Chem Phys 2014;215:2135–7. [Google Scholar]
- [20].Becker G, Wurm FR. Functional biodegradable polymers via ring-opening polymerization of monomers without protective groups. Chem Soc Rev 2018;47:7739–82. [DOI] [PubMed] [Google Scholar]
- [21].Tardy A, Nicolas J, Gigmes D, Lefay C, Guillaneuf Y. Radical ring-opening polymerization: scope, limitations, and application to (bio)degradable materials. Chem Rev 2017;117:1319–406. [DOI] [PubMed] [Google Scholar]
- [22].Liang Y, Sun H, Thompson MP, Gianneschi NC. Degradable polyphosphoramidate via ring-opening metathesis polymerization. ACS Macro Lett 2020;9:1417–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fu CK, Xu JT, Boyer C. Photoacid-mediated ring opening polymerization driven by visible light. Chem Commun 2016;52:7126–9. [DOI] [PubMed] [Google Scholar]
- [24].Zhao W, Lv YF, Li J, Feng ZH, Ni YH, Hadjichristidis N. Fast and selective organocatalytic ring-opening polymerization by fluorinated alcohol without a cocatalyst. Nat Commun 2019;10:3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dong P, Sun H, Quan DP. Synthesis of poly(L-lactide-co-5-amino-5-methyl-1, 3-dioxan-2-ones) [P(L-LA-co-TAc)] containing amino groups via organocatalysis and post-polymerization functionalization. Polymer 2016;97: 614–622. [Google Scholar]
- [26].Grazon C, Salas-Ambrosio P, Ibarboure E, Buol A, Garanger E, Grinstaff MW, et al. Aqueous ring-opening polymerization-induced self-assembly (ROPISA) of N-carboxyanhydrides. Angew Chem Int Ed 2020;59:622–6. [DOI] [PubMed] [Google Scholar]
- [27].Dechy-Cabaret O, Martin-Vaca B, Bourissou D. Controlled ring-opening polymerization of lactide and glycolide. Chem Rev 2004;104:6147–76. [DOI] [PubMed] [Google Scholar]
- [28].Shieh P, Nguyen HVT, Johnson JA. Tailored silyl ether monomers enable backbone-degradable polynorbornene-based linear, bottlebrush and star copolymers through ROMP. Nat Chem 2019;11:1124–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Elling BR, Su JK, Xia Y. Degradable polyacetals/ketals from alternating ring-opening metathesis polymerization. ACS Macro Lett 2020;9:180–4. [DOI] [PubMed] [Google Scholar]
- [30].Feist JD, Xia Y. Enol ethers are effective monomers for ring-opening metathesis polymerization: synthesis of degradable and depolymerizable Poly(2,3-dihydrofuran). J Am Chem Soc 2020;142:1186–9. [DOI] [PubMed] [Google Scholar]
- [31].Steinbach T, Alexandrino EM, Wurm FR. Unsaturated poly(phosphoester)s via ring-opening metathesis polymerization. Polym Chem 2013;4:3800–6. [Google Scholar]
- [32].Perrier S 50th Anniversary perspective: RAFT polymerization-a user guide. Macromolecules 2017;50:7433–47. [Google Scholar]
- [33].Guegain E, Zhu C, Giovanardi E, Nicolas J. Radical ring-opening copolymerization-induced self-assembly (rROPISA). Macromolecules 2019;52:3612–24. [Google Scholar]
- [34].Smith RA, Fu GY, McAteer O, Xu MZ, Gutekunst WR. Radical approach to thioester-containing polymers. J Am Chem Soc 2019;141:1446–51. [DOI] [PubMed] [Google Scholar]
- [35].Bingham NM, Roth PJ. Degradable vinyl copolymers through thiocarbonyl addition-ring-opening (TARO) polymerization. Chem Commun 2019;55:55–8. [DOI] [PubMed] [Google Scholar]
- [36].Pesenti T, Nicolas J. 100th Anniversary of macromolecular science viewpoint: degradable polymers from radical ring-opening polymerization: latest advances, new directions, and ongoing challenges. ACS Macro Lett 2020;9:1812–35. [DOI] [PubMed] [Google Scholar]
- [37].Hill MR, Kubo T, Goodrich SL, Figg CA, Sumerlin BS. Alternating radical ring-opening polymerization of cyclic ketene acetals: access to tunable and functional polyester copolymers. Macromolecules 2018;51:5079–84. [Google Scholar]
- [38].Ogba OM, Warner NC, O’Leary DJ, Grubbs RH. Recent advances in ruthenium-based olefin metathesis. Chem Soc Rev 2018;47:4510–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hoveyda AH, Zhugralin AR. The remarkable metal-catalysed olefin metathesis reaction. Nature 2007;450:243–51. [DOI] [PubMed] [Google Scholar]
- [40].Blum AP, Kammeyer JK, Gianneschi NC. Activating peptides for cellular uptake via polymerization into high density brushes. Chem Sci 2016;7:989–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wright DB, Touve MA, Thompson MP, Gianneschi NC. Aqueous-phase ring-opening metathesis polymerization-induced self-assembly. ACS Macro Lett 2018;7:401–5. [DOI] [PubMed] [Google Scholar]
- [42].Church DC, Pokorski JK. Cell engineering with functional poly(oxanorbornene) block copolymers. Angew Chem Int Ed 2020;59:11379–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Moatsou D, Nagarkar A, Kilbinger AFM, O’Reilly RK. Degradable precision polynorbornenes via ring-opening metathesis polymerization. J Polym Sci Polym Chem 2016;54:1236–42. [Google Scholar]
- [44].Bielawski CW, Grubbs RH. Living ring-opening metathesis polymerization. Prog Polym Sci 2007;32:1–29. [Google Scholar]
- [45].Chen ZX, Mercer JAM, Zhu XL, Romaniuk JAH, Pfattner R, Cegelski L, et al. Mechanochemical unzipping of insulating polyladderene to semicon-ducting polyacetylene. Science 2017;357:475–8. [DOI] [PubMed] [Google Scholar]
- [46].Elacqua E, Gregor M. Poly(arylenevinylene)s through ring-opening metathesis polymerization of an unsymmetrical donor-acceptor cyclophane. Angew Chem Int Ed 2019;58:9527–32. [DOI] [PubMed] [Google Scholar]
- [47].Tan XY, Lu H, Sun YH, Chen XY, Wang DL, Jia F, et al. Expanding the materials space of DNA via organic-phase ring-opening metathesis polymerization. Chem 2019;5:1584–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Varlas S, Lawrenson SB, Arkinstall LA, O’Reilly RK, Foster JC. Self-assembled nanostructures from amphiphilic block copolymers prepared via ring-opening metathesis polymerization (ROMP). Prog Polym Sci 2020;107:101278. [Google Scholar]
- [49].Wolfe PS, Wagener KB. An ADMET route to unsaturated polyacetals. Macromol Rapid Commun 1998;19:305–8. [Google Scholar]
- [50].Fishman JM, Kiessling LL. Synthesis of functionalizable and degradable polymers by ring-opening metathesis polymerization. Angew Chem Int Ed 2013;52:5061–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Shearouse WC, Lillie LM, Reineke TM, Tolman WB. Sustainable polyesters derived from glucose and castor oil: building block structure impacts properties. ACS Macro Lett 2015;4:284–8. [DOI] [PubMed] [Google Scholar]
- [52].Tee HT, Lieberwirth I, Wurm FR. Aliphatic long-chain polypyrophosphates as biodegradable polyethylene mimics. Macromolecules 2019;52:1166–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lv A, Cui Y, Du FS, Li ZC. Thermally degradable polyesters with tunable degradation temperatures via postpolymerization modification and intramolecular cyclization. Macromolecules 2016;49:8449–58. [Google Scholar]
- [54].Mutlu H, Barner-Kowollik C. Green chain-shattering polymers based on a self--immolative azobenzene motif. Polym Chem 2016;7:2272–9. [Google Scholar]
- [55].Fraser C, Hillmyer MA, Gutierrez E, Grubbs RH. Degradable cyclooctadiene acetal copolymers - versatile precursors to 1,4-hydroxytelechelic polybutadiene and hydroxytelechelic polyethylene. Macromolecules 1995;28:7256–61. [Google Scholar]
- [56].Chang CC, Emrick T. Functional polyolefins containing disulfide and phosphoester groups: synthesis and orthogonal degradation. Macromolecules 2014;47:1344–50. [Google Scholar]
- [57].Mallick A, Xu Y, Lin YC, He JX, Chan-Park MB, Liu XW. Oxadiazabicy-clooctenone as a versatile monomer for the construction of pH sensitive functional polymers via ROMP. Polym Chem 2018;9:372–7. [Google Scholar]
- [58].Gutekunst WR, Hawker CJ. A general approach to sequence-controlled polymers using macrocyclic ring opening metathesis polymerization. J Am Chem Soc 2015;137:8038–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Fu LB, Sui XL, Crolais AE, Gutekunst WR. Modular approach to degradable acetal polymers using cascade enyne metathesis polymerization. Angew Chem Int Ed 2019;58:15726–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Bhaumik A, Peterson GI, Kang C, Choi TL. Controlled living cascade polymerization to make fully degradable sugar-based polymers from D-glucose and D-galactose. J Am Chem Soc 2019;141:12207–11. [DOI] [PubMed] [Google Scholar]
- [61].Sui X, Zhang T, Pabarue AB, Fu L, Gutekunst WR. Alternating cascade metathesis polymerization of enynes and cyclic enol ethers with active ruthenium fischer carbenes. J Am Chem Soc 2020;142:12942–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Schrock RR, Feldman J, Cannizzo LF, Grubbs RH. Ring-opening polymerization of norbornene by a living tungsten alkylidene complex. Macromolecules 1987;20:1169–72. [Google Scholar]
- [63].ODonoghue MB, Schrock RR, LaPointe AM, Davis WM. Preparation of well-defined, metathetically active oxo alkylidene complexes of tungsten. Organometallics 1996;15:1334–6. [Google Scholar]
- [64].Schrock RR, Murdzek JS, Bazan GC, Robbins J, Dimare M, Oregan M. Synthesis of molybdenum imido alkylidene complexes and some reactions involving acyclic olefins. J Am Chem Soc 1990;112:3875–86. [Google Scholar]
- [65].Schwab P, France MB, Ziller JW, Grubbs RH. A series of well-defined metathesis catalysts - synthesis of [RuCl2(=Chr’)(Pr(3))(2)] and its reactions. Angew Chem Int Ed 1995;34:2039–41. [Google Scholar]
- [66].Schwab P, Grubbs RH, Ziller JW. Synthesis and applications of RuCl2(=CHR’)(PR(3))(2): the influence of the alkylidene moiety on metathesis activity. J Am Chem Soc 1996;118:100–10. [Google Scholar]
- [67].Scholl M, Ding S, Lee CW, Grubbs RH. Synthesis and activity of a new generation of ruthenium-based olefin metathesis catalysts coordinated with 1,3-dimesityl-4,5-dihydroimidazol-2-ylidene ligands. Org Lett 1999;1:953–6 [DOI] [PubMed] [Google Scholar]
- [68].Love JA, Morgan JP, Trnka TM, Grubbs RH. A practical and highly active ruthenium-based catalyst that effects the cross metathesis of acrylonitrile. Angew Chem Int Ed 2002;41:4035–7. [DOI] [PubMed] [Google Scholar]
- [69].Kingsbury JS, Harrity JPA, Bonitatebus PJ, Hoveyda AH. A recyclable Ru-based metathesis catalyst. J Am Chem Soc 1999;121:791–9. [Google Scholar]
- [70].Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. Efficient and recyclable monomeric and dendritic Ru-based metathesis catalysts. J Am Chem Soc 2000;122:8168–79. [Google Scholar]
- [71].Atallah P, Wagener KB, Schulz MD. ADMET: the future revealed. Macromolecules 2013;46:4735–41. [Google Scholar]
- [72].Schulz MD, Wagener KB. Precision polymers through ADMET polymerization. Macromol Chem Phys 2014;215:1936–45. [Google Scholar]
- [73].Hopkins TE, Pawlow JH, Koren DL, Deters KS, Solivan SM, Davis JA, et al. Chiral polyolefins bearing amino acids. Macromolecules 2001;34:7920–2. [Google Scholar]
- [74].Turunc O, Meier MAR. Thiol-ene vs. ADMET: a complementary approach to fatty acid-based biodegradable polymers. Green Chem 2011;13:314–20. [Google Scholar]
- [75].Parkhurst RR, Balog S, Weder C, Simon YC. Synthesis of poly(sulfonate ester)s by ADMET polymerization. RSC Adv 2014;4:53967–74. [Google Scholar]
- [76].Steinbach T, Alexandrino EM, Wahlen C, Landfester K, Wurm FR. Poly(phosphonate)s via olefin metathesis: adjusting hydrophobicity and morphology. Macromolecules 2014;47:4884–93. [Google Scholar]
- [77].Vert M Aliphatic polyesters: great degradable polymers that cannot do everything. Biomacromolecules 2005;6:538–46. [DOI] [PubMed] [Google Scholar]
- [78].Cameron DJA, Shaver MP. Aliphatic polyester polymer stars: synthesis, properties and applications in biomedicine and nanotechnology. Chem Soc Rev 2011;40:1761–76. [DOI] [PubMed] [Google Scholar]
- [79].Sun H, Choi W, Zang N, Battistella C, Thompson MP, Cao W, et al. Bioactive peptide brush polymers via photoinduced reversible-deactivation radical polymerization. Angew Chem Int Ed 2019;58:17359–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Zhu JL, Sun H, Callmann CE, Thompson MP, Battistella C, Proetto MT, et al. Paclitaxel-terminated peptide brush polymers. Chem Commun 2020;56:6778–81. [DOI] [PubMed] [Google Scholar]
- [81].Zhou XF, Li ZB. Advances and biomedical applications of polypeptide hydrogels derived from alpha-amino Acid N-carboxyanhydride (NCA) polymerizations. Adv Healthc Mater 2018;7:1800020. [DOI] [PubMed] [Google Scholar]
- [82].Song ZY, Fu HL, Wang J, Hui JS, Xue TR, Pacheco LA, et al. Synthesis of polypeptides via bioinspired polymerization of in situ purified N-carboxyanhydrides. Proc Natl Acad Sci USA 2019;116:10658–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Fan JW, Borguet YP, Su L, Nguyen TP, Wang H, He X, et al. Two-dimensional controlled syntheses of polypeptide molecular brushes via N-carboxyanhydride ring-opening polymerization and ring-opening metathesis polymerization. ACS Macro Lett 2017;6:1031–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Ulery BD, Nair LS, Laurencin CT. Biomedical applications of biodegradable polymers. J Polym Sci Polym Phys 2011;49:832–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Liu B, Thayumanavan S. Substituent effects on the pH sensitivity of acetals and ketals and their correlation with encapsulation stability in polymeric nanogels. J Am Chem Soc 2017;139:2306–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Khaja SD, Lee S, Murthy N. Acid-degradable protein delivery vehicles based on metathesis chemistry. Biomacromolecules 2007;8:1391–5. [DOI] [PubMed] [Google Scholar]
- [87].Jessen HJ, Ahmed N, Hofer A. Phosphate esters and anhydrides - recent strategies targeting nature’s favoured modifications. Org Biomol Chem 2014;12:3526–30. [DOI] [PubMed] [Google Scholar]
- [88].Elzeny H, Zhang FW, Ali EN, Fathi HA, Zhang SY, Li RC, et al. Polyphosphoester nanoparticles as biodegradable platform for delivery of multiple drugs and siRNA. Drug Des Dev Ther 2017;11:483–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Zhang SY, Zou J, Elsabahy M, Karwa A, Li A, Moore DA, et al. Poly(ethylene oxide)-block-polyphosphester-based paclitaxel conjugates as a platform for ultra-high paclitaxel-loaded multifunctional nanoparticles. Chem Sci 2013;4:2122–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Steinbach T, Wurm FR. Poly(phosphoester)s: a new platform for degradable polymers. Angew Chem Int Edit 2015;54:6098–108. [DOI] [PubMed] [Google Scholar]
- [91].Pearce AK, Foster JC, O’Reilly RK. Recent developments in entropy–driven ring-opening metathesis polymerization: mechanistic considerations, unique functionality, and sequence control. J Polym Sci Polym Chem 2019;57:1621–34. [Google Scholar]
- [92].Xue Z, Mayer MF. Entropy-driven ring-opening olefin metathesis polymerizations of macrocycles. Soft Matter 2009;5:4600–11. [Google Scholar]
- [93].Monfette S, Fogg DE. Equilibrium ring-closing metathesis. Chem Rev 2009;109:3783–816. [DOI] [PubMed] [Google Scholar]
- [94].Gao W, Hagver R, Shah V, Xie WC, Gross RA, Ilker MF, et al. Glycolipid polymer synthesized from natural lactonic sophorolipids by ring-opening metathesis polymerization. Macromolecules 2007;40:145–7. [Google Scholar]
- [95].Weiss RM, Short AL, Meyer TY. Sequence-controlled copolymers prepared via entropy-driven ring-opening metathesis polymerization. ACS Macro Lett 2015;4:1039–43. [DOI] [PubMed] [Google Scholar]
- [96].Nowalk JA, Fang C, Short AL, Weiss RM, Swisher JH, Liu P, et al. Sequence-controlled polymers through entropy-driven ring-opening metathesis polymerization: theory, molecular weight control, and monomer design. J Am Chem Soc 2019;141:5741–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Behrendt FN, Schlaad H. Metathesis polymerization of cystine-based macrocycles. Polym Chem 2017;8:366–9. [Google Scholar]
- [98].Behrendt FN, Hess A, Lehmann M, Schmidt B, Schlaad H. Polymerization of cystine-derived monomers. Polym Chem 2019;10:1636–41. [Google Scholar]
- [99].Lin YJ, Kouznetsova TB, Craig SL. Mechanically Gated Degradable Polymers. J Am Chem Soc 2020;142:2105–9. [DOI] [PubMed] [Google Scholar]
- [100].Lin YJ, Kouznetsova TB, Chang CC, Craig SL. Enhanced polymer mechanical degradation through mechanochemically unveiled lactonization. Nat Commun 2020;11:4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Fishman JM, Zwick DB, Kruger AG, Chemoselective KLL. Postpolymerization modification of bioactive, degradable polymers. Biomacromolecules 2019;20:1018–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Debsharma T, Behrendt FN, Laschewsky A, Schlaad H. Ring-opening metathesis polymerization of biomass-derived levoglucosenol. Angew Chem Int Ed 2019;58:6718–21. [DOI] [PubMed] [Google Scholar]
- [103].McGuire TM, Perale C, Castaing R, Kociok-Kohn G, Buchard A. Divergent catalytic strategies for the cis/trans stereoselective ring-opening polymerization of a dual cyclic carbonate/olefin monomer. J Am Chem Soc 2019;141:13301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Hsu TG, Zhou JF, Su HW, Schrage BR, Ziegler CJ, Wang JP. A polymer with “locked” degradability: superior backbone stability and accessible degradability enabled by mechanophore installation. J Am Chem Soc 2020;142:2100–4. [DOI] [PubMed] [Google Scholar]
- [105].Boadi FO, Zhang JL, Yu XX, Bhatia SR, Sampson NS. Alternating ring-opening metathesis polymerization provides easy access to functional and fully degradable polymers. Macromolecules 2020;53:5857–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Stayshich RM, Meyer TY. New insights into poly(lactic-co-glycolic acid) microstructure: using repeating sequence copolymers to decipher complex NMR and thermal behavior. J Am Chem Soc 2010;132:10920–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Schleyer PV, Williams JE, Blanchard KR. Evaluation of strain in hydrocarbons - strain in adamantane and its origin. J Am Chem Soc 1970;92:2377–86 2377-+. [Google Scholar]
- [108].Neary WJ, Kennemur JG. Polypentenamer renaissance: challenges and opportunities. ACS Macro Lett 2019;8:46–56. [DOI] [PubMed] [Google Scholar]
- [109].Song K, Kim K, Hong D, Kim J, Heo CE, Kim HI, et al. Highly active ruthenium metathesis catalysts enabling ring-opening metathesis polymerization of cyclopentadiene at low temperatures. Nat Commun 2019;10:3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Neary WJ, Kennemur JG. Variable temperature ROMP: leveraging low ring strain thermodynamics to achieve well-defined polypentenamers. Macromolecules 2017;50:4935–41. [Google Scholar]
- [111].Jung ME, Piizzi G. gem-Disubstituent effect: theoretical basis and synthetic applications. Chem Rev 2005;105:1735–66. [DOI] [PubMed] [Google Scholar]
- [112].Hilf S, Grubbs RH, Kilbinger AFM. Sacrificial synthesis of hydroxy-functionalized ROMP polymers: an efficiency study. Macromolecules 2008;41:6006–11. [Google Scholar]
- [113].Hilf S, Berger-Nicoletti E, Grubbs RH, Kilbinger AFM. Monofunctional metathesis polymers via sacrificial diblock copolymers. Angew Chem Int Ed 2006;45:8045–8. [DOI] [PubMed] [Google Scholar]
- [114].Haider T, Shyshov O, Suraeva O, Lieberwirth I, von Delius M, Wurm FR. Long-chain polyorthoesters as degradable polyethylene mimics. Macromolecules 2019;52:2411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Elling BR, Xia Y. Living alternating ring-opening metathesis polymerization based on single monomer additions. J Am Chem Soc 2015;137:9922–6. [DOI] [PubMed] [Google Scholar]
- [116].Sanford MS, Love JA, Grubbs RH. Mechanism and activity of ruthenium olefin metathesis catalysts. J Am Chem Soc 2001;123:6543–54. [DOI] [PubMed] [Google Scholar]
- [117].Hejl A, Scherman OA, Grubbs RH. Ring-opening metathesis polymerization of functionalized low-strain monomers with ruthenium-based catalysts. Macromolecules 2005;38:7214–18. [Google Scholar]
- [118].Tuba R, Grubbs RH. Ruthenium catalyzed equilibrium ring-opening metathesis polymerization of cyclopentene. Polym Chem 2013;4:3959–62. [Google Scholar]
- [119].Neary WJ, Isais TA, Kennemur JG. Depolymerization of bottlebrush polypentenamers and their macromolecular metamorphosis. J Am Chem Soc 2019;141:14220–9. [DOI] [PubMed] [Google Scholar]
- [120].Park H, Choi TL. Fast tandem ring-opening/ring-closing metathesis polymerization from a monomer containing cyclohexene and terminal alkyne. J Am Chem Soc 2012;134:7270–3. [DOI] [PubMed] [Google Scholar]
- [121].Park H, Lee HK, Choi TL. Tandem ring-opening/ring-closing metathesis polymerization: relationship between monomer structure and reactivity. J Am Chem Soc 2013;135:10769–75. [DOI] [PubMed] [Google Scholar]
- [122].Park H, Kang EH, Muller L, Choi TL. Versatile tandem ring-opening/ring-closing metathesis polymerization: strategies for successful polymerization of challenging monomers and their mechanistic studies. J Am Chem Soc 2016;138:2244–51. [DOI] [PubMed] [Google Scholar]
- [123].Chen CC, Dai LH, Ma LX, Guo RT. Enzymatic degradation of plant biomass and synthetic polymers. Nat Rev Chem 2020;4:114–26. [DOI] [PubMed] [Google Scholar]
- [124].Choi W, Sun H, Battistella C, Berger O, Vratsanos MA, Wang MM, et al. Biomolecular densely grafted brush polymers: oligonucleotides, oligosaccharides and oligopeptides. Angew Chem Int Ed 2020;59:19762–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Blum AP, Kammeyer JK, Yin J, Crystal DT, Rush AM, Gilson MK, et al. Peptides displayed as high density brush polymers resist proteolysis and retain bioactivity. J Am Chem Soc 2014;136:15422–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Joo W, Chen CH, Moerdyk JP, Deschner RP, Bielawski CW, Willson CG. Photoinitiated ring-opening metathesis polymerization. J Polym Sci Polym Chem 2019;57:1791–5. [Google Scholar]
- [127].Tonge CM, Sauve ER, Cheng S, Howard TA, Hudson ZM. Multiblock bottlebrush nanofibers from organic electronic materials. J Am Chem Soc 2018;140:11599–603. [DOI] [PubMed] [Google Scholar]
- [128].Martin B, Posseme F, Le Barbier C, Carreaux F, Carboni B, Seiler N, et al. (Z)-1,4-diamino-2-butene as a vector of boron, fluorine, or iodine for cancer therapy and imaging: Synthesis and biological evaluation. Biorg Med Chem 2002;10:2863–71. [DOI] [PubMed] [Google Scholar]
- [129].Higman CS, Lummiss JAM, Fogg DE. Olefin metathesis at the dawn of implementation in pharmaceutical and specialty-chemicals manufacturing. Angew Chem Int Edit 2016;55:3552–65. [DOI] [PubMed] [Google Scholar]
- [130].Maynard HD, Grubbs RH. Purification technique for the removal of ruthenium from olefin metathesis reaction products. Tetrahedron Lett 1999;40:4137–40. [Google Scholar]
- [131].Cho JH, Kim BM. An efficient method for removal of ruthenium byproducts from olefin metathesis reactions. Org Lett 2003;5:531–3. [DOI] [PubMed] [Google Scholar]
- [132].Ahn YM, Yang K, Georg GI. A convenient method for the efficient removal of ruthenium byproducts generated during olefin metathesis reactions. Org Lett 2001;3:1411–13. [DOI] [PubMed] [Google Scholar]
- [133].Ogawa KA, Goetz AE, Boydston AJ. Metal-free ring-opening metathesis polymerization. J Am Chem Soc 2015;137:1400–3. [DOI] [PubMed] [Google Scholar]
- [134].Yasir M, Liu P, Tennie IK, Kilbinger AFM. Catalytic living ring-opening metathesis polymerization with Grubbs’ second- and third-generation catalysts. Nat Chem 2019;11:488–94 [DOI] [PubMed] [Google Scholar]