

Abstract
Human regulatory T cells (Tregs) are crucial regulators of tissue repair, autoimmune diseases, and cancer. However, it is challenging to inhibit the suppressive function of Tregs for cancer therapy without affecting immune homeostasis. Identifying pathways that may distinguish tumor-restricted Tregs is important, yet the transcriptional programs that control intratumoral Treg gene expression, and that are distinct from Tregs in healthy tissues, remain largely unknown. We profiled single-cell transcriptomes of CD4+ T cells in tumors and peripheral blood from patients with head and neck squamous cell carcinomas (HNSCC) and those in nontumor tonsil tissues and peripheral blood from healthy donors. We identified a subpopulation of activated Tregs expressing multiple tumor necrosis factor receptor (TNFR) genes (TNFR+ Tregs) that is highly enriched in the tumor microenvironment (TME) compared with nontumor tissue and the periphery. TNFR+ Tregs are associated with worse prognosis in HNSCC and across multiple solid tumor types. Mechanistically, the transcription factor BATF is a central component of a gene regulatory network that governs key aspects of TNFR+ Tregs. CRISPR-Cas9–mediated BATF knockout in human activated Tregs in conjunction with bulk RNA sequencing, immunophenotyping, and in vitro functional assays corroborated the central role of BATF in limiting excessive activation and promoting the survival of human activated Tregs. Last, we identified a suite of surface molecules reflective of the BATF-driven transcriptional network on intratumoral Tregs in patients with HNSCC. These findings uncover a primary transcriptional regulator of highly suppressive intratumoral Tregs, highlighting potential opportunities for therapeutic intervention in cancer without affecting immune homeostasis.
INTRODUCTION
Regulatory T cells (Tregs) are essential for immune homeostasis. The development, maintenance, and function of Tregs depend on the transcription factor forkhead box protein 3 (FOXP3) (1). Individuals who lack FOXP3 expression develop immune dysregulation, polyendocrinopathy, enteropathy, and X-linked (IPEX) syndrome, a severe autoimmune lymphoproliferative disease that is fatal without bone marrow transplantation (2). However, in the tumor microenvironment (TME), tumor-infiltrating lymphocyte Tregs (herein referred to as TILTregs) exert immune suppressive functions and subsequently inhibit immune-mediated tumor clearance, suggesting that immunotherapies should inhibit the suppressive function of TIL Tregs specifically to enhance antitumor immunity without affecting immune homeostasis (3, 4).
An elevated frequency of Tregs within a tumor is associated with an adverse prognosis across many cancers (5–9). The current methods of targeting Tregs in the clinic are direct Treg depletion, targeting costimulatory or coinhibitory receptors, halting Treg migration into the TME, and inducing Treg fragility (10–14). However, immune-related adverse effects such as pneumonitis and colitis were seen in clinical studies of Treg-targeted therapies (15–17). A major limitation underlying the autoimmune toxicity and limited efficacy observed with Treg therapies in the clinic is an inability to target TIL Tregs selectively, with unwanted side effects resulting from inhibition of Tregs in the periphery or because the function of antitumor effector T cells is also impaired (12, 18).
We recently reported on the immune landscape of head and neck squamous cell carcinoma (HNSCC) (19). We found that TIL Tregs are phenotypically heterogeneous in the HNSCC TME and that only a small subset of TIL Tregs exhibited effector-like phenotypes. Identifying genes that are specific to TIL Treg, but that are not present in peripheral Tregs nor effector T cells, is a strategy toward identifying molecules that could be potentially targeted for therapies aiming to modulate immunosuppressive Tregs in the TME specifically. FOXP3, alone or in combination with the interleukin-2 (IL-2) receptor α-chain (IL2RA/CD25), is commonly used to detect TIL Tregs in tumor samples by immunohistochemistry and flow-based studies, but these approaches may be limited by the ability to incorporate an extended number of additional markers that allow identification of functionally active Tregs in the TME (20–22) or other distinctive gene expression signatures. In this study, we leveraged single-cell RNA sequencing (scRNA-seq) technology to provide a detailed view of the heterogeneity of Tregs in the TME by profiling CD4+ T cells from tumors and peripheral blood mononuclear cells (PBMCs) taken from patients with HNSCC and comparing them with CD4+ T cells isolated from inflamed tonsil tissues from patients with tonsilitis and from non-inflamed tonsil tissues from patients with sleep apnea and with healthy donor (HD) PBMCs. We identified a subset of activated TIL Tregs that expressed multiple tumor necrosis factor receptor (TNFR) member genes, including TNFRSF4 (OX40), TNFRSF9 (4–1BB), and TNFRSF18 (GITR) (herein referred to as TNFR+ Tregs). TNFR+ Tregs were highly enriched in a variety of tumor types compared with nontumor tissues and are associated with worse prognosis across solid tumors. Using single-cell analysis approaches and graph-based modeling, we generated a comprehensive map of gene regulatory networks (GRNs) that govern TIL Treg phenotypes. We found that basic leucine zipper ATF-like transcription factor (BATF), an activator protein-1 (AP-1) superfamily transcription factor (TF), is a central component of a GRN that controls the transcriptional signature of TNFR+ Tregs. Knockout (KO) of BATF in cultured human activated Tregs with CRISPR-Cas9 in conjunction with immunophenotyping corroborated the central role of BATF in regulating activation and function of activated TIL Tregs. Bulk RNA sequencing (RNA-seq) and in vitro functional assays further interrogated the roles of BATF in human activated Tregs. The role of BATF in TNFR+ Tregs was further corroborated in human Tregs under continuous T cell receptor (TCR) stimulation and hypoxic conditions (i.e., conditions that mimic the persistent antigenic stimulation and metabolic stress in the TME). Our analyses highlighted the regulatory roles of BATF with several surface markers differentially expressed on HNSCC TIL Tregs, including 4–1BB, OX40, GITR, CD74, CD96, and CD39. These findings revealed a distinct transcriptional signature of an intratumoral Treg subpopulation that is associated with worse prognosis, identified distinct surface markers that define functionally suppressive Tregs within the TME, and provide insights into immunotherapeutic targets for the treatment of solid tumors.
RESULTS
HNSCC TIL Tregs have distinct transcriptional signatures compared with CD4+ conventional T cells and peripheral Tregs
We recently reported on the immune landscape of HNSCC (19). To identify gene signatures and molecular pathways that are specific to human TIL Tregs over peripheral Tregs and effector T cells, CD4+ conventional T cells (Tconv, CD4+CD25lowCD127high) and CD4+ Tregs (CD4+CD25highCD127low) were sorted from nontumor inflamed tonsils from patients with tonsilitis and HD PBMCs and profiled by scRNA-seq (fig. S1). We integrated these data with the raw sequencing data obtained from TIL CD4+ T cells from the previously published HNSCC scRNA-seq dataset (19) as well as the sequencing data for CD4+ T cells isolated from non-inflamed tonsils from patients with sleep apnea (Fig. 1A and fig. S2, A and B). After filtering for quality control and data integration by Seurat v4 pipeline, we were able to obtain 51,195 CD4+ T cells from 26 patients with HNSCC, 6 patients with tonsilitis, 5 patients with sleep apnea, and 10 HDs (data file S1, tab data S1) (23). After applying nonlinear dimension reduction using uniform manifold approximation and projection (UMAP) and Louvian graph–based clustering, CD4+ T cells from various origins were partitioned into 19 clusters (fig. S2A). Most cells were grouped together on the basis of their cell types and cell origins, whereas several clusters retained both Tconv and Tregs from the same tissue source. Moreover, some clusters aggregated one of the cell types from different cell origins, suggesting that there were tissue-intrinsic signatures and phenotypic overlap between CD4+ T cells (Fig. 1, B and C, and fig. S2B). For instance, both Tconv and Tregs from HD PBMCs expressed high levels of TCF7 and S1PR1, and Tconv and Tregs from both inflamed and non-inflamed tonsils were enriched in FOS and DUSP1 (fig. S2C and data file S1, tab data S1). We detected relatively higher levels of IL7R and TCF7 expression in tonsil Tconv from patients with sleep apnea compared with those from patients with tonsilitis. In addition, higher expression of CTLA4, TNFRSF1B, and IFITM1 in tonsil Tconv from patients with tonsilitis, compared with the non-inflamed controls, suggested a more inflamed signature of CD4+ Tconv in patients with tonsilitis (fig. S3, A to E). This inflamed tissue signature was further confirmed by enrichment of pathways involving CD28 costimulation signaling, nuclear factor κB (NF-κB) signaling, and IL-12 family signaling in CD4+ Tconv from patients with tonsilitis (fig. S3, F to I). However, although we detected the expression of TNFRSF4 and TNFRSF18 in HNSCC PBMC Tregs, they were increased in HNSCC TIL Tregs compared with HNSCC PBMC Tregs and nontumor tonsil tissue Tregs (fig. S2C).
Fig. 1. Intratumoral CD4+ T cells have distinct transcriptional signatures.
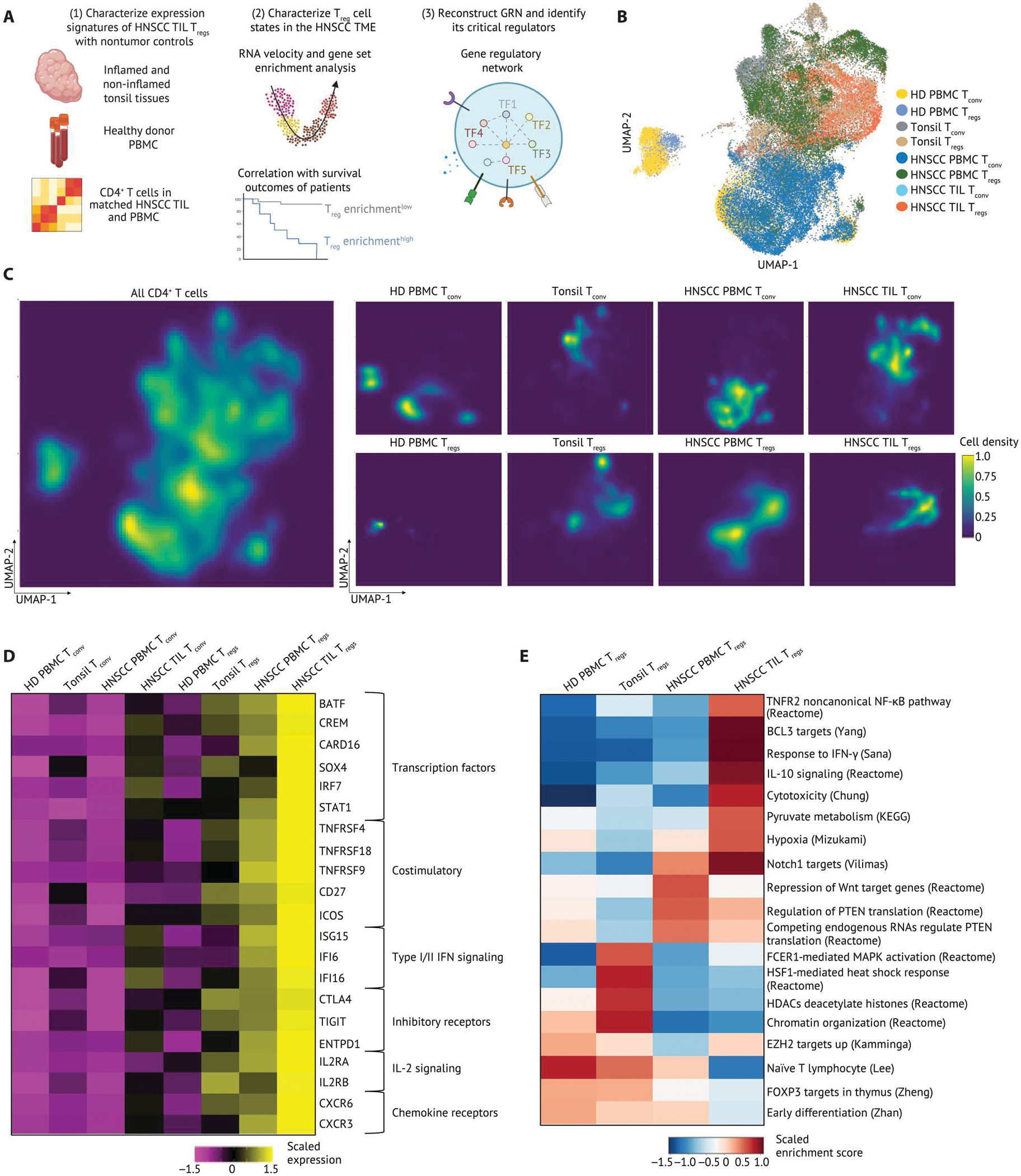
(A) Schematic of the experimental setup. Live Tconv and Tregs from HD blood and tonsil tissues were sequenced using 10X Genomics 3′ based scRNA-seq. scRNA-seq data were integrated with data from a previous study (19). We leveraged multiple bioinformatic approaches to identify cell types and states, infer drivers of differentiation, and reconstructed GRNs using SCENIC and causal MGM. (B) UMAP embedding and clustering of 51,195 CD4+ T cells from matched TILs and PBMCs from patients with HNSCC (n = 26), tonsil tissues from patients with tonsilitis (n = 5), tonsil tissues from patients with sleep apnea (n = 6), and HD PBMCs (n = 10). UMAP embedding of 51,195 CD4+ T cells were color-coded by cell type and cell origin. (C) Two-dimensional galaxy plots of CD4+ T cells were grouped by cell origins, and lighter color is indicative of a higher density of cells across sample groups. (D) A heatmap of gene signatures differentially expressed in HNSCC TIL Tregs compared with all other cells. The expression is scaled by transforming the gene expression in each population to zero mean, and unit SD is shown in the heatmap. (E) Selected gene sets that were highly enriched in subsets of Tregs were visualized on a heatmap. Tregs were grouped on the basis of the cell origin. The z-score of gene sets in each Treg subset was calculated by R package Singleseqgset. Similarly, the enrichment score is scaled by row. KEGG, Kyoto Encyclopedia of Genes and Genomes. PTEN, phosphatase and tensin homolog.
To characterize TIL Treg-specific signatures further, we examined differentially expressed genes and published gene sets in HNSCC TIL Tregs compared with Tregs in nontumor tissue, in peripheral blood from patients with HNSCC and Tconv from all sites. In particular, HNSCC TIL Tregs expressed ICOS; CD27; and TNFR members TNFRSF4, TNFRSF9 (the gene encoding 4–1BB), and TNFRSF18, which reflected a specific activated Treg phenotype. These TIL Tregs were also enriched for a gene set associated with noncanonical TNFR2-NF-κB pathway (Fig. 1, D and E and data file S1, tab data S2) (21, 24). In addition to up-regulation of IL-10 signaling, we also detected selective expression of CTLA4, TIGIT, and ENTPD1 (gene for CD39) in HNSCC TIL Tregs, highlighting a potentially more suppressive phenotype of Tregs in the HNSCC TME compared with tissue Tregs and peripheral Tregs (Fig. 1, D and E). Type I/II interferon (IFN) response signatures, including ISG15, IFI6, IFI16 (Fig. 1D), and a gene set associated with response to IFN-γ (Fig. 1E) were observed in HNSCC TILTregs, implying that Tregs undergo functional specialization after activation in the TME. Through cross-checking all differentially expressed genes in HNSCC TIL Tregs with a published human TF database (25), we identified six TFs highly expressed in HNSCC TIL Tregs, including BATF, CREM, CARD16, SOX4, IRF7, and STAT1 (Fig. 1D). By contrast, HD PBMC Tregs were characterized by naïve Treg signatures including high levels of TCF7 and S1PR1 (Fig. 1D) as well as gene sets associated with EZH2, FOXP3, early differentiation, and naïve T cell pathways (Fig. 1E and fig. S2C). Tonsil tissue Tregs exhibited an early activation phenotype by up-regulating FOS and gene sets associated with histone modifications, chromatin organization, and mitogen-activated protein kinase (MAPK) activation (Fig. 1E and fig. S2C). In addition, a heat shock response gene set and associated genes were highly enriched in Tregs from tonsils (Fig. 1E and fig. S2C). Although we detected mRNA expression of BATF in tonsil Tregs from patients with tonsilitis, it was increased in HNSCC TIL Tregs compared with tonsil tissue Tregs from patients with tonsilitis or sleep apnea (fig. S3J). We observed similar gene expression profiles in nontumor tonsil Tregs from patients with sleep apnea or tonsilitis and distinct from those in the HNSCC TME, suggesting a specific gene expression signature of TIL Tregs compared with inflamed and non-inflamed tissues (fig. S3, K to P). These analyses provided an overview of expression signatures differentially enriched in HNSCC TIL Tregs versus Tregs in nontumor tissues and the periphery and the distinct transcriptional signatures from Tconv from various tissue sites.
TNFR+ Tregs are a distinct differentiation state and correlate with worse prognosis in patients with HNSCC
To characterize the heterogeneity of Tregs within the HNSCC TME and understand the context-dependent mechanisms modulating different Treg subsets in the HNSCC TME, we refined the Treg analysis by partitioning them into 13 subclusters (Fig. 2A; fig. S4, A and B; and data file S1, tab data S3). We also performed pseudotemporal modeling of differentiation using RNA velocity (26). The inferred pseudotemporal ordering indicated a differentiation trajectory across Treg clusters (Fig. 2B and fig. S4C). On the basis of the pseudotime trajectory, canonical marker genes and gene sets that associated with distinct T cell state transitions defined 13 clusters, characterized as naïve/memory Tregs (clusters 1 to 5), early activated Tregs (clusters 6 to 8), TNFR+ Tregs (clusters 9 to 12), and IFN+ Tregs (cluster 13) (Fig. 2, B to D, and fig. S4, C and D). For instance, Treg clusters 1 to 5 were identified as naïve/memory Tregs by up-regulation of SELL (gene encoding L-selectin), CCR7, and TCF7 as well as gene sets associated with ribosome and naïve T cell signatures (Fig. 2D and fig. S4D). In addition to enhanced expression of FOS, JUN, and BCL2, gene sets associated with T cell activation including phosphorylation of β-actin, activation of AP-1 family signaling, and MAPK3/ERK activation were highly enriched in Treg clusters 6 to 8. We found two Treg subsets that are highly activated and characterized by the up-regulation of gene sets associated with coinhibitory receptor programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) signaling and TNFRSF-induced noncanonical NF-κB pathway. These signatures were confirmed by expression levels of TNFR super family members [TNFRSF4, TNFRSF18, and TNFRSF1B (gene encoding TNFR2)] and coinhibitory/stimulatory receptors [ICOS, CTLA4, and PDCD1 (gene encoding PD-1)] (Fig. 2D and fig. S4D). However, genes and gene sets associated with IFN-stimulated responses (IFIT3, ISG15, and IFI6) were exclusively enriched in cluster 13 (IFN+ Tregs) (Fig. 2D and fig. S4D). In addition, cells within cluster 13 highly expressed genes associated with T helper cell type 1 (TH1) phenotype, including TBX21 (gene encoding T-bet), CCL4, CCL5, and IFNG (gene encoding IFN-γ), suggesting a specialized function of Tregs for TH1-related and antiviral immune responses (fig. S4D) (27–29). We also detected augmented expression of CD39 and CD27 in Treg clusters 9 to 12 (TNFR+ Tregs), which highlighted different programs regulating activated Treg function in the TME. A pseudotemporal ordering by RNA velocity further revealed that the TNFR+ Tregs and IFN+ Tregs were the most terminally differentiated Treg subpopulations (fig. S4E). Most cells identified as TNFR+ Tregs and IFN+ Tregs were predominantly enriched in the HNSCC TME, whereas nontumor tonsil tissues were enriched in naïve/memory and early activated Tregs, demonstrating both tumor-specific and overlapping phenotypes of Tregs in the TME versus nontumor tissues (fig. S4F).
Fig. 2. Intratumoral Tregs are heterogeneous in the HNSCC TME and regulated by distinct transcriptional programs.
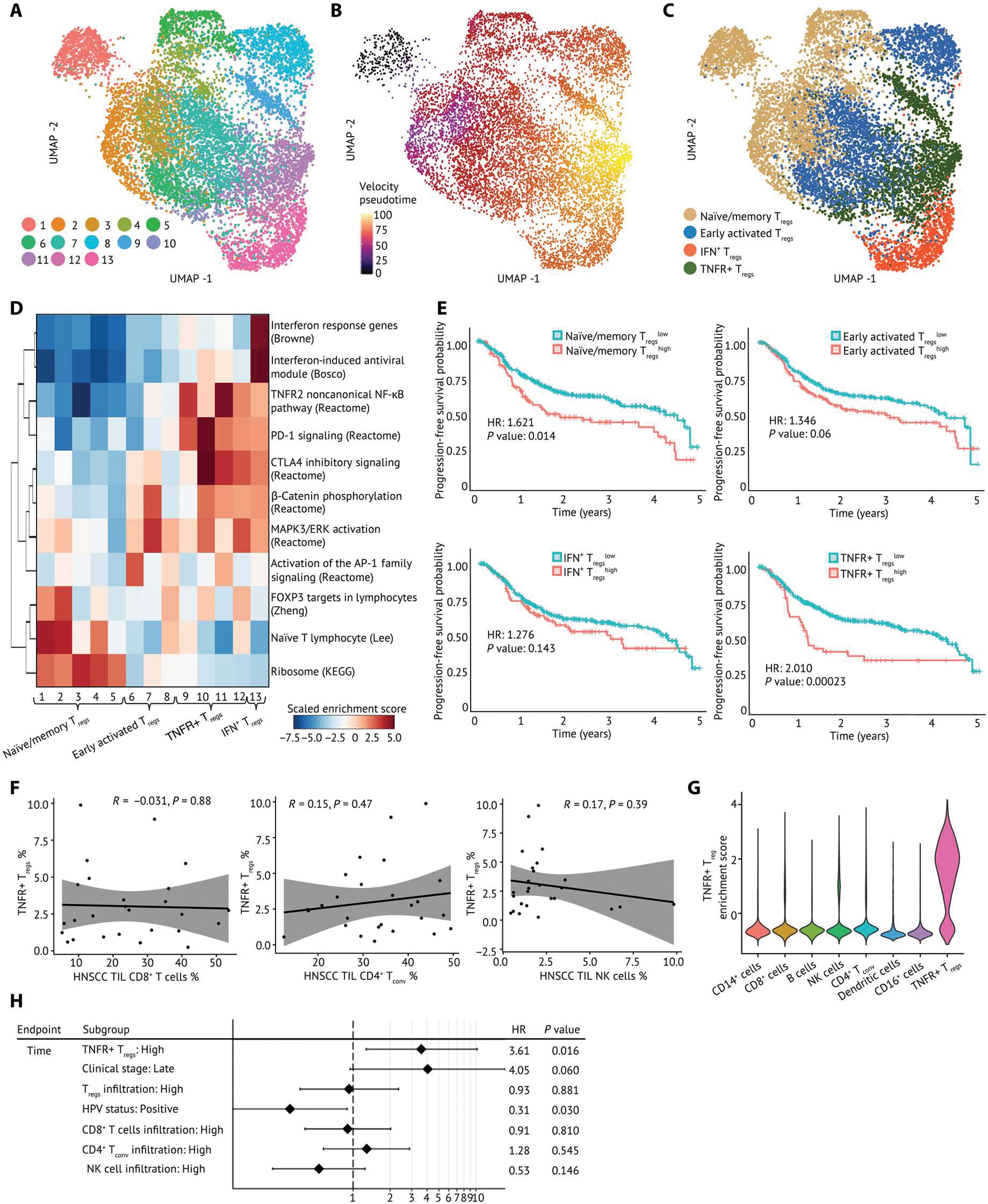
(A) A UMAP embedding of 9688 Tregs across all samples. Thirteen clusters were identified by Louvian graph–based unbiased clustering. (B) Pseudotime was derived from RNA velocity and visualized on a UMAP embedding, which revealed a differentiation process across Treg clusters. (C) Tregs were annotated by cell state and visualized in a UMAP embedding. (D) Gene set enrichment analysis across Treg clusters identified distinct phenotypes of Tregs. Specifically, clusters 1 to 5 are associated with naïve/memory Treg phenotype, clusters 6 to 8 have an early activated Treg phenotype, clusters 9 to 13 exhibit an activated Treg phenotype, clusters 9 to 12 are enriched for TNFR member genes, and cluster 13 has IFN response genes and a TH1-like expression signature. The enrichment score is scaled by row. (E) Association of the enrichment of each Treg subpopulation with survival outcomes of patients with HNSCC was calculated and visualized on Kaplan-Meier curves. Hazard ratio (HR) is calculated by monovariate Cox proportional regression, and the P value is calculated by likelihood ratio test. (F) Scatterplots of correlation between the frequency of TNFR+ Tregs and CD8+ T cells, CD4+ Tconv, and NK cells. Pearson correlation coefficient (R) and P values (P) are calculated between TNFR+ Tregs and each cell type. The proportion of each type among all lymphocytes in the HNSCC TME is shown. (G) Violin plot of the TNFR+ Treg signature in other cell types in the HNSCC TME. (H) Multivariable analysis of the enrichment of TNFR+ Tregs and PFS outcomes.
To determine whether the enrichment of Treg subpopulations was related to survival outcomes of patients, we performed survival analysis using clinical data and bulk RNA-seq expression data from the Cancer Genome Atlas (TCGA). We scored each patient for the enrichment of Treg signatures derived from HNSCC scRNA-seq data and used the enrichment score as a reference profile to impute Treg enrichment in bulk RNA-seq datasets by CIBERSORTx (30). Gene sets representing Treg enrichment were derived from the top 200 differentially expressed genes in each Treg subpopulation. We found that high enrichment of TNFR+ Tregs in patients with HNSCC was associated with worse progression-free survival (PFS), suggesting that this TIL Treg subpopulation suppresses antitumor immunity in HNSCC (Fig. 2E). By contrast, the enrichment of early activated Tregs or IFN+ Tregs did not show association with survival (Fig. 2E). Although the enrichment of naïve/memory Tregs was also associated with poorer survival of patients, these naïve/memory Treg signatures were not tumor specific because they were observed in the periphery and nontumor tissues (Fig. 2E and fig. S4F). The frequency of TNFR+ Tregs was not correlated with CD8+ T cells, CD4+ Tconv, or natural killer (NK) cells in the HNSCC TME (Fig. 2F). In addition, the enrichment of each Treg subpopulation did not show correlation with each other (fig. S5A). Fisher’s exact test indicated that there was no association between the enrichment of TNFR+ Tregs with overall Treg infiltration, patients’ clinical stage, or human papillomavirus (HPV) status of patients with HNSCC (fig. S5B). To assess whether the TNFR+ Treg signature used in the survival association analysis is shared by other cell types, we calculated the enrichment score for the TNFR+ Treg signature in other immune cell types present in the HNSCC TME. Minimal enrichment scores were identified in other immune cell types, further suggesting that the association of TNFR+ Tregs and survival outcomes of patients with HNSCC was not confounded by the gene signatures of other Treg subpopulations or other cell types. (Fig. 2G). After correcting the clinical stage, overall Treg infiltration, HPV status of patients, tumor infiltrations of CD8+ T cells, CD4+ Tconv or NK cells as covariates, the association between worse PFS and high TNFR+ Treg enrichment remained in a multiple variable survival analysis, indicating that the association of TNFR+ Tregs and patient survival outcomes was not confounded by tumor infiltration of other cell types or clinical factors (Fig. 2H).
GRN inference revealed critical transcriptional circuit controlling TNFR+ Treg subpopulation
Cell state transitions during T cell activation are governed by TFs and their associated cofactors, which work together to regulate gene expression and cell function. To determine transcriptional regulators critical to TIL Tregs at each cell state, we systematically assessed the genes that are likely to be governed by TFs in each Treg subpopulation by applying SCENIC (31) to infer TF activity and downstream target genes (Fig. 3A and data file S1, tab data S4). Naïve/memory Tregs were highly enriched with well-known TFs mediating T cell homeostasis including KLF2 and FOXP1 (32, 33), whereas regulons governed by EGR1 and AP-1 family TFs (FOSB and FOS), known as important regulators of T cell activation and early response to TCR engagement, were detected in early activated Tregs (34). In activated Treg subsets, enrichment of regulons IRF2, TBX21, and EOMES was increased in IFN+ Tregs, reflecting the specialized Treg function for helper T cell responses (28, 35, 36). Regulons governed by BATF, EPAS1, and noncanonical NF-κB pathway genes including NFKB2, RELB, and BCL3 showed enhanced activities in TNFR+ Tregs (Fig. 3A). NF-κB family genes are well-known for regulating Treg function and development (37–39). EPAS1, which encodes the hypoxia-inducible factor 2A, has been recently described for its role in moderating the development and function of Tregs (40, 41). Although BATF was identified as a driver of key regulons in Tregs, it is unknown what transcriptional regulation(s) or Treg subpopulations are governed by expression of genes controlled by this TF.
Fig. 3. An integrated BATF transcriptional network regulates key phenotypes of TNFR+ Tregs.
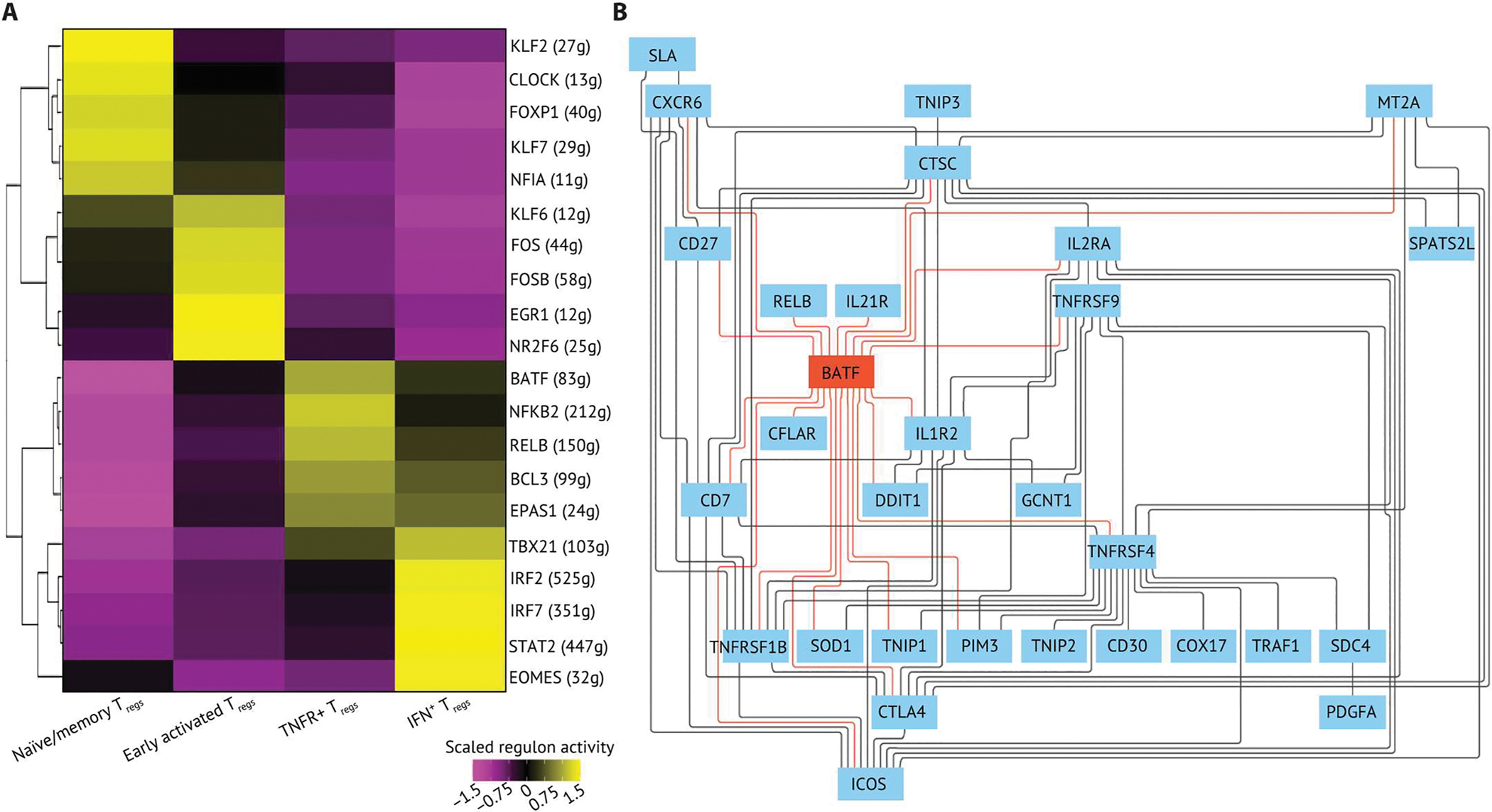
(A) The top five regulons differentially expressed in each Treg state were ranked by log2 fold change and visualized on a heatmap. The regulons were generated by using SCENIC with all Tregs (n = 9736) in the dataset. The regulon score was calculated by using AUCell in R, and the regulon score of each Treg subpopulation is scaled by row. The top five regulons were determined by their log2 fold change. (B) The GRN of TNFR+ Tregs. The GRN was constructed using directed MGM and FCI-MAX modeling with TFs and downstream targets from the top five regulons in TNFR+ Tregs. The direct connections between BATF and target genes are colored in red.
Limitations of scRNA-seq technologies, such as dropouts, stochastic variation of gene expression at the single-cell level, and the relatively low expression of some TFs, can hamper the detection of TFs orchestrating cellular identity and function. Despite the minimal gene expression of NFKB2, RELB, and BCL3, regulons governed by these TFs were enriched with several TNFR members and showed differential enrichment in TNFR+ Tregs (fig. S6, A and B). Moreover, we found that multiple downstream target genes were shared by top enriched regulons in each Treg subpopulation, suggesting a synergistic regulation of TFs controlling Treg identity, activation, and function in the TME. We integrated the top five regulons into combined GRNs and discovered distinct signaling pathways that are specific to each Treg subpopulation (figs. S7 and S8, A to D). Reactome pathway analysis showed that forkhead box class O (FOXO)–mediated signaling and RUNX3-mediated signaling were enriched in the GRN identified in naïve/memory Tregs (fig. S8A). In early activated Tregs, cell cycle checkpoint, cellular response to stimuli, and NOTCH-mediated signaling in the GRN represented the pathways vital for Treg proliferation and activation (fig. S8B). In addition to TNFR members mediating noncanonical NF-κB pathway, we also observed that the GRN from TNFR+ Tregs are highly involved in cell apoptosis and IL-4/IL-13 and IL-10 signaling pathways, and the GRN from IFN+ Tregs plays important roles in type I/II IFN responses, cell death, and antigen presentation signaling (fig. S8, C and D).
We next used directed mixed graphical modeling (MGM) and fast causal inference-Max (FCI-MAX) (42) to define the central regulatory circuit controlling the GRN of TNFR+ Tregs. Genes with near-zero variance and low overall nonzero values were filtered out, and normalized gene expression data from TNFR+ Treg were used as input. All downstream targets of the top five enriched regulons in TNFR+ Tregs were used to construct an undirected network by using MGM. The central regulatory network was imputed using FCI-MAX by connecting the key transcriptional regulators and downstream targets within the undirected network. We found that BATF is central to the TNFR+ Treg GRN by having the most connections compared with other genes (Fig. 3B). In addition, BATF was predicted to regulate multiple key signatures of HNSCC TIL Tregs as previously defined. For instance, BATF is directly linked to costimulatory/inhibitory receptors TNFRSF4, TNFRSF9, TNFRSF1B, CD27, ICOS, IL2RA, and CTLA4, which have been shown to be important in TIL Treg activation and function. Furthermore, we found that the chemokine receptor CXCR6 and T cell apoptosis signaling molecules CFLAR (gene encoding c-FLIP) and PIM3 were directly connected to BATF in the regulatory circuit, demonstrating a potential role for BATF in controlling TIL Treg migration and survival (Fig. 3B) (43–45). Together, we identified that TNFR+ Tregs were a distinct differentiation state compared with other Treg subpopulations, showed distinct gene expression signatures, and that this activated subpopulation was associated with worse prognosis of patients with HNSCC. By constructing the regulatory networks from scRNA-seq, we identified specific GRNs that govern Treg phenotypes in HNSCC across cell states and inferred a transcriptional circuit centered around BATF that is central to TNFR+ Tregs in the TME.
TNFR+ Tregs are enriched in multiple solid tumor types and regulated by similar transcriptional programs
To validate whether this TNFR+ Treg subpopulation is distinct to the HNSCC TME, we isolated Tregs from matched tumors and blood from four patients with non–small cell lung cancer (NSCLC), one patient with small cell lung cancer (SCLC), and four patients with melanoma (Fig. 4A and fig. S9A). Unbiased clustering revealed six Treg clusters that were evenly mixed with Tregs from melanoma and lung cancer and that exhibited distinct enrichment from Tregs from TILs or peripheral blood (Fig. 4B and fig. S9B). Cell states of Tregs were characterized by using RNAvelocity to evaluate differentiation and identifying genes associated with each Treg subpopulations (Fig. 4C and fig. S9, C and D). We found that clusters 4 to 6 were predominantly enriched with TIL Tregs and expressed Treg activation–related immune markers, including ICOS, CD27, and TIGIT (fig. S9, E to G). Cluster 6 was highly enriched with TNFR+ Treg signatures identified in the HNSCC TME, whereas cells in cluster 5 expressed IFN+ Treg signatures (Fig. 4, D and E, and fig. S9, G to N). Next, we quantified the activity for each regulon derived from HNSCC TNFR+ Tregs in the TIL Treg analyses in this dataset (46). Consistent with our analysis of HNSCC Tregs, the regulon governed by BATF was up-regulated in TNFR+ Tregs (cluster 6) in the tumors from patients with lung cancer and melanoma (Fig. 4F).
Fig. 4. TNFRSF-activated Tregs are highly enriched in solid TME compared with nontumor tissues and associated with worse prognosis across solid tumors.
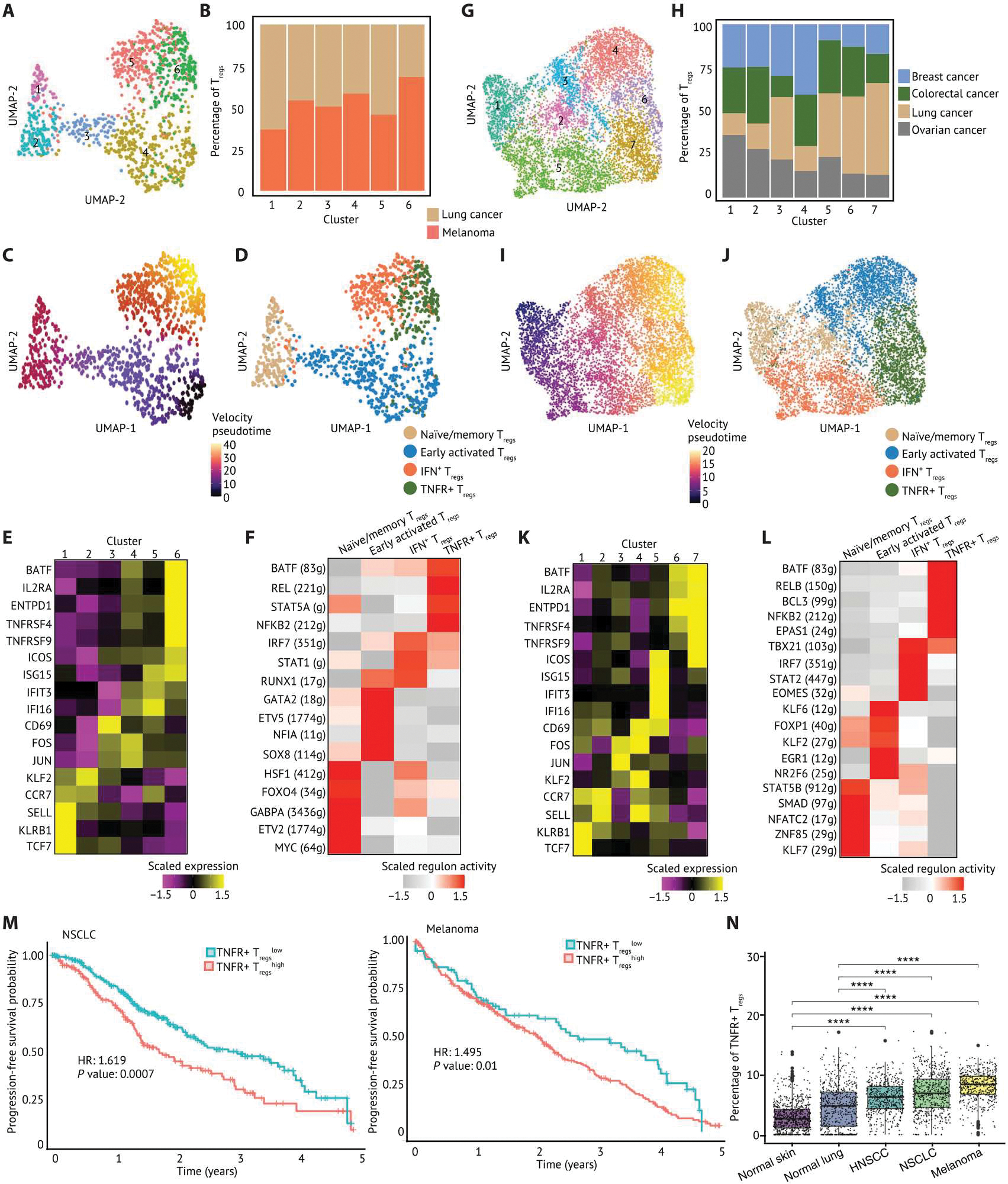
(A) A UMAP embedding of 1294 Tregs in TILs and PBMCs from patients with melanoma (n = 4), NSCLC (n = 4), and SCLC (n = 1). Six clusters were identified by Louvian graph–based unbiased clustering. (B) A stacked barplot of percentage of Tregs in each cluster showed that Tregs from lung cancer and melanoma were distributed across six clusters. (C) Pseudotime derived from RNA velocity was visualized in a UMAP embedding, suggesting that Tregs in cluster 6 were at later pseudotime and more terminally differentiated. (D) Tregs were annotated by cell state and visualized in a UMAP embedding based on the cell state–related canonical marker genes, gene sets, and pseudotime inference. (E) The relative expression of selected canonical marker genes is visualized on a heatmap. (F) The top five regulons differentially expressed in each Treg cluster ranked by log2 fold change were visualized on a heatmap. (G) A UMAP embedding of 7045 Tregs in TIL from patients with ovarian cancer (n = 5), lung cancer (n = 8), breast cancer (n = 14), and colorectal cancer (n = 7). Seven clusters were identified by unbiased clustering. (H) A stacked barplot of percentage of Tregs in each cluster highlights the distribution of Tregs from each tumor type. Clusters 6 and 7 were mixed with Tregs across tumor types. (I) Pseudotime inferred by Slingshot was visualized in a UMAP embedding. (J) Tregs were annotated by cell state and visualized in a UMAP embedding. (K) The relative expression of selected canonical marker genes is visualized on a heatmap. (L) The top five regulons differentially expressed in each Treg cluster ranked by log2 fold change were visualized on a heatmap. The gene expression and enrichment score are scaled by transforming the expression or enrichment score in each population to zero mean, and unit SD is shown in the heatmap. (M) Associations of the enrichment of each Treg subpopulation with survival outcomes of patients with NSCLC and melanoma were calculated and visualized on Kaplan-Meier curves. Hazard ratio is calculated by monovariate Cox proportional hazard regression, and the P value is calculated by likelihood ratio test. (N) The enrichment of TNFR+ Tregs visualized in box plots was inferred using Cibersortx on datasets from the TCGA and GTEx. The inferred proportion of TNFR+ Tregs among all cells within each tissue score is shown. A pairwise comparison between each tissue source is calculated by a nonparametric Wilcoxon signed rank test. P values: ****P ≤ 0.0001.
In addition, we isolated Tregs from a previously published dataset (47) involving breast cancer, colorectal cancer, lung cancer, and ovarian cancer and interrogated their gene expression signatures and pseudotime trajectory (Fig. 4, G and H, and fig. S10A). We found that Tregs in clusters 6 and 7 expressed TNFR members and BATF, in agreement with our analyses of other tumor types (Fig. 4, I to K, and fig. S10B). By contrast, TIL Treg cluster 5 expressed high levels of IFN-stimulated genes (ISF15, IFI16, and IFI44L) as well as markers associated with T cell activation (ICOS and CD69) (Fig. 4K and fig. S10B). Consistent with our previous analyses, we observed that regulons governed by BATF, NFKB2, BCL3, RELB, and EPAS1 showed the highest activity scores in TNFR+ Tregs (Fig. 4L).
To investigate whether TNFR+ Tregs are present in both squamous cell carcinoma (SCC) and non-SCC histology types, we grouped TIL Tregs by histological types of cancers (fig. S11A). We found that TNFR+ Tregs were enriched in patients with adenocarcinoma or SCC (fig. S11B). In addition to a similar expression level of BATF, we also observed comparable BATF regulon enrichment and TNFR+ Treg enrichment scores in TNFR+ Tregs from patients with adenocarcinoma or SCC (fig. S11, C and D). The expression of BATF, BATF regulon activity, and TNFR+ Treg enrichment scores were also further confirmed in TNFR+ Tregs in patients with invasive ductal carcinoma or large cell carcinoma, suggesting that TNFR+ Tregs and the associated BATF regulatory network were not cancer subtype specific (fig. S11, E to G).
The enrichment of TNFR+ Tregs was correlated with worse PFS for patients with NSCLC and melanoma (Fig. 4M). By acquiring data from the Genotype-Tissue Expression Project (GTEx) and imputing the enrichment of Treg subpopulations within normal tissues, we found higher enrichment of TNFR+ Tregs in solid tumors compared with normal lung and normal skin (sun exposed) (Fig. 4N). These observations show that at least a subset of activated TIL Tregs have a distinct transcriptional signature versus Tregs present in normal tissues. The transcriptional programs modulating TNFR+ Treg function revealed by GRN construction are not specific to the HNSCC TME but are also present in other cancer types. These findings confirm that TNFR+ Tregs are enriched in multiple cancers compared with normal tissues, inferring an immunosuppressive role in the TME.
BATF functions as a transcriptional nexus of human activated Tregs
Our system approach can identify key TFs that modulate cellular functions in TIL Tregs, but it is crucial to experimentally validate the inferred GRNs. To investigate the impact of BATF on the expression of regulon members in the TNFR+ Tregs and validate our network prediction from scRNA-seq analyses, we used a CRISPR-Cas9 ribonucleoprotein (CRISPR RNP) KO approach that was developed to specifically target cultured primary human Tregs (fig. S12, A and B) (48). Given the challenge of isolating an adequate number of activated TIL Tregs from patients with cancer and the limitations of current CRISPR-Cas9 editing technology for targeting low numbers of human T cells, we used ex vivo expanded human primary Tregs isolated from umbilical cord blood as a surrogate to interrogate the impact of BATF deletion at the transcriptional, protein, and functional levels. CRISPR RNPs targeting BATF or scrambled control RNPs (functional RNPs loaded with scrambled guide RNAs) were electroporated into expanded human Tregs. After a 3-day TCR stimulation post-electroporation, the expression levels of BATF, surface and intracellular proteins, and cytokines were evaluated by multiparameter flow cytometry. A robust reduction in the BATF protein level was observed in BATF KO activated Tregs compared with the scrambled control (Fig. 5, A and B). These data suggested that the CRISPR RNP KO approach successfully altered BATF RNA and protein levels and allowed us to examine the effects of BATF disruption in human primary activated Tregs.
Fig. 5. CRISPR/Cas9-RNP KO reveals that BATF regulates human activated Tregs by multiple signaling pathways.
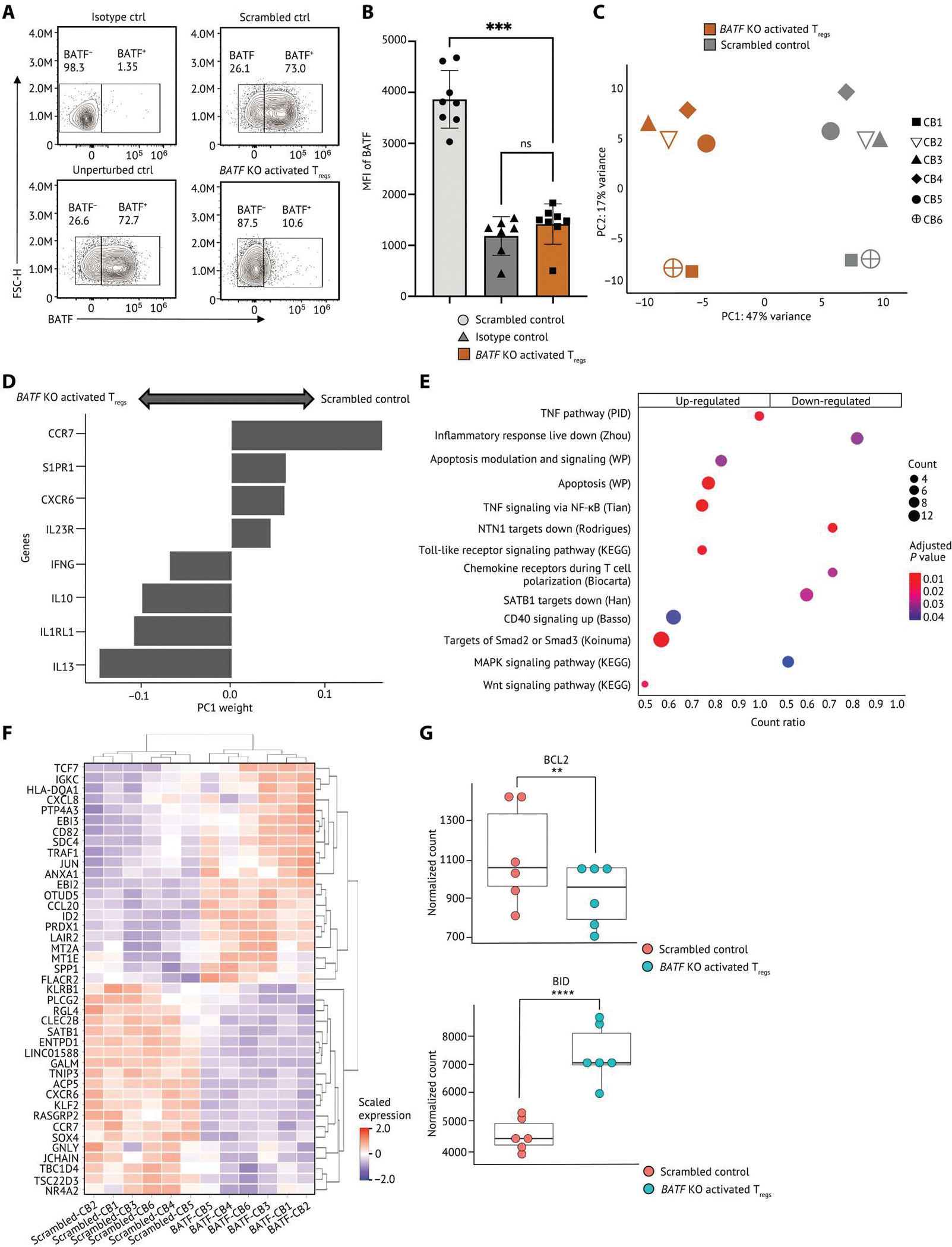
(A and B) Human primary Tregs isolated from cord blood were CRISPR-edited and then cultured with TCR stimulation for 3 days. (A) Representative flow staining of Tregs showing BATF protein level in nontargeting scrambled control and unperturbed control Tregs. (B) The BATF expression by median fluorescence intensity (MFI) in Tregs (n = 8) is shown. Each dot indicates an individual replicate. Bars indicate the median of expression, and error bars represent 1 SD. (C to G) RNA-seq was conducted on BATF KO activated Tregs (n = 6) and scrambled control from the same donor. (C) PCA of the transcriptome of the scrambled control and BATF KO activated Tregs. Human activated Tregs were stratified by PC1. (D) Weightings of the genes that were the strongest drivers of PC1 were visualized on a bar plot. (E) Selected gene sets that were enriched in human BATF KO activated Tregs were visualized on a dot plot. Genes are differentially expressed in both TNFR+ Tregs from scRNA-seq and BATF KO activated Tregs in bulk RNA-seq selected as input to the gene set enrichment analysis. Dots are colored by the false discovery rate (FDR)–adjusted P value, and dot sizes are scaled by the number of significantly up-regulated genes within each gene set (<0.1% FDR). (F) The relative expression of the top 20 up/down-regulated genes differentially expressed in both TNFR+ Tregs and BATF KO activated Tregs were visualized on a heatmap. The gene expression is scaled by row. (G) Dot plots showing the expression levels of BCL2 and BID in BATF KO activated Tregs and scrambled controls from eight individual replicates. Data in (B) were analyzed by a ratio paired t test, and the FDR P values in (G) were calculated by Wald test. P values: **P < 0.01; ***P < 0.001, ****P < 0.0001. ns, not significant.
To dissect the phenotypic and functional effects of BATF in human activated Tregs, we performed RNA-seq of BATF KO activated Tregs and scrambled controls from six cord blood samples. Results from principal components (PC) analysis revealed that PC1 stratified Tregs by BATF KO versus scramble control (Fig. 5C and fig. S12C). Genes associated with T cell migration (CCR7, CXCR6, and S1PR1), cytokines (IFNG, IL10, and IL13), and cytokine receptors (IL23R and IL1RL1) were the strongest drivers of PC1 observed between BATF KO activated Tregs and the scrambled control, suggesting an important role of BATF in activated Treg trafficking and function (Fig. 5D). To further characterize the functions of BATF in HNSCC intratumoral TNFR+ Tregs, we evaluated all differentially expressed genes of TNFR+ Tregs from the HNSCC scRNA-seq dataset and interrogated their transcriptional changes in BATF KO activated Tregs. In addition to the reduction of CXCR6 and CCR7, we observed gene sets associated with Treg migration down-regulated in BATF KO activated Tregs compared with scrambled control, including chemokine receptors signaling during T cell polarization and NTN1 signaling (Fig. 5E). NTN1 encodes Netrin-1, which is a neuronal guidance molecule that was shown to increase Treg infiltration to lung tissues of mice with lung ischemia-reperfusion (49). These data suggested a potential role of BATF in Treg infiltration into the TME. We also detected a complex impact of BATF deletion in human activated Tregs, including up-regulation of gene sets associated with signaling of SMAD2/3, WNT signaling, and the TNFRSF/NF-κB pathway, as well as the down-regulation of gene sets associated with special AT-rich sequence-binding protein1 (SATB1) and MAPK signaling pathways (Fig. 5E). Moreover, BATF KO activated Tregs exhibited an increase in TCR signaling–related genes (HLA-DQA1 and CD82) and NF-κB signaling–related gene TRAF1, which further indicated the important role of BATF in controlling Treg activation (Fig. 5F). In addition to a reduction of BCL2 expression in BATF KO activated Tregs, we found gene sets associated with apoptosis highly up-regulated in BATF KO activated Tregs along with increased expression of BID, an antagonist of BCL2, suggesting that BATF is essential for activated Treg survival (Fig. 5, E to G). Together, by interrogating the impact of BATF disruption on genes that are selectively modulated in intratumoral TNFR+ Tregs, we found that BATF is a critical regulator mediating multiple signaling pathways in human activated Tregs. These data suggested that BATF functions to limit excessive activation of human activated Tregs and promotes activated Treg survival and trafficking.
BATF regulates the suppressive function of human activated Tregs
Given these observations, we hypothesized that BATF might also modulate the suppressive function of human activated Tregs. Although the protein level of FOXP3 and CD25 remained unchanged with BATF deletion, we observed an increase of suppressive function of BATF KO activated Tregs, demonstrating that human activated Tregs maintained their identity with BATF deletion and that the impact on Treg-suppressive function is independent of FOXP3 signaling (Fig. 6, A to C). Consistent with these results, BATF KO Tregs exhibited an increased activated phenotype exemplified by increased expressions and proportions of cells expressing the costimulatory receptors such as ICOS, OX40, and 4–1BB on Tregs (Fig. 6, D to F). We also detected an increased proportion of human activated Tregs expressing coinhibitory receptor LAG3, inhibitory cytokine IL-35 subunit EBI3, and IL-10, highlighting the increased suppression after BATF deletion in activated Tregs (Fig. 6, G to I, and fig. S12D). The number of human activated Tregs expressing NRP1 and the per-cell expression level of NRP1+ Tregs were enhanced by BATF ablation, indicating an important role for BATF in regulating Treg stability (Fig. 6J and fig. S12E) (50). These results were consistent with their transcript levels in BATF KO Tregs from bulk RNA-seq (fig. S12F). Together, these findings revealed a critical role of BATF in modulating the stability of activated Tregs and functioning independently of FOXP3 to control activated Treg-suppressive function.
Fig. 6. BATF modulates the suppressive function of human activated Tregs.
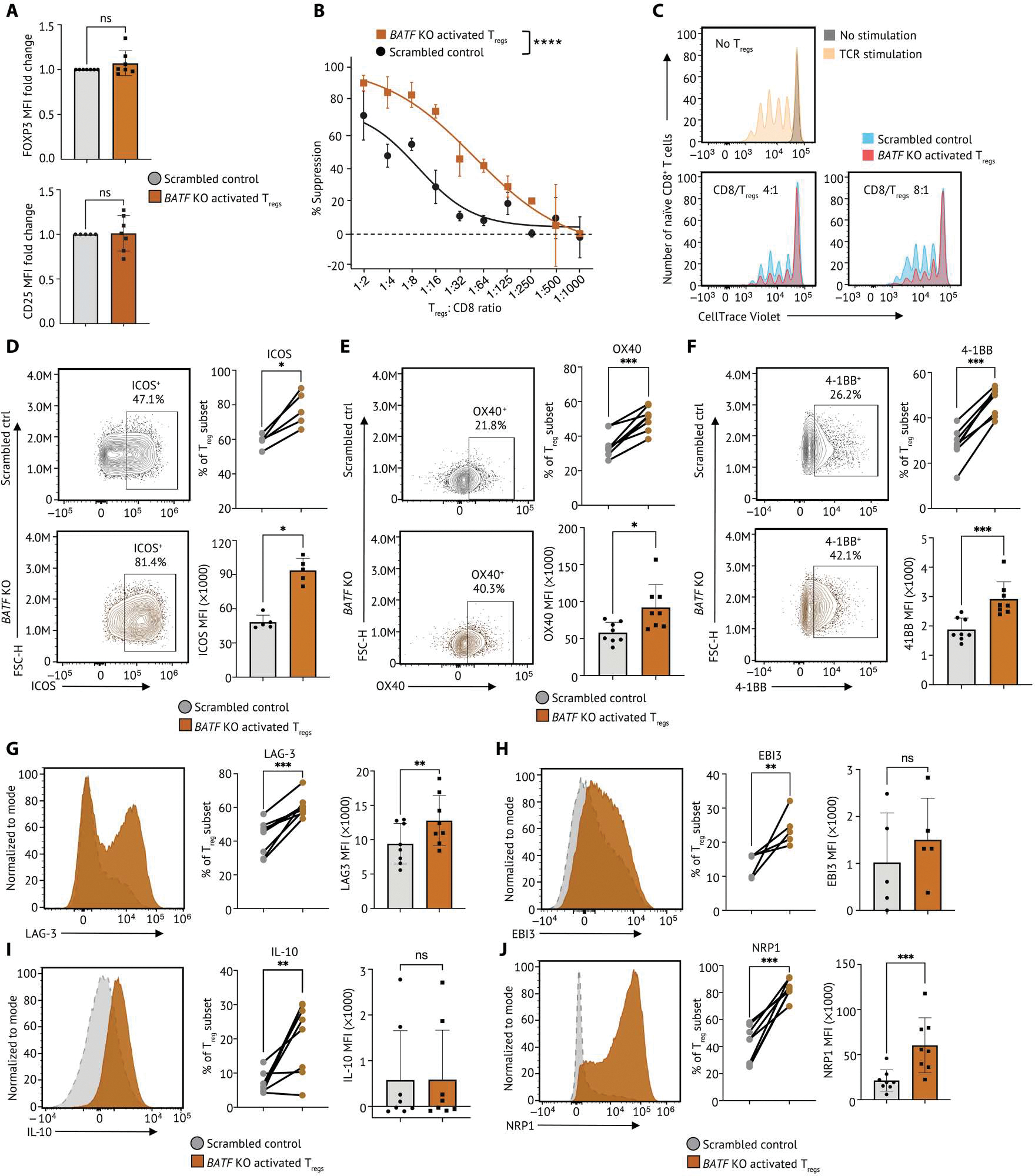
(A) Tabulation of FOXP3 (top) and CD25 expression (bottom) in BATF KO activated Tregs (n = 7) and scrambled control from the same donor. Data were reported as fold change normalized to marker MFI of nontargeting scrambled control groups. Each dot indicates an individual replicate. Bars indicate the median of expression, and error bars represent 1 SD. (B) In vitro microsuppression assay comparing BATF KO activated Tregs (n = 6) and scrambled control from the same donor are shown. (C) Representative CD8+ T cell proliferation in Fig. 6B at different conditions: no TCR stimulation (gray); 5-day TCR stimulation without coculture with Tregs (yellow); 5-day TCR stimulation with CD8:Treg ratios as 4:1 and 8:1. Cell number at each condition was normalized to the highest cell number in the plot. (D) The expression of ICOS on BATF KO activated Tregs and scrambled control were determined by flow cytometry. The percentage of Tregs expressing ICOS was summarized on a dot plot, and the MFI of ICOS on Tregs was summarized on a bar plot. Data were pooled from five individual replicates. (E) The expression of OX40 on BATF KO activated Tregs and scrambled control were visualized by represented flow cytometry plots from eight individual replicates. The percentage of Tregs expressing OX40 was summarized on a dot plot, and the MFI of OX40 on Tregs (n = 8) was summarized on a bar plot. (F) The expression of 4–1BB on BATF KO Tregs and scrambled control were visualized by represented flow cytometry plots from seven individual replicates. The percentage of Tregs expressing 4–1BB was summarized on a dot plot, and the MFI of 4–1BB in Tregs was summarized on a bar plot. (G to J) The expression of (G) LAG-3, (H) EBI3, (I) IL-10, and (J) NRP1 on BATF KO activated Tregs and scrambled control were visualized by represented flow cytometry plots from five individual replicates. The percentage of Tregs expressing marker genes and the MFI were summarized on a dot plot and a bar plot, respectively. Samples from the same donor were connected by solid lines. Each dot indicates an individual replicate. Bars indicate the median of expression, and error bars represent 1 SD. Data were pooled from five to eight individual replicates. Markers that have fewer replicates than others were subsequently incorporated into the flow panel as the study progressed. Data in (A) and (D) to (J) were analyzed by a ratio paired t test, and data in (B) were analyzed by two-way ANOVA test. P values: ns: P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001, ****P ≤ 0.0001.
Tregs given continuous TCR stimulation under hypoxia mirror intratumoral TNFR+ Tregs and are regulated by BATF
A recent study has shown that continuous TCR stimulation under hypoxia (low oxygen levels) resulted in severe T cell dysfunction consistent with a T cell exhaustion phenotype (51). We then adapted this in vitro system to mimic the persistent antigenic stimulation and metabolic stress in the TME and interrogate the role of BATF in Tregs under these conditions. Briefly, Tregs were isolated from cord blood and stimulated with magnetic beads coated with anti-CD3/anti-CD28 for 24 hours in the presence of IL-2, then washed, and split into four conditions (fig. S13A). Cells were expanded with IL-2 only (“acute” TCR stimulation) or cocultured with anti-CD3/anti-CD28–coated beads and IL-2 (“continuous” TCR stimulation). These Tregs under acute or continuous TCR activation were cultured in either normoxic (18.6% O2) or hypoxic (1.5% O2) gas atmosphere conditions for 10 days. We observed an increase in BATF in Tregs after continuous TCR stimulation under hypoxia compared with Tregs exposed to continuous TCR stimulation or hypoxia alone (Fig. 7, A and B). In addition, we detected a higher frequency of TNFR family members (4–1BB and GITR) on Tregs given continuous TCR stimulation under hypoxia compared with other conditions (Fig. 7, C and D). We also observed that LAG-3 and PD-1 were highly expressed on Tregs given continuous TCR stimulation under hypoxia, consistent with the phenotype of TNFR+ Treg in our scRNA-seq analyses above (Fig. 7, E and F, and fig. S4D). In addition, we also detected a substantive increase in molecules associated with T cell exhaustion (TIM-3 and TOX) in Tregs given continuous TCR stimulation under hypoxia (Fig. 7, G and H). Although KI67 was increased in Tregs given continuous TCR stimulation, there was no difference between normoxic and hypoxic conditions (Fig. 7I). Together, human Tregs cultured in this in vitro system mirrored many of the features of intratumoral TNFR+ Tregs, suggesting that this system provides an opportunity to interrogate the role of BATF in TNFR+ Tregs.
Fig. 7. Human Tregs cultured with continuous TCR stimulation under hypoxia mimic intratumoral BATF-driven TNFR+ Treg phenotypes.
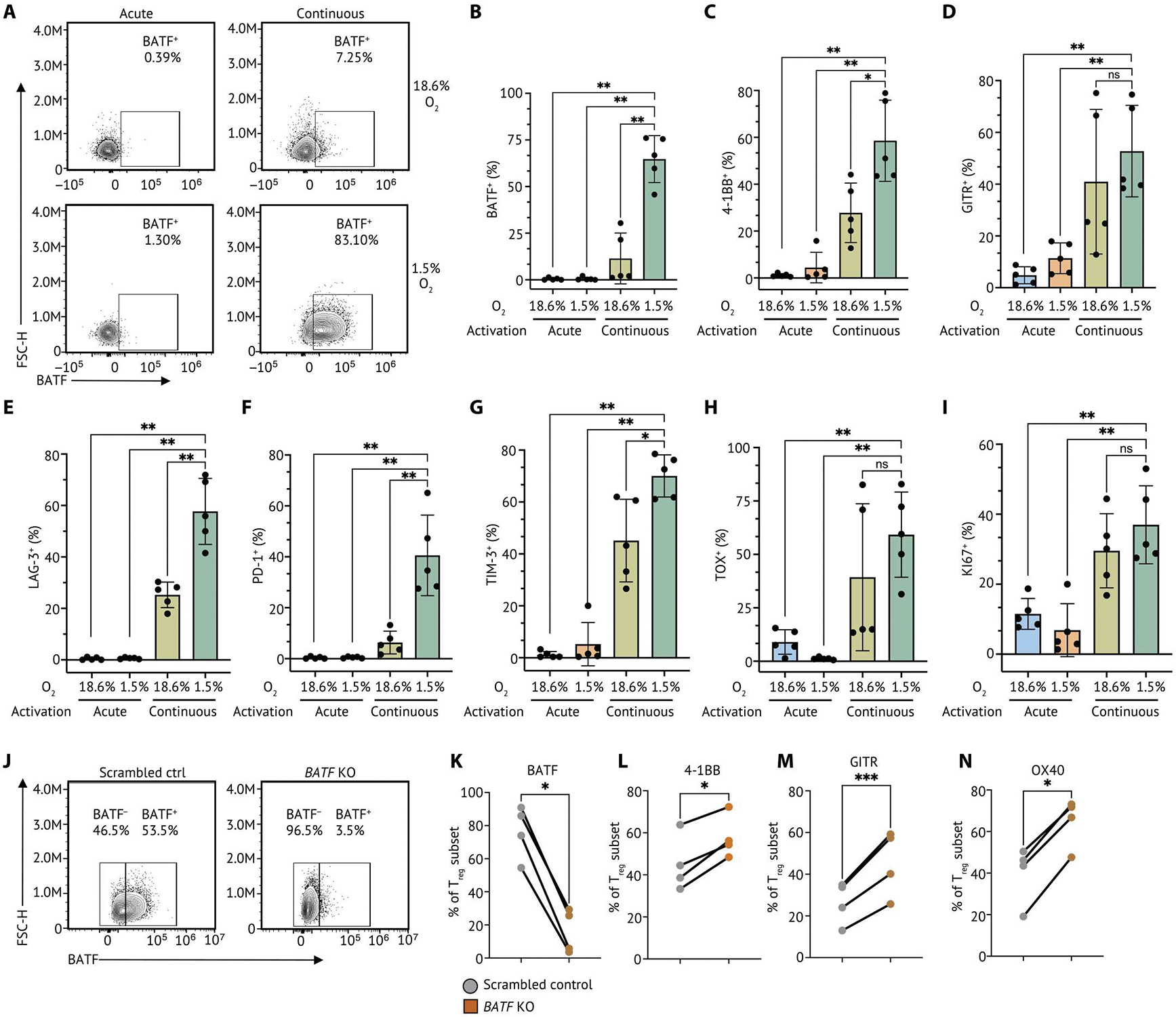
(A) Human primary Tregs isolated from cord blood were cultured in acute TCR stimulation (acute) in normoxia (20% O2), continuous TCR stimulation (continuous) in normoxia, acute stimulation in hypoxia (1.5% O2), and continuous stimulation in hypoxia. Representative flow cytometry plots of five individual replicates showing BATF protein level in Tregs with continuous TCR stimulation under normoxia and hypoxia. (B) A bar plot summarizing the percentage of Tregs expressing BATF identified in Tregs in each culture condition. The percentage of Tregs expressing 4–1BB (C), GITR (D), LAG-3 (E), PD-1 (F), TIM-3 (G), TOX (H), and KI67 (I) identified in TNFR+ Tregs (n = 5) was summarized on a bar plot. Each dot indicates an individual replicate. Bars indicate the median of expression, and error bars represent 1 SD. (J) BATF deletion was conducted by CRISPR RNP KO in human Tregs with continuous TCR stimulation under hypoxia at day 10. Representative flow cytometry plots of four individual replicates showing the proportion of Tregs expressing BATF after 48-hour repeated TCR stimulation. (K) Tabulation of the percentage of Tregs expressing BATF from four individual replicates. (L to N) The percentage of Tregs expressing 4–1BB, GITR, and OX40 was summarized on a dot plot. Samples from the same donor were connected by solid lines, and each dot indicates an individual replicate. Data were pooled from four individual replicates. Data in (B) to (I) were analyzed by a nonparametric Mann-Whitney U test, and data in (K) to (N) were analyzed by a ratio paired t test. P values: ns: P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
To further assess whether BATF serves as a nexus for modulating TNFR+ Tregs, BATF deletion was conducted by CRISPR RNP KO in human Tregs with continuous TCR stimulation under hypoxia at day 10 (fig. S13, A and B). After 48-hour repeated TCR stimulation in hypoxia post-electroporation, a substantive reduction of BATF was observed after CRISPR targeting compared with the scrambled control (Fig. 7, J and K). Consistent with our previous analyses, we detected increased expression of 4–1BB, GITR, and OX40 in BATF KO Tregs, further confirming the important roles of BATF in modulating the activation of TNFR+ Tregs (Figs. 6, F and G, and 7, L to N).
BATF regulates the cell surface phenotype of TNFR+ Tregs
TNFR member genes have been reported in TIL Tregs and are associated with a late differentiated state found in various cancer types (19, 52). Although several agonistic antibodies of TNFR members are under development to target Tregs in the TME, these therapeutic targets may also modulate circulating Tregs and cause immune-relative adverse events (12, 53, 54). Identifying additional surface markers expressed on TNFR+ Tregs is thus warranted to aid future development of immunotherapies with activity specifically in the TME. We were able to detect multiple genes that were differentially expressed on TNFR+ Tregs compared with other Treg subpopulations, including CD96, CD39, and CD74 and TNFR super family genes (TNFRSF9 and TNFRSF4) (Fig. 8A). We observed reduction of CD96 and CD39 in BATF KO activated Tregs and a higher frequency of GITR, CD83, and CD74 (Fig. 8B). The differential expression (DE) changes of CD39 and CD83 were further confirmed in BATF KO Tregs with continuous TCR stimulation in hypoxia (fig. S13, C and D). We next sought to evaluate whether these cell surface markers were reflective of the BATF-driven TNFR+ Tregs in patients with cancer. To assess this, we interrogated PBMCs and TILs from six patients with HNSCC and found higher frequency and increased expression of BATF, OX40, 4–1BB, and GITR on TIL Tregs compared with Tregs in HNSCC PBMCs and HD PBMCs (fig. S14A). The preferential expression of BATF, OX40, 4–1BB, and GITR in HNSCC TIL Tregs remained when comparing with Tregs in the periphery as well as CD8+ T cells and CD4+ Tconv in the HNSCC TME (Fig. 8, C to G, and fig. S14, B to D). In addition, OX40, 4–1BB, and GITR were coexpressed with BATF in HNSCC TIL Tregs, which supported the notion that BATF modulated the identity and activation of TNFR+ Tregs (Fig. 8, E to G, and fig. S14, B to D). We also observed that CD96, CD39, and CD74 were highly expressed in HNSCC TIL Tregs compared with peripheral Tregs, CD8+ T cells and CD4+ Tconv in the HNSCC TME and showed a coexpression signature with BATF in HNSCC TIL Tregs (Fig. 8, H to J, and fig. S14, E to G). These findings revealed a distinct cell surface phenotype of activated Tregs regulated by BATF in the HNSCC TME, which provides additional opportunities for therapeutic intervention targeting intratumoral functionally suppressive Tregs.
Fig. 8. TNFR+ Tregs express BATF-regulated surface markers preferentially in the HNSCC TME.
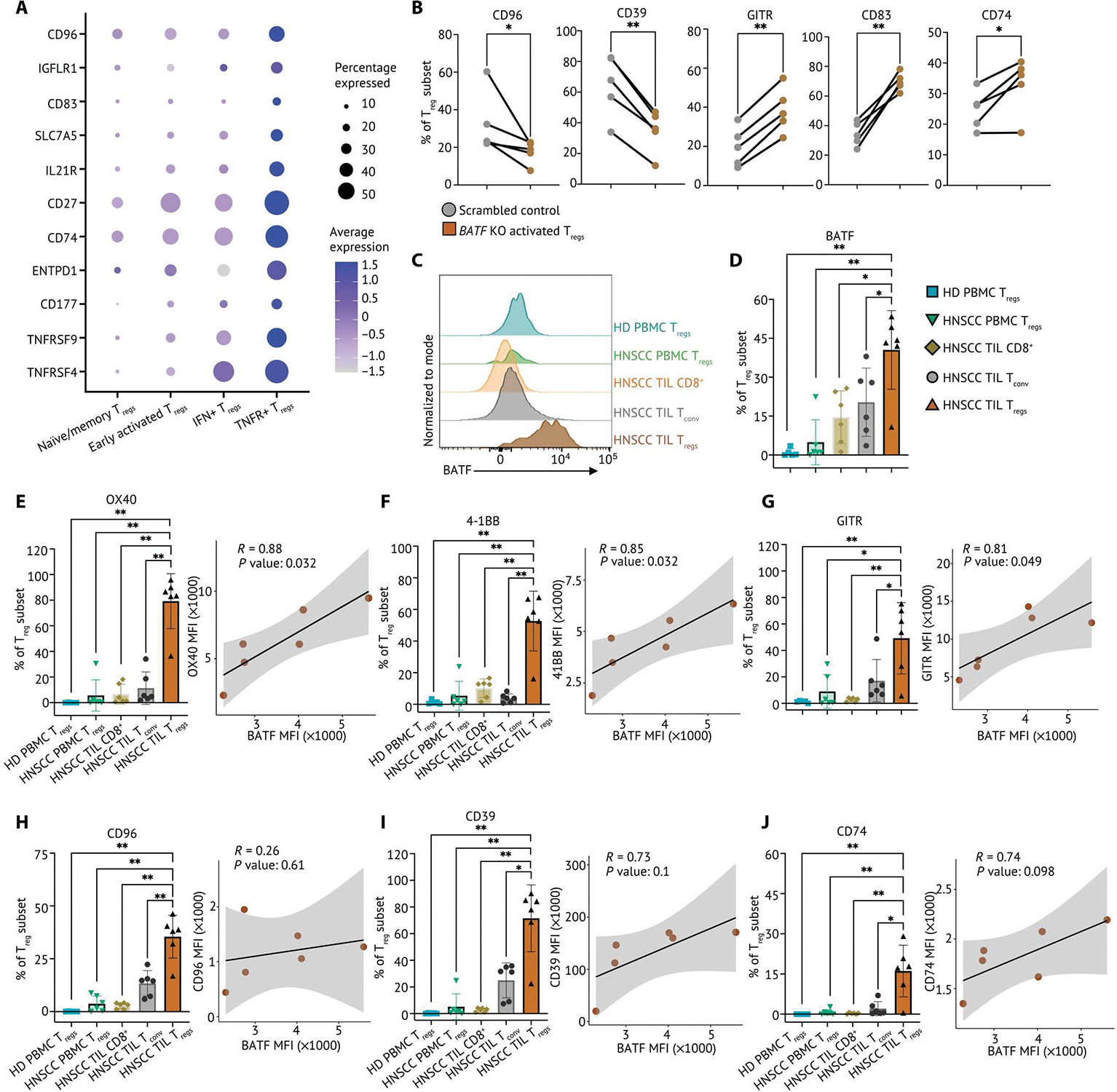
(A) Relative expression of surface markers differentially expressed on TNFR+ Tregs and percentage of Treg subset in the single-cell HNSCC dataset were visualized on a dot plot. (B) Frequency of CD96, CD39, GITR, CD83, and CD74 expression determined by flow cytometry from control and BATF-null human activated Tregs from cord blood (n = 5). Samples from the same donor were connected by solid lines, and each dot indicates an individual replicate. (C) Matched TILs and PBMCs from patients with HNSCC (n = 6) and HD PBMC (n = 5) were stained for flow cytometry phenotyping. Representative flow plot of BATF expression in TILs and PBMCs from patients with HNSCC and HD PBMCs, normalized to mode scales as a percentage of the maximum count. (D) Tabular summary of percentage of cells expressing BATF from patients with HNSCC (n = 6) and HD PBMCs (n = 5). (E to J) The percentage of Tregs expressing surface markers identified in TNFR+ Tregs from matched TILs and PBMCs from patients with HNSCC (n = 6) and HD PBMCs (n = 5) is summarized on a bar plot. Each dot indicates an individual replicate. Bars indicate the median of expression, and error bars represent 1 SD. The coexpression of BATF expression and marker genes by MFI in HNSCC TIL Tregs was shown in scatterplots. The Pearson correlation coefficient (R) between the expression BATF and marker genes was calculated by the MFI in HNSCC TIL Tregs. The significance of Pearson correlation coefficient (P) was calculated by t test. Data in (B) were analyzed by a ratio paired t test, and data in (D) to (J) were analyzed by a nonparametric Mann-Whitney U test. P values: *P ≤ 0.05; **P ≤ 0.01.
DISCUSSION
Tregs are important regulators of immune homeostasis and tissue repair but suppress antitumor immunity (1, 3, 10, 29, 55–58). A previous study revealed conserved effector Treg phenotypes that are shared between normal tissues and melanoma (45). In contrast, we found a Treg subpopulation that was restricted to the TME and that expressed high levels of genes encoding TNFR family member. This Treg subpopulation was associated with worse survival in HNSCC, melanoma, and lung cancer. These TNFR+ Tregs exhibited a highly suppressive phenotype compared with all other Treg subpopulations and had a distinct transcriptional program compared with all Tconv. By further characterizing the heterogeneity of Tregs within the tumor, our results revealed the diversity of cell states that TIL Tregs can acquire, highlighting the urgency of understanding the cell-intrinsic signaling that modulates the phenotypic state of functionally suppressive Tregs. Our work provides a comprehensive view of gene networks coordinating signaling mechanisms essential for key Treg phenotypes at each state.
The reconstruction of GRN by SCENIC and directed MGM allowed us to identify critical TFs and target genes modulating the activation, migration, and function of Tregs that are functionally suppressive in HNSCC TME, specifically intratumoral TNFR+ Tregs. Validation using an additional dataset that included Tregs from lung cancer and melanoma as well as an independent dataset that included Tregs from breast cancer, colorectal cancer, lung cancer, and ovarian cancer (47) further confirmed the presence of intratumoral TNFR+ Tregs. These findings underscore the importance of these intratumoral TNFR+ Tregs and the GRN that governs this cell state across solid tumors. The conservation of phenotypes and the GRN governing them across distinct solid tumor types suggests that modulation of Tregs could be broadly used to therapeutically enhance antitumor immunity in a wide variety of tumor types.
BATF showed the highest gene expression level among all TFs in TIL Tregs and was enriched in TNFR+ Tregs. Although previous studies supported an accumulation of BATF expression during Treg activation within the TME in several cancers (47, 59), the role for BATF in controlling the function and activation of differentiated and activated Tregs in the TME was unclear. BATF is one of the AP-1 family members that has shown important roles in many T cell lineages (60). Within the CD4+ T cell lineage, BATF is required for the differentiation of IL-17–producing helper T cells and plays an important role in the development of follicular helper T cells (61, 62). Furthermore, BATF is required for differentiation of effector CD8+ T cells (60). Studies of CD8+ T cells suggest that BATF positively regulates lineage-specific TFs including TBX21 and PRDM1 during effector cell differentiation while negatively regulating downstream effector molecules such as IFN-γ, perforin, and granzyme B (60). Conversely, increased expression of BATF in exhausted CD8+ T cells suppresses their effector function (63). Such counterintuitive roles of BATF indicate distinct regulatory functions at different T cell states and cell subtypes. In Tregs, BATF has been found to regulate differentiation and accumulation of tissue Tregs and modulate the development and maintenance of adipose tissue resident Tregs in murine Foxp3-mutant and Batf-deficient mouse models (64, 65). A recent study suggested that BATF epigenetically promotes activation of TIL Tregs, and BATF deficiency in Tregs resulted in inhibited tumor growth in murine models (58). Consistent with previous studies, we found that BATF and the BATF integrated network play important roles in controlling key gene signatures that regulate the differentiation and activation of TIL Tregs. However, conditional deletion in Batf-deficient mice occurs before Treg differentiation, complicating the evaluation of transcriptional and functional changes in activated intratumoral Tregs. We hypothesized that BATF exhibits distinct roles at different Treg states and plays an especially important role in highly activated Tregs. Understanding the function of BATF in activated TIL Tregs is necessary to uncover the distinct transcriptional regulations within the highly suppressive Treg subpopulation. Our unbiased network analyses characterized the high expression of BATF as characteristic of highly activated Tregs in the TME and revealed the potential role of BATF in modulating Treg migration, activation, and function within the GRN by regulating CXCR6, TNFRSF4, TNFRSF9, RELB, ICOS, and CTLA4, among others. Consistent with results from other T cell subsets, BATF plays a complex role in governing human activated Treg function. We found that BATF promotes activated Treg survival and cellular trafficking while simultaneously limiting excessive activation of Tregs. Perturbation of BATF signaling in Tregs results in substantial changes of cell surface phenotypes and suppressive function.
Our CRISPR-Cas9 RNP KO approach allowed us to experimentally validate the BATF-driven GRN identified in our scRNA-seq analyses and to determine the functional consequences of modulating this network. We leveraged an in vitro system to mimic the persistent antigenic stimulation and metabolic stress Tregs experience in the TME. These Tregs appeared to mimic features of exhaustion, raising the possibility that this might be a relevant phenotype for intratumoral Tregs, highlighting the value for this system to mimic conditions within the TME. The disruption of BATF in human primary activated Tregs and Tregs with continuous TCR stimulation under hypoxia resulted in distinct phenotypic changes, reflecting the importance of the BATF-driven GRN in intratumoral TNFR+ Tregs. CRISPR-editing results revealed an enhancement of immunosuppression and activation in BATF KO activated Tregs accompanied with increased expression of 4–1BB, OX40, ICOS, LAG3, NRP1, and BCL2. We also observed a reduction of CD39 and CD96, indicating that BATF functions as a transcriptional nexus in human activated Tregs that is essential for Treg activation, function, stability, and survival. Although we went to considerable effort to try to KO BATF in human Tregs isolated by HNSCC samples using current CRISPR technology, we were unable to recover enough cells with acceptable viability. Future studies should seek to disrupt BATF in activated TIL Tregs specifically to interrogate the roles of BATF modulating the activation and function of activated Tregs from patients with cancer.
Therapeutic targeting of TIL Tregs in cancer without affecting immune homeostasis has been challenging. We show that TNFR+ Tregs are frequently found and are associated with worse prognosis across numerous solid tumors. In addition, several surface markers including CD96, CD39, and CD74 were identified on TNFR+ Tregs that are preferentially expressed by HNSCC TIL Tregs, suggesting a possible path to selectively targeting suppressive TIL Tregs without causing overt autoimmunity in normal tissues. A deeper understanding of transcriptional networks that govern Treg function will likely provide opportunities for the treatment of cancer and may identify ways to promote Treg function for the treatment of autoimmunity and inflammatory diseases.
MATERIALS AND METHODS
Study design
The objective of this study was to reconstruct transcriptional networks of suppressive TIL Tregs in the TME and identify key regulators that control TIL Treg function and activation. Tumors from patients with HNSCC were acquired under the University of Pittsburgh Cancer Institute Institutional Review Board (IRB)–approved protocol 99-069, with written informed consent obtained from each patient. Nontumor tonsil tissues from patients with sleep apnea or tonsilitis were collected on the same IRB-approved protocol. Tumors from patients with lung cancer were acquired under IRB–approved protocol MODCR19060269-008. HD peripheral blood was collected by venipuncture, using EDTA as the anticoagulant.
Data file S1, tab data S5, summarizes the clinical characteristics of participants, including their sex and age. scRNA-seq was performed to characterize distinct transcriptional signatures and cell states of TIL Tregs compared with nontumor tissue Tregs, peripheral Tregs, and all Tconv. CIBERSORTx and TCGA survival analyses were performed to identify subpopulations of TIL Tregs that were associated with patient survival outcomes. SCENIC, directed MGM, and FCI-MAX were performed to construct the GRN of TNFR+ Tregs. Bioinformatic analyses on two independent scRNA-seq datasets were performed to validate the findings in the HNSCC TME. CRISPR-Cas9 RNP KO was performed to perturb BATF signaling in human primary activated Tregs. Bulk RNA-seq was performed to globally dissect the transcriptional changes in human activated Tregs with BATF deletion. In vitro microsuppression assays were performed to interrogate the functional changes of BATF KO activated Tregs. Flow cytometry staining with TIL and PBMC from patients were performed to validate and identify additional surface markers that characterize TNFR+ Tregs. Detailed method is included in the Supplementary Materials.
Blood and tissue sample processing
PBMCs were isolated from whole blood by density gradient centrifugation in Ficoll/Hypaque for 20 min at 400g with the brake off. Carryover red blood cells were lysed with BD Pharm Lyse, and samples were then resuspended in staining buffer. Single-cell suspensions from tonsil tissues were generated by mechanical disruption followed by enzymatic digestion in serum-free RPMI media. After initial isolation from tissue, cells were passed through a 100-μm filter and spun down to yield single-cell suspensions. Cells were stained and sorted for live CD4+ Tconv (CD4+CD25− CD127high) and Tregs (CD4+CD25highCD127low) by fluorescence-activated cell sorting (FACS) on the Sony MA900 at the Hillman Cancer Center Cytometry Facility. Data file S1, tab data S6, summarizes the detailed information of antibodies used for cell sorting.
scRNA-seq library preparation and sequencing
Immediately after sorting, Tconv and Tregs were centrifuged for 5 min at 500g and were resuspended in phosphate-buffered saline (PBS) with 0.04% bovine serum albumin. Cells were then counted using the Cellometer Auto2000 (Nexcelom) and loaded into the 10X Controller (10X Genomics) targeting a recovery of 2000 cells per sample. The RNA capture, barcoding, cDNA, and library preparation were performed according to the manufacturer’s recommendations. The 10x libraries were pooled and sequenced on either a NextSeq500 at the Health Sciences Sequencing Core at Children’s Hospital of Pittsburgh or on a NovaSeq6000 at the University of Pittsburgh Medical Center (UPMC) Genome Core. Data file S1 (tab data S1) summarizes the final cell number per participant analyzed in the study after QC.
Treg isolation and expansion
Human umbilical cord samples were collected from the umbilical vein immediately after vaginal delivery by the Obstetric Specimen Procurement Unit at UPMC Magee-Womens Research Institute. PBMC isolation followed the same procedures as described above. Tregs from cord blood PBMCs were enriched by a kit and purified again by FACS. Freshly isolated Tregs were cultured in complete RPMI. Tregs were expanded for 7 days. Data file S1 (tab, Data S6) summarizes the detailed information of antibodies used for cell sorting and T cell activation.
In vitro human Treg culture with TCR stimulation in hypoxia
Tregs from cord blood were isolated as shown above. Tregs were then activated at 20,000 cells per 96-well round-bottom plates with an equivalent number of Dynabeads in the presence of IL-2 (1000 U/ml) in 200 μl of cRPMI. After 24-hour activation, Tregs were expanded with IL-2 only (acute TCR stimulation) or cocultured with 10-fold Dynabeads and IL-2 (continuous TCR stimulation). Tregs under acute or continuous TCR activation were cultured in either normoxic (18.6% O2) or hypoxic (1.5% O2) gas atmosphere conditions for 10 days.
Cas9 RNP assembly and electroporation
crRNA and trans-activating RNA (tracrRNA) were mixed in a 1:1 ratio and incubated for 30 min at 37°C to generate crRNA–tracrRNA CRISPR duplex. Cas9 protein (Thermo Fisher Scientific) was mixed with the crRNA–tracrRNA duplex and incubated for 15 min at 37°C to generate Cas9 RNP. Expanded human Tregs were pelleted and resuspended in P3 buffer. Then, Cas9 RNP and electroporation enhancer (Integrated DNA Technologies) were added directly to the cells and transferred to a 16-well reaction cuvette (Lonza). Tregs were electroporated using program EH-115 on the Amaxa 4D-Nucleofector (Lonza). Prewarmed cRPMI was immediately added to each well after electroporation. CRISPR-edited Tregs were then rested with cRPMI supplemented with human IL-2 (200 UI/ml) for 48 hours at 37°C. CRISPR-edited Tregs and scrambled control Tregs were then activated for 72 hours with TCR stimulation. Data file S1, tab data S7, summarizes the sequences of guides used in the experiments.
In vitro microsuppression assay
Naïve CD8+ T cells were isolated by a T cell isolation kit, and antigen-presenting cells (APCs) were isolated by FACS from HD PBMCs. Isolated naïve CD8+ T cells were then labeled with Cell-Trace Violet (CTV; Invitrogen) for 10 min at 37°C. After a 72-hour TCR restimulation, CRISPR-treated Tregs were purified again by FACS. Two thousand CRISPR-edited Tregs in 50 ml of cRPMI were seeded in the first column of a round-bottom 96-well plate. A serial twofold dilution of Tregs was conducted for nine columns. Two thousand APCs and 2000 CTV-labeled naïve CD8+ T cells in 50 ml of cRPMI were added separately to all wells to change the Tregs:CD8+ T cell ratio from 1:2 to 1:1000. Fifty milliliters of cRPMI supplemented with anti-CD3 (2 μg/ml) were then added to all wells. Cells were cultured for 5 days at 37°C with 5% CO2. Then, cells were spun down and stained for flow cytometry analysis.
Surface and intracellular antibody staining
CRISPR-edited Tregs, scrambled controls, and unperturbed controls were resuspended in staining buffer and labeled with antibodies at 1:100 for 25 min at 4°C, followed by viability staining using fixable dye (eFluor 780 viability dye at 1:4000 or Zombie NIR at 1:2000 ratio). Cells were then spun down at 500g for 5 min and washed with PBS. Foxp3/Transcription Factor Staining Buffer (eBioscience) was added into cells for 60 min at room temperature for cell fixation. Cells were then washed by permeabilization buffer (eBioscience) and labeled with antibodies for 60 min at room temperature for intracellular staining. Cells were washed with permeabilization buffer and FACS buffer. The BD LSRFortessa II flow cytometer or Cytek Aurora was used for acquiring flow cytometry readouts, and FlowJo V10 was used for data analysis. Data file S1, tab data S6, summarizes the detailed information of antibodies used for flow cytometry.
Bulk RNA-seq
CRISPR-edited Tregs and scrambled controls were double-sorted (purity, >99.5%) directly into individual wells of a 96-well plate containing 2 μl of lysis buffer. The plate was spun down at 2000g for 2 min, and reverse transcription was then performed. An addition of 15-cycle complementary DNA amplification was performed after cDNA synthesis by the KAPA Hot Start II High-Fidelity DNA Polymerase. cDNA size was verified with TapeStation D5000 and quantified by the Qubit (Thermo Fisher Scientific). Sequencing libraries were prepared from 1 ng of cDNA using the Nextera XT DNA Library Prep kit (Illumina FC-131-1096), following the manufacturer’s instructions. cDNA libraries were quantified by the KAPA library quantification kit (KAPA KK4854), and size was verified on a TapeStation D1000. Ten diluted libraries were pooled and sequenced with the NextSeq 500/550 High Output v.2 kit.
Bulk RNA-seq data analysis
We used the R package DESeq2 (1.40.1) for downstream bulk RNA-seq analysis (66). The batch effects between donors were corrected by ComBat-seq (v3.36.0) (67). Genes were filtered as not expressed if their read count was less than 10. Differential gene analysis was done by DESeq function in DESeq2 (1.40.1). Log2 fold changes were added into the analysis by lfcShrink function in DESeq2 (1.40.1). Gene set enrichment analysis was performed by using R package clusterProfiler (4.8.1) (68).
scRNA-seq data integration and clustering
We used the R package Seurat v4 for downstream scRNA-seq analysis. The dataset was split by patient and normalized using SCTransform v1 (23). The most variable genes in each patient were identified, and the top 2000 variable genes overlapped across groups were selected for integration. Then, we scaled the integrated dataset to remove confounding sources of variation by regressing out the percentage of mitochondrial genes expressed per cell and the number of genes per cell. Further, we used principal components analysis (PCA) to dimensionally reduce the dataset to 30 dimensions and chose 1 to 20 PCs that explained the most variance in the dataset for visualization and cell clustering. We next performed Louvain graph–based clustering and applied a nonlinear dimensional reduction method UMAP to visualize the dataset. Tconv and Tregs were identified on the basis of the original paper and previous sorting results.
DE analysis and gene set enrichment analysis
We next used FindAllMarkers function in Seurat to run DE analysis between different groups of T cell subpopulations depending on the comparison. The top DE genes in each subpopulation were ranked on the basis of the log2 fold change. We then performed the gene set enrichment analysis to Tregs in the dataset by using the R package singleseqgset as previously described (19). The log2 fold change in gene expression was calculated for all genes across Treg clusters and used as input for a variance inflation–corrected Wilcoxon rank sum test to calculate whether the gene sets were up-regulated in a concerted manner within a cluster.
GRN reconstruction
To reconstruct the GRNs within Treg subpopulations, we used SCENIC to infer regulons with normalized data matrices as input. The regulons were generated, and the regulon activities were calculated following the pySCENIC (0.11.2) pipeline (46). We identified the differentially activated regulons in each Treg subpopulation by the Wilcoxon rank sum test against all the other cells. To identify GRNs central to TNFR+ Treg subpopulation, we combined the target genes and regulators in the top five regulons ranked by log2 fold change and used normalized data metrices as input for the causal MGM (42). We used MGM to build a skeleton network and then FCI-MAX to refine the network and determine correlation (69). Signaling pathways were analyzed by Reactome (70).
Pseudotime analysis
To infer Treg developmental trajectory, we applied the RNAvelocity algorithm to infer pseudotime for each Treg (26, 71). The differentiation trajectory and pseudotime inference were performed by scanpy (version 1.6.1) and scVelo (v0.2.4) (26). To infer the developmental trajectory of Tregs from the online published datasets, we applied the Slingshot (72) algorithm to infer pseudotime for each Treg. Briefly, we took the first three PCs from the PCA generated during cell clustering procedure (as described above) and normalized the dataset for input into the R package Slingshot. We used the predefined clustering results to identify the global lineage structure by a cluster-based minimal spanning tree, fitted principal curves to describe each lineage, and lastly aligned cells on a pseudotime trajectory.
Survival analysis using TCGA
To determine whether the enrichment of Treg subpopulations was associated with survival outcomes of patients with cancer, we used bulk RNA-seq data of patients with HNSCC, NSCLC, and melanoma available through TCGA. Similarly, bulk RNA-seq data of normal lung and normal skin (sun exposed) were obtained from GTEx. The enrichment score for each Treg subpopulation from each patient was calculated as previously described (73, 74). The gene set was defined by using the top 200 genes that were differentially expressed in each Treg subpopulation. Enrichment scores were calculated and used as a reference profile to deconvolute bulk RNA-seq data from TCGA and GTEx by CIBERSORTx (30). We then performed monovariate and multivariable analyses based on patients’ PFS.
Statistical analysis
Specific statistical analyses including Wilcoxon rank sum test for DE gene analysis, Cox proportional hazard regression for monovariate and multivariable analyses, and Pearson’s correlation for gene coexpression and Treg signature analysis were performed using R-4.3.1. Statistical analyses, including two-way analysis of variance (ANOVA) test, nonparametric Mann-Whitney U test, and ratio paired t test for flow cytometry analyses, were performed using GraphPad/Prism v9.
Supplementary Material
Acknowledgments:
We thank members of the Vignali, Bruno, and Benos Labs for constructive comments and feedback. We thank the Delgoffe Lab for support of the in vitro T cell culture with TCR stimulation in hypoxia. We thank C. Sander and E. Rush and the Kirkwood lab for assistance in providing clinical samples from patients with melanoma. We thank J. Ward for assistance in providing clinical samples from patients with lung cancer. We thank D. Liu for assistance in coordination and collection of lung cancer specimens. We thank the Hillman Cytometry Facility for assistance with flow cytometry and the Immunology Flow Core for cell sorting. We thank Obstetric Specimen Procurement Unit at UPMC Magee-Womens Research Institute for cord blood samples. This research was supported in part by the University of Pittsburgh Center for Research Computing through the resources provided. Graphical abstract and experimental schematics were designed using Biorender.com.
Funding:
This work was supported by the National Institutes of Health {R35 CA263850, P50 CA254865, and P50 CA097190 [D.A.A.V.], R01 HL159805 and R01 HL157879 [P.V.B.], the Cancer Immunology Training Program T32 [T32 CA082084 (D.A.A.V.), awarded to A.R.C. and D.Y.], and Hillman Postdoctoral Fellowship for Innovative Cancer Research [A.R.C.]}.
Footnotes
Competing interests: D.A.A.V. is cofounder and stockholder of Novasenta, Potenza, Tizona, and Trishula; stockholder of Oncorus and Werewolf; has patents licensed and royalties from BMS and Novasenta; scientific advisory board member of Tizona, Werewolf, F-Star, Bicara, Apeximmune, and T7/Imreg Bio; is a consultant for BMS, Incyte, Regeneron, Ono Pharma, and Avidity Partners; and obtained research funding from BMS and Novasenta. T.C.B. receives research funding for Alkermes and Pfizer and is a consultant for Walking Fish Therapeutics, iTeos Therapeutics, and BeSpoke Therapeutics. A.R.C. is a consultant for AboundBio. A.P. is an investigator in a research grant to UPMC from Novasenta. J.D.L. is an investigator in a research grant to UPMC from Novasenta. All other authors declare that they have no competing interests.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Codes for all custom algorithms are publicly available in GitHub repositories at: https://github.com/shf43/BATF_Treg_Network. The sequencing datasets generated in this study are deposited at the Gene Expression Omnibus: GSE239750. The previously published datasets discussed in this publication are GSE139324, E-MTAB-8107, E-MTAB-6149, and E-MTAB-6653.
REFERENCES AND NOTES
- 1.Vignali DA, Collison LW, Workman CJ, How regulatory T cells work. Nat. Rev. Immunol. 8, 523–532 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD, The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 27, 20–21 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Liu C, Workman CJ, Vignali DAA, Targeting regulatory T cells in tumors. FEBS J. 283, 2731–2748 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Scott EN, Gocher AM, Workman CJ, Vignali DAA, Regulatory T cells: Barriers of immune infiltration into the tumor microenvironment. Front. Immunol. 12, 702726 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu S, Foulkes WD, Leung S, Gao D, Lau S, Kos Z, Nielsen TO, Prognostic significance of FOXP3+ tumor-infiltrating lymphocytes in breast cancer depends on estrogen receptor and human epidermal growth factor receptor-2 expression status and concurrent cytotoxic T-cell infiltration. Breast Cancer Res. 16, 432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shang B, Liu Y, Jiang S-J, Liu Y, Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 5, 15179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W, Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 10, 942–949 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Leffers N, Gooden MJM, de Jong RA, Hoogeboom BN, ten Hoor KA, Hollema H, Boezen HM, van der Zee AGJ, Daemen T, Nijman HW, Prognostic significance of tumor-infiltrating T-lymphocytes in primary and metastatic lesions of advanced stage ovarian cancer. Cancer Immunol. Immunother. 58, 449–459 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.West NR, Kost SE, Martin SD, Milne K, deLeeuw RJ, Nelson BH, Watson PH, Tumour-infiltrating FOXP3+ lymphocytes are associated with cytotoxic immune responses and good clinical outcome in oestrogen receptor-negative breast cancer. Br. J. Cancer 108, 155–162 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shan F, Somasundaram A, Bruno TC, Workman CJ, Vignali DAA, Therapeutic targeting of regulatory T cells in cancer. Trends Cancer 8, 944–961 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ Jr., Colligon TA, Trosko JA, Leinbach LI, Pletcher CH, Tweed CK, DeMichele A, Fox KR, Domchek SM, Riley JL, Vonderheide RH, CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci. Transl. Med. 4, 134ra162 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zappasodi R, Sirard C, Li Y, Budhu S, Abu-Akeel M, Liu C, Yang X, Zhong H, Newman W, Qi J, Wong P, Schaer D, Koon H, Velcheti V, Hellmann MD, Postow MA, Callahan MK, Wolchok JD, Merghoub T, Rational design of anti-GITR-based combination immunotherapy. Nat. Med. 25, 759–766 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirschhorn-Cymerman D, Rizzuto GA, Merghoub T, Cohen AD, Avogadri F, Lesokhin AM, Weinberg AD, Wolchok JD, Houghton AN, OX40 engagement and chemotherapy combination provides potent antitumor immunity with concomitant regulatory T cell apoptosis. J. Exp. Med. 206, 1103–1116 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dépis F, Hu C, Weaver J, McGrath L, Klebanov B, Buggé J, Umiker B, Fregeau C, Upadhyay D, Singh A, Xu CA, Spaulding V, Priess M, Wong M, Naheed S, Zhang Y, Legendre K, Stack EC, Mora A, Willer M, Meetze K, Gostissa M, Meehl MA, Shaffer DR, Abstract 4532: Preclinical evaluation of JTX-1811, an anti-CCR8 antibody with enhanced ADCC activity, for preferential depletion of tumor-infiltrating regulatory T cells. Cancer Res. 80, 4532–4532 (2020). [Google Scholar]
- 15.Friedman C, Ascierto P, Davar D, O’Hara M, Shapira-Frommer R, Dallos M, Khemka V, James L, Fischer B, Demes S, Li L, Kozicki M, Ravindran P, Xu K, Kollia G, Shoukry J, Yunan M, Massey A, Gutierrez M, 393 First-in-human phase 1/2a study of the novel nonfucosylated anti–CTLA-4 monoclonal antibody BMS-986218 ± nivolumab in advanced solid tumors: Initial phase 1 results. J. Immunother. Cancer 8, A239 (2020). [Google Scholar]
- 16.Infante JR, Hansen AR, Pishvaian MJ, Chow LQM, McArthur GA, Bauer TM, Liu SV, Sandhu SK, Tsai FYC, Kim J, Stefanich E, Li CC, Gilbert H, MCCALL B, Anderson MS, Huseni M, Rhee IP, Siu LL, Gordon MS, A phase Ib dose escalation study of the OX40 agonist MOXR0916 and the PD-L1 inhibitor atezolizumab in patients with advanced solid tumors. J. Clin. Oncol. 34, 101–101 (2016). [Google Scholar]
- 17.Diab A, Hamid O, Thompson JA, Ros W, Eskens FALM, Doi T, Hu-Lieskovan S, Klempner SJ, Ganguly B, Fleener C, Wang X, Joh T, Liao K, Salek-Ardakani S, Taylor CT, Chou J, el-Khoueiry AB, A Phase I, Open-Label, Dose-Escalation Study of the OX40 Agonist Ivuxolimab in Patients with Locally Advanced or Metastatic Cancers. Clin. Cancer Res. 28, 71–83 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacobs JFM, Punt CJA, Lesterhuis WJ, Sutmuller RPM, Brouwer HM-LH, Scharenborg NM, Klasen IS, Hilbrands LB, Figdor CG, de Vries IJM, Adema GJ, Dendritic cell vaccination in combination with anti-CD25 monoclonal antibody treatment: A phase I/II study in metastatic melanoma patients. Clin. Cancer Res. 16, 5067–5078 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Cillo AR, Kürten CHL, Tabib T, Qi Z, Onkar S, Wang T, Liu A, Duvvuri U, Kim S, Soose RJ, Oesterreich S, Chen W, Lafyatis R, Bruno TC, Ferris RL, Vignali DAA, Immune Landscape of Viral- and Carcinogen-Driven Head and Neck Cancer. Immunity 52, 183–199.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, Maeda Y, Hamaguchi M, Ohkura N, Sato E, Nagase H, Nishimura J, Yamamoto H, Takiguchi S, Tanoue T, Suda W, Morita H, Hattori M, Honda K, Mori M, Doki Y, Sakaguchi S, Two FOXP3+CD4+ T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med. 22, 679–684 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, Parizot C, Taflin C, Heike T, Valeyre D, Mathian A, Nakahata T, Yamaguchi T, Nomura T, Ono M, Amoura Z, Gorochov G, Sakaguchi S, Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 30, 899–911 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Chen X, Subleski JJ, Kopf H, Howard OMZ, Männel DN, Oppenheim JJ, Cutting edge: Expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+ T regulatory cells: Applicability to tumor-infiltrating T regulatory cells. J. Immunol. 180, 6467–6471 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hao Y, Hao S, Andersen-Nissen E, Mauck III WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, McElrath JM, Blish CA, Gottardo R, Smibert P, Satija R, Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vasanthakumar A, Liao Y, Teh P, Pascutti MF, Oja AE, Garnham AL, Gloury R, Tempany JC, Sidwell T, Cuadrado E, Tuijnenburg P, Kuijpers TW, Lalaoui N, Mielke LA, Bryant VL, Hodgkin PD, Silke J, Smyth GK, Nolte MA, Shi W, Kallies A, The TNF receptor superfamily-NF-κB axis is critical to maintain effector regulatory t cells in lymphoid and non-lymphoid tissues. Cell Rep. 20, 2906–2920 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Lambert SA, Jolma A, Campitelli LF, das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, Weirauch MT, The Human Transcription Factors. Cell 172, 650–665 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Bergen V, Lange M, Peidli S, Wolf FA, Theis FJ, Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408–1414 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Sjaastad LE, Owen DL, LaRue RS, Farrar MA, Interferons in Treg development and function. J. Immunol. 204, 228.17 (2020). [Google Scholar]
- 28.Levine AG, Mendoza A, Hemmers S, Moltedo B, Niec RE, Schizas M, Hoyos BE, Putintseva EV, Chaudhry A, Dikiy S, Fujisawa S, Chudakov DM, Treuting PM, Rudensky AY, Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature 546, 421–425 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gocher-Demske AM, Cui J, Szymczak-Workman AL, Vignali KM, Latini JN, Pieklo GP, Kimball JC, Avery L, Cipolla EM, Huckestein BR, Hedden L, Meisel M, Alcorn JF, Kane LP, Workman CJ, Vignali DAA, IFNγ-induction of TH1-like regulatory T cells controls antiviral responses. Nat. Immunol. 24, 841–854 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D, Diehn M, Alizadeh AA, Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 37, 773–782 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J, van den Oord J, Atak ZK, Wouters J, Aerts S, SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Konopacki C, Pritykin Y, Rubtsov Y, Leslie CS, Rudensky AY, Transcription factor Foxp1 regulates Foxp3 chromatin binding and coordinates regulatory T cell function. Nat. Immunol. 20, 232–242 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sebzda E, Zou Z, Lee JS, Wang T, Kahn ML, Transcription factor KLF2 regulates the migration of naive T cells by restricting chemokine receptor expression patterns. Nat. Immunol. 9, 292–300 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Zemmour D, Zilionis R, Kiner E, Klein AM, Mathis D, Benoist C, Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat. Immunol. 19, 291–301 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Z, Zheng Y, Hou C, Yang L, Li X, Lin J, Huang G, Lu Q, Wang CY, Zhou Z, DNA methylation impairs TLR9 induced Foxp3 expression by attenuating IRF-7 binding activity in fulminant type 1 diabetes. J. Autoimmun. 41, 50–59 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Roessner PM, Llaó Cid L, Lupar E, Roider T, Bordas M, Schifflers C, Arseni L, Gaupel AC, Kilpert F, Krötschel M, Arnold SJ, Sellner L, Colomer D, Stilgenbauer S, Dietrich S, Lichter P, Izcue A, Seiffert M, EOMES and IL-10 regulate antitumor activity of T regulatory type 1 CD4+ T cells in chronic lymphocytic leukemia. Leukemia 35, 2311–2324 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruan Q, Chen YH, Nuclear factor-κB in immunity and inflammation: The Treg and Th17 connection. Adv. Exp. Med. Biol. 946, 207–221 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Ronin E, Lubrano di Ricco M, Vallion R, Divoux J, Kwon HK, Grégoire S, Collares D, Rouers A, Baud V, Benoist C, Salomon BL, The NF-κB RelA Transcription Factor Is Critical for Regulatory T Cell Activation and Stability. Front. Immunol. 10, 2487 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grinberg-Bleyer Y, Caron R, Seeley JJ, de Silva NS, Schindler CW, Hayden MS, Klein U, Ghosh S, The Alternative NF-κB Pathway in Regulatory T Cell Homeostasis and Suppressive Function. J. Immunol. 200, 2362–2371 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh Y, Garden OA, Lang F, Cobb BS, MicroRNAs regulate T-cell production of interleukin-9 and identify hypoxia-inducible factor-2α as an important regulator of T helper 9 and regulatory T-cell differentiation. Immunology 149, 74–86 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsu T-S, Lin YL, Wang YA, Mo ST, Chi PY, Lai ACY, Pan HY, Chang YJ, Lai MZ, HIF-2α is indispensable for regulatory T cell function. Nat. Commun. 11, 5005 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sedgewick AJ, Buschur K, Shi I, Ramsey JD, Raghu VK, Manatakis DV, Zhang Y, Bon J, Chandra D, Karoleski C, Sciurba FC, Spirtes P, Glymour C, Benos PV, Mixed graphical models for integrative causal analysis with application to chronic lung disease diagnosis and prognosis. Bioinformatics 35, 1204–1212 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plaza-Sirvent C, Schuster M, Neumann Y, Heise U, Pils MC, Schulze-Osthoff K, Schmitz I, c-FLIP Expression in Foxp3-Expressing Cells Is Essential for Survival of Regulatory T Cells and Prevention of Autoimmunity. Cell Rep. 18, 12–22 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Beharry Z, Mahajan S, Zemskova M, Lin YW, Tholanikunnel BG, Xia Z, Smith CD, Kraft AS, The Pim protein kinases regulate energy metabolism and cell growth. Proc. Natl. Acad. Sci. U.S.A. 108, 528–533 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miragaia RJ, Gomes T, Chomka A, Jardine L, Riedel A, Hegazy AN, Whibley N, Tucci A, Chen X, Lindeman I, Emerton G, Krausgruber T, Shields J, Haniffa M, Powrie F, Teichmann SA, Single-Cell Transcriptomics of Regulatory T Cells Reveals Trajectories of Tissue Adaptation. Immunity 50, 493–504.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van de Sande B, Flerin C, Davie K, De Waegeneer M, Hulselmans G, Aibar S, Seurinck R, Saelens W, Cannoodt R, Rouchon Q, Verbeiren T, De Maeyer D, Reumers J, Saeys Y, Aerts S, A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 15, 2247–2276 (2020). [DOI] [PubMed] [Google Scholar]
- 47.Qian J, Olbrecht S, Boeckx B, Vos H, Laoui D, Etlioglu E, Wauters E, Pomella V, Verbandt S, Busschaert P, Bassez A, Franken A, Bempt MV, Xiong J, Weynand B, van Herck Y, Antoranz A, Bosisio FM, Thienpont B, Floris G, Vergote I, Smeets A, Tejpar S, Lambrechts D, A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 30, 745–762 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schumann K, Raju SS, Lauber M, Kolb S, Shifrut E, Cortez JT, Skartsis N, Nguyen VQ, Woo JM, Roth TL, Yu R, Nguyen MLT, Simeonov DR, Nguyen DN, Targ S, Gate RE, Tang Q, Bluestone JA, Spitzer MH, Ye CJ, Marson A, Functional CRISPR dissection of gene networks controlling human regulatory T cell identity. Nat. Immunol. 21, 1456–1466 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Z, Chen Y, Zhou J, Li Y, Gong C, Wang X, Netrin-1 reduces lung ischemia-reperfusion injury by increasing the proportion of regulatory T cells. J. Int. Med. Res. 48, 300060520926415 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delgoffe GM, Woo SR, Turnis ME, Gravano DM, Guy C, Overacre AE, Bettini ML, Vogel P, Finkelstein D, Bonnevier J, Workman CJ, Vignali DAA, Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature 501, 252–256 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scharping NE, Rivadeneira DB, Menk AV, Vignali PDA, Ford BR, Rittenhouse NL, Peralta R, Wang Y, Wang Y, DePeaux K, Poholek AC, Delgoffe GM, Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 22, 205–215 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng L, Qin S, Si W, Wang A, Xing B, Gao R, Ren X, Wang L, Wu X, Zhang J, Wu N, Zhang N, Zheng H, Ouyang H, Chen K, Bu Z, Hu X, Ji J, Zhang Z, Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 374, abe6474 (2021). [DOI] [PubMed] [Google Scholar]
- 53.Lam JH, Hong M, Koo SL, Chua CWL, Lim KL, Wee F, Wan WK, Leow WQ, Yeo JG, Tan IBH, Yeong J, Lim TKH, Lim TS, CD30+OX40+ Treg is associated with improved overall survival in colorectal cancer. Cancer Immunol. Immunother. 70, 2353–2365 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sukumar S, Wilson DC, Yu Y, Wong J, Naravula S, Ermakov G, Riener R, Bhagwat B, Necheva AS, Grein J, Churakova T, Mangadu R, Georgiev P, Manfra D, Pinheiro EM, Sriram V, Bailey WJ, Herzyk D, McClanahan TK, Willingham A, Beebe AM, Sadekova S, Characterization of MK-4166, a Clinical Agonistic Antibody That Targets Human GITR and Inhibits the Generation and Suppressive Effects of T Regulatory Cells. Cancer Res. 77, 4378–4388 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Tanaka A, Sakaguchi S, Targeting Treg cells in cancer immunotherapy. Eur. J. Immunol. 49, 1140–1146 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Boothby IC, Cohen JN, Rosenblum MD, Regulatory T cells in skin injury: At the crossroads of tolerance and tissue repair. Sci. Immunol. 5, eaaz9631 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Traxinger BR, Richert-Spuhler LE, Lund JM, Mucosal tissue regulatory T cells are integral in balancing immunity and tolerance at portals of antigen entry. Mucosal Immunol. 15, 398–407 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Itahashi K, Irie T, Yuda J, Kumagai S, Tanegashima T, Lin YT, Watanabe S, Goto Y, Suzuki J, Aokage K, Tsuboi M, Minami Y, Ishii G, Ohe Y, Ise W, Kurosaki T, Suzuki Y, Koyama S, Nishikawa H, BATF epigenetically and transcriptionally controls the activation program of regulatory T cells in human tumors. Sci. Immunol. 7, eabk0957 (2022). [DOI] [PubMed] [Google Scholar]
- 59.Yan M, Hu J, Yuan H, Xu L, Liao G, Jiang Z, Zhu J, Pang B, Ping Y, Zhang Y, Xiao Y, Li X, Dynamic regulatory networks of T cell trajectory dissect transcriptional control of T cell state transition. Mol. Ther. Nucleic Acids 26, 1115–1129 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kurachi M, Barnitz RA, Yosef N, Odorizzi PM, DiIorio MA, Lemieux ME, Yates K, Godec J, Klatt MG, Regev A, Wherry EJ, Haining WN, The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat. Immunol. 15, 373–383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ise W, Kohyama M, Schraml BU, Zhang T, Schwer B, Basu U, Alt FW, Tang J, Oltz EM, Murphy TL, Murphy KM, The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat. Immunol. 12, 536–543 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schraml BU, Hildner K, Ise W, Lee WL, Smith WAE, Solomon B, Sahota G, Sim J, Mukasa R, Cemerski S, Hatton RD, Stormo GD, Weaver CT, Russell JH, Murphy TL, Murphy KM, The AP-1 transcription factor Batf controls TH17 differentiation. Nature 460, 405–409 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Quigley M, Pereyra F, Nilsson B, Porichis F, Fonseca C, Eichbaum Q, Julg B, Jesneck JL, Brosnahan K, Imam S, Russell K, Toth I, Piechocka-Trocha A, Dolfi D, Angelosanto J, Crawford A, Shin H, Kwon DS, Zupkosky J, Francisco L, Freeman GJ, Wherry EJ, Kaufmann DE, Walker BD, Ebert B, Haining WN, Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat. Med. 16, 1147–1151 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hayatsu N, Miyao T, Tachibana M, Murakami R, Kimura A, Kato T, Kawakami E, Endo TA, Setoguchi R, Watarai H, Nishikawa T, Yasuda T, Yoshida H, Hori S, Analyses of a Mutant Foxp3 Allele Reveal BATF as a Critical Transcription Factor in the Differentiation and Accumulation of Tissue Regulatory T Cells. Immunity 47, 268–283.e9 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R, Kawamoto S, Fagarasan S, Mielke LA, Afshar-Sterle S, Masters SL, Nakae S, Saito H, Wentworth JM, Li P, Liao W, Leonard WJ, Smyth GK, Shi W, Nutt SL, Koyasu S, Kallies A, The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat. Immunol. 16, 276–285 (2015). [DOI] [PubMed] [Google Scholar]
- 66.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Y, Parmigiani G, Johnson WE, ComBat-seq: Batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2, lqaa078 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, Fu X, Liu S, Bo X, Yu G, clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb) 2, 100141 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Raghu VK, Ramsey JD, Morris A, Manatakis DV, Sprites P, Chrysanthis PK, Glymour C, Benos PV, Comparison of strategies for scalable causal discovery of latent variable models from mixed data. Int. J. Data Sci. Anal. 6, 33–45 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gillespie M, Jassal B, Stephan R, Milacic M, Rothfels K, Senff-Ribeiro A, Griss J, Sevilla C, Matthews L, Gong C, Deng C, Varusai T, Ragueneau E, Haider Y, May B, Shamovsky V, Weiser J, Brunson T, Sanati N, Beckman L, Shao X, Fabregat A, Sidiropoulos K, Murillo J, Viteri G, Cook J, Shorser S, Bader G, Demir E, Sander C, Haw R, Wu G, Stein L, Hermjakob H, D’Eustachio P, The reactome pathway knowledgebase 2022. Nucleic Acids Res. 50, D687–D692 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, Fan J, Borm LE, Liu Z, van Bruggen D, Guo J, He X, Barker R, Sundström E, Castelo-Branco G, Cramer P, Adameyko I, Linnarsson S, Kharchenko PV, RNA velocity of single cells. Nature 560, 494–498 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Street K, Risso D, Fletcher RB, das D, Ngai J, Yosef N, Purdom E, Dudoit S, Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Q, Chikina M, Szymczak-Workman AL, Horne W, Kolls JK, Vignali KM, Normolle D, Bettini M, Workman CJ, Vignali DAA, LAG3 limits regulatory T cell proliferation and function in autoimmune diabetes. Sci. Immunol. 2, eaah4569 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ruffin AT, Cillo AR, Tabib T, Liu A, Onkar S, Kunning SR, Lampenfeld C, Atiya HI, Abecassis I, Kürten CHL, Qi Z, Soose R, Duvvuri U, Kim S, Oesterrich S, Lafyatis R, Coffman LG, Ferris RL, Vignali DAA, Bruno TC, B cell signatures and tertiary lymphoid structures contribute to outcome in head and neck squamous cell carcinoma. Nat. Commun. 12, 3349 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Codes for all custom algorithms are publicly available in GitHub repositories at: https://github.com/shf43/BATF_Treg_Network. The sequencing datasets generated in this study are deposited at the Gene Expression Omnibus: GSE239750. The previously published datasets discussed in this publication are GSE139324, E-MTAB-8107, E-MTAB-6149, and E-MTAB-6653.