

Abstract
We disclose the development of a Cu-catalyzed C–O coupling method utilizing a new N1,N2-diarylbenzene-1,2-diamine ligand, L8. Under optimized reaction conditions, structurally diverse aryl and heteroaryl bromides underwent efficient coupling with a variety of alcohols at room temperature using an L8-based catalyst. Notably, the L8-derived catalyst exhibited enhanced activity when compared to the L4-based system previously disclosed for C–N coupling, namely the ability to functionalize aryl bromides containing acidic functional groups. Mechanistic studies demonstrate that C–O coupling utilizing L8•Cu involves rate-limiting alkoxide transmetallation, resulting in a mechanism of C–O bond formation that is distinct from previously described Pd-, Cu-, or Ni-based systems. This lower energy pathway leads to rapid C–O bond formation; a 7-fold increase relative to what is seen with other ligands. The results presented in this report overcome limitations in previously described C–O coupling methods and introduce a new ligand that we anticipate may be useful in other Cu-catalyzed C–heteroatom bond-forming reactions.
Keywords: Cu Catalysis, C–O Coupling, Ligand Design
Graphical Abstract
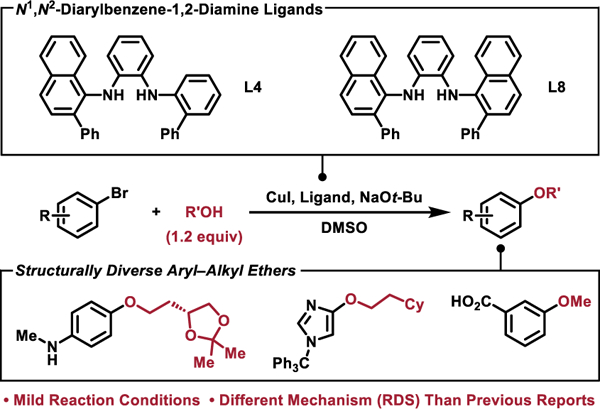
A room-temperature Cu-catalyzed C–O coupling method has been developed utilizing a new N1,N2-diarylbenzene-1,2-diamine ligand. These reactions feature mild reaction conditions, no need to use excess alcohol, and a unique mechanism that does not feature rate-determining reductive elimination.
Introduction
Aryl–alkyl ether bond construction is one of the most frequently employed reactions in the pharmaceutical industry (Figure 1A).[1–4] These linkages are often formed via classical substitution chemistry, such as the Williamson ether synthesis,[5,6] Mitsunobu reaction,[7,8] or by nucleophilic aromatic substitution.[9,10] Alternative promising methods involve sulfonate ester transfer[11] or the use of diaryliodonium salts.[12–14] Despite the utility of these techniques, only specific and/or limited classes of substrates can undergo efficient C–O bond formation. As a result, transition-metal-catalyzed approaches to form C–O bonds have been explored to improve the functional group tolerance and substrate scope of these reactions, while simultaneously utilizing milder reaction conditions.[15]. Our group and others have developed various phosphine ligands that have enabled the Pd-catalyzed coupling of many classes of aryl halide electrophiles with alcohol nucleophiles.[3,16–21] In particular, Pd catalysts derived from bulky ligands such as RockPhos,[20] AdCyBrettPhos,[22,23] and t-BuBrettPhos[24] enable efficient C–O bond formation by facilitating the rate-determining reductive elimination from a proposed Pd(II) intermediate (Figure 1B).[25,26] In the absence of bulky ancillary ligands, many Pd-catalyzed protocols suffer from competitive β-hydride elimination of the Pd(II)-bound alkoxide, as a consequence of slow reductive elimination, to form undesired carbonyl byproducts and LPd(Ar)H, which leads to the overall protodehalogenation of the aryl halide.[27–29] In addition to the inefficient reductive elimination of C–O bonds, the cost and issues of removal of Pd have motivated the use of alternative metal catalysts.[30,31] In particular, Ni-catalyzed methods to form aryl–alkyl ethers have received recent attention. MacMillan has reported metallophotoredox conditions to affect these transformations.[32] Additionally, the use of (Cy)PAd-DalPhos-based Ni-precatalysts have been employed by Stradiotto to effectively promote C –O couplings without the use of photoredox catalysis (Figure 1B).[33,34] Baran has disclosed the Ni-electrocatalytic O–arylation of alcohols that enabled the coupling of substrates that were challenging using Pd- or Cu-catalyzed methods (Figure 1B).[35] Despite the diminished cost of Ni-based catalysts their toxicity remains a primary concern,[36,37] especially in the preparation of active pharmaceutical ingredients (APIs) where metal-catalyzed coupling reactions are often utilized.[38–40] Additionally, these Ni-catalyzed methods necessitate the use, with few exceptions, of ≥2 equiv of alcohols, which complicates their use in late-stage pharmaceutical development or when non-commercial alcohols are employed.
Figure 1.
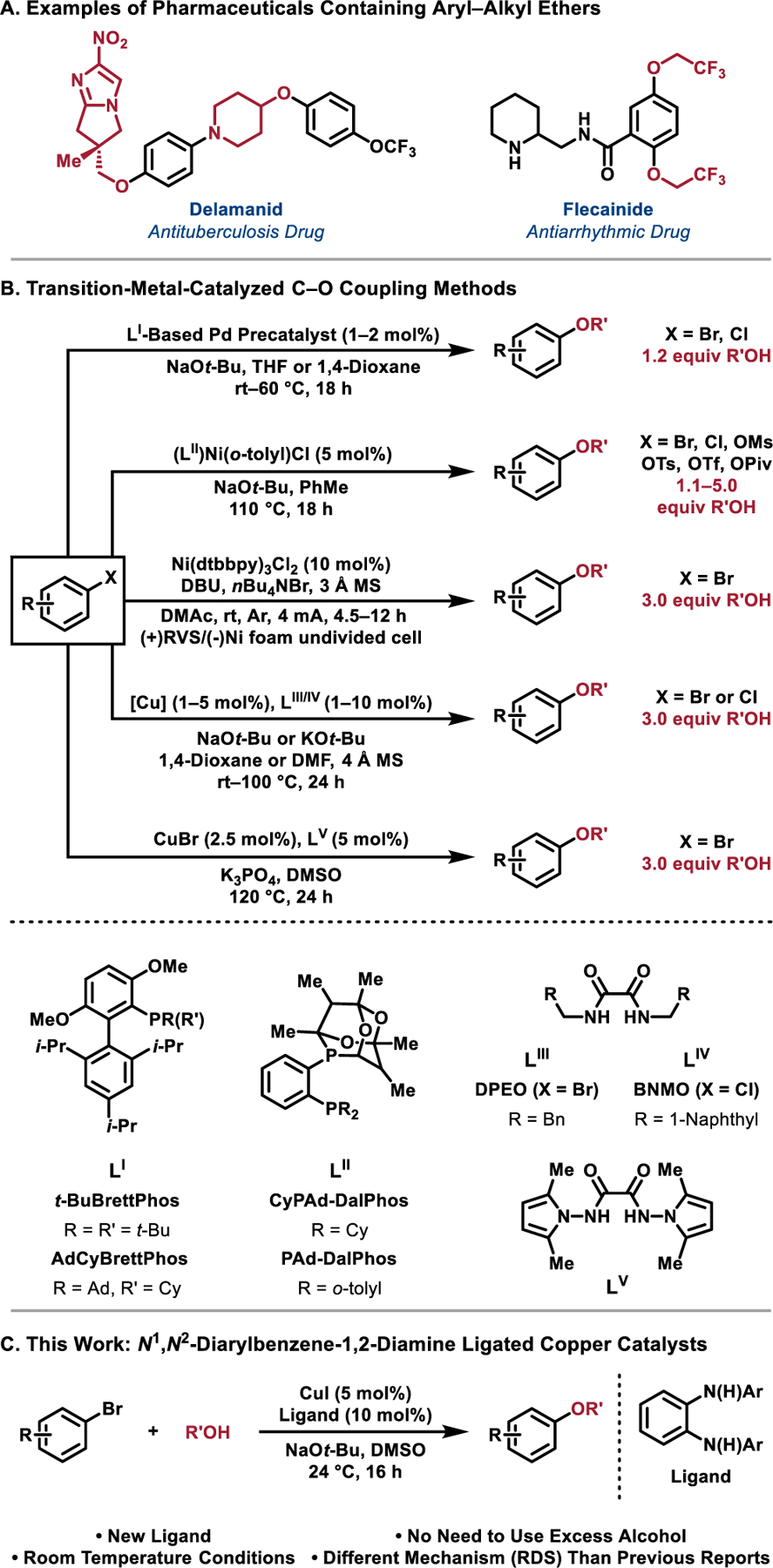
Transition-metal-catalyzed C–O bond forming reactions. (A) Selected examples of pharmaceuticals containing aryl–alkyl ethers. (B) Transition-metal-catalyzed C –O coupling methods employing Pd-, Ni-, or Cu-based catalysts. (C) This work in which N1,N2-diarylbenzene-1,2-diamine ligands are utilized to construct C–O bonds under mild reaction conditions and with minimal excess of alcohol.
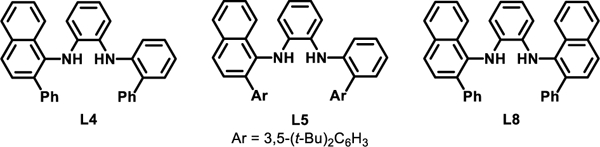
In light of the limitations of Pd- and Ni-catalyzed methods, we sought to develop improved transition-metal-catalyzed C–O coupling methods. In particular, we considered Cu-based catalysts promising candidates to offer complimentary reactivity to these systems. Cu-based catalysts rarely undergo β-hydride elimination without an external redox mediator[41–44] and are known to catalyze cross-coupling reactions involving aryl halide electrophiles via rapid reductive elimination of Cu(III) intermediates.[45–47] Utilizing neutral ligands on Cu, such as 1,10-phenanthroline,[48] BINAM,[49] or diol-based systems,[50] several reports on the coupling of aryl halides and aliphatic alcohols have emerged. Many of these methods rely on harsh reaction conditions to promote efficient oxidative addition of the aryl halide to the Cu catalyst and were either performed in neat alcohol or with a significant molar excess of alcohol. In developing new Cu-catalyzed methods it became apparent that minimizing the reliance on excess alcohol would enable a broader adoption of the method, particularly in the preparation of highly functionalized aryl–alkyl ethers where using a large molar excess of alcohol is not practical nor desirable. In analogous C–N coupling methods, anionic ligands accessed through deprotonation under the basic reaction conditions have demonstrated the ability to lower the energetic barrier to oxidative addition, relative to what it observed with neutral ligands.[51] These advances have resulted in C–N coupling methods that utilize milder reaction conditions and display improved functional group tolerance.[52–58] Building on methods utilizing N,N-dimethylglycine as the ancillary ligand on Cu,[59] Ma pioneered the use of oxalamide ligands for the coupling of various aryl halides with aliphatic alcohols and phenol derivatives (Figure 1B).[58,60–63] Anionic ligands based on the oxalohydrazide[64] and oxalamide[65] scaffold have also been employed by Hartwig for the Cu-catalyzed preparation of a wide variety of diaryl ethers, along with select examples of aryl–alkyl ethers (Figure 1B). Recently, we disclosed the DFT-guided development of N1,N2-diarylbenzene-1,2-diamine ligands that promoted the Cu-catalyzed amination of aryl bromides under mild reaction conditions.[51] Through the combined effects of increasing the electron density on Cu and stabilizing the active anionic catalyst via non-covalent interactions, these ligands enabled these C–N coupling reactions to proceed efficiently at room temperature, with minimal excess of the amine coupling partner. In an effort to further expand the chemistry enabled by N1,N2-diarylbenzene-1,2-diamine ligands, we aimed to develop an analogous method that would enable the C–O coupling of aryl bromides with aliphatic alcohols.
Results and Discussion
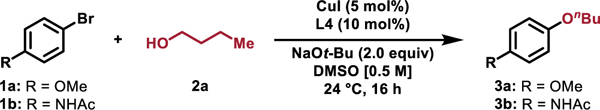
We began our investigations by optimizing the reaction of 4-bromoanisole (1a) with n-butanol (2a, 1.2 equiv), to afford n-butyl ether 3a (Table 1, Entry 1). Utilizing the L4-derived Cu catalyst previously reported for C–N coupling reactions,[51] 3a was formed in quantitative yield at room temperature with NaOt-Bu as the base and DMSO as the solvent ([1a] = 0.5 M). Previously, we reported that L5, a variant of L4 possessing bis-3,5-t-Bu-phenyl groups, in some cases led to enhanced reactivity in the C–N coupling of aryl bromides and alkyl amines.[51] However, using an L5-derived catalyst resulted in the minimal formation of 3a (Table 1, Entry 2), likely due to the previously described inefficient formation of the active L5-based catalyst when NaOt-Bu was employed.[51] Consistent with the required deprotonation of the N–H bonds in L4 to form the active catalyst, the use of weaker inorganic bases, such as K2CO3, resulted in 0% conversion (Table 1, Entry 3). However, the use of NaOTMS, a mild base that is still able to deprotonate L4,[51] also resulted in no reaction (Table 1, Entry 4). Together, these observations suggest that the deprotonation of 2a is likely not facilitated by coordination to Cu, in contrast to what is observed in many Pd- or Ni-catalyzed methods.[22,23,34] In addition, the use of alternative alkoxide bases with diminished steric profiles, such as NaOi-Pr and NaOMe (Table 1, Entry 5), resulted in poor yields of 3a due to the competitive formation of isopropyl and methyl ether products, respectively. Unfortunately, utilizing the L4-derived catalyst provided little or no yield of product when aryl bromides containing acidic functional groups were utilized. As an example, attempts to couple N-(4-bromophenyl)acetamide (1b) with n-butanol 2a resulted in no formation of the desired ether 3b under a variety of reaction conditions including at elevated reaction temperatures.[66] We next examined several new N1,N2-diarylbenzene-1,2-diamine ligands and identified L8 as a potential ligand. Using the initially optimized reaction conditions with L8, 3b was formed in 33% yield (Table 1, Entry 9). Increasing the reaction temperature to 40 °C and increasing both the quantity of CuI and L8 further increased the yield to 76% (Table 1, Entry 10). The use of L8 was also successful for other challenging substrates including carboxylic acid and aryl amine-containing aryl bromides.[66] Further optimizing the coupling of 1a and 2a by surveying alternative Cu-sources identified that using either Cu(I) or Cu(II) salts led to excellent yields of 3a (Table 1, Entries 12 and 13).
Table 1.
Optimization of Cu-catalyzed C–O coupling[a]
| |||
|---|---|---|---|
|
| |||
| Entry | ArBr | Deviation from Standard | Yield (%) |
| 1 | 1a | None | 99% |
| 2 | 1a | Ligand = L5 | 3% |
| 3 | 1a | Base = K2CO3 | 0% |
| 4 | 1a | Base = NaOTMS | 0% |
| 5 | 1a | Base = NaOMe | 25% |
| 6 | 1a | Solvent = 50% THF; 50% DMSO | 95% |
|
| |||
| 7 | 1b | None | 0% |
| 8 | 1b | Temperature = 40 °C | 0% |
| 9 | 1b | Ligand = L8 | 33% |
| 10 | 1b | 10 mol% CuI; 20 mol% L8; 40 °C | 76% |
|
| |||
| 11 | 1a | Ligand = L8 | 99% |
| 12 | 1a | Ligand = L8; [Cu] = CuBr | 98% |
| 13 | 1a | Ligand = L8; [Cu] = CuBr2 | 96% |
| 14 | 1a | Ligand = L8; [Cu] = CuBr; Air | 0% |
| 15 | 1a | No CuI | 0% |
| 16 | 1a | No L8 | 0% |
|
| |||
| |||
Standard reaction conditions: 4-bromoanisole (1a, 0.10 mmol, 1.0 equiv), n-butanol (2a, 0.12 mmol, 1.2 equiv), NaOt-Bu (0.20 mmol, 2.0 equiv), CuI (5 mol%), L4 (10 mol%), DMSO (0.2 mL), 24 °C, 16 h. Yields were determined by 1H NMR spectroscopy of the crude reaction mixtures using 1,1,2,2-tetrachloroethane as an internal standard.
Recently, Hartwig reported an elegant study on the coupling of aryl halides and phenols that proceeded through oxidative addition to a Cu(II) complex, aided by electron delocalization onto the oxalamide ligand backbone.[65] Despite the utility of Cu(II) salts in facilitating the formation of 3a, attempts to run the C–O coupling reaction open to the air resulted in 0% conversion (Table 1, Entry 14). The lack of air stability, along with the observation of an induction period in the reaction kinetics when Cu(II) salts were utilized, is consistent with an active Cu(I) catalyst accessed through rapid reduction of the Cu(II) source.[66] Furthermore, DFT calculations highlight that a lower energy barrier to oxidative addition is accessed when proceeding through an active Cu(I) catalyst (23.5 kcal/mol) as opposed to an active Cu(II) catalyst (28.9 kcal/mol) analogous to that reported in the Hartwig paper. Omitting either CuI or L8 from the reaction mixture resulted in 0% conversion (Table 1, Entries 15 and 16), an observation that is inconsistent with trace Pd from the preparation of L8 (solely) responsible for the observed catalysis.
With optimized reaction conditions in hand, we determined the scope of compatible aryl bromides and alcohols in C–O coupling reactions utilizing the catalyst derived from L8. The reaction was found to proceed at room temperature for a range of aryl bromides. Both electron-rich and electron-poor substrates were readily coupled with structurally diverse primary (1a, 1c, 1d, 1e, 1g, 1h, 1i, 1j, 1l, 1o, 1p) and secondary alcohols (1b) in good-to-excellent yields. The reaction exhibited tolerance for many common functional groups as shown in Scheme 1, most notably a tert-butyl ester (1a), pendant heterocycle (1b), N-aryl amine with an acidic proton (1d), carboxylic acid (1f), nitrile (1g), Boc-protected secondary amine (1g), secondary amide (1l), and an alkyl fluoride (1o). For aryl bromides containing non-tert-butyl esters, transesterification with the alcohol coupling partner was observed.[66] However, this limitation could be circumvented in a few cases by matching the identity of the ester to the alcohol nucleophile, such as in the case of 1i, which formed in excellent yield. The presence of ortho substituents on the aryl bromide, such as a methyl group (1e), and even an isopropyl group (1c), as well as an aryl chloride (1j) were well tolerated. Chromane (1k) was also prepared in excellent yield via an intramolecular C–O coupling. Lastly, the coupling of tert-butanol with 4-bromoanisole was facilitated by the L8-derived catalyst in moderate yield (1m). Despite this observation, attempts to utilize alternative tertiary alcohols resulted in mixtures of the desired aryl–alkyl ether and the tert-butyl ether arising from reductive elimination of the NaOt-Bu base with the oxidative addition complex. Efforts to optimize the coupling of tertiary alcohols are underway in our laboratory. Collectively, the ability to prepare a variety of structurally diverse aryl–alkyl ethers in good to excellent yield highlights the utility of this protocol.
Scheme 1.
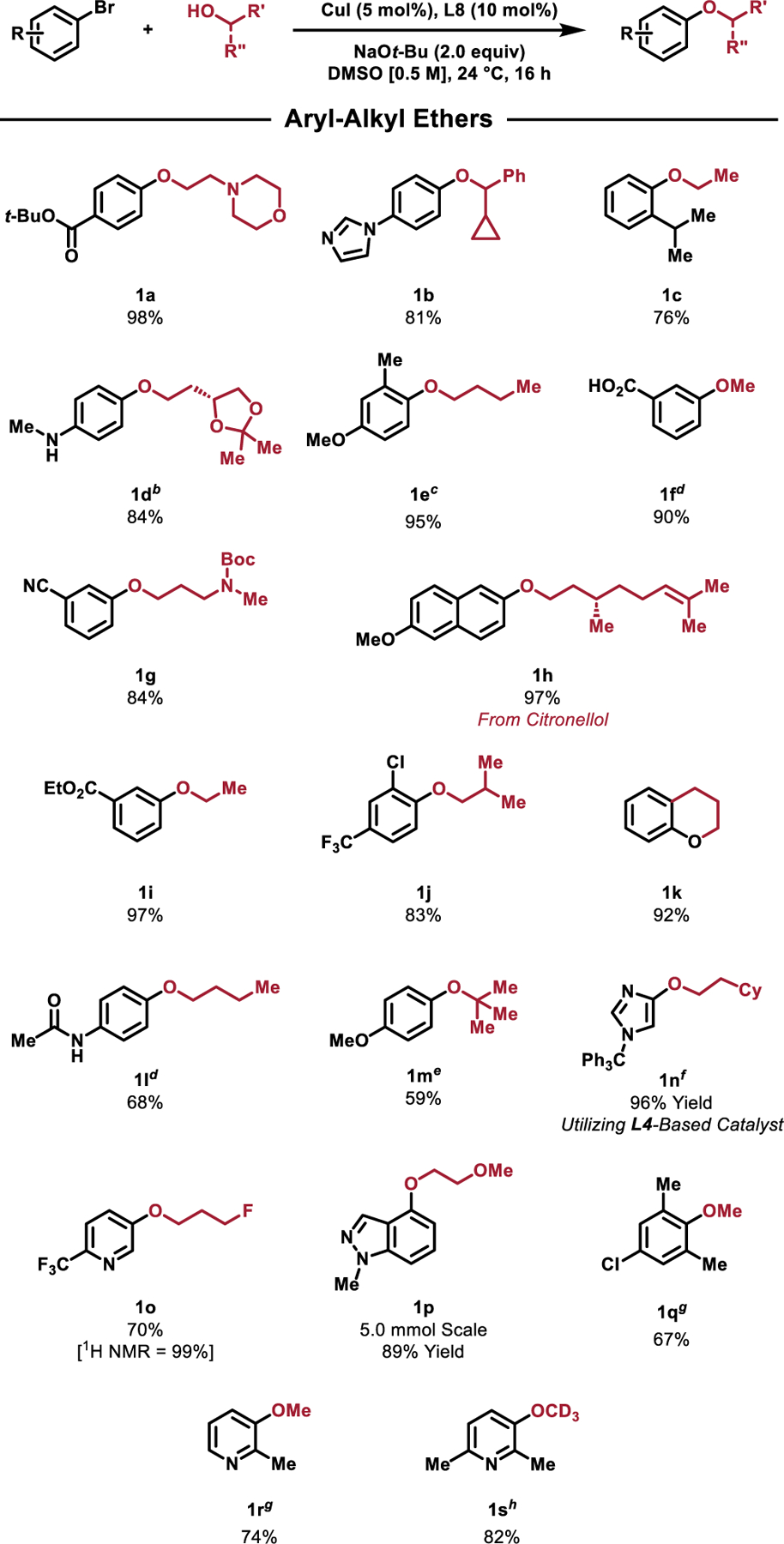
Substrate scope of the Cu-catalyzed C–O coupling between aliphatic alcohols and aryl bromidesa
aAll yields represent the average of two isolated yields. Standard reaction conditions: aryl bromide (1.0 mmol), alcohol (1.2 mmol, 1.2 equiv), NaOt-Bu (2.0 mmol, 2.0 equiv), CuI (5.0 mol%), L4 (10 mol%), DMSO (2.0 mL), 24 °C, 16 h. bReaction temperature = 40 °C cCuI (10.0 mol%), L4 (20 mol%). dCuI (10.0 mol%), L4 (20 mol%), 40 °C. e CuI (10.0 mol%), L8 (20 mol%), t-BuOH (2.4 mmol, 2.4 equiv). fL4 is used in place of L8. Solvent = 1:1 THF/DMSO. gBase = NaOMe hBase = NaOt-Bu.
A variety of heteroaryl bromides also underwent coupling with primary alcohols to yield diverse heteroaryl–alkyl ethers in good-to-excellent yields. Heterocycles which often present difficulties in metal-catalyzed coupling reactions,[67] due to their propensity to coordinate to and subsequently deactivate the metal catalyst, were found to be suitable substrates in reactions with primary and primary allylic alcohols. In this way, C–O bonds were formed utilizing pyridine (1o, 1r, 1s), indazole (1p), and imidazole (1n) based heteroaryl bromides. In the case of coupling 1-trityl-4-bromoimidazole with 2-cyclohexylethan-1-ol we note that the use of the L4-derived catalyst resulted in the formation of 1n in excellent yield, while the L8-derived catalyst yielded poor conversion of the initial aryl bromide (33%). We currently have no explanation for this unusual result. The ability to form 1n in excellent yield is particularly noteworthy, as 1,3-azoles unsubstituted at the 2-position are often prone to decomposition by strong alkoxide bases.[68,69] The newly developed protocol involving the L8-based catalyst system was found to be amenable to preparative-scale synthesis. Using standard Schlenk techniques, the coupling of 4-bromoindazole and 2-methoxyethanol was scaled to 5.0 mmol to provide 1p in excellent yield (89%, 0.92 g). Unsurprisingly, aryl–methyl ethers could be prepared utilizing methanol/NaOMe. Additionally, the coupling of methanol with 2-bromo-5-chloro-1,3-dimethylbenzene resulted exclusively in the formation of 1q, thereby highlighting the chemoselectivity of this coupling protocol. The use of d4-deuterated methanol enabled access to the isotopically enriched anisole derivates in good yield (1s). These products are of interest due to their enhanced metabolic stability and have been featured in several pharmaceuticals and clinical candidates.[70–73] Limitations include reactions with arenes containing unprotected phenols or enolizable ketones that led to little or no formation of the desired product. As was observed in the previously reported C–N coupling reaction that utilized L4,[51] heterocycles featuring an unprotected N–H heterocycle, such as 5-bromoindole, were unreactive, presumably as a result of catalyst deactivation by the deprotonated heterocycle.[66]
To elucidate the basis for the increased reactivity of the L8-based catalyst relative to the L4-based system we investigated the mechanisms of C–O coupling with these ligands. Empirical rate laws for the coupling of 4-bromoanisole and n-butanol catalyzed by either L4- or L8-ligated Cu were determined using initial rate measurements (Figure 2A-C). For the model reaction using the L4-based catalyst, a zero-order dependence on the concentration of alcohol was observed. In addition, the reaction was found to exhibit a positive first-order dependence on the concentrations of both 4-bromoanisole and CuI/L4. Finally, an inverse first-order dependence on the concentration of NaOt-Bu was recorded. Based on previous DFT calculations, [51] we propose that in situ formed [Cu(Ot-Bu)2]− deprotonates one of the N–H bonds of the diamine ligand to form a ligated complex [LCuI(Ot-Bu)]− that represents the resting state of the catalytic cycle. Before productive catalysis can take place, tert-butoxide must dissociate sequentially with the second N–H deprotonation of the ligand to form the active catalyst, consistent with the inverse first-order dependence of the reaction rate on the concentration of NaOt-Bu. Taken together, these kinetic data imply that the reaction catalyzed by the L4-based catalyst proceeds via a resting state featuring a Cu-bound tert-butoxide and a rate-limiting sequence involving dissociation of the alkoxide followed by oxidative addition to the aryl bromide. In addition to probing the reaction kinetics, a Hammett analysis of the reaction rates of various 4-substituted bromobenzene derivatives revealed a positive ρ value (+0.31) which is consistent with oxidative addition being at least partially rate-limiting and is in line with observations made in related Cu-catalyzed transformations including our previously reported amination reactions employing the same ligand (Figure 2E).[51,74,75] In contrast to the observations made for reactions catalyzed by Cu•L4, the reaction rate for the coupling of 4-bromoanisole and n-butanol utilizing the L8-based catalyst was, unexpectedly, found to be independent of the concentration of 4-bromoanisole. However, the reaction exhibited a positive first-order dependence on the concentration of n-butanol (Figure 2B). In analogy to reactions utilizing L4, reactions with L8 exhibited a positive first-order dependence on the concentration of CuI and L8 and an inverse first-order dependence on the concentration of NaOt-Bu. Finally, a Hammett analysis of the reaction rates of various 4-substituted bromobenzene derivatives displayed no discernable trend. These observations are consistent with (1) a change in mechanism between reactions with catalysts derived from L4 and L8 to invert the order of alkoxide transmetallation and oxidative addition or (2) a change in the rate-limiting step of the transformation. Given the bidentate and bisanionic nature of L8, oxidative addition to a Cu(III) alkoxide intermediate is not mechanistically reasonable. Collectively, these results are consistent with a post-oxidative addition, rate-limiting alkoxide transmetallation when L8 is employed (Figure 2F). Notably, this mechanistic discrepancy is accompanied by a 7-fold rate increase when utilizing L8 in place of L4 (Figure 2D). One explanation for the observed rate enhancement when using the catalyst derived from L8 is that it is more stable than the L4-based analog. This enhanced stability could lead to an increased concentration of the active catalyst. One pathway that commonly results in deactivation of ligands within this class is ligand arylation arising from reductive elimination of the ligand and arene after oxidative addition. DFT calculations predict that the energy barrier to this deactivation process is substantially higher in the L8 system (20.1 kcal/mol, Figure 2H) relative to the L4 system (14.3 kcal/mol, Figure 2G). The change in mechanism when the L8-based catalyst is used is particularly noteworthy as it represents one of the few examples in which reductive elimination is not rate-determining in transition-metal-catalyzed C–O coupling.
Figure 2.
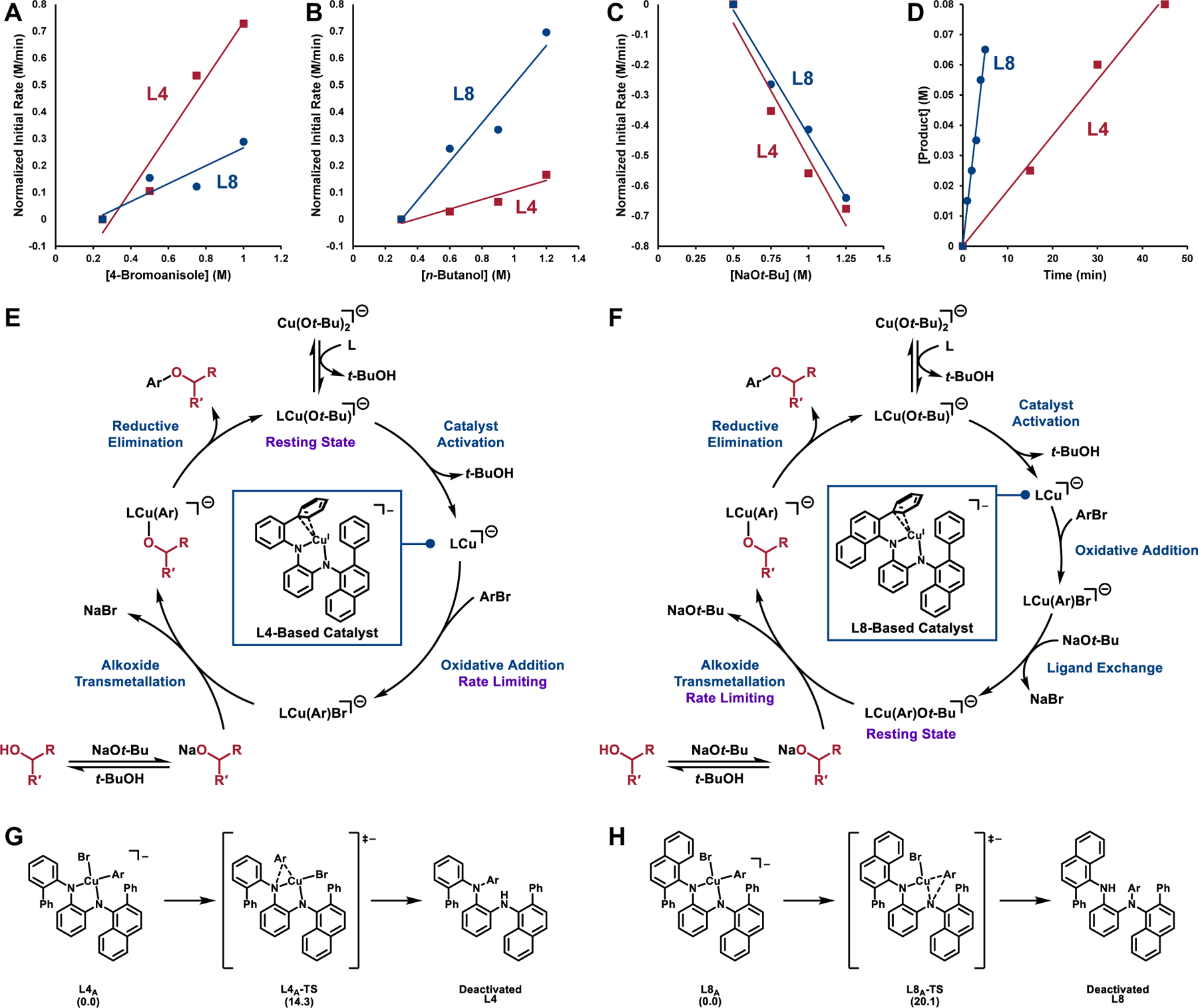
Mechanistic analyses of the Cu-catalyzed C–O bond-forming reactions utilizing L4 and L8. Comparison of the normalized initial rates of the coupling of 4-bromoanisole and n-butanol employing L4 (red) and L8 (blue) in the presence of varying equivalents of (A) 4-bromoanisole, (B) n-Butanol, and (C) NaOt-Bu. (D) Product formation as a function of time in the coupling of 4-bromoanisole and n-butanol utilizing L4- (red) or L8-based (blue) catalysts. (E) Proposed mechanism for C–O bond formation when L4 is used. (F) Proposed mechanism for C–O bond formation when L8 is used. (G) Energy barrier to catalyst deactivation via ligand arylation in an L4-based catalyst. All energies are expressed in kcal/mol. (H) Energy barrier to catalyst deactivation via ligand arylation in an L8-based catalyst. All energies are expressed in kcal/mol.
In addition to studying the mechanisms of reactions utilizing L4 and L8, we sought to understand the ability of L8-based catalysts to catalyze the etherification of substrates bearing acidic protons (i.e., secondary amides, carboxylic acids, and aniline derivatives). To probe the effects these substrates have on reactions catalyzed by the L4-based catalyst, the coupling of 4-bromoanisole and n-butanol was first allowed to proceed for 90 minutes. After this time, 1-butoxy-4-methoxybenzene was formed in 25% yield (as estimated by 1H NMR) and N-(4-bromophenyl)acetamide (1.0 equiv in 0.1 mL DMSO) was added to the reaction mixture that was subsequently allowed to stir for an additional 14.5 hours. Analysis of the reaction mixture revealed no additional product formation during this time, consistent with inhibition of the catalyst by the deprotonated amide (expected yield without addition of 4a = 99%, Figure 3). In an analogous experiment using the catalyst derived from L8, no catalyst inhibition was observed upon addition of 4a. In order to monitor the structure of the ligand that was employed throughout the reaction, fluorinated analogous of both ligands (L4F and L8F) were prepared. Utilizing these ligands, the reaction of 4-bromoanisole and n-butanol was monitored using 19F NMR spectroscopy. In both cases, a single major peak was observed, which we attribute to the resting state of the catalyst.[66] Upon addition of N-(4-bromophenyl)acetamide (1.0 equiv in 0.1 mL DMSO) to these reaction mixtures, a new major species was formed in the case of L4, and no change in the spectrum of the L8•Cu-catalyzed reaction was observed. Taken together, these observations are consistent with the proposal that the L8-based catalyst features rate-limiting alkoxide transmetallation and exhibits a stronger interaction with Cu that renders the catalyst more resistant to deactivation via ligand displacement.
Figure 3.
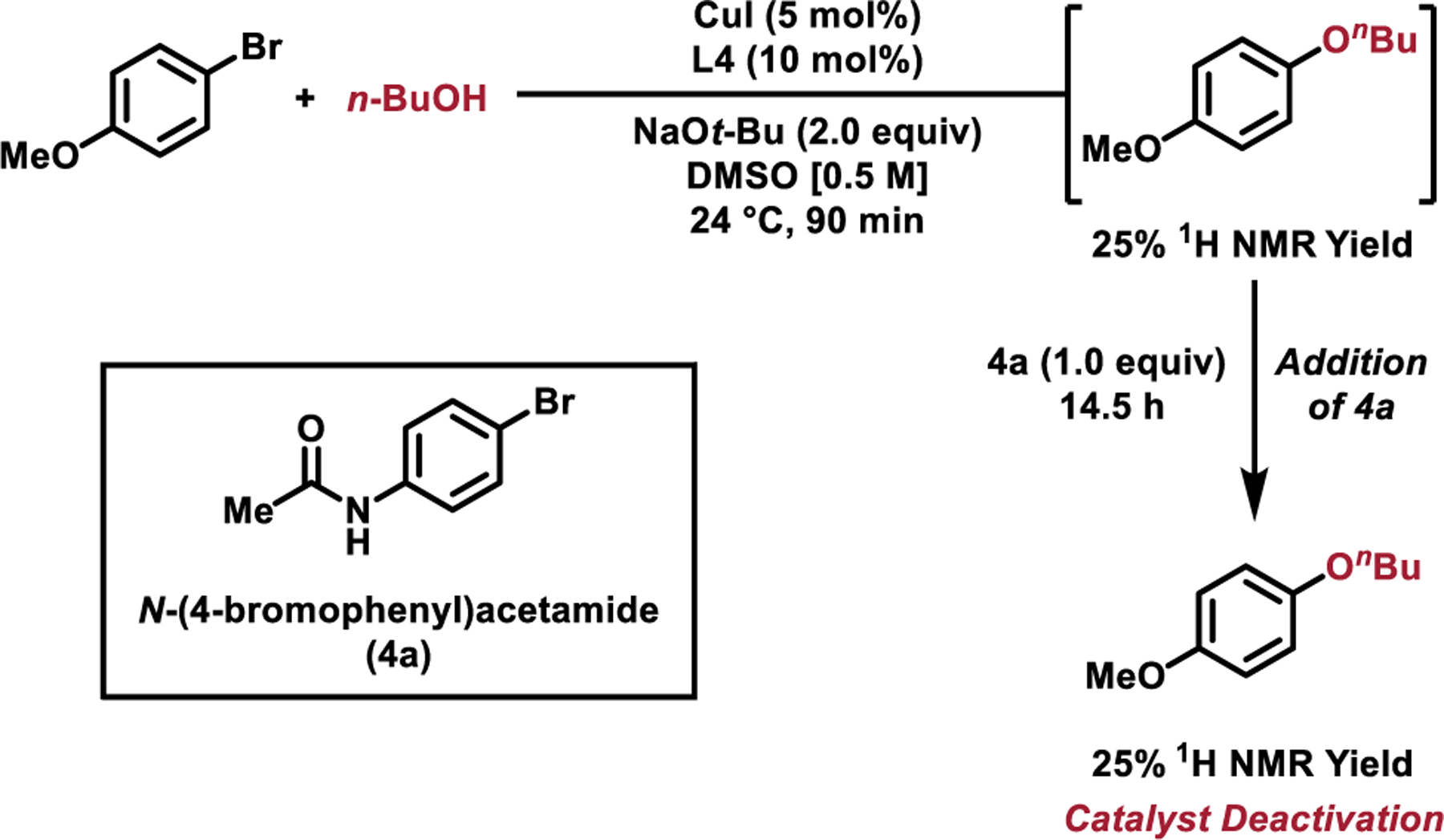
Reactions utilized to monitor catalyst deactivation in response to amide-containing substrates. (A) Model reaction in which amide-containing substrate (4a) was added to the coupling of 4-bromoanisole and n-butanol after 90 min of reaction time. No further reaction was observed following the addition of 4a.
Conclusion
We have developed a practical and robust Cu-catalyzed etherification of aryl bromides at room temperature utilizing N1,N2-diarylbenzene-1,2-diamine ligands. This method was successfully applied to the formation of structurally diverse C–O bonds from aryl and heteroaryl bromides and aliphatic alcohols. In developing this method, a new ligand in this family, L8, exhibited enhanced reactivity. The use of an L8-supported Cu catalyst enabled the efficient etherification of a wide variety of aryl bromides, including those possessing acidic functional groups, with various alcohol nucleophiles. Mechanistic analyses demonstrate that reactions utilizing L4 and L8 obey fundamentally different rate laws and that a 7-fold rate acceleration provided by L8 is likely due to the introduction of a new lower energy pathway in which alkoxide transmetallation is rate-limiting and an increased concentration of the active catalyst. This mechanistic phenomenon results in a mechanism of C–O coupling that is distinct from various Pd-, Cu-, and Ni-catalyzed methods. We anticipate that the results of this study and the introduction of a new ligand will motivate further explorations into Cu-catalyzed C–heteroatom bond-forming reactions.
Supplementary Material
Acknowledgements
This work was supported by the NIH (Grant No. R35-GM122483) and Bristol Myers Squibb. M.J.S. was supported by an NIH Postdoctoral Fellowship under Grant No. F32GM146391–01. Any opinions, findings, conclusions, or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the NIH. We thank MilliporeSigma for the generous donation of ligands and precatalysts which were used in the preparation of some of the ligands presented in this manuscript. We thank Prof. Marisa Kozlowski (University of Pennsylvania), Drs. Hanjun Ai (MIT), Nicola Webb (BMS), Jon Jaworski (BMS), and Yong-Jin Wu (BMS) for helpful discussions. We are grateful to Drs. Dennis Kutateladze, Simon Rössler, and Christine Nguyen (MIT) for their help in editing this manuscript. We thank the referees for their helpful comments in the revision of this manuscript.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Roughley SD, Jordan AM, J. Med. Chem. 2011, 54, 3451–3479. [DOI] [PubMed] [Google Scholar]
- [2].Brown DG, Boström J, J. Med. Chem. 2016, 59, 4443–4458. [DOI] [PubMed] [Google Scholar]
- [3].Enthaler S, Company A, Chem. Soc. Rev. 2011, 40, 4912–4924. [DOI] [PubMed] [Google Scholar]
- [4].Evano G, Wang J, Nitelet A, Org. Chem. Front. 2017, 4, 2480–2499. [Google Scholar]
- [5].Wang Z, Comprehensive Organic Name Reactions and Reagents 2010, 3026–3030. [Google Scholar]
- [6].Fuhrmann E, Talbiersky J, Org. Process Res. Dev. 2005, 9, 206–211. [Google Scholar]
- [7].Fletcher S, Org. Chem. Front.2015, 2, 739–752. [Google Scholar]
- [8].Swamy KCK, Kumar NNB, Balaraman E, Kumar KVPP, Chem Rev 2009, 109, 2551–2651. [DOI] [PubMed] [Google Scholar]
- [9].Caron S, Ghosh A, Practical Synthetic Organic Chemistry: Reactions, Principles, and Techniques 2011, 237–253. [Google Scholar]
- [10].Henderson AS, Medina S, Bower JF, Galan MC, Org. Lett. 2015, 17, 4846–4849. [DOI] [PubMed] [Google Scholar]
- [11].Sach NW, Richter DT, Cripps S, Tran-Dubé M, Zhu H, Huang B, Cui J, Sutton SC, Org. Lett. 2012, 14, 3886–3889. [DOI] [PubMed] [Google Scholar]
- [12].Lindstedt E, Stridfeldt E, Olofsson B, Org. Lett. 2016, 18, 4234–4237. [DOI] [PubMed] [Google Scholar]
- [13].Sundalam SK, Stuart DR, J. Org. Chem. 2015, 80, 6456–6466. [DOI] [PubMed] [Google Scholar]
- [14].Lindstedt E, Ghosh R, Olofsson B, Org. Lett. 2013, 15, 6070–6073. [DOI] [PubMed] [Google Scholar]
- [15].Evano G, Wang J, Nitelet A, Org. Chem. Front. 2017, 4, 2480–2499. [Google Scholar]
- [16].Rangarajan TM, Brahma R, Ayushee A. K. Prasad, Verma AK, Singh RP, Tetrahedron Lett 2015, 56, 2234–2237. [Google Scholar]
- [17].Palucki M, Wolfe JP, Buchwald SL, J. Am. Chem. Soc. 1997, 119, 3395–3396. [Google Scholar]
- [18].Cheung CW, Buchwald SL, Org. Lett. 2013, 15, 3998–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gowrisankar S, Neumann H, Beller M, Chem. Eur. J. 2012, 18, 2498–2502. [DOI] [PubMed] [Google Scholar]
- [20].Sawatzky RS, Hargreaves BKV, Stradiotto M, Eur. J. Org. Chem. 2016, 2016, 2444–2449. [Google Scholar]
- [21].Rangarajan TM, Singh R, Brahma R, Devi K, Singh RP, Singh RP, Prasad AK, Chem. Eur. J. 2014, 20, 14218–14225. [DOI] [PubMed] [Google Scholar]
- [22].Zhang H, Ruiz-Castillo P, Schuppe AW, Buchwald SL, Org. Lett. 2020, 22, 5369–5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang H, Ruiz-Castillo P, Buchwald SL, Org. Lett. 2018, 20, 1580–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wu X, Fors BP, Buchwald SL, Angew. Chem. Int. Ed. 2011, 50, 9943–9947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Widenhoefer RA, Buchwald SL, J. Am. Chem. Soc. 1998, 120, 6504–6511. [Google Scholar]
- [26].Hartwig JF, Inorg. Chem. 2007, 46, 1936–1947. [DOI] [PubMed] [Google Scholar]
- [27].Jones WD, Kuykendall VL, Inorg. Chem. 1991, 30, 2615–2622. [Google Scholar]
- [28].Palucki M, Wolfe JP, Buchwald SL, J. Am. Chem. Soc. 1997, 119, 3395–3396. [Google Scholar]
- [29].Gillie A, Stille JK, J. Am. Chem. Soc. 1980, 102, 4933–4941. [Google Scholar]
- [30].Garrett CE, Prasad K, Adv. Synth. Catal. 2004, 346, 889–900. [Google Scholar]
- [31].Chatzopoulou M, Madden KS, Bromhead LJ, Greaves C, Cogswell TJ, Da Silva Pinto S, Galan SRG, Georgiou I, Kennedy MS, Kennett A, Apps G, Russell AJ, Wynne GM, ACS Med. Chem. Lett. 2022, 13, 262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Terrett JA, Cuthbertson JD, Shurtleff VW, MacMillan DWC, Nature 2015, 524, 330–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Morrison KM, McGuire RT, Ferguson MJ, Stradiotto M, ACS Catal. 2021, 11, 10878–10884. [Google Scholar]
- [34].Macqueen PM, Tassone JP, Diaz C, Stradiotto M, J. Am. Chem. Soc. 2018, 140, 5023–5027. [DOI] [PubMed] [Google Scholar]
- [35].Zhang H-J, Chen L, Oderinde MS, Edwards JT, Kawamata Y, Baran PS, Zhang H-J, Chen L, Kawamata Y, Baran PS, Oderinde MS, Bristol JTE, Squibb M, Angew. Chem. Int. Ed. 2021, 133, 20868–20873. [Google Scholar]
- [36].Egorova KS, Ananikov VP, Angew. Chem. Int. Ed. 2016, 55, 12150–12162. [DOI] [PubMed] [Google Scholar]
- [37].Das KK, Reddy RC, Bagoji IB, Das S, Bagali S, Mullur L, Khodnapur JP, Biradar MS, J Basic Clin. Physiol. Pharmacol. 2019, 30, 141–152. [DOI] [PubMed] [Google Scholar]
- [38].Garrett CE, Prasad K, Adv. Synth. Catal. 2004, 346, 889–900. [Google Scholar]
- [39].Magano J, Transition Metal-Catalyzed Couplings in Process Chemistry: Case Studies From the Pharmaceutical Industry 2013, 313–355. [Google Scholar]
- [40].Roughley SD, Jordan AM, J. Med. Chem. 2011, 54, 3451–3479. [DOI] [PubMed] [Google Scholar]
- [41].Hoover JM, Steves JE, Stahl SS, Nature Protocols 2012, 7, 1161–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hoover JM, Stahl SS, J. Am. Chem. Soc. 2011, 133, 16901–16910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hoover JM, Ryland BL, Stahl SS, J. Am. Chem. Soc. 2013, 135, 2357–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Xu B, Lumb JP, Arndtsen BA, Angew. Chem. Int. Ed. 2015, 54, 4208–4211. [DOI] [PubMed] [Google Scholar]
- [45].Editors Bert Klein Gebbink G, van Koten G, Albrecht M, Lindner MM, Bossi G, Putignano E, Rigo P, Baratta W, Huffman LM, Stahl SS, Yuan D, Tang H, Xiao L, Vinh Huynh H, Dalton Transactions 2011, 40, 8796–8799.21725566 [Google Scholar]
- [46].Huffman LM, Casitas A, Font M, Canta M, Costas M, Ribas X, Stahl SS, Chem. Eur. J. 2011, 17, 10643–10650. [DOI] [PubMed] [Google Scholar]
- [47].Monnier F, Taillefer M, Angew. Chem. Int. Ed. 2009, 48, 6954–6971. [DOI] [PubMed] [Google Scholar]
- [48].Wolter M, Nordmann G, Job GE, Buchwald SL, Org. Lett. 2002, 4, 973–976. [DOI] [PubMed] [Google Scholar]
- [49].Naidu AB, Sekar G, Tetrahedron Lett. 2008, 49, 3147–3151. [Google Scholar]
- [50].Naidu AB, Jaseer EA, Sekar G, J. Org. Chem. 2009, 74, 3675–3679. [DOI] [PubMed] [Google Scholar]
- [51].Kim S-T, Strauss MJ, Cabré A, Buchwald SL, J. Am. Chem. Soc. 2023, 145, 6966 –6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yang C-T, Fu Y, Huang Y-B, Yi J, Guo Q-X, Liu L, Yang C, Fu Y, Huang Y, Yi J, Guo Q, Liu L, Angew. Chem. Int. Ed. 2009, 48, 7398–7401. [DOI] [PubMed] [Google Scholar]
- [53].Kwong FY, Buchwald SL, Org. Lett. 2003, 5, 793–796. [DOI] [PubMed] [Google Scholar]
- [54].Fan M, Zhou W, Jiang Y, Ma D, Org. Lett. 2015, 17, 5934–5937. [DOI] [PubMed] [Google Scholar]
- [55].Gao J, Bhunia S, Wang K, Gan L, Xia S, Ma D, Org. Lett. 2017, 19, 2809–2812. [DOI] [PubMed] [Google Scholar]
- [56].Zhou W, Fan M, Yin J, Jiang Y, Ma D, J. Am. Chem. Soc. 2015, 137, 11942–11945. [DOI] [PubMed] [Google Scholar]
- [57].Zhang Y, Yang X, Yao Q, Ma D, Org. Lett. 2012, 14, 3056–3059. [DOI] [PubMed] [Google Scholar]
- [58].Yang Q, Zhao Y, Ma D, Org. Process Res. Dev. 2022, 26, 1690–1750. [Google Scholar]
- [59].Zhang H, Ma D, Cao W, Synlett 2007, 2007, 243–246. [Google Scholar]
- [60].Chen Z, Jiang Y, Zhang L, Guo Y, Ma D, J. Am. Chem. Soc. 2019, 141, 3541–3549. [DOI] [PubMed] [Google Scholar]
- [61].Bhunia S, Pawar GG, Kumar SV, Jiang Y, Ma D, Angew. Chem. Int. Ed.2017, 56, 16136–16179. [DOI] [PubMed] [Google Scholar]
- [62].Fan M, Zhou W, Jiang Y, Ma D, Angew. Chem. Int. Ed. 2016, 55, 6211–6215. [DOI] [PubMed] [Google Scholar]
- [63].Zhai Y, Chen X, Zhou W, Fan M, Lai Y, Ma D, J. Org. Chem. 2017, 82, 4964–4969. [DOI] [PubMed] [Google Scholar]
- [64].Ray R, Hartwig JF, Angew. Chem. Int. Ed.2021, 60, 8203–8211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Delaney CP, Lin E, Huang Q, Yu IF, Rao G, Tao L, Jed A, Fantasia SM, Püntener KA, Britt RD, Hartwig JF, Science 2023, 381, 1079–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66]. For more details see the supporting information document.
- [67].Sather AC, Martinot TA, Org. Process Res. Dev. 2019, 23, 1725–1739. [Google Scholar]
- [68].Kemp DS, Casey ML, J. Am. Chem. Soc. 1973, 95, 6670–6680. [Google Scholar]
- [69].Casey ML, Kemp DS, Paul KG, Cox DD, J. Org. Chem. 1973, 38, 2294–2301. [Google Scholar]
- [70].Khoury R, Marx C, Mirgati S, Velury D, Chakkamparambil B, Grossberg GT, Expert. Opin. Pharmacother. 2021, 7, 783–795. [DOI] [PubMed] [Google Scholar]
- [71].Garay RP, Grossberg GT, Expert Opin. Investig. Drugs. 2017, 1, 121–132. [DOI] [PubMed] [Google Scholar]
- [72].Schmidt C, Nat Biotechnol 2017, 35, 493–494. [DOI] [PubMed] [Google Scholar]
- [73].Frank S, Testa CM, Stamler D, Kayson E, Davis C, Edmondson MC, Kinel S, Leavitt B, Oakes D, O’Neill C, Vaughan C, Goldstein J, Herzog M, Snively V, Whaley J, Wong C, Suter G, Jankovic J, Jimenez-Shahed J, Hunter C, Claassen DO, Roman OC, Sung V, Smith J, Janicki S, Clouse R, Saint-Hilaire M, Hohler A, Turpin D, James RC, Rodriguez R, Rizer K, Anderson KE, Heller H, Carlson A, Criswell S, Racett BA, Revilla FJ, Nucifora F, Margolis RL, Ong MJ, Mendis T, Mendis N, Singer C, Quesada M, Paulsen JS, Brashers-Krug T, Miller A, Kerr J, Dubinsky RM, Gray C, Factor SA, Sperin E, Molho E, Eglow M, Evans S, Kumar R, Reeves C, Samii A, Chouinard S, Beland M, Scott BL, Hickey PT, Esmail S, Fung WLA, Gibbons C, Qi L, Colcher A, Hackmyer C, McGarry A, Klos K, Gudesblatt M, Fafard L, Graffitti L, Schneider DP, Dhall R, Wojcieszek JM, La Faver K, Duker A, Neefus E, Wilson-Perez H, Shprecher D, Wall P, Blindauer KA, Wheeler L, Boyd JT, Houston E, Farbman ES, Agarwal P, Eberly SW, Watts A, Tariot PN, Feigin A, Evans S, Beck C, Orme C, Edicola J, Christopher E, JAMA 2016, 316, 40–50. [DOI] [PubMed] [Google Scholar]
- [74].Casitas A, Ribas X, Chem. Sci. 2013, 4, 2301–2318. [Google Scholar]
- [75].Ribas X, Güell I, Pure and Applied Chemistry 2014, 86, 345–360. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.