

Abstract
Raine syndrome (RNS) is a rare autosomal recessive osteosclerotic dysplasia. RNS is caused by loss-of-function disease-causative variants of the FAM20C gene that encodes a kinase that phosphorylates most of the secreted proteins found in the body fluids and extracellular matrix. The most common RNS clinical features are generalized osteosclerosis, facial dysmorphism, intracerebral calcifications and respiratory defects. In non-lethal RNS forms, oral traits include a well-studied hypoplastic amelogenesis imperfecta (AI) and a much less characterized gingival phenotype. We used immunomorphological, biochemical, and siRNA approaches to analyze gingival tissues and primary cultures of gingival fibroblasts of two unrelated, previously reported RNS patients. We showed that fibrosis, pathological gingival calcifications and increased expression of various profibrotic and pro-osteogenic proteins such as POSTN, SPARC and VIM were common findings. Proteomic analysis of differentially expressed proteins demonstrated that proteins involved in extracellular matrix (ECM) regulation and related to the TGFβ/SMAD signaling pathway were increased. Functional analyses confirmed the upregulation of TGFβ/SMAD signaling and subsequently uncovered the involvement of two closely related transcription cofactors important in fibrogenesis, Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ). Knocking down of FAM20C confirmed the TGFβ-YAP/TAZ interplay indicating that a profibrotic loop enabled gingival fibrosis in RNS patients. In summary, our in vivo and in vitro data provide a detailed description of the RNS gingival phenotype. They show that gingival fibrosis and calcifications are associated with, and most likely caused by excessed ECM production and disorganization. They furthermore uncover the contribution of increased TGFβ–YAP/TAZ signaling in the pathogenesis of the gingival fibrosis.
Keywords: FAM20C, FAM20A, Secretome, Proteome, Raine syndrome, TGF beta, YAP/TAZ, Gingival fibroblast, Ectopic mineralization, Gingival fibromatosis, Fibrosis, Collagen, Periostin
Subject terms: Cell adhesion, Cell signalling, Mechanisms of disease, Cell biology, Diseases
Introduction
Family with sequence similarity 20 (FAM20) is a gene family of atypical secreted protein kinases comprising three members in vertebrates, FAM20A, FAM20B and FAM20C. FAM20B, the ancestral template protein for the FAM20 family is a xylosylkinase that phosphorylates xylose residues in the glycosaminoglycan-protein linkage regions of proteoglycan and promotes their production1. Mutations in FAM20B cause severe neonatal short-limb dysplasia2. FAM20A is a pseudokinase; it lacks active site residues and cannot hydrolyze ATP but binds to and allosterically activates FAM20C3. Disease-causative variants in FAM20A lead to enamel renal syndrome (ERS), a rare hereditary disorder combining AI, gingival fibrosis and nephrocalcinosis4–10. FAM20C conserved from nematodes to humans, is the genuine Golgi casein kinase that phosphorylates the S-x-E/pS motif of secreted proteins11–13. It is a ubiquitously expressed, 584 amino acids protein. FAM20C contains a catalytic pocket, a FAM20A binding domain and three distinct N-glycosylation sites necessary for protein folding and secretio14–16. FAM20C is required for the phosphorylation of ECM proteins involved in the biomineralization of bone and teeth17–20. Other FAM20C functions include the regulation of endoplasmic reticulum homeostasis, coagulation and cardiac function21–26.
Loss-of-function variants in the FAM20C gene result in RNS (OMIM #259775) a very rare autosomal recessive disorder with an estimated prevalence of < 1 in 100000027–31. The phenotypic spectrum encompasses neonatal lethal osteosclerotic bone dysplasia with respiratory distress and nonlethal forms of various severity. Typical features of the latter comprise hypophosphatemia, neurological disorders, midface hypoplasia, exophthalmos, depressed nasal bridge and various orodental anomalies including AI, gingival overgrowth and ectopic calcifications32–35. To date, 42 FAM20C disease-causative variants, 22 lethal and 20 nonlethal, have been described29,36. They include whole gene deletion, microdeletions, missense, nonsense and splice-site variants. Variants that affect protein stability and/or secretion or abolish the kinase activity are generally lethal whereas residual activity is compatible with life15,37.
We previously reported the systemic and orodental features of two consanguineous Brazilian families both presenting with AI, facial dysmorphism and hypophosphatemia32. Genetic analysis identified two distinct homozygous disease-causative variants in FAM20C, confirming nonlethal forms of RNS. The P496L substitution (Patient RNS-1) probably disrupting the FAM20C activation loop was associated with hypophosphatemic rickets, intracranial calcifications, visual impairment and an oral phenotype characterized by AI, dentinal anomalies and gingival hyperplasia with calcifications. The W202Cfs* splice site variant (Patient RNS-2) most likely produced a truncated protein of 239 amino acids and low levels of the wild-type protein presumably creating a hypomorphic allele. Mildly dysmorphic facies, intracranial calcifications, hearing impairment, AI, impaired dentin formation and gingival calcifications were observed32. Among the orodental features AI is the best studied in nonlethal RNS; moreover the specific role of FAM20C in amelogenesis has been established in murine models and in vitro32,34,38–41. On the contrary much less is known about the alterations of the RNS gingiva: gingivitis, gingival hypertrophy and gingival hyperplasia are the terms used in the literature to describe the gingival phenotype of the patients32,34,35.
Using conditioned media from primary gingival fibroblast (GF) cultures we previously characterized the secretome of ERS-derived GFs42. We identified TGFβ as a pathogenetic factor for ERS gingival fibrosis and showed that FAM20A and FAM20C were co-expressed in the human gingiva8,42. Given the described FAM20A/FAM20C interactions3 we wondered whether FAM20C dysfunction would also result in similar molecular and signaling modifications and/or gingival phenotype. To provide a precise description of the RNS-associated gingival phenotype and gain insight into the underlying pathogenetic mechanism(s) we here combined immunomorphological and biochemical analyses of RNS gingiva and RNS gingiva-derived GFs (RNS-GFs). We used a liquid chromatography tandem-mass-spectrometry-based label-free quantitative proteomic approach (LC–MS/MS) to analyze the proteome of control and RNS-GF lysates and furthered our investigation of the secretome of the same cell types. The protein signature of RNS-1 and RNS-2 cellular proteomes and secretomes were differentially compared to control samples. Variant-specific, albeit subtle differences were observed in the number of dysregulated proteins and cellular functions. ECM disorganization and impaired ossification were common findings in RNS-1 and RNS-2 whereas cytoskeletal modifications were mainly associated with RNS-2. Subsequent RT-PCR and biochemical analyses confirmed the proteomic data and revealed that TGFβ signaling was abnormally increased in the mutant gingival tissue and cells. Increased TGFβ signaling promoted YAP/TAZ signaling most likely creating a profibrotic loop consistent with persistent gingival fibrosis. The TGFβ-YAP/TAZ-FAM20C interplay was further confirmed in FAM20C- depleted human GFs. In sum, our data clearly define the gingival defects of RNS patients, provide the first to our knowledge proteomic analysis of RNS-GFs and uncover the previously unrecognized role of TGFβ–YAP/TAZ signaling in the pathogenesis of the gingival fibrosis in RNS.
Results
FAM20A and FAM20C expression in the control and RNS gingivas
The co-expression of FAM20A and FAM20C in the human gingival keratinocytes, endothelial cells, macrophages and fibroblasts was previously reported8. In agreement with these data, FAM20C and FAM20A were readily detected and displayed a similar pattern in the control gingival keratinocytes and connective tissue (Fig. 1A,D,G and Supplementary Fig. 1A). The FAM20C signal was vesicular and perinuclear especially in the RNS-1 gingival cells (Fig. 1B,E,C,F). FAM20A distribution was not modified in the mutant tissues (Fig. 1G–I and Supplementary Fig. 1A). In vitro, FAM20C displayed a scattered vesicular pattern; in the mutant GFs a perinuclear distribution of the signal was also observed (Fig. 1J–L and Supplementary Fig. 1B). Whereas in control GFs FAM20C could be seen in HPA-positive cis-Golgi vesicles (Fig. 1J), no co-localization between FAM20C and HPA was evidenced in the RNS gingivas (Fig. 1K,L). We further analysed the subcellular distribution of FAM20C using the endoplasmic reticulum (ER) marker concanavalin A, the nuclear marker lamin-B1, the lysosomal marker Lamp1 and the ubiquitin–proteasome system associated protein UCHL1 known to be localized in the cytosol and the nucleus43 (Fig. 1 M–O and Supplementary Fig. 1C–K). Fam20C was readily identified in ER structures especially in the patients’ GFs. No co-localization could be seen with the nuclear or lysosomal markers either in control or patients’ GFs.
Figure 1.
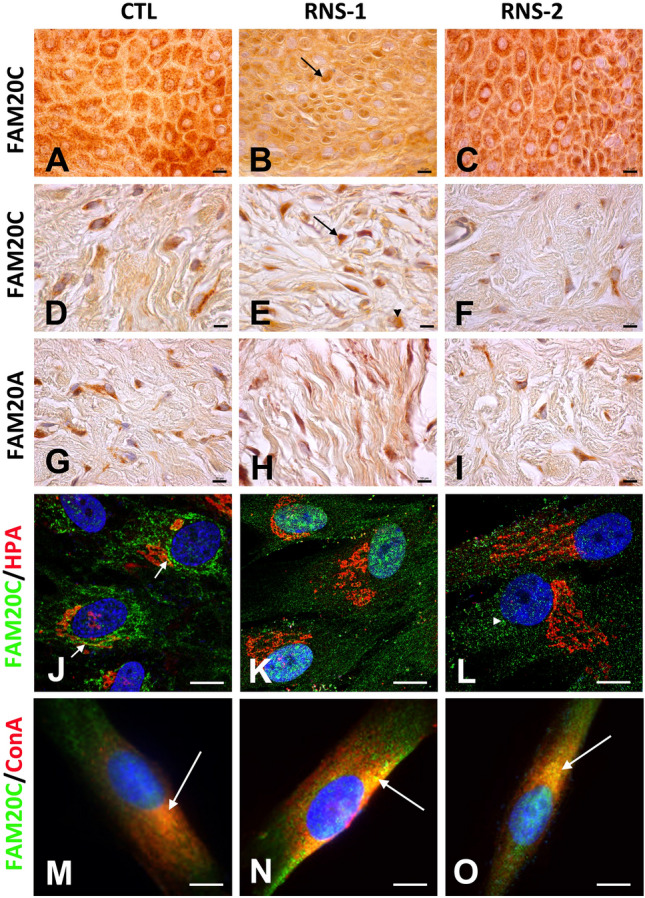
Distribution of FAM20C and FAM20A in control and RNS mutant gingivas. (A,D) In control gingiva, FAM20C immunoreactivity was found in large cytoplasmic vesicles of keratinocytes (A) and fibroblasts (D). (B,E) In RNS-1 gingiva, FAM20C immunoreactivity was localized around the nuclei of keratinocytes (arrow, (B)) and fibroblasts (arrow, (E)). Diffuse cytoplasmic immunoreactivity was also observed in some cells (arrowhead). (C,F) In RNS-2 gingiva, FAM20C was normally found in the cytoplasm of both keratinocytes (C) and fibroblasts (F), albeit with a weaker staining. (G–I) FAM20A was expressed in the cytoplasm of fibroblasts of control (G) as well as RNS-1 (H) and RNS-2 (i) gingivas. (J) In GFs FAM20C expression (green) was found in the endoplasmic reticulum and at the interface with the cis-Golgi in HPA-positive (red) compartment (arrows). (K) In RNS-1 GFs, the staining was perinuclear. (L) In RNS-2 GFs, the staining was weaker and appeared in small cytoplasmic aggregates (arrowhead). (M–O) Co-staining of control, RNS-1 and RNS-2 GFs with Concanavalin A. (M) Limited co-localization (arrow) of FAM20C (green) and the ER marker Concanavalin A in control GFs. (N) Extensive colocalization (arrow) of FAM20C and Concanavalin A in RNS-1 GFs. (O) Extensive colocalization (arrow) of FAM20C and Concanavalin A in RNS-2 GFs. Scale bars: (A–I) = 30 μm; (J–O) = 12 μm.
Immunohistological analysis of RNS gingival tissue
The presence of gingival calcifications in the patients was previously reported32. Hematoxylin–eosin staining showed epithelial acanthosis and inflammatory infiltrates exclusively in RNS-1 and RNS-2 samples (Fig. 2A–C; Supplementary Fig. 2A–C). The vascular network was overdeveloped, particularly in RNS-1 (Fig. 2B and Supplementary Fig. 2B) and the connective tissue contained disorganized collagen fibre bundles (Fig. 2A–C). Picrosirius red polarizing staining confirmed this observation and showed that thick orange-red, presumably Collagen I, fibers were irregularly distributed in the mutant connective tissues (Fig. 2D–F). Collagen distribution was further evidenced by immunostaining (Supplementary Fig. 2D–F). Small, calcified aggregates were seen in the connective tissue in both RNS-1 and RNS-2 samples (Fig. 2G,H) but not the control gingivas.
Figure 2.

Histologic features of RNS variants. (A–C) Hematoxylin–eosin staining showed numerous tortuous blood vessels (bv) with an abnormally large diameter in the papillary layer and associated extensive inflammatory infiltrates in RNS-1 gingiva (B). Abnormally large and shredded collagen bundles running in all directions were found in RNS-2 gingiva (C). (D–F) Picrosirius red polarizing staining. (D) Fibers of collagen organized in bundles of fixed diameter running in perpendicular pathways. (E,F) In RNS-1 and RNS-2, collagen fibers did not organize in packed bundles, but appeared shredded (arrow) and organized in a mesh network. (G,H) Alizarin red staining identified calcium aggregates (arrow) in the reticular layer of the lamina propria of RNS-1 (G) and RNS-2 (H) mutants. Scale bars: (A–C), (G,H) = 200 μm; (D–F) = 50 μm.
Cell proliferation, cell death and ectopic calcifications in RNS-GFs
Primary GF cultures were obtained from unrelated controls and the above described RNS patients. Western blot analysis of control and mutant cell extracts showed similar expression levels of PCNA and Annexin A1 in control and RNS- GFs indicating that cell proliferation and apoptosis were not affected (Supplementary Fig. 3).
For mineralization assays, cells were cultured at passage 2 for a total period of 21 days in control and mineralization-inducing media as previously described ERS8. Figure 3 shows control, RNS-1- and RNS-2-derived GFs cultured in standard and mineralization-inducing media for 21 days. Alizarin red staining showed deposits of calcium aggregates in the mutant fibroblasts (Fig. 3E,F). In control GFs deposits were occasionally seen (Fig. 3D). Morphometric analysis showed a time dependent trend; calcium deposition significantly increased between 7 and 21 days of culture (Fig. 3I). This set of results suggested that FAM20C dysfunction may favor the osteogenic modifications of the mutant gingiva and ectopic calcification.
Figure 3.

Mineralization in vitro. (A–D) Representative bright field images of normal (A), and FAM20C RNS (B,C) deficient GFs grown for 21 days in standard medium colored with Alizarin Red. (D–F) Representative bright field images of normal (D) and FAM20C (E,F) deficient GFs grown for 21 days in a mineralization-inducing medium colored with Red Alizarin. Arrowheads indicate rare and small calcium deposits in controls GFs. (I) Quantitative analysis of Alizarin Red expression by morphometric analysis on the pooled results of three separate experiments; the value represents the means ± SD. ***p < 0.001. Scale bars: (A–F) = 20 μm.
Mass spectrometry analysis
The GFs were cultured in standard conditions as described in the “Methods” section. Nano-LC–MS/MS was performed on conditioned media and cell lysates for secretome, and cellular proteome analysis respectively. To evaluate the reproducibility of the experiments, different linear regressions were performed by plotting the logarithm of protein intensities for the different samples of the same group (as mentioned in the “Methods” section). The averaged regression coefficient measured to evaluate the robustness of the technical scenario between biological replicates for the different groups of samples was estimated as follows: for cellular proteomes RCtls = 0.9954 and RRNS1/RNS2 = 0.9994; for secretome RRCtls = 08,835, RRNS1/RNS2 = 0.7938. The high regression coefficient of the samples analyzed allowed a reliable comparison of the control and mutant secretomes and cellular proteomes. For each comparison, a Principal Component Analysis on all peptidic ions showed a good discrimination of the 2 populations (Fig. 4A,B). By providing different statistical filters (peptide ions filters and proteins filters), the number of identified and quantified proteins was mentioned. A volcano plot, highlighting up- (orange circle) and down- (blue circle) regulated proteins, is presented for each comparison with a particular focus on proteins discussed and tested by RT-PCR (see further down).
Figure 4.

Mass spectrometry analysis. (A) Differential quantitative analysis between RNS1 (n = 3), RNS2 (n = 3) and Control (n = 3) on secretome. (B) Differential quantitative analysis between RNS1 (n = 3), RNS2 (n = 3) and Control (n = 3) on both cell proteome. By providing different statistical filters (Peptidic ions filters and proteins filters), the number of identified and quantified proteins are mentioned. A volcano plot, highlighting down- (blue circle) and up- (orange circle) regulated proteins, is presented for each comparison.
In the secretome, 577 (RNS-1) and 498 (RNS-2) differentially expressed proteins were identified (Fig. 4A and Supplementary Table S1). Using criteria described in material and methods (at least two different peptides found; Anova p-value < 0.05; and power > 0.8), 268 (RNS-1) and 231 (RNS-2) proteins were significantly differentially expressed (Supplementary Table S2) compared with the controls. In RNS-1, 114 were underrepresented and 75 were overrepresented; in RNS-2, 114 were underrepresented and 48 were overrepresented (Supplementary Table S2). Taken together, 65 proteins were found in common between RNS-1 and RNS-2, among them 47 were underrepresented and 18 overrepresented (Supplementary Fig. 4, Supplementary Table S2). Further analysis showed that 55 of the 65 dysregulated proteins were ECM components (Supplementary Table S2).To identify how these up- and down-regulated proteins assembled into functional networks we used the Markov cluster algorithm http://www.micans.org/mcl/ with a 3.2 inflation parameter provided by the string-db.org (Supplementary Fig. 5 and Supplementary Table S3). We identified a major cluster of 44 COL1-associated secreted ECM proteins (Supplementary Fig. 5). This cluster was centered on the overrepresented COL1A1, COL1A2, COL5A2, IL-6, POSTN, SPARC, VIM, MMP3, SERPINE1, THSB1 and THSB2 (nodes in blue/grey circles) and the underrepresented FN1, COL12A1, BGN, COL15A1, LAMA4, LAMB2, LAMC1, TIMP1 and MMP1 (nodes in red/rose circles). ECM organization, regulation of cell-substrate adhesion and response to wound healing (Table 1) were the most enriched biological processes. GO analysis of the molecular functions showed a very strong enrichment in ECM structural constituents. Molecular functions related to the collagen-containing ECM were significantly enriched (Table 1). We used the Monarch Initiative, an integrative data and analytic platform connecting phenotypes to genotypes to integrate our data into functions (Table 2). Not surprisingly, in this group of proteins, most Monarch annotations corresponded to genes associated with skin wound healing or bone morphology and density (Table 2). GO disease analysis showed that among diseases, connective tissue disease was highly significant (Table 2). To identify specific functions that could be masked by a high number of genes, we also performed the same clustering analysis with individual RNS secretomes (Supplementary Tables S4, S5, S6 and S7). Cluster analysis of the dysregulated proteins in RNS-1 showed that 4 clusters of more than 6 proteins were formed (Supplementary Table S4). GO, Monarch analysis of the main cluster of 83 proteins indicated almost the same annotations as those mentioned above (Supplementary Fig. 6 and Supplementary Table S5). Cluster analysis of the proteins dysregulated in RNS-2 identified 4 clusters of more than 6 proteins (Supplementary Fig. 7 and Supplementary Table S6). As above, the main cluster of 63 proteins displayed similar annotations (Supplementary Table S7).
Table 1.
Most enriched GO terms in RNS secretomes: biological process, molecular function, cellular component.
| Name | p value | Genes from input | Genes in annotation | |
|---|---|---|---|---|
| Biological process | ||||
| GO:0030198 | Extracellular matrix organization | 1.85e–26 | 24 | 3338 |
| GO:0010810 | Regulation of cell-substrate adhesion | 5.96e–09 | 11 | 212 |
| GO:0051093 | Negative regulation of developmental process | 5.96e–09 | 18 | 983 |
| GO:0051241 | Negative regulation of multicellular organismal process | 1.32e–08 | 19 | 1231 |
| GO:0009611 | Response to wounding | 1.63e–08 | 14 | 532 |
| GO:0042060 | Wound healing | 2.17e–08 | 13 | 439 |
| GO:0030155 | Regulation of cell adhesion | 4.04e–08 | 15 | 712 |
| GO:0032963 | Collagen metabolic process | 2.41e–07 | 7 | 63 |
| GO:0050793 | Regulation of developmental process | 2.41e–07 | 24 | 2448 |
| GO:0030334 | Regulation of cell migration | 4.25e–07 | 15 | 865 |
| Molecular function | ||||
| GO:0005201 | Extracellular matrix structural constituent | 6.76e–17 | 14 | 119 |
| GO:0005198 | Structural molecule activity | 2.90e–11 | 17 | 635 |
| GO:0005518 | Collagen binding | 3.32e–07 | 7 | 68 |
| GO:0061134 | Peptidase regulator activity | 1.74e–06 | 9 | 226 |
| GO:0030020 | Extracellular matrix structural constituent conferring tensile strength | 6.59e–06 | 5 | 28 |
| GO:0005539 | Glycosaminoglycan binding | 3.77e–05 | 8 | 240 |
| GO:0050840 | Extracellular matrix binding | 0.00011 | 5 | 56 |
| GO:0002020 | Protease binding | 0.00032 | 6 | 138 |
| GO:0004866 | Endopeptidase inhibitor activity | 0.00098 | 6 | 181 |
| GO:0004867 | Serine-type endopeptidase inhibitor activity | 0.00098 | 5 | 98 |
| Cellular component | ||||
| GO:0031012 | Extracellular matrix | 3.29e–35 | 31 | 527 |
| GO:0062023 | Collagen-containing extracellular matrix | 1.88e–31 | 27 | 396 |
| GO:0005576 | Extracellular region | 2.42e–23 | 42 | 4166 |
| GO:0005615 | Extracellular space | 1.57e–21 | 38 | 3195 |
| GO:1903561 | Extracellular vesicle | 3.62e–13 | 27 | 2121 |
| GO:0005788 | Endoplasmic reticulum lumen | 1.40e–12 | 14 | 308 |
| GO:0005604 | Basement membrane | 6.24e–12 | 10 | 96 |
| GO:0070062 | Extracellular exosome | 2.19e–11 | 25 | 2099 |
| GO:0031982 | Vesicle | 9.84e–11 | 31 | 3879 |
| GO:0005581 | Collagen trimer | 9.72e–06 | 6 | 88 |
Table 2.
Most enriched MONARCH and disease terms in RNS secretomes.
| Name | pValue | Genes from input | Genes in annotation | |
|---|---|---|---|---|
| Monarch | ||||
| HP:0001075 | Atrophic scars | 4.44e–10 | 8 | 35 |
| HP:0000977 | Soft skin | 1.97e–08 | 7 | 35 |
| HP:0002808 | Kyphosis | 1.97e–08 | 13 | 406 |
| HP:0100699 | Scarring | 1.97e–08 | 10 | 162 |
| HP:0001058 | Poor wound healing | 3.95e–08 | 6 | 20 |
| HP:0000592 | Blue sclerae | 4.01e–08 | 8 | 83 |
| HP:0004348 | Abnormality of bone mineral density | 4.01e–08 | 13 | 469 |
| HP:0008780 | Congenital bilateral hip dislocation | 7.08e–08 | 5 | 8 |
| HP:0002645 | Wormian bones | 7.62e–08 | 7 | 54 |
| HP:0002644 | Abnormality of pelvic girdle bone morphology | 1.21e–07 | 12 | 419 |
| HP:0002814 | Abnormality of the lower limb | 1.27e–07 | 19 | 1509 |
| HP:0000974 | Hyperextensible skin | 1.33e–07 | 7 | 63 |
| HP:0011025 | Abnormal cardiovascular system physiology | 1.63e–07 | 17 | 1168 |
| HP:0001030 | Fragile skin | 2.01e–07 | 6 | 34 |
| HP:0001374 | Congenital hip dislocation | 2.60e–07 | 7 | 72 |
| Disease | ||||
| DOID:65 | Connective tissue disease | 1.58e–12 | 19 | 715 |
| DOID:17 | Musculoskeletal system disease | 7.68e–11 | 20 | 1074 |
| DOID:13359 | Ehlers-Danlos syndrome | 5.95e–08 | 6 | 23 |
| DOID:0080001 | Bone disease | 1.17e–06 | 12 | 523 |
| DOID:7 | Disease of anatomical entity | 1.17e–06 | 29 | 4452 |
| DOID:12347 | Osteogenesis imperfecta | 8.66e–06 | 5 | 30 |
In the cellular proteome 980 (RNS-1) and 893 (RNS-2) differentially expressed proteins were identified compared with the controls (Fig. 4B and Supplementary Table S8); of these proteins 424 (in RNS-1) and 446 (in RNS-2) were significantly modified (Supplementary Table S9). 188 dysregulated proteins were found in common in RNS-1 and RNS-2. Out of these proteins 25 were more abundant and 163 less abundant (Supplementary Fig. 4 and Supplementary Table S10) compared with the controls. All over- and underrepresented proteins were further analyzed using the Markov Cluster algorithm http://www.micans.org/mcl/ with a 3.2 inflation parameter provided by string-db.org. This analysis failed to give any relevant association; the statistical power of GO annotated processes, molecular functions or compartments was indeed low (Supplementary Table S10). Individual, variant-specific analysis was performed using the same algorithm. Cluster analysis of the dysregulated proteins in RNS-1 indicated 15 clusters of more than 6 proteins (Supplementary Table S11). These clusters essentially concerned mRNA translation, mRNA splicing, carboxylic acid metabolic process (Table S12). We focused on three clusters associated with cell-to-cell, cell-ECM interactions, collagen and actin cytoskeleton organization (Tables 3, 4; Supplementary Table S12). The first one was centered on the overrepresented VIM and THBS1 and the underrepresented FN1 and MMP1 (nodes in blue/grey circles in Fig. 5A–C). The second one comprised the overrepresented ACTA, TWF1, TPM3 and the underrepresented FSCN1 and TAGLN2 (nodes in red/rose circles in Fig. 5A–C). The third one was centered on the overrepresented COL1A1 and COL6A3 and the underrepresented COL12A1, COL6A1 and COL6A2.
Table 3.
Most enriched GO terms in RNS-1 cellular proteome concerning extracellular matrix cluster: biological process, molecular function, cellular component.
| Name | pValue | Genes from input | Genes in annotation | |
|---|---|---|---|---|
| Biological process | ||||
| GO:0009653 | Anatomical structure morphogenesis | 8.11e–11 | 18 | 2165 |
| GO:0022610 | Biological adhesion | 1.84e–10 | 14 | 931 |
| GO:0048646 | Anatomical structure formation involved in morphogenesis | 2.19e–09 | 13 | 883 |
| GO:0007155 | Cell adhesion | 2.94e–09 | 13 | 925 |
| GO:0030198 | Extracellular matrix organization | 9.71e–08 | 9 | 338 |
| GO:0048856 | Anatomical structure development | 1.93e–07 | 20 | 5402 |
| GO:0031589 | Cell-substrate adhesion | 1.03e–06 | 7 | 182 |
| GO:0001525 | Angiogenesis | 1.09e–06 | 8 | 315 |
| GO:0030154 | Cell differentiation | 1.67e–06 | 17 | 3702 |
| GO:0016043 | Cellular component organization | 3.23e–06 | 19 | 5447 |
| Molecular function | ||||
| GO:0005178 | Integrin binding | 7.04e–15 | 11 | 147 |
| GO:0050839 | Cell adhesion molecule binding | 2.66e–14 | 14 | 538 |
| GO:0044877 | Protein-containing complex binding | 3.01e–08 | 13 | 1216 |
| GO:0005102 | Signaling receptor binding | 5.80e–07 | 13 | 1581 |
| GO:0005518 | Collagen binding | 0.00062 | 4 | 68 |
| GO:0005515 | Protein binding | 0.0021 | 18 | 7026 |
| GO:0005201 | Extracellular matrix structural constituent | 0.0038 | 4 | 119 |
| GO:0050840 | Extracellular matrix binding | 0.0134 | 3 | 56 |
| GO:0005198 | Structural molecule activity | 0.0141 | 6 | 635 |
| GO:0098634 | Cell–matrix adhesion mediator activity | 0.0141 | 2 | 7 |
| Name | pValue | Genes from input | Genes in annotation | |
|---|---|---|---|---|
| GO:0005925 | Focal adhesion | 4.69e–14 | 13 | 405 |
| GO:0005576 | Extracellular region | 9.58e–08 | 18 | 4166 |
| GO:0009897 | External side of plasma membrane | 1.92e–07 | 8 | 331 |
| GO:0098552 | Side of membrane | 2.67e–07 | 9 | 531 |
| GO:0009986 | Cell surface | 5.19e–07 | 10 | 824 |
| GO:0031252 | Cell leading edge | 2.29e–05 | 7 | 425 |
| GO:0031012 | Extracellular matrix | 8.97e–05 | 7 | 527 |
| GO:0030027 | Lamellipodium | 0.00021 | 5 | 202 |
| GO:0062023 | Collagen-containing extracellular matrix | 0.00026 | 6 | 396 |
| GO:0034668 | Integrin alpha4-beta1 complex | 0.00088 | 2 | 3 |
Table 4.
Most enriched GO terms in RNS-1 cellular proteome concerning the actin cytoskeleton cluster: biological process, molecular function, cellular component.
| Name | p value | Genes from input | Genes in annotation | |
|---|---|---|---|---|
| Biological process | ||||
| GO:0007015 | Actin filament organization | 1.39e–13 | 10 | 254 |
| GO:0008154 | Actin polymerization or depolymerization | 4.50e–07 | 5 | 57 |
| GO:1902743 | Regulation of lamellipodium organization | 3.65e–05 | 4 | 47 |
| GO:0030042 | Actin filament depolymerization | 0.00010 | 3 | 11 |
| GO:0006928 | Movement of cell or subcellular component | 0.00065 | 8 | 1501 |
| GO:0010591 | Regulation of lamellipodium assembly | 0.0016 | 3 | 35 |
| GO:0032956 | Regulation of actin cytoskeleton organization | 0.0016 | 5 | 348 |
| GO:1902745 | Positive regulation of lamellipodium organization | 0.0016 | 3 | 33 |
| GO:0120032 | Regulation of plasma membrane bounded cell projection assembly | 0.0029 | 4 | 181 |
| GO:0008064 | Regulation of actin polymerization or depolymerization | 0.0030 | 4 | 189 |
| Molecular function | ||||
| GO:0003779 | Actin binding | 7.31e–12 | 10 | 438 |
| GO:0051015 | Actin filament binding | 1.05e–10 | 8 | 199 |
| GO:0005515 | Protein binding | 0.0030 | 12 | 7026 |
| Component | ||||
| GO:0015629 | Actin cytoskeleton | 4.12e–14 | 11 | 477 |
| GO:0070161 | Anchoring junction | 2.39e–06 | 8 | 820 |
| GO:0005925 | Focal adhesion | 2.91e–05 | 6 | 405 |
| GO:0030027 | Lamellipodium | 2.91e–05 | 5 | 202 |
| GO:0032432 | Actin filament bundle | 2.91e–05 | 4 | 73 |
| GO:0043292 | Contractile fiber | 4.43e–05 | 5 | 238 |
| GO:0005884 | Actin filament | 5.87e–05 | 4 | 101 |
| GO:0070062 | Extracellular exosome | 5.87e–05 | 9 | 2099 |
| GO:0005938 | Cell cortex | 7.37e–05 | 5 | 292 |
| GO:0031982 | Vesicle | 0.00042 | 10 | 3879 |
Figure 5.
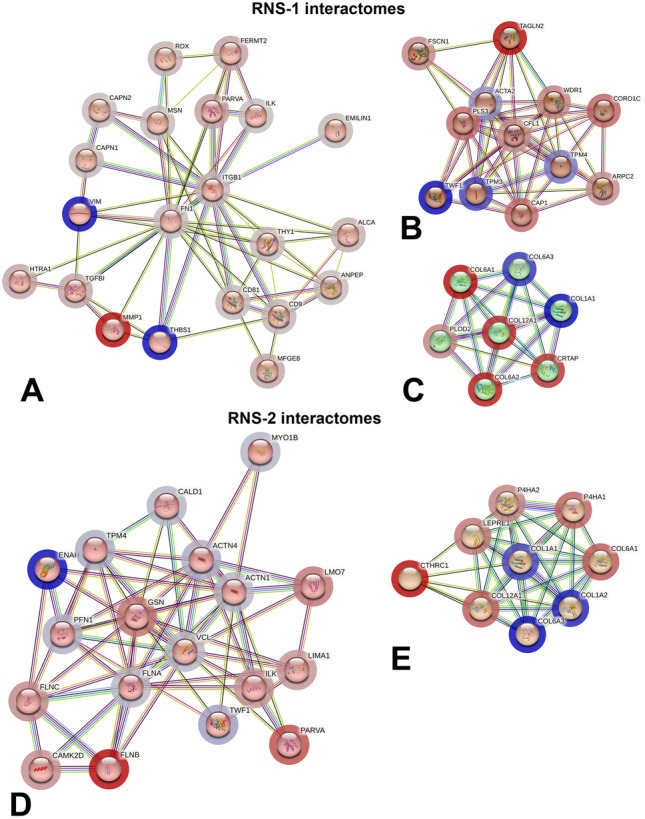
Protein–protein association network using String analysis performed with the differentially selected clustered proteins, over-expressed and under-expressed in the RNS proteomes. (A–C) Main interactomes of RNS-1 compared to controls, concerning extracellular matrix organization (A), actin filament organization (B) and collagen fibril organization (C). (D,E) Main interactomes of RNS-2 compared to controls, concerning actin skeleton organization (D) and collagen fibril organization (E). Nodes in blue/grey circles stand for overrepresented proteins; Nodes in red/grey circles stand for underrepresented proteins. Code color clusterisation, protein names, identifications and descriptions are provided in Tables S11 and S13.
Cluster analysis of the dysregulated proteins in RNS-2 identified 15 clusters of more than 6 proteins (Supplementary Table S13). As in RNS-1, clusters of proteins involved in mRNA splicing and carboxylic acid metabolic processes were seen (Supplementary Table S14). We focused on two clusters; the first one was involved in actin filament organization and comprised the overrepresented ENAH, TWF1, TPM4, ACTN4 and ACTN1 (nodes in blue/gray circles in Fig. 5D,E) and the underrepresented GELS, FLNB and PARVA (nodes in red/rose circles in Fig. 5D,E). The second one was centered on the overrepresented COL1A1, COL1A2, COL6A3 and the underrepresented COL6A1 and COL12A1 (Fig. 5E, Tables 5, 6, and Supplementary Table S14).
Table 5.
Most enriched GO terms in RNS-2 cellular proteome concerning actin cytoskeleton cluster: biological process, molecular function, cellular component.
| Name | pValue | Genes from Input | Genes in Annotation | |
|---|---|---|---|---|
| Biological process | ||||
| GO:0030029 | Actin filament-based process | 1.96e–12 | 13 | 592 |
| GO:0030036 | Actin cytoskeleton organization | 1.34e–11 | 12 | 516 |
| GO:0007015 | Actin filament organization | 2.32e–09 | 9 | 254 |
| GO:0097435 | Supramolecular fiber organization | 1.03e–08 | 10 | 480 |
| GO:0032970 | Regulation of actin filament-based process | 7.50e–05 | 7 | 390 |
| GO:0030048 | Actin filament-based movement | 0.00015 | 5 | 121 |
| GO:0034329 | Cell junction assembly | 0.00024 | 6 | 280 |
| GO:0034330 | Cell junction organization | 0.00024 | 7 | 493 |
| GO:0051017 | Actin filament bundle assembly | 0.00028 | 4 | 55 |
| GO:0000904 | Cell morphogenesis involved in differentiation | 0.00046 | 7 | 566 |
| Molecular function | ||||
| GO:0003779 | Actin binding | 5.72e–19 | 15 | 438 |
| GO:0008092 | Cytoskeletal protein binding | 3.59e–16 | 16 | 973 |
| GO:0050839 | Cell adhesion molecule binding | 3.67e–12 | 12 | 538 |
| GO:0045296 | Cadherin binding | 4.40e–09 | 9 | 334 |
| GO:0051015 | Actin filament binding | 4.40e–09 | 8 | 199 |
| GO:0005515 | Protein binding | 0.00018 | 17 | 7026 |
| GO:0044877 | Protein-containing complex binding | 0.00019 | 9 | 1216 |
| GO:0005546 | phosphatidylinositol-4,5-bisphosphate binding | 0.00034 | 4 | 78 |
| GO:0003785 | Actin monomer binding | 0.0010 | 3 | 28 |
| Name | pvalue | Genes from input | Genes in annotation | |
|---|---|---|---|---|
| GO:0005925 | Focal adhesion | 9.53e–20 | 15 | 405 |
| GO:0015629 | Actin cytoskeleton | 4.18e–15 | 13 | 477 |
| GO:0030016 | Myofibril | 6.80e–15 | 11 | 227 |
| GO:0005856 | Cytoskeleton | 2.37e–11 | 16 | 2221 |
| GO:0099512 | Supramolecular fiber | 4.30e–10 | 12 | 939 |
| GO:0030017 | Sarcomere | 1.14e–09 | 8 | 207 |
| GO:0001725 | Stress fiber | 4.65e–09 | 6 | 65 |
| GO:0005903 | Brush border | 5.36e–08 | 6 | 106 |
| GO:0030018 | Z disc | 1.72e–07 | 6 | 131 |
| GO:0005938 | Cell cortex | 4.02e–07 | 7 | 292 |
Table 6.
Most enriched GO terms in RNS-2 cellular proteome concerning ECM cluster: biological process, molecular function, cellular component.
| Name | pvalue | Genes from input | Genes in annotation | |
|---|---|---|---|---|
| Biological process | ||||
| GO:0030198 | Extracellular matrix organization | 2.91e–05 | 6 | 338 |
| GO:0030199 | Collagen fibril organization | 2.91e–05 | 4 | 46 |
| GO:0060351 | Cartilage development involved in endochondral bone morphogenesis | 2.91e–05 | 4 | 43 |
| GO:0060348 | Bone development | 3.66e–05 | 5 | 199 |
| GO:0001501 | Skeletal system development | 4.82e–05 | 6 | 499 |
| GO:0003429 | Growth plate cartilage chondrocyte morphogenesis | 0.00011 | 3 | 17 |
| GO:0009887 | Animal organ morphogenesis | 0.00061 | 6 | 967 |
| GO:0032963 | Collagen metabolic process | 0.0014 | 3 | 63 |
| GO:0071230 | Cellular response to amino acid stimulus | 0.0017 | 3 | 69 |
| GO:0001503 | Ossification | 0.0018 | 4 | 265 |
| Molecular function | ||||
| GO:0030020 | Extracellular matrix structural constituent conferring tensile strength | 4.02e–09 | 5 | 28 |
| GO:0019798 | Procollagen-proline dioxygenase activity | 8.69e–06 | 3 | 7 |
| GO:0048407 | Platelet-derived growth factor binding | 1.97e–05 | 3 | 11 |
| GO:0031418 | L-ascorbic acid binding | 6.39e–05 | 3 | 20 |
| GO:0004656 | Procollagen-proline 4-dioxygenase activity | 0.0011 | 2 | 4 |
| GO:0005506 | Iron ion binding | 0.0077 | 3 | 147 |
| Name | pvalue | Genes from input | Genes in annotation | |
|---|---|---|---|---|
| GO:0005788 | Endoplasmic reticulum lumen | 6.37e–11 | 8 | 308 |
| GO:0005581 | Collagen trimer | 7.36e–10 | 6 | 88 |
| GO:0062023 | Collagen-containing extracellular matrix | 3.26e–06 | 6 | 396 |
| GO:0005584 | Collagen type I trimer | 0.00032 | 2 | 2 |
| GO:0005589 | Collagen type VI trimer | 0.00046 | 2 | 3 |
We used cell lysates of GFs cultured under standard conditions to analyze the transcript levels of several strongly dysregulated proteins including POSTN, SPARC, TWF1, MMP3, ENAH, FNDC1, a protein which contains a conserved protein domain of FN1 and IL6. Several of these proteins are known TGFβ targets and have previously been associated with fibrotic modifications44. We therefore also analyzed the levels of the TGFβ signaling actors TGFB1, TGFB2 and TGFBR2 as well as FAM20A and FAM20C.
Consistent with the data of dysregulated proteins in secretomes and cellular proteomes, the mRNA levels for SPARC, POSTN, IL6, TWF1and ENAH were significantly albeit differentially increased in RNS-1 and RNS-2 samples (Fig. 6A–E). FNDC1 synthesis was almost sixfold upregulated in RNS-2 but not significantly modified in RNS-1 despite very low protein levels in both secretomes (Fig. 6F). The TGFB1 transcript was strongly upregulated in RNS-1- and RNS-2-GFs (Fig. 6G) whereas TGFB2 was significantly increased in RNS-2-GFs (Fig. 6H). No significant differences were evident for MMP3, TGFBR2, FAM20C and FAM20A mRNA levels (Fig. 6I–L).
Figure 6.

Real time RT-PCR analysis of candidate genes corresponding to proteins with differential abundance characterized in proteomic analysis and to proteins of the TGFβ pathway: Osteonectin ((A), SPARC), Periostin ((B), POSTN), Interleukin-6 ((C), IL6), Twinfillin-1 ((D), TWF1), Protein Enabled Homolog ((E), ENAH), Fibronectin type III domain-containing protein 1 ((F), FNDC1), TGFB1 (G), TGFB2 (H), MMP3 (I), TGFBR2 receptor (J), FAM20A (K) and FAM20C (L). Control values correspond to the mean of 3 independent experiments in triplicates of three control cultures. RNS values correspond to the mean of 3 independent experiments in triplicates of RNS patient cultures. Data represent mean fold gene expressions ± s.d. Data were analyzed via one-way ANOVA with Bonferroni multiple comparisons test (*p < 0.05, **p < 0.01, ***p < 0.001: #not significant).
Taken together, the above data suggested that biological processes involved in ECM, actin cytoskeleton and stress fiber organization, response to wound healing and connective tissue disease were modified in the mutant GFs. These data were consistent with the fibrotic phenotype of the RNS gingiva shown in Fig. 2 and published data involving FAM20C in connective tissue disease15,45,46. They also strongly suggested that modifications in the TGFβ pathway may result from FAM20C dysfunction in the gingival tissue. We therefore subsequently investigated the expression of selected dysregulated proteins in vivo and analyzed their involvement in fibrotic processes.
In vivo expression of selected dysregulated proteins
The secretome data showed that the expression of several TGFβ signaling targets including the profibrotic proteins POSTN, COL5A, VIM, SPARC and FN1 was significantly modified in RNS-GFs. To confirm the in vivo relevance of these data, we investigated the expression of the above proteins as well as of α-SMA, a key marker of fibroblast-to-myofibroblast differentiation and known TGFβ target42,44,47–50 in gingival tissues.
The expression of POSTN, COL5A, VIM, α-SMA, SPARC and FN1 in RNS gingivas is shown in Fig. 7 (Fig. 7 and Supplementary Fig. 6). POSTN, a matricellular protein upregulated in remodeling tissues51 displayed a vesicular pattern in the control fibroblasts; in the RNS gingivas POSTN accumulated in the surrounding ECM (Fig. 7A–C). COL5A was similarly distributed in control and mutant tissues but VIM a key factor in fibrotic processes, was strongly expressed in the fibroblasts and/or myofibroblasts throughout the RNS gingivas (Fig. 7D–F).
Figure 7.
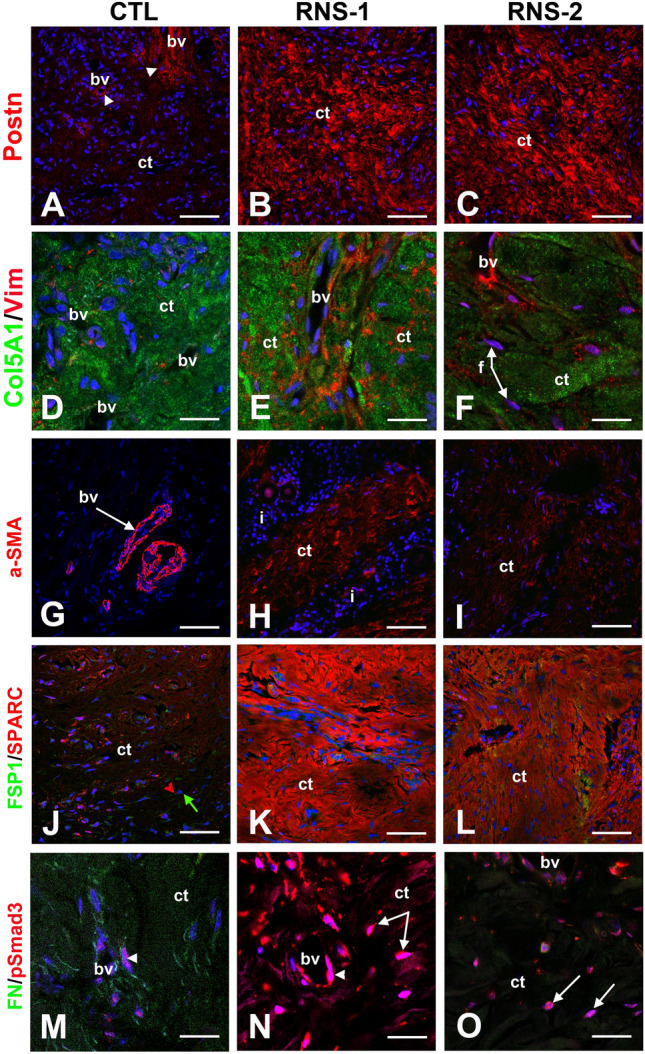
Immunocytochemical features of gingival connective tissues in the gingiva of RNS variants. (A) In normal gingiva, POSTN is localized in a loose extracellular network around blood vessels (arrowheads; bv) whereas in both RNS-1 (B) and RNS-2 (C) mutants POSTN extracellular expression is localized along thick and disorganized fibers in the connective tissue (ct). (D) In normal gingiva, Collagen 5 alpha-1 (green; Col5A1) is secreted and deposited along collagen 1 fibers and Vimentin (red; Vim) is expressed by endothelial cells. (E,F) Increased expression of Col5A1 is detected in the connective tissue (ct) of RNS-1 (E) and RNS-2 (F) mutants.Vim is normally localized in endothelial cells and abnormally in fibroblasts (F). (G) Alpha-SMA (α-SMA) is a specific marker of contractile endothelial cells of bv. (H,I) In RNS mutants, α-SMA is also expressed by fibroblasts in ct. Note the presence of inflammatory infiltrates (i) in RNS-1 gingiva. (J) In normal gingiva, Fibroblast-specific protein 1 (green; FSP1) is expressed by gingival fibroblasts (green arrow). Osteonectin (red; SPARC) is expressed in discrete spots by fibroblasts (red arrowhead) and endothelial cells. (K,L) FSP-1 is normally expressed in ct. SPARC expression and SPARC deposition along fibers are dramatically increased in RNS mutant connective tissues. (M) Fibronectin (green; FN) is specifically expressed by fibroblasts. Phospho-SMAD3 (red; pSMAD3) is mainly localized in nuclei of vascular cells (arrowhead). (N,O) In RNS-derived gingiva FN expression is dramatically decreased. pSMAD3 expression is dramatically increased in both vascular cells (arrowhead) and fibroblasts (arrows) nuclei. Scale bars: (A–C), (G–L) = 100 μm; (D–F), (M–O) = 30 μm.
Alpha-SMA, a hallmark of mature myofibroblasts is also normally expressed by vascular smooth muscle cells and/or pericytes52. A perivascular staining was observed for α-SMA in control and mutant gingivas (Fig. 7G-I). In addition, a particularly intense signal was seen in the mutant connective tissue, most likely associated with myofibroblasts (Fig. 7H,I). SPARC modulates cell–matrix interactions and is generally found in tissues undergoing remodeling53. SPARC was expressed by gingival fibroblasts, also positive for the fibroblastic marker FSP1 (Fig. 7J–L). In the RNS gingivas the staining was particularly strong and SPARC was seen both in the cytoplasm and the surrounding ECM (Fig. 7K,L).
A typical mediator of TGFβ signaling is Smad3; the phosphorylation and subsequent nuclear localization of pSmad3 indicate activation of the pathway54. In control and RNS gingivas pSmad3 was detected in vascular and fibroblastic cells (Fig. 7M–O). The nuclear localization of pSmad3 specifically in the RNS fibroblasts indicated hyperactivation of the TGFβ pathway in this cell type (Fig. 7M–O). In agreement with the proteomic data, FN1 was not detectable in the RNS gingivas (Fig. 7M–O).
FAM20C dysfunction or knockdown increase TGFβ and YAP/TAZ signaling
To further investigate the above data and highlight the potential role of the TGFβ pathway in the RNS-associated gingival phenotype we treated control and RNS GFs with TGFβ 1 (5 ng/ml for 6 h). pSmad3 was readily detected in the nuclei of untreated RNS-1- and RNS-2-GFs and only occasionally in control GFs (Fig. 8A–C and Supplementary Fig. 7). The addition of TGFβ 1 for 6 h resulted in nuclear localization of pSmad3 in the controls and further enhanced nuclear pSmad3 accumulation in the mutants (Fig. 8D–F and Supplementary Fig. 7).
Figure 8.

SMAD3 activation in untreated RNS GFs and treated control and RNS GFs. Immunocytochemical staining of control (A,D) and RNS (B,C,E,F) GFs cultured without TGFβ1 (A–C) or with 5 ng/ml TGFβ1 (D–F) for 6 h. Cells were fluorescently labeled for p-SMAD3 (green), nuclei (blue), and the specific F-actin marker, phalloidin (red). Co-localization of p-SMAD3 and nuclei indicated nuclear translocation of p-SMAD3 (green arrow). (A) In control untreated GFs, p-SMAD3 immunoreactivity is detected at low levels in some nuclei. (B,C) In RNS untreated GFs, p-SMAD3 was increased in intensity in all nuclei. (D–F) TGFβ1 induced an increase in p-SMAD3 nuclei in normal (D) and mutant GFs (E,F). (G) Western blot were performed on cell lysates. P-YAP (Ser 397) protein levels were decreased in RNS GFs without or with TGFβ1 compared to Control. TAZ protein levels were increased in RNS GFs compared to normal GFs. Densitometric analysis of Phospho-YAP and TAZ bands normalized to corresponding GAPDH bands. Data represent mean fold change in band intensity ± s.d. relative to GAPDH of 3 independent experiments in triplicates. Data were analyzed by one-way ANOVA with Bonferroni multiple comparisons test (**p < 0.01, ***p < 0.001). (H) Western blots were performed on cell lysates. Alpha-SMA protein levels were increased in controls GFs cultured with TGFβ1. Alpha-SMA protein levels were increased in RNS GFs cultured without or with TGFβ1 compared to control. Densitometric analysis of a-SMA bands normalized to corresponding GAPDH levels. Scale bars: (A-F) = 20 µm.
It was shown that fibroblast responsiveness to TGFβ was favored by the Hippo pathway targets, the transcription cofactors YAP/TAZ55–58. YAP/TAZ activity is regulated by phosphorylation and subcellular localization. YAP phosphorylation on Ser397 promotes protein degradation59 whereas the dephosphorylated/active form translocates to the nucleus where it allows transcription of target genes.
Under control conditions the levels of YAP/TAZ were higher in mutant GFs (Fig. 8G). The addition of TGFβ 1 for 6 h increased YAP/TAZ levels in both control and mutant cells (Fig. 8G) suggesting activation of YAP signaling. This was also confirmed by the amount of pYAP (S397) which was dramatically decreased in both untreated and TGFβ 1-induced RNS-GFs (Fig. 8G).Activation of YAP/TAZ and TGFβ signaling favor myofibroblast differentiation and fibrotic remodeling60. The myofibroblast marker α-SMA was indeed strongly upregulated in untreated RNS-GFs and this was confirmed at the protein and mRNA levels (Fig. 8H). Treatment with TGFβ 1 for 6 h increased α-SMA protein expression in control and slightly in RNS-GFs (Fig. 8H). This set of data indicates that activation of YAP/TAZ and TGFβ signaling contributes to the fibrotic remodeling of the mutant GFs.
To confirm the interaction loop between FAM20C and TGFβ actors and targets we used a siRNA approach on primary human GFs to deplete FAM20C. We used the same approach to comparatively study the impact of FAM20A depletion. The knockdown was efficient for both FAM20C and FAM20A (Fig. 9A–F). Compared with the control siRNA the FAM20C- and FAM20A-depleted cells had a more elongated morphology (Fig. 9B,E). Both FAM20C and FAM20A siRNAs but not the control one resulted in a strong upregulation of TGFB1 (Fig. 9G). TGFB2 was significantly upregulated in FAM20C-depleted cells (Fig. 9H) whereas TGFBR2 was strongly upregulated in both (Fig. 9I). Consistent with TGFβ signaling activation the depletion of FAM20C or FAM20A led to a significant upregulation of the established TGFβ target and fibrotic remodeling marker POSTN (Fig. 9J). We further investigated whether the YAP/TAZ pathway was activated in the knockdown cells. Immunostaining revealed a predominantly nuclear localization of YAP/TAZ in both FAM20C- and FAM20A-depleted GFs (Fig. 9K–N and Supplementary Fig. 8) confirming the activation of YAP/TAZ signaling.
Figure 9.

FAM20C and FAM20A silencing. (A–G) GFs were transfected with siRNAs targeted either FAM20C, FAM20A or scrambled siRNAs used as controls. Three days after transfection, cells were analyzed for FAM20C or FAM20A expression using immunocytochemistry (A,B,D,E) or QPCR (C,F). (A) FAM20C protein was localized to endoplasmic reticulum in GFs transfected with scrambled siRNAs. (B) This staining was not observed in GFs transfected with siRNAs targeted FAM20C. (C) Using real time RT-PCR, a 90% decrease in mRNA to FAM20C was observed. (D) FAM20A protein was localized in large vesicles in GFs transfected with scrambled siRNAs. (E) This staining was absent in GFs transfected with siRNAs targeting FAM20A. (F) Using real time RT-PCR, a significant 80% decrease in mRNA to FAM20A was observed. (G–J) Real Time RT-PCR analysis of candidate genes corresponding to TGFB1 (G), TGFB2 (H), the receptor TGFBR2 (I) and Periostin POSTN (J). Scrambled siRNAs, siFAM20C or siFAM20A values correspond to the mean of 3 independent experiments in triplicates of three GFs cultures. Datas represent mean fold gene expressions ± s.d. Data was analyzed via one-way ANOVA with Bonferroni multiple comparisons test (*p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.001). (K–M) YAP-TAZ immunocytochemistry showed some lightly immunoreactive nuclei in GFs transfected with scrambled siRNAs (K). In GFs transfected with siFAM20C or siFAM20A a dramatic increase in the staining was observed in all nuclei (L,M). (N) Graph representation of the relative fluorescence intensity of YAP-TAZ in GFs transfected with scrambled siRNAs, siFAM20C or siFAM20A. Statistically significant differences are shown as ***P < 0.001. Scale bars: (A,B,D,E) = 20 µm; (K–M) = 50 µm.
The above data clearly showed that dysfunction or knockdown of FAM20C similarly activated the TGFβ pathway, the primary mediator of the fibrogenesis process. Our results also uncovered the contribution of YAP/TAZ in FAM20C-associated gingival fibrosis confirming published data on the YAP/TAZ-TGFβ cross-talk55.
Discussion
In this study we characterized the gingival phenotype of RNS patients and showed for the first time that FAM20C dysfunction impaired TGFβ and YAP/TAZ signaling pathways in the fibrotic RNS gingiva.
Using proteomic and biochemical approaches we found that the major dysregulated proteins in RNS-GFs were associated with ECM, actin filament and collagen fibril organization as well as bone development and ossification. The immunomorphological in vivo and in vitro data confirmed the fibrotic and osteogenic potential of the RNS-GFs, showed that the TGFβ cascade was strongly activated in these cells and revealed that TGFβ hyperactivation was associated with increased YAP/TAZ signaling. Subsequent FAM20C depletion from control cells resulted in a similar activation of the TGFβ-YAP/TAZ pathway, confirming the interaction of FAM20C with the profibrotic TGFβ-YAP/TAZ network.
FAM20C is a ubiquitous protein kinase phosphorylating numerous secreted proteins including the ones involved in calcium binding and biomineralization. Pathogenic variants that abolish the kinase activity result in a usually perinatal lethality whereas partial activity is compatible with life. RNS-1 and RNS-2 patients were previously described32. The homozygous missense variant in RNS-1 was predicted to result in a residual kinase activity whereas in the homozygous splice site mutation in RNS-2 some wild-type sized FAM20C transcript was detected and the clinical phenotype was milder. In this study, the histopathological analysis showed that compared to RNS-2, the RNS-1 gingival connective tissue appeared more disorganized, much more vascularized and contained less calcified deposits indicating that the gingival modifications were to some extent variant-specific. The in vitro RNS-GFs data were consistent with the genetic, clinical and histological variability, they also suggested that the specific FAM20C mutations could result in altered distribution and/or processing and/or expression level of the mutant proteins. We did not identify significantly modified FAM20C mRNA levels in the patients’ GFs. This piece of result is consistent with the nature of the variants and resulting proteins, i.e. the P496L substitution (RNS-1) is thought to decrease the kinase activity of the mutant protein but not protein synthesis whereas in the RNS-2 patient normal protein synthesis albeit at lower levels, is expected to occur. In terms of protein distribution however, both mutant forms of FAM20C abnormally accumulated in the ER. A similar distribution has previously been reported in HEK293T cells overexpressing mutant FAM20C constructs31; to our knowledge this is the first time that a similar result is reported for native RNS-derived GFs.
FAM20C is a Golgi-localized secretory kinase and its activity is optimal in the Golgi milieu. Nevertheless, it is possible that FAM20C is active during its transit across the ER and several ER/Golgi localized proteins are FAM20C targets. Ero1α, as well as proteins involved in intracellular calcium storage handling, including the histidine rich calcium binding protein (HRC), calreticulin and calsequestrin-2 are phosphorylated by FAM20C24,61,62. The role of FAM20C in ER protein phosphorylation is further supported by the observation that STIM1, mainly localized in the ER is a FAM20C target24. The mutant forms of FAM20C described here are thought to retain some kinase activity32. A prolonged residence of mutant FAM20C protein in the ER and aberrant phosphorylation of its local targets could thus modify their function, disrupt calcium regulation and contribute to ER stress. In addition, ER accumulation of FAM20C would prevent the S1P-mediated proteolysis of FAM20C in the Golgi and block further secretion and autophosphorylation63. Although the significance of the last modification for FAM20C activity is unclear, blocking the secretion could impair biomineralization processes and favor ectopic calcification63.
Because of the broad substrate repertoire of FAM20C it is difficult to define singular events dictating FAM20C functions. However, the role of FAM20C in ER calcium storage regulation and protein secretion strongly suggests that FAM20C may be broadly regulating ER calcium associated processes and ER stress64. ER stress has previously been described in cells overexpressing mutant FAM20C forms65. Our data showing subtle differences in cytosolic and nuclear distribution of UCHL1 in RNS-GFs may suggest impaired protein turnover66 but additional experiments are required to test this hypothesis.
ER stress favors the release of pro-fibrotic factors, the activation of molecular pathways such as TGFβ signaling and contributes to the pathogenesis of idiopathic pulmonary, renal and cardiac fibrosis67–69. Our observations clearly support the idea that FAM20C modulates TGFβ signaling: we showed that FAM20C dysfunction or silencing similarly resulted in hyperactivation of the TGFβ pathway. Our data thus indicate that FAM20C is required for normal TGFβ activity and GF homeostasis, indirectly preventing fibrotic remodeling and ectopic calcifications.
Protein kinases including FAM20C, phosphorylate numerous functionally distinct proteins, changing thus the activity of the substrate, mediating transcriptional regulation, and modulating protein secretion in health and disease45,70. Several groups have indeed shown that FAM20C is essential not only in ER homeostasis but also in coagulation, hormonal regulation, cardiac health and biomineralization. We here broaden the spectrum of FAM20C target tissues and show that gingival health also requires intact FAM20C activity.
In vitro, mineralization-inducing conditions resulted in the formation of calcium deposits in RNS-GFs. This result was compatible with the reported human phenotype but contrasted with murine in vitro and in vivo studies using Fam20C KO mice or cells19,30,32,36,39,71. Ectopic calcification was observed exclusively in the eye of the Fam20C null mutant39 and the formation of mineralized nodules was low in murine Fam20C-deficient osteoblasts19. This discrepancy can be explained by the histological and anatomical differences between human and murine gingiva, the different cell types used and the presence or not of a residual FAM20C activity. We previously showed that human GFs expressed minute amounts of osteogenic genes and this expression remained low even under mineralization-inducing conditions8. Consistent with the previous and current data the formation of calcified nodules was very limited in the control GFs but more pronounced in RNS-GFs implicating FAM20C in aberrant soft tissue mineralization.
Fibrosis and ectopic mineralization were intertwined in the RNS gingiva. The pathogenesis of these processes was highlighted by the biochemical analyses indicating excessive ECM protein production together with dysregulated matrix remodeling.
Myofibroblasts, generally α-SMA positive72–74, are essential in fibrotic processes and absent from normal gingiva. Myofibroblasts synthesize most of the structural and matricellular ECM proteins and contribute to matrix remodeling by producing various proteases including MMPs and their inhibitors TIMPs75–78. We found that the expression of α-SMA, was strongly increased in RNS-GFs and this was correlated with increased levels of IL-6, known to promote differentiation of fibroblasts into myofibroblasts and trigger collagen synthesis79. Enhanced IL-6 production was indeed previously associated with gingival overgrowth80. The expression of POSTN and VIM thought to be myofibroblast markers in multiple tissues81,82 was also particularly high and was accompanied by the abundantly expressed type I collagen and the matricellular proteins SPARC and TSP2, all involved in fibrosis progression including in drug-induced gingival fibrosis83–88.
In addition to the above-described excessive ECM production, matrix remodeling was defective in RNS-GFs. High levels of the serine protease inhibitor SERPINE1, generally observed in fibrotic tissues89 and of MMP3 involved in the degradation of laminins and other basement membrane proteins90 could exaggerate the accumulation of ECM and fibrosis. Downregulation of the collagenase MMP1, specific for collagens I–III, and its main inhibitor TIMP1 could further perturb gingival tissue remodeling. Decreased MMP1, but not TIMP1, production was indeed previously associated with hereditary gingival fibromatosis (HGF), a genetically heterogeneous condition characterized by pathological fibrosis of gingival tissue and abundant production of type I collagen and VIM91. FN1 regulates matrix metalloproteinase expression and was functionally associated with gingival overgrowth in HGF92. Low amounts of FN1 were rather surprising but could be attributed to increased protein degradation including by MMP393. The same could stand for the discrepancy between FNDC1 protein and transcript levels. FNDC1 and FN1 are functionally and structurally close and thought to be coregulated; indeed, FNDC1-dependent FN1 expression was observed in prostate cancer94,95. It is currently unknown whether a similar regulatory pathway operates in RNS-GFs but the decrease in FNDC1 protein could be compatible with perturbed ossification96 and the rather low abundance of calcified nodules in RNS gingiva.
It is unclear why for some proteins including the above mentioned FNDC1 and MMP3 the mRNA and protein levels are discordant. In view of the regulatory role of FAM20C on protein secretion45 we propose that increased secretion of MMP3 despite normal synthesis, or intracellular retention of FNDC1 despite normal (RNS1) or increased (RNS2) levels may explain these results. Alternatively, as indicated above, low protein levels may result from increased protein degradation.
Our findings, i.e. increased ECM protein synthesis, a shift in the protease/antiprotease balance towards a matrix-preserving process and increased production of TGF-regulated factors suggested that this pathway played a key role in RNS gingival fibrosis. The central role of TGFβ in fibrotic disease is established including in hereditary and acquired gingival fibromatosis49,51,97,98. We indeed recently showed that this pathway was hyperactivated in ERS GFs and tissues expressing mutant forms of FAM20A42.
We here showed that a similar hyperactivation occurs in RNS-derived gingival tissue and cells. Our present results revealed that Smad3, the crucial TGFβ-signaling mediator in fibrosis was intrinsically activated/phosphorylated in RNS gingiva and GFs, which synthesized excessive amounts of collagen I-associated ECM. The levels of TGFB1 mRNA were strongly increased in the RNS GFs; a similar upregulation was previously associated with the activation of YAP/TAZ signaling in the dermis99. Nuclear YAP/TAZ promotes TGFβ signaling via retaining activated Smad3 in the nucleus100. The level of YAP/TAZ was higher in RNS Gfs compared to control GFs and the amount of the inactive phospho-YAP dramatically decreased suggesting a shift towards YAP/TAZ activation. Addition of TGFβ1 further increased active YAP/TAZ levels consistent with converging TGFβ and YAP/TAZ signaling in RNS GFs. We investigated the FAM20C/TGFβ/YAP/TAZ link by knocking down FAM20C and indeed showed that silencing of FAM20C was sufficient to promote POSTN upregulation and nuclear YAP/TAZ localization. Nuclear YAP/TAZ localization was also observed in the control cells. It is very likely that the culture conditions, i.e. cells grown on a hard substrate like plastic, were sufficient to trigger YAP/TAZ nuclear translocation101. It is beyond the scope of this work to detail the mechanical regulation of YAP/TAZ expression; we assume that the mechanical stress information caused by the culture conditions was similar in control and mutant cells. Nevertheless, the nuclear YAP/TAZ signal was much stronger after FAM20C silencing suggesting a stronger activation in the depleted cells and providing a direct link between FAM20C function and TGFβ/YAP/TAZ signaling. It is very interesting to note that we obtained the same results by knocking down FAM20A. The interdependence between FAM20A and FAM20C concerning the kinase activity of the latter has been described16. Our finding that both FAM20C and FAM20A may be involved in the same signaling pathways suggests additional, yet to be uncovered cellular functions of these proteins.
Published data on FAM20C and FAM20A dysfunction in humans and mice19,102–104 described increased and/or attenuated BMP signaling in ectopic intrapulpal calcifications, AI, root defects and salivary gland development. Our previous results, in ERS GFs, and present proteomic analyses did not show any modification in BMP actors indicating that this pathway may not be directly involved in the fibrotic/osteogenic modifications of the gingiva.
In summary, our in vivo and in vitro data provide a novel and detailed description of the RNS gingival phenotype. We show that excessive ECM production and impaired matrix remodeling are the imperatives for gingival fibrosis and calcification in RNS patients. Our finding that both TGFβ and YAP/TAZ signaling are activated is completely new and highlights new aspects of FAM20C activity in signaling regulation.
Methods
Ethics—patients recruitment
Patients were referred for oral rehabilitation at the Reference Center of rare dental diseases (Oral Center for Inherited Diseases, University Hospital of Brasília, Brasil). Diagnosis of RNS was based on clinical and radiological features5,8. Patients and healthy age-matched controls (n = 3) were recruited following informed consent in accordance with the principles outlined in the declaration of Helsinki. Written informed consent was obtained from probands for the publication of any potentially identifiable images or data included in this article. This project was approved by the Research Ethics Committee of the Faculty of Medicine of the University of Brasília (CAAE: 34149814.1.0000.5558). Samples from probands and controls were harvested during oral rehabilitation and were prepared for histological or cell culture analyses (authorization CODECOH DC-2018-3382).
Immunocytochemistry and histology
Approximately, 1 cm3 gingival samples from patients were fixed for 24 h at 4 °C in 4% paraformaldehyde and then embedded in paraffin wax.
Histology
Sections were stained with routine protocols for Haematoxylin and Eosin (HE), Picro-Sirius Red, and Alizarin Red stains.
Immunohistochemistry
Epitope retrieval was achieved by heat. Sections were incubated for 2 h at room temperature or overnight at 4 °C with primary antibodies; rabbit anti-collagen V alpha-1 (1/1000; NBP-38162; Novus biologicals), rabbit anti-FAM20A (1/250; OACD03385; Aviva), rabbit anti-Fam20C (1/250; OAAB01003; Aviva), rabbit anti-Periostin (1/500; ab14041; Abcam), rabbit anti-phospho-SMAD3 (1/500; ab52903; Abcam), rabbit anti-SPARC (1/2000; ab255733; ABCAM), mouse anti-alpha Smooth Muscle Actin (1/1000; 1A4; ab7817; Abcam), mouse anti-Fibronectin (1/1000; P1H11; DSHB), mouse anti-FSP1 (1/2000; ab218512; ABCAM), mouse anti-Vimentin (1/1000; RV203; NBP-19767; Novus biologicals) and rat anti-Procollagen type I (1/500; M-58; ab64409; Abcam), mouse anti-Ubiquitin (UCHL1; 1/400; 66230-1-Ig; Proteintech), goat anti-Lamin B1(1/200; sc6216), Concanavalin A-Alexa 633 conjugate (70 mg/ml; 2616059; Thermo Fischer). Secondary antibodies used were Alexa 488- or Cy3-conjugated donkey anti-rabbit (Jackson Immunoresearch Laboratories, West Grove, PA; 1:500), Alexa 488- or Cy3-conjugated donkey anti-mouse (Invitrogen, Eugene, OR; 1:500), and Alexa 488- or Cy3-conjugated donkey anti-goat (1:500). Nuclear staining was achieved by 20 min incubation at room temperature in Hoechst 33,342 (Invitrogen, Eugene, OR). Images were collected by confocal microscopy (Zeiss LSM8) and processed using ZEN version 3,8(Carl Zeiss Microscopy GmbH, Jena, Germany) and ImageJ software (http://rsweb.nih.gov/ij/index.html).
DAB (3,3’-Diaminobenzidine) staining was achieved using standard protocol (donkey anti-rabbit biotin conjugated, ab7082; streptavidin-HRP, ab64269; DAB substrate kit, ab64238; all products, Abcam).
Fibroblast cell culture
Controls and proband gingival fibroblasts were established by plating small pieces of excised gingival on plastic dishes. Cells, particularly gingival fibroblasts, migrate out of the explant and colonize the petri dish. The flasks were filled with Dulbecco’s modified Eagle’s medium–low glucose (DMEM) containing 20% fetal calf serum (FCS), 1% non-essential amino acid, penicillin/streptomycin (100 mg/mL) and amphotericin B (2 ng/mL). The flasks are then placed in an incubator programmed at 37 °C in a humidity atmosphere at 5% CO2 and the cell culture medium was changed twice a week until the confluence of the cells (90% after about 3 weeks). Once at confluence, the gingival fibroblasts were trypsinized (Trypsin–EDTA, GIBCO®, 1 mL at 0.05%) and single-cell suspensions were seeded in 25 cm2 flasks containing DMEM 10% of FCS, passaged by splitting when they reached confluence, and frozen in liquid nitrogen until use. Cells at passages 3 to 6 were used in all experiments. We checked each cell culture for the morphology and the marker of fibroblasts (fibroblast-specific protein 1 [FSP1]; ab27957; Abcam, Cambridge, UK). We confirmed that all cell cultures did not exhibit the morphological changes during the passages, and that FSP1 was clearly detected in these cells (data not shown). Each experiment using these cells was repeated at least three times.
For the experimentations, GFs were seeded and cultured in DMEM 10% FBS for three days and then cultured in a BSA/FBS free medium for two days. These conditioned supernatants were used to perform the secretome analysis. Cultured supernatants were analysed by LC–MS/MS. The remaining cell lysates were used to the proteome analysis. Using the culture conditions, cells were collected for RT-qPCR and Western blot analyses. Cells were seeded at 7000 cells per cm2 surface area in 6 well plates for RT-qPCR and in dishes (100 mm diameter) for western blot experiments.
TGF-β1 treatment
For TGF-β1 treatment, recombinant human TGF-β1 at 5 ng/ml was used (R&D Systems, MN, USA). Prior to TGF-β1 treatment, nearly confluent cells were serum-starved in low-glucose DMEM for 24 h and washed with serum-free DMEM. Immediately after, cells were treated with TGF-β1 for 6 h.
For RT-qPCR and Western blot analyses cells were seeded at 7000 cells per cm2 surface area in 6 well plates and in 100 mm diameter dishes respectively. For immunofluorescence cells chamber slides (Nunc Lab-Tek, Thermofisher) were used. GFs from two controls and RNS subjects were used for all the experiments. Three replicates of each experiment were performed for each test to ensure reproducibility.
Si RNA transfection
The loss of function analysis for FAM20A and FAM20C was performed using small interfering RNA (siRNA). Three unique siRNAs per gene (Flexitube siRNA, Hs_FAM20A_3, Hs_FAM20C_3; Qiagen) were used along with a control scrambled siRNA (AllStars Negative Control siRNA, Qiagen).
Gingival fibroblasts, exhibiting a confluence of 50–70%, were pretreated with 10% FCS (fetal calf serum) supplemented DMEM (Dulbecco’s Modified Eagle Medium) without antibiotics. Prior to transfection siRNAs were complexed with LipofectamineTM RNAi REAGENT (InVitroGen). Gingival fibroblasts were transfected for 6 h following the protocol outlined by Thermo Fisher Scientific. The subsequent incubation was carried out at 37 °C with a 5% CO2 environment for a period ranging from 1 to 3 days.
For RT-qPCR cells were seeded at 7000 cells per cm2 surface area in 6 well plates. For immunofluorescence cells chamber slides (Nunc Lab-Tek, Thermofisher) were used. Three replicates of each experiment were performed for each test to ensure reproducibility.
Mass spectrometry
A high-resolution mass spectrometry (MS)-based approach was used to perform differential quantitative analysis on both secreted and cell lysate proteins from GFs cultures. Independent GFs cultures (control and RS) were seeded and cultured in triplicates, in low glucose DMEM 10% FBS for three days and then serum-deprived DMEM for two additional days. Each serum free cell supernatant (Control, n = 3; RNS-1, n = 3; RNS-2, n = 3) and remaining cell lysate (Control, n = 3; RNS-1, n = 3; RNS-2, n = 3) was analyzed in a single-run of LC–MS/MS.
Proteomic analysis
Sample preparation for proteomic mass spectrometry
Samples were prepared as previously described42. Sample preparation was different for secretome or cell lysate. For secretome analysis, each sample was precipitated with DOC/TCA (0.1%/10%) to concentrate the protein pool. After precipitation, proteins were resuspended in a homemade solubilization buffer. For cell proteome analysis, cell lysate was harvested using FastPrep Technologies (MP Biomedicals) and resuspended in a solubilization buffer. Then protein concentration of all samples was estimated using Bradford Assay (Biorad), according to the manufacturer’s instructions. Based on Bradford results, 25 µg of proteins of each sample were loaded into a 7% polyacrylamide gel (Acrylamide/Bis-Acrylamide 30% [29:1], Sigma Aldrich) and a migration was performed in a short period (90 min at 10–20 mA/gel) to stack all proteins in in a small piece of gel. After Coomassie blue staining, the revealed protein bands were excised. Proteins were reduced with 5 mM dithiothreitol for 40 min followed by alkylation with 20 mM iodoacetamide for 40 min in the dark (all products from Sigma Aldrich). After washing steps with water and acetonitrile (Sigma Aldrich), gel bands were submitted to protein digestion by 1 µg of trypsin (Promega). After overnight incubation at 37 °C, several steps of peptide extraction were performed with of 0.1% formic acid (FA) in water and acetonitrile solutions. Finally, for each sample, peptide fractions were combined and dried.
Nano LC–MS/MS analysis
For each sample, peptide fractions were solubilized in FA 0.1% (v/v) and analyzed on a LTQ-Orbitrap Elite apparatus coupled to an Easy nanoLC II system (Thermo Scientific). An amount of 0.2 µg of peptides was injected onto an enrichment column (C18 Pepmap100, Thermo Scientific). The separation was carried out with an analytical column needle (NTCC-360/100-5-153, Nikkyo-Technos). The flow rate was 300 nL/min and the mobile phase composed of H2O/0.1% FA (buffer A) and ACN/0.1% FA (buffer B). The elution gradient duration was 120 min: 0-106 min, 2–40% B; 106-110 min, 40–100% B; 110-120 min, 100% B. The mass spectrometer was operated in positive mode with CID fragmentation. For mass spectrometry settings, the capillary voltage was 1.5 kV and the temperature of the capillary was 275 °C. The m/z detection range was 400–1800 in MS scan at a resolution of 60 000. The 20 most intense peptide ions were selected and the fragmentation occurred with a normalized collision energy of 35. Dynamic exclusion of already fragmented precursor ions was applied for 30 s.
Quantification and statistical analysis
After MS analysis, raw data were imported into Progenesis QI v3.0 software(waters corporation,nonlinear dynamics, (https://www.nonlinear.com/progenesis/qi/v3.0/download/). To perform quantification, the peptide maps alignment and normalization had to be done. Briefly, one sample was set as a reference to align retention times of all other samples and a normalization step managed by the software (based on the idea that much of the peptides remain with unmodified abundances between the experiments, and so the quantitative abundance ratio of these majority of features should equal to 1), allowed us to determine a scalar factor for each sample, thereafter used for normalization. Statistical analysis was performed using the in-built Progenesis statistical box ‘one-way ANOVA’. MS/MS spectra were then exported for peptide identification with Mascot (Matrix Science, version 2.6.0). Database searches were performed with the following parameters: taxonomy: human (22,244 sequences); 1 missed cleavage; variable modification: carbamidomethyl of cysteine and oxidation of methionine. Mass tolerances for precursor and fragment ions were 10 ppm and 0.35 Da respectively. False discovery rates were calculated using a decoy-fusion approach in Mascot. Identified spectrum matches with -10logP value of 20 or higher were kept, at a FDR threshold of 5%. Mascot search results were imported into Progenesis. For each condition, the total cumulative abundance of protein was calculated by summing the abundance of peptides. All proteins identified with at least 2 peptides were considered for further analysis. A protein was considered differentially expressed if the p value is inferior to 0.05 and the statistical value of Power superior to 0.8.
Network biology and systems level analysis
Proteins identified by MS were entered in STRING database (https://string-db.org/) to create a protein–protein association network105. Network nodes represent all the proteins produced by a single protein-coding gene locus noting that splice isoforms or post-translational modifications are collapsed. Edges represent protein–protein associations that are specific and meaningful, such as proteins that jointly contribute to a shared function (note that this does not necessarily mean they physically bind each other). Thickness of network edges indicates the strength of data support based on textmining, experiments, databases, co-expression, neighbourhood, gene fusion and co-occurrence. The minimum required interaction score was set to a high confidence level of 0.7. Predicted interactome was evaluated using String software based on annotation enrichment strategy. The process of String to attach biological information relied on a series of enrichment-based tools widely used for the analysis of genes that are differentially expressed including Gene Ontology Resource (GO; geneontology.org), PANTHER16.0 (pantherdb.org), KEGG, or DAVID.
Western blotting
Western blotting was performed as previously described8. In brief, GFs were washed twice with PBS and protein was isolated in RIPA buffer (Sigma Aldrich) containing protease (Roche Diagnostics) and phosphatase inhibitors (Calbiochem) cocktails. Protein concentration was determined by Pierce® BCA Protein assay kit (Pierce; Waltham, MA, USA). A 25 µg quantity of protein from each sample was separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. Membranes were washed with Tris-buffered saline containing 0.05% Tween-20 (TBS-T) and blocked with 5% dried milk in TBS-T. Primary antibodies for anti-alpha Smooth Muscle Actin (1/1000; 14,968; Cell Signaling Technologies), phospho-YAP1Ser397 (1/2,000; 29018-1-AP; Proteintech), YAP-TAZ (1/1000; 93,622; Cell Signaling Technologies) and GAPDH (MAB374; Millipore; 1:2000) were used to incubate the membranes for 12 h. Detection was with appropriate peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch; West Grove, PA, USA; 1:2000), which were developed with SuperSignal Western Pico Chemiluminescence Substrate (Pierce).
Quantitative RT-qPCR
Total RNA was isolated using commercially available kits according to manufacturer guidelines (RNeasy Mini, Qiagen) and measured (Nanodrop, Peqleb). One μg was used in a reverse transcription reaction (SuperScript First strand synthesis, Thermofisher). Quantitative-PCR was performed using Quantifast SYBR Green PCR Kit (Qiagen), reactions were performed in triplicate. Transcript levels were calculated using the standard curves generated using serial dilutions of cDNA obtained by reverse transcription of control RNA samples then normalized to HPRT. Primer sequences were listed in supplementary Table S15. Amplification specificities were assessed by melting curve analyses and amplicons were sequenced. Values correspond to the mean of 3 independent experiments in triplicates of three control cultures and the RNS patient cultures. Data represent mean fold gene expressions ± s.d. relative to control. Data were analyzed via two-way ANOVA with Bonferroni multiple comparisons test (*p < 0.05, **p < 0.01, ***p < 0.001).
Immunocytochemistry in vitro
Cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and blocked with 1% bovine serum albumin. Primary antibodies for phospho-SMAD3 (1/500; ab52903; Abcam) and YAP-TAZ (1/1000; 93,622; Cell Signaling Technologies) were incubated one hour. Phalloidin staining was achieved simultaneously (Alexa 594 Phalloidin, A12381, ThermoFisher Scientific) Secondary antibodies used were Alexa 488- donkey anti-rabbit (Jackson Immunoresearch Laboratories, West Grove, PA; 1:500). Nuclear staining was achieved by 20 min incubation at room temperature in Hoechst 33,342 (ThermoFisher Scientific). No cellular autofluorescence and no nonspecific labeling were detected in these conditions. Images were collected by confocal microscopy (Zeiss LSM8) and processed using ZEN (Zeiss) and ImageJ software.
Image acquisition
Images were obtained on a Zeiss LSM800 confocal microscope (Carl Zeiss Microscopy). In the only step involving operator discretion, all image parameters including pinhole size, detector gain, amplifier offset, amplifier gain and laser intensity were first set for the DAPI and specific-antigen channels using normal control tissue, and the same setting used for all samples imaged on a given day. Frame size, scan speed and averaging were the same for all images. For each sample four non-overlapping images for each channel were acquired and stored as 16-bit fluorescent images (TIFF) for analysis.
Image analysis
Image analysis was performed in the ImageJ software program.
Fluorescence
The original DAPI and specific antigen images are opened; an intensity threshold for the DAPI signal is chosen by the user. Pixels with signals above this threshold constitute DAPI regions of interest (ROI). ROI are converted into a binary mask where areas of interest have a pixel intensity value of 1 and background has a pixel intensity value of 0. In order to eliminate background noise, ROI areas smaller than 3 × 3 contiguous pixels are removed from the image by an erosion step, leaving areas of equal to or greater than 3 × 3 contiguous pixels that are presumed to define contiguous nuclear membrane. The resulting image is dilated to restore positive regions. The dilated image is used to create a DAPI mask. The DAPI mask is overlaid on to the DAPI image and intensities are quantified. The DAPI mask is overlaid onto the specific-antigen image and intensities are quantified. The Excel file contains values for average specific-antigen intensity, average DAPI intensity, and a specifc-antigen/DAPI ratio for each image pair analysed. Average specific-antigen intensity was divided by average DAPI intensity to give a specific-antigen/DAPI ratio. These values were recorded for each of the four images per sample. The four specific-antigen/DAPI ratio values were themselves averaged to generate an overall specific-antigen/DAPI ratio for each sample.
DAB
In brief, for color deconvolution of IHC images, DAB and hematoxylin staining were digitally separated using ImageJ Fiji software (https://imagej.net/software/fiji/) with a color deconvolution plugin. Deconvoluted images with DAB staining were subjected to measurement of mean gray values, with the lower and upper thresholds set at 120 and 220 for FAM20C and FAM20A. The number of nuclei in the same field was determined from the hematoxylin staining by analyzing particle numbers using ImageJ Fiji software. The mean gray values of FAM20C and FAM20A were then normalized by the number of cell nuclei. The expression levels of FAM20C and FAM20A were determined as the average of the gray values normalized by nuclei number from 4 images per gingival tissue determined.
Statistical analysis
Statistical analysis was by one-way or two-way ANOVA, as appropriate, followed by Bonferroni multiple comparisons test with Graphpad Software version 5 (Graphpad Software; La Jolla, CA, USA) (https://www.graphpad.com/support/prism-5-updates/) (p < 0.05 was considered significant). Data are expressed as the mean ± standard deviation of 3 or 5 individual experiments with independent primary cultures from different subjects. Individual experiments included three replicates.
Institutional review board statement
This project was approved by the Research Ethics Committee of the Faculty of Medicine of the University of Brasília (CAAE: 34149814.1.0000.5558). The studies involving human gingival fibroblast cultures were reviewed and approved by CODECOH DC-2018-3382 INSERM.
Informed consent statement
Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Supplementary Information
Author contributions
PC, ACA and RK: study conception and design—writing original draft. CRRC, RC,OC, AmB, GC, VSE, LA, BA, CLL, VC: performing experiments. PMY, VSE, CG, LA, AB and CC: formal analysis. MB participated in histological study. All authors contributed to review and editing of the article and approved the submitted version.
Funding
This study was supported by TETECOU Filière (RK), FHU-DDS ParisNet (RK) (Université de Paris, AP-HP, INSERM), ECOS-Nord (RK) (C21501) and CAPES/COFECUB (ACA,CRRC,AB) (#918/2018), DPI University of Brasilia (UnB) (ACA),Rouen Normandy University and Normandy Region (PC,RK). Europe gets involved in Normandy with European Regional Development Fund (PC).This study contributes to the IdExUniversité de Paris ANR-18-IDEX-0001.
Data availability
The proteomic datasets used during the current study available from the corresponding author on reasonable request. All data generated during this study are included in this published article [and its supplementary information files].
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Cláudio Rodrigues Rezende Costa, Rym Chalgoumi, Amina Baker and Clément Guillou.
These authors jointly supervised this work: Pascal Cosette, Renata Kozyraki and Ana Carolina Acevedo.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-59713-0.
References
- 1.Koike T, Izumikawa T, Tamura J-I, Kitagawa H. FAM20B is a kinase that phosphorylates xylose in the glycosaminoglycan-protein linkage region. Biochem. J. 2009;421:157–162. doi: 10.1042/BJ20090474. [DOI] [PubMed] [Google Scholar]
- 2.Kuroda Y, et al. A novel gene (FAM20B encoding glycosaminoglycan xylosylkinase) for neonatal short limb dysplasia resembling Desbuquois dysplasia. Clin. Genet. 2019;95:713–717. doi: 10.1111/cge.13530. [DOI] [PubMed] [Google Scholar]
- 3.Cui J, et al. A secretory kinase complex regulates extracellular protein phosphorylation. Elife. 2015;4:e06120. doi: 10.7554/eLife.06120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martelli-Júnior H, et al. Case reports of a new syndrome associating gingival fibromatosis and dental abnormalities in a consanguineous family. J. Periodontol. 2008;79:1287–1296. doi: 10.1902/jop.2008.070520. [DOI] [PubMed] [Google Scholar]
- 5.Jaureguiberry G, et al. Nephrocalcinosis (enamel renal syndrome) caused by autosomal recessive FAM20A mutations. Nephron. Physiol. 2012;122:1–6. doi: 10.1159/000349989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang S-K, et al. FAM20A mutations can cause enamel-renal syndrome (ERS) PLoS Genet. 2013;9:e1003302. doi: 10.1371/journal.pgen.1003302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kantaputra PN, et al. Enamel-renal-gingival syndrome and FAM20A mutations. Am. J. Med. Genet. A. 2014;164A:1–9. doi: 10.1002/ajmg.a.36187. [DOI] [PubMed] [Google Scholar]
- 8.Simancas Escorcia V, et al. Lack of FAM20A, ectopic gingival mineralization and chondro/osteogenic modifications in enamel renal syndrome. Front. Cell Dev. Biol. 2020;8:605084. doi: 10.3389/fcell.2020.605084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho SH, et al. Novel FAM20A mutations in hypoplastic amelogenesis imperfecta. Hum. Mutat. 2012;33:91–94. doi: 10.1002/humu.21621. [DOI] [PubMed] [Google Scholar]
- 10.Nitayavardhana I, et al. Four novel mutations of FAM20A in amelogenesis imperfecta type IG and review of literature for its genotype and phenotype spectra. Mol. Genet. Genom. 2020;295:923–931. doi: 10.1007/s00438-020-01668-8. [DOI] [PubMed] [Google Scholar]
- 11.Tagliabracci VS, et al. Secreted kinase phosphorylates extracellular proteins that regulate biomineralization. Science. 2012;336:1150–1153. doi: 10.1126/science.1217817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bingham EW, Farrell HM, Basch JJ. Phosphorylation of casein. Role of the golgi apparatus. J. Biol. Chem. 1972;247:8193–8194. doi: 10.1016/S0021-9258(20)81827-4. [DOI] [PubMed] [Google Scholar]
- 13.Lietz CB, et al. Phosphopeptidomics reveals differential phosphorylation states and novel SxE phosphosite motifs of neuropeptides in dense core secretory vesicles. J. Am. Soc. Mass Spectrom. 2018;29:935–947. doi: 10.1007/s13361-018-1915-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nalbant D, et al. FAM20: An evolutionarily conserved family of secreted proteins expressed in hematopoietic cells. BMC Genom. 2005;6:11. doi: 10.1186/1471-2164-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tagliabracci VS, et al. A single kinase generates the majority of the secreted phosphoproteome. Cell. 2015;161:1619–1632. doi: 10.1016/j.cell.2015.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H, et al. Structure and evolution of the Fam20 kinases. Nat. Commun. 2018;9:1218. doi: 10.1038/s41467-018-03615-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du E-X, et al. Characterization of Fam20C expression in odontogenesis and osteogenesis using transgenic mice. Int. J. Oral Sci. 2015;7:89–94. doi: 10.1038/ijos.2014.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, et al. FAM20C plays an essential role in the formation of murine teeth. J. Biol. Chem. 2012;287:35934–35942. doi: 10.1074/jbc.M112.386862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu C, et al. FAM20C regulates osteoblast behaviors and intracellular signaling pathways in a cell-autonomous manner. J. Cell. Physiol. 2018;233:3476–3486. doi: 10.1002/jcp.26200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma P, et al. The importance of serine phosphorylation of ameloblastin on enamel formation. J. Dent. Res. 2016;95:1408–1414. doi: 10.1177/0022034516661513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, et al. Secretory kinase Fam20C tunes endoplasmic reticulum redox state via phosphorylation of Ero1α. EMBO J. 2018 doi: 10.15252/embj.201798699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu J, et al. Phosphorylation switches protein disulfide isomerase activity to maintain proteostasis and attenuate ER stress. EMBO J. 2020;39:e103841. doi: 10.15252/embj.2019103841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hecht TK-H, et al. Fam20C regulates protein secretion by Cab45 phosphorylation. J. Cell. Biol. 2020;219:e201910089. doi: 10.1083/jcb.201910089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pollak AJ, et al. A secretory pathway kinase regulates sarcoplasmic reticulum Ca2+ homeostasis and protects against heart failure. Elife. 2018;7:e41378. doi: 10.7554/eLife.41378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ben Djoudi Ouadda A, et al. Ser-phosphorylation of pcsk9 (proprotein convertase subtilisin-kexin 9) by Fam20C (family with sequence similarity 20, member C) kinase enhances its ability to degrade the LDLR (low-density lipoprotein receptor) Arterioscler. Thromb. Vasc. Biol. 2019;39:1996–2013. doi: 10.1161/ATVBAHA.119.313247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tagliabracci VS, et al. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc. Natl. Acad. Sci. U. S. A. 2014;111:5520–5525. doi: 10.1073/pnas.1402218111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raine J, Winter RM, Davey A, Tucker SM. Unknown syndrome: Microcephaly, hypoplastic nose, exophthalmos, gum hyperplasia, cleft palate, low set ears, and osteosclerosis. J. Med. Genet. 1989;26:786–788. doi: 10.1136/jmg.26.12.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simpson MA, et al. Mutations in FAM20C are associated with lethal osteosclerotic bone dysplasia (Raine syndrome), highlighting a crucial molecule in bone development. Am. J. Hum. Genet. 2007;81:906–912. doi: 10.1086/522240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simpson MA, et al. Mutations in FAM20C also identified in non-lethal osteosclerotic bone dysplasia. Clin. Genet. 2009;75:271–276. doi: 10.1111/j.1399-0004.2008.01118.x. [DOI] [PubMed] [Google Scholar]
- 30.Fradin M, et al. Osteosclerotic bone dysplasia in siblings with a Fam20C mutation. Clin. Genet. 2011;80:177–183. doi: 10.1111/j.1399-0004.2010.01516.x. [DOI] [PubMed] [Google Scholar]
- 31.Ishikawa HO, Xu A, Ogura E, Manning G, Irvine KD. The Raine syndrome protein FAM20C is a Golgi kinase that phosphorylates bio-mineralization proteins. PLoS One. 2012;7:e42988. doi: 10.1371/journal.pone.0042988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Acevedo AC, et al. Variability of systemic and oro-dental phenotype in two families with non-lethal Raine syndrome with FAM20C mutations. BMC Med. Genet. 2015;16:8. doi: 10.1186/s12881-015-0154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheth J, Bhavsar R, Gandhi A, Sheth F, Pancholi D. A case of Raine syndrome presenting with facial dysmorphy and review of literature. BMC Med. Genet. 2018;19:76. doi: 10.1186/s12881-018-0593-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elalaoui SC, et al. Non lethal Raine syndrome and differential diagnosis. Eur. J. Med. Genet. 2016;59:577–583. doi: 10.1016/j.ejmg.2016.09.018. [DOI] [PubMed] [Google Scholar]
- 35.Mameli C, et al. Natural history of non-lethal Raine syndrome during childhood. Orphanet J. Rare Dis. 2020;15:93. doi: 10.1186/s13023-020-01373-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palma-Lara I, et al. FAM20C overview: Classic and novel targets, pathogenic variants and raine syndrome phenotypes. Int. J. Mol. Sci. 2021;22:8039. doi: 10.3390/ijms22158039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hung CY, et al. A novel FAM20C mutation causes a rare form of neonatal lethal Raine syndrome. Am. J. Med. Genet. A. 2019;179:1866–1871. doi: 10.1002/ajmg.a.61291. [DOI] [PubMed] [Google Scholar]
- 38.Mamedova E, et al. Non-lethal raine syndrome in a middle-aged woman caused by a novel FAM20C Mutation. Calcif. Tissue Int. 2019;105:567–572. doi: 10.1007/s00223-019-00599-w. [DOI] [PubMed] [Google Scholar]
- 39.Vogel P, et al. Amelogenesis imperfecta and other biomineralization defects in Fam20a and Fam20c null mice. Vet. Pathol. 2012;49:998–1017. doi: 10.1177/0300985812453177. [DOI] [PubMed] [Google Scholar]
- 40.Wang X, et al. The specific role of FAM20C in amelogenesis. J. Dent. Res. 2013;92:995–999. doi: 10.1177/0022034513504588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dong C, et al. The phosphorylation of serine55 in enamelin is essential for murine amelogenesis. Matrix Biol. 2022;111:245–263. doi: 10.1016/j.matbio.2022.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simancas Escorcia V, et al. Pathogenesis of enamel-renal syndrome associated gingival fibromatosis: A proteomic approach. Front. Endocrinol. (Lausanne) 2021;12:752568. doi: 10.3389/fendo.2021.752568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Osaka H, et al. Ubiquitin carboxy-terminal hydrolase L1 binds to and stabilizes monoubiquitin in neuron. Hum. Mol. Genet. 2003;12:1945–1958. doi: 10.1093/hmg/ddg211. [DOI] [PubMed] [Google Scholar]
- 44.Shibata S, Ishiyama J. Secreted protein acidic and rich in cysteine (SPARC) is upregulated by transforming growth factor (TGF)-β and is required for TGF-β-induced hydrogen peroxide production in fibroblasts. Fibrogenes. Tissue Repair. 2013;6:6. doi: 10.1186/1755-1536-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Worby CA, Mayfield JE, Pollak AJ, Dixon JE, Banerjee S. The ABCs of the atypical Fam20 secretory pathway kinases. J. Biol. Chem. 2021;296:100267. doi: 10.1016/j.jbc.2021.100267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dzamukova M, et al. Mechanical forces couple bone matrix mineralization with inhibition of angiogenesis to limit adolescent bone growth. Nat. Commun. 2022;13:3059. doi: 10.1038/s41467-022-30618-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chung Y, et al. Role of Shh and TGF in cyclosporine-enhanced expression of collagen and α-SMA by gingival fibroblast. J. Clin. Periodontol. 2015;42:29–36. doi: 10.1111/jcpe.12333. [DOI] [PubMed] [Google Scholar]
- 48.Marconi GD, et al. Transforming growth factor-beta1 and human gingival fibroblast-to-myofibroblast differentiation: Molecular and morphological modifications. Front. Physiol. 2021;12:676512. doi: 10.3389/fphys.2021.676512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wright HJ, Chapple IL, Matthews JB. TGF-beta isoforms and TGF-beta receptors in drug-induced and hereditary gingival overgrowth. J. Oral Pathol. Med. 2001;30:281–289. doi: 10.1034/j.1600-0714.2001.300505.x. [DOI] [PubMed] [Google Scholar]
- 50.Wang Z, et al. Periostin: An emerging activator of multiple signaling pathways. J. Cell Commun. Signal. 2022;16:515–530. doi: 10.1007/s12079-022-00674-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim SS, Jackson-Boeters L, Darling MR, Rieder MJ, Hamilton DW. Nifedipine induces periostin expression in gingival fibroblasts through TGF-beta. J. Dent. Res. 2013;92:1022–1028. doi: 10.1177/0022034513503659. [DOI] [PubMed] [Google Scholar]
- 52.Shinde AV, Humeres C, Frangogiannis NG. The role of α-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2017;1863:298–309. doi: 10.1016/j.bbadis.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trombetta-Esilva J, Bradshaw AD. The function of SPARC as a mediator of fibrosis. Open Rheumatol. J. 2012;6:146–155. doi: 10.2174/1874312901206010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Massagué J, Chen YG. Controlling TGF-beta signaling. Genes Dev. 2000;14:627–644. doi: 10.1101/gad.14.6.627. [DOI] [PubMed] [Google Scholar]
- 55.Varelas X, et al. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat. Cell. Biol. 2008;10:837–848. doi: 10.1038/ncb1748. [DOI] [PubMed] [Google Scholar]
- 56.Piersma B, Bank RA, Boersema M. Signaling in fibrosis: TGF-β, WNT, and YAP/TAZ converge. Front. Med. (Lausanne) 2015;2:59. doi: 10.3389/fmed.2015.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Szeto SG, et al. YAP/TAZ are mechanoregulators of TGF-β-Smad signaling and renal fibrogenesis. J. Am. Soc. Nephrol. 2016;27:3117–3128. doi: 10.1681/ASN.2015050499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiang D, et al. Physalin D attenuates hepatic stellate cell activation and liver fibrosis by blocking TGF-β/Smad and YAP signaling. Phytomedicine. 2020;78:153294. doi: 10.1016/j.phymed.2020.153294. [DOI] [PubMed] [Google Scholar]
- 59.Zhao B, Li L, Tumaneng K, Wang C-Y, Guan K-L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP) Genes Dev. 2010;24:72–85. doi: 10.1101/gad.1843810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mannaerts I, et al. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J. Hepatol. 2015;63:679–688. doi: 10.1016/j.jhep.2015.04.011. [DOI] [PubMed] [Google Scholar]
- 61.Zhang J, et al. Secretory kinase Fam20C tunes endoplasmic reticulum redox state via phosphorylation of Ero1α. EMBO J. 2018;37:e98699. doi: 10.15252/embj.201798699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pollak AJ, et al. Phosphorylation of serine96 of histidine-rich calcium-binding protein by the Fam20C kinase functions to prevent cardiac arrhythmia. Proc.. Natl. Acad. Sci. U. S. A. 2017;114:9098–9103. doi: 10.1073/pnas.1706441114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen X, et al. Proteolytic processing of secretory pathway kinase Fam20C by site-1 protease promotes biomineralization. Proc. Natl. Acad. Sci. U. S. A. 2021;118:e2100133118. doi: 10.1073/pnas.2100133118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sehgal P, et al. Inhibition of the sarco/endoplasmic reticulum (ER) Ca2+-ATPase by thapsigargin analogs induces cell death via ER Ca2+ depletion and the unfolded protein response. J. Biol. Chem. 2017;292:19656–19673. doi: 10.1074/jbc.M117.796920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kinoshita Y, Hori M, Taguchi M, Fukumoto S. Functional analysis of mutant FAM20C in Raine syndrome with FGF23-related hypophosphatemia. Bone. 2014;67:145–151. doi: 10.1016/j.bone.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 66.Saigoh K, et al. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat. Genet. 1999;23:47–51. doi: 10.1038/12647. [DOI] [PubMed] [Google Scholar]
- 67.Mohanan A, Washimkar KR, Mugale MN. Unraveling the interplay between vital organelle stress and oxidative stress in idiopathic pulmonary fibrosis. Biochim. Biophys. Acta Mol. Cell Res. 2024;1871:119676. doi: 10.1016/j.bbamcr.2024.119676. [DOI] [PubMed] [Google Scholar]
- 68.Yuan Q, et al. MiR-185-5p ameliorates endoplasmic reticulum stress and renal fibrosis by downregulation of ATF6. Lab. Investig. 2020;100:1436–1446. doi: 10.1038/s41374-020-0447-y. [DOI] [PubMed] [Google Scholar]
- 69.Wang H, et al. Diosmetin-7-O-β-D-glucopyranoside suppresses endothelial-mesenchymal transformation through endoplasmic reticulum stress in cardiac fibrosis. Clin. Exp. Pharmacol. Physiol. 2023;50:789–805. doi: 10.1111/1440-1681.13802. [DOI] [PubMed] [Google Scholar]
- 70.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 71.Al-Mane K, Al-Dayel F, McDonald P. Intracranial calcification in Raine syndrome: Radiological pathological correlation. Pediatr. Radiol. 1998;28:820–823. doi: 10.1007/s002470050473. [DOI] [PubMed] [Google Scholar]
- 72.Tarbit E, Singh I, Peart JN, RoseMeyer RB. Biomarkers for the identification of cardiac fibroblast and myofibroblast cells. Heart Fail. Rev. 2019;24:1–15. doi: 10.1007/s10741-018-9720-1. [DOI] [PubMed] [Google Scholar]
- 73.Venugopal H, Hanna A, Humeres C, Frangogiannis NG. Properties and functions of fibroblasts and myofibroblasts in myocardial infarction. Cells. 2022;11:1386. doi: 10.3390/cells11091386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang H, Zhou Y, Wen D, Wang J. Noncoding RNAs: Master regulator of fibroblast to myofibroblast transition in fibrosis. Int. J. Mol. Sci. 2023;24:1801. doi: 10.3390/ijms24021801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014;71:549–574. doi: 10.1007/s00018-013-1349-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li L, Fu H, Liu Y. The fibrogenic niche in kidney fibrosis: Components and mechanisms. Nat. Rev. Nephrol. 2022;18:545–557. doi: 10.1038/s41581-022-00590-z. [DOI] [PubMed] [Google Scholar]
- 77.Nikoloudaki G, Snider P, Simmons O, Conway SJ, Hamilton DW. Periostin and matrix stiffness combine to regulate myofibroblast differentiation and fibronectin synthesis during palatal healing. Matrix Biol. 2020;94:31–56. doi: 10.1016/j.matbio.2020.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Farbehi N, et al. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife. 2019;8:e43882. doi: 10.7554/eLife.43882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gallucci RM, Lee EG, Tomasek JJ. IL-6 modulates alpha-smooth muscle actin expression in dermal fibroblasts from IL-6-deficient mice. J Invest Dermatol. 2006;126:561–568. doi: 10.1038/sj.jid.5700109. [DOI] [PubMed] [Google Scholar]
- 80.Morton RS, Dongari-Bagtzoglou AI. Regulation of gingival fibroblast interleukin-6 secretion by cyclosporine A. J. Periodontol. 1999;70:1464–1471. doi: 10.1902/jop.1999.70.12.1464. [DOI] [PubMed] [Google Scholar]
- 81.Kanisicak O, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016;7:12260. doi: 10.1038/ncomms12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li C, Kuemmerle JF. The fate of myofibroblasts during the development of fibrosis in Crohn’s disease. J. Dig. Dis. 2020;21:326–331. doi: 10.1111/1751-2980.12852. [DOI] [PubMed] [Google Scholar]
- 83.Gagliano N, et al. Differential effect of cyclosporin A and FK506 on SPARC mRNA expression by human gingival fibroblasts. Biomed. Pharmacother. 2005;59:249–252. doi: 10.1016/j.biopha.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 84.Takeuchi H, et al. Nicotine-induced CCN2: From smoking to periodontal fibrosis. J. Dent. Res. 2010;89:34–39. doi: 10.1177/0022034509353403. [DOI] [PubMed] [Google Scholar]
- 85.Kiuchi M, et al. An assessment of mast cells and myofibroblasts in denture-induced fibrous hyperplasia. J. Oral Pathol. Med. 2014;43:53–60. doi: 10.1111/jop.12072. [DOI] [PubMed] [Google Scholar]
- 86.Riley HJ, et al. SPARC production by bone marrow-derived cells contributes to myocardial fibrosis in pressure overload. Am. J. Physiol. Heart Circ. Physiol. 2021;320:H604–H612. doi: 10.1152/ajpheart.00552.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kozumi K, et al. Transcriptomics identify thrombospondin-2 as a biomarker for NASH and advanced liver fibrosis. Hepatology. 2021;74:2452–2466. doi: 10.1002/hep.31995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li FJ, et al. Citrullinated vimentin mediates development and progression of lung fibrosis. Sci. Transl. Med. 2021;13:eaba2927. doi: 10.1126/scitranslmed.aba2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ghosh AK, Vaughan DE. PAI-1 in tissue fibrosis. J. Cell. Physiol. 2012;227:493–507. doi: 10.1002/jcp.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bauer A, Habior A. Concentration of serum matrix metalloproteinase-3 in patients with primary biliary cholangitis. Front. Immunol. 2022;13:885229. doi: 10.3389/fimmu.2022.885229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gawron K, et al. TIMP-1 association with collagen type I overproduction in hereditary gingival fibromatosis. Oral Dis. 2018;24:1581–1590. doi: 10.1111/odi.12938. [DOI] [PubMed] [Google Scholar]
- 92.Strzelec K, et al. Clinics and genetic background of hereditary gingival fibromatosis. Orphanet J. Rare Dis. 2021;16:492. doi: 10.1186/s13023-021-02104-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang X, Chen CT, Bhargava M, Torzilli PA. A comparative study of fibronectin cleavage by MMP-1, -3, -13, and -14. Cartilage. 2012;3:267–277. doi: 10.1177/1947603511435273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Das DK, et al. miR-1207-3p regulates the androgen receptor in prostate cancer via FNDC1/fibronectin. Exp. Cell Res. 2016;348:190–200. doi: 10.1016/j.yexcr.2016.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Das DK, Ogunwobi OO. A novel microRNA-1207-3p/FNDC1/FN1/AR regulatory pathway in prostate cancer. RNA Dis. 2017;4:e1503. [PMC free article] [PubMed] [Google Scholar]
- 96.Ibelli AMG, et al. Downregulation of growth plate genes involved with the onset of femoral head separation in young broilers. Front. Physiol. 2022;13:941134. doi: 10.3389/fphys.2022.941134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chin Y-T, et al. Antioxidants protect against gingival overgrowth induced by cyclosporine A. J Periodontal. Res. 2021;56:397–407. doi: 10.1111/jre.12832. [DOI] [PubMed] [Google Scholar]
- 98.Meng X-M, Nikolic-Paterson DJ, Lan HY. TGF-β: the master regulator of fibrosis. Nat. Rev. Nephrol. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 99.Lee M-J, Byun MR, Furutani-Seiki M, Hong J-H, Jung H-S. YAP and TAZ regulate skin wound healing. J. Investig. Dermatol. 2014;134:518–525. doi: 10.1038/jid.2013.339. [DOI] [PubMed] [Google Scholar]
- 100.Beyer TA, et al. Switch enhancers interpret TGF-β and Hippo signaling to control cell fate in human embryonic stem cells. Cell Rep. 2013;5:1611–1624. doi: 10.1016/j.celrep.2013.11.021. [DOI] [PubMed] [Google Scholar]
- 101.Aragona M, et al. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell. 2013;154:1047–1059. doi: 10.1016/j.cell.2013.07.042. [DOI] [PubMed] [Google Scholar]
- 102.Wang S-K, et al. FAM20A mutations and transcriptome analyses of dental pulp tissues of enamel renal syndrome. Int. Endod. J. 2023;56:943–954. doi: 10.1111/iej.13928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yin M, et al. The effect and mechanism of gene Fam20a on the development and function of salivary glands in mice. Arch. Oral Biol. 2022;137:105367. doi: 10.1016/j.archoralbio.2022.105367. [DOI] [PubMed] [Google Scholar]
- 104.Li L, et al. Ablation of FAM20C caused short root defects via suppressing the BMP signaling pathway in mice. J. Orofac. Orthop. 2022 doi: 10.1007/s00056-022-00386-7. [DOI] [PubMed] [Google Scholar]
- 105.Szklarczyk D, et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The proteomic datasets used during the current study available from the corresponding author on reasonable request. All data generated during this study are included in this published article [and its supplementary information files].