

Abstract
Huntington’s disease (HD) is a progressive neurodegenerative disorder with clinical presentations of moderate to severe cognitive, motor, and psychiatric disturbances. HD is caused by the trinucleotide repeat expansion of CAG of the huntingtin (HTT) gene. The mutant HTT protein containing pathological polyglutamine (polyQ) extension is prone to misfolding and aggregation in the brain. It has previously been observed that copper and iron concentrations are increased in the striata of post-mortem human HD brains. Although it has been shown that the accumulation of mutant HTT protein can interact with copper, the underlying HD progressive phenotypes due to copper overload remains elusive. Here, in a Drosophila model of HD, we showed that copper induces dose-dependent aggregational toxicity and enhancement of Htt-induced neurodegeneration. Specifically, we found that copper increases mutant Htt aggregation, enhances the accumulation of Thioflavin S positive β-amyloid structures within Htt aggregates, and consequently alters autophagy in the brain. Administration of copper chelator D-penicillamine (DPA) through feeding significantly decreases β-amyloid aggregates in the HD pathological model. These findings reveal a direct role of copper in potentiating mutant Htt protein-induced aggregational toxicity, and further indicate the potential impact of environmental copper exposure in the disease onset and progression of HD.
Introduction
Huntington’s disease (HD) is an autosomal dominant, adult-onset, and progressive rare neurodegenerative disorder caused by the unstable CAG repeat expansion in exon 1 of the HTT gene [1]. It encodes a stretch of polyglutamine (polyQ) residues at the N-terminus of the huntingtin (HTT) protein, in which more than 35 repeats causes disease [2]. Over the course of 10-20 years before death, the most common symptoms individuals manifest include progressive motor impairments such as chorea and gait disturbances, as well as neuropsychiatric symptoms such as depression, dementia, anxiety, and loss of self and spatial awareness [3–5]. Endogenous HTT is widely expressed in the nervous system and in all peripheral tissues [6, 7]. Specifically, it is found in neural cell bodies, dendrites, and it is enriched in nerve terminals. It acts as a scaffold and is involved in vesicular transport, neurotransmitter release, cell signaling, endocytosis, autophagy, and transcriptional regulation [5, 7–10]. The first brain region affected in HD is the basal ganglia (caudate nucleus and putamen) where the mutant HTT protein causes the disruption of several downstream pathways, resulting in neuronal death and glial activation [11]. Moreover, human HTT with expanded polyQ repeats have been shown to misfold and form soluble monomeric and oligomeric proteins, as well as insoluble aggregates in vitro and in vivo [12–14].
Essential elements such as copper, zinc, and manganese are essential for life, but they are only required in trace levels since excessive metal accumulation in the brain is deleterious and may cause several detrimental effects that lead to neurodegeneration, such as inducing oxidative stress, mitochondrial dysfunction, and protein misfolding [15–19]. Brain is the organ with the second largest copper accumulation and is particularly susceptible to metal toxicity [17]. Importantly, abnormal iron, copper, and manganese homeostasis has been observed in the brains of HD patients and in animal models [12, 20–22]. In contrast, it has been shown that there is no difference of Cu levels in the blood of HD patients [15, 23]. However, this could be due to the fact that copper is available to brain cells from cerebrospinal fluid (CSF). The normal range of serum copper concentrations in adults is 5-25 μM, of which approximately 95% is bound to ceruloplasmin (Cp), and the concentration of copper in the CSF is approximately 70-80 μM, which is at least three-fold higher compared to the serum, therefore increasing the possibility of specific copper signaling in the brain [17, 23].
Humans obtain their daily source of 1mg of copper through diet, including fruits, whole grains, vegetables, chocolate, mushrooms, seafood, and nuts [17, 22, 24–26]. In addition, major commercial uses of copper include electrical wiring, pipes, valves, coins, and cooking utensils [24, 27]. Biologically, copper is involved in respiration, oxidative stress protection, pigmentation, iron homeostasis, and neurotransmitter biosynthesis [16, 18, 19, 26, 28]. Most of these enzymatic reactions rely on the ability of copper to undergo redox transitions between the reduced Cu(I) cuprous ion and the oxidized Cu(II) cupric ion states [16, 17, 28, 29]. Therefore, copper has the potential to promote altered mutant huntingtin conformation, aggregation, and redox activity [30, 31]. Although it has been shown that the accumulation of mutant HTT protein can interact with copper, the underlying HD progressive phenotypes due to copper overload remains elusive. We previously optimized a Drosophila model for HD and discovered an age-dependent, compartment-specific accumulation of mutant Htt aggregates that are Thioflavin S positive (ThS+) and display amyloid-like adhesive properties that lead to clustering of mitochondria and synaptic components, which contribute to mutant Htt-induced neurotoxicity [32]. In this study, we specifically assess the effect of copper in Htt aggregation and neurodegeneration in a Drosophila model of HD, reveal the interaction between Cu and Htt protein aggregation in potentiating Htt protein-induced neurodegeneration, and further indicate the potential impact of environmental copper exposure in the disease pathology of HD.
Results
Copper enhances Htt-induced synaptic degeneration
Previous studies have reported the general toxicity of Cu exposure in wild type flies where 1mM Cu2+ exposure resulted in a significant decrease in survival [26], as well as in a HD model with 93 polyglutamine expansion (Htt-Q93) where 0.25mM CuCl2 feeding resulted in reduced survival rate [12]. To assess the effect of dietary copper exposure in a Drosophila model of HD, we established a copper dose-dependent feeding paradigm consisting of feeding 0 mM, 0.25 mM, or 0.50 mM CuCl2 from eclosion, and analyzed the cellular and biochemical properties at specific time points and ages (Figure 1A). To determine if there was a difference in feeding due to the presence of copper in the food, a measurement of food intake was performed. Flies of 2-5 DAE were starved for 2 hours and then allowed to feed from a 5% sucrose and 3% Coomassie brilliant blue dye solution. The amount of blue dye in the abdomen would correlate with the amount of food intake. The measurement of the concentration of blue dye present in the tissue extract showed no significant difference in feeding among CuCl2 concentrations, however, interestingly, a genotype-specific significant increase of food intake was observed in flies expressing Htt-Q138 when compared to that of Htt-Q15 (Figure 1B).
Figure 1: Copper enhances Htt-induced synaptic degeneration.
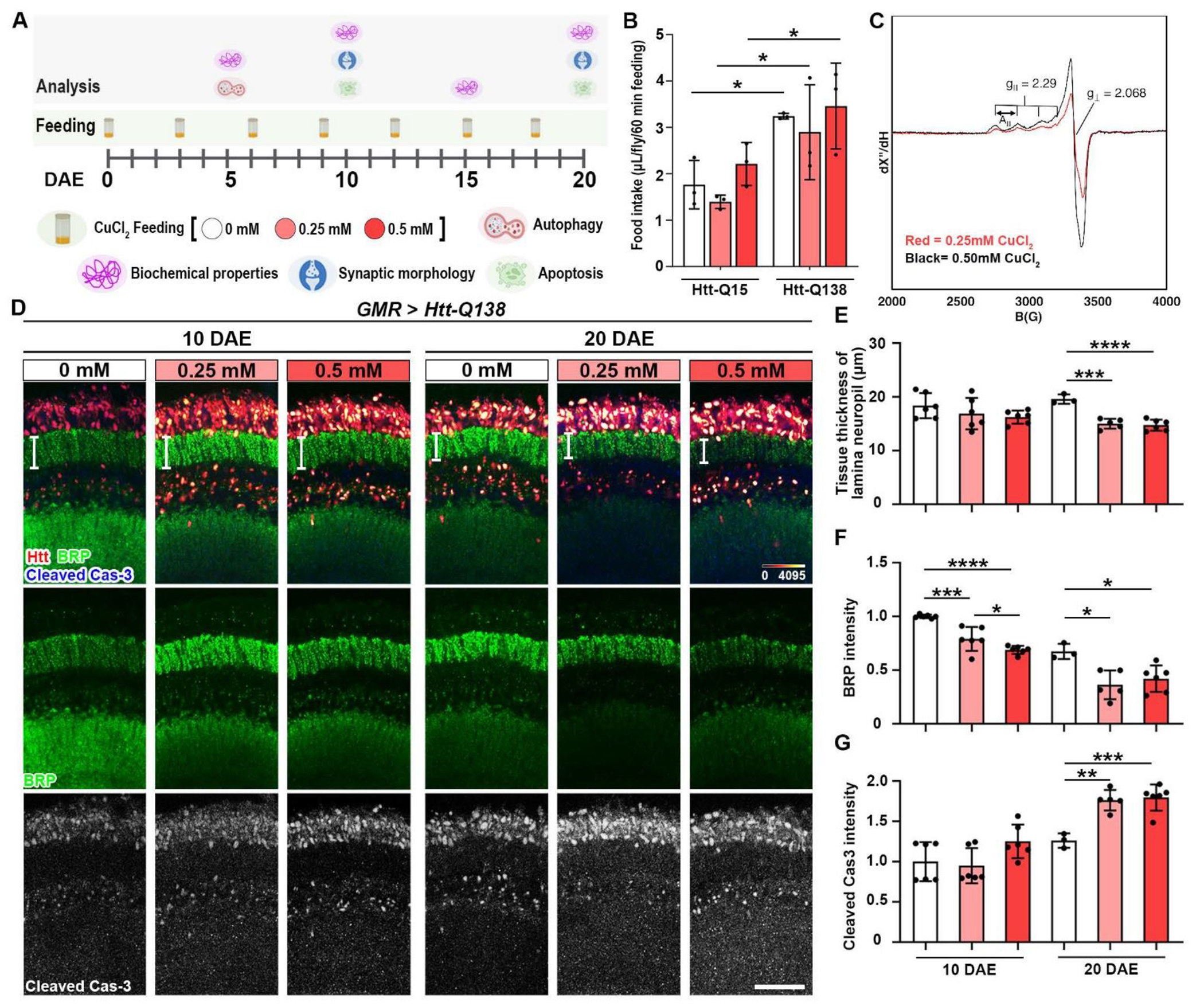
(A) CuCl2 feeding paradigm. Pathological Htt-Q138 flies were driven by GMR-GAL4 and fed with different concentrations of CuCl2 until 20 DAE and assessed cellular and biochemical properties. (B) Measurement of food intake (μL/fly/60 minutes feeding) of GMR>Htt-Q15 and GMR>Htt-Q138 flies during a 1hr feeding session following 2hrs starvation. n=3 biological replicates with 4-5 flies per group. (C) X-band (9.63 GHz microwave frequency) EPR spectra for samples prepared as 0.25 mM (red) and 0.5 mM (black) CuCl2. Data were recorded at 77 K, using 6.325 mW microwave power. Markers for g-values and hyperfine splitting (A‖) are also shown in the figure. (D) Lamina structures at 10 DAE and 20 DAE probed for BRP (green) and cCas3 (gray). Scale bar = 30μm. (E) Quantification of tissue thickness of lamina neuropil (white bars shown on panel C). (F) Quantification of BRP intensity. (G) Quantification of cleaved caspase-3 intensity. Data are presented as mean ± SD. n=3-5, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
To demonstrate the accuracy of the concentration of copper after CuCl2 is incorporated into the food, we determined the concentration of Cu2+ in the food using electron paramagnetic resonance spectroscopy (EPR). EPR is a sensitive spectroscopic technique that can be used to characterize the spin states and local environments of paramagnetic centers, such as Cu2+, Fe3+, and Mn2+ among others [33, 34]. Although inductively coupled plasma mass spectrometry (ICP-MS) is a powerful tool to determine the concentrations of trace metals, EPR is uniquely suited for determination of Cu2+ concentrations (into the low micromolar concentration range), as well as paramagnetic organic radicals that may be present in the sample. Furthermore, oxidation state specific analysis is important, as Cu+/Cu2+ speciation may offer the potential for side reactions that are accessible in biological systems. Since CuCl2 was added during preparation of the Drosophila food, EPR quantitation provides an accurate measure of the sample, in a more comparable state, as opposed to other analytical methods. In this experiment, 3 food vials of each concentration were used to measure accuracy between vials and copper concentration between groups. Quantitative analysis of copper concentration is performed via double integration of the EPR spectrum to determine the area under the curve and comparing this value to that for a spin standard of known concentration. The experimental data are plotted with magnetic field (in Gauss) on the x-axis. The curves show characteristic EPR signals for Cu2+, each with four hyperfine features centered around g=2.00. Double integration of the experimental EPR spectra shown in (Figure 1C) relative to a spin standard enabled quantitative determination of copper concentration in each food vial. From this analysis we calculated a final concentration of 0.103±0.004 mM copper in the 0.25 mM CuCl2 food sample, and 0.187±0.007 mM for the 0.50 mM CuCl2 fly food sample (Figure 1C). The experimentally-determined Cu(II) concentrations (0.103 and 0.187 mM) were lower than expected (0.25 and 0.50 mM), likely due to the fact that EPR measures Cu(II) EPR-active species, thus suggesting that there were likely other copper species present in the Drosophila media that were EPR silent under the measurement conditions. For this manuscript, we use 0.25 and 0.50 mM to indicate the reconstituted copper concentration.
Copper modulates synaptic activity as well as excitotoxic cell death and neurotrophic induced signaling cascades, therefore it is important for a variety of neuronal functions [17]. Previous studies have shown HD mice models with significant brain atrophy and loss of striatal volume, as well as caspase cleavage of mutant Htt being required for striatal volume loss [35, 36]. To analyze copper induced morphology and synaptic integrity changes, we took advantage of the Drosophila visual system, where the photoreceptor neurons are highly organized in parallel columns and make synapses with lamina monopolar cells. Age-dependent and copper concentration-dependent changes to the lamina were assessed by analyzing active zone marker bruchpilot (BRP) and apoptosis marker cleaved caspase-3 (cCas3) (Figure 1D). As a result of both 0.25 mM CuCl2 and 0.50 mM CuCl2 feeding, a decrease of tissue thickness of the lamina neuropil is shown (Figure 1E), as well as a decrease of BRP intensity at both 10 DAE and 20 DAE when compared to the 0 mM CuCl2 group, indicating synaptic degeneration (Figure 1F).
Apoptosis occurs normally during development and aging as a homeostatic mechanism to maintain cell populations in tissues [37]. To investigate the state of neuronal cell death, an apoptosis marker cleaved caspase-3 (cCas3) was used [37]. A significant increase in cCas3 intensity was observed at 20 DAE with feeding of CuCl2 (Figure 1G), suggesting exacerbated apoptosis by Cu feeding. To dissect and extract potential HD-specific toxicity from the general copper toxicity, we examined the effect of copper on flies expressing the Htt protein with non-pathological Q15 expansion [38]. Htt-Q15 flies were fed in the same conditions and assessed at 10 DAE and 20 DAE for synaptic phenotypes (Figure 2A). Interestingly, no difference in tissue thickness was observed (Figure 2B), indicating that synaptic integrity was largely intact with copper feeding. However, there was an age-dependent decrease in BRP intensity with 0.50 mM CuCl2 feeding (Figure 2C), suggesting a loss of active zone protein. In addition, cleaved caspase-3 intensity was increased at 20 DAE with 0.50 mM CuCl2 (Figure 2D). Collectively, these results indicate that copper has a mild effect on synaptic integrity in a non-pathological model, however, the effects of copper are significantly substantiated in a pathological model of HD.
Figure 2: Copper mildly decreases active zone protein BRP in a non-pathological model.
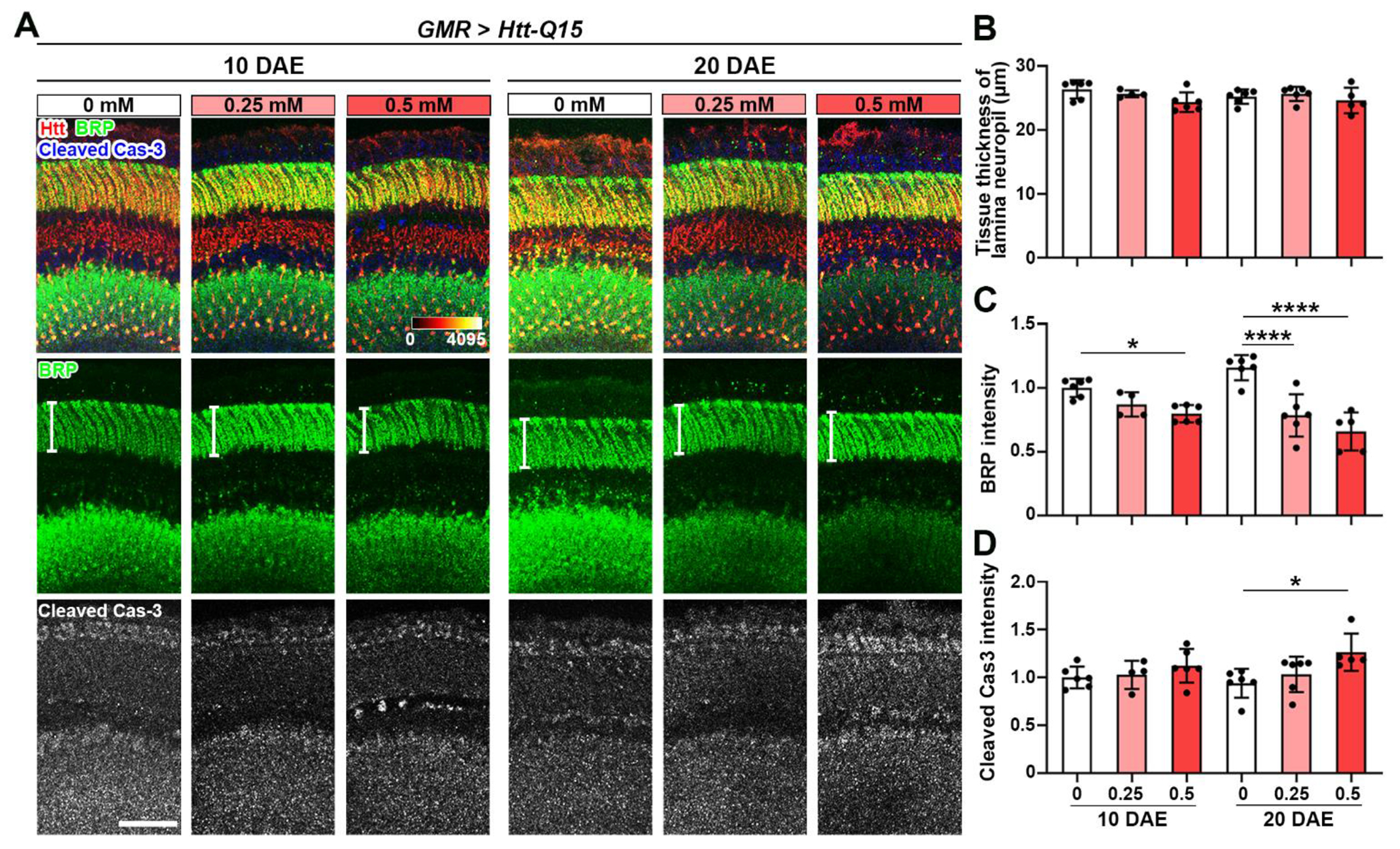
(A) Non-pathological Htt-Q15 flies were driven by GMR-GAL4 and fed with different concentrations of CuCl2 until 10 DAE and 20 DAE. Lamina structures at 10 DAE and 20 DAE were stained for BRP (green) and cCas3 (gray). Scale bar = 30μm. (B) Quantification of tissue thickness of lamina neuropil (white bars shown on panel A). (C) Quantification of BRP intensity. (D) Quantification of cleaved caspase-3 intensity. Data are presented as mean ± SD. n=3-5, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Copper increases Thioflavin S positive (ThS+) Htt aggregates
Aggregational toxicity is associated with Huntington’s Disease. Previous studies indicated that polyQ aggregation is an ordered process resembling amyloid fibril formation in both assembly kinetics and aggregate structures [39]. To analyze the properties of the aggregates in our model, we used Thioflavin S (ThS), which is a fluorescent dye used to detect β-amyloid sheet formation [40]. Fly brains from 5 DAE, 10 DAE, and 15 DAE fed with 0 mM, 0.25 mM, or 0.50 mM CuCl2, were dissected and probed for ThS (Figure 3A). Copper feeding at all ages increased aggregate intensity, compared to the 0 mM feeding group (Figure 3B). Specifically, at 5 DAE and 10 DAE, copper feeding at 0.50 mM CuCl2 significantly increased the number of aggregates per lamina (Figure 3C). To further assess the toxicity of these aggregates, we determined the number and percent of ThS+ aggregates per lamina. At 15 DAE with 0.50 mM CuCl2, there is a significant increase in both number (Figure 3D) and percentage (Figure 3E) of ThS+ aggregates per lamina. Lastly, to identify any specific potential effect of copper on size of the aggregates, we determined the changes of aggregate population based on size. Interestingly, copper had no effect on the number of large aggregates (>10 μm2) and a minor effect on the number of medium aggregates (5-10 μm2), but there was a significant increase on the number of small aggregates (<5 μm2) at 5 DAE and 10 DAE (Figure 3F). These results show that copper promotes the formation of β-amyloid structures in aggregates and specifically enhances the formation of small aggregates.
Figure 3: Copper increases ThS+ Htt aggregates.
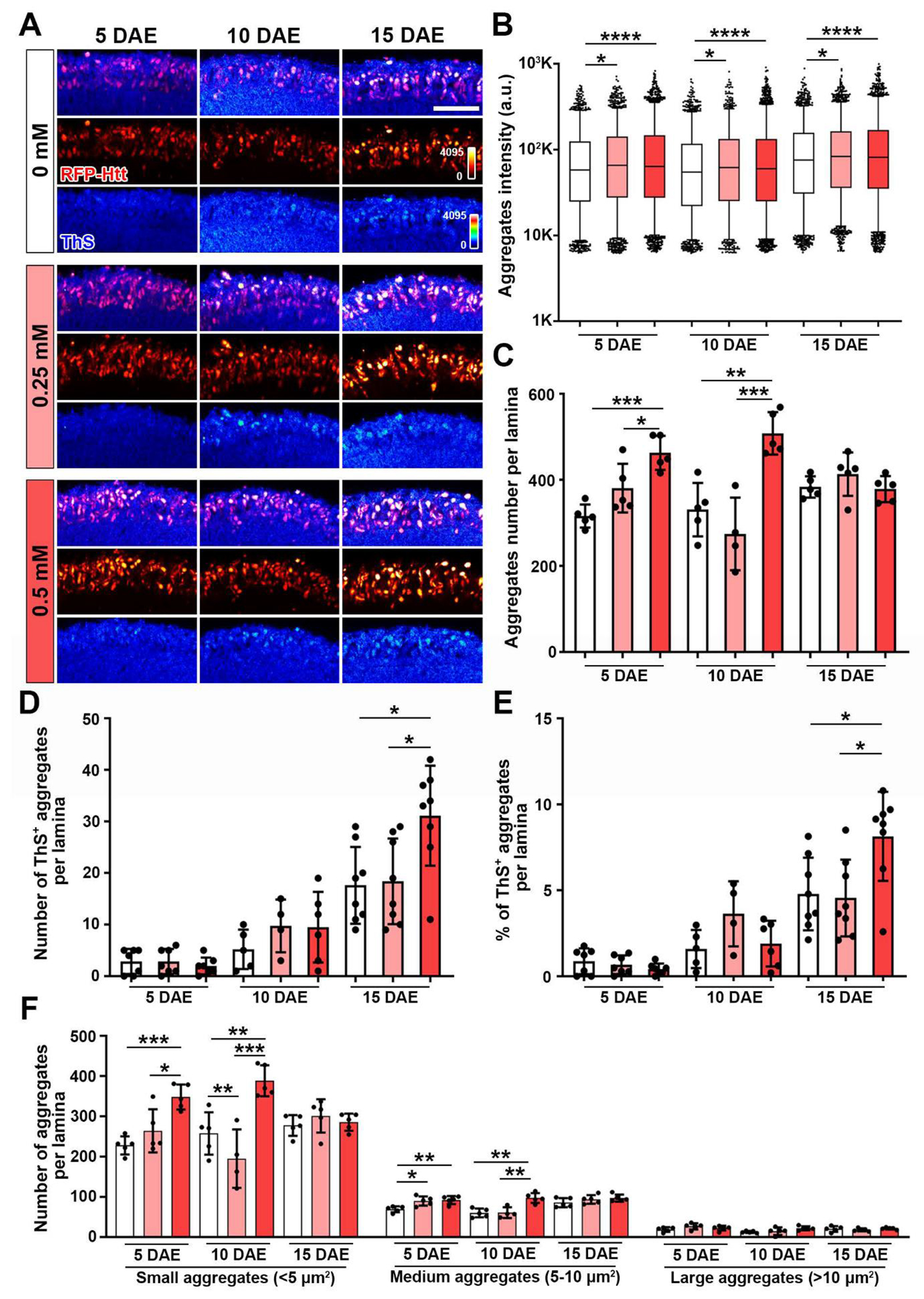
(A) ThS staining of the lamina layer (ThS and RFP-Htt heatmaps 0-4095) at 5 DAE, 10 DAE, and 20 DAE. Scale bar: 30μm (B) Quantification of Htt aggregates intensity. Data are presented as box and whiskers plot, 5-95 percentile. (C) Quantification of Htt aggregates number per lamina. Data are presented as mean ± SD. (D) Number of ThS+ aggregates per lamina. Data are presented as mean ± SD. (E) Percentage of ThS+ aggregates per lamina. Data are presented as mean ± SD. (F) Quantification of number of aggregates per lamina separated by size: Small (<5 μm2), Medium (5-10 μm2) and Large (>10 μm2). Data are presented as mean ± SD. n=3-8, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Copper enhances the association of autophagy initiation factor Ref(2)P with Htt aggregates
The ubiquitin-proteasome system (UPS) and autophagy are the major mechanisms for removing unfolded proteins. Autophagy plays a particularly important role in promoting the health of neuronal cells, which accumulate ubiquitinated protein aggregates when autophagy is impaired [41]. It has been previously shown that autophagy plays a key role in degrading mutant Htt [42]. The initiation of autophagy begins with Ref(2)P, which is the Drosophila homolog of human p62/SQSTM1, an adaptor protein that tethers ubiquitinated Htt for degradation [32, 43]. We have previously observed that Ref(2)P forms a ring-like structure surrounding large Htt aggregates [32, 43]. To assess how copper affects the interaction between Htt aggregates and Ref(2)P, an immunofluorescence approach was used to assess endogenous Ref(2)P expression and localization. Htt-Q138 was overexpressed in flies using elav-GAL4, a pan-neuronal driver, and fed with the three different concentrations of CuCl2. The data shows the co-localization between Htt and Ref(2)P (Figure 4A). To further analyze these aggregates, we quantified them by total size, size distribution, and intensity. We found that the concentration of 0.50 mM CuCl2 feeding increased total Htt aggregates size (Figure 4B). Interestingly, consistent with our aggregates analysis in Figure 3, when we grouped the aggregates by size, we found that a significant increase in the number of only small aggregates (<5 μm2) but not in the number of medium (5-10 μm2) or large aggregates (>10 μm2) suggesting that copper specifically promotes the formation of small aggregates (Figure 4C). In addition, copper feeding with either 0.25 mM or 0.50 mM CuCl2 increased total intensity of Htt aggregates when compared to the 0 mM control group (Figure 4D).
Figure 4: Copper increases Ref(2)P+ Htt aggregates.
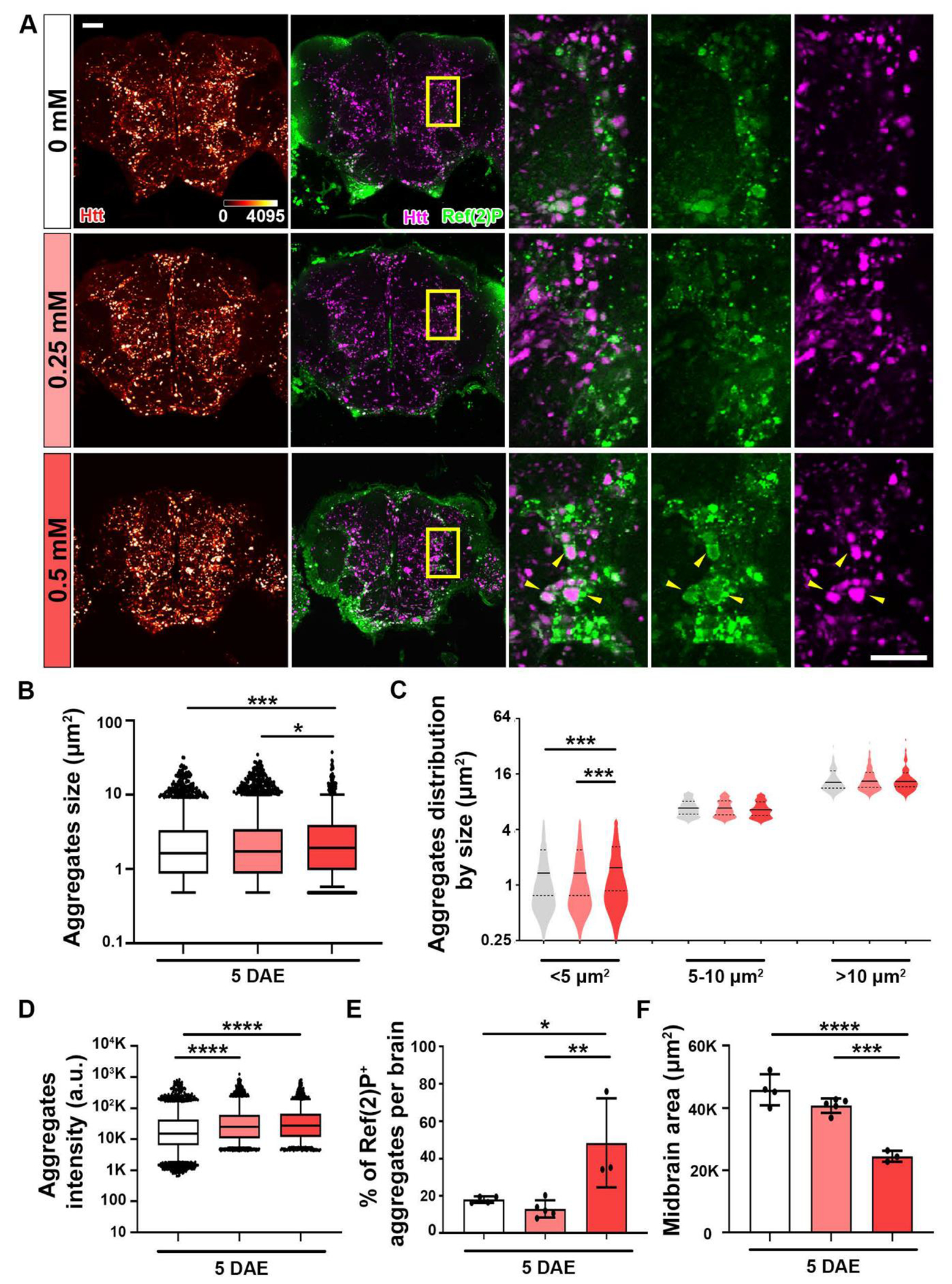
Pathological Htt-Q138 flies driven by elav-GAL4 were fed with different CuCl2 concentrations until 5 DAE. (A) Midbrain shown with accumulation of Htt aggregates (heatmap 0-4095). Merged image of Htt aggregates (magenta) and Ref(2)P (green) staining. Yellow boxed areas are shown in higher magnification. Yellow arrows pointing at co-localization. Scale bar = 30 μm. (B) Quantification of total aggregate size. Data are presented as box and whiskers plot, 5-95 percentile. (C) Quantification of size distribution. Small aggregates are classified as <5 μm2, medium aggregates as 5-10 μm2, and large aggregates as >10 μm2. Data are presented as violin plot with quartiles. (D) Quantification of total aggregate intensity. Data are presented as box and whiskers plot, 5-95 percentile. (E) Quantification of percentage of Ref(2)P+ aggregates per lamina. Data are presented as mean ± SD. (F) Quantification of midbrain. Data are presented as mean ± SD. n=3-5, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
To start to evaluate the mechanism underlying the effect of copper on Htt aggregation, we examine the relationship between Ref(2)P and Htt aggregates. We determined the percentage of Ref(2)P+ Htt aggregates per brain, and we observed an increase in the 0.50 mM CuCl2 group (Figure 4E). The increased colocalization between Ref(2)P and Htt aggregates is likely a consequence of increased Htt aggregation. Association with Ref(2)P indicates the initiation of autophagy process but not the clearance or degradation of the aggregates. The increased colocalization between Ref(2)P and Htt aggregates could also be the result of a block of downstream autophagic flux and an accumulation of the phagophore at Ref(2)P associated state. This can also be due to the Ref(2)P/p62 protein level increases after oxygen radical stress [43]. Lastly, while previous research shows that Htt-Q138 overexpression reduces brain size [32], the results from this experiment reveal an exacerbated phenotype of significantly reduced brain size due to copper feeding at 5 DAE with 0.50 mM CuCl2 (Figure 4F).These results reveal that copper is enhancing Htt induced aggregation and toxicity by enhancing the association between Ref(2)P and Htt aggregates.
Chelation agent D-penicillamine decreases ThS+ Htt aggregates
The HD treatment currently available is limited to suppressing chorea and battling the mood-altering aspect of the disorder [3, 4, 44]. Administration of D-penicillamine (DPA) is mainly used as a copper chelating agent in copper poisoning and in the treatment of Wilson’s Disease, demonstrating reduction of copper and increase in urine excretion [25, 45, 46]. DPA is a thiol with a sulfhydryl group that binds copper, therefore it is proposed to be able to remove the accumulated copper. [25]. Administration of DPA has also been observed to prolong survival of wild-type flies and protect against Cu2+ induced mortality [26]. DPA has the ability to function as a mono-, bi-, or tridentate ligand [26]. To determine whether feeding DPA would ameliorate the Htt-induced aggregate toxicity, Htt-Q138 flies were fed with 0.50 mM CuCl2 from eclosion and switched at 10 DAE to food containing DPA at concentrations of 0 mM, 0.05 mM, or 0.10 mM for 5 days (Figure 5A). Then, at 15 DAE, the amyloid-like properties of the Htt aggregates by ThS staining were assessed (Figure 5B). Feeding 0.10 mM DPA significantly decreased the number of ThS+ Htt aggregates per lamina (Figure 5C). In addition, flies fed with either 0.05 mM or 0.10 mM DPA showed decreased aggregate intensity (Figure 5D) and aggregate size (Figure 5E). Collectively, these results indicate the beneficial effect of copper chelation with D-penicillamine, where DPA feeding reduced mutant Htt aggregation and the accumulation of β-amyloid-like structures.
Figure 5: Chelation decreases ThS+ Htt aggregates.
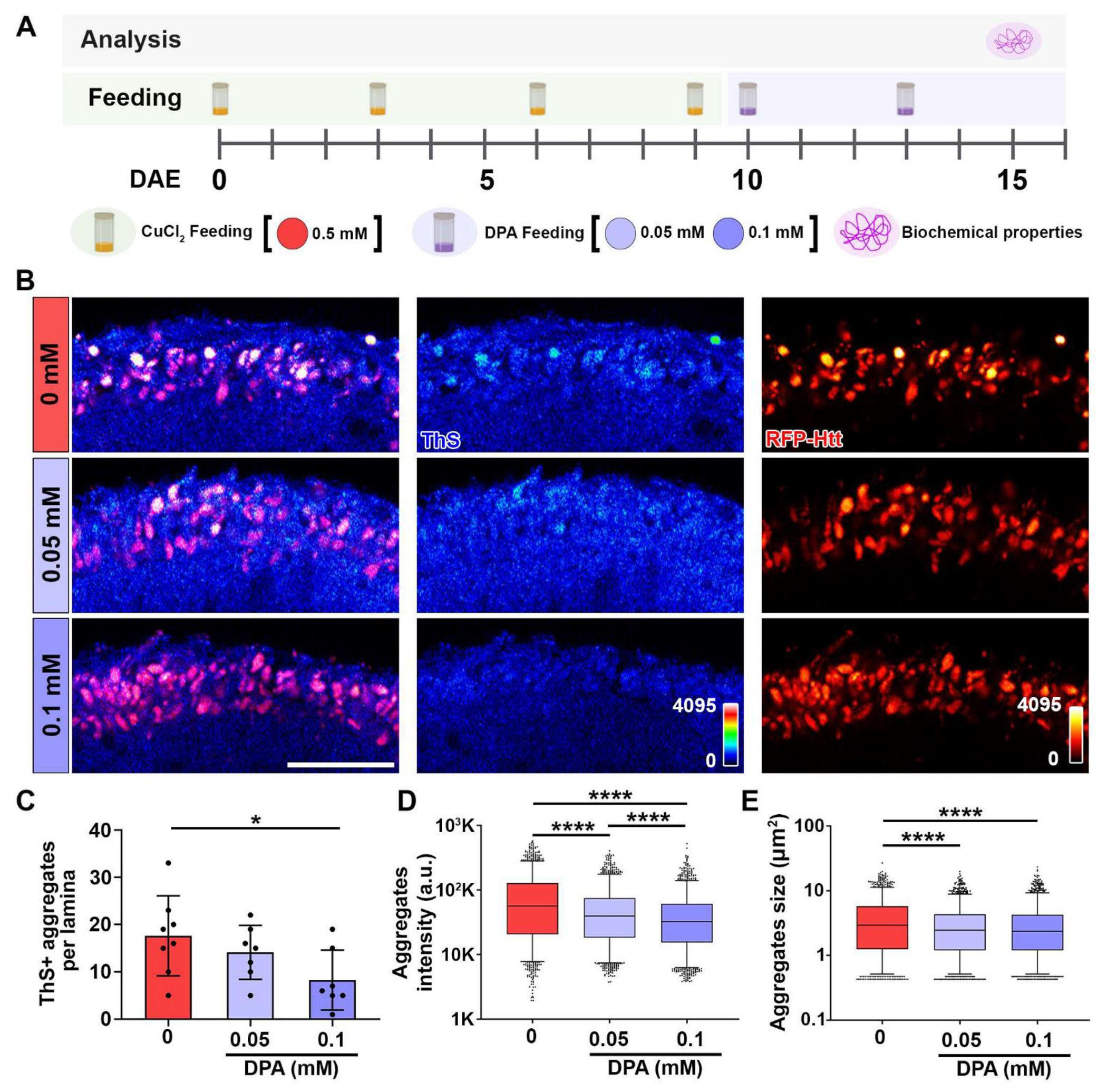
(A) DPA feeding paradigm. Pathological Htt-Q138 flies driven by GMR-GAL4 were fed with 0.50 mM CuCl2 until 10 DAE, then switched to DPA feeding. Biochemical properties of aggregates were assessed at 15 DAE. (B) ThS staining of lamina layer (ThS and RFP-Htt heatmaps 0-4095) at 15 DAE. Scale bar: 30 μm. (C) Quantification of ThS+ aggregates per lamina. Data is shown as mean ± SD. (D) Quantification of aggregates intensity. Data is shown as box and whiskers plot, 5-95 percentile. (E) Quantification of aggregates size. Data is shown as box and whiskers plot, 5-95 percentile. n=6-8, *p<0.05, ****p<0.0001.
Discussion
In this study, we examined the effect of copper on the toxicity of mutant Htt protein with polyglutamine expansion in a Drosophila model of HD. We discovered that dietary copper increased cytotoxicity of mutant Htt aggregates in a dose-dependent manner. Specifically, copper promoted the formation of small aggregates, enhanced the accumulation of ThS+ amyloid-like structures, and increased the association of autophagy initiation factor Ref(2)P with the Htt aggregates. As a result, synaptic degeneration was exacerbated by dietary copper, evidenced by increased caspase-mediated apoptosis activity, tissue thinning of lamina neuropil, and reduced brain size. Strikingly, copper chelation with D-penicillamine feeding reversed the impact of copper, reduced the number of aggregates, and decreased the accumulation of β-amyloid-like structures within aggregates. This work proposes a mechanistic model of the potential impact of dietary copper in HD pathology and further suggests the promising effect of copper chelation as a therapeutic venue (Figure 6).
Figure 6: Copper enhances aggregational toxicity of mutant huntingtin.
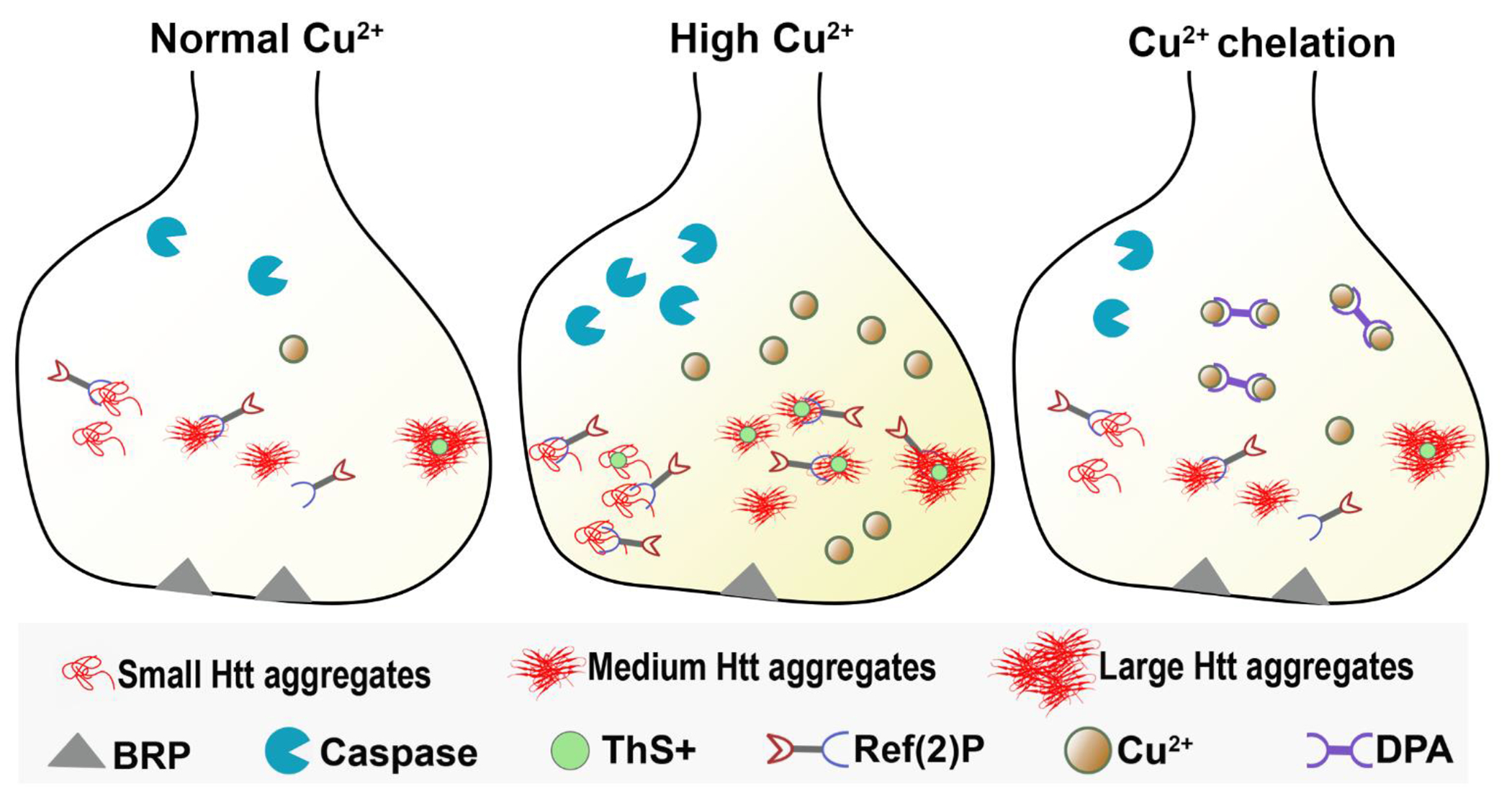
Schematic model of synapse showing that under normal copper concentration, there is cytotoxicity of mutant Htt aggregates, and some co-localization of Ref(2)P with the aggregates. Under high Cu exposure, there is increased Ref(2)P clustering with Htt aggregates, increased cell death, and reduced BRP levels at the synapse. After Cu2+ chelation by DPA feeding, there is reduced Htt aggregates clustering.
Copper is essential for nervous system development [47], mitochondrial activity, synaptic transmission, and defense against oxidative stress [17]. Copper enters the brain through the Cu transporter located at the brain barriers in a controlled manner. The blood-brain barrier (BBB) and blood-cerebrospinal fluid barrier (BCB) regulate copper homeostasis in the brain [48]. Altered metal accumulation has been implicated directly or indirectly in the pathogenesis of multiple neurological diseases including Alzheimer’s Disease (AD), Parkinson’s Disease (PD), Amyotrophic Lateral Sclerosis (ALS), Menkes Disease, Wilson’s Disease, and Huntington’s Disease [17, 31]. The neurotoxicity of excessive copper is not only by promoting protein aggregation, but also related to the production of reactive oxygen species (ROS) [49]. The brain is especially vulnerable to attack by ROS [48]. In AD, serum copper levels were approximately 54% higher in patients than those in controls, and the presence of APOε4 gene is associated with higher serum copper concentrations [17, 31]. Aberrant copper-protein interactions with amyloid precursor protein, β-amyloid peptide and α-synuclein have been implicated in AD and PD, respectively [17, 50, 51]. For example, Aβ aggregates can produce ROS in the presence of copper ions.[48]. In Wilson’s Disease, there is impairment in the ability of copper to be incorporated into ceruloplasmin, which contains 95% of blood copper [31]. Other evidence suggests that copper contributes to ALS pathogenesis through enhanced free radical generation that leads to increased neuronal damage [52]. In addition, it has been previously shown that N171 fragment of huntingtin reduced Cu2+ in vitro, indicating a very redox active state [13, 30]. Hence, tight regulation of copper metabolism is necessary to ensure normal function without leading to toxic effects that could enhance disease progression. A previous study using a cell culture model of α-synuclein aggregation detected a connection between metals and the formation of aggregates where cells with increased sensitivity to Cu toxicity showed increased aggregation, and reduction in cellular Cu resulted in a dramatic decrease in aggregate formation [27, 53]. Mutant HTT protein has been shown to interact with copper by oxidizing the polyglutamine-containing N171 fragment of huntingtin and promoting aggregation [13, 30], although the underlying mechanisms of how copper overload promotes HD progression remains elusive. It has been shown that mutant Htt proteins with polyglutamine expansion exert cytotoxicity through disruption of nuclear cytoplasmic transport [54–56], blocking trafficking [57], and sequestering of mitochondria and synaptic proteins due to increased age-dependent adhesiveness [32]. We hypothesize that accumulation of copper, or reduced copper clearance, is an indirect consequence of Htt protein dysfunction, that in turn, produces a feedback loop that exacerbates disease pathology. Our study here showed that copper specifically promoted the formation of amyloid-like, ThS+ structures that are responsible for the adhesiveness property, suggesting exacerbated aggregational toxicity by copper overload.
Metal-binding proteins fold in cellular environments where their cognate metals are present free in solution or bound to delivery proteins [58]. Excess copper in the brain can be released into the cerebrospinal fluid (CSF) and is taken up by the cells that form the blood-cerebrospinal fluid barrier (BCB) [48]. Formation of dimers or high order oligomeric forms may influence the protein misfolding and accumulation [59]. The process of amyloid fiber formation involves the protein monomer forming a nucleating ‘seed’ that is then followed by a more rapid self-templated growth where the ends of existing fibers recruit protein monomer and so extend fiber length [59]. It is known that small diffusible oligomers of Aβ in Alzheimer’s Disease are more neurotoxic than the mature amyloid fibers [59]. Interestingly, our study found that copper selectively increased the number of small aggregates without affecting the population of medium or large aggregates. This observation suggests that copper likely interacts with Htt monomers and oligomers and promotes the nucleation of the Htt aggregates. These results also correlate with a previous study indicating that copper can interact with huntingtin, and that excess copper may be contributing to a toxic microenvironment, through catalyzing toxic cellular reactions [30]. Our in vivo observation suggests an active role of copper in promoting the formation of small aggregates and supports the connection between copper and more aggressive protein aggregation.
The current HD treatment available is limited to suppressing chorea and battling the mood-altering aspect of the disorder. In this study, copper chelator D-penicillamine (DPA) was tested as a potential treatment to reduce HD-relevant molecular phenotypes. DPA functions by mobilizing tissue copper stores and its long-term use is associated with normalization of body copper balance [25]. DPA is currently used for Wilson’s Disease (WD), cystinuria, and rheumatoid arthritis, as well as for heavy metal toxicity [60]. In Wilson’s Disease specifically, clinical studies showed that DPA improved symptoms in 78% of all WD patients and in 56.3% of neurological WD patients [61]. The lower improvement rate with DPA treatment observed in neurological WD patients might be due to its low penetrance through the blood-brain barrier and fast elimination rate from the blood [60]. In this study, we took advantage of the high penetrance of the primitive hemolymph-brain barrier blood-brain barrier in Drosophila [62] and administered DPA through diet. We found that DPA feeding in copper-exposed HD pathological models resulted in a remarkable decrease in mutant Htt aggregation as well as the accumulation of toxic β-structures that are predominantly associated with pathological adhesiveness and sequestration of cellular organelles. These results highlight the beneficial effects of DPA in HD. It is encouraging to note that there have been efforts in developing methods including using nanoparticles to prolong the half-life of D-penicillamine in the body as well as enhance brain uptake [60]. Overall, our study underscores the risk of copper overexposure and the therapeutic potential of DPA, thus leading us to conclude that limiting free copper exposure may provide neuroprotective effects in Huntington’s Disease.
Methods
Electron Paramagnetic Resonance (EPR) spectroscopy measurement:
To prepare the EPR samples, 0.25 mM and 0.50 mM food samples were redissolved in distilled water. Glycerol was added as a glassing agent at a final 10% v/v ratio. To liquify the samples and transfer them into the EPR tubes, both samples were heated in a 70-80 °C water bath for 45 min. The dissolved samples were filtered using 0.22 μm syringe filters to remove undissolved particulates. Samples were then frozen in liquid nitrogen until they could be measured. The spectra presented here were recorded at 77K using a cryogen-free, variable temperature Bruker EMXplus X-band CW EPR spectrometer. All data were collected under non-saturating conditions and with a fixed microwave frequency of 9.65 GHz. Data were visualized and analyzed using the SpinCount software package from Professor Michael Hendrich (Carnegie Mellon University). To quantitatively determine the concentration of copper in each sample, we calculated the double integrals of the experimental data and compared these values to the double integrated curves for our standard solutions (0.25 mM and 0.50 mM CuCl2 spin standards of known concentration). The expression to calculate/quantify the unknown concentration is given below.
where, CDBLI= Corrected Double integral. A quantitative analysis of this type allows for detection into the low micromolar concentration range and offers the ability to assess purity of the metal speciation in the food samples. Additionally, because the samples are frozen, they can be thawed and recovered after recording the experimental spectra.
Drosophila stocks and experimental procedures:
The following fly strains were used in this study: UAS-RFP-HttQ15 and UAS-RFP-HttQ138 obtained from Dr. J. Troy Littleton [38], GMR-GAL4, and elav-GAL4 obtained from Bloomington Drosophila Stock Center. Flies were reared on cornmeal-molasses-yeast medium at 22°C, 65% humidity, with 12 h light / 12 h dark cycles. For feeding experiments, CuCl2 was dissolved in water and then mixed into 5 mL fly food with respective concentrations. 100mg of D-penicillamine was dissolved in 10 mL water and then mixed into 5 mL fly food with respective concentrations. An equal amount of water was mixed into the fly food as a control. The vials were dried at room temperature for 12 h before feeding.
Measurement of Drosophila food intake:
Feeding protocol was adapted from [63]. To examine food intake, 2-5 DAE flies were starved for 2 hours at room temperature in vials containing only a filter paper with water. After starvation, flies were immediately transferred into vials with a Kimwipe containing 5% sucrose and 3% Coomassie Brilliant Blue dye (Fluka C#27816) and were allowed to feed for 1 hour at room temperature. Flies were then frozen, homogenized, and centrifuged. The absorbance of the supernatant was read at 595 nm on a FluoStar Omega plate reader. The volume of solution ingested was calculated by comparing the net absorbance against a standard curve.
Drosophila CNS immunostaining, confocal imaging, and analysis:
Adult brains were dissected in phosphate-buffered saline (PBS, pH 7.4) followed by fixing in 4% formaldehyde for 10 min as previously described [64]. Samples were then washed in PBS containing 0.4% v/v Triton X-100 (PBTX) and incubated with primary antibodies followed by incubation with secondary antibodies. All antibodies were diluted in 0.4% PBTX with 5% normal goat serum and incubated 4°C overnight. DAPI (1:330, Invitrogen Ca# D1306) staining was performed at room temperature for 10min before mounting. The samples were mounted on glass slides with VECTASHIELD Antifade Mounting Medium (Vector Laboratories). The following primary antibodies were used: mouse anti-BRP antibody (1:250, Developmental Studies Hybridoma Bank Ca# NC82,), rabbit anti-cleaved Caspase-3 antibody (1:250, Cell Signaling Ca# 9661), and rabbit anti-Ref(2)P antibody (1:250, abcam Ca# ab178440). The following secondary antibodies were used: Alexa Fluor 488-conjugated anti-mouse secondary antibody (1:300, Invitrogen Ca# A21121) and Alexa Fluor Cy5-conjugated anti-rabbit secondary antibody (1:300, Rockland Ca# 611-110-122). Slides were imaged using an Olympus IX81 confocal microscope with 40x or 60x oil immersion objective lens with a scan speed of 8.0 μs per pixel and spatial resolution of 1,024x1,024 pixels. Images were processed using FluoView 10-ASW (Olympus). Quantification was carried out using ImageJ/Fiji (1.53q).
Thioflavin S (ThS) staining:
Adult brains with lamina were dissected in PBS (pH 7.4) and then fixed in 4% formaldehyde for 15 min, permeabilized with PBTX for 10 min (3 times), and washed with PBS for 5 min. 400 μL of 1mg/mL ThS (Sigma-Aldrich) diluted in distilled water was added to the samples and incubated for 30 min at room temperature. Lastly, samples were washed 3 times with distilled water, and mounted on glass slides with VECTASHIELD Antifade Mounting Medium.
Statistical analyses:
Biological sample size (n) and p values are indicated in the corresponding figure legends. One-way ANOVA or Two-way ANOVA with Tukey post-hoc test was applied to compare multiple groups. p < 0.05 was considered statistically significant. All statistical analyses were performed in GraphPad Prism software (version 10.0).
Highlights.
Copper enhances huntingtin aggregational toxicity and synaptic degeneration.
Copper increases Thioflavin S positive amyloid-like huntingtin aggregates.
Copper enhances binding of autophagy factor Ref(2)P with huntingtin aggregates.
Chelation agent D-penicillamine significantly decreases amyloid-like structures.
Acknowledgments
We thank Zoraida Diaz-Perez for technical assistance. This work was supported in part by the Frost Junior Fellows Program and a CTSI KL2 award UL1TR002736 (KKM), and NIH R33AT010408 (RGZ). The project described was supported in part by the National Center For Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR002736. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests:
Authors declare that they have no competing interests.
References
- 1.Bates G, Huntingtin aggregation and toxicity in Huntington’s disease. The Lancet, 2003. 361(9369): p. 1642–1644. [DOI] [PubMed] [Google Scholar]
- 2.Finkbeiner S, Huntington’s Disease. Cold Spring Harb Perspect Biol, 2011. 3(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Labbadia J and Morimoto RI, Huntington’s disease: underlying molecular mechanisms and emerging concepts. Trends Biochem Sci, 2013. 38(8): p. 378–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bachoud-Levi AC, et al. , International Guidelines for the Treatment of Huntington’s Disease. Front Neurol, 2019. 10: p. 710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barron JC, Hurley EP, and Parsons MP, Huntingtin and the Synapse. Front Cell Neurosci, 2021. 15: p. 689332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johri A and Beal MF, Antioxidants in Huntington’s disease. Biochim Biophys Acta, 2012. 1822(5): p. 664–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jurcau A, Molecular Pathophysiological Mechanisms in Huntington’s Disease. Biomedicines, 2022. 10(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harjes P and Wanker EE, The hunt for huntingtin function: interaction partners tell many different stories. Trends in Biochemical Sciences, 2003. 28(8): p. 425–433. [DOI] [PubMed] [Google Scholar]
- 9.Imarisio S, et al. , Huntington’s disease: from pathology and genetics to potential therapies. Biochem J, 2008. 412(2): p. 191–209. [DOI] [PubMed] [Google Scholar]
- 10.Jimenez-Sanchez M, et al. , Huntington’s Disease: Mechanisms of Pathogenesis and Therapeutic Strategies. Cold Spring Harb Perspect Med, 2017. 7(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jansen AH, et al. , Frequency of nuclear mutant huntingtin inclusion formation in neurons and glia is cell-type-specific. Glia, 2017. 65(1): p. 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiao G, et al. , Huntington disease arises from a combinatory toxicity of polyglutamine and copper binding. Proc Natl Acad Sci U S A, 2013. 110(37): p. 14995–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fox JH, et al. , Cysteine oxidation within N-terminal mutant huntingtin promotes oligomerization and delays clearance of soluble protein. J Biol Chem, 2011. 286(20): p. 18320–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wen Yang, D. JR, Andrews Richard B. and Wetzel Ronald, Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Human Molecular Genetics, 2002. 11: p. 2905–2917. [DOI] [PubMed] [Google Scholar]
- 15.Squadrone S, et al. , Trace elements profile in the blood of Huntington’ disease patients. J Trace Elem Med Biol, 2020. 57: p. 18–20. [DOI] [PubMed] [Google Scholar]
- 16.Giampietro R, et al. , The Pivotal Role of Copper in Neurodegeneration: A New Strategy for the Therapy of Neurodegenerative Disorders. Mol Pharm, 2018. 15(3): p. 808–820. [DOI] [PubMed] [Google Scholar]
- 17.Gromadzka G, et al. , Copper Dyshomeostasis in Neurodegenerative Diseases-Therapeutic Implications. Int J Mol Sci, 2020. 21(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaler V.D.a.S.G., Role of copper in human neurological disorders. American Society for Nutrition, 2008. 88: p. 855S–858S. [DOI] [PubMed] [Google Scholar]
- 19.Bakkar N, et al. , The M1311V variant of ATP7A is associated with impaired trafficking and copper homeostasis in models of motor neuron disease. Neurobiol Dis, 2021. 149: p. 105228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosas HD, et al. , Alterations in brain transition metals in Huntington disease: an evolving and intricate story. Arch Neurol, 2012. 69(7): p. 887–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joshi P, et al. , Huntington’s disease associated resistance to Mn neurotoxicity is neurodeveiopmentai stage and neuronal lineage dependent. Neurotoxicology, 2019. 75: p. 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agrawal S, et al. , Brain mitochondrial iron accumulates in Huntington’s disease, mediates mitochondrial dysfunction, and can be removed pharmacologically. Free Radio Biol Med, 2018. 120: p. 317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pfalzer AC, et al. , Alterations in metal homeostasis occur prior to canonical markers in Huntington disease. Sci Rep, 2022. 12(1): p. 10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Organization, W.H., Copper in Drinking-water. 2004. [Google Scholar]
- 25.Aggarwal A and Bhatt M, Advances in Treatment of Wilson Disease. Tremor Other Hyperkinet Mov (N Y), 2018. 8: p. 525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abolaji AO, et al. , D-Penicillamine prolongs survival and lessens copper-induced toxicity in Drosophila melanogaster. Toxicol Res (Camb), 2020. 9(4): p. 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaetke LM, Chow-Johnson HS, and Chow CK, Copper: toxicological relevance and mechanisms. Arch Toxicol, 2014. 88(11): p. 1929–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balamurugan K, et al. , Copper homeostasis in Drosophila by complex interplay of import, storage and behavioral avoidance. EMBO J, 2007. 26(4): p. 1035–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kardos J, et al. , Copper signalling: causes and consequences. Cell Commun Signal, 2018. 16(1): p. 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fox JH, et al. , Mechanisms of copper ion mediated Huntington’s disease progression. PLoS One, 2007. 2(3): p. e334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Desai V and Kaler SG, Role of copper in human neurological disorders. Am J Clin Nutr, 2008. 88(3): p. 855S–8S. [DOI] [PubMed] [Google Scholar]
- 32.Zhu Y, et al. , Nmnat restores neuronal integrity by neutralizing mutant Huntingtin aggregate-induced progressive toxicity. Proc Natl Acad Sci U S A, 2019. 116(38): p. 19165–19175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Donoso JP, et al. , Electron Paramagnetic Resonance Study of Copper–Ethylenediamine Complex Ion Intercalated in Bentonite. The Journal of Physical Chemistry C, 2013. 117(45): p. 24042–24055. [Google Scholar]
- 34.Arioz C and Wittung-Stafshede P, Folding of copper proteins: role of the metal? Q Rev Biophys, 2018. 51: p. e4. [DOI] [PubMed] [Google Scholar]
- 35.Slow EJ, et al. , Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet, 2003. 12(13): p. 1555–67. [DOI] [PubMed] [Google Scholar]
- 36.Graham RK, et al. , Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell, 2006. 125(6): p. 1179–91. [DOI] [PubMed] [Google Scholar]
- 37.Elmore S, Apoptosis: A Review of Programmed Cell Death. Toxicol Pathol, 2007. 35(4): p. 495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weiss KR, et al. , Huntingtin aggregation kinetics and their pathological role in a Drosophila Huntington’s disease model. Genetics, 2012. 190(2): p. 581–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Songming Chen VB, Bradley Hamilton J, O’Nuallain Brian, and Wetzel Ronald, Amyloid-like Features of Polyglutamine Aggregates and Their Assembly Kinetics. Biochemistry, 2002. 41: p. 7391–7399. [DOI] [PubMed] [Google Scholar]
- 40.Chiang HC, et al. , PI3 kinase signaling is involved in Abeta-induced memory loss in Drosophila. Proc Natl Acad Sci U S A, 2010. 107(15): p. 7060–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mauvezin C, et al. , Assays to monitor autophagy in Drosophila. Methods, 2014. 68(1): p. 134–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jeong H, et al. , Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell, 2009. 137(1): p. 60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bjorkoy G, et al. , p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol, 2005. 171(4): p. 603–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Potkin KTPSG, New directions in therapeutics for Huntington disease. Future Neurology, 2018. 13(2): p. 101–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.M VAN CAILLIE-BERTRAND HJD, LUIJENDIJK I, BOUQUET J, AND SINAASAPPEL M, Wilson’s disease: assessment of D-penicillamine treatment. Archives of Disease in Childhood, 1985. 60: p. 652–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walshe JM, Penicillamine, a New Oral Therapy for Wilson’s Disease. American Journal of Medicine, 1956: p. 487–495. [DOI] [PubMed] [Google Scholar]
- 47.Gitlin M.L.S.a.J.D., Copper Homeostasis in the CNS. Molecular Neurobiology, 2006. 33: p. 81–90. [DOI] [PubMed] [Google Scholar]
- 48.An Y, et al. , The Role of Copper Homeostasis in Brain Disease. Int J Mol Sci, 2022. 23(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leal SS, Botelho HM, and Gomes CM, Metal ions as modulators of protein conformation and misfolding in neurodegeneration. Coordination Chemistry Reviews, 2012. 256(19-20): p. 2253–2270. [Google Scholar]
- 50.Rasia Rodolfo M., B. CW, Marsh Derek, Hoyer Wolfgang, Cherny Dmitry, Zweckstetter Markus, Griesinger Christian, Jovin Thomas M., and Fernandez Claudio O., Structural characterization of copper(II) binding to α-synuclein: Insights into the bioinorganic chemistry of Parkinson’s disease. PNAS, 2005. 102: p. 4294–4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dingwall C, A copper-binding site in the cytoplasmic domain of BACE1 identifies a possible link to metal homoeostasis and oxidative stress in Alzheimer’s disease. Biochemical Society Transactions, 2007. 35: p. 571–573. [DOI] [PubMed] [Google Scholar]
- 52.Watanabe S, et al. , Increased affinity for copper mediated by cysteine 111 in forms of mutant superoxide dismutase 1 linked to amyotrophic lateral sclerosis. Free Radic Biol Med, 2007. 42(10): p. 1534–42. [DOI] [PubMed] [Google Scholar]
- 53.Wang X, et al. , Copper binding regulates intracellular alpha-synuclein localisation, aggregation and toxicity. J Neurochem, 2010. 113(3): p. 704–14. [DOI] [PubMed] [Google Scholar]
- 54.Woerner Andreas C., F. F, Hornburg Daniel, Feng Li R., Meissner Felix, Patra Maria, Tatzelt Jörg, Mann Matthias, Winklhofer Konstanze F., Hartl F. Ulrich, Hipp Mark S., Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science, 2016. 351(6269): p. 173–176. [DOI] [PubMed] [Google Scholar]
- 55.Grima JC, et al. , Mutant Huntingtin Disrupts the Nuclear Pore Complex. Neuron, 2017. 94(1): p. 93–107 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gasset-Rosa F, et al. , Polyglutamine-Expanded Huntingtin Exacerbates Age-Related Disruption of Nuclear Integrity and Nucleocytoplasmic Transport. Neuron, 2017. 94(1): p. 48–57 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wyan-Ching Mimi Lee MY, and Troy Littleton J, Cytoplasmic aggregates trap polyglutaminecontaining proteins and block axonal transport in a Drosophila model of Huntington’s disease. PNAS, 2004. 101: p. 3224–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Palm-Espling ME, Niemiec MS, and Wittung-Stafshede P, Role of metal in folding and stability of copper proteins in vitro. Biochim Biophys Acta, 2012. 1823(9): p. 1594–603. [DOI] [PubMed] [Google Scholar]
- 59.Viles J, Metal ions and amyloid fiber formation in neurodegenerative diseases. Copper, zinc and iron in Alzheimer’s, Parkinson’s and prion diseases. Coordination Chemistry Reviews, 2012. 256(19-20): p. 2271–2284. [Google Scholar]
- 60.Cui Z, et al. , Novel D-penicillamine carrying nanoparticles for metal chelation therapy in Alzheimer’s and other CNS diseases. Eur J Pharm Biopharm, 2005. 59(2): p. 263–72. [DOI] [PubMed] [Google Scholar]
- 61.Tang S, et al. , Comparison of the Effectiveness and Safety of d-Penicillamine and Zinc Salt Treatment for Symptomatic Wilson Disease: A Systematic Review and Meta-Analysis. Front Pharmacol, 2022. 13: p. 847436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Brown Natasha M., P. SJ, and Gu Chenghua, Bridging barriers: a comparative look at the blood–brain barrier across organisms. GENES & DEVELOPMENT, 2018. 32: p. 466–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu S, et al. , WIDE AWAKE mediates the circadian timing of sleep onset. Neuron, 2014. 82(1): p. 151–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brazill JM, et al. , Cuantitative Cell Biology of Neurodegeneration in Drosophila Through Unbiased Analysis of Fluorescently Tagged Proteins Using ImageJ. J Vis Exp, 2018(138). [DOI] [PMC free article] [PubMed] [Google Scholar]