

Abstract
Tissue adhesives capable of achieving strong and tough adhesion in permeable wet environments are useful in many biomedical applications. However, adhesion generated through covalent bond formation directly with the functional groups of tissues (i.e., -COOH and -NH2 groups in collagen), or using non-covalent interactions can both be limited by weak, unstable, or slow adhesion. Here, we show that by combining pH-responsive bridging chitosan polymer chains and a tough hydrogel dissipative matrix one can achieve unprecedented ultra-tough adhesion to tissues (>2000J/m2) in 5–10mins without covalent bond formation. The strong non-covalent adhesion was shown to be stable under physiologically relevant conditions and strongly influenced by chitosan molecular weight, molecular weight of polymers in the matrix, and pH. The adhesion mechanism relies primarily on the topological entanglement between the chitosan chains and the permeable adherends. To further expand the applicability of the adhesives, adhesion time can be decreased by dehydrating the hydrogel matrix to facilitate rapid chitosan interpenetration and entanglement (>1000J/m2 in ≤1min). The unprecedented adhesive properties presented in this study open opportunities for new strategies in the development of non-covalent tissue adhesives and numerous bio-applications.
Keywords: hydrogel, adhesive, bioinspiration, biomaterial, slug
Graphical Abstract
Tissue adhesives capable of achieving strong and tough adhesion in wet environments are useful in many biomedical applications. We show that combining pH-responsive bridging chitosan polymer chains and a tough hydrogel dissipative matrix can achieve unprecedented ultra-tough adhesion to tissues (>2000J/m2) without covalent bond formation. These unprecedented properties open opportunities for new strategies in the development of non-covalent tissue adhesives.
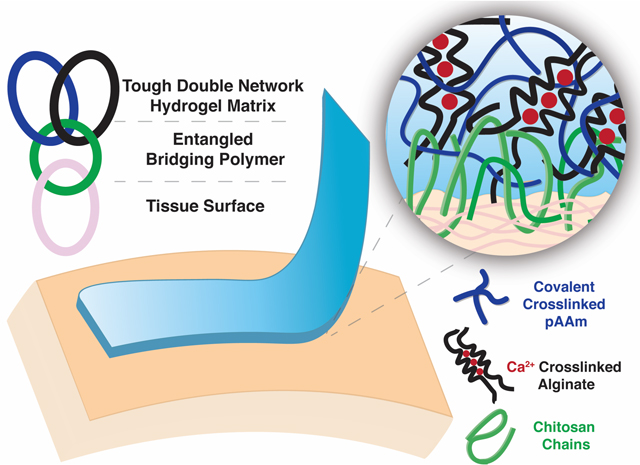
Introduction
Tissue adhesives have received increased attention in the last decades because of their potential applications as sealants,[1, 2] wound dressings,[3] and drug delivery systems,[4, 5] among others.[6, 7] Tissue adhesives approved for use inside the body are indicated as an adjunct to suturing and staples, but one day may replace, or at least be a viable alternative, to traditional staples and sutures.[2] Recently, advances in hydrogel technologies[8, 9] have highlighted their versatility, biocompatibility, and tunability as tissue adhesives. Additionally, several groups have recently highlighted the current progress in hydrogel adhesives technologies, including their applications and challenges.[7, 10, 11]
Hydrogel-based adhesives, however, suffer from several limitations, including the need of strong covalent bonds directly with tissue proteins to generate adhesion.[11] Cyanoacrylate-based adhesion involves the diffusion of reactive monomers at tissue sites and subsequent in-situ crosslinking, but this often releases toxic unreacted monomers into the bloodstream or generates reactive radical species.[12] Another strategy for direct covalent bond based adhesives is the use of a bridging layer and coupling reagents to facilitate bond formation between a gel and tissue. For example, carbodiimide coupling reactions combined with a tough hydrogel matrix achieve adhesion energies of more than 1000J/m2.[1] However, this strategy depends on covalent bond junctions and the need for specific and complementary reactive functional groups in both the hydrogel and tissue. Other systems involving direct bond formation between the gel and tissue have also been reported, achieving strong adhesion in a few minutes, but again requiring reactive species such as activated NHS-esters or aldehydes, among other reactive groups, for bond formation.[13, 14] Relying on specific functional groups can be a limitation under physiological and clinically relevant environments where blood and other fluids can dilute and interfere with the chemical reactions responsible for bond and adhesion formation.[2]
Tissue adhesives relying only on non-covalent bonding (~1–50kT for a single H-bond)[15] are typically weak, fragile, and slow to adhere.[16, 17] Adhesives using direct electrostatics and hydrogen bonding interactions between gel and tissue were recently reported. Non-covalent adhesion was achieved instantly but it was weak (<200J/m2).[17] Topological wet adhesion between two permeable adherends is another strategy which can be achieved merely based on topology and chain entanglements between the two adherends.[18, 19] As with most other adhesives relying on physical interactions, tissue adhesion was weak (<200J/m2) and time consuming (>1h to achieve peak strength), which may make biomedical application challenging. However, the mechanism of topological adhesion remains promising as it does not rely on specific functional groups and could be further harnessed in engineering new biomaterials.
In this study, we investigate adhesion to tissues and design an alginate-polyacrylamide tough adhesive (TA) that generates ultra-tough (>2000J/m2) and unprecedented topological tissue adhesion within minutes, without the need of covalent bond formation. The TA relies on a tough double network hydrogel as an energy dissipation matrix and a bridging polymer that bonds the gel and tissue together. The chitosan bridging layer used in this study is proposed to act as a stimuli-responsive polymer by forming strong intermolecular H-bonds upon a change in pH. This property allows the chains to diffuse and form an internal network between two permeable adherends, in this case, gel and tissue. We also explore how the properties of the tough gel matrix and the bridging polymer, including molecular weight, viscosity, and pH influence adhesion. Furthermore, we develop a strategy to accelerate chain diffusion and entanglement in the hydrogel to tune adhesion time and strength. Strong, rapid adhesion of >1500J/m2 was generated. The strong and fast adhesion reported in this study relies on a specific combination of mechanics and network topologies without the need for covalent bond formation. Given the unprecedented adhesive properties obtained with these biomaterials, multiple biomedical applications are possible.
Results
Chitosan enables rapid and strong tissue adhesion
The ability of chitosan to mediate adhesion to skin tissue was first investigated (Figure 1a). After application of the hydrogel and bridging chitosan-only adhesive to skin tissue, the adhesion energy increased rapidly and exceeded 1000J/m2 by 3 minutes, and 2000J/m2 by 10 minutes before reaching a steady state (Figure 1b). Peak forces during adhesive removal exceeded 20N, and periodic cohesive hydrogel failure during removal was observed (Figure S1). Notably, strong adhesion due to chitosan did not require the addition of coupling reagents mediating covalent bond formation, as addition of EDC and sNHS led to only a moderate increase in adhesion energy (Figure 1c). In addition to skin, strong adhesion using chitosan alone was also observed with tendon and heart tissue (Figure 1d). Other bridging polymers containing primary amines such as polyallylamine (PAA), polyethyleneimine (PEI), and carboxymethyl chitosan (CMC) were tested but no adhesion was obtained (Figure S2).
Figure 1 |. Chitosan enables rapid and strong adhesion without the need of covalent bonds.

(a) Schematic of tough double network hydrogel matrix adhered to tissue surfaces via a bridging polymer (chitosan) that is proposed to yield topological entanglement with the hydrogel and underlying tissue. The double network is composed of covalently crosslinked polyacrylamide (pAAm) and calcium crosslinked alginate. (b) The effect of time on chitosan-only adhesion. Data shown as mean ± s.d. as evaluated by a one-way ANOVA with post hoc t-tests with Bonferroni corrections (n=4–6 samples/group). (c) Adhesion energy to skin in the presence and absence of EDC/sNHS coupling agents. Data shown as mean ± s.d. as evaluated by a Student’s t-test. (d) The effect of tissue type on chitosan-only mediated adhesion. Data shown as mean ± s.d. as evaluated by one-way ANOVA with post hoc t-tests with Bonferroni corrections (n=6–8 samples/group). Adhesion tests were performed using tough gels prepared with alginate D and pAAm, and chitosan A as the bridging polymer. See tables S1 and S2 for additional specifications.
Adhesion is stable and robust to blood
To examine the versatility of this adhesive approach, testing was completed comparing performance with and without addition of blood and following incubation in DMEM at 37°C for 24h to achieve a fully swollen state. Non-covalent adhesion enabled strong adhesion to skin tissue, regardless of the presence of blood (Figure 2a) and it occurred independent of addition of covalent coupling reagents (Figure S3a). Adhesion to skin was maintained following incubation in DMEM for 24h at 37°C (Figure 2b) and was again independent of the addition of covalent coupling agents (Figure S3b).
Figure 2 |. Chitosan non-covalent adhesion is stable to blood and incubation in DMEM.

(a) The effect of blood contaminating skin on the adhesion energy was evaluated. Data shown as mean ± s.d. as evaluated by Student’s T-tests (n=6 samples/group). (b) The effect of incubation in DMEM after placement on the resulting adhesion energy was nonsignificant. Data shown as mean ± s.d. as evaluated by a Student T-tests (n=6–9 samples/group). Adhesion tests were performed using tough gels prepared with alginate D and pAAm, and chitosan A as the bridging polymer. See tables S1 and S2 for additional specifications.
Adhesion strength depends on pH, bridging polymer concentration, and viscosity
Previous studies have suggested pH-dependent topological adhesion between two hydrogels [20], and several factors were next investigated to explore if this mediates the strong tissue adhesion observed here (Figure 3a). The adhesion energy was increased with increased pH of skin, beyond the pKa of chitosan (Figure 3b). The adhesion energy was also dependent on the concentration of chitosan, showing a maximum at approximately 2wt/vol% (Figure 3c). Tough adhesion was affected by the MW of chitosan (Figure 3d), but not the degree of deacetylation (Figure 3e). Although similar bands in the FTIR spectra of ultrapure and non-ultrapure chitosan were observed, differences in thermal decomposition were detected (Figure S4) in concert with reported differences in MW/purity and degree of deacetylation (Table S1). However, the chitosan molecular weight of these chitosan samples resulted in significantly different viscosities, separated by at least one order of magnitude (Figure 3f). Topological adhesion with other chitosans with different molecular weights, purity, and degrees of deacetylation was also tested and similar trends were observed (Figure S5 and Table S1). Changes to the properties of the chitosan bridging polymer resulted in differences in the interfacial adhesive layer. Increased chitosan concentration resulted in a greater thickness of the bridging polymer layer and decreased penetration into skin tissue (Figure 4a–c). Similarly, increasing chitosan molecular weight resulted in a thicker interfacial chitosan layer and trended decrease in skin penetration (Figure 4d–f). Over time, chitosan penetration into skin increased (Figure 4g–i).
Figure 3 |. Chitosan topological adhesion depends on pH, polymer concentration, and viscosity.

(a) Proposed mechanism of topological entanglement by interchain interactions through H-bonds. (b) The effect of skin pH on the adhesion energy to skin tissue. Data shown as mean ± s.d., as evaluated by a one-way ANOVA with post hoc T-tests with Bonferroni corrections (n=4group). (c) The effect of chitosan concentration on the adhesion energy to skin tissue. Data shown as mean ± s.d., as evaluated by a one-way ANOVA with post hoc T-tests with Bonferroni corrections (n=4–5/group). (d,e) The effect of chitosan MW and degree of deacetylation on the adhesion energy. Data shown as mean ± s.d., as evaluated by a Student’s t-test (n=4–7/group). (f) The effect of chitosan MW/purity and concentration on chitosan viscosity. Data shown as mean ± s.d., as evaluated by a two-way ANOVA with post hoc T-tests with Bonferroni corrections (n=10 samples/group). Adhesion tests were performed using tough gels prepared with alginate D and pAAm, and chitosan A as the bridging polymer. For panels d and f chitosan D was used as the high MW bridging polymer. For panel e, chitosan A was used as the lower DDA. See tables S1 and S2 for additional specifications.
Figure 4 |. Chitosan concentration, chitosan molecular weight and time affect penetration.

Effect of (a-c) chitosan concentration (chitosan A), (d-f) chitosan molecular weight (chitosans D, E), and (g-i) time on bridging polymer thickness and penetration into skin (chitosan A). P-values reported after Student’s t-test. Data shown as mean ± s.d. with N=4–5 samples/group. Adhesion tests were performed using tough gels prepared with alginate D and pAAm, and fluorescently labelled chitosans. See tables S1 and S2 for additional specifications. Chitosan was labeled with FITC, and the skin was visualized with polarized light and DAPI (cells).
Gels with high molecular weight alginate facilitate stronger topological adhesion
The effect of different alginates (Table S2 and Figure 5) on hydrogel mechanics and adhesion energy were next evaluated to address the hypothesis that topological entanglement may be affected by molecular weight of polymers in the dissipative matrix. Hydrogel toughness was affected by alginate molecular weight (Figures 5a and S6), while modulus was affected by both alginate molecular weight and purity (Figures 5b and S6). Additionally, the adhesion energy was affected by alginate molecular weight regardless of whether ultrapure or non-ultrapure polymers were used in gel fabrication (Figure 5c). Indeed, the adhesion energy was strongly and positively correlated with alginate molecular weight (Figure 5d). The adhesion energy was also correlated to hydrogel toughness (R2 = 0.27) and modulus (R2 = 0.47) (Figure S7). Hydrogel toughness was correlated to hydrogel modulus (R2=0.68) (Figure S7). Alginates with higher G-content, the residue involved in ionic crosslinking, did not result in higher toughness, stretch, maximum stretch, modulus, or adhesion energy (Figure S8). Although the combination of high and low molecular weight alginates has been suggested to result in elevated toughness [9], adhesion energies did not follow this similar trend, with the highest observed in gels of higher molecular weight alginates only (Figure S9).
Figure 5 |. High molecular weight alginates facilitate stronger adhesion.

(a-c) The effect of alginate molecular weight on hydrogel toughness, modulus, and the adhesion energy was evaluated using both ultrapure (UP) and non-ultrapure (Non-UP) polymers. For the high MW alginates A and C were used, while alginates C and F were used as the low MW alginates. Data shown as mean ± s.d., as evaluated by a one-way ANOVA with post hoc t-tests with Bonferroni corrections (n=4–6 samples/group). (d) The adhesion energy was strongly correlated with alginate molecular weight (R2 = 0.7166, P<0.001). Adhesion tests on panels a-c were performed using tough gels with alginates A or C as the UP materials, and alginates E and F as the Non-UP. All tests were performed using chitosan A. See tables S1 and S2 for additional specifications.
Dehydrated gels generate near instantaneous adhesion to tissues by accelerating interpenetration and entanglement of the chitosan chains.
It was next examined whether adhesion could be accelerated by promoting rapid infiltration of chitosan into dehydrated gels. Chitosan is added to the hydrogel prior to application on tissue. After chitosan is applied on the hydrogel surface it diffuses into the dissipative matrix faster when it is in the dehydrated state (Figure 6a). While hydrogels require approximately 24h to dehydrate, they rehydrate to their original mass within ~30 minutes and then continue to swell over incubation in a large pool of buffer (Figure 6b), highlighting the fast hydration process. Dehydrated gels demonstrated more rapid, initial swelling than a gel that was not initially dehydrated (3x swelling rate over first 12 minutes). A similar final swelling equilibrium state was observed in vitro, regardless of initial gel dehydration state. Implantation of dehydrated gels also led to similar in vivo swelling as non-dehydrated, implanted gels over 2 weeks, but the swelling of both types of gels was considerably lower than observed in vitro (Figure S10). Dehydrating the tough gels to a film before adhesion also resulted in a 50,000-fold increase in linear modulus and a 1,000-fold increase in failure stress (Figure S11). Strikingly, the adhesion energy of the dehydrated tough gel exceeded 1000J/m2 within 1 minute of application and shear stresses exceeding 100kPa, in contrast to much lower values with the hydrated gels (Figure 6c). Confocal imaging of FITC labeled chitosan revealed that there was a noticeably greater chitosan penetration into dehydrated gels within 1-minute, as compared to hydrated tough gels (Figure 6d).
Figure 6 |. Dehydrated tough gels generate near instantaneous adhesion.

(a) Schematic of proposed mechanism for rapid adhesion. (b) The effect of time on tough hydrogel dehydration, rehydration, and swelling. “Regular” indicates swelling behavior of gels as prepared. “TG Films” indicates swelling behavior of dehydrated gels. (c) The effect of tough hydrogel dehydration on the adhesion energy and shear strength to porcine skin after 1-minute of application. Data shown as mean ± s.d., as evaluated by a Student’s t-test (n=3–4 samples/group). (d) The effect of tough hydrogel hydration and time on chitosan interpenetration at 1 minute. Chitosan was labeled with FITC, and the skin was visualized with polarized light. Scale bar = 100μm. Data shown as mean ± s.d., as evaluated by a two-way ANVOA with post hoc t-tests with Bonferroni corrections (n=3–4 samples/group). Key: TG: tough gel; C: chitosan; S: skin. Adhesion tests were performed using tough gels prepared with alginate D and pAAm, and chitosan A as the bridging polymer.
Discussion
This study investigated the potential adhesion mechanisms of chitosan-mediated tissue adhesion of alginate-pAAm tough hydrogels. We demonstrate that strong topological adhesion to tissues can be achieved using tough hydrogels, without the need of covalent bond formation.[1, 13] Adhesion strengths achieved were >2000J/m2, higher than typically employed hydrogel adhesive materials which rely on direct bond formation with tissues (e.g., cyanoacrylates, catechol, aldehyde, or activated NHS carbonyls).[1, 13, 21] Tissue adhesives relying on non-covalent interactions are typically weak and not stable.[18] However, here using a combination of a hydrogel dissipative matrix with high molecular weight alginate and a dilute chitosan solution we achieved fast, strong, and stable adhesion through physical non-covalent interactions mediated by the bridging polymer. Unprecedented levels of adhesion were shown to be stable, physiologically relevant, and occur independent of coupling reagents while remaining unperturbed after placement in large excess of buffer and bloody settings.
The adhesion mechanism observed in this study was mediated by the bridging polymer. Chitosan has long been used as an adhesive because of its biocompatibility and pH responsiveness.[22] However, without a tough matrix, peak adhesion stress in the absence of covalent bonds is typically <100kPa.[23] Our results support the previously proposed pH dependent adhesive properties of chitosan; deprotonation of its amine groups drive strong interchain interactions by cooperative H-bonding. It thus forms an extensive chitosan network within the gel and tissue.[20] As chitosan’s pKa is ~6.5, tissues (pH>6.5) can induce its gelation upon deprotonation of the chitosan chains, generating an internal interpenetrating network. Since the pH of blood (~pH 7) is above the pKa of chitosan, it is possible that the chitosan amines will begin to deprotonate, triggering the formation of a chitosan entangled network prior to interacting with the tissue. However, application of compression displaces some of the blood and allows the hydrogel to contact the underlying tissue. In this setting, it is unlikely that there is sufficient time for significant network formation within the chitosan solution. For tissues with lower pH, the addition of coupling reagents to mediate strong adhesion might be needed, although other bridging polymers with lower pKa’s could potentially be used instead of chitosan. Although the physiologic pH of skin trends to be slightly acidic, particularly in wound healing models, we speculate that skin pH may be adjusted through rinsing with warm water prior to gel application. Furthermore, most applications of adhesive materials in vivo will be with tissues of neutral pH. Under neutral and high pH, the formation of the chitosan interpenetrating network between the adherends is stable and non-reversible, likely as a significant decrease in pH would be needed to disturb the interchain H-bonds.
Several important factors influencing adhesion include properties of alginate (MW), tissue (pH), and chitosan (concentration, viscosity, degree of deacetylation, and MW). Although the main driver of adhesion with chitosan was the pH of the adherends, adhesion was significantly affected by chitosan concentration and MW, likely due to their effect on chitosan chain diffusion and interactions. Both high MW chitosan chains and concentrated polymer solutions have high viscosity due to chain-chain overlap, intermolecular interactions, and entanglements affecting the ability of the chains to diffuse into the permeable gel and tissue.[24] Chitosan solutions of 1–2% (wt/vol) resulted in the highest adhesion energy, likely due to an optimal combination of viscosity and chain density. Small dilutions of chitosan in the presence of blood likely do not impact adhesion as the decreased chain density is offset by a lower viscosity and improved chitosan tissue penetration. In our system, we speculate that diffusion and entanglement of the chitosan chains with the adherends play a key role. Thus, as demonstrated here, lower MW chitosan is expected to yield stronger topological adhesion because of the faster diffusion of low MW polymers. By using ultrapure chitosan which exhibits significantly lower viscosity while maintaining a high degree of deacetylation and medium molecular weight, we can test at much higher concentrations than previously possible with non-UP chitosan.[1] Additionally, the range of chitosan MW tested in the previous study relied on non-UP materials and covered a narrower MW range. Regarding gel properties, the greatest impact on topological adhesion was the molecular weight of the alginate. Alginate molecular weight may increase the density of entanglements in the initial alginate/AAm solutions, especially since the formulation used is beyond the critical concentration of high MW alginate, ~0.6wt% (for 196kDa alginate).[25] A highly entangled hydrogel matrix likely facilitates chitosan chain entanglement within the matrix to form an interpenetrating network.[26] This possibility is supported by the strong positive linear correlation between alginate MW and adhesion strength. Thus, we show that by using high MW alginate in the dissipative matrix the adhesion can be significantly increased, to values even higher than what has been previously reported with a similar tough hydrogel system relying on covalent bond formation.
Chitosan diffusion and interpenetration into the permeable adherends seems a dominant factor in the adhesion mechanism as increasing properties like MW and concentration, which affect chitosan diffusion, also resulted in a decrease in adhesion and skin penetration. Previous studies examining chitosan diffusion into tissue [1, 4] have highlighted significant tissue penetration. These studies found that chitosan penetrates as deep as ~25um in both tendon and skin after 10min[4] and 1h[1] of compression respectively, in the presence of EDC/sNHS coupling reagents. A similar penetration into skin was observed here and it was affected by both chitosan MW and concentration. Chitosan’s semirigid backbone and its ability to form a robust network through intermolecular interactions between chains above chitosan’s pKa seems another key factor for topological adhesion, as other bridging polymers like polyallylamine with a high density of primary amines but with a highly flexible C-C backbone showed no adhesion.[27] Other bridging polymers like branched polyethylenimine and N,O-carboxymethyl chitosan, featuring a branched structure and a lower density of primary amines respectively, also exhibited no adhesion. Additionally, chitosan has been shown to enhance transdermal drug permeation in various therapeutics by reversibly loosening intercellular tight junctions, leading to a widening of the paracellular routes and higher permeability while allowing faster diffusion of hydrophilic macromolecules.[28] This enhanced tissue permeability likely contributes to the rapid penetration of chitosan chains into tissue, allowing for chain entanglement and rapid, robust topological adhesion.
As interpenetration and physical entanglement of chitosan chains is proposed to be the primary mechanism of adhesion, enhancing their diffusion would be expected to reduce the adhesion time. It has previously been demonstrated that dry hydrophilic polymeric materials can rapidly remove interfacial water and promote near instantaneous adhesion (~5s of compression) to tissues, but through the formation of covalent bonds.[13, 29] This enhanced initial adhesion is likely in part due to a change in the toughness of the gel due to partial dehydration. The dry polymers show coupled hydration and swelling upon contact with water, due to their hydrophilicity and high diffusivity of water.[30] Here, in the presence of a semi-dilute chitosan solution, dehydrated gels demonstrate rapid uptake of the solution.[31] This process may be enhanced by the electrostatic attractions between the negatively charged carboxylic groups in the hydrogel matrix and the positively charged amines in chitosan. Here, dry gels achieved strong adhesion with just one minute of compression, with rapid initial swelling of the gel and uptake of the chitosan into the gel without the need of creating covalent bonds.
In conclusion, the findings of this study demonstrate a new approach to achieve strong and fast adhesion via secondary interactions between a hydrogel and tissue, and reveal factors and mechanisms involved in this adhesion. This approach reduces the need for covalent crosslinking components and could further enable the development of new improved bioadhesives for emerging applications.
Methods
Hydrogel Synthesis
Hydrogels were synthesized by mixing one syringe containing a 14mL solution of 2.2% sodium alginate (summary of alginates in TableS2) and 13.5% acrylamide (Sigma, A8887) in HBSS (Gibco), 50.4μl of 2% N,N’-methylenebis(acrylamide) (Sigma, M7279), and 11.2μl of TEMED (Sigma, T7024), with a second syringe containing 316.4μl of 6.6% ammonium persulfate (Sigma, A9164), and 267.4μl of 0.75M calcium sulfate dihydrate (Sigma, 31221). Alginates with different molecular weights (see TableS2 for details) were obtained from NovaMatrix (MVG, LVG, LVM, VLVG) and FMC BioPolymer (LF10/60, LF20/40). Low molecular weight sodium alginate (LF20–40-5Mrad) was prepared by irradiating the high molecular weight sodium alginate (LF20/40) under γ- rays at a dose of 5 Mrad.[32] The gel was cast into glass molds (110×15×1.7mm3) sealed on both sides with glass and left to crosslink for 24h. After 24h, tough gel strips were removed from molds and stored in sealed plastic bags at 4°C.
To generate dehydrated films, hydrogels with LF10/60 alginate were placed over glass and left to dry at room temperature exposed to ambient air overnight. The obtained films were then transferred into sealed plastic bags to stop further dehydration.
Hydrogel Mechanical Properties
Pure shear tests were carried out to measure the matrix toughness. In brief, rectangular specimens (20×5×1.7 mm3) were tested in tension (Instron 3342, 10N load cell) at 100mm/min. From the stress-stretch curves, the matrix maximum stretch, maximum stress, and fracture toughness were calculated[8] using custom MATLAB code.
Adhesive Application and Adhesion Energy Measurement
Chitosan (0.5–6%wt/vol) with or without coupling reagents (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (Sigma E6383) and sulfated N-hydroxy-succinimide) (Thermofisher, PG82071) (12 mg/mL) were quickly mixed in water by vortexing. A final concentration of 12mg/ml EDC/sNHS was used in adhesion experiments with coupling reagents. A final pH of ~5.5 was obtained in all chitosan solutions either by direct dissolution or by tuning with the addition of 0.1M HCl or 0.1M NaOH. This mixture was applied to the surface of the tough gel (50μL/cm2) (0.5mL total/sample) and compressed to tissue surfaces. Other bridging polymers were tested following the same procedure. Ultrapure Chitosan HCl (54046 and 54039) with different degrees of deacetylation and N,O-carboxymethyl chitosan (44002) were obtained from Heppe Medical Chitosan (HMC), Halle, Germany (see Table S1 for details). High molecular weight chitosan (419419), poly(allylamine hydrochloride) (283223) and branched poly(ethyleneimine) (408700) were obtained from Sigma. A summary of chitosans used is provided in Table S1.
Adhesion energy was measured with 180º peeling tests (Instron 3342) under uniaxial tension (100mm/min). A tissue strip (~75×11mm2) was placed over the gel and compression was applied for 10 mins unless stated otherwise. Applied compression strain was kept at ~5.5% in all cases. The back of TA was also bonded to a rigid polyethylene terephthalate (PET) film with cyanoacrylate (Krazy Glue), in order to limit deformation to the crack tip, and thus all the work done by the machine would be equal to the energy dissipated at the crack tip. The free ends of TA and the substrate were attached to acrylic pieces, to which the machine grips were attached. A mechanical testing system (Instron 3342, 50N load cell) was used to apply unidirectional tension, while recording the force and the extension. The loading rate was kept constant at 100 mm/min. The adhesion energy was two times the peak value of the ratio of the force and width.[1]
To test the adhesive in the presence of blood, the surface of tissue strips were fully covered with 300μL of bovine blood per sample. The same procedure was then performed to compress the gel against the blood-covered tissue, and initiate adhesion. To study the effect of tissue pH on adhesion, porcine skin strips were equilibrated with buffers of different pH ranging from 4.0 to 8.5. First, MES (Sigma) buffer solutions with pH 4.0, 5.5, 7.0, and 8.5 were prepared by adding 0.1M NaOH and monitoring the solutions with a pH meter. Tissue strips (N=4 per group) were then placed in petri dishes filled with the corresponding buffering solutions and allowed to equilibrate for 20 minutes before testing. pH of the solutions was monitored with pH strips to make sure there was no pH change during the equilibration period. The skin samples were then removed from the buffers and tested following the same procedure as before.
Interpenetration of the chitosan chains and confocal imaging
The skin-gel adhesive interface and chitosan diffusion profiles for both the hydrated and dehydrated tough gels were studied by using confocal imaging and fluorescently labeled chitosan (FITC Chitosan). FITC Chitosan was synthesized by reacting fluorescein isothiocyanate (Sigma, 1245460250) with chitosan. Briefly, 1g of chitosan HCl was dissolved in 100mL of 0.1M acetic acid and 100mg of FITC was dissolved in 100mL of anhydrous methanol at 1.0mg/mL in separate flasks. The FITC solution was then slowly added to the chitosan solution with continuous stirring. After ~3h, the reaction was quenched by slowly adding NaOH (0.5M) to increase the pH to ~10, precipitating the fluorescently labelled chitosan. The solutions were then centrifuged, and dIH2O was added to the precipitate after discarding the supernatant. These purification steps were repeated several times until FITC was not observed in the supernatant. The final product was dialyzed against acidic water for ~2days and freeze-dried. The FITC Chitosan was transferred to a container with aluminum foil to protect it from light and stored at 4°C until further use.
To study the gel-adhesive interface, the adhesion procedure describe previously was repeated using a 1:5 mixture of FITC:no-FITC chitosan as the bridging polymer. Compression was applied for 1 or 10 minutes and the samples were then snap frozen in liquid nitrogen. Interpenetration of the bridging polymer into the tissue was assessed using confocal microscopy (LSM710, Zeiss, Oberkochen, Germany). Sections were then mounted (Prolong™ Gold Antifade Mountant, ThermoFisher) and imaged using a confocal microscope for cell nuclei (DAPI), chitosan (FITC, excitation laser 488nm) and collagen (polarized light).
Hydrogel Swelling
Swelling ratio and rates for the gel and its dehydrated counterpart were studied. Both gel and film were originally cut to 15×20×1.7mm3 dimensions. A 6-well plate was filled with 8mL of DMEM and heated to ~37°C. The gel and films (N=6) were placed in each well individually and their weights recorded over time until a swelling equilibrium was reached. The swelling ratio was then calculated as:
where is the original mass of the as-synthesized gel.
Gas Permeation Chromatography (GPC) of Alginate and Chitosan
The molecular weight distributions of the alginates were determined using size exclusion chromatography and software on the 1260 Infinity Multi-Detector GPC/SEC System. Two serial Viscotek A5000 300×8.0mm columns were used for the size separation and all samples eluted off the columns using a 0.1M sodium nitrate with 0.5% w/v azide in double filtered Milli-Q water at a flow rate of 0.75 ml min−1. All samples were dissolved in the above eluent between 2–3mg/ml and injected at a volume of 100ul. The system was calibrated under triple detection method using an Agilent polyethylene glycol standard (PL2083–2001).
For evaluation of chitosan MW, a buffer or 0.1M sodium nitrate, 0.01M sodium monobasic phosphate, with 0.5% w/v azide, pH=3 adjusted with phosphoric acid in double filtered Milli-Q water at a flow rate of 0.75 ml min−1 was utilized. The system was calibrated under triple detection using an Agilent pullulan kit (PL2090–0101).
Fourier Transform Infrared Spectroscopy
To study the chemical structures of the different chitosan, Fourier Transform Infrared (FTIR) spectra was obtained of the dried polymers. All samples were measured as chitosan HCl salts. To prepare the samples, chitosan was dissolved in water and the pH was adjusted to 5.5 by adding 0.1M HCl. After a stable pH was obtained, the samples were frozen at −80 °C and lyophilized to eliminate water. FTIR spectra were recorded on the dry samples between 4000 and 400cm−1 on a Bruker ATR FTIR spectrometer.
Rheology
Rheological measurements of the alginate and chitosan gel material (2% wt/vol) were carried out on a TA Instrument AR-G2 rotational rheometer. The measurements used a 20mm diameter cone at 25°C and plate geometry. Samples were placed on plate using a syringe at 75ul volume and the upper cone was lowered to 0.060mm gap distance. All samples were allowed to equilibrate for one minute before beginning the run. Flow curves were made using the flow step method with varying shear stresses, from 8.000E-3 Pa to 1.000 Pa over 600 secs at 25°C.
Thermogravimetric Analysis
Chitosan thermogravimetric analysis was done using a Discovery Series TGA (S/N; TGA1–0031) and TRIOS software. A Platinum-HT (P/N: 957571.901) sample pan and Tzero Pan (P/N; 901683.901) were used. To analyze samples, platinum pans were heated using a heat gun to remove any residues. The following parameters were used: Mass Flow 40.0 mL/min; Initial Temperature 35.00°C; Ramp 5.00°C/min to 250.00°C; Isothermal 75.00 min. Samples were run in triplicate (n=3/group).
Subcutaneous Implantation Model
All mouse experiments were conducted according to approved IACUC protocols. Balb/C mice at 20 weeks of age were anesthetized with isoflurane (2–2.5%) and given buprenorphine subcutaneously (0.5mg/kg) for pain management. Hair on the mouse dorsum was removed with clippers and depilatory cream prior to adding three separate washes of betadine and ethanol. Animals were then transferred to the sterile field and placed beneath a separate sterile fenestrated drape. A small 6mm incision was made through skin in the animal’s back perpendicular to the midline, and then a subcutaneous pocket was created using scissors. Four separate gels (D=6mm, th=1.5mm) were then implanted subcutaneously, and the skin was closed with 4–0 Vicryl suture. Animals were monitored daily and gel swelling analyzed using HFUS (see below).
High Frequency Ultrasound Imaging (HFUS)
HFUS (VisualSonics Vevo 770 and Vevo 3100; 35–50MHz) was used to evaluate gel swelling in vivo. Axial images (20um axial resolution) were acquired that captured the skin and hydrogel. Images were used to quantify the thickness of the hydrogel and surrounding capsule after 0, 7, and 14 days. Images were analyzed using ImageJ (NIH).
Statistical Analysis
Data normality was assessed with Shapiro Wilk tests (SPSS). One-way (time) or two-way (healing and TA implantation) ANOVAs with post hoc Student’s t-tests were used.
Supplementary Material
Acknowledgments
This work was supported by the Wyss Institute for Biologically Inspired Engineering at Harvard and the National Institute on Aging of the NIH (K99AG065495). J.C.C. was supported by the Harvard GSAS Research Scholar initiative (NIH NIGMS Grant) and the Wyss Institute.
Grant Support:
This study was supported by the National Institute on Aging at the NIH (K99AG065495) and the Wyss Institute for Biologically Inspired Engineering.
Competing Interests Statement
The authors receive grant support through the NIH and Wyss Institute. The views and opinions expressed in this article are those of the authors and do not necessarily reflect the position of the Wyss Institute for Biologically Inspired Engineering at Harvard University.
References
- [1].Li J, Celiz AD, Yang J, Yang Q, Wamala I, Whyte W, Seo BR, Vasilyev NV, Vlassak JJ, Suo Z, Mooney DJ, Science 2017, 357, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Taboada GM, Yang K, Pereira MJN, Liu SS, Hu Y, Karp JM, Artzi N, Lee Y, Nature Reviews Materials 2020. [Google Scholar]
- [3].Blacklow SO, Li J, Freedman BR, Zeidi M, Chen C, Mooney DJ, Sci Adv 2019, 5, eaaw3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Freedman BR, Kuttler A, Beckmann N, Nam S, Kent D, Schuleit M, Ramazani F, Accart N, Rock A, Li J, Kurz M, Fisch A, Ullrich T, Hast MW, Tinguely Y, Weber E, Mooney DJ, Nature Biomedical Engineering 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hu S, Pei X, Duan L, Zhu Z, Liu Y, Chen J, Chen T, Ji P, Wan Q, Wang J, Nat Commun 2021, 12, 1689; [DOI] [PMC free article] [PubMed] [Google Scholar]; Wright ZM, Holt BD, Sydlik SA, J Mater Chem B 2017, 5, 7743. [DOI] [PubMed] [Google Scholar]
- [6].Lazow SP, Labuz DF, Freedman BR, Rock A, Zurakowski D, Mooney DJ, Fauza DO, J Pediatr Surg 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]; Freedman BR, Uzun O, Luna NMM, Rock A, Clifford C, Stoler E, Ostlund-Sholars G, Johnson C, Mooney DJ, Adv Mater 2021, 33, e2008553; [DOI] [PMC free article] [PubMed] [Google Scholar]; Freedman BR, Mooney DJ, Adv Mater 2019, 31, e1806695; [DOI] [PMC free article] [PubMed] [Google Scholar]; Deng J, Yuk H, Wu J, Varela CE, Chen X, Roche ET, Guo CF, Zhao X, Nat Mater 2021, 20, 229. [DOI] [PubMed] [Google Scholar]
- [7].Ma Z, Bao G, Li J, Adv Mater 2021, 33, e2007663. [DOI] [PubMed] [Google Scholar]
- [8].Sun JY, Zhao X, Illeperuma WR, Chaudhuri O, Oh KH, Mooney DJ, Vlassak JJ, Suo Z, Nature 2012, 489, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Li J, Illeperuma WR, Suo Z, Vlassak JJ, ACS Macro Letters 2014, 3, 4. [DOI] [PubMed] [Google Scholar]
- [10].Ge L, Chen S, Polymers (Basel) 2020, 12; [DOI] [PMC free article] [PubMed] [Google Scholar]; Bovone G, Dudaryeva OY, Marco-Dufort B, Tibbitt MW, ACS Biomater Sci Eng 2021, 7, 4048. [DOI] [PubMed] [Google Scholar]
- [11].Nam S, Mooney D, Chem Rev 2021, 121, 11336. [DOI] [PubMed] [Google Scholar]
- [12].Hong Y, Zhou F, Hua Y, Zhang X, Ni C, Pan D, Zhang Y, Jiang D, Yang L, Lin Q, Zou Y, Yu D, Arnot DE, Zou X, Zhu L, Zhang S, Ouyang H, Nat Commun 2019, 10, 2060; [DOI] [PMC free article] [PubMed] [Google Scholar]; Konuray O, Fernandez-Francos X, Ramis X, Serra A, Polymers (Basel) 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yuk H, Varela CE, Nabzdyk CS, Mao X, Padera RF, Roche ET, Zhao X, Nature 2019, 575, 169. [DOI] [PubMed] [Google Scholar]
- [14].Wang DA, Varghese S, Sharma B, Strehin I, Fermanian S, Gorham J, Fairbrother DH, Cascio B, Elisseeff JH, Nat Mater 2007, 6, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Aakeroy CB, Epa K, Forbes S, Schultheiss N, Desper J, Chemistry 2013, 19, 14998; [DOI] [PubMed] [Google Scholar]; Kinloch AJ, Journal of Materials Science 1980, 15, 25. [Google Scholar]
- [16].Chen J, Wang D, Wang LH, Liu W, Chiu A, Shariati K, Liu Q, Wang X, Zhong Z, Webb J, Schwartz RE, Bouklas N, Ma M, Adv Mater 2020, 32, e2001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cui C, Wu T, Chen X, Liu Y, Li Y, Xu Z, Fan C, Liu W, Advanced Functional Materials 2020, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yang J, Bai R, Chen B, Zuo S, Advanced Functional Materials 2019, 27. [Google Scholar]
- [19].Yang J, Bai R, Li J, Yang C, Yao X, Liu Q, Vlassak JJ, Mooney DJ, Suo Z, ACS Appl Mater Interfaces 2019. [DOI] [PubMed] [Google Scholar]
- [20].Yang J, Bai R, Suo Z, Adv Mater 2018, 30, e1800671. [DOI] [PubMed] [Google Scholar]
- [21].Pandey N, Soto-Garcia LF, Liao J, Philippe Z, Nguyen KT, Hong Y, Biomater Sci 2020, 8, 1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mati-Baouche N, Elchinger P, Baynast H, Pierre G, Delattre C, Michaud P, European Polymer Journal 2014, 60, 14. [Google Scholar]
- [23].Frost SJ, Mawad D, Higgins MJ, Ruprai H, Kuchel R, Tilley RD, Myers S, Hook JM, Lauto A, NPG Asia Materials 2016, 8, e280; [Google Scholar]; Lauto A, Mawad D, Barton M, Gupta A, Piller SC, Hook J, BioMedical Engineering OnLine 2010, 9, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zimm BH, The Journal of Chemical Physics 1956, 24, 269; [Google Scholar]; Hwang JK, Shin HH, Korea-Australia Rheology Journal 2000, 12, 5; [Google Scholar]; Rouse PE, The Journal of Chemical Physics 1953, 21, 1272. [Google Scholar]
- [25].Bonino CA, Krebs MD, Saquing CD, Jeong SI, Shearer KL, Alsberg E, Khan SA, Carbohydrate Polymers 2011, 85, 111. [Google Scholar]
- [26].Pragya A, Mutalik S, Younas MW, Pang S-K, So P-K, Wang F, Zheng Z, Noor N, RSC Advances 2021, 11, 10710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Davydova VN, Yermak IM, Molecular Biophysics 2018, 63, 11. [Google Scholar]
- [28].Junginger HE, Verhoef JC, Pharmaceutical Science & Technology Today 1998, 1, 370. [Google Scholar]
- [29].Singla S, Amarpuri G, Dhopatkar N, Blackledge TA, Dhinojwala A, Nature Communications 2018, 9, 1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mao X, Yuk H, Zhao X, Journal of the Mechanics and Physics of Solids 2020, 137, 103863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Klossner RR, Queen HA, Coughlin AJ, Krause WE, Biomacromolecules 2008, 9, 2947. [DOI] [PubMed] [Google Scholar]
- [32].Kong HJ, Kaigler D, Kim K, Mooney DJ, Biomacromolecules 2004, 5, 1720. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.