

 This work is licensed under a
This work is licensed under a Abstract
Cardiovascular disease, the primary cause of human mortality globally, is predominantly caused by a progressive disorder known as atherosclerosis. Atherosclerosis refers to the process of accumulation of cholesterol-enriched lipoproteins and the concomitant initiation of inflammatory processes in the arterial wall, including the recruitment of immune cells. This leads to the formation of atherosclerotic plaques, initially causing a thickening of the arterial wall and narrowing of arteries. However, as plaque formation progresses, atherosclerotic plaques may become unstable and rupture, leading to a blood clot that blocks the affected artery or travels through the blood to block blood flow elsewhere. In the early 1990s, emerging gene editing methods enabled the development of apolipoprotein E knockout (Apoe−/− ) and low-density lipoprotein receptor knockout (Ldlr−/− ) mice. These mice have been instrumental in unraveling the complex pathogenesis of atherosclerosis. Around the same time, human APOE*3-Leiden transgenic mice were generated, which were more recently cross-bred with human cholesteryl ester transfer protein (CETP) transgenic mice to generate APOE*3-Leiden.CETP mice. This model appears to closely mimic human lipoprotein metabolism and responds to classic lipid-lowering interventions due to an intact ApoE–LDLR pathway of lipoprotein remnant clearance. In this review, we describe the role of lipid metabolism and inflammation in atherosclerosis development and highlight the characteristics of the frequently used animal models to study atherosclerosis, with a focus on mouse models, discussing their advantages and limitations. Moreover, we present a detailed methodology to quantify atherosclerotic lesion area within the aortic root region of the murine heart, as well as details required for scoring atherosclerotic lesion severity based on guidelines of the American Heart Association adapted for mice.
Keywords: atherosclerosis, hyperlipidemia, mouse models, quantification, aortic root
Introduction
Cardiovascular diseases (CVDs) are currently the leading cause of death globally. Alarmingly, the total number of CVD cases has nearly doubled from 271 million in 1990 to 523 million 2019 (1). The main underlying cause of CVDs is atherosclerosis, a progressive disorder characterized by a thickening of arterial walls and narrowing of the arteries due to the formation of atherosclerotic plaques, that is, accumulation of cholesterol, macrophages, and cell debris in the intima of the arteries (2, 3). The main risk factors for the development of atherosclerotic CVD (asCVD) are combined hyperlipidemia (i.e. high levels of plasma triglycerides and cholesterol) and inflammation (4).
In this review, we will first describe the pathogenesis of atherosclerosis, followed by an overview of different animal models to study atherosclerosis, with a focus on mouse models. We will conclude this review with a detailed description of an established method in our laboratory to characterize atherosclerosis within the aortic root area of the mouse heart.
Atherosclerosis development
Traditionally, atherosclerosis has been regarded as a lipid-driven disease, where the retention of lipoproteins in the intima of arteries was considered to be the main causal factor. However, later observations showed that circulating monocytes infiltrate the developing plaques, revealing an inflammatory component of the disease (5). In the following sections, we will describe the role of both aspects in the development of atherosclerotic plaques.
Lipoprotein metabolism
Lipoproteins are essential carriers of lipids, transporting them in the lymphatics and circulation between tissues and organs. They typically consist of a hydrophobic core of nonpolar lipids (i.e. triglycerides and cholesteryl esters), surrounded by a hydrophilic shell containing phospholipids, unesterified cholesterol, and apolipoproteins (6). At least four classes of lipoproteins can be distinguished based on their size, lipid composition, and apolipoprotein composition, namely chylomicrons, very-low-density lipoprotein (VLDL), low-density lipoprotein (LDL), and high-density lipoprotein (HDL) (6).
Chylomicrons are formed in the small intestine (Fig. 1A) (4, 6). Starting in the gastrointestinal tract, dietary triglycerides are broken down by gastric lipase and pancreatic lipase into 2-monoacylglycerols and fatty acids, which are absorbed by enterocytes. Here, they are readily reconverted into triglycerides with the aid of monoacylglycerol acyltransferase (MGAT) and diacylglycerol transferase (DGAT) (7). Simultaneously, dietary cholesterol is taken up by enterocytes and largely esterified into cholesteryl esters by acyl-CoA cholesterol acyltransferase (ACAT). Within the endoplasmic reticulum of enterocytes, microsomal triglyceride transfer protein (MTP) subsequently combines lipid droplets formed from triglycerides, cholesteryl esters, and phospholipids with apolipoprotein (Apo) B48 (ApoB48; i.e. 48% of the ApoB transcript) to form pre-nascent chylomicron particles (8, 9). Ultimately, chylomicrons are secreted into the lymph and enter the circulation via the thoracic duct, where they acquire exchangeable apolipoproteins primarily produced by hepatocytes. This includes ApoC2, which is an essential cofactor for lipoprotein lipase (LPL). When LPL is activated on the endothelial surface of capillaries, it catalyzes the hydrolysis of triglycerides within chylomicrons, allowing for the uptake of the liberated fatty acids by the underlying tissue as an energy source (10). During delipidation, the generated chylomicron remnants acquire increasing amounts of ApoE, which allows the remnants to be cleared from the circulation primarily by hepatocytes via binding of ApoE to the LDL-receptor (LDLR) and the LDLR-related protein 1 (LRP1), followed by endocytosis.
Figure 1.
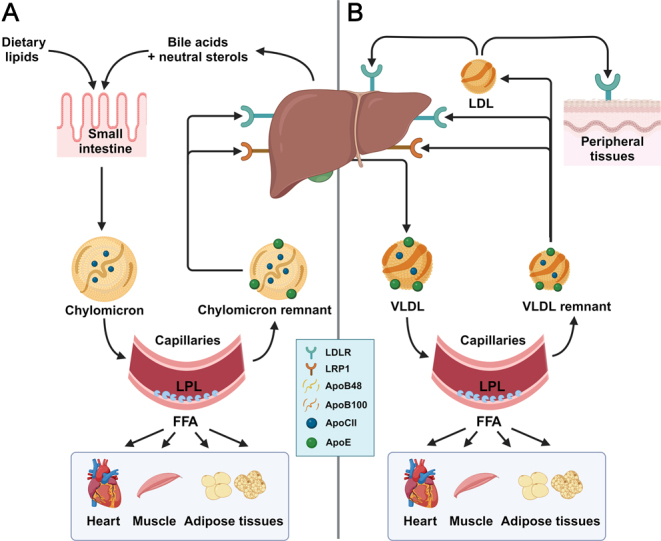
A schematic overview of ApoB-containing lipoprotein metabolism. See text for details. Apo, apolipoprotein; FFA, free fatty acid; LDL, low-density lipoprotein; LDLR, LDL receptor; LPL, lipoprotein lipase; LRP1, LDL receptor-related protein 1; VLDL, very-low-density lipoprotein.
The liver can use the lipids taken up from chylomicron remnants for oxidation, or use them together with de novo synthesized lipids and cholesterol to generate VLDL (Fig. 1B). Here, MTP facilitates the fusion of lipid droplets with ApoB100 (i.e. 100% of the ApoB transcript), and other apolipoproteins, including ApoA5, ApoC1, ApoC2, ApoC3, and ApoE, also associate with the newly produced VLDL (6, 11). Once in the circulation, LPL hydrolyzes the triglycerides within VLDL (similar to triglycerides within chylomicrons), after which the delipidated VLDL remnants are also cleared primarily via binding of ApoE to the LDLR and LRP1 on hepatocytes within the liver. Part of the VLDL remnants escape clearance by the liver and are further lipolyzed, leading to the formation of essentially triglyceride-free LDL (4). As LDL also no longer contains ApoE, it can only be cleared via interaction of ApoB100 with the LDLR on hepatocytes or on other organs that require cholesterol, such as the adrenals. As the affinity of ApoB100 for the LDLR is much lower than that of ApoE, and there is no interaction with LRP1, LDL has a much longer retention time in the circulation than VLDL and thus tends to accumulate. The metabolism of chylomicrons, VLDL, and LDL is summarized in Fig. 1.
Both the liver and the intestine synthesize and secrete lipid-poor ApoA1, the precursor of HDL (6). Once in the circulation, ApoA1 acquires excess phospholipids that are liberated from chylomicrons and VLDL during LPL-mediated lipolysis, mediated via the phospholipid transfer protein (PLTP) (12). This nascent pre-HDL also takes up cholesterol from peripheral organs via diffusion and via the ATP-binding cassette transporter A1 (ABCA1) and G1 (ABCG1), forming cholesterol-enriched pre-HDL. Subsequently, lecithin cholesterol acyltransferase (LCAT) catalyzes the esterification of free cholesterol into cholesteryl esters, producing mature HDL, which can interact with the scavenger receptor class B type 1 (SR-B1) on hepatocytes. This results in the selective uptake of cholesteryl esters from HDL by the liver (6, 12). Cholesteryl esters can also be transferred from HDL to ApoB-containing lipoproteins in exchange for triglycerides with the aid of cholesteryl ester transfer protein (CETP) (13, 14). Eventually, these cholesteryl esters partly end up in the liver via interaction of ApoE with the LDLR and LRP1, or ApoB100 with the LDLR. Regardless of the route, cholesteryl esters taken up by hepatocytes are hydrolyzed within lysosomes into cholesterol. Here, cholesterol can be temporarily stored in hepatocytes as cholesteryl esters after re-esterification or it can be secreted into the bile, either directly by transport via ABCG5 and ABCG8, or indirectly after conversion into bile acids, which is the major route of biliary cholesterol secretion (15, 16). Bile acids released into the gut facilitate the emulsification and absorption of dietary lipids, after which the majority (~95%) of the bile acids is reabsorbed (‘enterohepatic circulation’), with the remainder excreted via feces (17).
Dyslipidemia and inflammation: two risk factors in atherosclerosis development
Under certain conditions, such as metabolic syndrome, VLDL can be overproduced by the liver and/or the processing and removal of ApoB-containing lipoproteins (i.e. chylomicrons, VLDL, and LDL) can be impaired. This results in the accumulation of triglycerides and cholesteryl esters carried within these lipoproteins in the circulation, also known as combined hyperlipidemia, which poses a major risk for the development of coronary artery disease (CAD). ApoB-containing lipoproteins are pro-atherogenic because they can cross the endothelium of artery walls via paracellular transport (‘paracytosis’), thereby entering the arterial intima (Fig. 2) (4). Per particle, VLDL is considered to be more pro-atherogenic than LDL (18). Nonetheless, LDL likely contributes most to atherosclerosis development as it outnumbers VLDL in humans, and it can cross the endothelium not only via paracytosis but also via SR-B1-mediated transcytosis (19). Once in the intima, LDL can be oxidized by enzymatic or nonenzymatic reactions (20). In addition, LDL can aggregate within the intima, thereby increasing their affinity for arterial proteoglycans, promoting their retention (21). Interestingly, the susceptibility of LDL to aggregate has been shown to associate with future cardiovascular deaths (22).
Figure 2.

Schematic overview of atherosclerotic plaque development. See text for explanation. Green arrows indicate stimulation of the indicated process. LDL, low-density lipoprotein; LRP1, LDL receptor-related protein 1; SR-B1, scavenger receptor class B type 1; VLDL, very-low-density lipoprotein.
Besides hyperlipidemia, inflammatory pathways play an integral part in the initiation and progression of atherosclerosis. Although the triggers needed to start the inflammatory cascade in atherogenesis has/have not been fully elucidated, it is generally believed that one of the first steps involves the activation of endothelial cells and smooth muscle cells lining the arterial vessel wall. This leads to an increase in the expression of adhesion molecules, including intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1). Activated endothelial cells also start overexpressing chemoattractant proteins, such as monocyte chemoattractant protein 1 (MCP1), chemokine (C–C motif) ligand 5 (CCL5), interferon gamma-induced protein 10 (IP-10), and chemokine (C–X3–C motif) ligand 1 (CX3CL1) (23). As a result, immune cells, including monocytes, T-cells, and dendritic cells, are recruited into the intima (3, 23). The secretion of proinflammatory cytokines (e.g. macrophage-colony stimulating factor; MCSF and interleukin 1 beta; IL-1β) by endothelial cells additionally stimulates the newly recruited monocytes to differentiate into macrophages (24). In reaction to these various inflammatory cytokines (e.g. MCSF, MCP1, and IL-1β) the liver produces C-reactive protein (CRP), which in turn stimulates inflammatory reactions in the vascular endothelium, thereby further promoting plaque formation (25). In line with this, circulating levels of CRP have been shown to be predictive for the long-term outcome of patients with CAD and the risk of complications as a consequence of atherosclerosis (26, 27).
Once differentiated, macrophages in the intima upregulate the expression of scavenger receptors, such as scavenger receptor A (SRA) and cluster of differentiation 36 (CD36), to engulf mainly modified (i.e. oxidized or aggregated) LDL and lipoprotein remnants (3). In addition, smooth muscle cells can break through the internal elastic lamina where they can take up primarily aggregated LDL via LRP1 (28). The accumulated cholesterol in both macrophages and smooth muscle cells is stored as cholesteryl esters in cytoplasmic lipid droplets or in unesterified form in lysosomes and cell membranes. As the cholesterol levels in macrophages and smooth muscle cells continue to rise, the cells are converted into so-called foam cells.
Foam cells form the first basis of an atherosclerotic lesion, called a ‘fatty streak’, and accelerate lesion development by secreting proinflammatory cytokines and stimulating the retention of lipoproteins (3). Further accumulation of unesterified cholesterol in the foam cells ultimately leads to cell death and the formation of cholesterol crystals (29, 30, 31). In an early atherosclerotic lesion, apoptotic cells are cleared by macrophages and other phagocytic cells through a process called efferocytosis (32). However, as the plaque formation progresses, efferocytosis becomes impaired, leading to the accumulation of foam cells, apoptotic cells, and cell debris, resulting in the formation of a necrotic core within the atherosclerotic plaque (32). In parallel, an overlying matrix of collagen, proteoglycans, and smooth muscle cells, referred to as a ‘fibrous cap’, is formed, which is crucial to maintain lesion integrity (33). However, foam cells also release matrix metalloproteinases (MMPs) and other enzymes that degrade the extracellular matrix, thereby weakening the fibrous cap and increasing plaque vulnerability, specifically increasing the risk of rupture (34, 35). Upon rupture, the highly thrombotic material from the plaque’s interior is exposed to the circulation, thereby initiating a coagulation cascade (33), which may cause an acute thrombotic occlusion of a coronary or carotid artery, and may lead to a myocardial infarction or a stroke, respectively. In fact, plaque rupture is the most frequent finding in autopsy studies of patients with sudden cardiac death or myocardial infarction (36, 37).
Animal models to study atherosclerosis
What should be clear by now is that the pathophysiology of atherosclerosis is complex, multifactorial, and dependent on numerous immuno-metabolic interactions that cannot easily be studied in cell culture models. Furthermore, given the chronic, progressive character of the disease and the limited availability of visualization techniques, studying pathogenesis in humans, let alone studying the anti-atherogenic potential of novel strategies, is very challenging. For that reason, as early as 1908, Alexander Ignatowski established the first animal model to study atherosclerotic plaque formation using rabbits fed a diet enriched in fat and cholesterol through supplementation with egg yolk and milk (38). Since then, rabbits became a popular model within the atherosclerosis field and were essential for the discovery and description of the underlying pathology of atherosclerosis. For example, Nikolai Anichkov was the first to report in 1913 that hypercholesterolemia causes atheromatous changes in the vascular wall of rabbits (39). In addition, he was the first to observe multiple cell types present within atherosclerotic lesions, only later to be identified as macrophages, lymphocytes, and smooth muscle cells (40). Even though the initial suggestion that circulating lipids exist in complexes with proteins was made by Michel Machebouef in 1929 (41), it was not until the 1950s, when John W Gofman and Frank Lindgren were able to isolate different lipoprotein fractions using an ultracentrifuge protocol and showed that these lipoproteins build up in the vessel wall to form atherosclerotic lesions (42, 43).
In the early 1990s, emerging gene editing techniques enabled the generation of genetically modified mice as models to study atherosclerosis, and mice have ever since been the favored experimental animal to study atherosclerosis. The choice of mice as a model for atherosclerosis may be somewhat surprising, as no known inbred mouse strain exists that spontaneously develops atherosclerosis. This can be explained by the fact that mice have very low (VL)DL-cholesterol levels, and the majority of circulating cholesterol (approximately 80%) is found within HDL, while in humans most of the circulating cholesterol (approximately 75%) is within (V)LDL (44). Nevertheless, the use of mice in research has several advantages. For example, mice are small in size and thus have low housing costs, they rapidly reproduce, and if susceptible, they need a relatively short period of time to develop atherosclerosis (45, 46). With respect to the latter, Paigen et al. investigated ten different mouse inbred strains to show that C57BL/6 mice are most susceptible to developing diet-induced atherosclerosis (47). Given that the genetic map of inbred mouse strains was relatively well defined, this discovery even allowed for the identification of certain genetic links to atherosclerosis susceptibility in the years to follow (48, 49, 50). In mice, atherosclerotic plaques typically do not rupture. However, a stability index may be determined as a measure of plaque vulnerability (see section ‘Determining atherosclerotic lesion composition’). Also, the developmental stages of atherosclerotic plaques can be scored, ranging from early fatty streaks to clinically dangerous plaques, which is elaborated on in the section ‘Scoring atherosclerosis severity’.
ApoE knockout model
Among the first mouse models for atherosclerosis obtained through genetic engineering was the ApoE knockout model (Apoe−/−). Apoe−/− mice were generated through homologous recombination in embryonic stem cells, as reported in 1992 (51). As described in the section ‘Lipoprotein metabolism’, ApoE is essential for the uptake of triglyceride-rich lipoprotein remnants from the circulation via the LDLR and LRP1 present on hepatocytes (52, 53). Since hepatic uptake of remnants is thus abrogated in Apoe−/− mice, these mice exhibit hyperlipidemia with circulating total cholesterol levels up to 10–15 mM on a regular chow diet, and exceeding 25 mM on a cholesterol-containing Western-type diet (51, 54). In comparison, wildtype C57BL/6 mice do not exhibit cholesterol levels above 2.5 mM, of which the majority represents HDL cholesterol. As a result, Apoe−/− mice develop advanced atherosclerosis already at 2–3 months of age even on a regular chow diet (55).
In Apoe−/− mice, atherosclerotic lesions mainly develop in the valve sinus, including the coronary arteries, but also in the aortic root, aortic branches, the carotid artery, mesenteric artery, renal and pulmonary arteries (56, 57). Interestingly, strain-dependent susceptibility to atherosclerosis, as discussed before, translates into differences in the topography of lesion development. For example, Apoe−/− mice on a C57BL/6 background are more prone to develop atherosclerosis in the aortic root region, whereas a 129S6 background yields plaques mostly in the aortic arch, and on a DBA/2J background plaques develop in both locations (58). In addition, there seems to be a sex-dependent susceptibility to atherosclerosis with larger and more advanced lesions in young, but not old, female Apoe−/− mice compared with age-matched male mice on a normocholesterolemic diet (59). This is different from what is observed in humans, where males typically develop atherosclerotic lesions at a younger age than females in line with the atheroprotective effects of estrogen that have also been reported in Apoe−/− mice (60, 61).
Since their development, Apoe−/− mice have been widely used to discover new therapeutic drugs and drug targets for atherosclerosis, especially in the field of anti-inflammatory strategies. For example, ablation of IL-1β in Apoe−/− mice was found to reduce atherosclerosis development by approximately 30% (62). This finding appeared translationally relevant as the more recent Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) trial showed that canakinumab, a monoclonal antibody targeting IL-1β, lowers recurrent cardiovascular events in patients with a history of myocardial infarction (63). It should be noted, though, that immunomodulatory interventions do increase the risk for infections, as also became apparent with canakinumab (63); therefore, such strategies may only be suitable for specific subpopulations of patients, for example, those at a very high risk of recurrent cardiovascular events. ApoE also has functions beyond lipoprotein metabolism. For example, Apoe is expressed by hematopoietic cells, where it regulates proliferation rate (64). ApoE also suppresses the type I inflammatory responses, as concluded from elevated pro-inflammatory cytokine expression in lipopolysaccharide (LPS)-stimulated Apoe−/− mice but not in other hypercholesteremic models (65). Furthermore, ApoE in macrophages controls the cholesterol efflux to HDL, which contributes to the impaired reverse cholesterol transport as observed in Apoe−/− mice (66, 67).
LDLR knockout model
The genetic link between the LDLR and familial hypercholesterolemia was suggested already in 1974 when Brown and Goldstein showed that cells derived from patients with familial hypercholesterolemia lack high-affinity receptors for binding LDL (68). Therefore, by targeting the LDLR with homologous recombination of embryonic stem cells, an Ldlr−/− mouse model was created and reported in 1993 (69, 70). Ldlr−/− mice are hypercholesteremic on a regular chow diet, albeit to a lesser extent than observed in Apoe−/− mice, with total cholesterol levels of 5–8 mM (70). However, when fed a cholesterol-enriched diet, cholesterol levels can increase up to 40 mM, resulting in accelerated atherosclerosis development compared to the same mice on a regular chow diet (71). Nevertheless, the topography of plaques in the Ldlr−/− mouse on a C57BL/6 background is highly comparable to that in Apoe−/− mice on the same background (71, 72). As for Apoe−/− mice, there is surprisingly little information about sexual dimorphism in atherosclerosis development in Ldlr−/− mice and the published studies are typically not powered to address this issue. Nevertheless, a relatively large study suggested larger atherosclerotic lesions in female compared to male Ldlr−/− mice on a C57BL/6J background, but not on an FVB/NJ background (73, 74). Interestingly, a recent study suggested that the use of older Ldlr−/−mice more closely resembles the human situation with more advanced atherosclerotic lesions and the presence of age-associated T and B cells both in atherosclerotic plaques and in the circulation (75).
Similarly to Apoe−/− mice, the Ldlr−/− mice are widely employed to study atherosclerosis and have some advantages over the Apoe−/− mouse model. First, in contrast to Apoe−/− mice where cholesterol mainly accumulates within chylomicrons and VLDL, the Ldlr−/− mice accumulate cholesterol mostly as LDL particles, generating a more human-like lipid profile (51, 76). Secondly, the atherosclerotic phenotype of Ldlr−/− mice is in large caused by elevated lipid levels and is less affected by effects beyond those on lipoprotein metabolism, as is the case for Apoe−/− mice (77).
As many lipid-lowering drugs directly or indirectly rely on LDLR-mediated lipoprotein uptake, the effect of those drugs cannot be studied in Ldlr−/− mice, and this applies to some extent also for Apoe−/− mice as they lack the main ligand of the LDLR. These drugs include statins, which are used by more than 200 million people around the world to lower CVD risk. The reason for this is that, although inhibition of cholesterol biosynthesis via 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase is the primary target of statins, a large part of the cholesterol-lowering effect of statins is mediated via a compensatory hepatic upregulation of the LDLR (78, 79). Another example of a drug class that is not effective in the Ldlr−/− mouse is the recently developed proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors (78). PCSK9 is a protein that binds to the LDLR and facilitates its lysosomal degradation, thereby ultimately attenuating the hepatic clearance of lipoproteins remnants from the circulation (80). Inhibition of PCSK9 thus lowers LDL levels preventing the degradation of the LDLR. Altogether, this indicates that the Ldlr−/− mouse model is less suitable to test the efficacy, toxicity, or mechanism of action of newly developed lipid-lowering therapeutics (45).
APOE*3-Leiden.CETP model
Interestingly, even before the description of Apoe−/− and the Ldlr−/− mice, yet another mouse model for atherosclerosis was reported based on the discovery of the APOE*3-Leiden mutation in a Dutch family with a genetic form of dysbetalipoproteinemia (81). This APOE*3-Leiden mutation, characterized by a tandem repeat of seven amino acids just outside the LDLR binding domain (82), caused a decrease in the affinity of APOE*3 for the LDLR, resulting in a slower clearance of chylomicron and VLDL remnants from the circulation (71). A genomic construct containing APOE, APOC1, and all regulatory elements was isolated from a proband of this family and used to generate mice transgenic for human APOE*3-Leiden and APOC1 as described in 1991 (83).
A major advantage of the APOE*3-Leiden mice is that they still express endogenous ApoE, and thus have a functional ApoE–LDLR clearance pathway. Only when fed a cholesterol-containing Western-type diet, APOE*3-Leiden mice become hyperlipidemic and display total cholesterol levels of approximately 10–20 mM divided over (V)LDL and HDL (83). In 2006, the first report of a crossbreeding between APOE*3-Leiden mice and human CETP transgenic mice appeared, resulting in APOE*3-Leiden.CETP double-transgenic mice (84). While not expressed in wild-type mice, CETP mediates the transfer of cholesteryl esters from HDL to (V)LDL in exchange for triglycerides in humans, the main effect being a reduction in HDL-cholesterol and increasing levels of (V)LDL-cholesterol. Importantly, these mice appear to respond similar to humans when it comes to the lipid-lowering and atheroprotective effects of, for example, statins (85), fibrates (86), niacin (87), and the PCSK9 antibody alirocumab (88, 89) mainly due to the presence of the APOE*3-Leiden transgene, but also to the HDL-raising effects of the CETP inhibitors torcetrapib (86) and anacetrapib (90). Moreover, we have employed APOE*3-Leiden.CETP mice to show that newly developed experimental drugs for obesity and diabetes, such as a fibroblast growth factor 21 (FGF21) analogue (91), a glucagon-like peptide 1 (GLP-1) receptor agonist (92), and the combination of a GLP-1 receptor agonist with a glucose-dependent insulinotropic polypeptide (GIP) receptor agonist, also have lipid-lowering and atheroprotective effects (93).
Similar to the APOE*3-Leiden mice,APOE*3-Leiden.CETP mice are hyperlipidemic only when fed a cholesterol-containing Western-type diet, with total cholesterol levels reaching 10–30 mM depending on the amount of cholesterol in the diet (typically ranging between 0.10% and 0.25%). It should be noted, however, that in 10–15% of Western-type diet-fed APOE*3-Leiden and APOE*3-Leiden.CETP mice, total cholesterol levels remain below the threshold of approximately 7 mM to develop atherosclerosis after a typical run-in period of 4 weeks (often referred to as ‘low-responders’ or ‘non-responders’), and are therefore not included for randomization in experiments (94, 95). Also, cholesterol exposure (i.e. the area under the curve from circulating total cholesterol levels plotted against the time on a Western-type diet) is quite an accurate predictor for the average atherosclerotic lesion size in the aortic root region of APOE*3-Leiden and APOE*3-Leiden.CETP mice. We typically aim for a cholesterol exposure of 250–300 mM*weeks, corresponding with an average total atherosclerotic lesion size of approximately 1.0–1.5 × 105 µm2 per cross section of the aortic valve area in APOE*3-Leiden.CETP mice. In that situation, lesions of all severity types (as detailed in the section ‘Scoring atherosclerosis severity’) can be identified, allowing for studying both anti- and pro-atherogenic potential of (pharmacological) interventions (89, 91). In practice, this would require a treatment period of approximately 14 weeks, assuming a run-in period of 4 weeks and an average total cholesterol level of 15 mM. Concerning the topography of lesions, the different anatomical sites at which APOE*3-Leiden.CETP mice develop atherosclerosis have, to our knowledge, not been formally mapped. However, they have been mapped for the APOE*3-Leiden mice, which develops lesions in the aorta, proximal coronary arteries, aortic root, aortic arch, and large vessels upon a high-fat diet (96). We expect a comparable topography for APOE*3-Leiden.CETP mice.
A limitation of the APOE*3-Leiden.CETP mouse model is that only female mice develop atherosclerosis upon Western-type diet feeding. Although the exact reason remains unclear, male APOE*3-Leiden mice fail to increase hepatic VLDL production in response to a Western-type diet and typically have a more rapid VLDL clearance rate as compared to their female counterparts (97). As a consequence, in male mice, lipids tend to accumulate in the liver rather than in the circulation. This phenotypic feature, combined with the fact that male C57BL/6J mice are generally more prone to develop diet-induced obesity when compared to female mice (98, 99), actually makes male APOE*3-Leiden.CETP mice on their C57BL/6J background a very suitable model for studying lipid-modulating interventions in metabolic dysfunction-associated steatotic liver disease (MASLD), formerly known as nonalcoholic fatty liver disease (NAFLD). For example, we have shown that diet-induced MASLD in male APOE*3-Leiden.CETP mice can be prevented by long-acting FGF21 (100) and combined GLP1R/GIPR agonism (101).
With the identification of triglycerides as an independent risk factor for coronary heart disease (102), novel medication is being developed aimed at accelerating triglyceride removal from the circulation. To study the anti-atherogenic effects of triglyceride-lowering strategies, the presence of the APOE–LDLR clearance pathway appeared critical for the correct translation of findings to humans. For example, a recent study by Sui et al. showed that the clinically used β3-receptor agonist mirabegron markedly accelerates atherosclerotic plaque formation in Apoe−/− and Ldlr−/− mice as related to accelerated LPL-mediated triglyceride-derived fatty acid uptake by thermogenic adipose tissue, based on which the authors raised concerns about its use in patients (103). However, this effect is likely an artifact of the mouse models used as increased delipidation of triglyceride-rich lipoproteins leads to an accelerated formation of lipoprotein remnants, which are only pro-atherogenic when not cleared from the circulation. As anticipated, we therefore more recently found that mirabegron does not accelerate atherosclerosis development in APOE*3-Leiden.CETP mice, due to effective removal of VLDL remnants via the intact ApoE–LDLR pathway (104). Nonetheless, since thermogenic adipose tissue in humans primarily expresses the β2-adrenergic receptor rather than the β3-adrenergic receptor as in mice (105), mirabegron does not activate human thermogenic adipose tissue at the therapeutic dose of 50 mg (105), and 12 weeks of daily dosing therefore does not affect circulating cholesterol levels in humans (106).
The APOE*3-Leiden.CETP model thus appears to be a relevant model for evaluating the effects of modulation of lipid metabolism on atherosclerosis development. It is particularly relevant for the evaluation of compounds within the pipeline of pharmaceutical companies that are expected to directly or indirectly stimulate LPL activity.
Emerging models in atherosclerosis research
In the previous sections we have highlighted the most frequently used mouse models in atherosclerosis research, while there are many other available mouse models, for instance mice expressing both human APOB and CETP (107). Emerging genetic techniques furthermore allow for specific targeting of atherosclerosis-related genes without the need of germline alterations (108, 109). For instance, adeno-associated viruses (AAVs) have been used to overexpress PCSK9 in the liver and thereby stimulate hepatic LDLR degradation in mice (108), while others have exploited antisense oligonucleotides (ASOs) to directly reduce hepatic Ldlr expression (110). Other promising techniques, such as a vaccine against PCSK9, directly target atherosclerosis-related proteins by inducing an immune response. In APOE*3-Leiden.CETP mice, vaccination against PCSK9, resulted in a reduction of plasma lipids levels, lowered systemic and vascular inflammation, and less atherosclerotic lesions in the aorta (111).
While mice have been an essential tool in understanding the molecular mechanisms behind atherosclerotic plaque formation and progression, as well as in identifying novel treatment modalities, gene manipulation is required to induce atherosclerosis, and the absence of rupture events and sexual dimorphisms may limit their translational value. Other animal models are also employed to study atherosclerosis, such as zebrafish. Compared to mice, zebrafish have an even smaller body size, develop more rapidly, and produce a large number of offspring, allowing for affordable large-scale drug-screening experiments. This is why zebrafish have recently become a popular species in research, also in the field of atherosclerosis research, as reviewed by Tang et al. (112) However, while these models seem to work well to study the role of specific proteins in lipoprotein metabolism and can be used for drug screening, so far, atherosclerosis development seems to be limited to early lesions (113).
Lastly, recent advances have been made to model heart diseases in vitro, including the use of human organoids (i.e. heart-on-a-chip), and 3D-cell printing techniques (114, 115). These in vitro techniques are ideal for drug testing and determining therapies in personalized medicine approaches while simultaneously avoiding the use of animals in research. Moreover, they have already been employed to study more complex disease modalities such as arrhythmia (116), cardiac fibrosis (117), myocardial infarction, and ischemia and reperfusion injury (114, 118). However, they have not yet been used to study atherosclerosis development, as it remains difficult to model the interaction between metabolic organs, such as the adipose tissue and the liver, on atherosclerosis development, especially given that such metabolic organs are under neuronal and hormonal control.
Atherosclerosis quantification in the aortic root area of mouse hearts
Naturally, choosing the right mouse model is the first step in designing an experiment to study atherosclerosis. Equally important, however, is the type of quantification and analysis of atherosclerosis development, which can offer technical challenges. In the following sections, we will describe in detail key steps and considerations when collecting, processing, and analyzing atherosclerotic lesions within the aortic root area of mice. This includes a detailed method for the quantification of lesion size and composition as established in our laboratory. In addition, we provide a method for scoring atherosclerotic lesion severity based on guidelines of the American Heart Association adapted for mice.
Sample preparation
The first step in the quantification of atherosclerotic lesions is collecting and preparing samples. Before heart collection, hearts may be perfused with ice-cold phosphate buffered saline to remove any remaining blood cells from the heart. It is not advisable to use cervical dislocation for euthanasia as this would disturb the aortic root morphology. This is even more relevant when the aorta itself is also collected, for example, for en face atherosclerosis analysis within the aorta itself.
The collected hearts can be processed using standard methods for paraffin embedding, that is, the samples are fixed in a 4% paraformaldehyde for a duration of 24–48 h and subsequently dehydrated in 70% ethanol. The hearts can then be cut using a regular razor blade along the axis perpendicular to the aorta (Fig. 3A), after which the heart tissue containing the aortic root area (i.e. the top one-third) is embedded in paraffin, with the cutting surface positioned on the base of the mold. This ensures that all three (occasionally only two due to a genetic irregularity) aortic valve leaflets will be visible within the same histological cross sections.
Figure 3.

Schematic overview of the process of quantifying atherosclerotic lesion size within the aortic root area of the heart. (A) Collected hearts are cut along an axis perpendicular to the aorta, and are subsequently paraffin-embedded. (B) 120 serial cross sections (5 µm thickness) through the aortic root area are divided over 10 glass slides as depicted. (C) In this example, glass slide #2 from B was stained using hematoxylin–phloxine–saffron, and images of cross sections 12, 22, 32, 42, and 52 are shown. The three aortic valve regions are (arbitrarily) numbered; arrows depict the opening of aortic valve leaflets. In this example as explained in the main text, we would quantify atherosclerotic lesion size in cross sections 12, 22, 32, and 42 for aortic valve region #1, labeled 1a–d, and in cross section 22, 32, 42, and 52 for regions #2 and #3, labeled 2a–d and 3a–d, respectively. Combined with data of the other mice, (D) the absolute atherosclerotic lesion area can subsequently be plotted as a function of the distance from open aortic valves, from which (E) the average atherosclerotic lesion area can be derived. Data depicted in D and E are semi-randomly generated and serve as an example only.
In a recent statement, the American Heart Association recommended serial cross-sectioning of the aortic root area from the origin of the aortic valves to the ascending aorta (119). Although no further standard criteria are available, we typically prepare cross sections at a thickness of 5 µm throughout the aortic valve region. Next, starting with a cross section located roughly 50 µm before the appearance of the first open aortic valve leaflet, we recommend dividing 120 serial cross sections over ten separate glass slides in an arrangement where each adjacent cross section on a glass slide differs by 50 µm (Fig. 3B). This arrangement ensures that corresponding cross sections on adjacent glass slides differ by only 5 µm, allowing for a direct comparison of separate glass slides (e.g. to compare different stainings).
Quantification of atherosclerotic lesion size
After deparaffination, atherosclerotic lesions can be visualized on one glass slide of each mouse using a general staining, such as hematoxylin–phloxine–saffron. To keep atherosclerotic lesion quantification consistent, many research groups use the aortic valve leaflets as a landmark. To this end, the cross section at which that valve leaflet opens is identified. We do this for each aortic valve leaflet separately. Although the number of sections that are analyzed varies between research groups, we typically quantify the lesion sizes within the corresponding valve region in that cross section, and in the subsequent three cross sections, meaning that we are covering a total distance of 150 µm (Fig. 3C). The quantification itself can be done by manually drawing a region of interest around each lesion within each aortic valve region using open-source software such as ImageJ (National Institutes of Health), and determining the absolute lesion area in µm². Please note that in this process of manually delineating regions of interest, researchers should be blinded where possible, and images should be analyzed in a random order to minimize bias.
With the acquired data, one is able to express the absolute atherosclerotic lesion area as a function of the distance of the open aortic valves (i.e. the total lesion area of all aortic root areas combined at 0 µm, 50 µm, 100 µm, and 150 µm distance of open aortic valves; Fig. 3D). From this, the average atherosclerotic lesion area can be calculated by averaging the lesion area at the four distances (Fig. 3E).
Determining atherosclerotic lesion composition
Given the complexity and heterogeneity of atherosclerotic plaque, insight into compositional changes between groups provides additional insights to solely the lesion size. Atherosclerotic lesion composition (e.g. smooth muscle cell, collagen, and macrophage area %) can be determined using similar methods as described for the hematoxylin–phloxine–saffron staining. As mentioned before, ideally, glass slides adjacent to the general staining are used for such specific stainings as this allows for the most direct comparison with the general staining. After staining and digitalizing a glass slide, one can similarly manually draw regions of interest around the atherosclerotic lesions, again in four cross sections for each aortic valve region separately, starting at the opening of the corresponding aortic valve leaflet. An alternative approach may be to align images of each cross section to images of the corresponding cross section of the general staining, for example using OpenCV for Python (120). This would allow for the re-usage of the regions of interest that were delineated for the general staining with only minimal changes needed, which would save valuable time required for manually delineating entirely new regions of interest. We have recently published a Python script that uses OpenCV for such alignment that can be used for this purpose (121). Whichever method was used to acquire regions of interest around the atherosclerotic lesions for the specific staining, one can next determine the positively stained area within the atherosclerotic lesions, for example by manual or preferably automated color thresholding using software such as ImageJ. Subsequently, the positively stained area % can be obtained by dividing the total positively stained area by the total lesion area. These data may additionally be used to calculate a stability index as a measure for morphological plaque vulnerability. Such an index can be obtained per plaque by dividing the sum of smooth muscle cell area and collagen area by the macrophage area (91, 122).
Naturally, atherosclerotic lesion composition is highly dependent on natural compositional discrepancies between mild and severe lesions. When lesion size differs between treatment groups, it can therefore be insightful to first score atherosclerotic lesion severity, as described in the next section, and to stratify the lesion composition based on the severity. We recommend comparing the atherosclerotic lesion composition and stability index between groups within the compositionally heterogeneous type III lesions only.
Scoring atherosclerosis severity
In mice, atherosclerotic lesions can be categorized as mild lesions (type I–III) or severe lesions (type IV–V) based on the guidelines of the American Heart Association adapted for mice as originally described by Wong et al. (123). This provides insight into whether a treatment improves overall lesion severity, as related to, or independent from changes in atherosclerotic lesion size or composition. Here, the same hematoxylin–phloxine–saffron-stained lesions can be scored as were used for the quantification of atherosclerotic lesion size.
Mild lesions are early fatty streak-like lesions, characterized by the presence of foam cells within the intima. Here, type I and II lesions are defined as having a maximum or more than ten foam cells per cross section, respectively. Lesions are categorized as type III when foam cells are also localized within the media, and/or the lesion is covered by a fibrotic cap. In contrast, severe lesions are more progressive lesions. Specifically, type IV lesions have infiltrated into the media and are accompanied by fibrosis, but the overall architecture remains intact. However, in type V lesions, the media is severely damaged. In addition, elastic lamina are broken, and cholesterol crystals, mineralization, and/or necrosis can be present. Figure 4 depicts representative images of lesions categorized according to these criteria.
Figure 4.
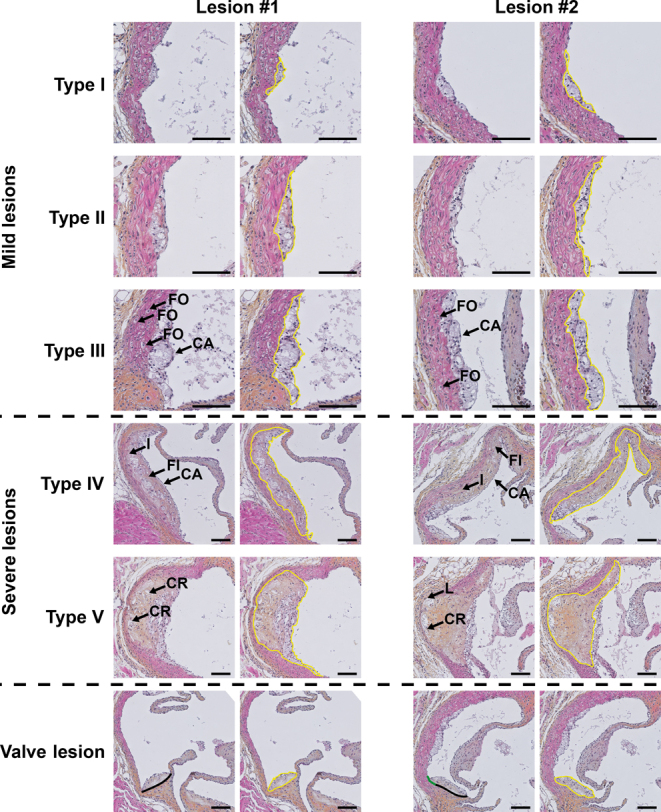
Representative images of aortic valve regions stained with hematoxylin–phloxine–saffron, depicting lesions classified in different severities. Atherosclerotic lesions are categorized into mild lesions (type I–III) and severe lesions (type IV–V). In addition, lesions can be categorized as ‘valve lesions’. Of each lesion type, two lesions are shown in duplicate; yellow lines indicate the circumference of each lesion. Type I, early fatty streak: a maximum of ten foam cells are visible within the intima per cross section. Type II, regular fatty streak: more than ten foam cells are visible within the intima per cross section. Type III, mild plaque: foam cells extend into the media (indicated with ‘FO’) and/or the lesion is covered by a fibrotic cap (indicated with ‘CA’). Type IV, moderate plaque: a progressively advancing lesion that infiltrates into the media (indicated with ‘I’), and is accompanied by fibrosis (indicated with ‘FI’) without architectural loss. Type V, severe plaque: the media is severely damaged. Elastic lamina are broken (indicated with ‘L’), and cholesterol crystals (indicated with ‘CR’), mineralization and/or necrosis can be present. Valve lesion, categorized as such when >50% of the base is located on the root of an aortic valve; black lines indicate (the part of) the base of the valve that is located on the aortic valve root, green lines indicate the remainder of the base of the valve, if any. Scale bars indicate 200 µm.
When scoring lesions according to abovementioned criteria, an additional class of lesions, so-called valve lesions, can be identified. Valve lesions are defined as having >50% of their base positioned on the root of a valve, as Fig. 4 exemplifies. It is relevant to distinguish valve lesions from other lesions when scoring atherosclerosis severity, as the morphology and development of these valve lesions usually differ from the other lesions. For that reason, we recommend treating valve lesions as a distinct group instead of scoring them as type I–V. However, it is important to note that we do not treat valve lesions differently in the quantification of atherosclerotic lesion size. Lastly, for each aortic valve region, the number of diseased-free cross sections (i.e. cross sections without any visible lesion or valve lesions) can be counted and reported.
Concluding remarks
Taken together, asCVD is caused by an interplay between lipids and inflammation. Given the significant burden that atherosclerosis forms on society, multiple animal models have been developed to study atherosclerosis development and to find novel therapeutic strategies to prevent the pathology. In this review, we have discussed the advantages and disadvantages of three most popular models. What should be clear is that it is of critical importance to choose the correct mouse model for correct interpretation of experiments. For example, the presence of the APOE–LDLR clearance pathway in mice is crucial to study the effects of cholesterol- and triglyceride-lowering medication on atherosclerosis development. Using Apoe-/- or Ldlr-/- mice to evaluate lipid-lowering strategies can therefore yield results that are opposite of what can be expected in humans. However, both models are very suitable to study inflammation in atherosclerosis development, while the APOE*3-Leiden.CETP model seems most suited to study the lipid-modulating effects of interventions on atherosclerosis development. Given the rapid development of novel genetic techniques and the emergence of zebrafish as experimental units, we anticipate large steps can be made in the field of atherosclerosis research in the coming years. While a suitable animal model to study atherosclerosis is essential, the quantification and analysis method is of equal importance. In this review, we have therefore provided a comprehensive description of all steps required to quantify atherosclerosis development within the aortic root region of the heart of mice, including a detailed description for scoring atherosclerosis severity based on the guidelines of the American Heart Association adapted for mice. This description may help standardize the way results of atherosclerosis experiments are presented in the literature.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the study reported.
Funding
This work was supported by the Royal Netherlands Academy of Sciences (CVON-GENIUS-II) to PCNR. SK is supported by the Dutch Heart Foundation (2017T016).
Authors contribution statement
JIVDV and RVE: writing – original draft; RVE, SK and PCNR: writing – review and editing, supervision, funding acquisition.
Acknowledgements
Figures were created with BioRender.com.
References
- 1.Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, Barengo NC, Beaton AZ, Benjamin EJ, Benziger CP, et al.Global burden of cardiovascular diseases and risk factors, 1990–2019: update from the GBD 2019 study. Journal of the American College of Cardiology 2020762982–3021. ( 10.1016/j.jacc.2020.11.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Panhuis WIH Kooijman S Brouwers B Verhoeven A Pronk ACM Streefland TCM Giera M Schrauwen P Rensen PCN & Schönke M. Mild exercise does not prevent atherosclerosis in APOE*3-Leiden.CETP mice or improve lipoprotein profile of men with obesity. Obesity 202028S93–S103. ( 10.1002/oby.22799) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Diepen JA Berbee JFP Havekes LM & Rensen PCN. Interac tions between inflammation and lipid metabolism: relevance for efficacy of anti-inflammatory drugs in the treatment of atherosclerosis. Atherosclerosis 2013228306–315. ( 10.1016/j.atherosclerosis.2013.02.028) [DOI] [PubMed] [Google Scholar]
- 4.Boren J, Chapman MJ, Krauss RM, Packard CJ, Bentzon JF, Binder CJ, Daemen MJ, Demer LL, Hegele RA, Nicholls SJ, et al.Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. European Heart Journal 2020412313–2330. ( 10.1093/eurheartj/ehz962) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Libby P. Inflammation in atherosclerosis. Nature 2002420868–874. ( 10.1038/nature01323) [DOI] [PubMed] [Google Scholar]
- 6.Feingold KR. Lipid and lipoprotein metabolism. Endocrinology and Metabolism Clinics of North America 202251437–458. ( 10.1016/j.ecl.2022.02.008) [DOI] [PubMed] [Google Scholar]
- 7.Shi YG & Cheng D. Beyond triglyceride synthesis: the dynamic functional roles of MGAT and DGAT enzymes in energy metabolism. American Journal of Physiology-Endocrinology and Metabolism 2009297E10–E18. ( 10.1152/ajpendo.90949.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lo CC & Coschigano KT. ApoB48 as an efficient regulator of intestinal lipid transport. Frontiers in Physiology 202011. ( 10.3389/fphys.2020.00796) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hooper AJ Burnett JR & Watts GF. Contemporary aspects of the biology and therapeutic regulation of the microsomal triglyceride transfer protein. Circulation Research 2015116193–205. ( 10.1161/CIRCRESAHA.116.304637) [DOI] [PubMed] [Google Scholar]
- 10.Larosa JC Levy RI Herbert P Lux SE & Fredrickson DS. A specific apoprotein activator for lipoprotein lipase. Biochemical and Biophysical Research Communications 19704157–62. ( 10.1016/0006-291X(7090468-7) [DOI] [PubMed] [Google Scholar]
- 11.Tiwari S & Siddiqi SA. Intracellular trafficking and secretion of VLDL. Arteriosclerosis, Thrombosis, and Vascular Biology 2012321079–1086. ( 10.1161/ATVBAHA.111.241471) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boon MR, Rensen PCN. Chapter 11: Vetten. Leerboek Voeding 1, pp. 145–159.Bohn Stafleu van Loghum, Netherlands, eds. 2023. [Google Scholar]
- 13.Mabuchi H Nohara A & Inazu A. Cholesteryl ester transfer protein (CETP) deficiency and CETP inhibitors. Molecules and Cells 201437777–784. ( 10.14348/molcells.2014.0265) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tall AR. Plasma cholesteryl ester transfer protein. Journal of Lipid Research 1993341255–1274. ( 10.1016/S0022-2275(2036957-1) [DOI] [PubMed] [Google Scholar]
- 15.Ouimet M Barrett TJ & Fisher EA. HDL and reverse cholesterol transport basic mechanisms and their roles in vascular health and disease. Circulation Research 20191241505–1518. ( 10.1161/CIRCRESAHA.119.312617) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu LQ Li-Hawkins J Hammer RE Berge KE Horton JD Cohen JC & Hobbs HH. Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol. Journal of Clinical Investigation 2002110671–680. ( 10.1172/JCI0216001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li TG & Chiang JYL. Bile acid signaling in metabolic disease and drug therapy. Pharmacological Reviews 201466948–983. ( 10.1124/pr.113.008201) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johansen MØ Vedel-Krogh S Nielsen SF Afzal S Smith GD & Nordestgaard BG. Per particle triglyceride-rich lipoproteins imply higher myocardial infarction risk than low-density lipoproteins: Copenhagen general population study. Atherosclerosis 2021331e39–e40. ( 10.1016/j.atherosclerosis.2021.06.113) [DOI] [PubMed] [Google Scholar]
- 19.Huang LZ, Chambliss KL, Gao XF, Yuhanna IS, Behling-Kelly E, Bergaya S, Ahmed M, Michaely P, Luby-Phelps K, Darehshouri A, et al.SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature 2019569565–569. ( 10.1038/s41586-019-1140-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stocker R & Keaney JF. Role of oxidative modifications in atherosclerosis. Physiological Reviews 2004841381–1478. ( 10.1152/physrev.00047.2003) [DOI] [PubMed] [Google Scholar]
- 21.Camejo G Fager G Rosengren B Hurtcamejo E & Bondjers G. Binding of low-density lipoproteins by proteoglycans synthesized by proliferating and quiescent human arterial smooth-muscle cells. Journal of Biological Chemistry 199326814131–14137. ( 10.1016/S0021-9258(1985218-3) [DOI] [PubMed] [Google Scholar]
- 22.Ruuth M, Nguyen SD, Vihervaara T, Hilvo M, Laajala TD, Kondadi PK, Gisterå A, Lähteenmäki H, Kittilä T, Huusko J, et al.Susceptibility of low-density lipoprotein particles to aggregate depends on particle lipidome, ismodifiable, and associates with future cardiovascular deaths. European Heart Journal 2018392562–2573. ( 10.1093/eurheartj/ehy319) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hansson GK & Hermansson A. The immune system in atherosclerosis. Nature Immunology 201112204–212. ( 10.1038/ni.2001) [DOI] [PubMed] [Google Scholar]
- 24.Smith JD Trogan E Ginsberg M Grigaux C Tian J & Miyata M. Decreased atherosclerosis in mice deficient in both macrophage-colony-stimulating factor (op) and apolipoprotein-E. PNAS 1995928264–8268. ( 10.1073/pnas.92.18.8264) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pasceri V Willerson JT & Yeh ETH. Direct proinflammatory effect of C-reactive protein on human endothelial cells. Circulation 20001022165–2168. ( 10.1161/01.CIR.102.18.2165) [DOI] [PubMed] [Google Scholar]
- 26.Anand IS, Latini R, Florea VG, Kuskowski MA, Rector T, Masson S, Signorini S, Mocarelli P, Hester A, Glazer R, et al.C-reactive protein in heart failure: prognostic value and the effect of valsartan. Circulation 20051121428–1434. ( 10.1161/CIRCULATIONAHA.104.508465) [DOI] [PubMed] [Google Scholar]
- 27.Ridker PM Rifai N Rose L Buring JE & Cook NR. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. New England Journal of Medicine 20023471557–1565. ( 10.1056/NEJMoa021993) [DOI] [PubMed] [Google Scholar]
- 28.Owsiany KM Alencar GF & Owens GK. Revealing the origins of foam cells in atherosclerotic lesions. Arteriosclerosis, Thrombosis, and Vascular Biology 201939836–838. ( 10.1161/ATVBAHA.119.312557) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lehti S, Nguyen SD, Belevich I, Vihinen H, Heikkila HM, Soliymani R, Käkelä R, Saksi J, Jauhiainen M, Grabowski GA, et al.Extracellular lipids accumulate in human carotid arteries as distinct three-dimensional structures and have proinflammatory properties. American Journal of Pathology 2018188525–538. ( 10.1016/j.ajpath.2017.09.019) [DOI] [PubMed] [Google Scholar]
- 30.Maxfield FR Steinfeld N & Ma CJ. The formation and consequences of cholesterol-rich deposits in atherosclerotic lesions. Frontiers in Cardiovascular Medicine 202310. ( 10.3389/fcvm.2023.1148304) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Varsano N, Beghi F, Elad N, Pereiro E, Dadosh T, Pinkas I, Perez-Berna AJ, Jin X, Kruth HS, Leiserowitz L, et al.Two polymorphic cholesterol monohydrate crystal structures form in macrophage culture models of atherosclerosis. PNAS 20181157662–7669. ( 10.1073/pnas.1803119115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yurdagul A Doran AC Cai BS Fredman G & Tabas IA. Mechanisms and Consequences of Defective efferocytosis in Atherosclerosis. Frontiers in Cardiovascular Medicine 20184. ( 10.3389/fcvm.2017.00086) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakakura K Nakano M Otsuka F Ladich E Kolodgie FD & Virmani R. Pathophysiology of atherosclerosis plaque progression. Heart Lung and Circulation 201423387–. ( 10.1016/j.hlc.2013.12.006) [DOI] [PubMed] [Google Scholar]
- 34.Dollery CM & Libby P. Atherosclerosis and proteinase activation. Cardiovascular Research 200669625–635. ( 10.1016/j.cardiores.2005.11.003) [DOI] [PubMed] [Google Scholar]
- 35.Sukhova GK Schonbeck U Rabkin E Schoen FJ Poole AR Billinghurst RC & Libby P. Evidence for increased collagenolysis by interstitial collagenases-1 and-3 in vulnerable human atheromatous plaques. Circulation 1999992503–2509. ( 10.1161/01.CIR.99.19.2503) [DOI] [PubMed] [Google Scholar]
- 36.Jia H, Abtahian F, Aguirre AD, Lee S, Chia S, Lowe H, Kato K, Yonetsu T, Vergallo R, Hu S, et al.In vivo diagnosis of plaque erosion and calcified nodule in patients with acute coronary syndrome by intravascular optical coherence tomography. Journal of the American College of Cardiology 2013621748–1758. ( 10.1016/j.jacc.2013.05.071) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Virmani R Burke AP Farb A & Kolodgie FD. Pathology of the vulnerable plaque. Journal of the American College of Cardiology 200647 (8) C13–C18. ( 10.1016/j.jacc.2005.10.065) [DOI] [PubMed] [Google Scholar]
- 38.Ignatowski A. Über die Wirkung des tierischen Eiweißes auf die aorta und die parenchymatösen Organe der Kaninchen. Virchows Archiv für Pathologische Anatomie und Physiologie und für Klinische Medizin 1909198248–270. ( 10.1007/BF01949591) [DOI] [Google Scholar]
- 39.Anitschkov NNC. S. Ueber experimentelle Cholesterinsteatose und ihre Bedeutung fuer die Entstehung einiger pathologischer Prozesse. Zentrbl Allg Pathol Pathol Anat 1913241–9. [Google Scholar]
- 40.Konstantinov IE Mejevoi N Anichkov NM & Nikolai N. Nikolai N. Anichkov and his theory of atherosclerosis. Texas Heart Institute Journal 200633417–423. [PMC free article] [PubMed] [Google Scholar]
- 41.Macheboeuf MA. Rec herches sur les phosphoaminolipides et les sterides du serum et du plasma sanguins. Bulletin de la Société de Chimie Biologique 192911268–293. [Google Scholar]
- 42.Gofman JW & Lindgren F. The role of lipids and lipoproteins in atherosclerosis. Science 1950111166–171. ( 10.1126/science.111.2877.166) [DOI] [PubMed] [Google Scholar]
- 43.Lindgren FT Elliott HA & Gofman JW. The ultracentrifugal characterization and isolation of human blood lipids and lipoproteins, with applications to the study of atherosclerosis. Journal of Physical Chemistry 19515580–93. ( 10.1021/j150484a010) [DOI] [PubMed] [Google Scholar]
- 44.Camus MC Chapman MJ Forgez P & Laplaud PM. Distribution and characterization of the serum-lipoproteins and apoproteins in the mouse, Mus-Musculus. Journal of Lipid Research 1983241210–1228. ( 10.1016/S0022-2275(2037904-9) [DOI] [PubMed] [Google Scholar]
- 45.Ilyas I Little PJ Liu Z Xu Y Kamato D Berk BC Weng J & Xu S. Mouse models of atherosclerosis in translational research. Trends in Pharmacological Sciences 202243920–939. ( 10.1016/j.tips.2022.06.009) [DOI] [PubMed] [Google Scholar]
- 46.Veseli BE Perrotta P De Meyer GRA Roth L Van der Donckt C Martinet W & De Meyer GRY. Animal models of atherosclerosis. European Journal of Pharmacology 20178163–13. ( 10.1016/j.ejphar.2017.05.010) [DOI] [PubMed] [Google Scholar]
- 47.Paigen B Morrow A Brandon C Mitchell D & Holmes P. Variation in susceptibility to atherosclerosis among inbred strains of mice. Atherosclerosis 19855765–73. ( 10.1016/0021-9150(8590138-8) [DOI] [PubMed] [Google Scholar]
- 48.Bennett BJ, Davis RC, Civelek M, Orozco L, Wu J, Qi H, Pan C, Packard RRS, Eskin E, Yan M, et al.Genetic architecture of atherosclerosis in mice: a systems genetics analysis of common inbred strains. PLoS Genetics 201511. ( 10.1371/journal.pgen.1005711) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith JD James D Dansky HM Wittkowski KM Moore KJ & Breslow JL. In silico quantitative trait locus map for atherosclerosis susceptibility in apolipoprotein E-deficient mice. Arteriosclerosis, Thrombosis, and Vascular Biology 200323117–122. ( 10.1161/01.ATV.0000047461.18902.80) [DOI] [PubMed] [Google Scholar]
- 50.Srivastava U Paigen BJ & Korstanje R. Differences in health status affect susceptibility and mapping of genetic loci for atherosclerosis (fatty streak) in inbred mice. Arteriosclerosis and Thrombosis Vas 2012322380–2386. ( 10.1161/ATVBAHA.112.255703) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Plump AS Smith JD Hayek T Aaltosetala K Walsh A Verstuyft JG Rubin EM & Breslow JL. Severe hypercholesterolemia and atherosclerosis in apolipoprotein-E-deficient mice created by homologous recombination in es cells. Cell 199271343–353. ( 10.1016/0092-8674(9290362-G) [DOI] [PubMed] [Google Scholar]
- 52.Alagarsamy J Jaeschke A & Hui DY. Apolipoprotein E in cardiometabolic and neurological health and diseases. International Journal of Molecular Sciences 202223. ( 10.3390/ijms23179892) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mineo C. Lipoprotein receptor signalling in atherosclerosis. Cardiovascular Research 20201161254–1274. ( 10.1093/cvr/cvz338) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Plump AS & Breslow JL. Apolipoprotein-E and the apolipoprotein E-deficient mouse. Annual Review of Nutrition 199515495–518. ( 10.1146/annurev.nu.15.070195.002431) [DOI] [PubMed] [Google Scholar]
- 55.Reddick RL Zhang SH & Maeda N. Atherosclerosis in mice lacking apo-E - evaluation of lesional development and progression (vol 14, Pg 141, 1994). Arteriosclerosis and Thrombosis 199414839. ( 10.1161/01.ATV.14.1.141) [DOI] [PubMed] [Google Scholar]
- 56.Coleman R Hayek T Keidar S & Aviram M. A mouse model for human atherosclerosis: long-term histopathological study of lesion development in the aortic arch of apolipoprotein E-deficient (E-0) mice. Acta Histochemica 2006108415–424. ( 10.1016/j.acthis.2006.07.002) [DOI] [PubMed] [Google Scholar]
- 57.Nakashima Y Plump AS Raines EW Breslow JL & Ross R. Apoe-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arteriosclerosis and Thrombosis 199414133–140. ( 10.1161/01.ATV.14.1.133) [DOI] [PubMed] [Google Scholar]
- 58.Kayashima Y & Maeda-Smithies N. Atherosclerosis in different vascular locations unbiasedly approached with mouse genetics. Genes 202011. ( 10.3390/genes11121427) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Caligiuri G Nicoletti A Zhou X Tornberg I & Hansson GK. Effects of sex and age on atherosclerosis and autoimmunity in apoE-deficient mice. Atherosclerosis 1999145301–308. ( 10.1016/S0021-9150(9900081-7) [DOI] [PubMed] [Google Scholar]
- 60.Meyrelles SS Peotta VA Pereira TM & Vasquez EC. Endothelial dysfunction in the apolipoprotein E-deficient mouse: insights into the influence of diet, gender and aging. Lipids in Health and Disease 201110211. ( 10.1186/1476-511X-10-211) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chiba T Ikeda M Umegaki K & Tomita T. Estrogen-dependent activation of neutral cholesterol ester hydrolase underlying gender difference of atherogenesis in apoE-/- mice. Atherosclerosis 2011219545–551. ( 10.1016/j.atherosclerosis.2011.08.051) [DOI] [PubMed] [Google Scholar]
- 62.Kirii H Niwa T Yamada Y Wada H Saito K Iwakura Y Asano M Moriwaki H & Seishima M. Lack of interleukin-1 beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arteriosclerosis, Thrombosis, and Vascular Biology 200323656–660. ( 10.1161/01.ATV.0000064374.15232.C3) [DOI] [PubMed] [Google Scholar]
- 63.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al.Antiinflammatory therapy with canakinumab for atherosclerotic disease. New England Journal of Medicine 20173771119–1131. ( 10.1056/NEJMoa1707914) [DOI] [PubMed] [Google Scholar]
- 64.Murphy AJ, Akhtari M, Tolani S, Pagler T, Bijl N, Kuo CL, Wang M, Sanson M, Abramowicz S, Welch C, et al.ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. Journal of Clinical Investigation 20111214138–4149. ( 10.1172/JCI57559) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ali K Middleton M Pure E & Rader DJ. Apolipoprotein E suppresses the type I inflammatory response in vivo. Circulation Research 200597922–927. ( 10.1161/01.RES.0000187467.67684.43) [DOI] [PubMed] [Google Scholar]
- 66.Langer C Huang Y Cullen P Wiesenhutter B Mahley RW Assmann G & von Eckardstein A. Endogenous apolipoprotein E modulates cholesterol efflux and cholesteryl ester hydrolysis mediated by high-density lipoprotein-3 and lipid-free apolipoproteins in mouse peritoneal macrophages. Journal of Molecular Medicine 200078217–227. ( 10.1007/s001090000096) [DOI] [PubMed] [Google Scholar]
- 67.Zanotti I Pedrelli M Poti F Stomeo G Gomaraschi M Calabresi L & Bernini F. Macrophage, but not systemic, apolipoprotein E is necessary for macrophage reverse cholesterol transport in vivo. Arteriosclerosis, Thrombosis, and Vascular Biology 20113174–80. ( 10.1161/ATVBAHA.110.213892) [DOI] [PubMed] [Google Scholar]
- 68.Brown MS & Goldstein JL. Familial hypercholesterolemia - defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase-activity. PNAS 197471788–792. ( 10.1073/pnas.71.3.788) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ishibashi S Brown MS Goldstein JL Gerard RD Hammer RE & Herz J. Hypercholesterolemia in low-density-lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. Journal of Clinical Investigation 199392883–893. ( 10.1172/JCI116663) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ishibashi S Goldstein JL Brown MS Herz J & Burns DK. Massive xanthomatosis and atherosclerosis in cholesterol-fed low-density-lipoprotein receptor-negative mice. Journal of Clinical Investigation 1994931885–1893. ( 10.1172/JCI117179) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Golforoush P Yellon DM & Davidson SM. Mouse models of atherosclerosis and their suitability for the study of myocardial infarction. Basic Research in Cardiology 2020115. ( 10.1007/s00395-020-00829-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Knowles JW & Maeda N. Genetic modifiers of atherosclerosis in mice. Arteriosclerosis, Thrombosis, and Vascular Biology 2000202336–2345. ( 10.1161/01.ATV.20.11.2336) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Robinet P Milewicz DM Cassis LA Leeper NJ Lu HS & Smith JD. Consideration of sex differences in design and reporting of experimental arterial pathology studies-statement from ATVB council. Arteriosclerosis, Thrombosis, and Vascular Biology 201838292–303. ( 10.1161/ATVBAHA.117.309524) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Teupser D Persky AD & Breslow JL. Induction of atherosclerosis by low-fat, semisynthetic diets in LDL receptor-deficient C57BL/6J and FVB/NJ mice: comparison of lesions of the aortic root, brachiocephalic artery, and whole aorta (en face measurement). Arteriosclerosis, Thrombosis, and Vascular Biology 2003231907–1913. ( 10.1161/01.ATV.0000090126.34881.B1) [DOI] [PubMed] [Google Scholar]
- 75.Smit V, de Mol J, Schaftenaar FH, Depuydt MAC, Postel RJ, Smeets D, Verheijen FWM, Bogers L, van Duijn J, Verwilligen RAF, et al.Single-cell profiling reveals age-associated immunity in atherosclerosis. Cardiovascular Research 20231192508–2521. ( 10.1093/cvr/cvad099) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang SH Reddick RL Piedrahita JA & Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein-E. Science 1992258468–471. ( 10.1126/science.1411543) [DOI] [PubMed] [Google Scholar]
- 77.Getz GS & Reardon CA. Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. Journal of Lipid Research 200950S156–S161. ( 10.1194/jlr.R800058-JLR200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ason B, van der Hoorn JWA, Chan J, Lee E, Pieterman EJ, Nguyen KK, Di M, Shetterly S, Tang J, Yeh W, et al.PCSK9 inhibition fails to alter hepatic LDLR, circulating cholesterol, and atherosclerosis in the absence of ApoE. Journal of Lipid Research 2014552370–2379. ( 10.1194/jlr.M053207) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zadelaar S Kleemann R Verschuren L de Vries-Van der Weij J van der Hoorn J Princen HM & Kooistra T. Mouse models for atherosclerosis and pharmaceutical modifiers. Arteriosclerosis, Thrombosis, and Vascular Biology 2007271706–1721. ( 10.1161/ATVBAHA.107.142570) [DOI] [PubMed] [Google Scholar]
- 80.Sundararaman SS Doring Y & van Der Vorst EPC. PCSK9: a multi-faceted protein that is involved in cardiovascular biology. Biomedicines 20219. ( 10.3390/biomedicines9070793) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Deknijff P Vandenmaagdenberg AMJM Stalenhoef AFH Leuven JAG Demacker PNM Kuyt LP Frants RR & Havekes LM. Familial dysbetalipoproteinemia associated with apolipoprotein E3-Leiden in an extended multigeneration pedigree. Journal of Clinical Investigation 199188643–655. ( 10.1172/JCI115349) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vandenmaagdenberg AMJM Deknijff P Stalenhoef AFH Leuven JAG Havekes LM & Frants RR. Apolipoprotein E*3-Leiden allele results from a partial gene duplication in Exon-4. Biochem Bioph Res Co 1989165851–857. ( 10.1016/S0006-291X(8980044-0) [DOI] [PubMed] [Google Scholar]
- 83.van den Maagdenberg AM Hofker MH Krimpenfort PJ de Bruijn I van Vlijmen B van der Boom H Havekes LM & Frants RR. Transgenic mice carrying the apolipoprotein-E3-Leiden gene exhibit hyperlipoproteinemia. Journal of Biological Chemistry 199326810540–10545. ( 10.1016/S0021-9258(1882232-3) [DOI] [PubMed] [Google Scholar]
- 84.Westerterp M van der Hoogt CC de Haan W Offerman EH Dallinga-Thie GM Jukema JW Havekes LM & Rensen PCN. Cholesteryl ester transfer protein decreases high-density lipoprotein and severely aggravates atherosclerosis in APOE* 3-Leiden mice. Arteriosclerosis, Thrombosis, and Vascular Biology 2006262552–2559. ( 10.1161/01.ATV.0000243925.65265.3c) [DOI] [PubMed] [Google Scholar]
- 85.de Haan W, de Vries-van der Weij J, van der Hoorn JWA, Gautier T, van der Hoogt CC, Westerterp M, Romijn JA, Jukema JW, Havekes LM, Princen HMG, et al.Torcetrapib does not reduce atherosclerosis beyond atorvastatin and induces more proinflammatory lesions than atorvastatin. Circulation 20081172515–2522. ( 10.1161/CIRCULATIONAHA.107.761965) [DOI] [PubMed] [Google Scholar]
- 86.Bijland S Pieterman EJ Maas ACE van der Hoorn JWA van Erk MJ van Klinken JB Havekes LM van Dijk KW Princen HMG & Rensen PCN. Fenofibrate increases very low density lipoprotein triglyceride production despite reducing plasma triglyceride levels in APOE*3-Leiden.CETP Mice. Journal of Biological Chemistry 201028525168–25175. ( 10.1074/jbc.M110.123992) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kuhnast S, Louwe MC, Heemskerk MM, Pieterman EJ, van Klinken JB, van den Berg SAA, Smit JWA, Havekes LM, Rensen PCN, van der Hoorn JWA, et al.Niacin reduces atherosclerosis development in APOE*3Leiden.CETP Mice Mainly by Reducing NonHDL-Cholesterol. PLoS One 20138e66467. ( 10.1371/journal.pone.0066467) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pouwer MG Pieterman EJ Worms N Keijzer N Jukema JW Gromada J Gusarova V & Princen HMG. Alirocumab, evinacumab, and atorvastatin triple therapy regresses plaque lesions and improves lesion composition in mice. Journal of Lipid Research 202061365–375. ( 10.1194/jlr.RA119000419) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhou E Li Z Nakashima H Choukoud A Kooijman S Berbee JFP Rensen PCN & Wang Y. Beneficial effects of brown fat activation on top of PCSK9 inhibition with alirocumab on dyslipidemia and atherosclerosis development in APOE*3-Leiden.CETP mice. Pharmacological Research 2021167105524. ( 10.1016/j.phrs.2021.105524) [DOI] [PubMed] [Google Scholar]
- 90.Kuhnast S, van der Tuin SJL, van der Hoorn JWA, van Klinken JB, Simic B, Pieterman E, Havekes LM, Landmesser U, Luscher TF, Willems van Dijk K, et al.Anacetrapib reduces progression of atherosclerosis, mainly by reducing non-HDL-cholesterol, improves lesion stability and adds to the beneficial effects of atorvastatin. European Heart Journal 20153639–50. ( 10.1093/eurheartj/ehu319) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu C, Schonke M, Zhou EC, Li Z, Kooijman S, Boon MR, Larsson M, Wallenius K, Dekker N, Barlind L, et al.Pharmacological treatment with FGF21 strongly improves plasma cholesterol metabolism to reduce atherosclerosis. Cardiovascular Research 2022118489–502. ( 10.1093/cvr/cvab076) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang Y, Parlevliet ET, Geerling JJ, van der Tuin SJL, Zhang H, Bieghs V, Jawad AHM, Shiri-Sverdlov R, Bot I, de Jager SCA, et al.Exendin-4 decreases liver inflammation and atherosclerosis development simultaneously by reducing macrophage infiltration. British Journal of Pharmacology 2014171723–734. ( 10.1111/bph.12490) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.van Eenige R, Ying ZX, Tramper N, Wiebing V, Siraj Z, de Boer JF, Lambooij JM, Guigas B, Qu H, Coskun T, et al.Combined glucose-dependent insulinotropic polypeptide receptor and glucagon-like peptide-1 receptor agonism attenuates atherosclerosis severity in APOE*3-Leiden.CETP mice. Atherosclerosis 202337219–31. ( 10.1016/j.atherosclerosis.2023.03.016) [DOI] [PubMed] [Google Scholar]
- 94.Paalvast Y, Gerding A, Wang Y, Bloks VW, van Dijk TH, Havinga R, et al.Male apoE*3-Leiden.CETP Mice on High-Fat High-Cholesterol Diet Exhibit a Biphasic Dyslipidemic Response, Mimicking the Changes in Plasma Lipids Observed through Life in Men. Physiol Rep 20175e13376. ( 10.14814/phy2.13376) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tarasco E Pellegrini G Whiting L & Lutz TA. Phenotypical heterogeneity in responder and nonresponder male ApoE*3Leiden.CETP mice. American Journal of Physiology. Gastrointestinal and Liver Physiology 2018315G602–G617. ( 10.1152/ajpgi.00081.2018) [DOI] [PubMed] [Google Scholar]
- 96.Lutgens E Daemen M Kockx M Doevendans P Hofker M Havekes L Wellens H & de Muinck ED. Atherosclerosis in APOE*3-Leiden transgenic mice: from proliferative to atheromatous stage. Circulation 199999276–283. ( 10.1161/01.CIR.99.2.276) [DOI] [PubMed] [Google Scholar]
- 97.van Vlijmen BJM vant Hof HB Mol MJTM van der Boom H van der Zee A Frants RR Hofker MH & Havekes LM. Modulation of very low density lipoprotein production and clearance contributes to age- and gender-dependent hyperlipoproteinemia in apolipoprotein E3-Leiden transgenic mice. Journal of Clinical Investigation 1996971184–1192. ( 10.1172/JCI118532) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Casimiro I Stull ND Tersey SA & Mirmira RG. Phenotypic sexual dimorphism in response to dietary fat manipulation in C57BL/6J mice. Journal of Diabetes and its Complications 202135107795. ( 10.1016/j.jdiacomp.2020.107795) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.van Beek L van Klinken JB Pronk ACM van Dam AD Dirven E Rensen PCN Koning F Willems van Dijk K & van Harmelen V. The limited storage capacity of gonadal adipose tissue directs the development of metabolic disorders in male C57BL/6J mice. Diabetologia 2015581601–1609. ( 10.1007/s00125-015-3594-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu C, Schonke M, Spoorenberg B, Lambooij JM, van der Zande HJP, Zhou E, Tushuizen ME, Andreasson AC, Park A, Oldham S, et al.FGF21 protects against hepatic lipotoxicity and macrophage activation to attenuate fibrogenesis in nonalcoholic steatohepatitis. eLife 202312. ( 10.7554/eLife.83075) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ying Z, van Eenige R, Ge X, van Marwijk C, Lambooij JM, Guigas B, Giera M, de Boer JF, Coskun T, Qu H, et al.Combined GIP receptor and GLP1 receptor agonism attenuates NAFLD in male APOE *3-Leiden. EBiomedicine 202393104684. ( 10.1016/j.ebiom.2023.104684) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sarwar N Danesh J Eiriksdottir G Sigurdsson G Wareham N Bingham S Boekholdt SM Khaw KT & Gudnason V. Triglycerides and the risk of coronary heart disease - 10. Circulation 2007115450–458. ( 10.1161/CIRCULATIONAHA.106.637793) [DOI] [PubMed] [Google Scholar]
- 103.Sui WH, Li HS, Yang YL, Jing X, Xue F, Cheng J, Dong M, Zhang M, Pan H, Chen Y, et al.Bladder drug mirabegron exacerbates atherosclerosis through activation of brown fat-mediated lipolysis. PNAS 201911610937–10942. ( 10.1073/pnas.1901655116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ying Z van Eenige R Beerepoot R Boon MR Kloosterhuis NJ van de Sluis B Bartelt A Rensen PCN & Kooijman S. Mirabegron-induced brown fat activation does not exacerbate atherosclerosis in mice with a functional hepatic ApoE-LDLR pathway. Pharmacological Research 2023187106634. ( 10.1016/j.phrs.2022.106634) [DOI] [PubMed] [Google Scholar]
- 105.Blondin DP, Nielsen S, Kuipers EN, Severinsen MC, Jensen VH, Miard S, Jespersen NZ, Kooijman S, Boon MR, Fortin M, et al.Human brown adipocyte thermogenesis is driven by β2-AR stimulation. Cell Metabolism 202032287–300.e7. ( 10.1016/j.cmet.2020.07.005) [DOI] [PubMed] [Google Scholar]
- 106.Finlin BS, Memetimin H, Zhu B, Confides AL, Vekaria HJ, El Khouli RH, Johnson ZR, Westgate PM, Chen J, Morris AJ, et al.The β3-adrenergic receptor agonist mirabegron improves glucose homeostasis in obese humans. Journal of Clinical Investigation 20201302319–2331. ( 10.1172/JCI134892) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Grass DS Saini U Felkner RH Wallace RE Lago WJP Young SG & Swanson ME. Transgenic mice expressing both human apolipoprotein-B and human CETP have a lipoprotein cholesterol distribution similar to that of normolipidemic humans. Journal of Lipid Research 1995361082–1091. ( 10.1016/S0022-2275(2039866-7) [DOI] [PubMed] [Google Scholar]
- 108.Bjorklund MM Hollensen AK Hagensen MK Dagnaes-Hansen F Christoffersen C Mikkelsen JG & Bentzon JF. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circulation Research 20141141684–1689. ( 10.1161/CIRCRESAHA.114.302937) [DOI] [PubMed] [Google Scholar]
- 109.De Giorgi M Jarrett KE de Aguiar Vallim TQ & Lagor WR. In vivo gene editing in lipid and atherosclerosis research. Methods in Molecular Biology 20222419673–713. ( 10.1007/978-1-0716-1924-7_42) [DOI] [PubMed] [Google Scholar]
- 110.Basu D, Hu YY, Huggins LA, Mullick AE, Graham MJ, Wietecha T, Barnhart S, Mogul A, Pfeiffer K, Zirlik A, et al.Novel reversible model of atherosclerosis and regression using oligonucleotide regulation of the LDL receptor. Circulation Research 2018122560–567. ( 10.1161/CIRCRESAHA.117.311361) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Landlinger C Pouwer MG Juno C van der Hoorn JWA Pieterman EJ Jukema JW Staffler G Princen HMG & Galabova G. The AT04A vaccine against proprotein convertase subtilisin/kexin type 9 reduces total cholesterol, vascular inflammation, and atherosclerosis in APOE*3Leiden.CETP mice. European Heart Journal 2017382499–2507. ( 10.1093/eurheartj/ehx260) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tang DD Geng F Yu CX & Zhang RL. Recent application of zebrafish models in atherosclerosis research. Frontiers in Cell and Developmental Biology 20219. ( 10.3389/fcell.2021.643697) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liu C Kim YS Kim J Pattison J Kamaid A & Miller YI. Modeling hypercholesterolemia and vascular lipid accumulation in LDL receptor mutant zebrafish. Journal of Lipid Research 201859391–399. ( 10.1194/jlr.D081521) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mourad O Yee R Li MY & Nunes SS. Modeling heart diseases on a chip: advantages and future opportunities. Circulation Research 2023132483–497. ( 10.1161/CIRCRESAHA.122.321670) [DOI] [PubMed] [Google Scholar]
- 115.St Hilaire C. Cardiovascular organoids/3D models review series: an introduction. Circulation Research 2023132481–482. ( 10.1161/CIRCRESAHA.123.322561) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Williams K Liang T Masse S Khan S Hatkar R Keller G Nanthakumar K & Nunes SS. A 3-D human model of complex cardiac arrhythmias. Acta Biomaterialia 2021132149–161. ( 10.1016/j.actbio.2021.03.004) [DOI] [PubMed] [Google Scholar]
- 117.Wang EY, Rafatian N, Zhao YM, Lee A, Lai BFL, Lu RX, Jekic D, Davenport Huyer L, Knee-Walden EJ, Bhattacharya S, et al.Biowire model of interstitial and focal cardiac fibrosis. ACS Central Science 201951146–1158. ( 10.1021/acscentsci.9b00052) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yadid M, Lind JU, Ardona HAM, Sheehy SP, Dickinson LE, Eweje F, Bastings MMC, Pope B, O’Connor BB, Straubhaar JR, et al.Endothelial extracellular vesicles contain protective proteins and rescue ischemia-reperfusion injury in a human heart-on-chip. Science Translational Medicine 202012. ( 10.1126/scitranslmed.aax8005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Daugherty A, Tall AR, Daemen MJAP, Falk E, Fisher EA, Garcia-Cardena G, Lusis AJ, Owens AP, Rosenfeld ME, Virmani R, et al.Recommendation on design, execution, and reporting of animal atherosclerosis studies: a scientific statement from the American Heart Association. Circulation Research 2017121e53–e79. ( 10.1161/RES.0000000000000169) [DOI] [PubMed] [Google Scholar]
- 120.OpenCV-Python. GIThub 2023 Available at: https://github.com/opencv/opencv-python#releases (Cited 6 September 2023) [Google Scholar]
- 121.Cross-section_aligner_v1.0.0. GitHub 10 October 2023. Available at: https://github.com/RvE54/Cross-section-aligner. (Cited 10 October 2023) [Google Scholar]
- 122.Zhou E, Hoeke G, Li Z, Eibergen AC, Schonk AW, Koehorst M, Boverhof R, Havinga R, Kuipers F, Coskun T, et al.Colesevelam enhances the beneficial effects of brown fat activation on hyperlipidaemia and atherosclerosis development. Cardiovascular Research 20201161710–1720. ( 10.1093/cvr/cvz253) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wong MC, van Diepen JA, Hu LH, Guigas B, de Boer HC, van Puijvelde GH, Kuiper J, van Zonneveld AJ, Shoelson SE, Voshol PJ, et al.Hepatocyte-specific IKK beta expression aggravates atherosclerosis development in APOE*3-Leiden mice. Atherosclerosis 2012220362–368. ( 10.1016/j.atherosclerosis.2011.06.055) [DOI] [PMC free article] [PubMed] [Google Scholar]