

Abstract
Dietary consumption serves as the primary source of iron uptake, while erythropoiesis acts as a major regulator of systemic iron demand. In addition to intestinal iron absorption, macrophages play a crucial role in recycling iron from senescent red blood cells. The kidneys are responsible for the production of erythropoietin (Epo), which stimulates erythropoiesis, while the liver plays a central role in producing the iron-regulatory hormone hepcidin. The transcriptional regulator hypoxia-inducible factor (HIF)2α has a central role in the regulation of Epo, hepcidin, and intestinal iron absorption and therefore plays a crucial role in coordinating the tissue crosstalk to maintain systemic iron demands. However, the precise involvement of Hif2α in macrophages in terms of iron homeostasis remains uncertain. Our study demonstrates that deleting Hif2α in macrophages does not disrupt the expression of iron transporters or basal erythropoiesis. Mice lacking Hif2α in myeloid cells exhibited no discernible differences in hemodynamic parameters, including hemoglobin levels and erythrocyte count, when compared to littermate controls. This similarity was observed under conditions of both dietary iron deficiency and acute erythropoietic demand. Notably, we observed a significant increase in the expression of iron transporters in the duodenum during iron deficiency, indicating heightened iron absorption. Therefore, our findings suggest that the disruption of Hif2α in myeloid cells does not significantly impact systemic iron homeostasis under normal physiological conditions. However, its disruption induces adaptive physiological changes in response to elevated iron demand, potentially serving as a mechanism to sustain increased erythropoietic demand.
Keywords: macrophage, hypoxia, HIF, iron, anemia, hemochromatosis
Introduction
Systemic iron homeostasis is tightly regulated as both excess and deficiency of iron have deleterious effects. Despite tight systemic regulation, more than one billion people worldwide suffer from iron related disorders 1; 2. Systemic iron homeostasis requires hetero-tissue communication. Diet is the sole source of iron, and it is actively absorbed in the small intestine through the transporter divalent metal transporter-1 (Dmt1; gene name Slc11a2) 3; 4. Iron is stored within enterocyte in an iron storage protein, ferritin (FTN) or exported out of the cell and into circulation through the only known mammalian iron exporter ferroportin (Fpn; gene name Slc40a1)5; 6. Expression of Fpn, on the basolateral membrane is regulated at post translational level by a hepatic hormone called hepcidin (gene name Hamp)7. Under high systemic iron levels or iron overload conditions expression of hepcidin is induced in hepatocytes. Increased hepcidin expression degrades or blocks Fpn-mediated iron export and limits iron absorption in the intestine 8; 9; 10; 11. Perturbations in this intestinal Fpn/hepatic hepcidin axis induce systemic iron overload or iron deficiency 12.
The vast majority of iron is used for synthesis of red blood cells (RBC) as an essential component of oxygen carrying protein hemoglobin. Iron and oxygen coordination are central to maintain the production of RBCs. Hypoxia-inducible factor (HIF)2α is an iron and oxygen regulatable transcription factor and a critical regulator of iron regulatory genes 13. Intestinal disruption of Hif2α abrogates dietary iron absorption 14. More, recently a heterocellular crosstalk between hepatic hepcidin and intestinal Hif2α was identified to be instrumental in maintaining iron flux through the duodenal epithelium 15. Hif2α also plays an essential role in regulating the expression of erythropoietin (Epo)16. Liver is the primary source of Epo embryonically, whereas the kidney is major tissue to maintain Epo levels in adults 17. Both hepatic and renal Epo expression is regulated by Hif2α 18. Mice lacking Hif2α in the kidney show lower circulating levels of Epo and are highly anemic 19. Lastly, Hif2α activation of Epo is a major repressor of hepatic hepcidin further increasing iron absorption to maintain RBC synthesis 20; 21. Activation of Hif2α by pharmacological drugs reduced hepcidin production and increase iron absorption in the intestine 22. Moreover, the untranslated region of Hamp contains Hif2α binding HRE elements and deletion of these HREs lead to loss to regulation of hepcidin by systemic iron cues 23.
Apart from the intestine, liver and kidney, macrophages are central to maintain systemic iron by recycling iron from senesced RBCs. Macrophage iron stores are mobilized in response to systemic iron levels through the Fpn-hepcidin axis very similar to that in the duodenum 24; 25. However, the role of Hif2α in regulating iron recycling is not well understood. Moreover, with clinically relevant agents that can increase Hif2α activity26.27 or inhibit its function28, it is critical to understand which cell types are essential to maintain cellular and systemic iron homeostasis following modulation of HIF2α.
In this study we identify that macrophage Hif2α does not play a role in regulating systemic iron homeostasis. We report no changes in systemic iron levels, erythrocytes count as well as hemoglobin levels under basal or iron deficient conditions. Moreover, Hif2α in macrophages was not essential in maintaining RBC levels following an erythropoietic stimuli. However, under iron deficiency, mice disrupted for Hif2α in the myeloid cells showed significantly increased expression of iron transporters involved in the intestinal iron absorption. We speculate this could be a compensatory adaptation to maintain erythropoiesis under potentially impaired iron recycling through the macrophages.
Methods
Animals and treatments.
For myeloid-specific disruption of Hif2α, mice floxed for Hif2α (Hif2αfl/fl) on a C57BL/6J background were crossed with C57BL/6J mice harboring Cre recombinase under the control of lysozyme 2 gene (Lyz2) (LysMCre) to generate LysMCre Hif2α fl/fl mice. WT littermates were used as controls for all animal studies (Hif2αfl/f), and analysis began on mice that were between 2 and 2.5 months of age for each of the respective experiments. Phz (Sigma-Aldrich) was administered via i.p. injection at a dose of 60 mg/kg BW, as described previously 34. All mice were fed ad libitum and maintained under a 12-hour light/12-hour dark cycle in a specific pathogen free environment. All mice were fed either a standard chow diet (Research Diets) or a purified AIN-93G iron-replete (350 ppm) or low-iron (<5 ppm) diet (Diets). All mice were housed in the Unit for Laboratory Animal Management (ULAM) at the University of Michigan.
Hematological and iron analysis.
The Unit for Laboratory Animal Medicine Pathology Core at The University of Michigan performed the complete blood count analysis. Serum iron was measured as described in Das et.al 2015 35.
Bone Marrow-derived macrophage (BMDM) isolation and culture
Male and female LysMCre Hif2α fl/fl mice or wild type littermates were euthanized by cervical dislocation and the femur bones were dissected. The extra muscles were removed, and the bone marrow was extracting by cutting the end of the bones and flushing with 10mL sterile PBS using a 25-gauge needle in a sterile petri-dish. The extracted bone marrow was passed through a 70μm cell strainer. The suspension was centrifuged at 250g for 5 minutes to pellet the cells. The pelleted cells were resuspended in 9mL of water to lyse all the erythroid progenitor cells, followed by addition of 1mL of 10X PBS immediately after. The remaining cells were counted and resuspended and plated at a cell density of 2x106 cells/10 mL in complete RPMI media containing 10%FBS, 1% antibiotic-antimycotic and 20ng/mL GM-CSF in a 10cm petri dish. The cells were cultured in 37°C incubator with 5% CO2 with media change every 3 days.
BMDMs culture and treatment
The BMDMs maintained in an M0 stage were plated on day 10 after the isolation and were treated with LPS 20ng/mL (Sigma L4391-1mg) or IL4 20ng/mL (Gibco 2141420UG) for 48 hours to polarize to M1 and M2, respectively.
Quantitative reverse transcription PCR.
Total RNA was extracted from the BMDMs, liver, kidney and small intestine using the Trizol reagent. 1ug of the total RNA was reverse transcribed to cDNA using SuperScriptTM III First-Strand Synthesis System (Invitrogen). mRNA was measured by quantitative reverse transcription PCR (qPCR) (Life Technologies, Thermo Fisher Scientific). The primers used are listed in Supplemental Table 1. Quantification cycle (Cq) values were normalized to β-actin and expressed as the fold change.
Inductively coupled plasma mass spectrometry (ICP-MS).
Total cellular iron was quantification was performed as previously described in Das et. 202236. BMDMs were isolated and cultured from the Male and female LysMCre Hif2α fl/fl mice or wild type littermates as described above and the BMDMs were digested with 2 ml/g total cell pellet weight nitric acid (BDH Aristar Ultra) for 24 h and then digested with 1 ml/g total weight hydrogen peroxide (BDH Aristar Ultra) for 24 h at room temperature. Specimens were preserved at 4 °C until quantification of metals. Ultrapure water was used for the final sample dilution. Samples were analyzed using a PerkinElmer Nexion 2000 ICP-MS. For iron-57Fe measurements, 57Fe was dissolved in 0.4 mol/l H2SO4 to achieve a total concentration of 22.85 g/l overnight at a 37 °C shaking incubator.
Flow Cytometry.
Blood, spleen, and bone-marrow were isolated from LysMCre Hif2αfl/fl mice and their WT littermates. The blood was lysed with RBC lysis buffer (Gibco), centrifuged, and washed twice with FACS buffer (PBS +2%FBS). Spleen was homogenized and filtered with 70μM filter after which the solution was centrifuged and washed with the FACS buffer. Bone-marrow cells were differentiated to BMDMs as described above under ‘Bone Marrow-derived macrophage (BMDM) isolation and culture’. BMDMs were harvested using PBS with 10mM EDTA. Blood cells, splenic cells and BMDMs were stained for 45 minutes on ice with CD45 Alexa-eFluor 780, 1:200 (eBioscience) , CD11b APC 1:250 (eBioscience), Ly6C V450 1:200 (BD Bioscience), F4/80 BV510 1:100 (BD Bioscience), 7AAD Percp Cy 5.5 1:300 (BD Bioscience) and ferroOrange dye 1 μmol/l (Invitrogen). The cells were washed twice with FACS buffer, resuspended in PBS and 50,000 events was acquired on Cytek Aurora Spectral Analyzer (Cytek Biosciences). FSC-SSC gating was done to remove debris after which live single cells (negative 7AAD staining) were gated, and then leukocytes were identified using CD45 positivity. From the leukocyte population, monocytes (Ly6C and CD11b positive) and macrophages (F4/80 and CD11b positive) cells were gated. The mean fluorescence intensity of FerroOrange on monocyte/macrophage gate was used to evaluate the level of labile iron.
Statistics.
Results are expressed as the mean ± SEM. Significance between 2 groups was tested using a 2-tailed, unpaired t test. Significance among multiple groups was tested using a 1-way or 2-way ANOVA followed by Tukey’s post hoc test for multiple comparisons. A P value of less than 0.05 was considered statistically significant. GraphPad Prism 9.0 was used to conduct the statistical analyses.
Study approval.
All animal procedures were approved by the IACUC of the University of Michigan.
Results
Hif2α deletion in macrophages does not alter expression of iron transporters.
To understand the role of Hif2α in maintaining macrophage and systemic iron levels, mice that express a constitutive Cre recombinase protein under the control of the lysozyme M promoter (LysM) were crossed with Hif2α floxed mice (Hif2αΔmφ) (Figure 1A). The Hif2αΔmφ mice are healthy, viable and indistinguishable from wild-type littermates (Hif2αfl/fl). Hif2α transcript was significantly decreased in BMDMs isolated from Hif2αΔmφ mice (Figure 1B). Consistent with prior work, Hif2α modulates macrophages cell states 37 and our data suggests that Hif2α regulates the anti-inflammatory response in the macrophages but not the pro-inflammatory response (Figure 1C). Hif2α is also established as a transcriptional regulator of intracellular iron uptake and subcellular distribution. However, BMDMs from Hif2αΔmφ mice did not show any significant alterations in the total cellular iron content measured by ICP-MS (Figure1D). Furthermore, macrophage expression of transferrin receptor (Tfrc), Slc11a2 and Slc40a1 were not altered in Hif2α Δmφ compared to wild-type littermates (Figure 1E). This data indicates that Hif2α does not play a significant role in regulating basal iron transporters in macrophages.
Figure 1. Hif2α deletion in macrophages does not alter expression of iron transporter.
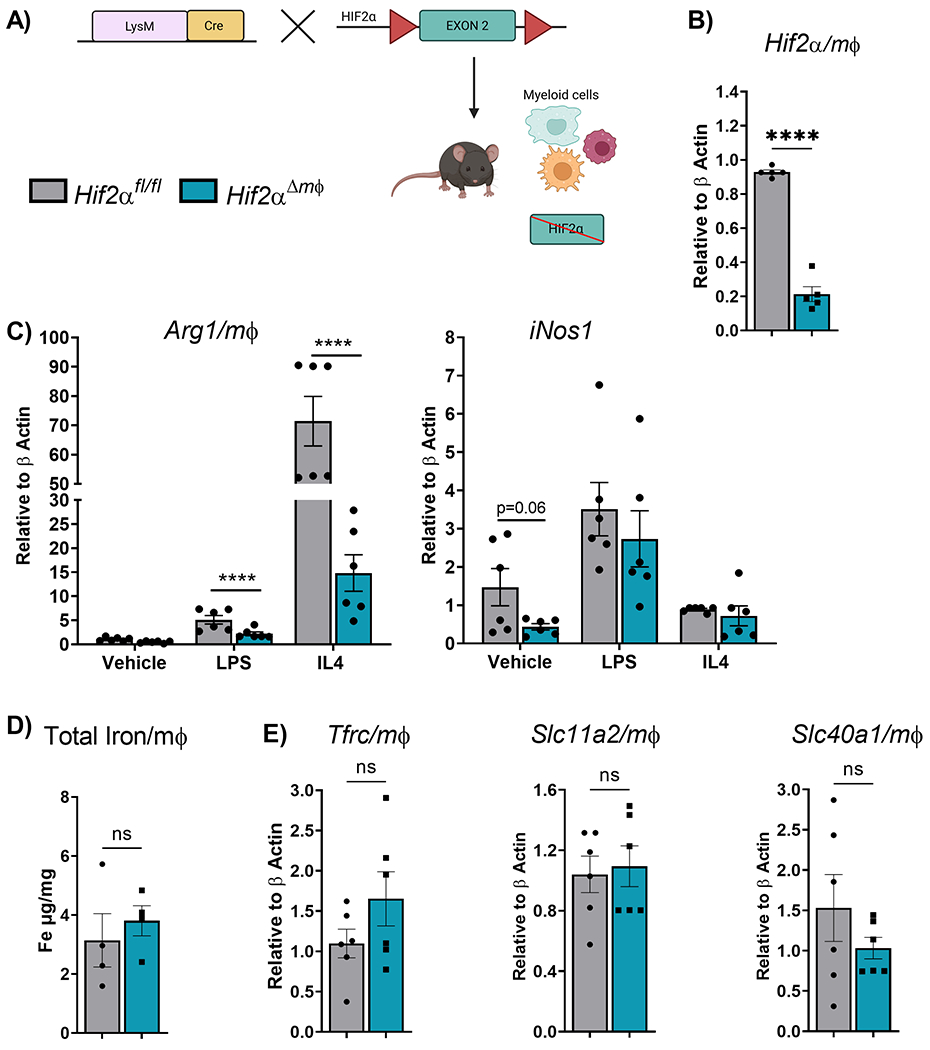
(A) Schematic representation of mice with disruption of Hif2α in the myeloid lineage cells. (B) qPCR analysis of macrophage Hif2α transcript expression levels (n=5-7 per group). (C) qPCR analysis of Arg 1 and iNos1 from Bone marrow derived macrophages (BMDMs) maintained in GMCSF (10ng/mL) for 5 days followed by treatment with IL4 (20ng/mL) and LPS (20ng/mL) for 48 hours (n=4-5 per group). (D) Total intracellular iron measured by Inductively coupled plasma mass spectrometry (ICP-MS). (E) qPCR analysis of macrophage Tfrc (Transferrin receptor), Slc11a2 (Dmt1), Slc40a (Fpn) transcript expression levels (n=5–7 per group). Data represent the mean ± SEM. Significance was determined by unpaired t test. *P < 0.05, ***P < 0.001, and ****P < 0.0001 versus the Hif2αfl/fl group.
Monocyte differentiation and intracellular labile iron are not altered following HIF2α in myeloid cells.
Macrophages are tissue resident immune cells and are integral to innate immune system. In the systemic circulation macrophages exist in the non-differentiated monocyte state and differentiate into activated macrophages within the tissues. To test whether Hif2αΔmφ mice exhibit any changes in the overall circulating/splenic monocyte population or BMDMs a flow cytometry-based approach was utilized.Hif2αΔmφ mice did not show any significant changes in the circulating monocytes, BMDMs, splenic monocytes and macrophages (Figure 2A–C). To assess the intracellular labile iron status (which is considered as accessible iron pool unlike total cellular iron which could be complexed with various macromolecules) a cell permeable labile iron dye specific to ferrous iron was utilized. No difference in the intracellular labile iron pool was observed in the Hif2αΔmφ mice as compared to wild-type littermate controls (Figure 2D–F). This data indicates that myeloid Hif2α is not essential for monocyte/macrophage differentiation and maintenance and does not contribute to intracellular iron homeostasis.
Figure 2. Monocyte differentiation and intracellular labile iron are not altered following HIF2α in myeloid cells.
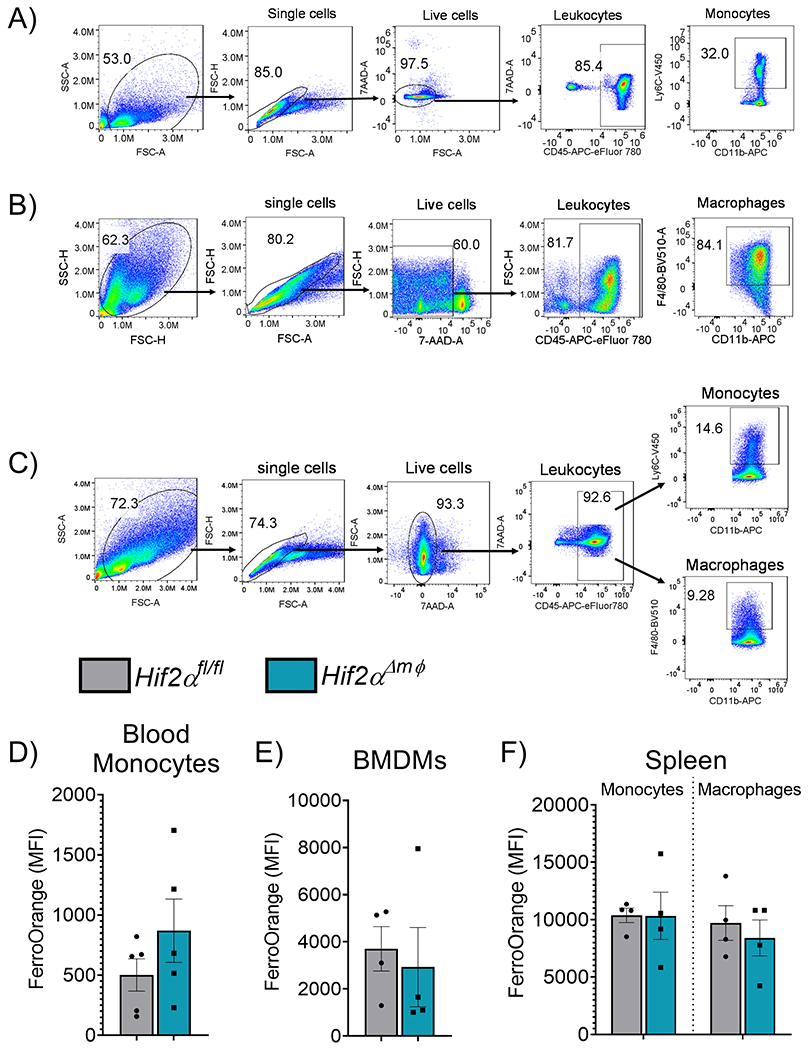
Representative Gating strategy for (A) circulating blood monocytes (n=5,each group) (B) bone marrow derived macrophages (BMDMs) (n=4, each group) and (C) splenic monocytes (n=4, each group) or macrophages (n=4, each group). Mean fluorescence intensity of FerroOrange gated on (D) blood monocytes. (E) BMDMs or (F) splenic monocytes and macrophages. Data represent the mean ± SEM. Significance was determined by unpaired t test. *P < 0.05, ***P < 0.001, and ****P < 0.0001 versus the Hif2αfl/fl group.
Hif2α deletion in macrophages is not required for basal erythropoiesis.
Alterations in the systemic iron levels either due to iron deficiency, malabsorption of dietary iron or inflammation often manifest as anemia. We did not observe any obvious phenotypic differences in the Hif2α Δmφ mice as compared to wild-type littermates in 10-12 week old mice. To understand if Hif2α deletion in macrophages alters the systemic iron levels and cause anemia in older mice, 8 month old Hif2α Δmφ mice were utilized. The total serum iron levels remain unchanged in Hif2α Δmφ mice as compared to the wild-type littermate controls (Figure 3A) Complete blood count (CBC) results revealed no statistically significant changes in any RBC parameters such as RBC count, mean corpuscular volume (MCV), Hemoglobin (HB) and Hematocrit (HCT) (Figure 3B). The Hif2α Δmφ mice showed normal total white blood, monocytes, neutrophils, and non-myeloid lymphocytes (Figure 3C). This data indicated that Hif2α is not essential in mobilizing iron stores from the macrophages under normal physiological conditions. Moreover, Hif2α deletion does not dysregulate hematopoiesis and maintenance of RBCs.
Figure 3. Hif2α deletion in macrophages is not required for basal erythropoiesis.
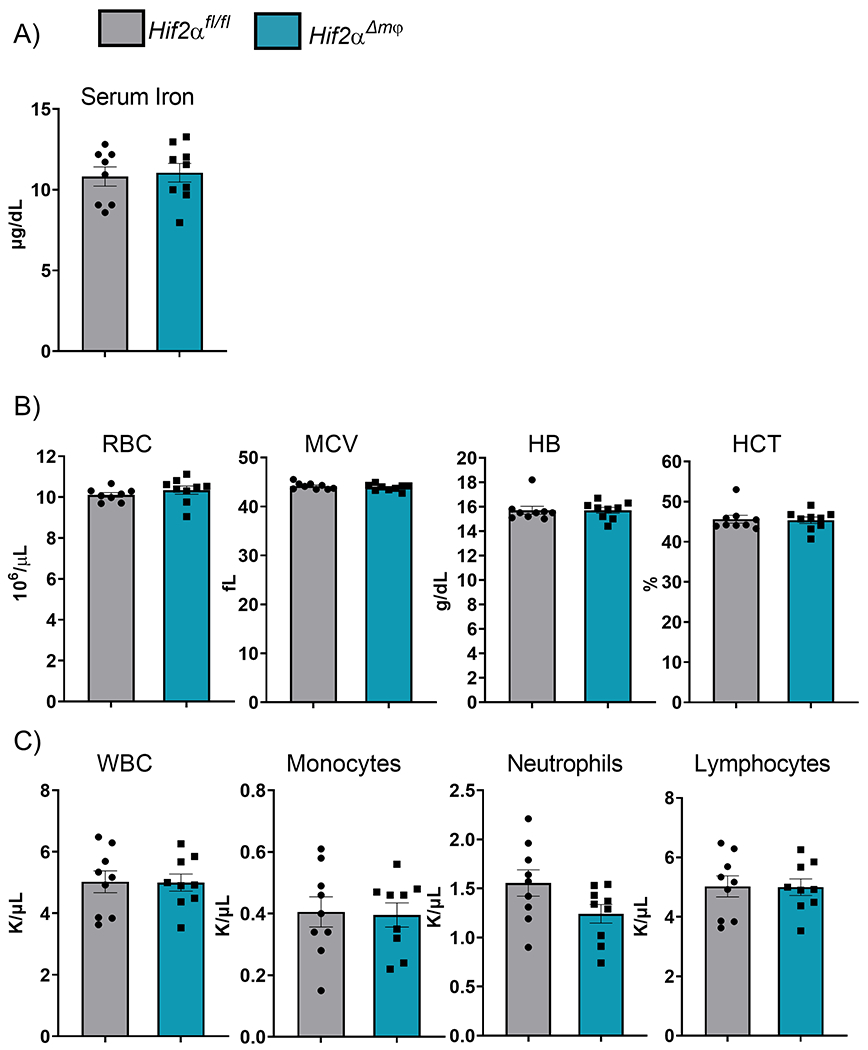
(A) Serum iron measured by colorimeteric iron assay. (B) Analysis of RBC, MCV, HB, and HCT from 48 month old Hif2αfl/fl and Hif2αΔmφ (n = 7-9 per group). (C) Analysis of WBC, Monocytes, Neutrophils, Lymphocytes from 8 month old Hif2αfl/fl and Hif2αΔmφ (n = 7–9 per group). Data represent the mean ± SEM. Significance was determined by unpaired t test. *P < 0.05, ***P < 0.001, and ****P < 0.0001 versus the Hif2αfl/fl group.
Deletion of Hif2α does not alter the systemic response to acute iron and erythropoietic demand.
Erythropoiesis is an essential physiological process and is highly dependent on systemic iron availability. An increase in erythropoietic demand leads to an adaptive response inducing iron mobilization from hepatocytes and macrophages and increase in intestinal iron absorption 34; 38; 39; 40 . Several studies have shown the role of Hif2α in coordinating the intestinal and hepatic response to iron deficiency or erythropoietic demand 16; 18; 41; 42; 43. To determine whether Hif2α plays a role in mobilization of iron stores in the macrophages under acute iron demand we utilized a phenyl hydrazine-induced hemolytic anemia model (Figure 4A). Phenyl hydrazine induce hemolytic anemia causes systemic inflammation due high levels of free hemoglobin and degraded products of hemoglobin from the lysed red corpuscles. Hif2α Δmφ and littermate controls showed signs of anemia, with reduced RBC numbers, HB, and HCT as compared to the mice treated with vehicle control (Figure 4B). Upon phenyl hydrazine treatment both groups showed increased MCV suggesting accumulation of circulating unhealthy erythrocytes (Figure 4B). However, no significant differences were observed within the Hif2α Δmφ and littermate controls. This data demonstrates that Hif2α is not essential for the mobilization of iron stores from macrophages under acute iron demand following hemolysis. The circulating immune cell levels upon Phenyl Hydrazine treatment also remain unchanged in both Hif2α Δmφ and the littermate controls (Figure 4C). Therefore, indicating that Hif2α in macrophages does not attenuate inflammatory response in hemolytic model.
Figure 4. Deletion of Hif2α does not alter the systemic response to acute iron and erythropoietic demand.
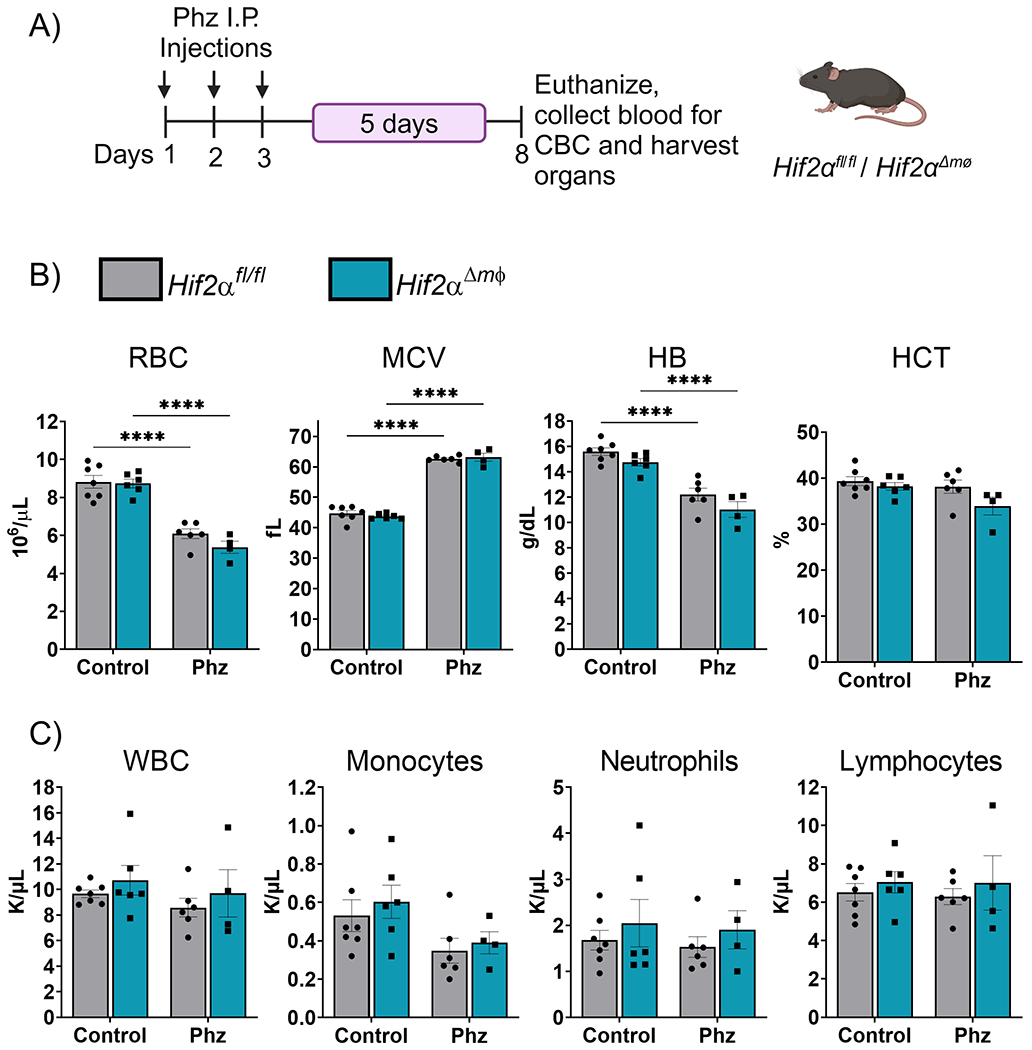
(A) Experimental design for Phz-induced hemolytic anemia model. (B) Analysis of RBC, MCV, HB, and HCT from Hif2αfl/fl and Hif2αΔmφ (n = 7–9 per group). (C) Analysis of WBC, Monocytes, Neutrophils, Lymphocytes from Hif2αfl/fl and Hif2αΔmφ (n = 7–9 per group). Data represent the mean ± SEM. Significance was determined by unpaired t test. *P < 0.05, ***P < 0.001, and ****P < 0.0001 versus the Hif2αfl/fl group.
The systemic response to iron deficiency is not altered under Hif2α deletion in the myeloid cells.
Macrophages play a crucial role in clearing senescent RBCs and releasing the erythrocytic iron back into circulation. To determine whether Hif2α is essential for the mobilization of macrophage iron stores under iron stress, we used a diet-induced iron deficiency model. This model causes mild to moderate iron deficiency in 2 weeks, with mice showing signs of anemia after 4 weeks. We placed 12-week old Hif2αΔmφ mice and littermate controls on standard (350ppm) or iron-deficient (5ppm) diet (Figure 5A). We regularly monitored the food intake of the mice to ensure that any observed differences were not due to variations in food consumption. At the end of the 2 weeks, the mice were euthanized and blood was collected to perform CBC and to assess serum iron levels. The serum iron did not show any significant differences in the Hif2αΔmφ mice and littermate controls or within the groups given standard (350ppm) or iron-deficient (5ppm) diet (Figure 5B) There were no significant differences in any of the hematological parameters measured such as RBC count, MCV, HB and HCT (Figure 5C). As mentioned previously, anemia is often a late symptom of chronic systemic iron deficiency. To account for any late stage effects of Hif2α deletion, we repeated the experiment in 8 month old mice. In the older mice, which had Hif2α deletion for a longer period of time, there were no statistically significant changes in the serum iron levels (Figure 5D). A similar trend was observed in RBC number, MCV, HB and HCT levels (Figure 5E). These results indicate that Hif2α in macrophages does not regulate the mobilization of iron stores iron deficiency and does not alter the physiological response to iron deficiency. However, long term effects of iron deficiency would need to be assessed with a long term dietary intervention.
Figure 5. The systemic response to iron deficiency is not altered under Hif2α deletion in the myeloid cells.
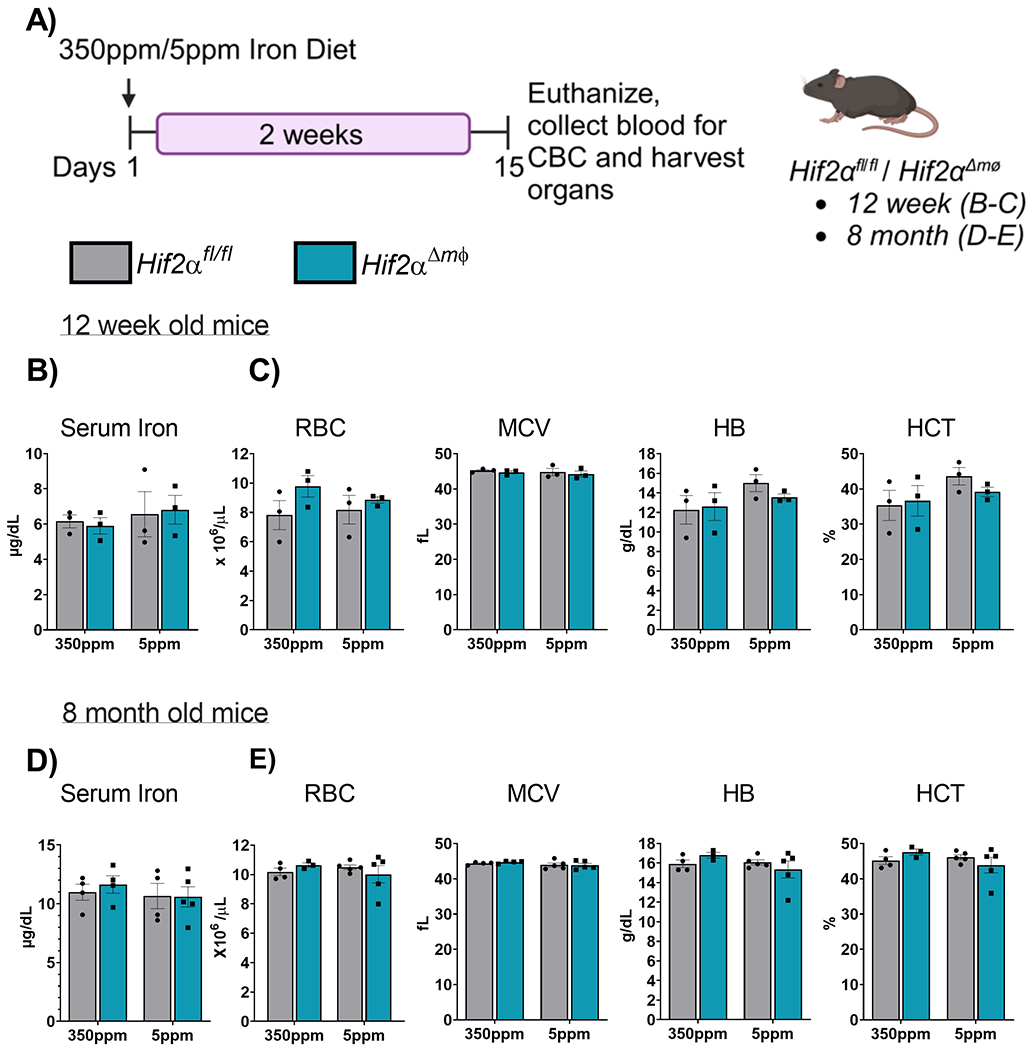
(A) Schematic of iron diet treatment 350ppm and <5ppm in Hif2αfl/fl and Hif2αΔmφ mice to induce iron deficiency anemia. (B) Serum iron measured in 12 week old Hif2αfl/fl and Hif2αΔmφ mice 2 week post dietary intervention (C) Analysis of RBC, MCV, HB, and HCT from 12 week old Hif2αfl/fl and Hif2αΔmφ (n = 7–9 per group). (D) Serum iron measured in 8 month old Hif2αfl/fl and Hif2αΔmφ mice 2 week post dietary intervention. (E) Analysis of RBC, MCV, HB, and HCT from 8 month old Hif2αfl/fl and Hif2αΔmφ (n = 7–9 per group). Data represent the mean ± SEM. Significance was determined by unpaired t test. *P < 0.05, ***P < 0.001, and ****P < 0.0001 versus the Hif2αfl/fl mice fed with 350ppm iron containing diet.
Hif2α deletion in the macrophages induce dietary iron absorption under iron deficiency.
Mice with a myeloid specific Hif2α deletion do not show any differences in terms of systemic iron homeostasis under normal physiology as well as iron deficiency and erythropoietic demand. Under systemic iron deficiency as described previously several physiological adaptive changes occur to maintain erythropoietic output. These physiological changes include drop in hepatic hepcidin to ramp up intestinal iron absorption, increased renal erythropoietin expression to stimulate maturation of erythroid progenitor cells in the bone marrow and finally increased expression of luminal iron transporters. To determine whether there are other compensatory adaptive responses which maintain the erythropoietic output under Hif2α deletion in the macrophages, we measured mRNA expression of hepatic hepcidin (Hamp), renal Epo and intestinal iron transporters. Dcytb (duodenal cytochrome b) is a luminal reductase expressed in the brush border cells of the duodenal epithelium which reduces ferric iron to ferrous form for efficient transport via Dmt1 (Slc11a2). Both Hif2αΔmφ mice and littermate controls showed a decrease in the hepcidin transcript under iron deficiency, consistent with the previous reports regarding the role of hepcidin is regulating systemic iron homeostasis (Figure 6A) 7. The levels of Epo in the kidney showed a moderate increase between the groups that received standard (350ppm) or iron-deficient (5ppm) diet however which was not statistically significant (Figure 6B). The expression of iron transporters Slc11a2, Slc40a1 and Tfrc were significantly induced in the Hif2αΔmφ mice under iron deficiency as compared littermate controls (Figure 6C–F). This data suggests that the transcriptional changes seen in the intestine correspond to increased intestinal absorption of the dietary iron which then leads to maintenance of RBC counts under iron deficiency.
Figure 6. Hif2α deletion in the macrophages induces dietary iron absorption under iron deficiency.
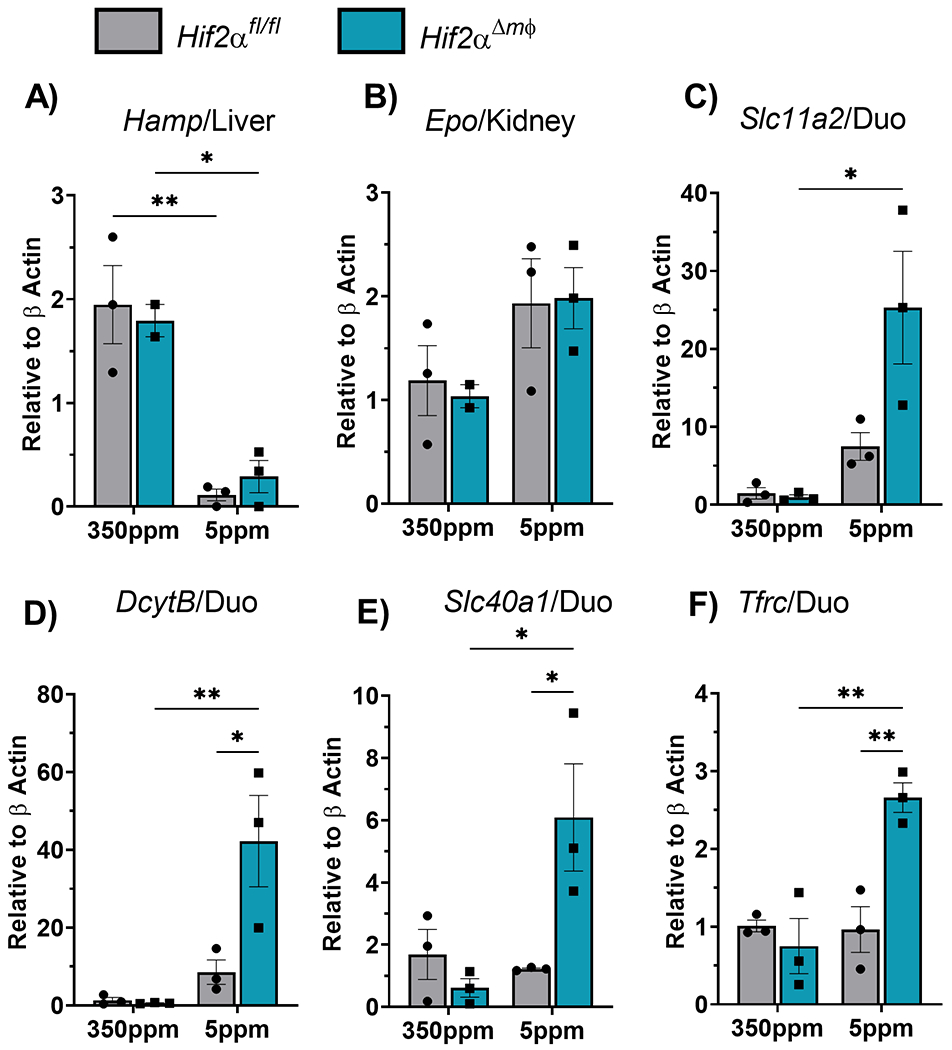
qPCR analysis of A) Liver Hepcidin, B) Renal Erythropoietin and C) duodenal Slc11a2 (Dmt1), (D) DcytB, (E) Slc40a1 (Fpn) and (F) Tfrc from mice fed 350ppm or 5ppm iron containing diet for 2 weeks (n=4-5 per group). Data represent the mean ± SEM. Significance was determined by unpaired t test. *P < 0.05, ***P < 0.001, and ****P < 0.0001 versus the Hif2αfl/fl mice fed with 350ppm iron containing diet.
Discussion
Systemic iron homeostasis is regulated through a multi-organ, heterocellular communication network between the intestine, liver, and kidney, in which Hif2α is thought to be a key transcriptional regulator 44; 45. In this study, we generated mice that were disrupted for Hif2α in macrophages via a myeloid specific Cre recombinase and assessed systemic iron homeostasis under various physiological conditions. We found that myeloid Hif2α does not significantly regulate systemic iron homeostasis in mice. Mice lacking Hif2α in macrophages did not develop symptoms associated with dysregulated iron homeostasis, such as anemia, and had normal red and white blood cell counts. These mice also responded to dietary iron deficiency similarly to wild type controls and did not have increased susceptibility to iron deficiency anemia. The disruption of Hif2α in macrophages during iron demand did not alter the systemic response, and there were no differences in RBC counts or hemoglobin levels observed. However, under iron deficiency mice lacking Hif2α in macrophages showed a significant increase in the expression of various iron transporters in the duodenum, associated with increased iron absorption.
Previous studies from our lab and other groups have shown that Hif2α is a major transcriptional regulator of iron homeostasis and induces expression of various iron transporters 13; 14; 34; 43; 46. However, we show that deletion of Hif2α did not alter the expression of the major cellular iron transporters. The expression of these transporters is also under the control of other iron sensing pathways through iron regulatory protein 1 and 2 which could be the dominant regulator in macrophages 47; 48.
Fpn is the only known mammalian iron transporter and is essential in mobilization of iron stores from macrophages and contribute to systemic homeostasis by regulating iron recycling from the senescent erythrocytes. Deletion of Fpn in the macrophages leads to mild iron deficiency anemia 49. Fpn deficient mice developed severe anemia upon PHZ treatment to induce acute hemolytic as well as under dietary iron deficiency, suggesting that iron recycling is essential for maintaining an adequate RBC count. This study also showed iron retention in liver and spleen suggesting impaired mobilization of iron stores which is previously shown to be dependent on Fpn. Similarly, under iron deficiency mice lacking Hif2α in macrophages showed a significant increase in the expression of various iron transporters in the duodenum, associated with increased iron absorption. This could be an adaptive response to maintain erythroid pool during iron deficiency when iron recycling is potentially disrupted upon Hif2α deletion.
Fpn is shown to be transcriptionally regulated by Hif2α in the intestine where iron deficiency in the enterocytes located on the intestinal epithelium stabilizes and activates Hif2α which then induces Fpn expression thereby ramping up iron absorption 15. Intracellular iron deficiency in the enterocytes indicate systemic iron deficiency and therefore increase in Fpn would be an ideal response to increasing iron absorption. However, in the macrophages, induction of Fpn under cellular iron deficiency via Hif2α signaling would lead to increase iron export and further depletion of cellular iron which can be detrimental for the macrophages. Interestingly, apart from the transcriptional regulation, Fpn protein stability and export function are also regulated by the hepatic hepcidin 7; 50. It is possible that iron export from macrophages through the Fpn pathway is primarily regulated by serum hepcidin rather than cellular Hif2α. This would ensure that iron mobilization from macrophages is better synchronized with systemic iron demands, rather than being influenced solely by cellular iron levels. Consequently, this could provide an explanation as to why we observe no alteration in the Fpn transcript when Hif2α is deleted in macrophages, along with other iron transporters.
In conclusion, our study demonstrates that myeloid-specific deletion of Hif2α in macrophages does not significantly impact systemic iron homeostasis in mice. Despite the established role of Hif2α as a key transcriptional regulator in the multi-organ communication network involved in systemic iron regulation, the absence of Hif2α in macrophages does not result in anemia or abnormal blood cell counts. Interestingly, while the disruption of Hif2α in macrophages does not affect the systemic response to iron deficiency, it leads to increased expression of various iron transporters in the duodenum, potentially promoting enhanced iron absorption. This response may serve as an adaptive mechanism to maintain the erythroid pool during iron deficiency when iron recycling is compromised due to Hif2α deletion.
Supplementary Material
Acknowledgments
The schematic diagram (Figure 1A, Figure 3A, Figure 4A) were created with BioRender.com.
This work was supported by National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grants R01DK095201, the Center for Gastrointestinal Research (DK034933) (Y.M.S.), University of Michigan, Rackham International student fellowship (C.J.). NIH/NCI grant R01CA269025 (K.E.L.), Crohn’s and Colitis Foundation Research Fellowship Award (1003279) (R.S.)
Footnotes
Conflict-of-interest disclosure: The authors declare no competing financial interests.
References
- 1.MCLEAN E et al. Worldwide prevalence of anaemia, WHO Vitamin and Mineral Nutrition Information System, 1993-2005. Public Health Nutr, v. 12, n. 4, p. 444–54, Apr 2009. ISSN 1368-9800. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/18498676 >. [DOI] [PubMed] [Google Scholar]
- 2.BRISSOT P et al. Haemochromatosis. Nat Rev Dis Primers, v. 4, p. 18016, Apr 05 2018. ISSN 2056-676X. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/29620054 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.FLEMING MD et al. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet, v. 16, n. 4, p. 383–6, Aug 1997. ISSN 1061-4036. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/9241278 >. [DOI] [PubMed] [Google Scholar]
- 4.GUNSHIN H et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature, v. 388, n. 6641, p. 482–8, Jul 31 1997. ISSN 0028–0836. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/9242408 >. [DOI] [PubMed] [Google Scholar]
- 5.AROSIO P; INGRASSIA R; CAVADINI P Ferritins: a family of molecules for iron storage, antioxidation and more. Biochim Biophys Acta, v. 1790, n. 7, p. 589–99, Jul 2009. ISSN 0006–3002. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/18929623 >. [DOI] [PubMed] [Google Scholar]
- 6.DONOVAN A et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab, v. 1, n. 3, p. 191–200, Mar 2005. ISSN 1550–4131. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/16054062 >. [DOI] [PubMed] [Google Scholar]
- 7.NEMETH E et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science, v. 306, n. 5704, p. 2090–3, Dec 2004. ISSN 1095–9203. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/15514116 >. [DOI] [PubMed] [Google Scholar]
- 8.GANZ T; NEMETH E Hepcidin and iron homeostasis. Biochim Biophys Acta, v. 1823, n. 9, p. 1434–43, Sep 2012. ISSN 0006–3002. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/22306005 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DRAKESMITH H; NEMETH E; GANZ T Ironing out Ferroportin. Cell Metab, v. 22, n. 5, p. 777–87, Nov 03 2015. ISSN 1932–7420. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/26437604 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.PIGEON C et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem, v. 276, n. 11, p. 7811–9, Mar 16 2001. ISSN 0021–9258. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/11113132 >. [DOI] [PubMed] [Google Scholar]
- 11.DE DOMENICO I et al. The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Mol Biol Cell, v. 18, n. 7, p. 2569–78, Jul 2007. ISSN 1059–1524. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/17475779 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.FERNANDES A et al. The molecular basis of hepcidin-resistant hereditary hemochromatosis. Blood, v. 114, n. 2, p. 437–43, Jul 09 2009. ISSN 1528–0020. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/19383972 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MASTROGIANNAKI M et al. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J Clin Invest, v. 119, n. 5, p. 1159–66, May 2009. ISSN 1558–8238. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/19352007 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.SHAH YM et al. Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab, v. 9, n. 2, p. 152–64, Feb 2009. ISSN 1932–7420. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/19147412 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.SCHWARTZ AJ et al. Hepatic hepcidin/intestinal HIF-2α axis maintains iron absorption during iron deficiency and overload. J Clin Invest, v. 129, n. 1, p. 336–348, Jan 02 2019. ISSN 1558–8238. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/30352047 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.KAPITSINOU PP et al. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood, v. 116, n. 16, p. 3039–48, Oct 21 2010. ISSN 1528–0020. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/20628150 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.JACOBSON LO et al. Role of the kidney in erythropoiesis. 1957. J Am Soc Nephrol, v. 11, n. 3, p. 589–90; discussion 589–91, Mar 2000. ISSN 1046–6673. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/10703684 >. [PubMed] [Google Scholar]
- 18.RANKIN EB et al. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest, v. 117, n. 4, p. 1068–77, Apr 2007. ISSN 0021–9738. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/17404621 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.SCORTEGAGNA M et al. HIF-2alpha regulates murine hematopoietic development in an erythropoietin-dependent manner. Blood, v. 105, n. 8, p. 3133–40, Apr 15 2005. ISSN 0006–4971. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/15626745 >. [DOI] [PubMed] [Google Scholar]
- 20.NICOLAS G et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest, v. 110, n. 7, p. 1037–44, Oct 2002. ISSN 0021–9738. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/12370282 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MASTROGIANNAKI M et al. Hepatic hypoxia-inducible factor-2 down-regulates hepcidin expression in mice through an erythropoietin-mediated increase in erythropoiesis. Haematologica, v. 97, n. 6, p. 827–34, Jun 2012. ISSN 1592–8721. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/22207682 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.PEYSSONNAUX C et al. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J Clin Invest, v. 117, n. 7, p. 1926–32, Jul 2007. ISSN 0021–9738. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/17557118 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.SCHWARTZ AJ et al. Hepcidin sequesters iron to sustain nucleotide metabolism and mitochondrial function in colorectal cancer epithelial cells. Nat Metab, v. 3, n. 7, p. 969–982, Jul 2021. ISSN 2522–5812. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/34155415 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.GANZ T Macrophages and systemic iron homeostasis. J Innate Immun, v. 4, n. 5–6, p. 446–53, 2012. ISSN 1662–8128. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/22441209 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.KNUTSON MD et al. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc Natl Acad Sci U S A, v. 102, n. 5, p. 1324–8, Feb 01 2005. ISSN 0027–8424. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/15665091 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.AKIZAWA T et al. Roxadustat Treatment of Chronic Kidney Disease-Associated Anemia in Japanese Patients Not on Dialysis: A Phase 2, Randomized, Double-Blind, Placebo-Controlled Trial. Adv Ther, v. 36, n. 6, p. 1438–1454, Jun 2019. ISSN 1865–8652. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/30953333 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.CHEN N et al. Roxadustat Treatment for Anemia in Patients Undergoing Long-Term Dialysis. N Engl J Med, v. 381, n. 11, p. 1011–1022, September 12 2019. ISSN 1533–4406. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/31340116 >. [DOI] [PubMed] [Google Scholar]
- 28.CHEN W et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature, v. 539, n. 7627, p. 112–117, Nov 03 2016. ISSN 1476–4687. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/27595394 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.RECALCATI S et al. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur J Immunol, v. 40, n. 3, p. 824–35, Mar 2010. ISSN 1521–4141. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/20039303 >. [DOI] [PubMed] [Google Scholar]
- 30.CORNA G et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica, v. 95, n. 11, p. 1814–22, Nov 2010. ISSN 1592–8721. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/20511666 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.GAETANO C; MASSIMO L; ALBERTO M Control of iron homeostasis as a key component of macrophage polarization. Haematologica, v. 95, n. 11, p. 1801–3, Nov 2010. ISSN 1592–8721. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/21037324 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.SINDRILARU A et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest, v. 121, n. 3, p. 985–97, Mar 2011. ISSN 1558–8238. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/21317534 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.KAEMPFER T et al. Extracellular hemoglobin polarizes the macrophage proteome toward Hb-clearance, enhanced antioxidant capacity and suppressed HLA class 2 expression. J Proteome Res, v. 10, n. 5, p. 2397–408, May 06 2011. ISSN 1535–3907. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/21405025 >. [DOI] [PubMed] [Google Scholar]
- 34.ANDERSON ER; XUE X; SHAH YM Intestinal hypoxia-inducible factor-2alpha (HIF-2alpha) is critical for efficient erythropoiesis. J Biol Chem, v. 286, n. 22, p. 19533–40, Jun 03 2011. ISSN 1083–351X. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/21498508 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DAS N et al. Intestine-specific Disruption of Hypoxia-inducible Factor (HIF)-2α Improves Anemia in Sickle Cell Disease. J Biol Chem, v. 290, n. 39, p. 23523–7, Sep 25 2015. ISSN 1083–351X. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/26296885 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DAS NK et al. Modulation of the HIF2α-NCOA4 axis in enterocytes attenuates iron loading in a mouse model of hemochromatosis. Blood, v. 139, n. 16, p. 2547–2552, April 21 2022. ISSN 1528–0020. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/34990508 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.TAKEDA N et al. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev, v. 24, n. 5, p. 491–501, Mar 01 2010. ISSN 1549–5477. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/20194441 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.LATUNDE-DADA GO et al. Tissue-specific changes in iron metabolism genes in mice following phenylhydrazine-induced haemolysis. Biochim Biophys Acta, v. 1690, n. 2, p. 169–76, Oct 14 2004. ISSN 0006–3002. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/15469906 >. [DOI] [PubMed] [Google Scholar]
- 39.SCHMID H; SCHIFFL H Erythropoiesis stimulating agents and anaemia of end-stage renal disease. Cardiovasc Hematol Agents Med Chem, v. 8, n. 3, p. 164–72, Jul 2010. ISSN 1875–6182. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/20443766 >. [DOI] [PubMed] [Google Scholar]
- 40.ANDERSON GJ; POWELL LW; HALLIDAY JW Transferrin receptor distribution and regulation in the rat small intestine. Effect of iron stores and erythropoiesis. Gastroenterology, v. 98, n. 3, p. 576–85, Mar 1990. ISSN 0016–5085. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/2298364 >. [DOI] [PubMed] [Google Scholar]
- 41.JELKMANN W Regulation of erythropoietin production. J Physiol, v. 589, n. Pt 6, p. 1251–8, Mar 15 2011. ISSN 1469–7793. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/21078592 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.FATRAI S et al. Identification of HIF2alpha as an important STAT5 target gene in human hematopoietic stem cells. Blood, v. 117, n. 12, p. 3320–30, Mar 24 2011. ISSN 1528–0020. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/21263150 >. [DOI] [PubMed] [Google Scholar]
- 43.TAYLOR M et al. Hypoxia-inducible factor-2α mediates the adaptive increase of intestinal ferroportin during iron deficiency in mice. Gastroenterology, v. 140, n. 7, p. 2044–55, Jun 2011. ISSN 1528–0012. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/21419768 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.SHAH YM; XIE L Hypoxia-inducible factors link iron homeostasis and erythropoiesis. Gastroenterology, v. 146, n. 3, p. 630–42, Mar 2014. ISSN 1528–0012. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/24389303 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.CAMASCHELLA C et al. The mutual crosstalk between iron and erythropoiesis. Int J Hematol, v. 116, n. 2, p. 182–191, Aug 2022. ISSN 1865–3774. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/35618957 >. [DOI] [PubMed] [Google Scholar]
- 46.RAMAKRISHNAN SK et al. Maternal intestinal HIF-2α is necessary for sensing iron demands of lactation in mice. Proc Natl Acad Sci U S A, v. 112, n. 28, p. E3738–47, Jul 2015. ISSN 1091–6490. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/26124130 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.MUCKENTHALER MU; GALY B; HENTZE MW Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu Rev Nutr, v. 28, p. 197–213, 2008. ISSN 0199–9885. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/18489257 >. [DOI] [PubMed] [Google Scholar]
- 48.GALY B et al. Iron regulatory proteins are essential for intestinal function and control key iron absorption molecules in the duodenum. Cell Metab, v. 7, n. 1, p. 79–85, Jan 2008. ISSN 1550–4131. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/18177727 >. [DOI] [PubMed] [Google Scholar]
- 49.ZHANG Z et al. Ferroportin1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood, v. 118, n. 7, p. 1912–22, Aug 18 2011. ISSN 1528–0020. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/21705499 >. [DOI] [PubMed] [Google Scholar]
- 50.NEMETH E et al. The N-terminus of hepcidin is essential for its interaction with ferroportin: structure-function study. Blood, v. 107, n. 1, p. 328–33, Jan 01 2006. ISSN 0006–4971. Disponível em: < https://www.ncbi.nlm.nih.gov/pubmed/16141345 >. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.