

Abstract
Eos (lkzf4) is a member of the Ikaros family of transcription factors and is preferentially expressed in T-regulatory (Treg) cells. However, the role of Eos in Treg function is controversial. One study using siRNA knock down of Eos demonstrated that it was critical for Treg suppressor function. In contrast, Treg from mice with a global deficiency of Eos had normal Treg function in vitro and in vivo. To further dissect the function of Eos in Tregs, we generated mice with a conditional knock out of Eos in Treg cells (lkzf4fl/fl X Foxp3YFP–cre, Eos cKO). Deletion of Eos in Treg resulted in activation of CD4+Foxp3− and CD8+ T cells at the age of 3 months, cellular infiltration in non-lymphoid tissues, hyperglobulinemia, and anti-nuclear antibodies. While Tregs from Eos cKO mice displayed normal suppressive function in vitro, Eos cKO mice developed severe Experimental Autoimmune Encephalomyletis (EAE) following immunization with myelin oligodendrocyte glycoprotein (MOG) and Eos cKO Treg were unable to suppress Inflammatory Bowel Disease (IBD). Eos cKO mice had decreased growth of the transplantable murine adenocarcinoma MC38 tumor accompanied by enhanced IFN-γ/TNF-α production by CD8+ T cells in tumor draining lymph nodes. Mice with a global deficiency of Eos or a deficiency of Eos only in T cells developed autoimmunity at a much older age (12 months or 7–8 months, respectively). Taken together, Eos appears to play an essential role in multiple aspects of Treg suppressor function, but also plays an as yet unknown role in the function of CD4+Foxp3− and CD8+ T cells and potentially in non-T cells.
Keywords: Eos, Transcription factor, Foxp3, EAE, T effector cells, T regulatory cells
1. Introduction
Eos (lkzf4) is a transcription factor and belongs to the Ikaros family of transcription factors, which also includes Helios, Aiolos and Pegasus [1]. The Ikaros family of transcription factors in general are involved in hematopoiesis and development of the immune system [1–3]. Eos was originally thought to be expressed and function only in the brain and nervous system [1,4]. Eos has two conserved zinc finger regions, one at the C-terminus responsible for homo- or heterodimerization with itself or other family members and the other at the N-terminus, which is important for DNA binding. It has been shown previously that targeted mutation of the Ikaros family members either at the DNA binding domain or the dimerization domain has wide ranging effects from lack of lymphoid lineage, to B cell lymphomas, to defects in Treg function [2,5–7]. Eos is preferentially expressed in T regulatory cells (Tregs) in the steady state and is one of a unique group of genes (including IRF4, Satb1, Lef1 and GATA-1) which act in concert with Foxp3 [8–10] to maintain the Treg signature.
Tregs are an important subset of T cells and are involved in maintaining the homeostasis of the immune system [8,11–15]. Defective functioning of Tregs leads to development of autoimmune diseases, whereas over-activation of these cells may suppress immune responses and result in impaired responses against pathogens and tumors [16–18]. The role of Eos in the function of Tregs remains controversial. Pan et al. [8] used an siRNA knock down approach in vitro and demonstrated that Eos interacts directly with Foxp3 and induces chromatin modifications that results in gene silencing in Tregs. Tregs deficient in Eos produced IL-2, lost their suppressive function in vitro and in vivo, and acquired the phenotype of effector T cells. In addition, in pulldown experiments, they demonstrated that Eos directly interacted with Foxp3 and was required for Foxp3-mediated down-regulation of gene expression. However, a comprehensive analysis of Foxp3 interacting partners using biochemical and mass spectrometric studies showed that Foxp3 forms large transcriptional complexes comprising several hundred partners, but failed to observe an interaction with Eos [19]. Sharma et al. [20] demonstrated that certain conditions of antigen priming in vivo resulted in loss of expression of Eos by a major subset of Tregs that were reprogrammed into T helper (Th) cells but maintained expression of Foxp3. In contrast, Rieder et al. [9] demonstrated that mice with a global deletion of Eos appeared to be completely normal and Tregs purified from these mice had normal suppressive function both in vivo and in vitro. In addition, this study also demonstrated that Eos was also expressed in CD4+Foxp3− T cells.
The goal of the present study was to further define the role of Eos, if any, in Treg cells by generating mice with a conditional deletion of Eos (Eos cKO) in Tregs using the Cre-lox system.
At 3-month-old age under steady state condition, we found that Eos cKO mice manifested signs of polyclonal T cell activation with a Th1 phenotype in the presence of an increase in the percentage of Treg cells. These mice manifested signs of both systemic and organ-specific autoimmunity with hyperglobulinemia, anti-nuclear antibodies, and marked lymphocyte infiltration in non-lymphoid organs. Eos cKO mice also had enhanced experimental autoimmune encephalomyelitis (EAE) and enhanced tumor immunity. Eos deficient Tregs were unable to suppress effector responses in the IBD cell transfer model, but had normal suppressive function in vitro. Mice with a global deletion of Eos or with a deletion of Eos only in T cells (CD4-Cre) also developed a similar phenotype, but at much older ages, indicating that Eos also plays a role in the function of CD4+ Foxp3− T cells, CD8+ T cells and non-T cells and that a defect in the activation of one or all of these cell types masks the defect in Treg function. Ultimately, the defective Treg phenotype dominates and autoimmune disease develops.
2. Methods
2.1. Mice
Eosfl/fl Mice were generated and donated by C.Benoit and D. Mathis, Harvard Medical School, Boston. Eos cKO Treg mice were generated by breeding Eosfl/fl mice with Foxp3-YFP Cre mice (Jackson Laboratory, Bar Harbor, ME). Mice with a global deletion of Eos were a generous gift of Dr. B. Morgan (Massachusetts General Hospital, Boston, MA). Mice with a deletion of Eos in all T cells were generated by breeding the Eosfl/fl mice to CD4-Cre mice (Jackson Laboratory). Wild type (WT) C57BL/6 and RAG−/− mice were obtained from Taconic Farms (Germantown, NY). All studies in this paper were approved by NIAID Animal Care and Use Committee. Animals of both sexes were used equally in these studies.
2.2. Flow cytometry and cell sorting
The following anti–mouse antibodies (from BD Bioscience or eBioscience): CD4 (RM4–5), CD8 (53–6.7), CD25 (7D4), CD44 (IM7), CD62L (MEL-14), PD-1(J43), CXCR-5 (SPRCL5), CD138 (281–2), CD19 (1D-3), CD38 (90), IgD (11–26c), GL-7, and CD95 (15A7) were used for surface staining. Single cell suspensions were made from the spleen, draining lymph nodes (dLNs), thymus and Peyer’s patches. Cells were surface stained and then permeabilized with FixPerm buffer (eBioscience) for intracellular staining. Cells were then washed and stained with antibodies against Foxp3 (FJK-16s), IFN-γ (XMG1.2), IL-17 (TC11–18H10.1), TNF-α (ΜP6-XT22), K-I67 (eBio17b7). For evaluation of intracellular cytokine production, cells were stimulated for 4 h with cell stimulation cocktail plus protein transport inhibitor (eBioscience) and then stained. We used anti-GFP antibody (Life Technologies, Grand Island, NY) during intracellular staining for the detection of YFP. All antibodies were used at 1:100 dilutions unless otherwise stated. The LSRII (BD Bioscience) was used for data acquiring and samples were analyzed using FlowJo V10 software (TreeStar). For cell sorting experiments, cells were labeled with different surface markers and sorted using a FACS Aria flow cytometer (BD Biosciences).
2.3. In vitro suppression assay
Naïve WT cells (CD4+CD25−CD62LhiCD44low) were FACS-sorted from spleen and lymph nodes. Cells (5 × 104) were co-cultured in 96-well plates with irradiated autoMACS-sorted T-depleted splenocytes (5 × 104) in the presence of soluble anti-CD3 (1 μg/ml). In some cultures, either sorted CD4+CD25+ T cells from WT or Eos deficient mice cells were added in indicated numbers for 72 h. Cultures were pulsed with [3H]-thymidine for the last 6 h of culture. In other experiments, cell proliferation was measured by labeling the naïve T cells with cell proliferation dye (eBioscience) according to manufacturer’s instructions.
2.4. Immunization
Mice were immunized intraperitoneally with 2 × 108 Sheep Red Blood Cells (SRBCs) purchased from Lampire Biological Laboratories (Pipersville, PA). Spleens were harvested and analyzed by flow cytometry 7 day post immunization.
2.5. EAE
EAE was induced by immunization of mice with Myelin Oligodendrocyte Glycoprotein (MOG) peptide (400 μg, MEVGWYRSP-FSRVVHLYRNGK) in Complete Freund’s Adjuvant (CFA) containing Mycobacterium tuberculosis strain H37Ra (400 μg, DIFCO). The mice were then injected with Pertussis toxin (200 ng/mouse, EMD) i.v. on d 0 and d 2. The following criteria were used to score mice: 0, no signs of disease; 1, complete tail paralysis; 2, hind limb paresis; 3, complete hind limb paralysis; 4, unilateral forelimb paralysis; and 5, moribund/death. Data presented are the mean clinical scores of six mice per group.
2.6. IBD
CD4+CD25−CD45RBhi naive T cells (4 × 105 cells) were sorted from WT mice and injected i.v. into Rag2−/− mice in combination with 2 × 105 CD4+CD25+ T cells from Eos cKO or Eosfl/fl mice (12 wk old and sex matched). Mice were observed daily and weighed weekly. Five weeks after cell transfer, the mice were euthanized for analysis.
2.7. Tumor immunity
Murine colon adenocarcinoma cells (MC38) cells were grown in complete DMEM medium [DMEM supplemented with 10% heat-inactivated FBS, l-glutamine (2 mM), sodium pyruvate (1 mM), HEPES (1 mM), non-essential amino acids (0.1 mM), 2-mercaptoethanol (50 mM), and penicillin and streptomycin (100 U/ml)]. Age-matched Foxp3YFP–cre and Eos cKO mice were injected with 2 × 105 MC38 cells subcutaneously (flank region). Tumor growth was measured and quantitated using digital calipers (Fisher Scientific), and tumor volumes were calculated by the formula: length × width × depth, (length × width2)/2. On day 18 post tumor implant, tumors were excised, and tumor infiltrating lymphocytes were prepared by mincing the tumor, and digestion with 1x HBSS containing collagenase type IV (0.5 mg/ml), Dnase I (0.1 mg/ml), and Hyaluronidase (2.5 units/ml) for 1 h at 37 °C. Following digestion, cell suspensions were washed, treated with ACK lysing buffer to lyse RBCs, and tumor infiltrating lymphocytes (TILs) were purified by density gradient centrifugation using buffered percoll (Sigma-Aldrich, 80%/40%).
2.8. ELISA
Anti-Nuclear Antibodies (ANA) were detected with the Total Ig kit from Alpha Diagnostic International (San Antonio, Texas). For the detection of different immunoglobulin subclasses, we used Mouse IgG ELISA Kit from eBioscience.
2.9. Histology and immunohistochemistry
H & E staining of sections of spleen, salivary glands, kidneys, lung, small intestine and liver from Eosfl/fl and Eos cKO mice were performed by HistoServ Labs Inc. (Gaithersburg, MD). Confocal images were taken in the Biological Imaging Section, NIAID, NIH. Histology scores were determined by an independent scorer.
Affixed cryostat sections were dried at 25 °C, fixed in ice-cold acetone for 10 min and dried again at 25 °C. Slides were rehydrated with 1x Tris-buffered saline (TBS) pH 7.6 and placed in a humidifier chamber. Sections were blocked for 2 h at 25 °C with IHC/ICC Blocking High Protein Buffer (eBioscience). Sections were stained overnight at 4 °C in blocking buffer with anti-IgD PerCP e-Fluor 710 (11–26c; eBioscience), anti-CD4 Alexa Fluor 647 (GK1.5; Biolegend), anti-GL7 PE (Biolegend). Slides were then washed in 1x TBS for 5 min with gentle agitation. Slides were mounted with Fluoromount-G with DAPI (eBioscience). Images were visualized and collected using a Leica SP8 inverted 5-channel confocal microscope (Leica) and analyzed in Imaris 8.1 (Bitplane, Oxford Instruments). Images were taken in the Biological Imaging core, NIAID, NIH.
2.10. Statistical analysis
All data are presented as the mean values ± SD. ANOVA with posthoc Turkey test was used for comparisons between groups (Graph Pad Prism, version 6) and also Student’s T test was used. Statistical significance was established at the levels of * P ≤ 0.05, **P ≤ 0.005, ***P ≤ 0.0005 and ****P ≤ 0.0001.
3. Results
3.1. Eos cKO mice develop lymphoproliferation and a Th1 phenotype as early as 3 months of age
When Eos cKO mice were examined at 2 months of age, they exhibited slight splenomegaly and lymphadenopathy. At 3 months of age, marked splenomegaly and lymphadenopathy were observed with an increase in the absolute number of total CD4+ and CD8+ T cells, as well as Tregs (Supplementary Fig. 1). All three T cell populations exhibited an activated phenotype with significantly elevated percentages of CD44hiCD62Llo cells (Fig. 1A–C). Elevated percentages of Tregs were also present in the cKO mice (Fig. 1D). Both the CD4+Foxp3− and CD8+ T cells from the Eos cKO mice displayed a Th1 phenotype with an increased capacity to secrete IFN-γ, but not IL-17 or IL-4 (Fig. 1E and data not shown). The phenotype seen in the Eos cKO mice is typical of mice with a defect in Treg suppressor function where higher percentages of activated Tregs are present in the animals, but they are incapable of controlling the development of the autoimmune phenotype [5]. The deletion of Eos was confirmed by standard genotyping (Supplementary Fig. 4).
Fig. 1.
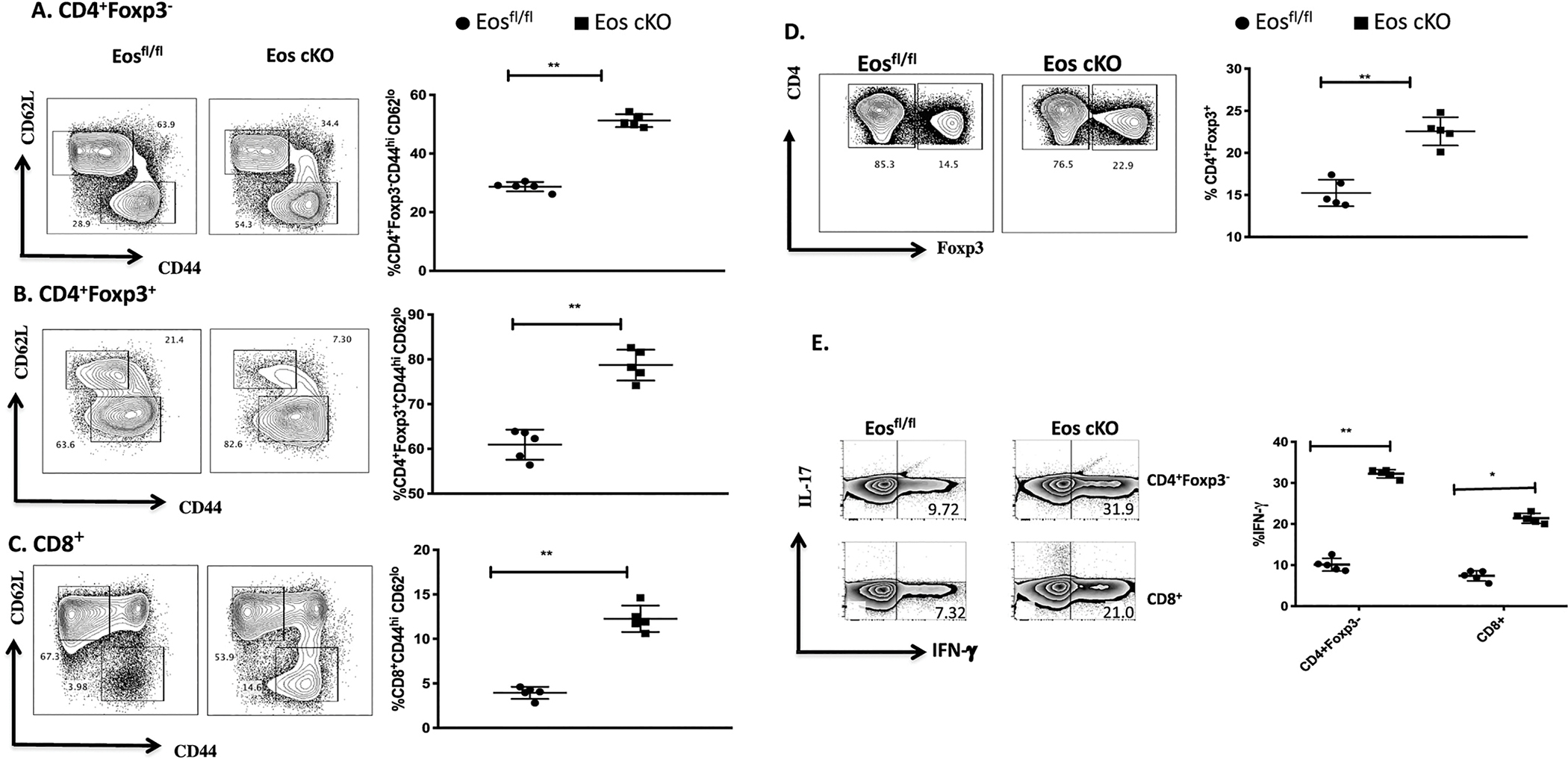
Eos cKO mice display an activated phenotype at 3 months of age. A) CD4+Foxp3−, B) CD4+Foxp3+, and C) CD8+ T cells in spleens from Eosfl/fl or Eos cKO mice were stained with anti-CD44 and anti-CD62L. D) Splenocytes were gated on total CD4+ T cells from Eosfl/fl (left) or Eos cKO(right) mice and percentages of Foxp3+ T cells were analyzed by flow cytometry. E) Splenocytes from Eosfl/fl and Eos cKO mice were cultured with cell stimulation cocktail for 4 h and then stained for IFN-γ and IL-17 production among CD4+Foxp3− and CD8+ T cells.
3.2. Eos cKO mice develop both organ-specific and systemic autoimmunity
Pathologic examination of multiple organs revealed a marked lymphoid infiltration in the lungs, liver, salivary glands and kidneys with evidence of glomerulonephritis (Fig. 2). The livers of the cKO mice also contained hepatic microgranulomas (Supplementary Fig. 2). Microvilli of the small intestine of the cKO mice were blunted with hyperplastic epithelium and increased lymphocyte infiltration along with some neutrophils in the lamina propria (Fig. 2).
Fig. 2.
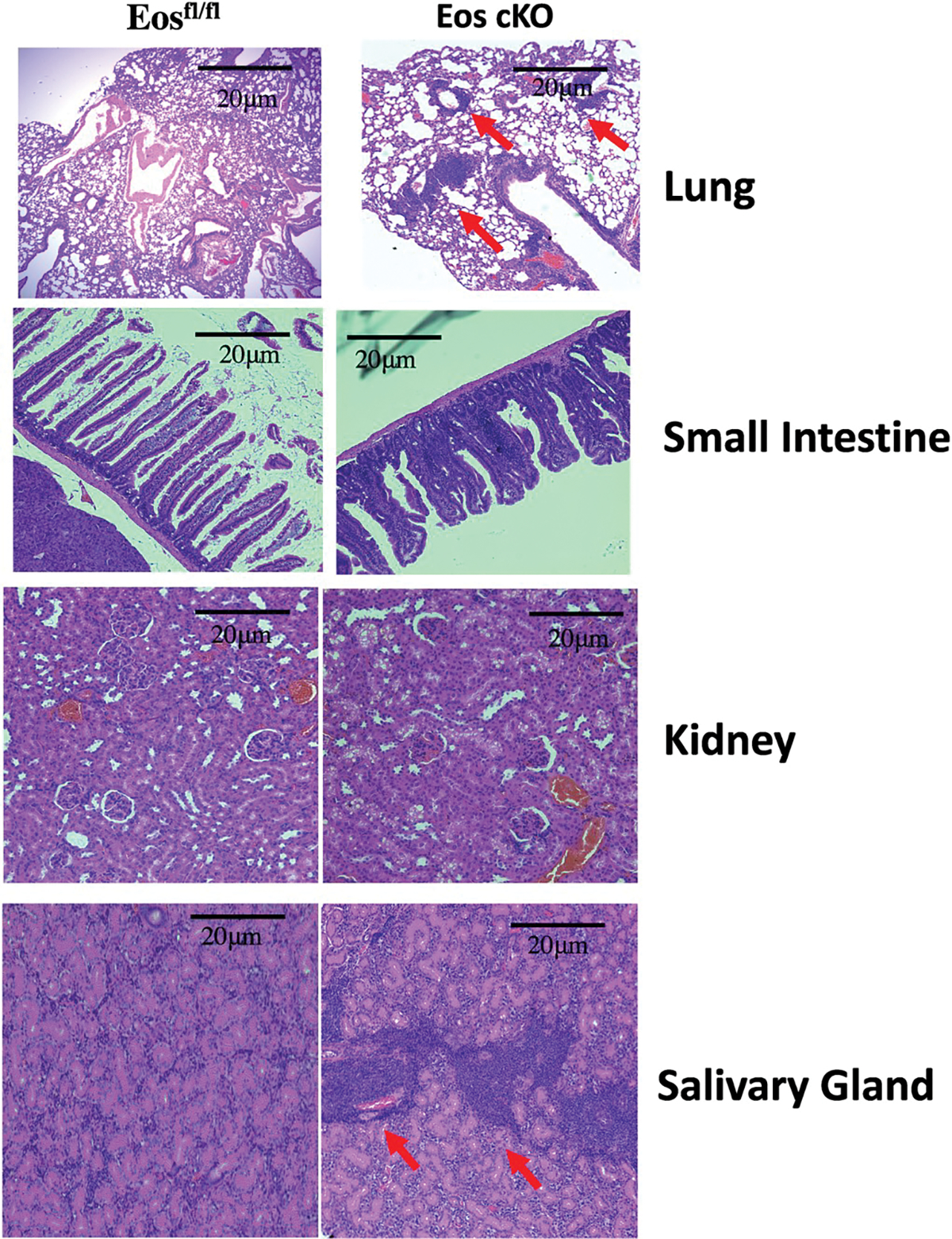
Eos cKO mice develop organ infiltration as early as 3 months of age. Lungs, small intestine, kidneys and salivary glands of 3 month old Eosfl/fl or Eos cKO mice were sectioned and stained with H&E. Scale bars are 20 and 30 μm, respectively.
Eos cKO mice had increased levels of all immunoglobulin isotypes as well as anti-nuclear antibodies (ANA) in their sera (Fig. 3A). The percentage of T follicular (Tf) cells as defined by expression of CXCR5 and PD-1 in unimmunized Eos cKO mice at steady state was significantly higher when compared to unimmunized controls. The increase in Tf cells was primarily made up of Tf helper (Tfh) cells as the percentage of Tf regulatory (Tfr) cells was markedly decreased consistent with a defect in the generation or function of Tfr cells. Following immunization with SRBCs both the control mice and the Eos cKO mice had an expansion of Tf cells, while Eos cKO mice had a greater decrease in the percentage of Tfr cells again consistent with a defect in Tfr function (Fig. 3B). The increased numbers of Tfh cells were accompanied by a significant increase in the percentages of germinal center (GC) B cells in the Eos cKO mice both pre- and post-immunization (Fig. 4A). Immunohistochemical staining of the spleen also revealed a marked increase in the number of follicles and germinal centers in the Eos cKO mice 7d post immunization (Fig. 4B).
Fig. 3.
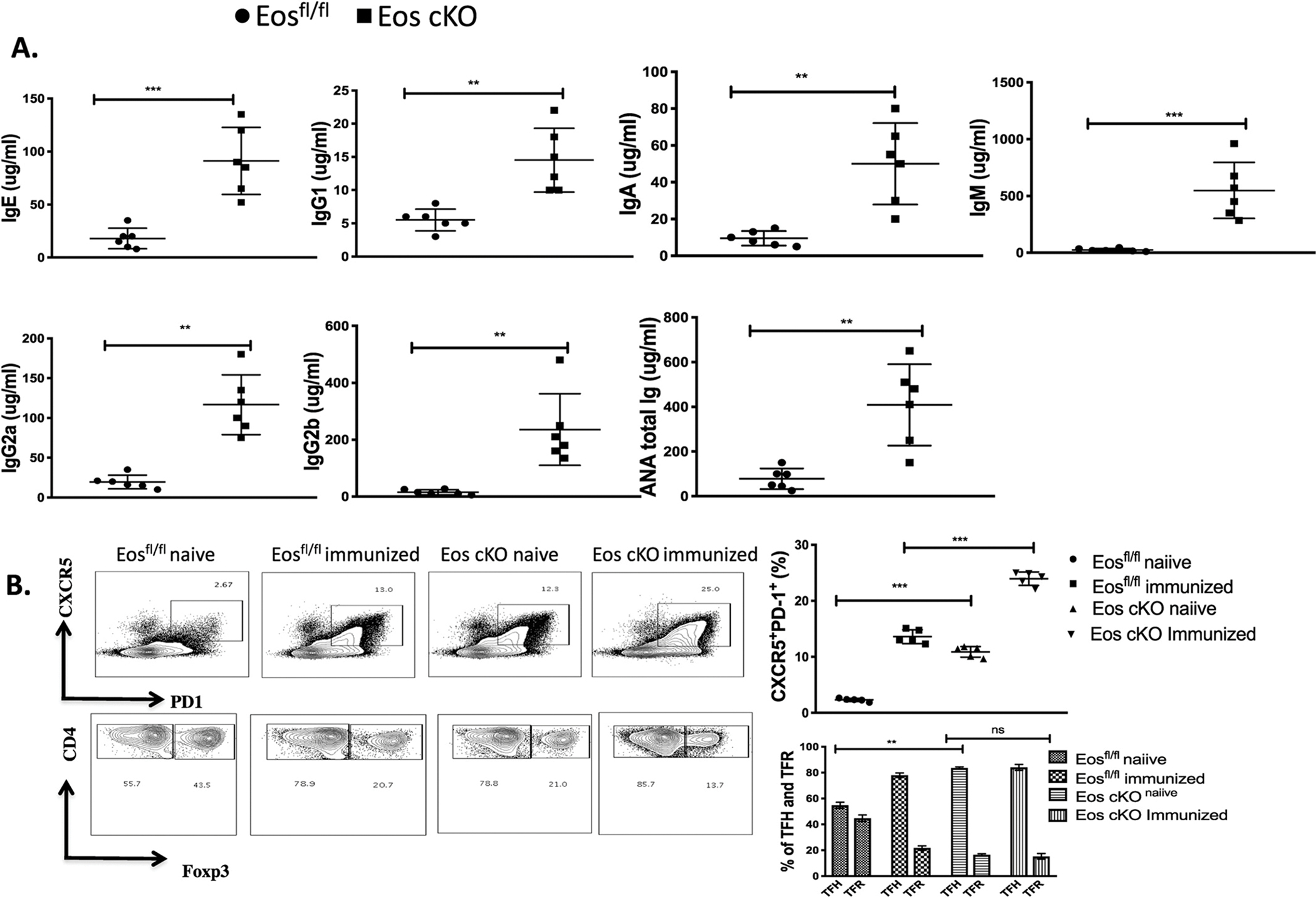
Eos cKO mice have hypergammaglobulinemia and decreased Tfr function. A) Sera from 3 month old Eosfl/fl and Eos cKO mice were analyzed by ELISA for the indicated Ig isotype and the presence of ANA. B) Eos cKO mice have increased percentages of Tfh cells and decreased percentages of Tfr cells both before and after immunization. Top: Eosfl/fl mice and Eos cKO mice were either not immunized or immunized with SRBCs and analyzed 7d post immunization. Splenocytes were gated on CD4+CD19− T cells and analyzed for CXCR5 and PD-1 expression. Bottom: CXCR5+PD-1+ cells (Tf cells) were then analyzed for Foxp3 expression to determine the percentages of Tfh and Tfr cells.
Fig. 4.

Eos cKO mice have enhanced percentages of germinal center B cells and larger and more numerous germinal centers. A) Eosfl/fl or Eos cKO mice were not immunized or immunized with SRBCs and stained for GL7+CD95+ GC B cells 7 days post immunization. B) 3-month old Eosfl/fl or cKO mice were unimmunized or immunized with SRBC. Spleen sections were frozen, sectioned, and stained for IgD (blue), CD4 (green), and GL7 (red). Quantitation of active germinal centers is shown in the right panel. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.3. Tregs from the Eos cKO mice display normal suppressive function in vitro, but defective suppressive function in vivo
Both CD4+ Foxp3− and CD8+ T cells from Eos cKO mice proliferated as well as CD4+Foxp3− T and CD8+ T cells from Eosfl/fl mice when stimulated with anti-CD3 and APC (Fig. 5A). Tregs from Eos cKO mice suppressed the proliferation of CD4+Foxp3− T cells from WT mice as efficiently as Tregs from Eosfl/fl mice (Fig. 5B). Similar results were observed when we co-cultured Tregs from global Eos KO and Eosfl/fl CD4cre mice with effectors from WT mice (Supplementary Fig. 3).
Fig. 5.
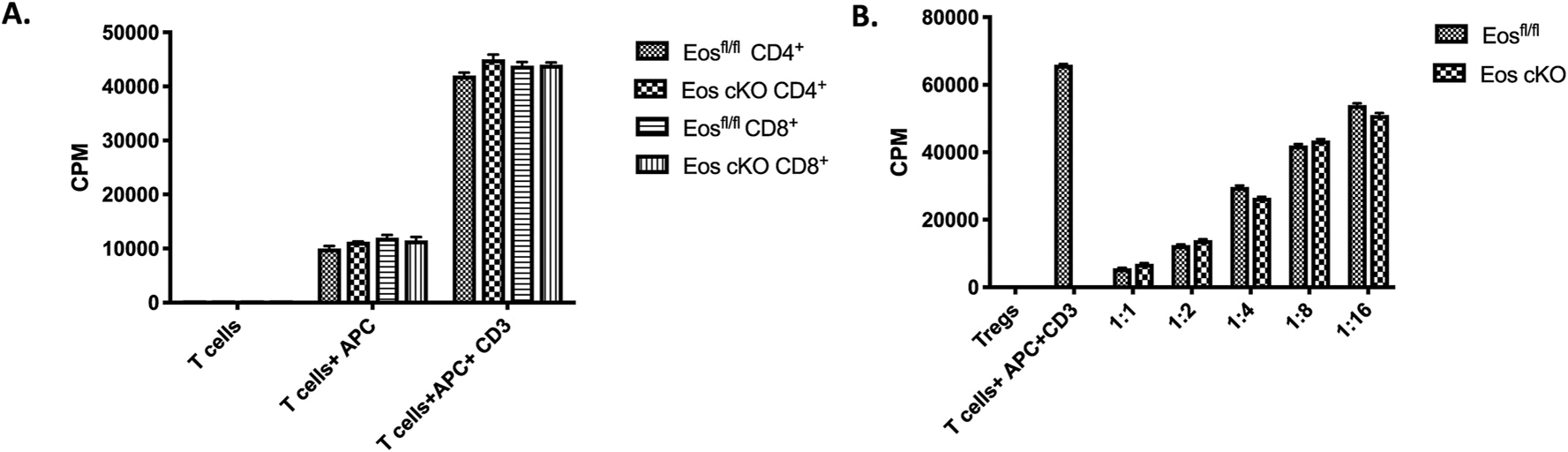
CD4+Foxp3− T cells from Eos cKO proliferate normally and Eos cKO Treg exhibit normal suppressive function in vitro. A) Eosfl/fl or Eos cKO naive CD4+Foxp3−CD44hiCD62Llo T cells (5 × 104) were stimulated with soluble anti-CD3 and irradiated T cell-depleted splenocytes (5 × 104) for 3 days. Cultures were pulsed with [3H]-thymidine for the last 6 h. B) Naïve Eosfl/fl T cells were stimulated with anti-CD3 and T cells-depleted splenocytes in the presence of graded numbers of CD4+CD25+ Treg from either Eosfl/fl or cKO mice. [3H]- thymidine was added for the last 6 h of culture.
To determine the suppressive capacity of Tregs from the Eos cKO mice in vivo, we induced IBD by the transfer of CD4+CD25−CD45RBhi T cells from WT mice to RAG−/− mice. Mice that received CD4+CD25−CD45RBhi T cells alone began losing body weight as early as week 1 and most of the mice lost more than 20% of their original body weight by week 5 (Fig. 6A); mice that received Eosfl/fl Tregs continued to gain weight. In contrast, mice that received Tregs from Eos cKO mice began to lose weight beginning at one week after transfer and continued to lose body weight during the course of the entire experiment indicating a marked defect in the suppressive function of Tregs from the Eos cKO mice (Fig. 6A). The production of single IFN-γ and IL-17 CD4+ T cells as well as double producer CD4+ T cells was greatly increased in the mesenteric LNs of mice that received the Eos cKO Tregs compared to control Tregs (Fig. 6B). The failure of the Tregs from the Eos cKO mice to protect was not secondary to a defect in cell survival or to the loss of Foxp3 expression (“ex-Tregs”) as the absolute number of Tregs recovered or their level of Foxp3 expression in mice that received Eos cKO Tregs was not statistically different from the number that received Tregs from Eosfl/fl controls (Fig. 6C and D). However, the percentages of transferred Tregs in the mesenteric LN of mice receiving Treg from the Eos cKO mice were less than those in the mice receiving control Tregs secondary to the enhanced numbers of T effector cells and the development of disease in the cKO mice (Fig. 6D).
Fig. 6.
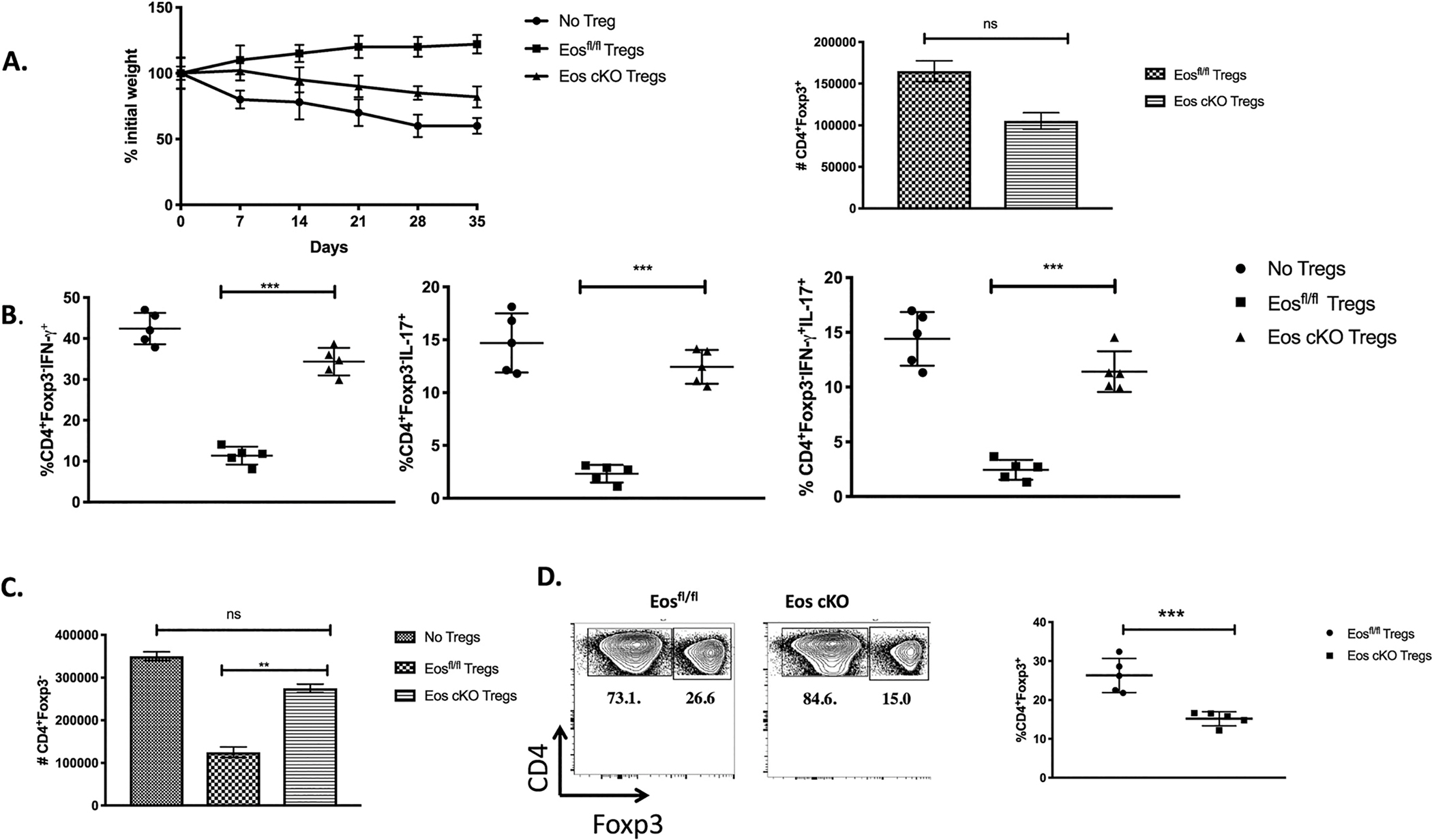
Eos cKO Treg fail to suppress IBD. A) CD4+CD25−CD45RBhi T cells from 8-week-old mice C57BL/6 mice were injected (4 × 105 cells/mouse) into 8–10-week-old RAG−/− recipients. CD4+CD25+ cells (2 × 105 cells/mouse) from 3-month old Eosfl/fl or Eos cKO mice were co-injected where indicated. Mice were monitored weekly for weight loss. Data is plotted as percent weight change from original weight and is representative of two independent experiments (n = 5 per group). Mice were sacrificed on day 35 post transfer and mesenteric lymph node cells analyzed for: B) percentages of CD4+Foxp3− producing IFN-γ, IL-17, and IFN-γ/IL-17 double producers, and C) absolute number of CD4+Foxp3− T cells, and D) percentage of CD4+Foxp3+ T cells were shown.
To determine if Eos cKO Tregs were dysfunctional in a second model of autoimmunity, we induced EAE by immunization of 3 month old Eos cKO mice with MOG35–55 in CFA. Both the Eos cKO mice and the control Eosfl/fl mice began to develop disease on day 8, but disease progression was more severe in the cKO mice and they did not show a decrease is disease severity even after day 21 (Fig. 7A). The percentages of CD4+Foxp3− T cells among total T cells were significantly higher in the spinal cord, brain, draining LNs and spleen of the Eos cKO mice compared to the controls, while the percentages of Tregs were lower in the Eos cKO mice (Fig. 7B). There was modest increase in the frequency of CD4+IL-17+ cells in the Eos cKO, but no difference in the CD4+IFN-γ+ cells, in the spinal cord, brain or the draining LNs (Fig. 7C–D). There was a modest increase in CD4+IFNγ+IL-17+ double producers in the spleen and draining LN of the cKO mice, but not in the spinal cord or brain (Fig. 7E).
Fig. 7.
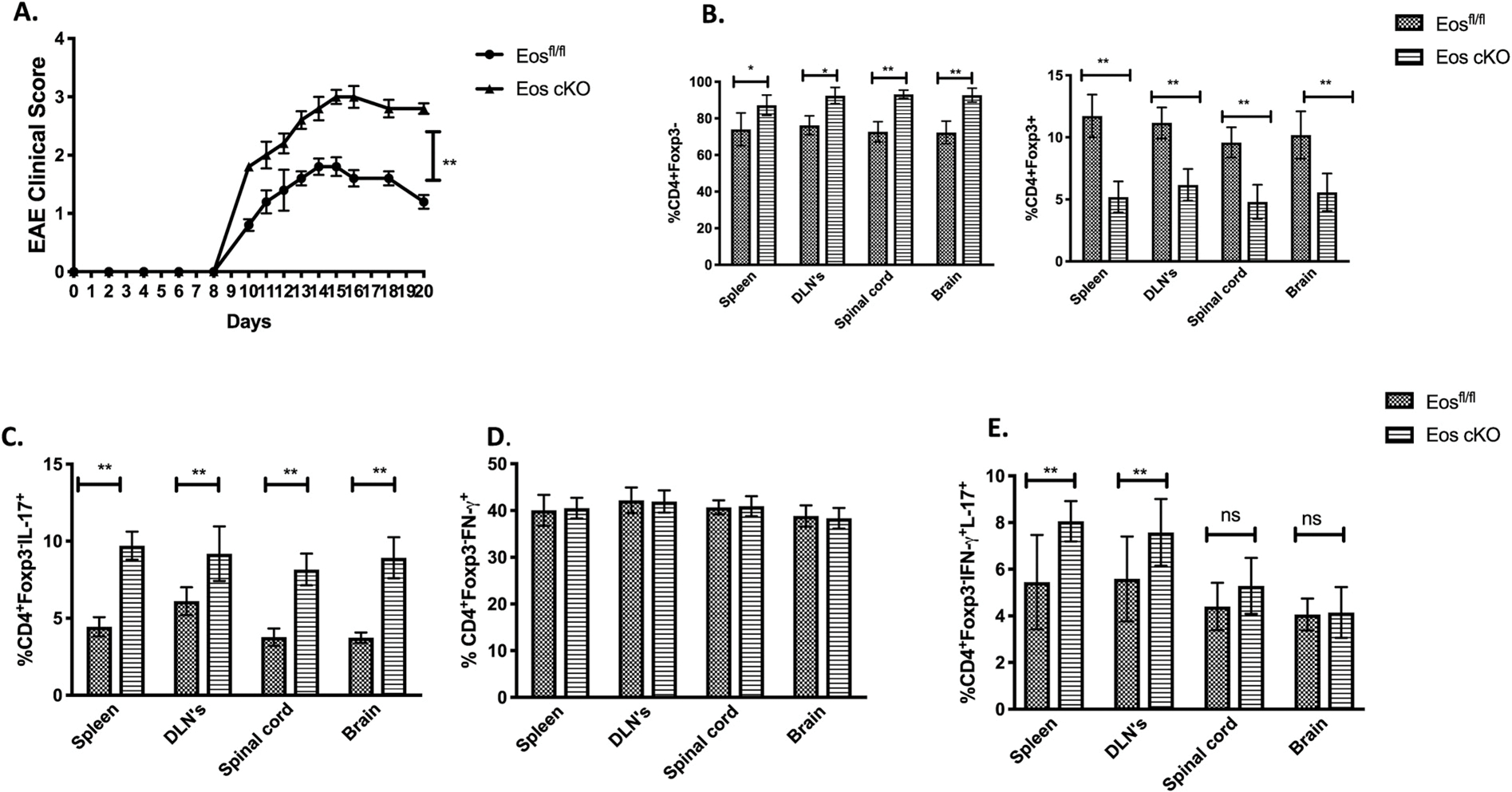
Eos cKO mice develop more severe EAE. A) Mice were immunized with MOG/CFA and injected with pertussis toxin on day 0 and 2. B) Percentages of CD4+Foxp3− (left panel) CD4+Foxp3+(right panel) T cells in the spleen, dLNs, spinal cord and brain on day 14 were shown. C) Percentage of CD4+IL-17+ D) CD4+IFN-γ+ T cells, and E) CD4+IFN-γ+IL-17+ T cells in the spleen, DLNs, spinal cord and brain of WT and Eos cKO mice on day 14 were shown. The cells were harvested and stimulated with PMA/Ionomycin for 4 h before measurement of cytokine production.
Lastly, we evaluated the suppressive capacity of the Eos cKO Treg in tumor immunity (Fig. 8A). When the colon adenocarcinoma, MC38 cells were transplanted into Eos cKO mice, tumors developed more slowly and were always smaller in size. The percentages of Treg in TIL were markedly reduced in the Eos cKO mice (Fig. 8B), while there was an increase in the percentages of CD8+ T cells resulting in an enhancement of the CD8/Treg ratio (Fig. 8C–D). CD8+ T cells from the TIL of the Eos cKO mice expressed higher amounts of IFN-γ and TNF-α than CD8+ T cells from TIL derived from control mice after restimulation with PMA/ionomycin (Fig. 8E).
Fig. 8.
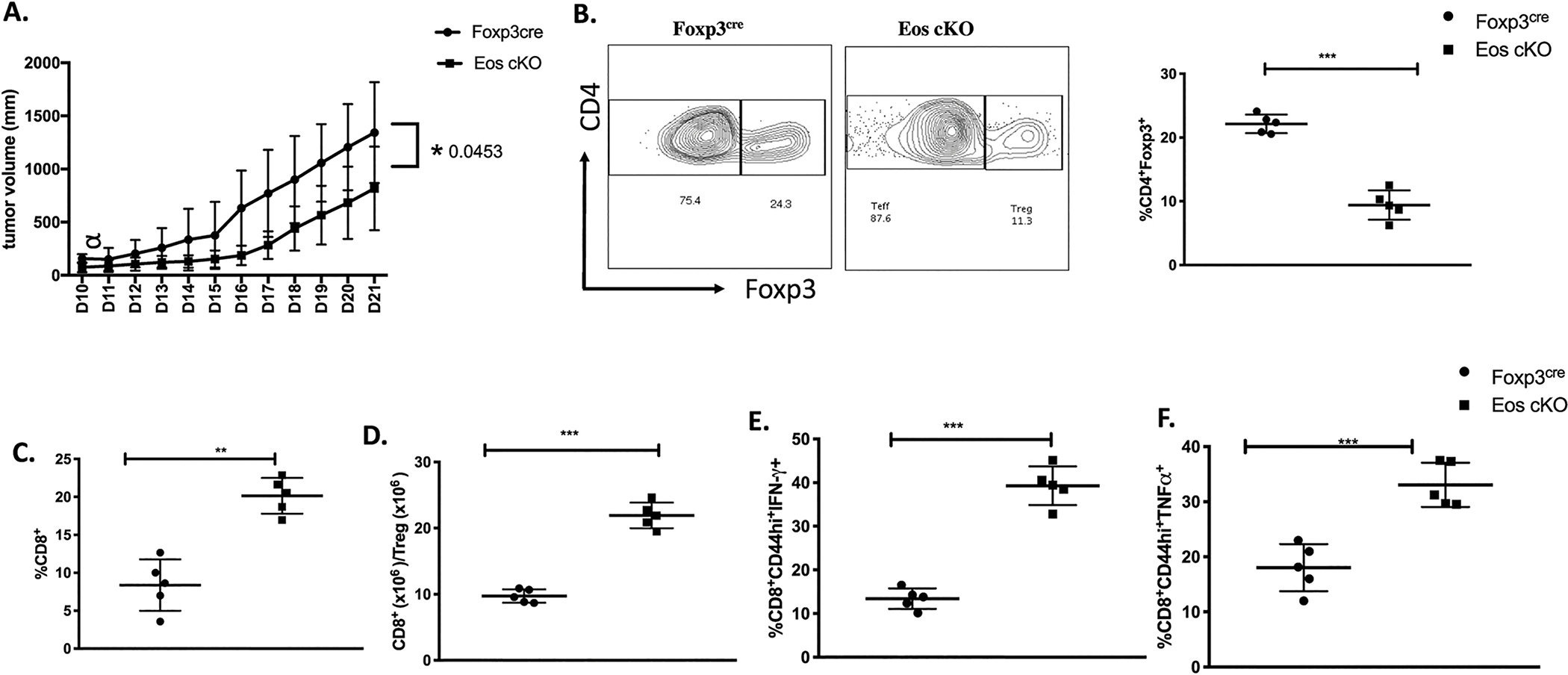
Eos cKO mice have enhanced tumor immunity. A) Foxp3YFP–cre and Eos cKO mice were injected with MC38 (2 × 105) cells subcutaneously; tumor volumes were determined daily. B) TIL were isolated on d18 after tumor implantation and the frequencies of CD4+Foxp3+ Tregs, C) CD8+ T cells and D) the ratio of CD8+ to CD4+Foxp3+ cells within TIL were determined. E) TIL from d18 post tumor implantation were stimulated with cell stimulation cocktail for 5 h at 37 °C. CD8+CD44hi T cells from TIL from d18 post tumor implants were stained for IFN-γ and TNF-α producing cells. *P < 0.05, **P < 0.01 and ***P < 0.0005 (unpaired two-tailed Student’s t-test). Data is a representative of two independent experiments (A-D) involving three to five mice per group (mean ± SEM).
3.4. Eos plays a role in the activation of CD4+Foxp3− T cells
We did not observe any signs of autoimmunity in our previous study of mice with a global deletion of Eos [9], but this study was restricted to relatively young mice (8–10 weeks of age). As the results of the present study on the cKO mice clearly demonstrate a marked defect in the suppressor function of Treg in the cKO mice, it was somewhat surprising that we did not detect any defects in the mice with the global deletion of Eos. As we had previously observed that Eos was also expressed in activated CD4+Foxp3− T cells, it remained possible that the defective Treg function in the globally deleted mice was balanced by defective activation of CD4+Foxp3− or CD8+ T cells. To explore this issue in greater depth, we generated mice with defective Eos expression in all T cells (lkzf fl/fl X CD4-Cre) and also examined the Eos global knock outs at older ages (Table 1). We first noted signs of generalized T cell activation in the lkzf fl/fl X CD4-Cre mice at 6–7 months of age, but it was not till 12 months of age that we observed T cell activation in the globally deleted mice. In both of these strains, the percentage of Foxp3+ T cells was elevated and the mice manifested cellular infiltrates in non-lymphoid organs (Table 1). Taken together, these findings are consistent with a critical role of Eos in the activation of CD4+Foxp3− and/or CD8+ T cells and the further delay in development of autoimmune disease in mice with the global deletion of Eos strongly suggest that Eos also plays a role in the activation of non-T cells or non-lymphoid cells.
Table 1.
Comparison of different Eos deficient mouse strains.
| Parameters | Eosfl/fl X Foxp3cre | Eosfl/fl X CD4cre | Eos global KO |
|---|---|---|---|
|
| |||
| Onset of Autoimmunity like symptoms | ~3 months | ~6–7 months | ~12 months |
| CD4+Foxp3+ | 22.6 | 23.2 | 22 |
| CD4+Foxp3+CD44hi | 80.4 | 80.6 | 86.2 |
| CD4+Foxp3−CD44hi | 51 | 42.8 | 67.9 |
| CD8+CD44hi | 12.3 | 15.6 | 45.7 |
| Cellular infiltration in non-lymphoid organs | Kidneys, Salivary glands, liver and lungs | Salivary glands, Lungs and Kidneys | Salivary glands, kidneys, liver and lungs |
| CXCR5+PD-1+ (Tf cells) | 11.8 | 9.5 | NA |
| Tfr Cells | 16.9 | 24.4 | NA |
| CD4+Foxp3−IFN-γ+ | 31.9 | 28.3 | NA |
| CD8+IFN-γ+ | 21 | 23.3 | NA |
The parameters are represented as average percentages in spleen tissue.
4. Discussion
The role of Foxp3+ Treg cells in the control of immune tolerance and immune homeostasis is now well established [11–14]. The mechanisms by which Treg mediate their suppressor functions remain poorly defined and include both cell contact-dependent, cytokine-dependent, and metabolic mechanisms [15,21]. Nevertheless, expression of Foxp3 is critical for maintenance of suppressive function. Foxp3 itself interacts with multiple molecular partners including numerous other transcription factors [19]. Eos, a member of the Ikaros transcription factor family has been shown to be expressed preferentially in Tregs and reduction in Eos expression using siRNA technology has been claimed not only to completely diminish Treg suppressor function, but also to convert Treg into T effector cells [8]. Other studies have shown that under certain conditions of antigen priming, Eos expression is labile in a subpopulation of Tregs, and that this labile subpopulation converts to T effector cells but maintains Foxp3 expression [20]. On the other hand, studies of mice with a global deletion of Eos have failed to demonstrate defective Treg function both in vitro and in vivo [9].
Our goal here was to define the role of Eos in Treg function by using mice with a conditional deletion of Eos in Treg cells and to compare the phenotype of these mice with mice with a deletion of Eos only in T cells and mice with a global deletion of Eos. We demonstrate that Eos is required for many, but not all, aspects of Treg suppressor function in vivo, and that Eos cKO mice do not develop a lethal autoimmune disease. Mice with a deficiency of Eos both in Treg and T effector cells as well as mice with a global deletion of Eos also develop an autoimmune phenotype similar to the cKO mice but at much older age suggesting that the defect in Treg function is counter-balanced by a requirement of Eos for the activation of all T cells and even non-T cells.
4.1. Eos is required for treg-mediated suppression
Eos cKO mice rapidly developed signs of generalized immune activation between 2 and 3 months of age. Between 3 and 6 months of age, the mice exhibited manifestations of both organ-specific autoimmunity with marked lymphocytic infiltration of non-lymphoid organs and signs of systemic autoimmunity characterized by hyperglobulinia, anti-nuclear antibodies, and enhanced germinal center formation. The enhanced antibody responses appeared secondary to a defect in the generation or function of Tfr cells. While Treg suppressor function was normal in vitro, we could demonstrate that Tregs from the cKO mice exhibited a cell intrinsic defect in suppressor function in vivo in the IBD transfer model and that the Eos cKO mice developed a more severe EAE than control strains, but also exhibited enhanced anti-tumor responses when challenged with a colon adenocarcinoma. Although we did not prove that the latter two phenotypes were definitively secondary to defective Treg suppressor function, the results are consistent with this possibility. Although these studies indicate that multiple aspects of Treg suppressor function are compromised in the absence of Eos, the mice did not exhibit weight loss and were viable for periods of as long as one year when we terminated the studies. Thus, Eos is critical for certain aspects of Treg-mediated suppression, but not others. The Eos cKO mice represent an excellent model for the study of the evolution and potential treatment of a chronic autoimmune disease.
4.2. Comparison of Eos cKO mice with helios cKO mice
We have previously characterized mice with a conditional deletion of a second member of the Ikaros family, Helios (lkzf2), in Treg [5,22]. There are both similar and different aspects of the phenotype of autoimmune diseases that develops in these two strains. Helios cKO mice develop signs of systemic autoimmunity with generalized T cell activation with a Th1 phenotype, hypergammaglobulinemia, and abnormal Tfr cell function in a manner similar to the Eos cKO mice, but at a much slower pace (between 4 and 6 months of age). Surprisingly, in contrast to the Eos cKO mice, they never develop signs of organ-specific infiltrates in liver, lung, bowel or kidney, but do develop an autoimmune lipodystrophy and the metabolic syndrome. While the Eos cKO mice live for the duration of the experiments, the Helios cKO require euthanasia between 8 and 12 months of age due to massive hepatomegaly secondary to the metabolic syndrome. The difference in disease phenotypes is not secondary to differences in the strains as both cKO have been bred for multiple generations on the C57BL/6 backgrounds. Both Eos and Helios can homodimerize as well as heterodimerize with other members of the Ikaros gene family [2] and it is likely that differences in this process account for the differences in disease phenotypes. In this regard, it would be of interest to breed the two cKO strains together as well as to mice with a deletion of Ikaros (lkzf1) in peripheral T cells [23].
4.3. Comparison of our results with earlier studies
It is difficult to reconcile our results with the findings of Pan et al. [8] who used siRNA technology to knock down Eos expression in Tregs resulting in a dramatic loss of Treg suppressor function in vitro and in vivo. One might have predicted from these experiments that a genetic loss of Eos expression in Treg would have resulted in mice with a phenotype similar to that of Scurfy mice that have a complete loss of Foxp3 expressing Treg and would have succumbed to autoimmune disease at 3–4 weeks of age. Pan et al. [8] also claimed that Eos is physically associated with Foxp3 which was not found in extensive pull down mass spectrometry studies reported by Rudra et al. [19]. Sharma et al. [20] reported that Treg in the tumor microenvironment lost expression of Eos, but maintained Foxp3 expression, in the presence of T effector cells stimulated by antigen in the presence of a TLR ligand resulting in conversion of the Treg that had lost Eos to a T helper-like phenotype. In our studies, we found no evidence for effector cytokine production by Eos deficient Treg cells and mice that developed an autoimmune phenotype had a higher percentage of Treg cells expressing normal levels of Foxp3 than controls consistent with a model in which the Eos deficient Treg cells were attempting to control the autoreactive effector cells by increasing their numbers, but failing in this attempt. One difficulty in interpreting the earlier studies on Eos expression in Treg is the lack of a mAb that would allow detection of Eos expression at a single cell level on the FACS. The studies of Pan et al. [8] and Sharma et al. [20] used polyclonal anti-Eos antibodies generated against peptides derived from Eos. In our hands, such reagents frequently reacted with cells from the Eos global knock out mice and most likely recognized other members of the Ikaros gene family or other unknown specificities. Further studies of the function of Eos at the cellular and molecular levels will require more specific reagents.
4.4. Role of Eos in non-treg cells
In addition to comparing our results with those of other groups, we addressed discrepancies between the present studies and those we previously published using the Eos global knock out mouse [8]. For this purpose, we also generated a mouse deficient in Eos in all T cells using CD4-cre. A comparison of all three of the strains revealed that mice with a deficiency of Eos in all T cells and in all cells do develop the same phenotype of autoimmunity as the Eos cKO mouse but with delayed kinetics. Previous studies [8,24] have reported that Eos expression is upregulated on the activated CD4+Foxp3− T cells. The simplest explanation for these findings is that Eos is required for the activation and development of pathogenic T effector cells and in the absence of Eos, effector T cells can still be suppressed by the defective Eos deficient Tregs. The delay in development of disease in the global knock out compared to the mouse with Eos deficiency only in T cells strongly suggests that Eos expression in non-T cells, perhaps in antigen presenting cells or cells of the innate immune system, plays an indirect role in the development of pathogenic T effector cells. One other aspect of this comparison of the different strains is that purified Tregs from young Eos global knock out mice suppressed the development of IBD induced by WT T effectors [9], while Tregs from somewhat older cKO mice were defective in the same assay. One intriguing interpretation of this result is that the defect in Treg suppressor function in the Eos cKO mice evolves with age. A similar gradual loss of suppressor function has been observed in mice with a Treg conditional knock out of Tcf1 and Lef1 [25]. Alternatively, some of the suppressor mechanisms may be intact and normally used in young mice, but these mechanisms are not used as the mouse age. It also remains possible that some of the Treg subpopulations that are present in young mice do not survive and are replaced by newly generated Treg that use different suppressor mechanisms. If defects in Treg function are one of the fundamental causal factors in the development of autoimmune disease as has been proposed in multiple studies [26,27], these defects in human Treg function may also be acquired over many years. Further study at the molecular and single cell level will be required to address this concept of acquired Treg cell deficiency, but it has multiple implications for approaches to the enhancement of Treg function for the treatment of autoimmunity by cellular immunotherapy, biologics, or small molecules.
5. Conclusions
Expression of Eos in Treg cells is required for their suppressive function in vivo. Mice with a conditional deletion of Eos in Treg develop both non-lethal organ-specific and systemic autoimmunity. However, some aspects of their suppressive function remain intact as the cKO mice can live as long as one year. Deletion of Eos in either all cells or all T cells also results in the development of a similar spectrum of autoimmune disease, but at much older age. The expression of Eos in both all T cells and non-T cells is likely to play a role in cellular activation.
Supplementary Material
Acknowledgments
Authors would like to thank and acknowledge Sundar Ganesan, Staff Scientist, Biological Imaging Facility at NIAID for his help in acquiring confocal images of the germinal centers and follicles in spleen; Debbie Glass for irradiation of DC’s; Ke Wang, Flow Cytometry Section, NIAID for the assistance in cell sorting and Dr. Sadiye Reider for her guidance. This work is supported by the Division of Intramural Research, NIAID. NIH. The authors declare no competing financial interests.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jaut.2019.06.011.
References
- [1].Honma Y, Kiyosawa H, Mori T, Oguri A, Nikaido T, Kanazawa K, et al. , Eos: a novel member of the Ikaros gene family expressed predominantly in the developing nervous system, FEBS Lett. 447 (1999) 76–80. [DOI] [PubMed] [Google Scholar]
- [2].Georgopoulos K, Winandy S, Avitahl N, The role of Ikaros in lymphocyte development and homeostasis, Annu. Rev. Immunol. 15 (1997) 155–176. [DOI] [PubMed] [Google Scholar]
- [3].Perdomo J, Holmes M, Chong B, Crossley M, Eos and pegasus, two members of the Ikaros family of proteins with distinct DNA binding activities, J. Biol. Chem. 275 (2000) 38347–383454. [DOI] [PubMed] [Google Scholar]
- [4].Bao J, Lin H, Ouyang Y, Lei D, Osman A, Kim TW, et al. , Activity-dependent transcription regulation of PSD-95 by neuregulin-1 and Eos, Nat. Neurosci. 7 (2004) 1250–1258. [DOI] [PubMed] [Google Scholar]
- [5].Sebastian M, Lopez-Ocasio M, Metidji A, Rieder SA, Shevach EM, Thornton AM, Helios controls a limited subset of regulatory T cell functions, J. Immunl. 196 (2016) 144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Quintana FJ, Lin H, Burns EJ, Nadeau M, Yeste A, Kumar D, et al. , Aiolos promotes TH17 diffentiation by directly silencing IL2 expression, Nat. Immunol. 13 (2012) 770–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wang JH, Avitahl N, Cariappa A, Friedrich C, Ikeda T, Renold A, et al. , Aiolos regulates B cell activation and maturation to effector state, Immunity 9 (1998) 543–553. [DOI] [PubMed] [Google Scholar]
- [8].Pan F, Yu H, Dang EV, Barbi J, Pan X, Grosso JF, et al. , Eos mediates Foxp3-dependent gene silencing in CD4+ regulatory T cells, Science 325 (2009) 1142–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rieder SA, Metidji A, Glass DD, Thornton AM, Ikeda T, Morgan BA, et al. , Eos is redundant for regulatory T cell function but plays an important role in IL-2 and Th17 production by CD4(+) conventional T cells, J. Immunol. 195 (2015) 553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fu W, Ergun A, Lu T, Hill JA, Haxhinasto S, Fassett MS, et al. , A multiply redundant genetic switch ‘locks in’ the transcriptional signature of regulatory T cells, Nat. Immunol. 13 (2012) 972–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sakaguchi S, Wing K, Yamaguchi T, Dynamics of peripheral tolerance and immune regulation mediated by Treg, Eur. J. Immunol. 39 (2009) 2331–23366. [DOI] [PubMed] [Google Scholar]
- [12].Sakaguchi S, Regulatory T cells: history and perspective, Methods Mol. Biol. 707 (2011) 3–17. [DOI] [PubMed] [Google Scholar]
- [13].Sakaguchi S, Wing K, Miyara M, Regulatory T cells - a brief history and perspective, Eur. J. Immunol. 37 (2007) S116–S623. [DOI] [PubMed] [Google Scholar]
- [14].Fontenot JD, Gavin MA, Rudensky AY, Foxp3 programs the development and function of CD4(+)CD25(+) regulatory T cells, Nat. Immunol. 4 (2003) 330–336. [DOI] [PubMed] [Google Scholar]
- [15].Shevach EM, Mechanisms of Foxp3(+) T regulatory cell-mediated suppression, Immunity 30 (2009) 636–645. [DOI] [PubMed] [Google Scholar]
- [16].Belkaid Y, Blank RB, Suffia I, Natural regulatory T cells and parasites: a common quest for host homeostasis, Immunol. Rev. 212 (2006) 287–300. [DOI] [PubMed] [Google Scholar]
- [17].Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL, CD4(+)CD25(+) regulatory T cells control Leishmania major persistence and immunity, Nature 420 (2002) 502–507. [DOI] [PubMed] [Google Scholar]
- [18].Facciabene A, Motz GT, Coukos G, T-regulatory cells: key players in tumor immune escape and angiogenesis, Cancer Res. 72 (2012) 2162–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rudra D, deRoos P, Chaudry A, Niec RE, Arvey A, Samstein RM, et al. , Transcription factor Foxp3 and its protein partners form a complex regulatory network, Nat. Immunol. 13 (2012) 1016–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sharma MD, Huang L, Choi JH, Lee EJ, Wilson JM, Lemos H, et al. , An inherently bifunctional subset of Foxp3+ T helper cells is controlled by the transcription factor eos, Immunity 38 (2013) 998–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Schmidt A, Oberle N, Krammer PH, Molecular mechanisms of treg-mediated T cell suppression, Front. Immunol. 3 (2012) 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. , Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3(+) T regulatory cells, J. Immunol. 184 (2010) 3433–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].de Ana CL, Arakcheeva K, Agnihotri P, Derosia N, Winandy S, Lack of Ikaros deregulates inflammatory gene programs in T cells, J. Immunol. 202 (2019) 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Campos-Mora M, Morales RA, Perez F, Gajardo T, Campos J, Catalan D, et al. , Neuropilin-1+ regulatory T cells promote skin allograft survival and modulate effector CD4+ T cells phenotypic signature, Immunol. Cell Biol. 93 (2015) 113–119. [DOI] [PubMed] [Google Scholar]
- [25].Xing S, Gai K, Shao P, Zheng Z, Zhao X, Zhao X, et al. , Tcf1 and Lef1 are required for the immunosuppressive function of regulatory T cells, J. Exp. Med. 216 (2019) 847–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kasper IR, Apostolidis SA, Sharabi A, Tsokos GC, Empowering regulatory T cells in autoimmunity, Trends Mol. Med. 22 (2016) 784–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Buckner JH, Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases, Nat. Rev. Immunol. 10 (2010) 849–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.