

Significance
Scramblases, proteins that equilibrate glycerophospholipids between the leaflets of membrane bilayers, are essential for the synthesis of new membranes for organellar maintenance or biogenesis. Yet, their identity is largely unknown. Biochemical and in silico data presented here indicate that another group of proteins that also functions in organelle maintenance and biogenesis, protein insertases that translocate peptide substrates across membranes, have the ability to scramble lipids. Cells lacking insertases are known to exhibit defects in membrane morphology or lipid metabolism consistent with a role for insertases as scramblases. These findings suggest insertases as among the elusive scramblases that participate in membrane dynamics.
Keywords: membrane biology, lipid transport, membrane proteins, molecular dynamics, cellular organelles
Abstract
Glycerophospholipids are synthesized primarily in the cytosolic leaflet of the endoplasmic reticulum (ER) membrane and must be equilibrated between bilayer leaflets to allow the ER and membranes derived from it to grow. Lipid equilibration is facilitated by integral membrane proteins called “scramblases.” These proteins feature a hydrophilic groove allowing the polar heads of lipids to traverse the hydrophobic membrane interior, similar to a credit card moving through a reader. Nevertheless, despite their fundamental role in membrane expansion and dynamics, the identity of most scramblases has remained elusive. Here, combining biochemical reconstitution and molecular dynamics simulations, we show that lipid scrambling is a general feature of protein insertases, integral membrane proteins which insert polypeptide chains into membranes of the ER and organelles disconnected from vesicle trafficking. Our data indicate that lipid scrambling occurs in the same hydrophilic channel through which protein insertion takes place and that scrambling is abolished in the presence of nascent polypeptide chains. We propose that protein insertases could have a so-far-overlooked role in membrane dynamics as scramblases.
A defining feature of eukaryotic cells is the presence of membrane bilayers that separate them from their environment and delineate intracellular organelles with specialized functions. Lipids, the main building blocks of membranes, are primarily synthesized in the cytosolic leaflet of the endoplasmic reticulum (ER) (1). From there, since most of them are unable to spontaneously translocate between leaflets due to the associated high energy barrier for interleaflet crossing, they are equilibrated to the ER’s luminal leaflet by integral membrane proteins called “scramblases,” allowing expansion of the ER membrane and vesicles that bud from it (2). Alternatively, lipids from the ER’s cytosolic leaflet can be transported to the cytosolic leaflet of another organelle by lipid transport proteins (3). At the receiving organelle, lipids must also be scrambled between membrane leaflets to allow for its membrane expansion. This is the case for autophagosomes (4–6), and likely for organelles disconnected from vesicle trafficking pathways, like mitochondria, that rely on protein-mediated transport rather than vesicle trafficking for both their protein and membrane lipid supply (4, 7).
While the role of scramblases in membrane biogenesis and homeostasis is widely accepted (8, 9), their identity is mostly unknown and only a handful of scramblases have been identified and characterized. These include mainly plasma membrane proteins, such as the well-studied TMEM16 (10–12) and XK families that scramble phosphatidylserine during apoptosis (9). Lipid scramblases of intracellular organelles have been more elusive (9), but recently discovered ER scramblases, VMP1 and TMEM41B, are proposed to work in combination with lipid transport proteins to facilitate lipid transport from the ER (4, 13, 14). A common feature of protein scramblases is the presence of a hydrophilic groove facing the hydrophobic membrane core which allows lipids to slide between hydrophilic membrane surfaces, much like a credit card through a reader (“credit-card model”) (15). Additionally, these proteins may facilitate scrambling by locally thinning the membrane, shortening the distance that lipid headgroups must traverse to cross the bilayer (16). Most likely, one or both of these features are shared by the still unidentified scramblases with roles in membrane biogenesis.
Similar structural features, i.e., the presence of a hydrophilic channel in the intermembrane space and the ability to locally thin membranes (17, 18) are shared by another family of integral membrane proteins that is localized in both the ER and organelles disconnected from vesicle trafficking, like mitochondria: protein insertases that translocate peptides across membranes. These structural analogies prompted us to investigate whether insertases could also function as lipid scramblases, thus playing a role not only in nonvesicular protein trafficking but also in nonvesicular lipid transfer. Here, we present in vitro and in silico evidence that lipid scrambling activity is a general feature of protein insertases. We propose that this class of proteins may be among the elusive scramblases with roles in membrane dynamics and expansion.
Results
In Vitro Investigation of Insertase Lipid Scrambling Function.
To investigate whether insertases have the ability to scramble lipids, we reconstituted a subset of known insertases into liposomes for use in a well-established fluorescence-based lipid scrambling assay (19–21) (Fig. 1A). In this assay, bovine serum albumin (BSA) is added to liposomes or proteoliposomes comprising a small percentage (0.5%) of short-chain nitrobenzoxadiazole (NBD)-labeled lipids distributed evenly between both bilayer leaflets. BSA extracts NBD-labeled lipids from the outer leaflet of liposomes, and because the fluorescence of NBD-lipids is reduced by ~50% upon binding by BSA, a ~25% decrease in fluorescence is observed. In the presence of a scramblase, over time, all NBD-lipids in the liposome bilayer become accessible to BSA, allowing for a larger fluorescence reduction of up to 50%, although in practice the reduction is often smaller (35 to 45%) because a fraction of liposomes is refractory to reconstitution or because some liposomes contain internal membranes. Note that for most known scramblases, the scrambling rate is faster than BSA diffusion/back extraction used to monitor the reaction (6, 21–23), so that the assay does not inform as to detailed kinetics. Note also that in contrast to a similar assay that uses dithionite to reduce surface-accessible NBD (21), the BSA back extraction assay can also be used with pore-forming proteins (since BSA is too large to enter the liposome lumen through the pore), and it is thus well suited to assay scramblase candidates of unknown structure or oligomeric state, including those whose pore-forming ability is unknown.
Fig. 1.

Multiple protein insertases have lipid scrambling activity in vitro. (A) Schematic of the BSA back extraction assay. (B–D) Members of the Oxa1 superfamily (YidC, Get1, and the Get1/2 complex) can scramble glycerophospholipids. (E and F) The β-barrel membrane protein insertase, Sam50 in complex with Sam35 and Sam37, and the bacterial ortholog of Sam50, BamA, have scrambling activity. (G) The outer mitochondrial membrane insertase MTCH2 scrambles. (H) Oxa1 itself scrambles. (I and J) Negative controls, GlpG and VAMP2, do not scramble. Proteoliposomes used in the assays were analyzed by SDS-PAGE (Insets) to confirm efficient reconstitution; approximate numbers for proteins/liposome were estimated assuming 50% recovery of lipids after reconstitution. (See Methods for exact liposome compositions, details of which varied according to experimentalist, and SI Appendix, Fig. S2 for size exclusion chromatographs and SDS PAGE analyses of the purified proteins). The experiments to monitor fluorescence were conducted using a platereader; breaks in the x (time)-axis, corresponding to BSA addition, are indicated. Since the scrambling reactions go to completion within this time window [except for Oxa1, in panel (H), reconstituted into liposomes at higher concentration], the assays are qualitative for scrambling. See SI Appendix, Fig. S4 for spectrofluorometer data, including data that Oxa1 reconstituted at lower concentration has robust scrambling activity.
We reconstituted a recently identified mitochondrial human insertase MTCH2 (24) as well as members of the well-studied Oxa1 (25) and Omp85 (26) superfamilies. Both MTCH2 and the Oxa1 proteins feature all-alpha-helical transmembrane (TM) domains, whereas the Omp85 proteins are beta-barrels (SI Appendix, Fig. S1). Among Oxa1 proteins, we investigated the inner mitochondrial membrane protein Oxa1 itself, the ER-resident Guided Entry of Tail-anchored proteins (GET) complex (WRB-CAML complex in metazoa), and the bacterial insertase YidC. In the Omp85 family, we investigated bacterial BamA as well as the Sorting and Assembly Machinery (SAM) complex of the outer mitochondrial membrane. The insertases were isolated in detergent, purified sequentially by affinity and size exclusion chromatography (SI Appendix, Fig. S2), and reconstituted into liposomes using the swelling method (27); then, the resulting mixture of liposomes and proteoliposomes was further purified by flotation in a density gradient, which allowed removal of unreconstituted proteins and defective liposomes (SI Appendix, Fig. S3). For those proteins for which the reconstitution into proteoliposomes was less efficient, we also discarded the protein-devoid liposomes in the very top-most fraction of the density gradient. YidC (Escherichia coli), the Get1 subunit of the GET complex (S. cerevisiae), the GET complex (comprising both Get1 and Get2 from S. cerevisiae), BamA (E. coli), the SAM complex (comprising Sam50, Sam35, and Sam37 from S. cerevisiae), and MTCH2 (Homo sapiens) all scrambled lipids robustly in the BSA back extraction assay (Fig. 1 B–G and SI Appendix, Fig. S4). Oxa1 (S. cerevisiae) also scrambled lipids robustly when reconstituted at lower concentrations (SI Appendix, Fig. S4), but rates were slower than BSA back extraction at higher concentrations (Fig. 1H and SI Appendix, Fig. S4). A likely explanation is that proteoliposomes reconstituted with higher concentrations of Oxa1 clustered (SI Appendix, Fig. S4), reducing BSA access and resulting in slowed back extraction. As reported previously, the TM protease GlpG did not scramble (4), nor did the SNARE VAMP2 (Fig. 1 I and J). Thus, these data support the hypothesis that scrambling activity might be a general property of insertases.
High-Throughput Coarse-Grain Molecular Dynamics Simulations Are Predictive for Scrambling Activity by Proteins.
To more broadly investigate lipid scrambling by insertases, we opted to use molecular dynamics (MD) simulations at the coarse-grain (CG) level of theory, since this methodology has been shown to reproduce well the activity of various scramblases (28, 29), and thus provides a cost-effective alternative to experimental approaches. In short, after in silico reconstitution of proteins into model lipid bilayers, the interleaflet dynamics of all lipid molecules in the system were followed over time, and transbilayer movement of individual lipids was quantified (Fig. 2A).
Fig. 2.

CG simulations recapitulate known activity of lipid scramblases. (A) Protocol used to quantify lipid scrambling in CG-MD simulations. For denoising purposes, the time traces for each final angle were smoothened by averaging over a 50-ns window and data points were collected each 25 ns. A scrambling event was counted when a lipid in the upper leaflet moved to an angle greater than 125°, and when a lipid in the bottom leaflet moved to an angle lower than 55°. These thresholds are highlighted by green lines. (B) CG-MD simulations reproduce lipid scrambling activity by known lipid scramblases of different structures and oligomerization states. (C) CG-MD simulations correctly reproduce lack of lipid scrambling activity by proteins that do not have scrambling activity in vitro. AlphaFold structures are denoted by the * symbol, oligomerization state is in parenthesis.
As a first step, we validated our approach by investigating lipid scrambling in silico for multiple known lipid scramblases, ranging over a diverse set of 3D structures, folds, oligomeric states, and organisms (Fig. 2B). Our dataset includes TMEM16F (12), TMEM41B (4, 14), VMP1 (4, 14), ATG9 (4–6), VDAC1 and VDAC2 (28), Rhodopsin (21), and MCP1 (30) (Fig. 1B). For all proteins, two replicates of 10 μs were run and multiple lipid scrambling events were observed during the MD trajectory, in agreement with the available experimental results. For several proteins, various oligomeric states as reported in the literature were tested (SI Appendix, Fig. S5), and the observed trends for lipid scrambling agree with available experimental data. These include, for example, the dimerization requirement for VDAC beta-barrels for proper lipid scrambling, or the higher activity for VDAC2 with respect to VDAC1 (28) (Fig. 2B and SI Appendix, Fig. S5).
Next, we tested our methodology for several negative controls, i.e., proteins that have been shown to not have lipid scrambling activity. In addition to pure lipid bilayers, where no scrambling was observed (Fig. 2C), we investigated three bona fide negative controls, the rhomboid protease GlpG (4), the lipid synthase DGGGp (14, 31) and the SNARE protein VAMP2 (this work). In addition, we tested proteins that are not supposed to work as lipid scramblases, such as two lysolipid flippases, Spns1 (32) and Mfsd2a (33), and six lipid flippases, ABCB1 (34), ABCB4 (35), ABCB11 (36), MsbA (37), PglK (38), and SERCA2b (39), including in different conformational states along the lipid flipping cycle (Fig. 2C). In all cases, no or negligible lipid transbilayer movement was observed (Fig. 2C). Finally, we tested the ability of our MD protocol to discriminate between known active (open) and inactive (closed) states of scramblases (40, 41). To this end, we tested different conformations (open vs. closed) of human TMEM16K and, in agreement with in vitro experiments (40, 41) and previous Coarse-Grained Molecular Dynamics (CG-MD) (40) and all-atom simulations (16) we observed significant scrambling activity exclusively in the open conformations (SI Appendix, Fig. S6).
Overall, our data suggest that CG-MD simulations can reproduce the experimentally characterized lipid scrambling activity of membrane proteins, including its dependency on protein conformation and oligomerization state.
Protein Insertase Complexes Have Scrambling Activity In Silico.
To further support our hypothesis that protein insertases could function as lipid scramblases, we first used CG-MD simulations to investigate lipid scrambling for several members of the Oxa1 family in their monomeric form. In addition to Get1, Oxa1, and YidC, as in the in vitro experiments above, we also investigated MisCB (Bacillus subtilis), OXA1L (H. sapiens), Cox18 (S. cerevisiae), Alb3 (Arabidopsis thaliana), Emc3 (S. cerevisiae), and TMCO1 (H. sapiens). Using our approach, we could indeed observe that all tested Oxa1 family insertases can scramble lipids in silico (Fig. 3A).
Fig. 3.
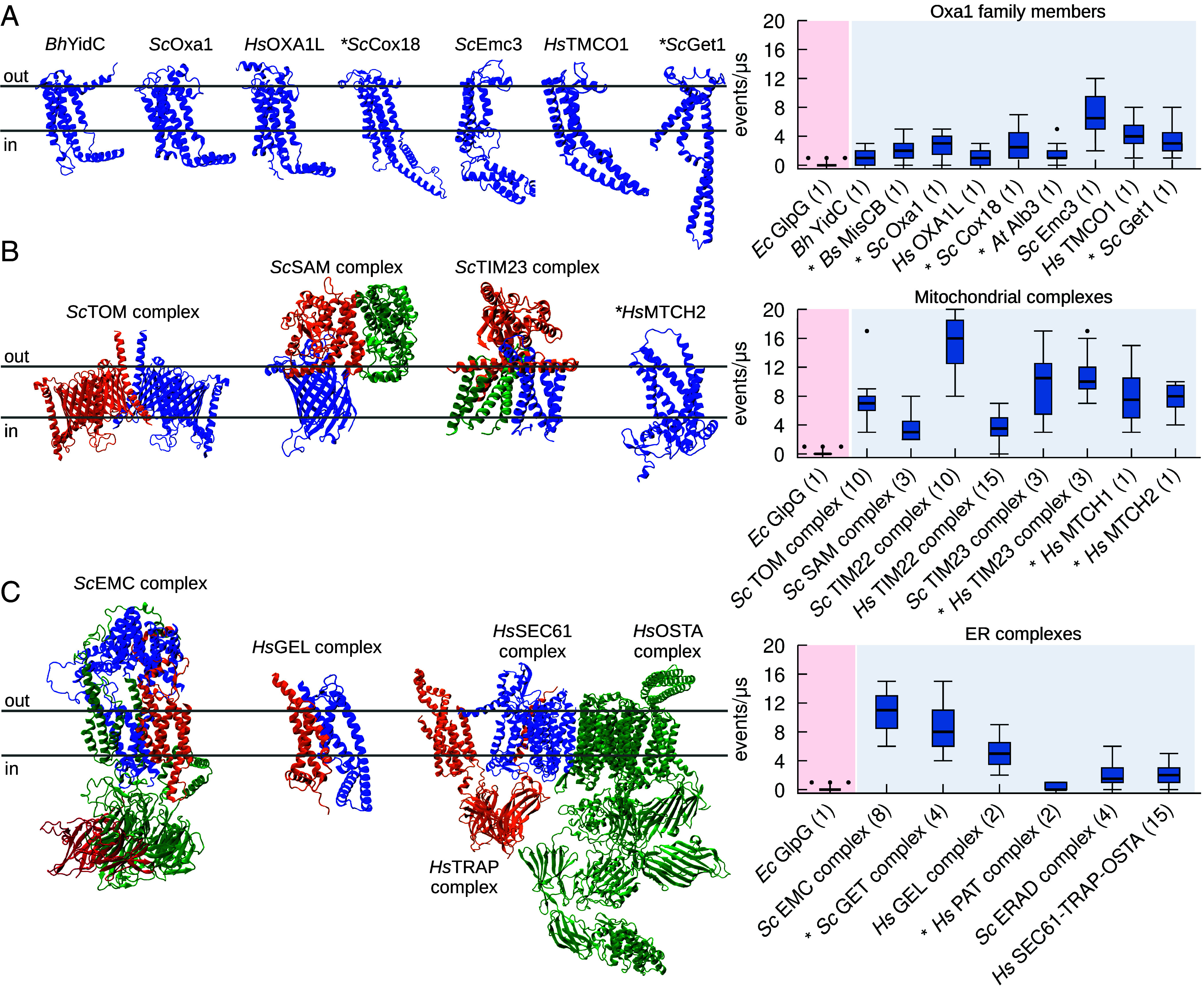
CG-MD simulations identify protein insertase complexes as lipid scramblases. (A) All members of the Oxa1 family of insertases have in silico lipid scrambling activity in their monomeric form. Left: 3D structure of selected members of the Oxa1 family. Right: In silico lipid scrambling quantification. The negative control EcGlpG is shown as reference. (B) Mitochondrial insertase complexes have in silico lipid scrambling activity. Left: 3D structure of selected mitochondrial insertase complexes. Right: In silico lipid scrambling quantification. (C) ER insertase complexes have in silico lipid scrambling activity. Left: 3D structure of selected ER insertase complexes. Right: In silico lipid scrambling quantification. AlphaFold structures are denoted by the * symbol, number of proteins in the complex is in parenthesis.
Next, since several Oxa1 family proteins, such as ScGet1, ScEmc3, and HsTMCO1 are subunits of larger dedicated protein insertion complexes, such as the GET- (42), ER membrane protein- (EMC) (43), and GET- and EMC-like- (GEL) (44) complexes, respectively, we extended our simulations to all the major protein insertase complexes. In addition to the SAM complex and MTCH2 studied biochemically (Fig. 1), we investigated the mitochondrial Translocase of the Outer Membrane (TOM) (45), Translocase of the Inner Membrane 22 (TIM22) (46, 47), and Translocase of the Inner Membrane 23 (TIM23) (48) complexes, (Fig. 3B). Our results indicate that all these mitochondrial complexes which engage in protein insertion, translocation or assembly into the membrane have clear scramblase activity in silico (Fig. 3B). In addition, insertases in the SoLute Carrier (SLC) family such as MTCH1 and MTCH2 also presented scrambling activity (Fig. 3B and SI Appendix, Figs. S7–S9).
Next, we focused on the major ER protein insertion complexes. In addition to the GET complex studied in vitro, we examined GEL, EMC, Protein Associated with Translocon (PAT) (49), ER-Associated protein Degradation (ERAD) (50), Back Of Sec (BOS) complex (51), SEC61, TRanslocon-Associated Protein (TRAP), and OligoSaccharylTransferase A (OSTA) complexes (52) (Fig. 3C). Again, all these ER complexes display lipid scrambling activity in silico (Fig. 3C). Interestingly, even for the only insertase complex showing low activity in our simulations (PAT, Fig. 3C), we were able to identify a component with high lipid scrambling activity: Asterix (SI Appendix, Figs. S10 and S12). A caveat is that in the absence of any experimental structure, we relied entirely on the AlphaFold-derived structures of both Asterix and the complex. In the AlphaFold structure, which is consistent with the cryo-EM structure for PAT in a multipass translocon (51), Asterix lipid scrambling ability is inhibited by its interaction with its binding partner CCDC47 (SI Appendix, Figs. S10 and S12), but we cannot exclude that the AlphaFold prediction for the complex is inaccurate, making a conclusion that Asterix does not scramble in the PAT complex premature.
Notably, our results show that lipid scrambling activity is promoted by specific proteins in the complexes, and that not all components of these complexes scramble lipids (SI Appendix, Figs. S7, S10, and S13). Interesting examples in this context are the mitochondrial ScTIM23 complex and the ER ScEMC. ScTIM23 is formed by three chains, two of which are integral TM proteins (Tim17 and Tim23) and one exposed to the mitochondrial matrix (Tim44) (SI Appendix, Figs. S7 and S9). The TM chain reported to be directly involved in protein insertion is Tim17, while Tim23 was suggested not to be involved. In agreement, we observed lipid scrambling exclusively for Tim17 (SI Appendix, Fig. S7). One additional component suggested to be a part of ScTIM23, Mgr2, was proposed to act as a seal/cap for Tim17, in relation to the insertion of specific substrates (48); notably, when present in our simulations (SI Appendix, Fig. S7) Mgr2 reduces lipid scrambling by Tim17 significantly, in agreement with what was previously proposed regarding its role in the ScTIM23 complex. The EMC, on the other hand, is composed of eight chains (Emc1-7 and Emc10, SI Appendix, Figs. S10 and S11), of which at least five are TM (Emc1, Emc3, Emc4, Emc5 and Emc6). Our results are consistent with the fact that both Emc3 and Emc4 are part of the vestibule for protein insertion (43), as we observed lipid scrambling only for these two components (Fig. 3A and SI Appendix, Figs. S10 and S11).
Taken together, our CG-MD simulations confirm and extend our in vitro observation (Fig. 1) that insertase proteins have the ability to scramble lipids. The extent of lipid scrambling depends on protein conformation, oligomerization, and interaction with other members of the insertase complex.
Lipid Scrambling and Protein Insertion Share Similar Pathways.
As all insertase complexes tested have lipid scrambling activity (Figs. 1 and 3), we next wondered whether, as hypothesized, lipid scrambling is facilitated by the same hydrophilic groove that promotes membrane protein insertion. Analysis of our trajectories indicates that the main lipid scrambling pathway is localized in the same protein region where protein insertion has been described to take place, and that lipid movement follows a “credit card-like” motion (Fig. 4A). In detail, the mechanism by which lipids are scrambled is mediated by direct interactions between the lipids’ polar heads and protein polar residues in the insertion cavity, thus preventing unfavorable contacts between the polar head of the lipid and the hydrophobic interior of the membrane, and in turn avoiding the interaction between the polar residues located in the insertion region and the hydrophobic body of the membrane. In Oxa1 family proteins, for example, lipid scrambling happens at the hydrophilic interface between three highly conserved α-helices in this family (Fig. 4A). Similarly, correlation between insertion and scrambling was preserved in our simulations for HsMTCH2, ScTim22, and ScTim17, where scrambling occurs in the described insertion region (Fig. 4A).
Fig. 4.

Lipid scrambling takes place via the same mechanistic pathway as protein insertion. (A) In silico lipid scrambling pathway (orange) in selected protein insertases. The position of the lipid polar head at different times along the scrambling pathway is depicted with orange spheres. Regions involved in protein insertion are shown in green, blue, and cyan for Oxa1 family proteins, while residues involved in protein insertion are shown in blue for HsMTCH2, ScTim17 and ScTim22. (B) Protein scramblases induce limited (0.2 nm on average) membrane thinning. (C) Membrane thickness has minimal correlation with lipid scrambling activity in silico. (D) Mutants proposed to decrease protein insertion activity also reduce lipid scrambling. (E) The presence of a nascent polypeptide inside the protein hydrophilic cavity abolishes lipid scrambling. Left: HsMCTH2. Right: HsOXA1L. (F) Different conformations of Sec61 (lateral gate open, partially open, and closed) have different lipid scrambling activity. AlphaFold structures are denoted by the * symbol. The number of proteins in the system is in parenthesis.
To further validate this observation, we tried to abrogate lipid scrambling by replacing polar residues in the hydrophilic cavity with hydrophobic ones (Leu), and specifically in the insertase Get1 (SI Appendix, Fig. S14). We observed that lipid scrambling activity is very robust, and we could abrogate lipid scrambling in silico in Get1 only after 10 mutations were introduced in its hydrophilic channel (SI Appendix, Fig. S14). However, the use of an elastic network in our CG simulations does not allow us to investigate the effect of potential mutations-induced protein conformational changes, and even after 10 mutations, our simulations are representative of a protein conformation with an open cavity for protein insertion, which may not correctly describe what happens in vitro. Unfortunately, we were unable to produce and test in vitro Get1 with such a high number of mutations as the protein does not fold correctly, and we are thus unable to confirm the importance of the hydrophilic channel via in vitro experiments. Further efforts will be required to find key residues that suppress lipid scrambling by insertases in a similar manner as has been done with single mutations to inactivate and/or activate lipid scrambling in TMEM16 members (53, 54).
In addition to promoting lipid scrambling by favorable interactions with membrane-buried polar and charged residues, we also observed that our dataset of “in silico” scramblases also moderately thin (by 0.2 nm on average) the membrane bilayer in its local (R = 1 nm) proximity (Fig. 4B), as previously proposed for lipid scramblases (16, 55). However, we observed only marginal correlation between lipid scrambling and membrane thinning (Fig. 4C), suggesting that while scramblases do indeed thin the membrane, this does not appear to be the main molecular mechanism responsible for lipid scrambling, at least for the dataset of positive lipid scramblases (including protein insertases) we tested.
Overall, our results suggest that lipid scrambling might employ the same mechanistic pathway used in protein membrane insertion. To further test this hypothesis, we performed simulations of insertase mutants that have been shown to reduce protein insertion [HsMTCH2 (24), HsAsterix (51), HsTim17 and ScTim17 (48), and BhYidC (56)]. In all cases, we observe reduced scrambling by these protein mutants (Fig. 4D). We further performed MD simulations of two distinct insertases, HsOXA1L and HsMTCH2, in the presence of nascent peptides in the insertion cavity (24, 57) (Fig. 4E). In both cases, we observed that when the nascent peptide stays in the cavity, no lipid scrambling occurs (Fig. 4E). Finally, for the specific case of Sec61, we tested its ability to scramble lipids in its “closed” and “open” states (52, 58, 59), and our results indicate that when the Sec61 lateral gate is partially closed or closed, lipid scrambling is strongly reduced or abolished, respectively (Fig. 4F). Since the state of the lateral gate has been shown to correlate with protein insertion (60), this result further suggests that lipid scrambling uses the same mechanistic pathway as protein membrane insertion.
Discussion
Our key finding is that proteins with the capability to insert polypeptide chains into lipid bilayers can also act as lipid scramblases, i.e., they can facilitate lipid translocation from one leaflet to the other. To reach this conclusion, we first used the most reliable approach to investigate lipid scrambling (27): biochemical reconstitution of membrane proteins into liposomes together with an in vitro scrambling assay. Even though this method is labor intensive, we succeeded in purifying, reconstituting, and assaying seven insertase proteins/complexes. For all of them, we consistently observed lipid scrambling activity in vitro.
Next, using the in vitro data, including both our results as well as previous reports (4–6, 12, 14, 21, 28, 30), as a reference, we established MD simulations as a robust tool for the assessment of lipid scrambling activity. Despite intrinsic limitations of this methodology, and specifically its established overestimation of lipid scrambling rates due to the smoother free energy profile by 2 to 3 orders of magnitudes compared to in vitro experiments (28, 54, 61–63), this approach allowed us to delve more deeply into the molecular mechanisms underlying lipid scrambling, and its correlation with membrane thinning and protein insertion. Moreover, by leveraging the wealth of 3D structures available for insertases and insertase complexes from structural studies (46–48, 52) and AlphaFold predictions (64, 65), MD simulations allow for large high-throughput screening of proteins and protein complexes in a relatively inexpensive, fast and accurate manner, outcompeting the limited scope of biochemical reconstitution approaches. Specifically, we directly tested in silico more than 150 distinct proteins and/or complexes, extending and generalizing our in vitro observations. A key advantage of MD simulations in this context is the ability to establish correlations between lipid scrambling activity and specific protein conformations (e.g., open vs. closed states) and protein complex assembly.
From a mechanistic perspective, a plausible hypothesis is that protein insertion into the membrane bilayer requires lipid rearrangements, both within and between membrane leaflets. In other words, the scrambling of lipids between leaflets might result in a local decrease of lipid packing, hence lowering barriers for protein insertion. Our observation that lipid scrambling happens in the same groove in which protein insertion takes place suggests that the two mechanisms are unlikely to be simultaneous. Rather, scrambling might precede protein translocation, promoting a poorly packed membrane environment conducive for protein insertion, or follow it to re-equilibrate the membrane bilayer. Overall, even if we were not able to test in vitro whether scrambling is required for insertion activity, because peptide insertion activity is more sensitive to mutation than scrambling activity, our results indicate that lipid scrambling could potentially be a general mechanism for local remodeling of membranes. Indeed, in support of our notion that lipid scrambling could alter membrane properties, a recent study indicates lipid scrambling as a mechanism to lower membrane bending stiffness (66).
Critically, lipid scrambling can take place independently of protein insertion and, as such, can be considered as a distinct activity. Thus, we propose that protein insertases might have a major, previously overlooked, function in addition to protein insertion, namely as lipid scramblases. Notably, insertases are localized in those organelles that require scrambling activity for membrane maintenance or expansion. These include not only the ER, where most integral membrane proteins enter the secretory pathway and most lipids are synthesized, but also organelles that are disconnected from vesicular trafficking and rely on both protein-mediated protein and lipid delivery, like mitochondria. Our data support a model where a single class of proteins, insertases, plays two roles in nonvesicular trafficking: in protein trafficking, their currently recognized function, and also in lipid transfer. Intriguingly, and consistent with this notion, it has been suggested that certain protein insertases might localize at membrane contact sites, where protein-mediated lipid transport also takes place (67, 68).
That insertases function more broadly in membrane lipid dynamics to participate in membrane growth and expansion would help to rationalize a number of puzzling in cellulo observations connecting lipid metabolism and transport with protein insertases. These include the role of the EMC in protein-mediated lipid transport between the ER and mitochondria (69); abnormal mitochondrial and lipid droplet morphologies in ERAD-defective brown adipocytes (70); the hypersensitivity to saturated fatty acids of GET complex deletion mutants (71); failed thylakoid compartment biogenesis in the absence of the Oxa1 family insertase Alb3 (72); mitochondrial morphology defects in the absence of MTCH2 (73) or upon Sam50 depletion (74, 75). Because scrambling and protein insertion make use of the same hydrophilic groove but scrambling activity is more robust than protein insertion (it is easier to move a lipid headgroup than an entire peptide across the membrane), it is challenging to uncouple these activities to definitively attribute such lipid metabolism or membrane morphology defects to a defect in scrambling. Indeed, because protein insertion was the only known function of these proteins, membrane defects arising from their dysfunction were ascribed primarily to misfolding or mislocalization of the insertases’ protein substrates. However, their null phenotypes are similar to those observed for ATG9, VMP1, or TMEM41B scramblases thought to function in membrane and organelle biogenesis, which include abnormal lipid distributions in cells or failures in organelle formation (5, 6, 13, 76, 77); and the observed phenotypes equally well reflect dysfunctional membrane dynamics.
While the molecular mechanisms underlying protein-mediated lipid transport are not yet well understood, an emerging model posits a partnership between bridge-like lipid transfer proteins and scramblases (4, 7). Specifically, for a membrane bilayer to expand, lipids delivered to its cytosolic leaflet by lipid transport proteins must be scrambled between the leaflets of the bilayer. In this context, several bridge-like lipid transport proteins are reported to interact with insertases of the mitochondria: Mdm10 in the ERMES complex (78), which mediates glycerophospholipid transport between the ER and mitochondria in yeast, interacts with the SAM complex via its Sam50 component (79). ATG2, a lipid transporter mediating diverse functions including autophagosome biogenesis (80–82), is reported to interact with the TOM complex (83). And both MTCH2 and TOM reportedly associate with the bridge-like lipid transporters VPS13A and VPS13D based on high throughput proteomics (84, 85). Scramblases in the ER may also be required to re-equilibrate its membrane bilayer as transport proteins extract lipid cargo from its cytosolic leaflet only. Thus, ERMES interacts with the EMC in the ER, and the interaction is required for phosphatidylserine transport to mitochondria (69). Hence, the protein insertases Sam50, TOM, MTCH2, and the EMC could support lipid transport as scramblases. As noted before, the scramblases that participate in lipid transport systems have for the most part not been identified, and our results indicate known insertases in the ER and mitochondria as attractive candidates.
Since there are many insertases in the ER and in mitochondria, each with different substrate preferences for insertion, this implies the existence of multiple scramblases in these membranes. However, our results in no way suggest that insertases are the only scramblases, and it is almost certain that still other classes of proteins [but clearly not all integral membrane proteins as per our in silico data and previous studies (86)] might also harbor scrambling activity. Scramblases may well be exceptions to the current paradigm of one protein-one function. For example, the TMEM16 proteins and VDAC1/2 in the mitochondria were well characterized as ion channels (87–89) and were subsequently shown also to scramble lipids (12, 28). Hence, the potential lipid scrambling activity of other proteins or classes of proteins will need to be examined on a case-by-case basis. For those proteins with other functions additional to scrambling, whether these functions are simultaneous with scrambling or happen independently as a result of different physiological clues (e.g., protein localization, post-translational modifications, protein-protein interactions, etc.) is a promising future research area.
Overall, lipid scramblases have been elusive players in membrane homeostasis, and their identities are only recently starting to emerge (9). Our study more than doubles the number of known lipid scramblases, describing tens of new ones. Our results highlight that integral membrane proteins could have additional functions beyond those currently known and suggest protein insertases as players in membrane dynamics, including in nonvesicular lipid transport. We expect that this concept will turn out to be particularly helpful not only in the interpretation of numerous existing observations, and especially genetic and physical interactions, but will also open future research directions by blurring the lines between the fields of membrane and protein homeostasis.
Methods
Materials.
All the lipids, including POPC (Cat. #850457C), POPE (Cat. #850757C), Soy PI (Cat. #840044), NBD-PE (Cat. #810151P), NBD-PC (Cat. #810122), and NBD-PG (Cat. #810161P) were purchased from Avanti Polar Lipids. All detergents were purchased from Anatrace. Bio-Beads™ SM2 Adsorbent Medium was purchased from BIO-RAD (Cat. #152-3920). The anti-FLAG M2 resin (Cat. #A2220), EDTA-free Roche cOmplete protease inhibitor cocktail (Cat. #4693159001), and Optiprep density gradient medium (Cat. #D1556) were purchased from Sigma Aldrich. Fatty acid-free BSA was purchased from AmericanBio. The Expi293™ Expression System Kit was purchased from Thermo Fisher Scientific (Cat. #A14635). The powdered Luria Broth and Terrific Broth were from RPI (Cat. #L24060 and T15100), Teknova, and Fischer Scientific (BP9723).
In vitro Studies.
Plasmids.
The sequence encoding full-length S. cerevisiae Get1 was cloned into the pET-Duet vector with an N-terminal His6-tag. The Get2-1sc-His6 construct was a gift from M. Mariappan*. The sequences encoding full-length E. coli YidC and BamA (including the N-terminal signal sequence of the latter) were cloned into the pET-29 vector with C-terminal His6-tags. Residues 43-402 of S. cerevisiae Oxa1, corresponding to the mature protein, were also cloned into the pET-29 vector with a C-terminal His6-tag. The Get2-1sc mutant (T421L/K428L/K433L/W459L/Y461L/S495L/G497L/W501L/N505L/N508L in Get1) sequence was synthesized by Genewiz. Condon-optimized sequences encoding full-length S. cerevisiae SAM50 (N-terminally 3xFlag tagged), SAM35 (no tag), SAM37 (N-terminally Strep tagged), and human MTCH2 (N-terminally 3xFlag tagged) were individually cloned into pCMV-10 vector. The GlpG expression plasmid was a gift from Y. Ha*. The glycerol stock of Rosetta2 cells containing the pTW2-Vamp2-His6 plasmid was generously provided by the laboratory of J. Rothman*.
Expression and purification of proteins.
His6-Get1, Get2-1sc-His6, and the Get2-1sc mutant.
Proteins were expressed and purified as previously described with modifications (90). The Get2-1sc WT and mutant constructs were transformed to LOBSTR BL21 E. coli cells (Kerafast) and Get1 was expressed in E. coli Ros2(DE3)/pLysS (Novagen). The overnight culture was inoculated into homemade TB medium and cultured at 200 rpm, 37 °C until OD600 reached 0.6. Protein expression was induced with 0.4 mM IPTG at 17 °C for 18 h for Get2-1sc and 37 °C for 18 h for Get1. Cells were harvested, resuspended in buffer A (500 mM NaCl, 50 mM HEPES, pH 8.0, 10% glycerol, and 1 mM TCEP), and lysed by five passes through the Emulsiflex-C5 microfluidizer. The crude lysate was centrifuged at 10,000 g for 20 min and the supernatant was further spun at 40,000 rpm for 2 h in a Ti45 rotor. The membrane pellet was homogenized in buffer A supplemented with 1% Anapoe-C12E9 (Anatrace) or n-Dodecyl-N, N-Dimethylamine-N-Oxide (LDAO, Anatrace), and incubated at 4 °C for 2 h with constant mixing. The suspension was centrifuged at 15,000 rpm for 30 min in a JA-20 rotor. The supernatant that contains the extracted proteins was incubated with Ni-NTA resin at 4 °C for 30 min. The resin was then drained in a gravity column and washed with buffer A supplemented with 20 mM imidazole and 0.02% C12E9 or 0.1% LDAO. The protein was eluted with buffer A supplemented with 250 mM imidazole and 0.02% C12E9 or 0.1% LDAO. The elution was concentrated in a 50 K MWCO Amicon concentrator and loaded onto the Superdex 200 10/300 column equilibrated with buffer B (200 mM NaCl, 50 mM HEPES, pH7.4, 5% glycerol, and 1 mM TCEP) supplemented with 0.02% C12E9 or 0.1% LDAO (SI Appendix, Fig. S2). The peak fractions were pooled and concentrated, and aliquots were frozen and stored at −80 °C until use.
3xFlag-MTCH2 and the SAM complex.
First, 200 μg constructs encoding 3xFlag-MTCH2 or 1:1:1 mixture of SAM35, Strep-SAM37, and 3xFlag-SAM50 were transfected with Expitransfectamine (Gibco) to 200 mL Expi293 cells at a density of 2.5 million cells/mL. Then, cells were enhanced after 18 h of transfection and harvested after 48 h of transfection. The cell pellet was resuspended and homogenized in buffer B supplemented with 2 mM MgCl2 and 1× protease inhibitor (Roche). The protein was extracted by incubating with 1% GDN (Anatrace) on a rotator for 2 h at 4 °C. The cell suspension was centrifuged at 15,000 rpm in a JA20 rotor for 30 min, and the supernatant was incubated with Flag resin for 2 h at 4 °C. The resin was washed twice with 10 mL of buffer B supplemented with 0.02% GDN and incubated overnight with buffer B supplemented with 1 mM ATP and 2 mM MgCl2 and 0.02% GDN. The resin was further washed two times and eluted with buffer B supplemented with 0.02% GDN and 0.2 mg/mL Flag peptide five times with 20 min of incubation in between each elution step. The eluted protein was then loaded onto the Superdex 200 10/300 column that was equilibrated with buffer B supplemented with 0.02% GDN (SI Appendix, Fig. S2). Peak fractions were pooled and concentrated, and aliquots were frozen and stored at −80 °C until use.
His6-GlpG.
The construct was transformed into C43 E. coli cells. Protein expression was induced with 0.2 mM IPTG when OD600 reached 0.9, and the cells were cultured at 22 °C for 18 h. Proteins were purified in buffer A as described for Get1, except that n-Dodecyl-β-D-Maltopyranoside (DDM, Anatrace) was used for protein extraction and throughout the purification process. The protein was buffer-exchanged into buffer B supplemented with 0.1% LDAO by loading it onto the Superdex 200 10/300 column (SI Appendix, Fig. S2).
VAMP2-His6.
Protein expression was induced with 0.5 mM IPTG when OD600 reached 0.8, and the cells were cultured at 37 °C for 4 h. After harvesting, the cells were resuspended in buffer C (25 mM HEPES, pH 7.4, 400 mM KCl, 10% glycerol, and 0.2 mM TCEP), supplemented with 1 mM PMSF and 4%Triton X-100. The cells were lysed and ultracentrifuged at 35,000 rpm in a Ti45 rotor for 30 min. The supernatant was collected and incubated with Ni-NTA resin for 2 h at 4 °C. The resin was then washed with buffer C supplemented with 1% Triton X-100 and 50 mM imidazole, followed by buffer C with 1% n-octyl-β-D-glucopyranoside (OG) and 50 mM imidazole. The protein was eluted with buffer C supplemented with 500 mM Imidazole and 1%OG. The eluted protein was loaded onto the Superdex 200 10/300 column equilibrated with buffer B with 0.1% LDAO (SI Appendix, Fig. S2).
YidC-His6 and BamA-His6.
Proteins were expressed and purified as previously described (91, 92) with modifications. Both constructs were transformed into C43 E. coli cells. The overnight cultures were inoculated into LB medium for YidC or TB medium for BamA, and cultured at 200 rpm, 37 °C, until OD600 reached approximately 0.7. Protein expression was induced at 37 °C for 2.5 h with 0.5 mM IPTG for YidC, and for 4 h with 1 mM IPTG for BamA. Cells were harvested, resuspended in buffer E (200 mM NaCl, 25 mM HEPES, pH 8.0, 10% glycerol, and 0.5 mM TCEP) supplemented with protease inhibitor (Roche); for BamA, protease inhibitors were included at approximately 2× final concentration and lysozyme was also present. Cells were lysed using the Emulsiflex-C5 microfluidizer, the crude lysate was centrifuged at low speed for 30 min and the supernatant was further spun at 40,000 rpm for 90 min in a Ti45 rotor. The membrane pellet was homogenized in buffer E supplemented with 1% n-Decyl-β-D-Maltopyranoside (DM, Anatrace) for YidC or n-Dodecyl-N, N-Dimethylamine-N-Oxide (LDAO, Anatrace) for BamA, and incubated at 4 °C for 2 h with constant mixing. Additionally, for BamA, the membranes were diluted to at least 25 mL per liter initial cell volume prior to solubilization. The solubilized membranes were incubated with Ni-NTA resin at 4 °C for at least 30 min. The resins were then drained in a gravity column and washed with buffer E supplemented with 20 to 30 mM imidazole and 0.2% DM or 0.1% LDAO respectively; in some cases, the detergent for BamA was exchanged on the resin to 0.02% DDM and used for all later steps. The proteins were eluted with buffer E supplemented with 300 to 330 mM imidazole and 0.2% DM or 0.1% LDAO respectively. The elutions were concentrated in 30 K (YidC) or 100 K (BamA) MWCO Amicon concentrators and loaded onto the Superdex 6 10/300 column equilibrated with buffer E supplemented with 0.2% DM or 0.1% LDAO respectively (SI Appendix, Fig. S2). The peak fractions were pooled and concentrated with new 100 K MWCO Amicon concentrators, and aliquots were frozen and stored at −80 °C until use.
Oxa1-His6.
Proteins were expressed and purified as previously described with modifications (93). Both constructs were transformed to BL21(DE3) codon+ E. coli cells. An overnight culture was inoculated in TB medium, which was subsequently cultured at 200 rpm, 37 °C until OD600 reached approximately 0.7. The cells were placed in a 4 °C cold room for approximately 30 min, and protein expression was induced at 25 °C overnight with 0.5 mM IPTG. Cells were harvested, resuspended in buffer F (500 mM NaCl, 25 mM HEPES, pH 7.0, 12% glycerol, and 0.5 mM TCEP) supplemented with protease inhibitor (Roche, 1.66× final concentration) and lysozyme. Cells were lysed using the Emulsiflex-C5 microfluidizer, the crude lysate was centrifuged at low speed for 30 min and the supernatant was further spun at 40,000 rpm for 90 min in a Ti45 rotor. The membrane pellet was diluted with buffer F to 40 mL per liter initial cell volume, supplemented with 1% n-Dodecyl-β-D-Maltopyranoside (DDM, Anatrace), and incubated at 4 °C for 2 h with constant mixing. The solubilized membranes were incubated with Ni-NTA resin at 4 °C for at least 30 min. The resin was then drained in a gravity column and washed with buffer F supplemented with 30 mM imidazole and 0.1% DDM. The proteins were eluted with buffer F supplemented with 540 mM imidazole and 0.1% DDM. The elution was concentrated in a 30 K MWCO Amicon concentrator and loaded onto the Superdex 200 10/300 column equilibrated with buffer F supplemented with 0.1% DDM (SI Appendix, Fig. S2). The peak fractions were pooled and concentrated with a new 100 K MWCO Amicon concentrator to approximately 40 μM, and aliquots were frozen and stored at −80 °C until use. Immediately prior to reconstitution, the thawed 40 μM aliquots were diluted twofold with buffer F, yielding final concentrations of 20 μM Oxa1 and 0.05% DDM, which were further diluted 10-fold when added to the reconstitution mixture (along with a matched buffer).
Liposome preparation.
For YidC, BamA, and Oxa1, 90% POPC (w/w%), 9.5% POPE, and 0.5% NBD-PE (or NBD-PG for BamA) were solubilized in chloroform dried under a nitrogen stream, and further dried under vacuum for at least 1 h. For all other proteins, 88% POPC (w/w%), 9.5% POPE, 2% Soy PI, and 0.5% NBD-lipid (NBD-PE or NBD-PC) were solubilized in chloroform, dried under a nitrogen stream, and further dried under vacuum overnight. The resulting lipid films were rehydrated in buffer D (200 mM NaCl, 25 mM HEPES, pH 7.5 to 8, and 1 mM TCEP) to generate a 10.5 mM lipid stock. The mixture was incubated at 37 °C for 60 min with intermittent vortexing every 20 min. The sample was subjected to seven freeze-thaw cycles and extruded 31 times against a 200 nm polycarbonate filter in the Avanti Mini-Extruder.
Proteoliposome preparation.
As described previously (27), for a standard 250 μL reaction, 125 μL of the extruded liposomes were mixed with the reconstitution buffer (200 mM NaCl, 25 mM HEPES, pH 7.5, and 1 mM TCEP) and Triton X-100 to a final volume of 225 μL. The concentration of Triton X-100 was determined by the swelling assay, typically ranging around 3.4 to 4.5 mM. After 1 to 2 h of destabilization, 25 μL of purified proteins at normalized concentrations were added to the mixture and incubated on a rotator for additional 1 to 2 h at room temperature. To remove detergents, prewashed Bio-Beads were added stepwise: The sample was mixed with an aliquot of Bio-Beads (22 to 28 mg) at room temperature for 1 h, followed by replacement with a new aliquot (22 to 28 mg) and mixing for 2 h. Finally, the sample was transferred to a new tube containing fresh Bio-Beads (44 to 56 mg) and rotated at 4 °C for 16 to 21 h. One hundred fifty microliter of the recovered sample was mixed with 150 μL of 60% Optiprep and layered with 200 μL of 10% Optiprep and 150 μL of the reconstitution buffer in a 0.8 mL tube (Beckman Coulter Cat. #344090). The tube was centrifuged at 40,000 rpm for 90 min in a SW-55 rotor. The floated liposomes were recovered with a final volume of 150 μL and used immediately. For YidC, Oxa1, and BamA, proteoliposomes were selectively harvested from the region directly below the 10 to 0% Optiprep interface, which was found to be protein-rich.
TEM characterization of liposomes or proteoliposomes.
Negative stain analysis was performed using 400 mesh carbon-coated copper grids (Electron Microscopy Sciences). Grids were glow discharged for 35 s at 25 mA using an easiGlow glow discharger (PELCO). The grid was floated in 40 µL of 0.25 mM liposomes for 1 min, rinsed twice in 40 µL of buffer, followed by floating in 40 µL of 1% uranyl acetate solution for 30 s before blotting to complete dryness. Negative stain images were collected using the FEI Tecnai T12 transmission electron microscope operating at 120 kV. Images were collected at a nominal magnification of 15,000×, corresponding to 7.48 Å/pixel at the specimen level.
BSA back extraction assay.
The scramblase assay was carried out in 96-well plates at 30 °C. In a triplicate setup, 5 μL of either protein-free liposomes or proteoliposomes were added to 95 μL of the reconstitution buffer. NBD fluorescence was measured using the Synergy H1 Hybrid Multi-Mode Reader (BioTek) with excitation/emission wavelengths set to 460/538 nm. To establish the 100% fluorescence baseline, the measurements were taken for 10 min until a steady fluorescence signal was achieved. Subsequently, 5 μL of 60 mg/mL fatty acid-free BSA was added to each well, mixed thoroughly, and the fluorescence was measured for 10 min. Finally, 0.5% Triton X-100 and 5 mM sodium dithionite were added to each well to ensure that the background signal was small enough (~3%) so that it did not affect the fluorescence readings. For data processing, each fluorescence reading was divided by the corresponding fluorescence baseline value in each sample.
For some samples, scrambling measurements were also collected with the FluoroMax Plus Spectrofluorometer (HORIBA Scientific). For each sample, 50 μL of the proteoliposomes were added to 1,950 μL of the reconstitution buffer (200 mM NaCl, 25 mM HEPES, pH 7.5, and 1 mM TCEP). The sample was vigorously stirred and measured for fluorescence at 460/538 nm for 50 to 70 s to establish a stable baseline. Then, 50 μL of 60 mg/mL fatty acid-free BSA was added, and the fluorescence data were collected for another 200 to 300 s.
Molecular Simulations.
3D structural modeling.
All proteins simulated in this work were obtained from either the Protein Data Bank (https://www.rcsb.org/) or AlphaFold (64) predicted models available from UniProt (https://www.uniprot.org/). For systems containing more than one chain and for which no structure was available, prediction was performed using AlphaFold-Multimer (94) which is implemented in ColabFold (65); for these cases, a total of 24 recycles were used. The various complexes investigated in the text are described below:
Mitochondrial complexes.
The monomer of the TOM complex consists of five chains (Tom5-Tom6-Tom7-Tom22-Tom40); thus, its dimer (10 chains) consists of two copies of each subunit from the monomer.
The SAM complex consists of three chains (Sam50, Sam35 and Sam37, the last two being nontransmembrane proteins).
The yeast TIM22 complex consists of four transmembrane subunits (Tim18, Tim22, Tim54, and Sdh3) and six helical proteins that form a structure like a ring in the intermembrane space (IMS); this structure serves as chaperone to conduct the substrate from the TOM complex to TIM22 complex (47). Similar is the case of the human TIM22 complex, where 14 chains form a double ring-like structure in the IMS and only Tim22 is the transmembrane part.
The yeast TIM23 complex is formed by three chains, two transmembranes (Tim17 and Tim23) and one exposed to the mitochondrial matrix (Tim44). One additional component, Mgr2, was suggested to be a part of ScTIM23 and to act as a seal/cap for Tim17 (48).
The human TIM23 complex is composed of three chains (Tim17-Tim23 and Tim50) as its yeast homolog.
ER complexes.
The EMC is composed of eight chains (Emc1-7 and Emc10), of which five chains are transmembrane (Emc1, Emc3, Emc4, Emc5, and Emc6).
The GET complex is composed of two copies of Get1 and two copies of Get2, forming a heterotetramer.
The TRC, the human homolog of the ScGET complex, is composed of the WRB, CAML, and TRC40 subunits, the human counterparts of yeast Get1, Get2, and Get3, respectively.
The GEL is composed of TMCO1 (member of the oxa1 family) and C20orf24.
The PAT complex consists of two subunits, Asterix and CCDC47.
The ERAD complex consists of four chains, Hrd1, Usa1, Der1, and Hrd3, with Hrd1 and Der1 being the major transmembrane components.
The SEC61 complex is composed of its α, β, and γ units.
The TRAP complex is composed of its α, β, γ, and δ units.
The OSTA complex is composed of RPN1, RPN2, OST4, OST48, DAD1, STT3a, TMEM258, and OSTC.
The translocon complex is composed of the SEC61, TRAP, and OSTA complexes, and its structure was assembled according to ref. 52.
Set-up of peptide-bound OXA1 and MTCH2 simulations.
The initial configuration for the MTCH2-peptide structure was based on the work by Guna et al. (24). The dimeric structure between HsMTCH2 (Uniprot ID: Q9Y6C9) and the transmembrane part (25-residue fragment from Ile118 to Leu145) of one of its substrates tested in vitro, the HsOMP25 (Uniprot ID: P57105), was built using AF multimer and ColabFold (65, 94).
Similarly, we predicted the dimer structure using AF multimer of the HsOXA1L-peptide structure based on the work of ref. 57. This dimeric structure consisted of HsOXA1L (Uniprot ID: Q15070) and a 32-residue polyalanine peptide. It is worth mentioning that in both cases, the best prediction based on AF corresponded to the peptide located into the well-characterized insertion cavity of each protein.
All systems were subjected to a minimization step in vacuum for a maximum of 50,000 steps or until the maximum force on any atom was less than 100 kJ mol−1 nm−1. For this purpose, the steepest descent algorithm and the AMBER99SB-ILDN force field were used (95).
CG-MD simulations.
The minimized structures of each system were embedded in a DOPC membrane using the CHARMM-GUI web server (96). Subsequently, CG-MD simulations were carried out using the GROMACS software, version 2019.6 (97), and the Martini 3 force field (98). Elastic network was used to preserve the 3D structure of the proteins and the multimers, using a force constant of 500 kJ mol−1 nm−2. Two replicates of each system were carried out using different initial velocities for 10 μs, using a time step of 20 fs. The temperature was maintained at 310 K using the V-rescale thermostat (99) and the pressure at 1 bar using the Parrinello–Rahman barostat (100). Additional information for all simulated systems is shown in SI Appendix, Table S1.
Calculation of scramblase activity from CG-MD.
Our approach is based, briefly, on measuring the tilting of each lipid throughout the simulation. To do this, we first formed two vectors, which were obtained between the last bead of each acyl tail (C4A and C4B) and the bead of the phosphate (PO4). Then, the angle between each of these two vectors with respect to the z-axis was measured. The value for the final angle per each lipid is the average of the two angles obtained for each tail. For denoising purposes, the time traces for each final angle were smoothened by averaging over a 50-ns window and data points were collected each 25 ns. Thus, following our approach, lipids located in the upper leaflet will have angles around ~30°, while lipids located in the bottom leaflet will have angles around ~150° (Fig. 2A). We define a buffer region between 55° and 125°, in order to reduce the noise generated by lipid movement (or very slow movement) and thus not overestimate the events obtained. A scrambling event was counted when a lipid in the upper leaflet moved to an angle greater than 125°, and when a lipid in the bottom leaflet moved to an angle lower than 55°. These angles were calculated using the gmx_gangle tool and were measured every 1 ns. For all analyses, the first 2 μs were omitted, and the events were accumulated per microsecond, thus resulting in 16 data points per simulated system (8 data per replica, thus each data represents the number of events in 1 μs). These 16 data points were used to build the corresponding boxplots using GNUPLOT.
Calculation of the local thickness of the membrane.
We extracted the local thickness of the membrane from the curves of the 2D density diagrams for the hydrophobic body of the membrane, i.e., excluding both the head group and the phosphate group. These 2D densities were calculated with the gmx_density tool along the z-axis, and were calculated every 5 ns, omitting the first 2 μs of each trajectory. The densities were calculated considering the lipids that were at a distance of 1.0 nm from the protein (Fig. 3B).
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank T. Melia, W. Kukulski, and W. Prinz for comments regarding this manuscript as well as lab members from the Vanni lab and Reinisch lab. We gratefully acknowledge support from NIGMS (R35GM131715 to K.M.R.), the Swiss NSF (grant CRSII5_189996 to S.V.), and the European Research Council under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 803952, to S.V.). This work was supported by grants from the Swiss National Supercomputing Centre under projects ID s1176, s1030, and s1221.
Author contributions
D.L., C.R.-R., M.A.S., K.M.R., and S.V. designed research; D.L., C.R.-R., and M.A.S. performed research; D.L., C.R.-R., M.A.S., K.M.R., and S.V. analyzed data; and C.R.-R., K.M.R., and S.V. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission. G.H. is a guest editor invited by the Editorial Board.
Contributor Information
Karin M. Reinisch, Email: karin.reinisch@yale.edu.
Stefano Vanni, Email: stefano.vanni@unifr.ch.
Data, Materials, and Software Availability
MD simulations data have been deposited in Zenodo (https://doi.org/10.5281/zenodo.10475371) (101). All other data are included in the manuscript and/or SI Appendix.
Supporting Information
References
- 1.Vance D. E., Choy P. C., Farren S. B., Lim P. H., Schneider W. J., Asymmetry of phospholipid biosynthesis. Nature 270, 268–269 (1977). [DOI] [PubMed] [Google Scholar]
- 2.Vance J. E., Phospholipid synthesis and transport in mammalian cells. Traffic 16, 1–18 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Reinisch K. M., Prinz W. A., Mechanisms of nonvesicular lipid transport. J. Cell Biol. 220, e202012058 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghanbarpour A., Valverde D. P., Melia T. J., Reinisch K. M., A model for a partnership of lipid transfer proteins and scramblases in membrane expansion and organelle biogenesis. Proc. Natl. Acad. Sci. U.S.A. 118, e2101562118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maeda S., et al. , Structure, lipid scrambling activity and role in autophagosome formation of ATG9A. Nat. Struct. Mol. Biol. 27, 1194–1201 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matoba K., et al. , Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat. Struct. Mol. Biol. 27, 1185–1193 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Melia T. J., Reinisch K. M., A possible role for VPS13-family proteins in bulk lipid transfer, membrane expansion and organelle biogenesis. J. Cell Sci. 135, jcs259357 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holthuis J. C. M., Jahn H., Menon A. K., Mizushima N., An alliance between lipid transfer proteins and scramblases for membrane expansion. Fac. Rev. 11, 22 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakuragi T., Nagata S., Regulation of phospholipid distribution in the lipid bilayer by flippases and scramblases. Nat. Rev. Mol. Cell Biol. 24, 576–596 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brunner J. D., Lim N. K., Schenck S., Duerst A., Dutzler R., X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature 516, 207–212 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Suzuki J., Denning D. P., Imanishi E., Horvitz H. R., Nagata S., Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 341, 403–406 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Suzuki J., Umeda M., Sims P. J., Nagata S., Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468, 834–838 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Huang D., et al. , TMEM41B acts as an ER scramblase required for lipoprotein biogenesis and lipid homeostasis. Cell Metab. 33, 1655–1670.e8 (2021). [DOI] [PubMed] [Google Scholar]
- 14.Li Y. E., et al. , TMEM41B and VMP1 are scramblases and regulate the distribution of cholesterol and phosphatidylserine. J. Cell Biol. 220, e202103105 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pomorski T., Menon A. K., Lipid flippases and their biological functions. Cell. Mol. Life Sci. 63, 2908–2921 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bethel N. P., Grabe M., Atomistic insight into lipid translocation by a TMEM16 scramblase. Proc. Natl. Acad. Sci. U.S.A. 113, 14049–14054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rapoport T. A., Li L., Park E., Structural and mechanistic insights into protein translocation. Annu. Rev. Cell Dev. Biol. 33, 369–390 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Wu X., Rapoport T. A., Translocation of proteins through a distorted lipid bilayer. Trends Cell Biol. 31, 473–484 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang Q., Gummadi S. N., Menon A. K., Chemical modification identifies two populations of glycerophospholipid flippase in rat liver ER. Biochemistry 43, 10710–10718 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Kubelt J., Menon A. K., Müller P., Herrmann A., Transbilayer movement of fluorescent phospholipid analogues in the cytoplasmic membrane of Escherichia coli. Biochemistry 41, 5605–5612 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Menon I., et al. , Opsin is a phospholipid flippase. Curr. Biol. 21, 149–153 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malvezzi M., et al. , Out-of-the-groove transport of lipids by TMEM16 and GPCR scramblases. Proc. Natl. Acad. Sci. U.S.A. 115, E7033–E7042 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathiassen P. P. M., Pomorski T. G., A fluorescence-based assay for measuring phospholipid scramblase activity in giant unilamellar vesicles. Bio-Protoc. 12, e4366 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guna A., et al. , MTCH2 is a mitochondrial outer membrane protein insertase. Science 378, 317–322 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anghel S. A., McGilvray P. T., Hegde R. S., Keenan R. J., Identification of Oxa1 homologs operating in the eukaryotic endoplasmic reticulum. Cell Rep. 21, 3708–3716 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gentle I., Gabriel K., Beech P., Waller R., Lithgow T., The Omp85 family of proteins is essential for outer membrane biogenesis in mitochondria and bacteria. J. Cell Biol. 164, 19–24 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ploier B., Menon A. K., A fluorescence-based assay of phospholipid scramblase activity. J. Vis. Exp. 54635 (2016), 10.3791/54635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jahn H., et al. , Phospholipids are imported into mitochondria by VDAC, a dimeric beta barrel scramblase. Nat. Commun. 14, 8115 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siggel M., Bhaskara R. M., Hummer G., Phospholipid scramblases remodel the shape of asymmetric membranes. J. Phys. Chem. Lett. 10, 6351–6354 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Adlakha J., Hong Z., Li P., Reinisch K. M., Structural and biochemical insights into lipid transport by VPS13 proteins. J. Cell Biol. 221, e202202030 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ren S., et al. , Structural and functional insights into an archaeal lipid synthase. Cell Rep. 33, 108294 (2020). [DOI] [PubMed] [Google Scholar]
- 32.He M., et al. , Spns1 is a lysophospholipid transporter mediating lysosomal phospholipid salvage. Proc. Natl. Acad. Sci. U.S.A. 119, e2210353119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chua G.-L., et al. , Mfsd2a utilizes a flippase mechanism to mediate omega-3 fatty acid lysolipid transport. Proc. Natl. Acad. Sci. U.S.A. 120, e2215290120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nosol K., et al. , Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc. Natl. Acad. Sci. U.S.A. 117, 26245–26253 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olsen J. A., Alam A., Kowal J., Stieger B., Locher K. P., Structure of the human lipid exporter ABCB4 in a lipid environment. Nat. Struct. Mol. Biol. 27, 62–70 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Wang L., et al. , Cryo-EM structure of human bile salts exporter ABCB11. Cell Res. 30, 623–625 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Galazzo L., et al. , The ABC transporter MsbA adopts the wide inward-open conformation in E. coli cells. Sci. Adv. 8, eabn6845 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez C., et al. , Structure and mechanism of an active lipid-linked oligosaccharide flippase. Nature 524, 433–438 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y., et al. , Cryo-EM analysis provides new mechanistic insight into ATP binding to Ca 2+ -ATPase SERCA2b. EMBO J. 40, e108482 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bushell S. R., et al. , The structural basis of lipid scrambling and inactivation in the endoplasmic reticulum scramblase TMEM16K. Nat. Commun. 10, 3956 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feng Z., Alvarenga O. E., Accardi A., Structural basis of closed groove scrambling by a TMEM16 protein. bioRxiv [Preprint] (2023). 10.1101/2023.08.11.553029 (Accessed 4 November 2023). [DOI] [PubMed]
- 42.McDowell M. A., et al. , Structural basis of tail-anchored membrane protein biogenesis by the GET insertase complex. Mol. Cell 80, 72–86.e7 (2020). [DOI] [PubMed] [Google Scholar]
- 43.Bai L., You Q., Feng X., Kovach A., Li H., Structure of the ER membrane complex, a transmembrane-domain insertase. Nature 584, 475–478 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGilvray P. T., et al. , An ER translocon for multi-pass membrane protein biogenesis. eLife 9, e56889 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tucker K., Park E., Cryo-EM structure of the mitochondrial protein-import channel TOM complex at near-atomic resolution. Nat. Struct. Mol. Biol. 26, 1158–1166 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qi L., et al. , Cryo-EM structure of the human mitochondrial translocase TIM22 complex. Cell Res. 31, 369–372 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y., et al. , Structure of the mitochondrial TIM22 complex from yeast. Cell Res. 31, 366–368 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sim S. I., Chen Y., Lynch D. L., Gumbart J. C., Park E., Structural basis of mitochondrial protein import by the TIM23 complex. Nature 621, 620–626 (2023), 10.1038/s41586-023-06239-6 (Accessed 9 July 2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sundaram A., et al. , Substrate-driven assembly of a translocon for multipass membrane proteins. Nature 611, 167–172 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu X., et al. , Structural basis of ER-associated protein degradation mediated by the Hrd1 ubiquitin ligase complex. Science 368, eaaz2449 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smalinskaitė L., Kim M. K., Lewis A. J. O., Keenan R. J., Hegde R. S., Mechanism of an intramembrane chaperone for multipass membrane proteins. Nature 611, 161–166 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gemmer M., et al. , Visualization of translation and protein biogenesis at the ER membrane. Nature 614, 160–167 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Le T., et al. , An inner activation gate controls TMEM16F phospholipid scrambling. Nat. Commun. 10, 1846 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee B.-C., et al. , Gating mechanism of the extracellular entry to the lipid pathway in a TMEM16 scramblase. Nat. Commun. 9, 3251 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Falzone M. E., et al. , TMEM16 scramblases thin the membrane to enable lipid scrambling. Nat. Commun. 13, 2604 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumazaki K., et al. , Structural basis of Sec-independent membrane protein insertion by YidC. Nature 509, 516–520 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Itoh Y., et al. , Mechanism of membrane-tethered mitochondrial protein synthesis. Science 371, 846–849 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gérard S. F., et al. , Structure of the Inhibited State of the Sec Translocon. Mol. Cell 79, 406–415.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Itskanov S., et al. , A common mechanism of Sec61 translocon inhibition by small molecules. Nat. Chem. Biol. 19, 1063–1071 (2023), 10.1038/s41589-023-01337-y (Accessed 18 August 2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shao S., Protein biosynthesis at the ER: Finding the right accessories. Mol. Biol. Cell 34, pe1 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khelashvili G., Menon A. K., Phospholipid scrambling by G protein-coupled receptors. Annu. Rev. Biophys. 51, 39–61 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Watanabe R., Sakuragi T., Noji H., Nagata S., Single-molecule analysis of phospholipid scrambling by TMEM16F. Proc. Natl. Acad. Sci. U.S.A. 115, 3066–3071 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Malvezzi M., et al. , Ca2+-dependent phospholipid scrambling by a reconstituted TMEM16 ion channel. Nat. Commun. 4, 2367 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jumper J., et al. , Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mirdita M., et al. , ColabFold: Making protein folding accessible to all. Nat. Methods 19, 679–682 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang H., Levental K. R., Lorent J. H., Revathi A. A., Levental I., Lipid scrambling facilitates membrane vesiculation through decreasing membrane stiffness. Biophys. J. 122, 22a–23a (2023). [Google Scholar]
- 67.González Montoro A., et al. , Vps39 interacts with Tom40 to establish one of two functionally distinct vacuole-mitochondria contact sites. Dev. Cell 45, 621–636.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Koch C., Räschle M., Prescianotto-Baschong C., Spang A., Herrmann J. M., The ER-SURF pathway uses ER-mitochondria contact sites for protein targeting to mitochondria. bioRxiv [Preprint] (2023). 10.1101/2023.08.10.552816 (Accessed 17 August 2023). [DOI] [PMC free article] [PubMed]
- 69.Lahiri S., et al. , A conserved endoplasmic reticulum membrane protein complex (EMC) facilitates phospholipid transfer from the er to mitochondria. PLoS Biol. 12, e1001969 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou Z., et al. , Endoplasmic reticulum–associated degradation regulates mitochondrial dynamics in brown adipocytes. Science 368, 54–60 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ruggles K. V., et al. , A functional, genome-wide evaluation of liposensitive yeast identifies the “RE2 required for viability” (ARV1) gene product as a major component of eukaryotic fatty acid resistance. J. Biol. Chem. 289, 4417–4431 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sundberg E., et al. , ALBINO3, an Arabidopsis nuclear gene essential for chloroplast differentiation, encodes a chloroplast protein that shows homology to proteins present in bacterial membranes and yeast mitochondria. Plant Cell 9, 717–730 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Labbé K., et al. , The modified mitochondrial outer membrane carrier MTCH2 links mitochondrial fusion to lipogenesis. J. Cell Biol. 220, e202103122 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jian F., et al. , Sam50 regulates PINK1-parkin-mediated mitophagy by controlling PINK1 stability and mitochondrial morphology. Cell Rep. 23, 2989–3005 (2018). [DOI] [PubMed] [Google Scholar]
- 75.Li X., et al. , Mitochondria shed their outer membrane in response to infection-induced stress. Science 375, eabi4343 (2022). [DOI] [PubMed] [Google Scholar]
- 76.Bilal M., et al. , Terahertz optical Hall effect in p-type monolayer hexagonal boron nitride on fused silica substrate. Opt. Lett. 46, 2196 (2021). [DOI] [PubMed] [Google Scholar]
- 77.Morita K., et al. , Genome-wide CRISPR screen identifies TMEM41B as a gene required for autophagosome formation. J. Cell Biol. 217, 3817–3828 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kornmann B., et al. , An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325, 477–481 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Meisinger C., et al. , The mitochondrial morphology protein Mdm10 functions in assembly of the preprotein translocase of the outer membrane. Dev. Cell 7, 61–71 (2004). [DOI] [PubMed] [Google Scholar]
- 80.Osawa T., et al. , Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat. Struct. Mol. Biol. 26, 281–288 (2019). [DOI] [PubMed] [Google Scholar]
- 81.Tan J. X., Finkel T., A phosphoinositide signalling pathway mediates rapid lysosomal repair. Nature 609, 815–821 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Valverde D. P., et al. , ATG2 transports lipids to promote autophagosome biogenesis. J. Cell Biol. 218, 1787–1798 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tang Z., et al. , TOM40 targets Atg2 to mitochondria-associated ER membranes for phagophore expansion. Cell Rep. 28, 1744–1757.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Antonicka H., et al. , A high-density human mitochondrial proximity interaction network. Cell Metab. 32, 479–497.e9 (2020). [DOI] [PubMed] [Google Scholar]
- 85.Liu X., et al. , An AP-MS- and BioID-compatible MAC-tag enables comprehensive mapping of protein interactions and subcellular localizations. Nat. Commun. 9, 1188 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Menon A. K., Watkins W. E., Hrafnsdóttir S., Specific proteins are required to translocate phosphatidylcholine bidirectionally across the endoplasmic reticulum. Curr. Biol. 10, 241–252 (2000). [DOI] [PubMed] [Google Scholar]
- 87.Galietta L. J. V., The TMEM16 protein family: A new class of chloride channels? Biophys. J. 97, 3047–3053 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hartzell H. C., Yu K., Xiao Q., Chien L.-T., Qu Z., Anoctamin/TMEM16 family members are Ca2+-activated Cl- channels. J. Physiol. 587, 2127–2139 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mannella C. A., The “ins” and “outs” of mitochondrial membrane channels. Trends Biochem. Sci. 17, 315–320 (1992). [DOI] [PubMed] [Google Scholar]
- 90.Zalisko B. E., Chan C., Denic V., Rock R. S., Keenan R. J., Tail-anchored protein insertion by a single Get1/2 heterodimer. Cell Rep. 20, 2287–2293 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ni D., Huang Y., The expression, purification, and structure determination of BamA from E. coli. Methods Mol. Biol. Clifton NJ 1329, 169–178 (2015). [DOI] [PubMed] [Google Scholar]
- 92.Serek J., et al. , Escherichia coli YidC is a membrane insertase for Sec-independent proteins. EMBO J. 23, 294–301 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kohler R., et al. , YidC and Oxa1 form dimeric insertion pores on the translating ribosome. Mol. Cell 34, 344–353 (2009). [DOI] [PubMed] [Google Scholar]
- 94.Evans R., et al. , Protein complex prediction with AlphaFold-Multimer. bioRxiv [Preprint] (2023). 10.1101/2021.10.04.463034 (Accessed 9 July 2023). [DOI]
- 95.Lindorff-Larsen K., et al. , Improved side-chain torsion potentials for the Amber ff99SB protein force field: Improved protein side-chain potentials. Proteins Struct. Funct. Bioinforma. 78, 1950–1958 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jo S., Kim T., Iyer V. G., Im W., CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008). [DOI] [PubMed] [Google Scholar]
- 97.Abraham M. J., et al. , GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015). [Google Scholar]
- 98.Souza P. C. T., et al. , Martini 3: A general purpose force field for coarse-grained molecular dynamics. Nat. Methods 18, 382–388 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bussi G., Donadio D., Parrinello M., Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007). [DOI] [PubMed] [Google Scholar]
- 100.Parrinello M., Rahman A., Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981). [Google Scholar]
- 101.Li D., Rocha-Roa C., Schilling M. A., Reinisch K. M., Vanni S., Data for lipid scrambling is a general feature of protein insertases. Zenodo. 10.5281/zenodo.10475371. Deposited 9 January 2024. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
MD simulations data have been deposited in Zenodo (https://doi.org/10.5281/zenodo.10475371) (101). All other data are included in the manuscript and/or SI Appendix.