

Significance
Here, we provide insight into gene regulation at the C9orf72 locus that will directly impact the development of therapeutics for C9orf72-related diseases including frontotemporal dementia (FTD) and ALS (amyotrophic lateral sclerosis). First, we demonstrate that messenger RNAs (mRNAs) from the mutant C9orf72 allele, which harbor a large expansion of a six-nucleotide repeat, are selectively upregulated and can be correctly spliced (i.e., rid of the toxic repeat). Second, we show that motor neurons tolerate the loss of one C9orf72 allele and that removal of the repeat expansion corrects all of the pathological hallmarks of C9-FTD/ALS in cultured motor neurons. Overall our findings support excision of the mutant C9orf72 allele as a promising therapeutic CRISPR gene editing strategy.
Keywords: C9orf72, ALS, FTD, CRISPR, neurodegeneration
Abstract
Efforts to genetically reverse C9orf72 pathology have been hampered by our incomplete understanding of the regulation of this complex locus. We generated five different genomic excisions at the C9orf72 locus in a patient-derived induced pluripotent stem cell (iPSC) line and a non-diseased wild-type (WT) line (11 total isogenic lines), and examined gene expression and pathological hallmarks of C9 frontotemporal dementia/amyotrophic lateral sclerosis in motor neurons differentiated from these lines. Comparing the excisions in these isogenic series removed the confounding effects of different genomic backgrounds and allowed us to probe the effects of specific genomic changes. A coding single nucleotide polymorphism in the patient cell line allowed us to distinguish transcripts from the normal vs. mutant allele. Using digital droplet PCR (ddPCR), we determined that transcription from the mutant allele is upregulated at least 10-fold, and that sense transcription is independently regulated from each allele. Surprisingly, excision of the WT allele increased pathologic dipeptide repeat poly-GP expression from the mutant allele. Importantly, a single allele was sufficient to supply a normal amount of protein, suggesting that the C9orf72 gene is haplo-sufficient in induced motor neurons. Excision of the mutant repeat expansion reverted all pathology (RNA abnormalities, dipeptide repeat production, and TDP-43 pathology) and improved electrophysiological function, whereas silencing sense expression did not eliminate all dipeptide repeat proteins, presumably because of the antisense expression. These data increase our understanding of C9orf72 gene regulation and inform gene therapy approaches, including antisense oligonucleotides (ASOs) and CRISPR gene editing.
Heterozygous expansion of a GGGGCC repeat in the first intron of the C9orf72 gene is the most frequent known genetic cause of both frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) (1–3) (C9-FTD/ALS). The C9orf72 gene is complex: It has two alternative start sites and can be transcribed in both sense and antisense directions (1, 4). The repeat expansion lies between the two start sites, and is transcribed in both directions. This complexity has made it difficult to understand how the repeat expansion causes pathology, and hampered the design of effective gene-based therapy for C9-FTD/ALS.
The prevalent hypothesis is that C9-FTD/ALS pathology results from toxic products derived from expression of the C9orf72 repeat expansion itself. Indeed, sense or antisense transcripts that harbor a large repeat expansion produce toxic dipeptide repeat proteins (DPRs) through repeat-associated noncanonical (RAN) translation (4–13), and may disrupt RNA processing by sequestering RNA-binding proteins (9, 14–17). An alternative hypothesis is that the repeat expansion disrupts the sense transcription or translation of the mutant allele’s coding region and causes haplo-insufficiency (18–21). However, haplo-insufficiency is unlikely to be the major contributor to C9-FTD/ALS. The most compelling evidence against this hypothesis is that large-scale population sequencing (such as gnomAD) and clinical sequencing show that people with C9orf72 heterozygous loss-of-function mutations do not develop C9-FTD/ALS (22). Second, homozygous knock-out mouse models have an autoimmune phenotype but no neurologic disease (23–26). Nevertheless, loss of C9orf72 function may exacerbate the toxic gain-of-function (25, 27) caused by the repeat expansion. Disentangling these possibilities has been challenging because it has been hard to reliably detect C9orf72 RNA and protein products and to measure allele-specific function. In addition, traditional sequencing methods are inadequate to accurately size large repeat expansions and confirm that a genomic edit has eliminated it. As a result, experiments often compare cells from a disease-carrier to cells from an unrelated healthy donor, rather than to edited isogenic cells, which is problematic because genetic background can have a big effect on phenotypic outcomes.
To overcome these obstacles, we have recently developed a series of analytical tools (28) that we deployed in the present study. Our goal was to understand the effects of manipulating the C9orf72 locus on gene expression and on pathology at the cellular level. This type of proof-of-concept study is critical to the development of gene therapies, including gene editing and RNA targeting approaches. We engineered a series of excisions that targeted either the mutant (expanded) allele, the normal (unexpanded) allele, or both alleles in the same patient line. The resulting six isogenic lines allowed us to compare the effects of the genetic manipulations without the confounding effect of varying genetic background. We replicated the isogenic series (five additional cell lines) in a nondiseased control cell line to study the effects of the genetic changes on normal gene function. Each line was made clonal and only harbors one change to the genome.
We carried out these manipulations in human iPSCs and examined their impact on both the RNA and protein products of the C9orf72 locus, and on a cellular marker of C9/ALS pathology, TDP43, in motor neurons derived from the iPSCs. Our analysis reveals unexpected aspects of gene expression and regulation at the C9orf72 locus that should inform future therapeutic gene targeting approaches.
Results
Engineering 11 Isogenic iPSC Lines across Two Genetic Backgrounds.
The C9orf72 repeat region lies between two alternative start sites (1, 2), one in exon 1A, the other in exon 1B (Fig. 1A). Our patient line contains ~250 GGGGCC repeats (28) on the mutant allele, and two on the wild-type (WT) allele. The donor was a 37-y-old female patient of European descent who was asymptomatic at the time of donation. We chose this line because of a rare but advantageous single nucleotide polymorphism (SNP) in exon 2 that allows us to quantify gene expression from each allele independently (Fig. 1B). From here on out, we refer to this line as C9-unedited. Our control cell line was donated by a healthy 30-y-old subject of mixed European and Asian descent. It contains 10 repeats on one allele and two on the other and is referred to as WT-control.
Fig. 1.

1A-containing transcripts are upregulated from the mutant, but not WT, allele and sense expression is independently controlled by each allele. (A) The repeat region of the C9orf72 gene lies in the 5′ UTR, between alternative start sites that give rise to noncoding exons 1A (blue) and 1B (green). Translation starts in exon 2. Only the allele with the repeat expansion causes disease. We created a series of isogenic lines using CRISPR. REx: excision of the repeat region (~1,057 bp on the mutant allele). 1Ax: 227 bp biallelic excision of exon 1A. HET(Mut)x: 21 kb excision of the mutant allele made by targeting SNPs (orange circles) in cis with the mutation. HET(WT)x: 22 kb excision of the WT allele only. KO was made via biallelic excision starting 5′ to exon 1A and extending through exon 3 (21 to 22 kb; C9-patient line) or exon 2 (7 kb, WT-control line). The schematics of KO lines are shown in SF5 and SF10 (B) Schematic of expected 1A and 1B allele-specific transcripts. ddPCR probes were designed to distinguish mRNA transcripts starting at exon 1A (V3) vs. exon 1B (V2), and total C9orf72 mRNAs (probe spanning exons 2 to 3). A coding SNP allows the quantification of RNA from the WT allele (carrying a T, purple) vs. the mutant allele (carrying a C, yellow). Exon 1A normally splices onto exon 2, but the repeat expansion disrupts this splicing event. Exon 1A transcripts from the mutant (C-allele) are only detected if correctly spliced, due to amplification failure of the repeat region. (C and D) ddPCR quantification of exon 1A-containing RNA (blue), exon 1B-containing RNA (green), and total C9orf72 RNA (gray) in isogenic lines from a C9-patient (C) or WT control (D), normalized to expression of the UBE2D2 housekeeping gene. In all cell lines, the majority of C9orf72 mRNAs start at exon 1B, and mRNAs starting at exon 1A are 20 to 100 times less abundant than those starting at exon 1B. In lines harboring a repeat expansion (C9-unedited, HET(WT)x), the sum of exon-1A- + exon-1B-containing transcripts was significantly smaller than the amount of total transcripts (paired t test corrected for multiple tests, *P < 0.01). We hypothesize that this gap corresponds to improperly spliced 1A RNA from the mutant allele, since the presence of a repeat expansion would disrupt amplification between exon 1A and exon 2 as well as probe binding. (E and F) Proportion of 1A (E) and 1B (F) transcripts coming from the C vs. T allele in unedited and edited C9 lines, as determined by ddPCR. (G and H) Amounts of 1A (G) and 1B (H) transcripts relative to UBE2D2 transcripts in unedited and edited C9 lines, as determined by ddPCR. The mutant allele produces 10× more 1A transcripts (C9-unedited) than WT allele does. Excising the repeat expansion (REx) decreased expression of the 1A transcript from the mutant allele (G; Sidak’s multiple comparisons, *P < 0.0001) and increased expression of the 1B transcript from the mutant allele (H; Sidak’s multiple comparisons, **P < 0.01) without altering expression of either the 1A (Sidak’s multiple comparisons, P > 0.99) or 1B transcript from the WT allele (Sidak’s multiple comparisons, P > 0.99). Sense transcription of either allele is independently regulated, as expression of 1A- and 1B- transcripts from one allele was not altered by excision of the other allele (G, Sidak’s multiple comparison 1A-T-allele HET(Mut)x vs. C9-unedited P > 0.99; 1A-C-allele HET(Mut)x vs. C9-unedited P = 0.90; H, 1B-T-allele HET(Mut)x vs. C9-unedited P = 0.78; 1B-C-allele HET(Mut)x vs. C9-unedited P > 0.99). Excision of exon 1A eliminated expression of 1A-transcripts (G) and decreased expression of 1B transcripts from the WT (H: 1Ax, Sidak’s multiple comparison P < 0.05) but not mutant (H: 1Ax, Sidak’s multiple comparison P = 0.98) alleles. Homozygous KO eliminated both the 1A and 1B transcripts as expected (G and H). *P < 0.01, **P < 0.001, ***P < 0.0001. Error bars = SEM.
Before carrying out our edits, we engineered our C9 and WT iPSC lines to contain the hNIL transgene cassette with a TET-on system in the CLYBL safe-harbor locus (29, 30) (SI Appendix, Fig. S1). This well-characterized inducible system (29, 30) puts expression of three human transcription factors, NGN2, ISL1, and LHX3, under doxycycline control, which drives the iPSCs to differentiate into motor neurons. The neurons express high levels of motor neuron markers (HB9 and ChAT) compared to iPSCs at the time point investigated in our expression studies (2 wk of age) (SI Appendix, Fig. S1 E and F).
We then introduced Cas9 and gRNA pairs via electroporation into the WT and patient hNIL iPSC lines to generate a variety of excisions of the C9orf72 genomic locus. We used CRISPOR (31) or AlleleAnalyzer (32) to design gRNAs (SI Appendix, Table S1) with the fewest computationally predicted overall off-targets. The genetic change was confirmed by PCR and Sanger sequencing or single-molecule sequencing in sorted single cells or hand-picked clones. All clones had a normal karyotype (SI Appendix, Figs. S1–S10).
We first excised the noncoding region containing the repeat expansion (REx, Fig. 1A and SI Appendix, Fig. S2). Since gRNAs targeting the repeated motif have numerous predicted off-targets throughout the genome, it is not safe to cut within the repeat region itself; instead, we made cuts just 5′ and 3′ to the repeats (Fig. 1A REx, SI Appendix, Fig. S2). These repeat-flanking sequences are highly conserved and do not offer allele-specific gRNA binding sites. Therefore, this excision was expected to be biallelic. However, by chance it only occurred on the mutant allele in the clone we selected from our patient cell line (SI Appendix, Fig. S2), leaving intact the WT allele with its two native repeats. Our WT-control clone with the same edit was biallelic as expected (SI Appendix, Fig. S7).
Newer versions of Cas9 can distinguish between alleles that differ by a single nucleotide (33), which allowed us to use SNPs in cis with the mutation to specifically excise the mutant allele. We phased SNPs to the repeat expansion mutation in our patient line using single-molecule sequencing (28). We used AlleleAnalyzer (32) to design allele-specific gRNA pairs based on common heterozygous polymorphisms found in a reference dataset of over 2,500 human genomes from around the world (34). Using two allele-specific gRNAs, we excised 22.2 kb of the mutant allele in the patient line starting 12.3 kb upstream of exon 1A and stretching all the way through exon 3 (Fig. 1A HET(Mut)x, SI Appendix, Fig. S3). In addition, we made the equivalent 21 kb excision on the WT allele in both the patient and WT lines (Fig. 1A HET(WT)x, SI Appendix, Figs. S6 and S8) (35).
Our fourth edit, excision of exon 1A, was designed to determine the impact of this exon on C9orf72 expression and to test the hypothesis that silencing the mutation would suffice to eliminate C9orf72 pathology. Exon 1A includes a transcriptional start site and controls the expression of the C9orf72 sense-transcript harboring the mutation. We excised it on both alleles (1Ax) in both the patient (Fig. 1 1Ax, SI Appendix, Fig. S4) and WT-control lines (SI Appendix, Fig. S9). As additional controls, we also created homozygous knock-outs of the gene in our patient and WT lines, using biallelic excisions starting 21 kb upstream of exon 1A and ending in exon 3 (patient line; SI Appendix, Fig. S5) or 7 kb upstream of exon 1A and ending in exon 2 (WT line; SI Appendix, Fig. S10). We found no detectable off-targets in the edited clonal iPSC lines REx, HET(Mut)x, 1Ax by comparing Sanger sequencing across computationally predicted potential off-target sites (SI Appendix, Fig. S11).
Exon 1A-Transcription Is Upregulated on the Mutant Allele.
We evaluated the effect of each of the edits on C9orf72 RNA expression in induced motor neurons derived from the edited and unedited iPSC lines. The C9orf72 locus is known to produce at least three sense mRNAs: variant 1 (exon 1A-short through exon 5), variant 2 (exon 1B-exon 11), and variant 3 (exon 1A-long through exon 11) (1). Using ddPCR probes spanning either the exon 1A-exon 2 or exon 1B-exon 2 splice junctions, we quantified the two major splice forms of C9orf72, variant 3 and variant 2, which we refer to as 1A-transcript and 1B-transcript from here on (Fig. 1B and SI Appendix, Table S2). We were not able to detect variant 1 in our lines, consistent with its low to undetectable expression in human tissue (36, 37). We also quantified total C9orf72 mRNA using a probe targeting the exon 2-exon 3 junction.
Across all lines, most C9orf72 mRNAs contained exon 1B, while exon 1A-containing transcripts represented only a small proportion of total transcripts (Fig. 1 C and D). However, the sum of 1A and 1B transcripts was inferior to the total amount of C9orf72 transcripts in lines containing the repeat expansion (Fig. 1C; C9-unedited, HET(WT)x). This discrepancy was not observed in patient lines in which the mutation was excised (REx, HET(Mut)x) or silenced (1Ax) (Fig. 1C) or in any of the WT lines (Fig. 1D). We hypothesize the gap corresponds to sense 1A-transcripts that retain the repeat expansion instead of splicing it out; such transcripts cannot be detected by ddPCR, because the repeat expansion disrupts the binding of the ddPCR exon-exon spanning probe and puts too much distance between the primers’ binding sites for a successful PCR. If this hypothesis is correct, the actual proportion of sense 1A-transcripts could reach 30% or more of total C9orf72 transcripts in lines with the repeat expansion, up from <1% in corrected and WT motor neurons (Fig. 1 C and D). We report the amount of sense mutant transcript in human induced motor neurons. This estimate does not include antisense repeat-containing transcripts, which are known to occur but are not captured by our assay, and would further increase the total amount of repeat-containing transcripts. We cannot confirm this amount directly, because it is not yet possible to PCR or sequence transcripts harboring a large repeat expansion.
Interestingly, the number of 1A-transcripts (blue bar) decreased in the C9 lines after removal of the repeat region (REx) or excision of the mutant allele (HET(Mut)x) but not after excision of the WT allele (HET(WT)x) (Fig. 1C). The removal of the repeat expansion in the patient line (C9-REx and C9-HET(Mut)x) decreased the 1A transcript level below that of unedited WT (0.2 vs. 0.5% of UBE2D2 expression, respectively; Fig. 1C vs. Fig. 1D). We are cautious to compare expression from the two lines directly because differences in genetic background can influence baseline transcriptional levels. However, we wondered whether editing affected the methylation state which could influence transcriptional regulation. We compared methylation patterns (38) between C9-unedited and the C9-REx and C9-HET(Mut)x iPSCs. We found no differences in methylation between either lines within a 5 kb region around the repeat expansion and including exon 1A and 1B (SI Appendix, Fig. S12). In summary, these data suggest that 1A-transcription is upregulated in the diseased state, most probably from the mutant allele.
To test this hypothesis, we determined the proportion of transcripts coming from the WT vs. mutant alleles. We took advantage of a coding SNP (rs10757668) in the exon 2 splice acceptor of our patient line and phased it to the repeat expansion by single-molecule sequencing. Then, using ddPCR probes that targeted either variant of this SNP, we determined the fraction of 1A- and 1B-transcripts derived from each C9orf72 allele (Fig. 1 E and F) as well as the amount of 1A and 1B transcripts produced by each allele relative to a house-keeping transcript (Fig. 1 G and H). Once again and as expected, our assay only detected correctly spliced RNA transcripts and not those retaining the repeat expansion. Nevertheless, most (94%) of the exon 1A-containing transcripts we were able to amplify came from the mutant rather than the WT allele in the unedited patient line (Fig. 1 E and G, C9-unedited). The imbalance was corrected by repeat expansion excision, which reduced expression from the mutant allele without altering expression from the WT allele (Fig. 1 E and G, REx). These findings suggest that at least some of the mutant 1A-transcripts can undergo normal splicing. Furthermore, since the mutant and WT 1A transcripts we detect differ only at the SNP (Fig. 1B), the excess of mutant 1A transcripts in the unedited C9 line must reflect increased transcription from the mutant allele, rather than a difference in RNA stability. According to our quantification, 1A is at least 10-fold more active in the mutant than the WT allele (Fig. 1E, C9-unedited). This is likely an underestimate since we cannot currently measure 1A-transcripts retaining the repeat expansion.
In contrast to 1A transcripts, 1B transcripts came predominantly (>68%) from the WT allele in C9-unedited motor neurons (Fig. 1 F and H). The balance between the two alleles was fully restored by the allele-specific excision of the repeat expansion (REx), suggesting that the repeat expansion partially inhibits 1B-transcription on the mutant allele. Biallelic excision of exon 1A also restored equal production of 1B transcripts by the two alleles (Fig. 1F; 1Ax). However, it reduced expression from the WT allele (Fig. 1G; compare height of the purple bar (WT) between 1Ax and C9-unedited), suggesting a positive interaction between 1A and 1B on the WT allele.
Sense Transcription Is Controlled by Each Allele Independently.
Interestingly, removal of the repeat expansion did not alter the production of 1A and 1B transcripts by the WT allele, and neither did excision of the mutant allele (Fig. 1 G and H; compare the height of purple bars between C9-unedited, REx and HET(Mut)x). Similarly, removal of the WT allele did not alter expression from the mutant allele (Fig. 1 G and H, compare the height of yellow bars in HET(WT)x relative to C9-unedited). Thus, expression from either allele is independently regulated. As expected, excision of either allele resulted in elimination of any detectable transcript from that allele (HET(Mut)x and HET(WT)x, Fig. 1 E and H).
In total, these findings demonstrate that transcripts harboring the repeat expansion can be correctly spliced (i.e., rid of the toxic repeat), an exciting finding that could inspire an additional therapeutic avenue. Second, the repeat expansion increases the activity of the 1A start site and decreases the activity of the 1B start site on the mutant allele but not the WT allele. Finally, sense transcription is allele-independent as removal of either allele did not alter sense expression from the other allele.
Patient-Derived Lines Lacking Either Allele Produce Normal Levels of Full-Length C9orf72 Protein.
To quantify C9orf72 protein, we used the Simple Western system (WES) after validating antibody specificity using our knock-out line (39). None of the edits in the patient line reduced the C9orf72 protein levels in induced motor neurons (Fig. 2 A and C). In WT motor neurons, only exon 1A excision reduced C9orf72 expression (Fig. 2 B and D). Furthermore, unedited WT and C9-patient motor neurons produced equivalent amounts of C9orf72 proteins (Fig. 2E). These results indicate that the repeat expansion itself or large excisions of the C9orf72 locus (such as removal of an entire allele) do not greatly alter total protein levels in motor neurons.
Fig. 2.

C9orf72 protein abundance is not affected by excision of the repeat expansion or excision of either mutant or WT allele. Quantification of C9orf72 protein expression by WES relative to GAPDH in neurons derived from unedited and edited C9 (A–C) and WT (D and E) iPSCs 2 wk post induction of neuronal differentiation. C9orf72 protein levels were not affected by edits, except for biallelic 1A excision, which reduced protein levels in WT neurons, or homozygous gene KO, which abolished protein signal in both C9 and WT neurons. (1-way ANOVA: C9: F(5,12) = 94.81, P < 0.0001; WT: F(4,10) = 32.98, P < 0.0001; Dunnett’s multiple comparison test *P < 0.05, ****P < 0.0001). (C) There was no significant difference in the amount of C9orf72 protein produced from either the unedited WT or C9-patient line (P = 0.43; two-tailed t test). Error bars = SEM.
Removal of the Repeat Expansion Is Required to Eliminate Toxic DPRs.
C9orf72 is transcribed off of both the sense and anti-sense strands, in both normal and diseased cells (Fig. 3A) (7, 15, 40, 41). Our data suggest that sense transcription of the repeat region starts from exon 1A, since excision of exon 1A closed the gap in “undetectable” sense transcript (Fig. 1C). However, it is unresolved where anti-sense transcription initiates. Regardless, both sense and anti-sense transcripts harboring the repeat expansion are translated through noncanonical RAN translation to form five DPRs thought to be toxic (6, 7, 10). These DPRs are likely to be variable in size. They deposit in the brains of C9orf72 mutation carriers but not in nondiseased controls and are not found in other neurodegenerative conditions (i.e., DPRs are a specific C9-pathology) (4, 42). The absence of DPRs in WT cells may reflect the rapid splicing out of the repeat region from sense 1A transcripts, the inefficiency of noncanonical RAN translation when there are too few repeats, or a combination of both.
Fig. 3.
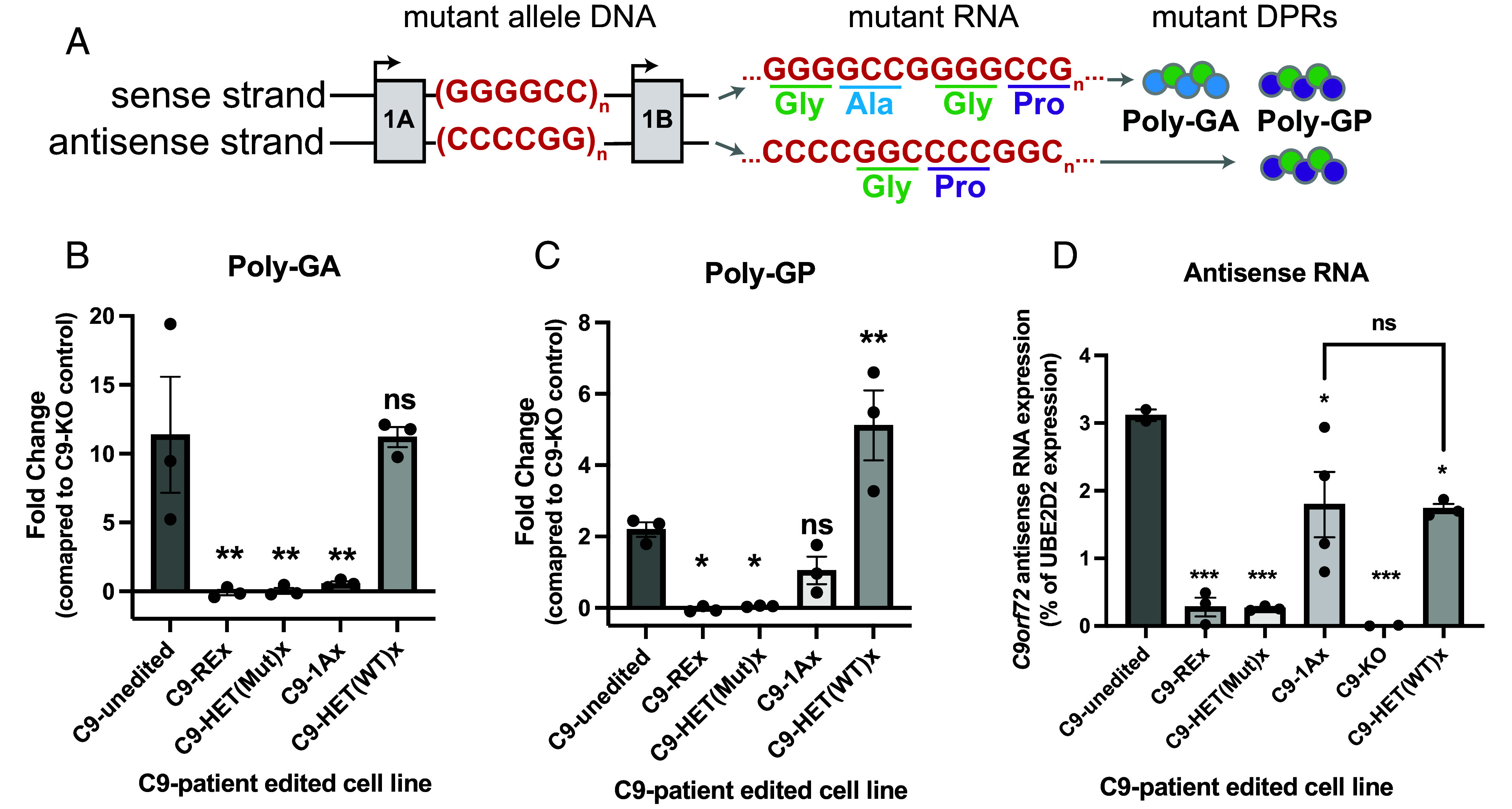
DPRs from sense RNA are eliminated by removal or silencing of the repeat expansion but antisense DPRs are only eliminated by removing the repeat expansion. (A) The repeat expansion is transcribed from the sense strand (starting at exon 1A) and the antisense strand (start site unknown) and gives rise to poly-GA and poly-GP peptides through noncanonical repeat-associated non-AUG (RAN) translation. Only sense transcription can give rise to a Poly-GA peptide, whereas Poly-GP can arise from the sense or antisense strands. (B and C) Quantification of Poly-GA (B) and Poly-GP (C) in neurons derived from unedited and edited C9 cell lines, relative to a baseline noise established by the C9 KO line, as measured via MSD ELISA. We measured DPRs in neurons 2 wk postinduction. Only excision of the repeat expansion or the mutant allele abolishes production of both Poly-GA (1-way ANOVA F(4,10) = 10.12, P < 0.001) and Poly-GP peptides (1-way ANOVA F(4,10) = 19.66, P < 0.0001). Excision of exon 1A only abolishes expression of Poly-GA, consistent with silencing of sense but not anti-sense transcription. Excision of the WT allele (HET(WTx)) more than doubled expression of Poly-GP. (D) Antisense RNA transcript measured by ddPCR is elevated in edited cell lines that retain the repeat expansion (C9-1Ax, C9-HET(WT)x) (1-way ANOVA F(5,11) = 13.07, P < 0.001). *P < 0.5, **P < 0.01 by Dunnett’s or Tukey’s multiple comparisons test sample vs. C9-unedited unless otherwise indicated by comparison bracket. Error bars = SEM.
We tested whether removing the repeat region could abrogate DPR production. We evaluated ten antibodies targeting each of these DPRs using Meso Scale Discovery’s (MSD) enzyme-linked immunosorbent assay (ELISA) immunoassay. We found two antibody combinations that could reliably detect the presence of poly-GA and poly-GP DPRs above the background level defined by our knock-out (KO) line (28, 39). The other antibodies had comparable signal in the KO line and the unedited C9-patient line, suggesting they were not specific to the C9orf72 DPRs. Using these two combinations, we measured poly-GA and poly-GP DPRs in 2-wk-old neurons derived from our C9-isogenic series.
The poly-GA is encoded exclusively on the sense strand, whereas the poly-GP can come from either strand (Fig. 3A). As expected, Poly-GA expression was eliminated by removal of the REx and excision of the mutant allele (HET(Mut)x), or by elimination of sense transcription through excision of exon 1A (1Ax) (Fig. 3B). Also as expected, all poly-GP was eliminated by removal of the repeat expansion or mutant allele. However, some poly-GP remained after excision of exon 1A (Fig. 3C, 1Ax). Since we know that sense 1A transcript is eliminated in the 1Ax line (Fig. 1 C, E, and G), the poly-GP we detect in this line must arise from antisense mutant transcript. We quantify DPR (poly-GP) expression specifically from the anti-sense transcript in patient-derived motor neurons rather than in artificial expression systems. Our data indicate that at least one-third to one-half of poly-GP derives from antisense transcription, and confirm that eliminating toxic DPRs requires the removal of the expanded repeat region. In total, these data also indicate that removal of the repeat expansion, rather than silencing sense expression, is required to completely eliminate DPRs.
Excision of the WT Allele Increased Poly-GP Expression.
A surprising finding is that excision of the WT allele more than doubled the amount of poly-GP expressed from the mutant allele (Fig. 3C, HET(WT)x). By contrast, poly-GA expression was not changed by excision of the WT allele (Fig. 3B, HET(WT)x), consistent with our earlier observation that sense transcription from one allele was unaffected by removal of the other allele (Figs. 1 E–H and 3B). Because poly-GP is translated from both sense and antisense transcripts, we asked whether an increase in antisense transcription from the loss of the WT allele could account for the increase in poly-GP expression in the HET(WT)x induced motor neuron. We measured intron-containing (antisense) RNA using ddPCR across the edited C9-induced motor neurons. Removal of the WT allele lowered antisense transcription by one-third in C9-HET(WT)x compared to C9-unedited (Fig. 3D). Additionally, elimination of sense transcription by excising exon 1A also lowered antisense transcription by one-third and was not statistically different than removal of the WT allele (Fig. 3D, 1Ax vs. HET(WT)x). Altogether, these findings suggest that the upregulation of poly-GP in HET(WT)x induced motor neurons (Fig. 3C) is not driven by an increase in either the sense or antisense transcripts we measured.
Biallelic and Allele-Specific Excisions Revert TDP-43 Pathology in 7-wk-old Motor Neurons Derived from Edited Patient iPSCs.
The pathological hallmark of C9-FTD/ALS is loss of nuclear TDP-43 and aggregation of cytoplasmic TDP-43 in affected neurons (43). These events are thought to be independent (43) and have been difficult to model in cellular and animal systems. Here we detected a clear loss of nuclear TDP-43 in 57% of TDP-43-positive motor neurons derived from our unedited patient iPSC line (Fig. 4A and SI Appendix, Fig. S13 pink arrows). This effect was age dependent, rising significantly 6 wk after induction of differentiation. The reason we could detect it is that we were able to maintain our motor neurons as a monoculture for 2 mo, after improving published protocols (29) for the generation of doxycycline-induced motor neurons by adding three growth factors (brain-derived neurotrophic factor (BDNF), glial cell line-derived neurotrophic factor (GDNF) and neurotrophin-3 (NT-3)). We quantified the loss of nuclear TDP-43 in the edited C9 lines that eliminated all RNA and DPR pathology (REx and HET(Mut)x) or silenced sense expression of the repeat expansion (1Ax) that at 7 wk postinduction. We found that on average, less than 30% of the TDP-43 positive cells lacked nuclear TDP-43 after removal of the repeat expansion (REx), the mutant allele (HET(Mut)x) or exon 1A (1Ax) (Fig. 4B). As the loss of nuclear TDP-43 was apparent only after aging motor neurons in culture for 7 wk postinduction, our system appears to recapitulate the interaction between age and genotype that is an important aspect of human C9-FTD/ALS. To our knowledge, TDP-43 pathology has not been detected in cultured human C9orf72 mutant cells so far and has only been detectable in two other cell culture systems [GRN mutant (44) and TDP-43 (45) mutant cell lines] without artificially altering the TDP-43 expression and in the absence of drug treatment.
Fig. 4.
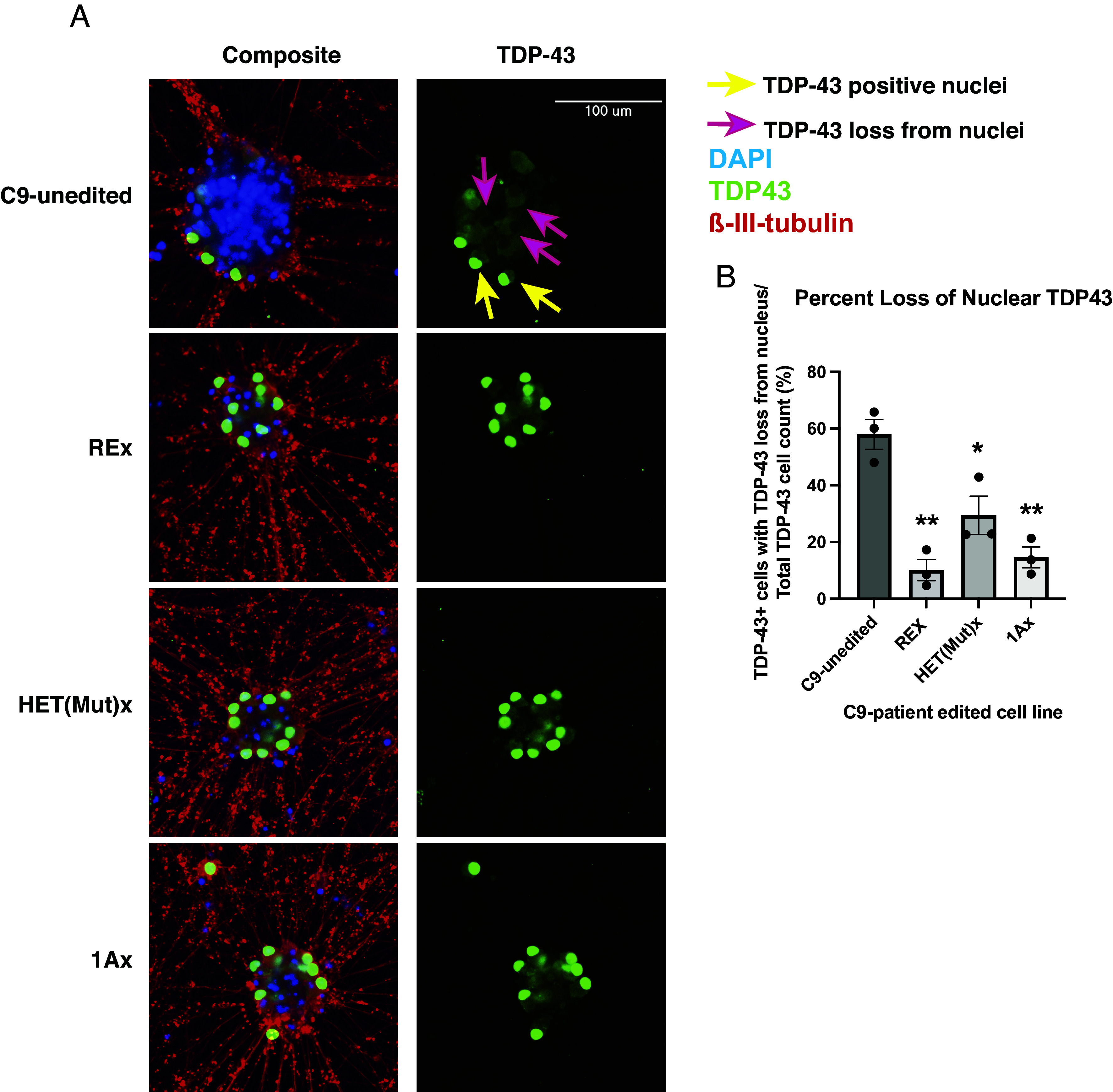
Three editing approaches ameliorate loss of nuclear TDP-43 in 7-wk-old neurons derived from C9 iPSC line. (A) Immunofluorescent images of neurons derived from unedited and edited C9 iPSCs. The neurons were grown for 7 wk and stained for TDP-43 (green), DAPI (blue) and beta-III-tubulin (red). Yellow arrow points to an example nuclei harboring TDP-43 and pink arrows to example TDP-43-positive cells whose nucleus are devoid of TDP-43. (B) Percentage of TDP-43-positive cells that lack nuclear TDP-43 (1-way ANOVA F(3,8) = 18.65; P < 0.001; *P < 0.05, **P < 0.01 by Tukey’s multiple comparison’s post hoc test between C9-unedited and each sample. REx, HET(Mut)x, and 1Ax were not statistically significantly different from one another). Each experiment contained three biologic replicates (separate wells). Error bars = SEM.
We noted that REx and HET(Mut)x cultures still harbored some motor neurons with nuclear loss of TDP-43, even though they lack the repeat expansion, and that removing ameliorated nuclear TDP-43 loss, even though it maintains antisense expression of the expanded repeat. Although a hallmark of C9-FTD/ALS, TDP-43 pathology is not specific to this gene mutation. It is also a classic pathological finding for other genetic and sporadic forms of FTD/ALS. In our cell culture system, we hypothesize the nuclear loss of TDP-43 is driven by the stress of aging to 7 wk, and exacerbated by the expression of the mutation. The take-away message is that the C9-unedited motor neurons had a greater pathological response to aging (i.e., more TDP-43-positive cells lost their nuclear TDP-43 expression) than did the edited cell motor neurons.
Biallelic and Allele-Specific Excisions Improve Electrophysiological Function in 2- and 6-wk-old Motor Neurons.
We next measured the electrophysiologic effect of removing the repeat expansion in motor neurons. We examined population-level electrophysiology using multielectrode arrays (MEAs) at 3 wk postinduction and single neuron function using whole-cell patch-clamp at 6 wk postinduction. The difference in timepoints used for these analyses was necessitated by differing rates of electrophysiological maturation in iPSC-derived neuronal cultures maintained at different culture densities; high densities for MEA recordings and low, subconfluent densities for whole-cell patch-clamp recordings.
On MEAs, burst firing is defined as a period of activity in which the time between detected depolarizing spikes (interspike interval) is less than 100 ms for a minimum of ten consecutive spikes. Increased burst activity is an indicator of increasing functional maturity in cultured neurons (46–48). Furthermore, network bursts indicate detection of simultaneous burst activity across multiple electrodes in a single array and suggest the formation of synaptic networks in cultured neurons.
The C9-unedited and 1Ax motor neurons showed no network burst activity at 3 wk of age; in contrast, both REx and HET(Mut)x motor neurons did demonstrate network burst activity (Fig. 5A), indicating that removing the repeat expansion, but not just silencing sense expression, enhances functional maturity and network forming capabilities in cultured motor neurons. Despite a lack of network activity, C9-unedited and 1Ax motor neurons were still spontaneously active (Fig. 5B). Spontaneous mean firing rate activity was nearly doubled (1.4 ± 0.17 Hz) in REx motor neurons and tripled (2.92 ± 0.48 Hz) in HET(Mut)x motor neurons compared to C9-unedited motor neurons (0.93 ± 0.11 Hz) (Fig. 5B).
Fig. 5.

Improved in vitro neuronal electrophysiological function after removal of the C9orf72 mutation. (A) C9-unedited and 1Ax 3-wk-old induced neurons showed minimal to no network bursting activity on MEAs (defined as burst events detected simultaneously from multiple electrodes within a single array). REx and HET(Mut)x showed network burst activity, with HET(Mut)x activity significantly increased compared to C9-unedited (1-way ANOVA F(3,84) = 17.3, P = 0.0003; ****P < 0.0001 by Tukey’s multiple comparison’s post hoc test). (B) Mean spontaneous firing rates weighted for electrodes showing activity on MEAs was significantly increased in HET(Mut)x compared to C9-unedited (1-way ANOVA F(3,89) = 13.3; P < 0.0001; ****P < 0.0001 by Tukey’s multiple comparison’s post hoc test). (C and D) Whole-cell patch clamp was used to identify the activity of individual neurons 40 d postinduction. Firing activity was measured during a 500 ms, 2 nA depolarizing current injection. Cells were binned into those firing a single action potential (single; D, red line), more than 1 action potential but with cessation of activity before 500 ms (adaptive; D, blue line), or repetitive firing that lasted the entire 500 ms (repetitive, D, black line). (C) Repetitive firing was increased, and adaptive firing decreased, in REx and HET(Mut)x compared to C9-unedited (χ2 test = 21.1 (6 degrees of freedom); P < 0.01). (E–G) There were no statistical differences between groups for individual action potential metrics, including (E) resting membrane potential (1-way ANOVA F(3,86) = 1.53; P = 0.2), (F) spike amplitudes (1-way ANOVA F(3,86) = 1.71; P = 0.17) and (G) action potential durations at 90% repolarization (APD90) (Kruskal–Wallis = 6.12; P = 0.11). Error bars = SEM.
It has been previously shown that C9orf72 mutant iPSC-derived motor neurons have a decreased capacity to fire repetitive action potential trains compared with controls (49). Such data indicate that poor firing capacity is a phenotypic identifier for C9-ALS neurons in culture and suggest that interventions that increase firing capacity may therefore have therapeutic benefit. Using whole-cell patch clamp, we found that 500 ms, 2 nA current injections led to an increase in the percentage of induced motor neurons capable of repetitive firing among motor neurons in which the mutant repeat had been removed compared to C9-unedited [Fig. 5C, dark gray, REx, HET(Mut)x)] but not among motor neurons in which sense transcription is silenced (Fig. 5C, 1Ax). This increase in repetitive firing capacity occurred at the single-cell level and was not dependent on network activity. Nor was it dependent on individual action potential characteristics, as the motor neurons in each group showed similar resting membrane potentials (Fig. 5E), spike amplitudes (Fig. 5F), and action potential durations (Fig. 5G).
We did not compare the C9-isogenic series to the WT-series on electrophysiology (or TDP-43 pathology) because the genetic backgrounds between the two series would be confounding. We have shown previously that baseline electrophysiology differs significantly between iPSC-derived motor neurons with different genotypes, even in the absence of disease-causing mutations (48). These differences necessitate electrophysiological comparisons be performed between isogenic groups. Previous work has shown that iPSC-derived motor neurons harboring C9orf72 and TDP-43 mutations exhibit reductions in repetitive firing behavior by patch (49) and burst firing on MEAs (45). Therefore, we interpret the capacity for targeted edits that remove the repeat expansion to improve these metrics in our C9-mutant line as an improvement in overall functional performance.
In summary, we found an improvement of in vitro electrophysiologic markers of neuronal functional capacity for network activity and repetitive firing after removal of the repeat expansion biallelically (REx) or in an allele-specific manner (HET(Mut)x). This effect highlights that deficits due to the C9orf72 mutation affect adaptive neuronal function, as the C9-unedited motor neurons demonstrated normal single-spike action potential properties at the single-neuron level.
Discussion
Expression of the C9orf72 locus is complex, with multiple transcription start sites and RNA splice forms, and translation from both the sense and antisense strands. Our eleven edited lines across two genetic backgrounds reveal important insights into the regulation of C9orf72. We demonstrate that transcription of 1A-transcripts is upregulated at least 10-fold on the mutant relative to the WT allele, using our quantitative allele-specific ddPCR methods. An upregulation of exon 1A-containing transcripts has previously been shown in postmortem human brain tissue (36), and our findings further this insight by demonstrating that upregulation is occurring from the mutant, but not WT, allele. Thus, transcriptional upregulation of the mutation is a possible biological driver of disease. We estimate that 30% or more of the total sense transcript from the C9orf72 locus contains the mutation (based on the gap in detectable RNA in Fig. 1C, C9-unedited). To this, we must add antisense mutant transcripts, which may also contain the repeat expansion. In all, our observations suggest that induced motor neurons harboring the mutation produce a high amount of mutant RNA.
While RNAs harboring large repeat expansions could be toxic in themselves, as well as via their associated DPRs, our experiments suggest that at least a portion of these transcripts splice out the repeat expansion. How efficient this splicing event is, and what becomes of the spliced-out repeats is unknown at this point. However, boosting this innate splicing event (14, 50, 51) could constitute a viable therapeutic approach.
Our work shows that C9orf72 is haplo-sufficient in motor neurons, since motor neurons containing a single copy of the gene produce the same amount of protein as do WT motor neurons. Our data on haplo-sufficiency of C9orf72 at the protein level in motor neurons is significant because a dominant view in the field is that C9orf72 is haplo-insufficient (18, 37, 52, 53). However, this possibility is not supported by population genetic (loss-of-function mutations exist in the human population but are not known to present with FTD/ALS) or by heterozygous and homozygous knock-out animal models (23, 24, 26, 54–56), which lack neurologic phenotypes. Now we have cellular confirmation of haplo-sufficiency in motor neurons. Nevertheless, artificially reducing C9orf72 function can exacerbate toxicity from the mutant allele (18, 21, 25), suggesting a protective role of WT C9orf72 gene expression. It is possible that downregulation of the C9orf72 RNA or protein in neurons or other cell types, or accelerated metabolism of either, occur in the context of age or disease state and exacerbate the toxic gain-of-function drivers of disease. This may explain why RNA (1, 36, 57, 58) and protein (53, 59–61) levels have been found to be low in patient tissues. But as a field we should be cautious in interpreting these observations as evidence that C9orf72 disease results from haplo-insufficiency, since the cumulative genetic, animal model and now human motor neuron cellular data instead support haplo-sufficiency.
Part of the confusion regarding haplo-sufficiency of C9orf72 may also be attributable to quantification of RNA rather than protein changes. We show that reduction in RNA by over half after removal of one allele (HET(Mut)x or HET(WT)x, Fig. 1 G and H) does not alter protein levels (Fig. 2 A–D) in motor neurons. A second potential explanation is that quantification of C9orf72 protein has been flawed because of nonspecific antibodies. We (28, 39) and others (62) have shown, using knock-out lines, that many of the antibodies commonly used in the field are not specific for C9orf72 despite showing signal in various assays. Given the nonspecificity of many antibodies for C9orf72 protein, it may be time to reexamine protein expression levels in postmortem and other human tissues.
Another major insight of our work is that sense transcription and the expression of sense RNA-derived poly-GA is allele-independent but the production of poly-GP dipeptide off the mutant allele is influenced by the WT allele. Approximately one-third of poly-GP protein is from the antisense strand, based on poly-GP production in the 1Ax motor neurons (Fig. 3C), in which there is no sense transcription (Fig. 1 C, E, and G). Removal of the WT allele increases the amount of poly-GP further, but without altering poly-GA levels (Fig. 3B). If loss of the WT allele altered the stability of dipeptide repeats globally, we would expect both poly-GA and poly-GP levels to rise in the HET(WT)x. Furthermore, the difference in poly-GP amounts between 1Ax and HET(WT)x is not due to a change in measurable antisense RNA levels, as antisense RNA levels were comparable between these two lines (Fig. 3D). We conclude that the upregulation of poly-GP reflects other cellular factors, such as preferential stabilization of the poly-GP peptides, increased translation of antisense transcripts or the presence of additional antisense transcripts not measured by our assay. Regardless of the underlying mechanisms, this surprising finding provides a potential explanation for the failure of ASOs to reverse pathology. ASOs target sense transcription, and are thought to suppress sense RNAs from both the WT and mutant alleles given their sequence homology to both alleles (15, 63, 64). In turn, suppression of sense RNAs by ASOs could facilitate the translation of antisense RNAs. Our observation merits further investigation but encourages caution when developing new therapeutics.
We show that TDP43 pathology can be revealed in iPSC-derived motor neurons carrying the C9orf72 mutation when they are aged; up to now, TDP43 was not detectable without directly manipulating the TDP43 pathway, and many thought it was not possible to see TDP43 pathology in cell culture. While we demonstrate an interaction between mutation and cellular age on the appearance of TDP-43 pathology, the other ingredient may be cellular stress induced by prolonged time in culture. We propose that our culture system may provide a means to investigate why patients who are born with the mutation and express it throughout the brain and body only develop the disease later in life.
An interesting observation is that while neither REx nor HET(Mut)x motor neurons have sense or antisense expression of the repeat expansion (which was removed in both lines), and both produce the same amount of C9orf72 protein, they differ in their ability to ability to rescue the TDP-43 (Fig. 4B) and electrophysiologic (Fig. 5 A–C) phenotypes. Biallelic excision of the intronic region containing the repeat expansion (REx) had a larger effect on TDP-43 pathology than did the 21 kb excision of the mutant allele in HET(Mut)x, which, by contrast, increased spontaneous and repetitive firing to a larger degree. We wonder whether the 21 kb excision removed regulatory regions on the mutant allele that normally limit firing capacity of the motor neurons. Our results suggest there is more to be learned about the influence of the intronic and regulatory regions of the C9orf72 gene in nondiseased and diseased states.
Our experimental design avoided a number of confounding factors that could have muddled the interpretation of our findings. One is the effect of genetic background, which we minimized by comparing three edits head-to-head in isogenic cell lines, all derived from the same patient’s iPSCs. The power of isogenic series to isolate the effects of specific genomic changes has been demonstrated particularly effectively by the iNDI project (65) to compare neurodegenerative disease–causing mutations on a single cell line. A limitation of this study is that our biological insight result primarily from a series of edits made in a single patient cell line. While our experiments will need to be replicated with cells from different patients and from lines with varying repeat lengths, isogenic comparisons are the current gold standard for head-to-head comparisons of genetic changes. Another confounding factor is the possibility that diseased cells influence the health of edited cells, for instance by secreting pathogenic factors such as DPRs. For this reason, we think another gold standard should be to compare genomic changes between clonal cell lines, as we have done here, rather than between unsorted cultures.
Our results have important therapeutic implications. Up to now, the C9orf72 locus has been challenging to edit in a manner compatible with clinical translation. Others have shown that CRISPR editing of the C9orf72 locus reduced pathologic burden in cells and mice (66, 67), but both approaches disrupted the normal allele in addition to the mutant allele (by biallelic excision of either exon 1A or exon 1B). This may be deleterious as homozygous knockout causes early lethality in mice (23–25) and as we have shown here that excision of exon 1A silences sense transcription but does not eliminate all pathology. In particular, it did not eliminate the production of toxic peptides from antisense transcripts and did not improve electrophysiologic measures. In contrast, our two other approaches (biallelic excision of the repeat expansion, or selective excision of the mutant allele) preserved WT protein levels, corrected RNA abnormalities, abrogated dipeptide repeat production, reduced TDP43 pathology, and improved adaptive electrophysiologic function in patient-derived iPSC-derived motor neurons. From these data, we can advance repeat expansion excision and allele-specific excision to further preclinical testing, including in postmitotic neurons and in vivo models.
Methods
Cell Line Generation and Editing.
We used a deidentified patient iPSC line harboring the C9orf72 mutation and a control cell line without mutation (WT-control). We first knocked in the inducible motor neuron transcription factor transgene cassette in the CLYBL safe-harbor locus. We then edited the lines using HiFi spCas9 (Macrolabs, UC Berkeley) and two gRNAs (SI Appendix, Table S1). We used PacBio single-molecule sequencing to size the repeat expansion, detect repeat expansion excision in C9-REx and detect methylation after editing across C9-patient lines. Further details on iPSC cell line generation (SI Appendix, Figs. S1–S11), maintenance, single-molecule sequencing, and motor neuron differentiation are detailed in SI Appendix, Supplemental Methods.
RNA Quantification by ddPCR.
500 ng of RNA from 2-wk-old induced motor neurons was run on the QX100 Droplet Reader (Bio-Rad 186-3002) with three technical replicates of each of three biologic replicates (independent wells of differentiated motor neurons) using primers and probes in SI Appendix, Table S2. See SI Appendix, Supplemental Methods for full details.
C9orf72 Protein Quantification by WES.
C9orf72 protein quantification by WES (Bio-Techne) was performed according to the manufacturer’s protocol and primary antibodies mouse anti-C9orf72 (GeneTex, GTX634482 at 1:100 and rabbit anti-GAPDH (AbCam, AB9485) at 1:1,000. Duplexed secondaries included 9.5 µL of mouse (ProteinSimple, DM-002) and 0.5 µL of 20× anti-rabbit (ProteinSimple, 043-426). See SI Appendix, Supplemental Methods for full details.
Dipeptide Repeat Quantification by MDS ELISA Immunoassay.
We used the Small Spot Streptavidin Plate (L45SA, MSD). Poly-GA was detected using anti-GA antibody (MABN889, Millipore) at 1 µg/mL (capture) and 2 µg/mL (detect) final concentration and 18 µg total protein per sample (blocking buffer A, solution PBS). Poly-GP was detected using anti-GP antibody (affinity purified TALS828.179 from TargetALS, purification lot A-I 0757 and stock concentration 1.39 mg/mL) at a final concentration of 2 µg/mL capture and 4 µg/mL detected with 18.5 µg total protein per sample (blocking buffer A, solution TBS). See SI Appendix, Supplemental Methods for full details.
TDP-43 Immunocytochemistry and Quantification.
TDP-43 immunocytochemistry and quantification was performed in fixed 7-wk-old induced motor neurons. Primary antibodies: rabbit anti-TDP43 (10782-2-AP, Proteintech) at 1:500, beta-III-tubulin (480011, Invitrogen) at 1:250 incubated overnight at 4 °C. Secondary antibodies: goat anti-rabbit Alexa Fluor 488 nm and Goat anti-mouse Alexa Fluor 594 nm incubated at room temperature for 1 h. DAPI (D1306, ThermoFisher Scientific) for 5 min at room temperature. See SI Appendix, Supplemental Methods for full details.
Electrophysiology.
Detailed protocols for induced motor neuron MEA and whole-cell patch clamp are available in SI Appendix, Supplemental Methods.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
C.D.C. is supported by NIH/NINDS K08-NS112330KO8, Alzheimer’s Association AACSF-17-531484, UCSF CTSI TL1 Fellowship 5TL1TR001871-04, Bright Focus Foundation A20201490F, Carol and Gene Ludwig Award for Early Career Research, Larry H. Hillblom Fellowship, Shupin Fellowship from the UCSF Neurology Endowment, Weill Institute for Neurosciences and UCSF Memory & Aging Center. C.D.C. is grateful to Nancy Sakamoto, Joe DiSabatao, the Wolfen Family Foundation, and Kathleen D. Mayhew and the Mayhew family for their generous support. B.R.C. and C.D.C. are supported by RF1-AG072052. O.A.A. is supported by NIH/NIA SC-00080. B.R.C. is supported by R01-HL130533, R01-HL13535801, P01-HL146366. B.R.C. acknowledges support through gifts from the Roddenberry Foundation, Keenon Werling and Pauline and Thomas Tusher. A.S.T.S. is supported by R03 TR004009 and an award from the University of Washington’s Royalty Research Fund. We thank F. Chanut, L. Judge, J. Perez-Bermejo, Z. Nevin, H. Sun, and M. Bernardi for technical advice and editorial comments.
Author contributions
B.R.C. and C.D.C. conceived the study; C.D.C. designed research; A.S., K.G., M.S., A.M.B., O.A.A., K.A.B., R.S.C., E.P., M.T.B., K.C.K., Y.-C.T., A.S.T.S., and C.D.C. performed research; H.L.W., K.C.K., and C.D.C. contributed new reagents/analytic tools; A.S., K.G., M.S., A.M.B., K.A.B., P.O.I.-L., K.C.K., Y.-C.T., A.S.T.S., and C.D.C. analyzed data; H.L.W. trained on analysis tools, provided feedback on assay development; and C.D.C. wrote the paper.
Competing interests
B.R.C. is a founder of Tenaya Therapeutics, a company focused on finding treatments for heart failure, including genetic cardiomyopathies. B.R.C. holds equity in Tenaya. C.D.C. is a founder, with equity, in Ciznor Co., a CNS therapeutics company. B.R.C. and C.D.C. are inventors on a patent application for this work. Y.-C.T. is a full-time employee at PacBio, a company developing single-molecule sequencing technologies. The other authors declare no competing interests.
Footnotes
This article is a PNAS Direct Submission. T.L. is a guest editor invited by the Editorial Board.
Contributor Information
Bruce R. Conklin, Email: bconklin@gladstone.ucsf.edu.
Claire Dudley Clelland, Email: claire.clelland@ucsf.edu.
Data, Materials, and Software Availability
Sequencing data have been deposited in NIH Bioproject; GEO (PRJNA1058535; GSE252200) (35, 38). All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.DeJesus-Hernandez M., et al. , Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Renton A. E., et al. , A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Majounie E., et al. , Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 11, 323–330 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ash P. E. A., et al. , Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gendron T. F., et al. , Cerebellar c9RAN proteins associate with clinical and neuropathological characteristics of C9ORF72 repeat expansion carriers. Acta Neuropathol. 130, 559–573 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mori K., et al. , The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Mori K., et al. , Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 126, 881–893 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Wen X., et al. , Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84, 1213–1225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zu T., et al. , RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. U.S.A. 110, E4968–E4977 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mackenzie I. R., et al. , Dipeptide repeat protein pathology in C9ORF72 mutation cases: Clinico-pathological correlations. Acta Neuropathol. 126, 859–879 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Mann D. M. A., et al. , Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol. Commun. 1, 68 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryan S., Rollinson S., Hobbs E., Pickering-Brown S., C9orf72 dipeptides disrupt the nucleocytoplasmic transport machinery and cause TDP-43 mislocalisation to the cytoplasm. Sci. Rep. 12, 4799 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nanaura H., et al. , C9orf72-derived arginine-rich poly-dipeptides impede phase modifiers. Nat. Commun. 12, 5301 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang S., et al. , Nuclear export and translation of circular repeat-containing intronic RNA in C9ORF72-ALS/FTD. Nat. Commun. 12, 4908 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lagier-Tourenne C., et al. , Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. U.S.A. 110, E4530–E4539 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Celona B., et al. , Suppression of C9orf72 RNA repeat-induced neurotoxicity by the ALS-associated RNA-binding protein Zfp106. eLife 6, e19032 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haeusler A. R., et al. , C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507, 195–200 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi Y., et al. , Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 24, 313–325 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ciura S., et al. , Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann. Neurol. 74, 180–187 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Balendra R., Isaacs A. M., C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 14, 544–558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shao Q., et al. , C9orf72 deficiency promotes motor deficits of a C9ALS/FTD mouse model in a dose-dependent manner. Acta Neuropathol. Commun. 7, 32 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harms M. B., et al. , Lack of C9ORF72 coding mutations supports a gain of function for repeat expansions in amyotrophic lateral sclerosis. Neurobiol. Aging 34, 2234.e13–2234.e19 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burberry A., et al. , Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci. Transl. Med. 8, 347ra93 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Rourke J. G., et al. , C9orf72 is required for proper macrophage and microglial function in mice. Science 351, 1324–1329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu Q., et al. , Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat. Neurosci. 23, 615–624 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sudria-Lopez E., et al. , Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol. 132, 145–147 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abo-Rady M., et al. , Knocking out C9ORF72 exacerbates axonal trafficking defects associated with hexanucleotide repeat expansion and reduces levels of heat shock proteins. Stem Cell Rep. 14, 390–405 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sckaff M., et al. , Two therapeutic CRISPR/Cas9 gene editing approaches revert FTD/ALS cellular pathology caused by a C9orf72 repeat expansion mutation in patient derived cells. bioRxiv [Preprint] (2022). 10.1101/2022.05.21.492887 (Accessed 21 May 2022). [DOI]
- 29.Fernandopulle M. S., et al. , TRanscription factor-mediated differentiation of human iPSCs into neurons. Curr. Protoc. Cell Biol. 79, e51 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feliciano C. M., et al. , Allele-specific gene editing rescues pathology in a human model of charcot-marie-tooth disease type 2E. Front. Cell Dev. Biol. 9, 723023 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haeussler M., et al. , Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 17, 148 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keough K. C., et al. , AlleleAnalyzer: A tool for personalized and allele-specific sgRNA design. Genome Biol. 20, 167 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee S. H., Park Y.-H., Jin Y. B., Kim S.-U., Hur J. K., CRISPR diagnosis and therapeutics with single base pair precision. Trends Mol. Med. 26, P337–P350 (2019), 10.1016/j.molmed.2019.09.008. [DOI] [PubMed] [Google Scholar]
- 34.Sudmant P. H., et al. , An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Issagholian-Lewin P. O., Clelland C. D., Reversal of C9orf72 mutation-induced transcriptional dysregulation and pathology in cultured human neurons by allele-specific excision (human). NIH Bioproject. https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1058535. Deposited 28 December 2023. [DOI] [PMC free article] [PubMed]
- 36.van Blitterswijk M., et al. , Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathol. 130, 863–876 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rizzu P., et al. , C9orf72 is differentially expressed in the central nervous system and myeloid cells and consistently reduced in C9orf72, MAPT and GRN mutation carriers. Acta Neuropathol. Commun. 4, 37 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Issagholian-Lewin P. O., Clelland C. D., C9P5 HET(WT)x. NCBI GEO. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE252200. Deposited 28 December 2023.
- 39.Salomonsson S. E., et al. , Validated assays for the quantification of C9orf72 human pathology. Sci. Rep. 14, 828 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gendron T. F., et al. , Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 126, 829–844 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mizielinska S., et al. , C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 126, 845–857 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Al-Sarraj S., et al. , p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 122, 691–702 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Vatsavayai S. C., Nana A. L., Yokoyama J. S., Seeley W. W., C9orf72-FTD/ALS pathogenesis: Evidence from human neuropathological studies. Acta Neuropathol. 137, 1–26 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valdez C., et al. , Progranulin-mediated deficiency of cathepsin D results in FTD and NCL-like phenotypes in neurons derived from FTD patients. Hum. Mol. Genet. 26, 4861–4872 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith A. S. T., et al. , Human induced pluripotent stem cell-derived TDP-43 mutant neurons exhibit consistent functional phenotypes across multiple gene edited lines despite transcriptomic and splicing discrepancies. Front. Cell Dev. Biol. 9, 728707 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chiappalone M., et al. , Burst detection algorithms for the analysis of spatio-temporal patterns in cortical networks of neurons. Neurocomputing 65–66, 653–662 (2005). [Google Scholar]
- 47.Kapucu F. E., et al. , Burst analysis tool for developing neuronal networks exhibiting highly varying action potential dynamics. Front. Comput. Neurosci. 6, 38 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith A. S. T., et al. , HDAC6 inhibition corrects electrophysiological and axonal transport deficits in a human stem cell-based model of charcot-marie-tooth disease (Type 2D). Adv. Biol. 6, e2101308 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Devlin A.-C., et al. , Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat. Commun. 6, 5999 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yin S., et al. , Evidence that C9ORF72 dipeptide repeat proteins associate with U2 snRNP to cause Mis-splicing in ALS/FTD patients. Cell Rep. 19, 2244–2256 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Niblock M., et al. , Retention of hexanucleotide repeat-containing intron in C9orf72 mRNA: Implications for the pathogenesis of ALS/FTD. Acta Neuropathol. Commun. 4, 18 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Su M.-Y., Fromm S. A., Zoncu R., Hurley J. H., Structure of the C9orf72 ARF GAP complex that is haploinsufficient in ALS and FTD. Nature 585, 251–255 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frick P., et al. , Novel antibodies reveal presynaptic localization of C9orf72 protein and reduced protein levels in C9orf72 mutation carriers. Acta Neuropathol. Commun. 6, 72 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koppers M., et al. , C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol. 78, 426–438 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiang J., et al. , Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 90, 535–550 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Atanasio A., et al. , C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci. Rep. 6, 23204 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Belzil V. V., et al. , Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 126, 895–905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Donnelly C. J., et al. , RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Viodé A., et al. , New antibody-free mass spectrometry-based quantification reveals that C9ORF72 long protein isoform is reduced in the frontal cortex of hexanucleotide-repeat expansion carriers. Front. Neurosci. 12, 589 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Waite A. J., et al. , Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol. Aging 35, 1779.e5–1779.e13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saberi S., et al. , Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol. 135, 459–474 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Laflamme C., et al. , Implementation of an antibody characterization procedure and application to the major ALS/FTD disease gene C9ORF72. eLife 8, e48363 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tran H., et al. , Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat. Med. 28, 117–124 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sareen D., et al. , Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 5, 208ra149 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramos D. M., Skarnes W. C., Singleton A. B., Cookson M. R., Ward M. E., Tackling neurodegenerative diseases with genomic engineering: A new stem cell initiative from the NIH. Neuron 109, 1080–1083 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Piao X., et al. , Dual-gRNA approach with limited off-target effect corrects C9ORF72 repeat expansion in vivo. Sci. Rep. 12, 5672 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meijboom K. E., et al. , CRISPR/Cas9-mediated excision of ALS/FTD-causing hexanucleotide repeat expansion in C9ORF72 rescues major disease mechanisms in vivo and in vitro. Nat. Comm. 13, 6286 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
Sequencing data have been deposited in NIH Bioproject; GEO (PRJNA1058535; GSE252200) (35, 38). All study data are included in the article and/or SI Appendix.