

Abstract
Cyclophilins exert both intracellular and extracellular activities related to immune responses and inflammation, which have been implicated in pathogenesis of atherosclerosis. Pan-inhibition of cyclophilins has both pro- and antiatherosclerotic properties, but specific contributions of extracellular and intracellular cyclophilins to these effects have not been characterized. Here, using selective inhibitor of extracellular cyclophilins, we investigated the role of these molecules in atherosclerosis. Apolipoprotein E–null mice fed a high-fat diet received intraperitoneal injections every second day of either vehicle or two analogs of cyclosporine A (CsA): [Melle]4-CsA (NIM811), a nonimmunosupressive cell-permeable inhibitor of both intracellular and extracellular cyclophilins; and [(4R)-4-[(6-carboxy-1H-benzo[d]imidazol-2-yl)-methyl]-4-methyl-l-threonine]1-CsA (MM284), cell-impermeable analog only inhibiting extracellular cyclophilins. Development of atherosclerosis and composition of plaques in aorta and innominate artery were studied. Both analogs increased abundance and cross-sectional size of the atherosclerotic plaques in aorta but did not affect development of atherosclerosis in innominate artery. Neither compound affected abundance of macrophages and amount of vascular cell adhesion molecule-1 or nitrotyrosine in the plaques of both arteries. Both compounds reduced the amount of collagen in innominate artery without affecting abundance of collagen in aortic sinus. MM284, but not NIM811, significantly reduced plasma concentration of tumor necrosis factor-α (TNFα); neither compound affected plasma concentrations of interleukin (IL)-6, IL-10 or monocyte chemoattractant protein-1. Ratio between different populations of immune cells in blood or isolated from lymph nodes and spleen as well as plasma lipoprotein profile were unaffected by both compounds. In conclusion, selective inhibition of extracellular cyclophilins reduced TNFα levels in plasma but increased atherosclerosis.
Introduction
Inflammation is an important element of pathogenesis of atherosclerosis, and cyclophilins are key mediators of inflammation (Kockx et al., 2010; Bukrinsky et al., 2013). Cyclophilins are expressed ubiquitously and participate in many intracellular inflammation-related pathways, including those relevant to the pathogenesis of atherosclerosis. For example, they stimulate expression of scavenger receptors (Nigro et al., 2011), activate platelets (Seizer et al., 2015), and regulate normal function of cholesterol transporter ATP-binding cassette transporter A1 (Le Goff et al., 2004). Additionally, cyclophilins are secreted both locally and into circulation; secreted extracellular cyclophilins also interfere with pathways relevant to atherosclerosis. Thus, extracellular cyclophilins were shown to activate endothelial cells (Jin et al., 2004), enhance secretion of matrix metalloproteinases by macrophages (Seizer et al., 2010), and proliferation of smooth muscle cells (Jin et al., 2000); they are also powerful chemotactic agents (Bukrinsky, 2002). Given that cyclophilins are involved in a considerable number of atherosclerosis-related pathways, both pro- and antiatherogenic, the overall effect of inhibition of cyclophilins on development of atherosclerosis is difficult to predict and experimental findings are contradictory. Thus, genetic ablation of CypA (cyclophilin A) in apolipoprotein E (apoE)–null mouse model of atherosclerosis was antiatherogenic (Nigro et al., 2011), whereas treatment of apoE-null mice with cyclosporine A (CsA) did not affect development of atherosclerosis (Moghadasian, 2006). In rabbits, treatment with CsA was proatherogenic in long-term treatment (Roselaar et al., 1995), but antiatherogenic in short-term experiments (Drew and Tipping, 1995). Such equivocal outcomes make pan-inhibition of cyclophilins a poor pharmacological approach for treatment of atherosclerosis and emphasize a need to separate pro- from antiatherogenic effects of cyclophilins.
In this study we tested a hypothesis that selective inhibition of extracellular activities of cyclophilins without affecting their intracellular activities, an approach proven beneficial in allergic lung inflammation (Balsley et al., 2010), may achieve separation of proatherogenic from antiatherogenic effects of cyclophilin inhibition. This anticipation was based on predominantly proinflammatory activities exerted by extracellular cyclophilins (Bukrinsky, 2015). We took advantage of availability of CsA derivative MM284 ([(4R)-4-[(6-carboxy-1H-benzo[d]imidazol-2-yl)-methyl]-4-methyl-l-threonine]1-CsA), a cell-impermeable cyclosporine derivative that only binds to extracellular cyclophilins (Malesevic et al., 2013; Seizer et al., 2015), and compared the effect of MM284 to the effect of NIM811 ([Melle]4-CsA), a cell-permeable nonimmunosuppressive cyclosporine A derivative, on development of atherosclerosis in an animal model of atherosclerosis. Surprisingly, while MM284 did exert anti-inflammatory activity, it exacerbated atherosclerosis.
Materials and Methods
Animal Studies.
Male 8-week-old apoE-deficient mice were placed on high-fat diet (SF-00219; Specialty Feeds, Glen Forrest, WA, Australia) containing 21% fat and 0.15% cholesterol, available ad libitum and separated into three groups of 10 mice. Where possible, each litter was equally split between treatment groups to reduce the possible effect of genetic drift or epigenetic differences between litters. The mice were treated with modified cyclosporine compounds MM284 and NIM811 dissolved in vehicle [15% ethanol in 15% cremophore EL (Sigma-Aldrich, St. Louis, MO)] at 6.6 mg/kg administered every second day by intraperitoneal injections. Control mice received the same volume of vehicle. After 6 weeks of treatment, the mice were euthanized by CO2 inhalation and blood was collected by cardiac puncture into EDTA tubes. The aorta, aortic sinus, and innominate artery were collected for analysis of plaque development. Spleen and iliac lymph nodes were collected for flow cytometry. All animal experiments were approved by Alfred Medical Research and Education Precinct ethics committee, were carried out in accordance with the Declaration of Helsinki, and conformed to the Australian code of practice for the care and use of animals for scientific purposes.
Histology.
Before collection, aortae were perfused with phosphate-buffered saline containing 2 mM EDTA. Excised aortae were stained for lipids with Sudan IV (ProSciTech Pty Ltd, Thuringowa Central, QLD, Australia), and stained periaortic fat was removed. Images of aorta were collected with Moticam 2500 camera on Motic 1100 microscope (Motic, Richmond, BC, Canada) and stitched with plug-in in Fiji imaging software (ImageJ; US National Institutes of Health, Bethesda, MD) (Preibisch et al., 2009).
Aortic sinus and innominate artery were imbedded in optimal cutting temperature (Sakura, Torrance, CA) and frozen for assessment of atherosclerotic plaques. Frozen tissue was cut on Microme HM550 (Zeiss, Jena, Germany); aortic sinus sections were collected when all three valves were apparent, innominate artery was sectioned from innominate bifurcation toward the aorta, sections were collected immediately after the merger of right subclavian and carotid arteries. Consecutive sections spanning 360 and 240 μm of aortic sinus and innominate artery, respectively, were collected. Lesion size was determined by staining with Oil Red O (Sigma-Aldrich) to determine the lesion size and with Masson’s trichrome to assess collagen content. Sections were also stained by standard immunohistochemistry for the abundance of vascular cell adhesion molecule (VCAM-1) (rat anti-mouse VCAM-1; BD Pharmingen, Franklin Lakes, NJ), nitrotyrosine (rabbit anti-nitrotyrosine; Millipore, Billerica, MA), and macrophage infiltration (rat anti-mouse CD68; BD Pharmingen) as previously described (Ditiatkovski et al., 2013).
The collected images were analyzed using ImagePro plus 6.0 (Media Cybernetics, Inc., Bethesda, MD) and Fiji (ImageJ) software. Data are presented as an absolute numbers in square millimeters or as a percentage of total plaque area as appropriate.
Analysis of Plasma Lipoproteins and Cytokines.
Blood collected by cardiac puncture after 6 weeks of treatment was centrifuged and plasma was collected. Plasma total cholesterol and triglyceride content were measured using colorimetric kits (Wako Pure Chemicals, Tokyo, Japan) per manufacturer’s instructions. Plasma high-density lipoprotein (HDL) contents were quantified with total cholesterol kit after apolipoprotein B depletion from plasma as per dextran sulfate and magnesium chloride method (Warnick et al., 1982). Low-density lipoprotein values were calculated using the Friedwald equation. Plasma levels of apolipoprotein A-I were calculated from SDS-PAGE followed by a standard Western blot, using an in-house anti–apolipoprotein A-I antibody and purified mouse HDL as a standard. Apolipoprotein B–depleted plasma was used to detect cytokines using BD cytometric bead array mouse inflammatory kit (BD Biosciences, Franklin Lakes, NJ) as per manufacturer’s specifications. Data were collected on FACSCanto II (Becton and Dickson, Franklin Lakes, NJ) and analyzed on FCAP Array software (BD Bioscience).
Immune Cells Analysis.
Spleen and lymph nodes were collected from the mice and placed into phosphate-buffered saline containing 1% heat inactivated fetal bovine serum and 2 mM EDTA (FACS buffer). Single cell suspensions were created with gentleMACS tissue dissociator (Myltenyi Biotec, Bergisch Gladbach, Germany) using C tubes per manufacturer’s instructions. Red blood cells were lysed by short incubation with 156 mM ammonium chloride, and cells were washed twice with FACS buffer. Cells were counted on a Coulter counter and 2 × 106 cells were aliquoted for staining. Nonspecific staining was blocked with Mouse BD Fc Block (BD Pharmingen), and the cell suspensions were labeled with anti-CD19 (1D3), -CD4 (RM4-5), -CD8a (53-6.7), -Gr-1 (RB6-8C5), -CD11b (M1/70) (BD Pharmingen), and -F4/80 (A3-1; AbD Serotec, Kidlington, UK). The antibodies were removed by two rounds of centrifugation and resuspended in FACS buffer. Data were acquired on FACSCanto II, and data were analyzed with Weasel software (The Walter and Eliza Hall Institute of Medical Research, Melbourne, VIC, Australia).
Statistics.
Mean ± S.E.M. are shown. Grubbs test was performed to check for significant outliers. Statistical significance of difference between groups was assessed with one way analysis of variance. Post hoc analysis was performed with Tukey's test when the data followed normal distribution; Dunn’s test was used for other cases.
Results
Compounds and Animal Model.
In this study we tested a cell-impermeable analog of CsA, MM284. MM284 is a CsA derivative that includes a negatively charged moiety coupled to presynthesized CsA (Malesevic et al., 2013; Seizer et al., 2015). The presence of this charged moiety prohibits passage of the compound through the plasma membrane, making it cell impermeable and capable of interacting with and inhibiting only extracellular pools of cyclophilins (Damsker et al., 2009). This analog was compared with a cell-permeable analog of CsA, NIM811 (Rosenwirth et al., 1994; Seizer et al., 2015), which probably blocks activity of most mammalian cyclophilins (Arora et al., 2005). NIM811 was chosen as a control for MM284, because both analogs are nonimmunosuppressive. Use of NIM811 instead of parent compound, cyclosporine A, allowed eliminating a confounding effect of immunosupression on atherosclerosis, limiting the effects of inhibition of cyclophilins. Each compound was administered every second day by intraperitoneal injection at a dose of 6.6 mg/kg; we previously found that this dose inhibited allergic lung inflammation (Balsley et al., 2010). Control mice were injected with vehicle (15% ethanol in 15% cremophore EL).
The compounds were tested in a well established mouse model of atherosclerosis, apoE−/− mice fed with high-fat diet. Development of atherosclerosis was assessed in two vessels, aorta, and innominate artery. Aortic sinus is an early site of atherosclerosis development with rapidly developing complex lesions providing an insight into underlying mechanisms of plaque formation. The innominate artery develops atherosclerosis at a slower rate; previous studies found that it closely represents the development and morphology of the human lesion (Rosenfeld et al., 2000).
Development of Atherosclerosis.
En face analysis of atherosclerosis in aortic arch and thoracic aorta demonstrated that, if anything, CsA analogs enhanced the development of atherosclerosis. Both compounds increased abundance of atherosclerotic plaques (Fig. 1, A–C). The effect was statistically significant in thoracic aorta (Fig. 1C) but not in aortic arch (Fig. 1B). There was little difference between the effects of NIM811 and MM284.
Fig. 1.

Effect of cyclophilin inhibition on atherosclerosis. (A–C) En face analysis of total aorta (A), aortic arch (B), and thoracic aorta (C). Lesion burden was calculated as a percentage of Sudan IV–stained area from total vessel area. (D) Cross-sectional analysis of total lesion area in the aortic sinus stained with Oil red O. (E) Representative micrographs of the aortic sinus stained with Oil red O. (F) Cross-sectional analysis of total lesion area in the innominate artery stained with Oil red O. (G) Representative micrographs of Oil red O–stained sections of the innominate artery. All graphs are presented as mean ± S.E.M. *P < 0.05 versus vehicle.
Analysis of sections of aortic sinus produced similar results: the area of atherosclerotic plaque increased after treatment with either compounds, for MM284 this increase was statistically significant (Fig. 1, D and E). There was no effect of either compound on the cross-sectional size of atherosclerotic plaque in innominate artery (Fig. 1, F and G). Thus, both compounds accelerated the development of atherosclerosis in apoE−/− mouse model.
Plaque Composition.
There was no effect of either NIM811 or MM284 on macrophage infiltration of the lesions (CD68-positive staining) in both aortic sinus (Fig. 2, A and B) and innominate artery (Fig. 2, C and D). Abundance of VCAM-1 in the lesions in aortic sinus (Fig. 2, E and F) and innominate artery (Fig. 2, G and H) was also unaffected by both compounds.
Fig. 2.
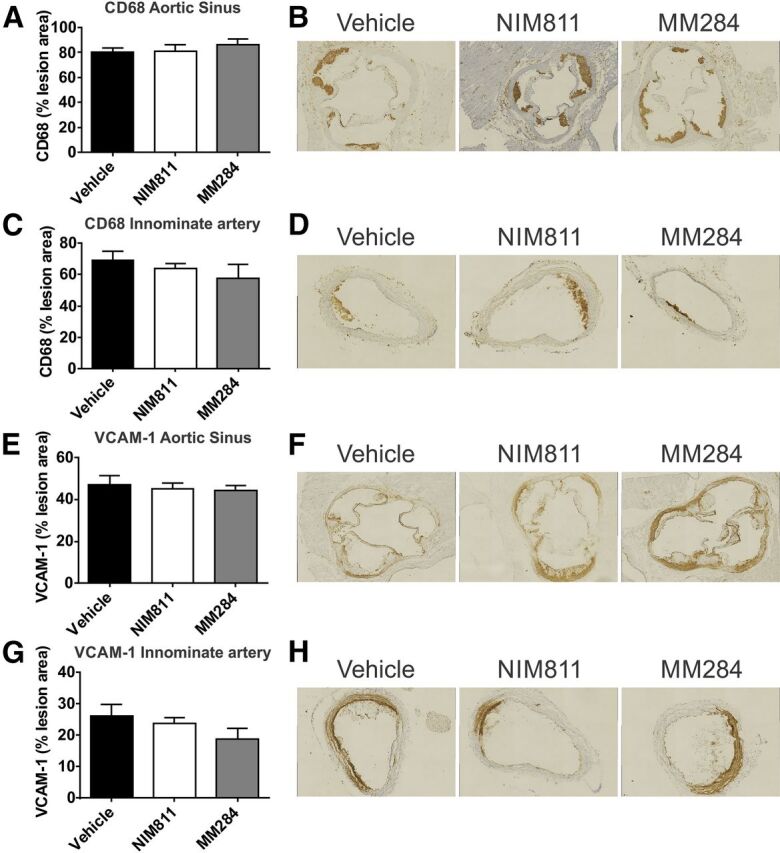
Cyclophilin inhibition and markers of inflammation in the atherosclerotic plaque. (A) Quantitation of CD68 staining for macrophages in the aortic sinus. Percentages of anti-CD68–stained area of total plaque area are shown. (B) Representative micrographs of the CD68 staining in the aortic sinus of mice treated with vehicle (left), NIM811 (center), and MM284 (right). (C) Quantitation of CD68 staining for macrophages in the innominate artery. (D) Representative micrographs of the innominate artery stained for CD68 of mice treated with vehicle (left), NIM811 (center), and MM284 (right). (E) Quantitation of VCAM-1 content of lesions in the aortic sinus. Percentages of anti-VCAM-1–stained area of total plaque area are shown. (F) Representative micrographs of VCAM-1 staining in the aortic sinus of mice treated with vehicle (left), NIM811 (center), and MM284 (right). (G) Quantitation of VCAM-1 content of lesions in the innominate artery. Percentages of anti-VCAM-1–stained area of total plaque area are shown. (H) Representative micrographs of the VCAM-1 staining in the innominate artery of mice treated with vehicle (left), NIM811 (center), and MM284 (right). All graphs are presented as mean ± S.E.M.
The abundance of collagen in the plaques was assessed using Masson's trichrome staining. There was no statistically significant effect of either compound on the abundance of collagen in the plaques in aortic sinus (Fig. 3, A and B). However, there was a statistically significant reduction of collagen abundance in innominate artery of animals treated with both compounds; there was no difference between the cell-permeable and -impermeable analogs (Fig. 3, C and D).
Fig. 3.
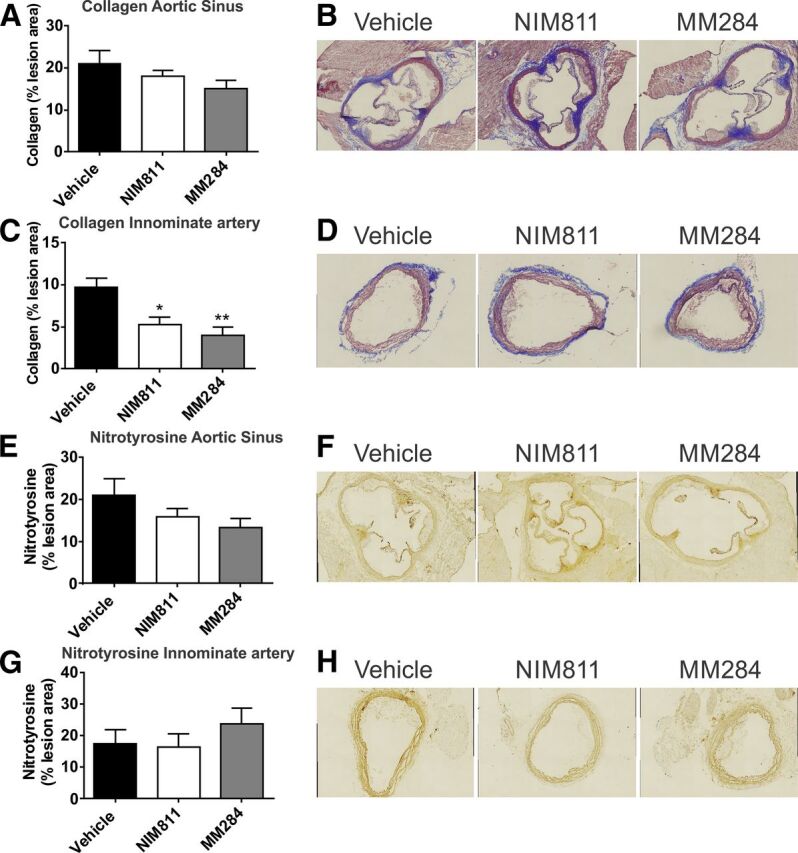
Effect of cyclophilin inhibition on markers of plaque stability and oxidation. (A) Quantitation of collagen content in of the aortic sinus after staining with Masson’s trichrome. Percentages of positive staining of total lesion area are shown. (B) Representative micrographs of staining of the aortic sinus with Masson's trichrome of mice treated with vehicle (left), NIM811 (center), and MM284 (right). (C) Quantitation of collagen content in the innominate artery. Percentages of positive staining of total lesion area are shown. (D) Representative micrographs of staining of the innominate artery with Masson's trichrome of mice treated with vehicle (left), NIM811 (center), and MM284 (right). (E) Quantitation of nitrotyrosine content in of the aortic sinus. Percentages of positive anti nitrotyrosine staining of total lesion area are shown. (F) Representative micrographs of staining of the aortic sinus for nitrotyrosine in mice treated with vehicle (left), NIM811 (center), and MM284 (right). (G) Quantitation of nitrotyrosine contents in the innominate artery. Percentages of positive anti nitrotyrosine staining of total lesion area are shown. (H) Representative micrographs of staining of the innominate artery for nitrotyrosine in mice treated with vehicle (left), NIM811 (center), and MM284 (right). All graphs are presented as mean ± S.E.M., *P < 0.05 versus vehicle; **P < 0.001 versus vehicle.
The abundance of nitrotyrosine, a marker of protein oxidation and nitric oxide availability, was not statistically significantly affected by either compound in both arteries (Fig. 3, E and F).
Thus, both cell-permeable and -impermeable analogs of CsA did not affect markers of inflammation and oxidation in the atherosclerotic plaque. Both analogs, however, similarly reduced the abundance of collagen in plaques of innominate artery, indicating possible stabilizing effect of cyclophilins on early atherosclerotic plaques.
Blood Markers of Inflammation and Plasma Lipoproteins.
When concentration of various cytokines in plasma was measured, MM284, but not NIM811, significantly reduced concentration of tumor necrosis factor-α (TNFα) (Fig. 4A). This effect is consistent with previously demonstrated induction of TNFα production in monocytes by extracellular cyclophilin (Yuan et al., 2010). However, neither compound affected plasma concentrations of other cytokines: levels of interleukin (IL)-6 (Fig. 4B), IL-10 (Fig. 4C), or monocyte chemoattractant protein-1 (Fig. 4D) remained unaffected by treatment. We then used flow cytometry to assess ratio between different populations of immune cells (CD8+ T cells, CD4+ T cells, B cells, GR1 lo monocytes, GR1 hi monocytes, neutrophils, and macrophages). No effect of either compound was found (Supplemental Table 1). We also analyzed the ratio between different populations of immune cells isolated from lymph nodes and spleen. Again, no effect of either compound was found (Supplemental Table 1).
Fig. 4.

Effect of cyclophilin inhibition on plasma cytokine levels. (A) TNFα; (B) IL-6; (C) IL-10; and (D) monocyte chemoattractant protein-1 (MCP-1). Mean ± S.E.M. are shown, *P < 0.05 versus vehicle.
Neither compound affected plasma concentrations of total cholesterol (Fig. 5A), low-density lipoproteins cholesterol (Fig. 5B), triglyceride (Fig. 5C), HDL cholesterol (Fig. 5D), nor apolipoprotein A-I (Fig. 5E).
Fig. 5.

Effect of cyclophilin inhibition on plasma lipid and lipoprotein levels. (A) Total cholesterol; (B) low-density lipoprotein (LDL)-C; (C) tryglycerides; (D) HDL-C; (E) apolipoprotein A-I (ApoAI). Mean ± S.E.M. are shown.
Discussion
In this study we investigated the role of extracellular cyclophilins in atherosclerosis. Our findings suggest that extracellular cyclophilins may have antiatherogenic activity. Depending on location of the atherosclerotic plaque, treatment with both permeable (NIM811) and impermeable (MM284) cyclosporine either did not affect or increased development of atherosclerosis and there was no difference in the effects of the two compounds. This finding is consistent with a number of previous studies using pan-inhibition of cyclophilins (for review, see Kockx et al., 2010; Bukrinsky et al., 2013). Previously, protection from atherosclerosis of apoE−/− mice with knocked out cyclophilin A gene was demonstrated (Nigro et al., 2011). Knockout of cypA gene eliminates both intracellular and extracellular CypA and should have been mimicked by NIM811. Instead, we observed the proatherogenic effect of NIM811, similar to that of MM284, which targets only extracellular cyclophilins. This finding suggests that the proatherogenic activity is exerted specifically by the intracellular CypA, whereas other intracellular cyclophilins, which are also inhibited by NIM811, may be essential for protection from atherosclerosis. Given that NIM811 targets both extracellular and intracellular cyclophilins, the fact that the effects of this compound on atherosclerosis were similar to the effects of MM284 suggests that inhibition of extracellular cyclophilins was responsible for proatherogenic effect. Consequently, separation of antiatherogenic from proathrogenic effects of cyclophilin inhibition could not be achieved by selective inhibition of extracellular cyclophilins.
The proatherogenic activity of MM284 seems to be inconsistent with limited anti-inflammatory activity of the compound evidenced by decreased levels of TNFα. However, the systemic level of TNFα in untreated animals was low, suggesting that systemic inflammation does not play a major role in pathogenesis of atherosclerosis in this model. The mechanisms behind the antiatherogenic activity of extracellular cyclophilins remain to be better characterized, but they are consistent with previously reported ability of extracellular CypA to attenuate oxidative stress and prevent apoptosis, thus protecting vascular smooth muscle cells (Jin et al., 2000). We did not observe any effect of MM284 or NIM811 on plaque composition, cellularity of the plaque, or markers of inflammation and oxidation nor did we find changes in plasma lipoproteins in drug-treated animals. Locally, consistent with previous findings (Kohjima et al., 2007; Seizer et al., 2010), both cyclosporines reduced stability of the plaques in innominate artery.
The doses and delivery mode of the CsA analogs were similar to our previous study (Balsley et al., 2010) where profound effects of the compounds were observed and slightly lower compared with those shown to have acute antithrombogenic effects (Seizer et al., 2015). This and several effects observed in this study make it unlikely that limited availability of the compounds was an issue. It is important to recognize, however, that both analogs of CsA tested in this study were nonimmunosuppressive (Billich et al., 1995); this property may have limited some anti-inflammatory effects of the compounds compared with CsA.
Abbreviations
- apoE
apolipoprotein E
- CsA
cyclosporine A
- CypA
cyclophilin A
- HDL
high-density lipoprotein
- IL
interleukin
- MM284
[(4R)-4-[(6-carboxy-1H-benzo[d]imidazol-2-yl)-methyl]-4-methyl-l-threonine]1-CsA
- NIM811
[Melle]4-CsA
- TNF
tumor necrosis factor
- VCAM
vascular cell adhesion molecule
Authorship Contributions
Participation in research design: Ditiatkovski, Bukrinsky, Sviridov.
Conducted experiments: Ditiatkovski, Neelisetti, Cui.
Contributed new reagents or analytic tools: Malesevic; Fischer.
Wrote or contributed to writing of the manuscript: Ditiatkovski, Bukrinsky, Sviridov.
Footnotes
This work was supported by National Health and Medical Research Council of Australia (NHMRC) [Grant GNT1036352]; and in part by the Victorian Government’s Operational Infrastructure Support Program. D.S. is a Fellow of the National Health and Medical Research Council of Australia [Grant GNT586607]. Photomicrographs were collected with equipment provided by Monash Micro Imaging (Monash University).
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Arora K, Gwinn WM, Bower MA, Watson A, Okwumabua I, MacDonald HR, Bukrinsky MI, Constant SL. (2005) Extracellular cyclophilins contribute to the regulation of inflammatory responses. J Immunol 175:517–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsley MA, Malesevic M, Stemmy EJ, Gigley J, Jurjus RA, Herzog D, Bukrinsky MI, Fischer G, Constant SL. (2010) A cell-impermeable cyclosporine A derivative reduces pathology in a mouse model of allergic lung inflammation. J Immunol 185:7663–7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billich A, Hammerschmid F, Peichl P, Wenger R, Zenke G, Quesniaux V, Rosenwirth B. (1995) Mode of action of SDZ NIM 811, a nonimmunosuppressive cyclosporin A analog with activity against human immunodeficiency virus (HIV) type 1: interference with HIV protein-cyclophilin A interactions. J Virol 69:2451–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukrinsky M. (2015) Extracellular cyclophilins in health and disease. Biochim Biophys Acta DOI: 10.1016/j.bbagen.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukrinsky M, Orekhov A, Ditiatkovski M, Sviridov D. (2013) Cyclophilins in atherosclerosis: a new therapeutic target? Curr Pharm Des 19:5904–5908. [DOI] [PubMed] [Google Scholar]
- Bukrinsky MI. (2002) Cyclophilins: unexpected messengers in intercellular communications. Trends Immunol 23:323–325. [DOI] [PubMed] [Google Scholar]
- Damsker JM, Okwumabua I, Pushkarsky T, Arora K, Bukrinsky MI, Constant SL. (2009) Targeting the chemotactic function of CD147 reduces collagen-induced arthritis. Immunology 126:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditiatkovski M, D’Souza W, Kesani R, Chin-Dusting J, de Haan JB, Remaley A, Sviridov D. (2013) An apolipoprotein A-I mimetic peptide designed with a reductionist approach stimulates reverse cholesterol transport and reduces atherosclerosis in mice. PLoS ONE 8:e68802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew AF, Tipping PG. (1995) Cyclosporine treatment reduces early atherosclerosis in the cholesterol-fed rabbit. Atherosclerosis 116:181–189. [DOI] [PubMed] [Google Scholar]
- Jin ZG, Lungu AO, Xie L, Wang M, Wong C, Berk BC. (2004) Cyclophilin A is a proinflammatory cytokine that activates endothelial cells. Arterioscler Thromb Vasc Biol 24:1186–1191. [DOI] [PubMed] [Google Scholar]
- Jin ZG, Melaragno MG, Liao DF, Yan C, Haendeler J, Suh YA, Lambeth JD, Berk BC. (2000) Cyclophilin A is a secreted growth factor induced by oxidative stress. Circ Res 87:789–796. [DOI] [PubMed] [Google Scholar]
- Kockx M, Jessup W, Kritharides L. (2010) Cyclosporin A and atherosclerosis—cellular pathways in atherogenesis. Pharmacol Ther 128:106–118. [DOI] [PubMed] [Google Scholar]
- Kohjima M, Enjoji M, Higuchi N, Kotoh K, Kato M, Takayanagi R, Nakamuta M. (2007) NIM811, a nonimmunosuppressive cyclosporine analogue, suppresses collagen production and enhances collagenase activity in hepatic stellate cells. Liver Int 27:1273–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Goff W, Peng D-Q, Settle M, Brubaker G, Morton RE, Smith JD. (2004) Cyclosporin A traps ABCA1 at the plasma membrane and inhibits ABCA1-mediated lipid efflux to apolipoprotein A-I. Arterioscler Thromb Vasc Biol 24:2155–2161. [DOI] [PubMed] [Google Scholar]
- Malesevic M, Gutknecht D, Prell E, Klein C, Schumann M, Nowak RA, Simon JC, Schiene-Fischer C, Saalbach A. (2013) Anti-inflammatory effects of extracellular cyclosporins are exclusively mediated by CD147. J Med Chem 56:7302–7311. [DOI] [PubMed] [Google Scholar]
- Moghadasian MH. (2006) Dietary phytosterols reduce cyclosporine-induced hypercholesterolemia in apolipoprotein E-knockout mice. Transplantation 81:207–213. [DOI] [PubMed] [Google Scholar]
- Nigro P, Satoh K, O’Dell MR, Soe NN, Cui Z, Mohan A, Abe J, Alexis JD, Sparks JD, Berk BC. (2011) Cyclophilin A is an inflammatory mediator that promotes atherosclerosis in apolipoprotein E-deficient mice. J Exp Med 208:53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preibisch S, Saalfeld S, Tomancak P. (2009) Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 25:1463–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roselaar SE, Schonfeld G, Daugherty A. (1995) Enhanced development of atherosclerosis in cholesterol-fed rabbits by suppression of cell-mediated immunity. J Clin Invest 96:1389–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld ME, Polinsky P, Virmani R, Kauser K, Rubanyi G, Schwartz SM. (2000) Advanced atherosclerotic lesions in the innominate artery of the ApoE knockout mouse. Arterioscler Thromb Vasc Biol 20:2587–2592. [DOI] [PubMed] [Google Scholar]
- Rosenwirth B, Billich A, Datema R, Donatsch P, Hammerschmid F, Harrison R, Hiestand P, Jaksche H, Mayer P, Peichl P, et al. (1994) Inhibition of human immunodeficiency virus type 1 replication by SDZ NIM 811, a nonimmunosuppressive cyclosporine analog. Antimicrob Agents Chemother 38:1763–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seizer P, Schönberger T, Schött M, Lang MR, Langer HF, Bigalke B, Krämer BF, Borst O, Daub K, Heidenreich O, et al. (2010) EMMPRIN and its ligand cyclophilin A regulate MT1-MMP, MMP-9 and M-CSF during foam cell formation. Atherosclerosis 209:51–57. [DOI] [PubMed] [Google Scholar]
- Seizer P, Ungern-Sternberg SN, Schönberger T, Borst O, Münzer P, Schmidt EM, Mack AF, Heinzmann D, Chatterjee M, Langer H, et al. (2015) Extracellular cyclophilin A activates platelets via EMMPRIN (CD147) and PI3K/Akt signaling, which promotes platelet adhesion and thrombus formation in vitro and in vivo. Arterioscler Thromb Vasc Biol 35:655–663. [DOI] [PubMed] [Google Scholar]
- Warnick GR, Benderson J, Albers JJ. (1982) Dextran sulfate-Mg2+ precipitation procedure for quantitation of high-density-lipoprotein cholesterol. Clin Chem 28:1379–1388. [PubMed] [Google Scholar]
- Yuan W, Ge H, He B. (2010) Pro-inflammatory activities induced by CyPA-EMMPRIN interaction in monocytes. Atherosclerosis 213:415–421. [DOI] [PubMed] [Google Scholar]