

Abstract
Angiotensin II (AngII) initiates cellular effects via its G protein-coupled angiotensin 1 (AT1) receptor (AT1R). Previously, we showed that AngII-induced expression of the prostanoid-producing enzyme cyclooxygenase 2 (COX-2) was dependent upon nuclear trafficking of activated AT1R. In the present study, mastoparan (an activator of G proteins), suramin (an inhibitor of G proteins), 1-[6-[[17β-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione (U73122; a specific inhibitor of phospholipase C), and sarcosine1-Ile4-Ile8-AngII (SII-AngII; a G protein-independent AT1R agonist) were used to determine the involvement of G proteins and AT1AR trafficking in AngII-stimulated COX-2 protein expression in human embryonic kidney-293 cells stably expressing AT1A/green fluorescent protein receptors and cultured vascular smooth muscle cells, respectively. Mastoparan alone stimulated release of intracellular calcium and increased COX-2 expression. Preincubation with mastoparan inhibited AngII-induced calcium signaling without altering AngII-induced AT1AR trafficking, p42/44 extracellular signal-regulated kinase (ERK) activation, or COX-2 expression. Suramin or U73122 had no significant effect on their own; they did not inhibit AngII-induced AT1AR trafficking, p42/44 ERK activation, or COX-2 expression; but they did inhibit AngII-induced calcium responses. SII-AngII stimulated AT1AR trafficking and increased COX-2 protein expression without activating intracellular calcium release. These data suggest that G protein activation results in increased COX-2 protein expression, but AngII-induced COX-2 expression seems to occur independently of G protein activation.
Angiotensin II (AngII), the vasoactive hormone of the renin-angiotensin system, initiates its cellular effects through activation of its cognate seven transmembrane-spanning G protein-coupled receptor, angiotensin type 1A (AT1A) receptor (AT1AR). Interaction of AngII with the AT1AR initiates conformational changes in the receptor, producing activation of its targeted G protein, Gq/11. Subsequent to receptor-mediated activation of Gq/11, a cascade of intracellular signaling events occurs, including acute activation of phospholipase C (PLC), release of intracellular stores of calcium, and subsequent activation of numerous kinases including p42/44 ERK (Millan et al., 1991; de Gasparo et al., 2000) and chronic cellular effects including induced protein synthesis of renin, angiotensinogen, and cyclooxygenase (COX)-2 (Eggena et al., 1993; Ohnaka et al., 2000; Morinelli et al., 2008).
Subsequent to G protein activation, G protein-coupled receptors (GPCRs) undergo β-arrestin-mediated internalization. In addition to serving as a means to interrupt cell signaling from the cell surface, receptor internalization also serves to continue the signaling cascade within the cell. This process, in which internalized endosomes containing desensitized receptors along with attached β-arrestins act as a scaffold, interacting with specific signaling proteins such as p42/44 ERK, has been described as a signalsome (Luttrell et al., 2001). These “signalsomes' are responsible for the prolonged activation of p42/44 ERK by GPCR agonists such as AngII.
In addition to the above-mentioned role of receptor trafficking in desensitization/recycling and cytoplasmic signaling, an additional pathway for GPCR trafficking has been proposed, namely, localization of the activated receptor to the nuclear area (Re et al., 1983; Eggena et al., 1993). Lu et al. (1998) demonstrated nuclear translocation of the AT1AR in response to cellular activation by AngII. We have shown previously that nuclear localization of the receptor may be dependent upon a putative nuclear localization sequence within the carboxyl tail and that localization to the nuclear area from the plasma membrane involves clathrin-coated pits and is associated with the ability of AngII to induce COX-2 protein expression (Morinelli et al., 2007, 2008).
The generation of prostanoids, via the activation of COX-1 and/or COX-2, is responsible for a plethora of physiological and pathological responses. The activity of constitutive COX-1 results in the generation of prostanoids used to maintain physiologic homeostasis. In rat aorta vascular smooth muscle cells (RASMC), AngII induces the transcription for COX-2 via involvement of nuclear factor-κB and mediation of several cytoplasmic kinases including Pyk2, MEKK4, and p38 (Ohnaka et al., 2000; Hu et al., 2002; Derbyshire et al., 2005). Thus, AngII has been implicated in the regulation of COX-2 and the activation of several chronic disease processes mediated by COX-2.
The present study was designed to elucidate further the pathway for nuclear localization of the AT1AR by testing the hypothesis that AngII-induced COX-2 expression in RASMC is not dependent upon the mediation of G proteins, in particular Gq. To test this hypothesis, we used mastoparan (an activator of G proteins), suramin (an inhibitor of G protein activation), U73122 (a specific inhibitor of PLC activation), and sarcosine1-Ile4-Ile8-AngII (SII-AngII), an AT1R agonist whose activation of cellular responses has been shown to be independent of G proteins (Wei et al., 2003).
Materials and Methods
Cell Culture. Primary culture of RASMC was performed as described previously (Morinelli et al., 2008). Cells were maintained in Dulbecco's modified Eagle's medium-high glucose supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic/antimycotic/amphotericin B (Fungizone; Invitrogen) and used between passages 3 and 8. HEK-293 cells (American Type Culture Collection, Manassas, VA) stably expressing a wild-type AT1AR/green fluorescent protein (AT1AR/GFP) construct were maintained using Ham's F-12 media supplemented with 10% FBS, 1% antibiotic/antimycotic/amphotericin B, and Geneticin (G418; Invitrogen, Carlsbad, CA) (400 μg/ml) (Morinelli et al., 2007). Cell culture media and supplements were obtained from Invitrogen.
Radioligand Binding Assays. Binding studies using 125I-AngII were performed as described previously (Morinelli et al., 2008). Confluent monolayers of RASMC in six-well plates were exposed to the various compounds for 30 min at 37°C. Subsequently, the growth medium containing the compounds was removed, and binding buffer containing 125I-AngII (∼200,000 cpm; ∼100 fmol) with or without the AT1R antagonist losartan (10 μM) was added to the cells, and incubation was carried out at 4°C for 90 min. Subsequently, cells were washed with ice-cold saline buffer to remove unbound radioligand and then solubilized in 0.1% SDS/0.1 M NaOH, and associated radioactivity was counted. Specific binding was determined, i.e., the difference between radioactivity associated with the cell lysates in the absence and presence of losartan, and corrected for total cell protein per well.
Laser Scanning Confocal Imaging. HEK-293 cells stably expressing the wild-type AT1AR/GFP construct were plated onto collagen-coated 25-mm glass coverslips in six-well plates and maintained in selection medium. Before study, cells were deprived of serum (0.1% FBS) for 24 to 48 h. On the day of study, compounds were added directly to the media and incubated for 30 min followed by addition of angiotensin II (100 nM) for 60 min and fixed with 4% paraformaldehyde solution in PBS for 15 min at room temperature. Cells were washed two times with PBS followed by addition of the DNA fluorescent dye DRAQ5 (2 μM; Alexis Laboratories, San Diego, CA). Confocal microscopy was performed using an LSM 510 META laser scanning microscope (Carl Zeiss, Thornwood, NY) equipped with a 60× objective, using the following laser wavelengths: GFP, excitation 488 nm and emission 505–530 nm; and DRAQ5, excitation 543 nm and emission 560–615 nm.
Calcium Measurements. RASMC were plated into 96-well clear-bottomed black plates at a density of 60,000 cells/well. The next day, media were changed to 0.1% bovine serum albumin/Dulbecco's modified Eagle's medium. Twenty-four to 48 h later, media were removed, and cells were incubated with the calcium-sensitive fluorescent probe Fluo-3 acetoxymethyl ester (2 μM; Invitrogen) in Hanks' balanced salt solution (HBSS), pH 7.4, containing 2.5 mM probenecid and 0.1% bovine serum albumin for 60 min at 37°C. At the end of the incubation, the cells were washed three times with HBSS and placed into a fluorometric imaging plate reader (Molecular Devices, Sunnyvale, CA) and exposed to the various compounds for 30 min, followed by AngII. Increases in intracellular free calcium (Cai) were reflected by increases in detected fluorescence.
Immunoblotting. Confluent monolayers of RASMC were serum-deprived (0.1% FBS) for 24 to 48 h. Cells, in cell culture media (COX-2 expression studies) or HBSS and 20 mM HEPES, pH 7.4 (p42/44 ERK activation assays) were exposed to vehicle, mastoparan (10 μM; BIOMOL Research Laboratories, Plymouth Meeting, PA), suramin (10 μM; Tocris Bioscience, Ellisville, MO), or U73122 (10 μM; Calbiochem, San Diego, CA) for 30 min at 37°C, followed by addition of AngII (Sigma-Aldrich, St. Louis, MO). At the end of the incubation period with AngII, the cells were washed two times with ice-cold PBS followed by addition of radioimmunoprecipitation assay buffer [50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, and 0.25% sodium deoxycholate plus protease inhibitor cocktail I; 500 mM 4-(2-aminoethyl)benzenesulfonyl fluoride, 150 nM aprotinin, 1 mM E-64, and 1 mM leupeptin; Calbiochem] for p42/44 ERK assays. For COX-2 assays, cells were lysed directly with 1× SDS-polyacrylamide gel electrophoresis buffer. Lysates (10–20 μg) were separated by SDS-polyacrylamide gel electrophoresis (4–20% gradient), transferred to nitrocellulose, and probed for the presence of phosphorylated p42/44 ERK (phospho-p42/44) and total p42/44 ERK (1:2000; Cell Signaling Technology Inc., Danvers, MA) according to the manufacturer's directions. Blots were stripped of antibodies between probing for phosphorylated and total proteins. In COX-2 expression studies, the COX-2 protein was detected using a rabbit polyclonal antibody (1:200; Millipore, Billerica, MA). Protein loading and transfer were corrected for by detection of β-actin (1:5000; Sigma-Aldrich). Detection of protein bands was performed by addition of CDP-Star reagent (New England Biolabs, Ipswich, MA) and visualized by exposure of the nitrocellulose to radiographic film (X-OMAT; Eastman Kodak, Rochester, NY). Quantitation of the visualized protein bands was performed by densitometric scanning of the exposed radiographic film (Kodak Molecular Imaging Software, Rochester, NY). The density ratio for phosphorylated protein to total protein was used as an indicator of kinase activation.
Statistics. Values shown are means ± S.E. from the indicated number of studies (n), and comparisons were made using Excel (Microsoft, Redmond, WA) data analysis package using analysis of variance with Fisher's post hoc t test or Kruskal-Wallis with Mann-Whitney test where indicated. Significance was tested at the 95% level.
Results
Activation of heterotrimeric G proteins, in particular Gq/11 and G12/13, by the AT1AR mediates cellular responses to AngII. Small molecular compounds such as suramin or naturally occurring peptides such as the wasp venom peptide mastoparan have been used to probe the involvement of G protein activation in cell signaling events. Suramin has been demonstrated to have multiple effects on G proteins, including the interruption of G protein/receptor interaction by blocking GDP to GTP exchange, and to promote epithelial cell proliferation. Mastoparan, conversely, has been shown to activate G protein signaling by promoting GDP to GTP exchange preferentially with Gi and Go, resulting in numerous cellular effects including enhanced GTPase activity, increased activation of phospholipase D activity, and AT1AR desensitization. AT1AR-induced activation of PLC with the resultant release of inositol 1,4,5-trisphosphate from membrane phospholipids results in release of calcium from intracellular storage sites. This activation of PLC is mediated by AT1AR activation of Gq. A pharmacological agent commonly used to demonstrate a role of Gq activation of PLC is the compound U73122, which inhibits PLC-dependent processes.
We first examined the ability of these compounds to alter the interaction of AngII with its cell surface receptor. Pretreatment of monolayers of RASMC with mastoparan, suramin, or U73122 had no significant effect on AngII binding to cell surface AT1ARs as detected by specific binding of 125I-AngII (Fig. 1).
To determine whether these compounds alter basal plasma membrane expression of AT1AR- or AngII-induced internalization and intracellular trafficking, AT1AR, AT1AR/GFP/HEK cells were examined by confocal microscopy after treatment with mastoparan, suramin, or U73122, followed by exposure to AngII (Fig. 2). This cell line has been characterized previously and is a useful cell model to visualize AT1AR trafficking (Morinelli et al., 2007). Addition of AngII to untreated cells produced characteristic localization of the receptor to the nuclear membrane area that has been observed previously (Morinelli et al., 2007). Exposure of cells to mastoparan, suramin, or U73122 alone did not alter basal distribution of the receptor, i.e., AT1AR/GFP-expressing cells maintained a plasma membrane distribution of the receptor similar to untreated cells. Subsequent addition of AngII to these cells produced the characteristic AT1AR internalization and nuclear membrane localization.
Fig. 1.

Effect of G protein-interacting compounds on AngII binding. RASMC were treated with vehicle (untreated), mastoparan (10 μM), suramin (10 μM), or U73122 (10 μM) for 30 min at 37°C. Media were removed, and 125I-angiotensin II radioligand binding was determined as described under Materials and Methods. Specific radioactivity associated with the cells was determined and corrected for total cell protein. Values shown are the average (± S.E.M.) from three studies.
Fig. 2.

Effect of G protein-interacting compounds on AngII-induced AT1AR intracellular trafficking. Representative laser scanning confocal microscope images from HEK-293 cells stably expressing AT1AR/GFP exposed to vehicle (untreated), mastoparan (10 μM), suramin (10 μM), or U73122 (10 μM) at 37°C for 30 min followed by stimulation with AngII (100 nM; 60′). Cells were fixed and prepared for imaging as described under Materials and Methods.AT1AR/GFP is seen as green, whereas nuclei are visualized with the DNA-specific dye DRAQ5 and seen as red. Corresponding differential interference contrast images also are shown.
One of the earliest events in the signaling cascade initiated by the AT1ARisGq-mediated activation of PLC with resultant increase in Cai. RASMC were treated with mastoparan, suramin, or U73122 before exposure to AngII (Fig. 3). Pretreatment with mastoparan caused an elevation in Cai, suggesting that mastoparan directly activated Gq and subsequently PLC, producing a release of calcium from intracellular stores. Addition of suramin or U73122 had no effect on Cai, as expected. Mastoparan treatment, subsequent to producing a direct effect on calcium, blocked the ability of AngII to increase Cai. Pretreatment with suramin also inhibited the ability of AngII to elevate Cai. U73221, an inhibitor of PLC, as expected, also blocked AngII-induced elevations of Cai. These results confirm the role of G proteins in AngII-induced elevations of Cai.
G protein-mediated activation of the mitogen-activated protein kinase pathway is a central element to the hypertrophy/hyperplasia response of many GPCRs. One of the key intermediary kinases in this pathway is the serine/threonine kinase p42/44 ERK. RASMC, when exposed to AngII, produce a rapid and reversible phosphorylation of p42/44 ERK. We examined the ability of mastoparan, suramin, and U73122 to influence the ability of AngII to activate this kinase. Mastoparan, an activator of heterotrimeric G proteins, activated p42/44 ERK (Fig. 4). This effect seemed to be additive to the stimulation produced by AngII. Suramin, a reported inhibitor of heterotrimeric G proteins, had no direct effects on p42/44 ERK and did not block the stimulation produced by AngII. U73122, an inhibitor of PLC, seemed to produce a small but not significant stimulatory effect on p42/44 ERK without effecting AngII-induced activation. These results support the concept that AngII-induced activation of mitogen-activated protein kinase signaling can occur by both G protein-dependent and -independent pathways.
Fig. 3.

Effect of G protein-interacting compounds on AngII-induced calcium elevations. A, RASMC were exposed to vehicle (untreated), mastoparan (10 μM), suramin (10 μM), or U73122 (10 μM), indicated by arrow at 0 min, and then monitored for changes in fluorescence as a measure of elevations in intracellular free calcium as described under Materials and Methods. Subsequently, these same cells were exposed to AngII (100 nM), indicated by arrow at approximately 22 min, and then monitored for changes in fluorescence. (RFU, relative fluorescence units). B, summary of maximum changes in intracellular fluorescence (Cai) in response to AngII after pre-exposure to the indicated compounds (average values ± S.E.M.; n = 6). *, p < 0.05 versus untreated (analysis of variance with Fisher's post hoc test).
Having seen the varied effects of G protein-interacting compounds on AngII-activated intracellular signaling, we next examined their effects on AngII-induced COX-2 expression. RASMC were pretreated with vehicle, mastoparan, suramin, or U73122 for 30 min, followed by addition of buffer or AngII for an additional 3 h, and expression of COX-2 was determined (Fig. 5). As seen previously, AngII increased COX-2 protein expression in a concentration-dependent manner, reaching approximately a 3-fold increase at 100 nM. Mastoparan, an activator of G proteins that produces elevations of Cai and activation of p42/44 ERK, also produced significant increases in the expression of COX-2. When AngII was added to mastoparan-pretreated RASMC, COX-2 expression was further increased. The G protein inhibitor suramin, which does not alter AT1AR trafficking or p42/44 ERK activation but inhibits AngII-induced Cai, had no effect on COX-2 expression on its own and suppressed slightly but not significantly the ability of AngII to increase COX-2 expression. U73122, an inhibitor of PLC, which does not alter AT1AR trafficking but inhibits elevations in AngII-induced Cai, did not inhibit AngII-induced COX-2 protein expression. The effects seen with mastoparan, suramin, and U73122 suggest that AngII-induced COX-2 expression depends partially on G protein activation and partially on AT1AR intracellular (nuclear) trafficking.
Fig. 4.

Effect of G protein-interacting compounds on AngII-induced activation of p42/44 ERK. A, representative immunoblots from lysates of RASMC exposed to vehicle (untreated; untr), mastoparan (MAST; 10 μM), suramin (SUR; 10 μM), or U73122 (U73; 10 μM) at 37°C for 30 min followed by stimulation with AngII (100 nM; 5′). Detection of activated p42/44 ERK was performed as described under Materials and Methods. Blot was stripped and reprobed with antibody for total p42/44 ERK as described under Materials and Methods. B, summary of densitometric scanning of immunoblots for detection of phospho-p42/44 ERK in response to stimulation by AngII after pre-exposure to the indicated compounds (average values ± S.E.M.; n = 3–5). *, p < 0.05 versus nonstimulated (-); +, p < 0.05 versus nonstimulated, untreated (-).
Because these data suggest that AngII activation of AT1ARs and subsequent COX-2 protein expression may not require the mediation of G proteins, we examined the ability of the G protein-independent AT1AR agonist SII-AngII to activate the receptor and induce expression of COX-2. SII-AngII is a ligand of the AT1AR that can induce receptor internalization and p42/44 ERK activation independently of G protein activation (Wei et al., 2003). Cells exposed to SII-AngII produced endocytosis of the AT1AR/GFP construct, which was inhibited by the AT1R antagonist losartan, and increased expression of the COX-2 protein without eliciting elevations in intracellular free calcium (Fig. 6).
Discussion
Our previous studies suggest that AngII-mediated increases in COX-2 protein expression depend on nuclear membrane localization of activated AT1AR subsequent to internalization through clathrin-coated pits (Morinelli et al., 2007, 2008). The present study investigated the role of heterotrimeric G proteins in this process, with data summarized in Table 1. Mastoparan, an activator of heterotrimeric G proteins, increased Cai, blocked AngII-stimulated increases in Cai, activated p42/44 ERK, increased the expression of COX-2, and enhanced AngII-induced expression of COX-2, without altering nuclear membrane trafficking of AT1ARs. Suramin, an inhibitor of heterotrimeric G proteins, inhibited AngII-induced Cai but had no effect on surface expression or nuclear membrane localization of AT1ARs and had no significant effect on AngII-induced COX-2 expression. U73122, an inhibitor of Gq-dependent PLC activation, inhibited AngII-induced Cai and activation of p42/44 ERK but had no inhibitory effects on AT1AR surface expression, nuclear membrane receptor localization, or AngII-induced COX-2 expression. SII-AngII, a heterotrimeric G protein-independent activator of AT1AR-signal transduction pathways, promoted nuclear membrane trafficking of AT1ARs and increased the expression of COX-2 protein without elevating intracellular free calcium, a Gq-dependent event.
TABLE 1.
Summary of effects of G protein-interacting compounds on AngII receptor binding and cell signaling
|
Agent |
G Protein |
Binding |
Trafficking |
Cai |
p42/44 ERK |
COX-2 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (–) |
(+) |
(–) |
(+) |
(–) |
(+) |
(–) |
(+) |
|||||||
| Mastoparan | Stimulate | No effect | No effect | Nuclear | Increase | Inhibit | Stimulate | No effect | Stimulate | Enhance | ||||
| Suramin | Inhibit | No effect | No effect | Nuclear | No effect | Inhibit | No effect | No effect | No effect | No effect | ||||
| U73122 | N.D. | No effect | No effect | Nuclear | No effect | Inhibit | No effect | Inhibit | No effect | No effect | ||||
| SII-AngII |
No effect |
Bind |
Nuclear |
N.D. |
No effect |
N.D. |
N.D. |
N.D. |
Stimulate |
N.D. |
||||
N.D., not determined. (–) and (+), in the absence and presence of AngII, respectively
Fig. 5.

Effect of G protein-interacting compounds on AngII-induced expression of COX-2. RASMC were pre-exposed to vehicle (control), mastoparan (10 μM), suramin (10 μM), or U73122 (10 μM). Thirty minutes later, cells were exposed to vehicle (0 M) or indicated concentrations of AngII for 3 h followed by cell lysis and immunoblotting to detect for the expression of COX-2. A, representative immunoblot showing increased expression of COX-2 after AngII treatment and effects of various compounds on this increased expression. Detection of β-actin used to correct for protein loading. B, summary of densitometric scanning of immunoblots showing average values ± S.E.M., n = 3to9. *, p < 0.05 versus unstimulated (0 M) control cells; +, p < 0.05 versus unstimulated cells (0 M).
The ability of cells to respond to AngII relies on the interaction of AngII with its cell surface G protein-coupled receptor. The major class of AngII receptors is the AT1 receptor, with the AT1A receptor being the subtype found in vascular smooth muscle cells. Activation of this receptor by AngII produces well characterized cellular effects related to its physiological/pathological activities, including cell contraction, protein synthesis, and cell proliferation. The G proteins Gq/11 mediate the majority of these cellular effects. Additional evidence suggests that AT1R under select conditions may also activate other G proteins such as G12/13 and possibly Gi/o (de Gasparo et al., 2000). Coincident with the activation of the above-mentioned signaling cascade, the activated AT1AR, as with other GPCRs, initiates a signal termination sequence producing activation of specific G protein receptor kinases, which produce phosphorylation of the receptor resulting in β-arrestin-mediated receptor desensitization, halting further activation of G proteins and directing the receptor into clathrin-coated pits along the surface of the membrane. The internalized pits are targeted to acidic endosomes (lysosomes), where the receptor complex is either dissociated as a result of dephosphorylation of the receptor produced by the acidic environment and rapidly recycled back to the plasma membrane (class A GPCRs) or, for class B GPCRs, held in the endosomes, with β-arrestin attached (Oakley et al., 2001; Luttrell and Lefkowitz, 2002).
Fig. 6.
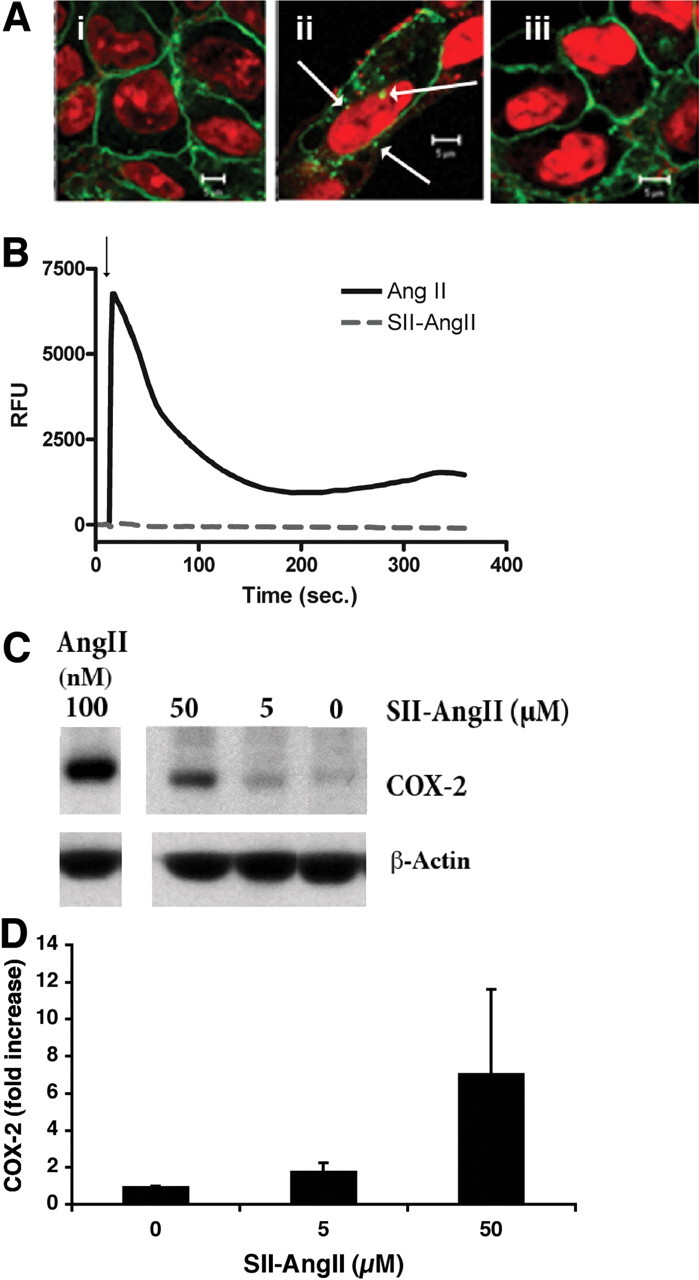
Effects of SII-AngII on AT1AR signaling and COX-2 protein expression. A, AT1AR/GFP internalization. Laser scanning confocal microscope imaging of HEK-293 cells stably expressing AT1AR/GFP were exposed to vehicle (i), SII-AngII (5 μM; ii), or losartan (10 μM) and then SII-AngII (iii) for 60′ at 37°C. Cells were fixed with formaldehyde as described under Materials and Methods and prepared for imaging. The specific DNA dye DRAQ5 was used to visualize nuclei. Arrows indicate nuclear localized receptors. Scale bar, 5 μm. Representative images from two similar studies. B, intracellular calcium response. AT1A R/GFP/HEK cells were loaded with the calcium-sensitive dye Fluo-3 acetoxymethyl ester, and changes in intracellular calcium determined as described under Materials and Methods. AngII (100 nM) or SII-AngII (5 μM) was added where indicated (arrow). C, immunoblot of COX-2 expression from lysates of RASMC exposed to various concentrations of SII-AngII or AngII (100 nM) for 3 h. Cell lysates prepared and detection of COX-2 was determined as described under Materials and Methods. D, summary of densitometric analysis of SII-AngII induced COX-2 expression in RASMC. Mean values ± S.E.M. from three similar studies.
More recent evidence points to an additional pathway for cellular responses to AngII involving the epidermal growth factor receptor (Shah and Catt, 2006). In this paradigm, activation of AT1R results in metalloproteinase-dependent release of surface bound epidermal growth factor and subsequent activation of the epidermal growth factor receptor followed by activation of its signaling pathway. Alternatively, non-G protein-dependent signaling pathways for AngII have been recently described in which the internalization of the receptor initiated by β-arrestin interaction permits subsequent prolonged activation of the MAP kinase pathway as a result of interaction of the kinases with the receptor/β-arrestin scaffold (Pierce et al., 2000).
To examine the involvement of heterotrimeric G protein activation in AngII-stimulated COX-2 protein expression, we used small, cell-permeable compounds that either stimulate or inhibit G proteins. Mastoparan, a peptide derived from wasp venom, activates G proteins by promoting the exchange of GTP for GDP, thus mimicking activation of G proteins by GPCR agonists (Higashijima et al., 1988). This tetradecapeptide has been shown to regulate numerous G protein signaling events, including Ca2+-ATPases, the monomeric G proteins rho and rac, and phospholipase D (Jones and Howl, 2006). To identify regions of the AT1AR involved in receptor desensitization, Tang et al. (1998) used mastoparan to desensitize the AT1AR. Treatment of Chinese hamster ovary cells expressing AT1AR with mastoparan resulted in desensitization of PLC to subsequent addition of AngII. The mastoparan-induced desensitization of the AngII response was comparable with that seen for pretreatment with AngII itself, i.e., homologous desensitization (Tang et al., 1998). In our present studies, mastoparan also produced a desensitization of the AngII-induced calcium response (a Gq-PLC-dependent signal) but did not inhibit AngII-induced AT1AR trafficking and COX-2 protein expression. Mastoparan, on its own, produced an increase in intracellular free calcium, activated p42/44 ERK, and also increased COX-2 protein expression. This supports published data showing G protein activation of PLC and the p42/44 ERK pathway and also implicates this pathway in COX-2 protein expression.
Another technique for exploring the role of G proteins in the AngII-induced expression of COX-2 protein is to use compounds that have been shown to inhibit the activity of G proteins. A family of small compounds that inhibit G proteins and thus inhibit the effects of GPCRs has been developed from the original suramin. Suramin inhibits G protein activity by interfering with the association of the Gα and Gβγ subunits, thus blocking the G protein-signaling pathway (Beindl et al., 1996; Freissmuth et al., 1996). The ability of suramin to block G protein-dependent signaling pathways has led to many studies documenting its anticancer effects. However, recent evidence suggests that suramin, in certain cell types such as Chinese hamster ovary cells and renal epithelial cells, may actually activate signaling pathways related to cell proliferation (Nakata, 2004; Zhuang and Schnellmann, 2005). In the present study, treatment of cells with suramin did not alter surface expression of AT1ARs, AngII-induced AT1AR trafficking, p42/44 ERK activation, or COX-2 expression. However, suramin did inhibit Gq-dependent PLC activation and increase of intracellular free calcium. Likewise, U73122, an inhibitor of PLC activity, did not alter any of the responses examined in this study except that for AngII-induced elevations of intracellular free calcium. These data indicate, as discussed above, that activation of G proteins by AT1AR is not essential for AngII to initiate nuclear membrane localization of its receptor and subsequently increase COX-2 expression. In addition, these data show that elevation of Cai does not necessarily mediate changes in COX-2 protein expression.
SII-AngII is a selective agonist for the AT1AR, in that this ligand can activate β-arrestin-mediated AT1AR internalization and p42/44 ERK activation without G protein activation. In our studies, use of SII-AngII promoted internalization and nuclear membrane localization of the AT1AR and also increased COX-2 protein expression in the absence of increases in intracellular free calcium, thus confirming lack of G protein activation in its signaling.
In summary, the present study examined the role of G protein activation in AngII-induced expression of the enzyme COX-2. Through the use of compounds that stimulate G proteins, inhibit G proteins, or inhibit an enzyme activated by a G protein, we conclude that the ability of AngII to increase COX-2 expression was dependent upon normal internalization and nuclear membrane trafficking of the AT1AR. G protein-dependent activation of PLC and subsequent elevations in intracellular free calcium is not required for this effect, because suramin or U73122 did not alter AT1AR trafficking and did not inhibit AngII-induced COX-2 expression. Previous studies implicated activation of the MAP kinase pathway in AngII-induced COX-2 expression (Ohnaka et al., 2000). The MAP kinase pathway may be a parallel pathway for activation of COX-2 expression, because mastoparan activated p42/44 ERK and also increased COX-2 expression. Last, the ability of the selective AT1AR agonist SII-AngII to promote AT1AR internalization, nuclear membrane localization, and COX-2 protein expression without causing an elevation in intracellular free calcium, a G protein-PLC-dependent event, again supports the concept that G protein activation is not a requirement for AngII to increase the expression of COX-2, whereas receptor internalization and nuclear localization may be required.
Footnotes
This work was supported by Dialysis Clinic Incorporated [Grant C-2342A] (research awards); and the Department of Veteran's Affairs Research Enhancement Award Program [Grant 0013].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.109.151829.
ABBREVIATIONS: AngII, angiotensin II; AT1, angiotensin type 1; AT1AR, angiotensin type 1A receptor; PLC, phospholipase C; ERK, extracellular signal-regulated kinase; COX, cyclooxygenase; GPCR, G protein-coupled receptor; RASMC, rat aorta smooth muscle cells; U73122, 1-[6-[[17β-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione; SII-AngII, sarcosine1-Ile4-Ile8-angiotensin II; FBS, fetal bovine serum; HEK, human embryonic kidney; GFP, green fluorescent protein; PBS, phosphate-buffered saline; HBSS, Hanks' balanced salt solution; Cai, intracellular free calcium; E-64, N-(trans-epoxysuccinyl)-l-leucine 4-guanidinobutylamide; p, phosphorylated; MAP, mitogen-activated protein.
References
- Beindl W, Mitterauer T, Hohenegger M, Ijzerman AP, Nanoff C, and Freissmuth M (1996) Inhibition of receptor/G protein coupling by suramin analogues. Mol Pharmacol 50: 415-423. [PubMed] [Google Scholar]
- de Gasparo M, Catt KJ, Inagami T, Wright JW, and Unger T (2000) International Union of Pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev 52: 415-472. [PubMed] [Google Scholar]
- Derbyshire ZE, Halfter UM, Heimark RL, Sy TH, and Vaillancourt RR (2005) Angiotensin II stimulated transcription of cyclooxygenase II is regulated by a novel kinase cascade involving Pyk2, MEKK4 and annexin II. Mol Cell Biochem 271: 77-90. [DOI] [PubMed] [Google Scholar]
- Eggena P, Zhu JH, Clegg K, and Barrett JD (1993) Nuclear angiotensin receptors induce transcription of renin and angiotensinogen mRNA. Hypertension 22: 496-501. [DOI] [PubMed] [Google Scholar]
- Freissmuth M, Boehm S, Beindl W, Nickel P, Ijzerman AP, Hohenegger M, and Nanoff C (1996) Suramin analogues as subtype-selective G protein inhibitors. Mol Pharmacol 49: 602-611. [PubMed] [Google Scholar]
- Higashijima T, Uzu S, Nakajima T, and Ross EM (1988) Mastoparan, a peptide toxin from wasp venom, mimics receptors by activating GTP-binding regulatory proteins (G proteins). J Biol Chem 263: 6491-6494. [PubMed] [Google Scholar]
- Hu ZW, Kerb R, Shi XY, Wei-Lavery T, and Hoffman BB (2002) Angiotensin II increases expression of cyclooxygenase-2: implications for the function of vascular smooth muscle cells. J Pharmacol Exp Ther 303: 563-573. [DOI] [PubMed] [Google Scholar]
- Jones S and Howl J (2006) Biological applications of the receptor mimetic peptide mastoparan. Curr Protein Pept Sci 7: 501-508. [DOI] [PubMed] [Google Scholar]
- Lu D, Yang H, Shaw G, and Raizada MK (1998) Angiotensin II-induced nuclear targeting of the angiotensin type 1 (AT1) receptor in brain neurons. Endocrinology 139: 365-375. [DOI] [PubMed] [Google Scholar]
- Luttrell LM and Lefkowitz RJ (2002) The role of {beta}-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci 115: 455-465. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, and Lefkowitz RJ (2001) Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds. Proc Natl Acad Sci U S A 98: 2449-2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MA, Jacobowitz DM, Aguilera G, and Catt KJ (1991) Differential distribution of AT1 and AT2 angiotensin II receptor subtypes in the rat brain during development. Proc Natl Acad Sci U S A 88: 11440-11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinelli TA, Raymond JR, Baldys A, Yang Q, Lee MH, Luttrell L, and Ullian ME (2007) Identification of a putative nuclear localization sequence within the angiotensin II AT1A receptor associated with nuclear activation. Am J Physiol Cell Physiol 292: C1398-C1408. [DOI] [PubMed] [Google Scholar]
- Morinelli TA, Walker LP, and Ullian ME (2008) Cox-2 expression stimulated by angiotensin II depends upon At1 receptor internalization in vascular smooth muscle cells. Biochim Biophys Acta 1783: 1048-1054. [DOI] [PubMed] [Google Scholar]
- Nakata H (2004) Stimulation of extracellular signal-regulated kinase pathway by suramin with concomitant activation of DNA synthesis in cultured cells. J Pharmacol Exp Ther 308: 744-753. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Barak LS, and Caron MG (2001) Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-β-arrestin complexes after receptor endocytosis*. J Biol Chem 276: 19452-19460. [DOI] [PubMed] [Google Scholar]
- Ohnaka K, Numaguchi K, Yamakawa T, and Inagami T (2000) Induction of cyclooxygenase-2 by angiotensin II in cultured rat vascular smooth muscle cells. Hypertension 35: 68-75. [DOI] [PubMed] [Google Scholar]
- Pierce KL, Maudsley S, Daaka Y, Luttrell LM, and Lefkowitz RJ (2000) Role of endocytosis in the activation of the extracellular signal-regulated kinase cascade by sequestering and nonsequestering G protein-coupled receptors. Proc Natl Acad Sci U S A 97: 1489-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Re RN, LaBiche RA, and Bryan SE (1983) Nuclear hormone mediated changes in chromatin solubility. Biochem Biophys Res Commun 110: 61-68. [DOI] [PubMed] [Google Scholar]
- Shah BH and Catt KJ (2006) TACE-dependent EGF receptor activation in angiotensin-II-induced kidney disease. Trends Pharmacol Sci 27: 235-237. [DOI] [PubMed] [Google Scholar]
- Tang H, Guo DF, Porter JP, Wanaka Y, and Inagami T (1998) Role of cytoplasmic tail of the type 1A angiotensin II receptor in agonist- and phorbol ester-induced desensitization. Circ Res 82: 523-531. [DOI] [PubMed] [Google Scholar]
- Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, and Lefkowitz RJ (2003) Independent {beta}-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci U S A 100: 10782-10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang S and Schnellmann RG (2005) Suramin promotes proliferation and scattering of renal epithelial cells. J Pharmacol Exp Ther 314: 383-390. [DOI] [PubMed] [Google Scholar]