

Abstract
Although the N-methyl-d-aspartate (NMDA) receptor (NMDAR) obligatory unit NMDAR1 is expressed in the vasculature and myocardium, the impact of peripheral NMDAR activation on blood pressure (BP) has received little attention. We demonstrate, for the first time, dose-related pressor responses elicited by systemic NMDA (125, 250, 500, and 1000 μg/kg) in conscious rats. The pressor response was peripheral NMDAR-mediated because: 1) it persisted after ganglion blockade (hexamethonium; 5 mg/kg i.v.); 2) it was attenuated by the selective NMDAR blocker dl-2-amino-5-phosphonopentanoic acid (5 mg/kg, i.v.) or the glycine/NMDAR antagonist R-(+)-3-amino-1-hydroxypyrrolid-2-one [R-(+)-HA-966; 10 mg/kg i.v.]; and 3) NMDA (1.25–10 mM) increased contractile force of rat aorta in vitro. It is noteworthy that ex vivo studies revealed enhanced nitric oxide (NO) and reactive oxygen species (ROS) generation in vascular tissues collected at the peak of the NMDAR-mediated pressor response. Pharmacological, ex vivo, and in vitro findings demonstrated attenuation of the NMDAR-mediated increases in BP and vascular NO and ROS by the nonselective NO synthase (NOS) inhibitor Nω-nitro-l-arginine methyl ester hydrochloride (10 mg/kg i.v.) or the neuronal NOS (nNOS) inhibitor Nω-propyl-l-arginine hydrochloride (150 μg/kg i.p.) but not by the endothelial NOS inhibitor N5-(1-iminoethyl)-l-ornithine (4 or 10 mg/kg i.v.). Furthermore, R-(+)-HA-966 attenuated NMDA-evoked generation of vascular NO and ROS. The findings suggest a pivotal role for enhanced vascular nNOS-derived NO in ROS generation and in the subsequent pressor response elicited by peripheral NMDAR in conscious rats.
Introduction
The N-methyl-d-aspartate (NMDA) receptor (NMDAR) constitutes one of three glutamate receptor subtypes that were initially identified within the CNS (Dingledine et al., 1999). The central NMDAR modulates cardiovascular function, because its activation elicits pressor or depressor response, depending on the targeted brainstem neuronal pool (el-Mas and Abdel-Rahman, 1993; Bonham and Chen, 2002). On the other hand, little is known about the role of peripheral NMDAR in blood pressure (BP) control despite its expression in the heart and vasculature (Leung et al., 2002). It is noteworthy that only one study, to our knowledge, has shown that systemic NMDA administration elicited a modest depressor response in anesthetized rats (Sitniewska et al., 2003). It is possible that the use of anesthesia influenced the blood pressure response in that study.
It is also not known whether systemically administered NMDA influences blood pressure by activating peripheral (vascular) or central NMDARs. Whereas NMDAR-mediated NO release within the CNS contributes to vasodilation (Alderton et al., 2001), NMDAR activation also enhances NADPH oxidase (Nox) activity (Brennan et al., 2009). It is noteworthy that the Nox family generates ROS within the cardiovascular system (Bedard and Krause, 2007), and the products of Nox (ROS) and NOS (NO) form peroxynitrite. Reported findings have implicated peroxynitrite in beneficial or detrimental cardiovascular effects, because it produces vasodilation (Katori et al., 2006) or contributes to various cardiovascular diseases (Pacher et al., 2007). Furthermore, NO contributes to ROS generation in the CNS after NMDAR activation (Girouard et al., 2009). Currently, to our knowledge, there are no studies on the effect of peripheral NMDAR activation on vascular NO and ROS and the impact of such responses on blood pressure in conscious rats. It is also imperative to note that vascular ROS generation mediates vasopressor responses (e.g., by angiotensin) via enhanced vascular Ca2+ (Touyz, 2005).
The first objective of this study was to investigate the effect of systemic NMDA on blood pressure in the absence of the confounding effects of anesthesia. Because our study was the first to demonstrate a dose-dependent pressor response after systemic (intravenous) NMDA in conscious unrestrained rats, it was important to verify the expression of the obligatory NMDAR1 subunit in the vasculature and heart in our model system and ascertain the role of peripheral NMDAR activation in such pressor responses. In the integrative studies, systemic (intravenous) NMDA was administered after ganglionic blockade (hexamethonium), peripheral selective NMDAR blockade (AP-5), or the glycine/NMDAR antagonist R-(+)-3-amino-1-hydroxypyrrolid-2-one [R-(+)-HA-966]; the latter has been used to determine the involvement of the glycine-sensitive component in NMDAR-mediated responses (Kemp and Leeson, 1993). The final objective was to elucidate the molecular mechanisms implicated in peripheral NMDAR-mediated pressor response; in this study, we conducted a series of integrative, ex vivo, and in vitro studies to elucidate the effect of peripheral NMDAR activation on vascular tone and vascular NO and ROS levels in the absence or presence of nonselective or selective NOS inhibitors. Our experimental approaches enabled us to determine the NOS isoform implicated in vascular NO generation and establish a causal role for NO-dependent ROS generation in the peripheral NMDAR-mediated pressor response.
Materials and Methods
Sprague-Dawley rats (Charles River Laboratories, Raleigh, NC), 11 to 12 weeks old, were used in this study. After vascular catheterization, the rats were housed individually in separate cages and allowed free access to Purina (St. Louis, MO) chow and water. The temperature was maintained at 22 ± 1°C, and in a 12-h light/dark cycle the lights automatically turned off at 7:00 PM. Surgical procedures were conducted in accordance with institutional animal use and care guidelines and the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996) as indicated by the National Institutes of Health.
Intravascular Catheterization
Femoral artery and vein catheterization was performed as described previously (Nassar et al., 2011). Animals received buprenorphine for analgesia (0.03 mg/kg) 30 min before surgery and were anesthetized with an injection of ketamine (9 mg/100 g i.p.) and xylazine (1 mg/100 g i.p.). Catheters consisting of 5-cm PE-10 tubing bonded to 15-cm PE-50 tubing were placed into the abdominal aorta and vena cava via the left femoral vessels for the measurement of arterial pressure and intravenous injections, respectively. Two venous catheters were inserted into a femoral vein to permit intravenous bolus administration and/or infusion of drugs. Catheters were tunneled subcutaneously and exteriorized at the back of the neck between the scapulae. Vascular catheters were flushed with heparinized saline and plugged by stainless-steel pins. Incisions were closed with surgical clips and swabbed with povidine-iodine solution. Postoperative care included buprenorphine (0.03 mg/kg) and penicillin G procaine (100,000 U/kg; Durapen; Vedco Inc., Overland Park, KS). The animals were allowed 2 days after surgery to recover before experiments were conducted.
On the day of the experiments, the arterial catheter was connected to a pressure transducer for the measurement of blood pressure in conscious freely moving rats. At least 30 min were allowed for the stabilization of blood pressure and heart rate at the beginning of an experiment. BP was recorded by a ML870 PowerLab 8/30 (ADInstruments, Colorado Springs, CO) and analyzed by LabChart Pro (version 6) software (ADInstruments). Heart rate was extracted from BP recording by the LabChart (version 6) blood pressure analysis module, and both variables were continuously recorded and stored for offline analysis.
Western Blot Analysis
After euthanasia, aortas, mesenteric arteries, and hearts were flash-frozen in 2-methylbutane (Sigma-Aldrich, St. Louis, MO) in dry ice and stored at −80°C until tissue processing for molecular studies. Tissues were homogenized on ice in lysis buffer containing 20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate, 1 mM activated sodium orthovanadate, and 1 μg/ml leupeptin with protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). After centrifugation (12,000g for 20 min), protein in the supernatant was quantified by using the Bio-Rad protein assay system (Bio-Rad Laboratories, Hercules, CA). Protein extracts for NMDAR1 from aorta, mesenteric arteries, or heart (50–60 μg/lane) or aorta for iNOS (60 μg/lane) were run on a NUPAGE 4 to 12% Bis-Tris polyacrylamide gel by electrophoresis at 100 V (Invitrogen, Carlsbad, CA). Gels were electroblotted to nitrocellulose membranes via a semidry transfer using the Bio-Rad TransBlot SD transfer cell (Bio-Rad Laboratories) at 25 V for 1 h. The nitrocellulose membranes were then incubated for 2 h in Odyssey Blocking Buffer (LI-COR Biosciences, Lincoln, NE). After blocking, membranes were incubated overnight at 4°C with mouse antibodies to NMDAR1 (1:100; Abcam Inc., Cambridge, MA) or iNOS (1:500; BD Biosciences, San Jose, CA) and a rabbit antibody to β-actin (1: 2000) diluted in Odyssey blocking buffer and 0.1% Tween. The blots were washed with 0.1% phosphate buffered saline-Tween buffer four times and incubated for 1 h at room temperature with fluorescently labeled secondary antibodies: anti-mouse for NMDAR1 and iNOS and anti-rabbit for β-actin (1:10,000, LI-COR Biosciences) in Odyssey Blocking Buffer (LI-COR Biosciences). After four additional washes, the nitrocellulose membrane was placed in phosphate-buffered saline, and imaging acquisition was performed via the Odyssey Infrared Imaging system (LI-COR Biosciences). Bands of NMDAR1 and iNOS were quantified by measuring the integrated density using Odyssey software (version 3.0; LI-COR Biosciences), normalized to β-actin, and expressed as a percentage of the control.
Quantification of Aortic Reactive Oxygen Species and NO
The 2′,7′-dichlorofluorescein (DCF) biochemical assay was used for quantification of ROS as reported previously (Zou et al., 2004) with the following modifications. Homogenization was performed by using Radnoti tissue grinders (Radnoti Glass Technology, Monrovia, CA) to increase protein yield, and kinetic readings were taken at 5-min intervals for 30 min at 37°C. ROS levels were calculated by relative DCF fluorescence per microgram of protein. For quantification of vascular NO, fresh unfixed aorta sections (10 μm) were incubated with 10 μM 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM) (Invitrogen) at 37°C in the presence of 5% CO2 in a moist chamber for 30 min. Positive and negative controls were used to validate the assay (see Experimental Groups and Protocols). Images were visualized with a Zeiss LSM 510 microscope (Carl Zeiss Inc., Thornwood, NY). Three to five images were acquired from four aortic sections for each experimental condition. Quantification was conducted by using ImageJ software (National Institutes of Health, Bethesda, MD), and changes in total fluorescence intensity, normalized to control, were calculated.
Measurement of Nitrite/Nitrate Level
Nitrite/nitrate (NOx) content was measured by using a colorimetric assay kit according to the manufacturer's instructions (Cayman Chemical, Ann Arbor, MI) and as described previously (Misko et al., 1993; El-Mas et al., 2009).
Experimental Groups and Protocols
Experiment 1: Effect of Ganglionic Blockade on Systemic NMDA-Evoked Pressor Response.
In this experiment, we examined the effect of ganglion blockade with hexamethonium (5 mg/kg i.v.) on NMDA-evoked cardiovascular responses. Because the hypotensive effect of hexamethonium (indicative of adequate ganglion blockade) lasted <20 min before BP started to recover, we compared the pressor responses elicited by intravenous NMDA (250 and 500 μg/kg) before and 10 min after hexamethonium in conscious male rats (n = 5–6). The two NMDA doses fell between the lowest and highest NMDA doses used to construct a dose-pressor response curve in our preliminary study (McGee and Abdel-Rahman, 2010) and experiment 2 in the next section.
Experiment 2: Effect of Peripheral NMDAR Blockade on Systemic NMDA-Evoked Dose-Related Pressor Responses.
In the first part of this experiment, four groups of conscious male rats (n = 4–6) were used to investigate the effect of peripheral NMDAR blockade with the selective blocker AP-5 on the cardiovascular responses elicited by systemic NMDA. However, as a first step, we opted to determine whether intravenous NMDA infusion could produce sustained elevation in BP. Preliminary experiments were conducted to determine an optimal dose/infusion rate of NMDA that produced an approximately 25 mm Hg increase in BP, which constituted approximately 50% of the maximal pressor response elicited by bolus intravenous NMDA (McGee and Abdel-Rahman, 2010). The rats were pretreated with saline (vehicle) or AP-5 (5 mg/kg i.v.) 30 min before NMDA (180 μg/kg/min) or saline infusion at a rate of 9 μl/min (Harvard compact pump model 975; Harvard Apparatus Inc., Holliston, MA) for 30 min. At the selected dose, AP-5 does not cross the blood-brain barrier (Dingledine et al., 1999). In the second part of the experiment, four groups of conscious male rats (n = 4–6) were used to investigate the effect of the glycine/NMDAR antagonist R-(+)-HA-966 on the NMDA-evoked pressor response. In two groups, we evaluated the blood pressure and heart rate effects of NMDA (125, 250, 500, or 1000 μg/kg i.v.) 30 min after administration of R-(+)-HA-966 (10 mg/kg i.v.) or its vehicle (saline). NMDA was administered at 15-min intervals to allow full recovery from the preceding dose except for the final (1000 μg/kg) dose where the animal was sacrificed immediately after the peak pressor response was achieved. The remaining two groups received intravenous R-(+)-HA-966 or equal volumes of saline. We conducted a preliminary experiment to identify an optimal R-(+)-HA-966 dose, from the reported dose (5–30 mg/kg) range, that exhibited negligible central effects known to be associated with the high (20–30 mg/kg) doses (McMillen et al., 2004). The selected dose (10 mg/kg i.v.) caused no behavioral effects (data not shown) and had no significant effect on blood pressure or heart rate. The vascular tissues were collected from drug- and vehicle-treated rats and stored for subsequent molecular studies.
Experiment 3: Effect of NMDA on Vascular Force Generation In Vitro.
Vascular force generation was measured in aortic rings as reported previously (Harris et al., 2010). Four male Sprague-Dawley rats were used to directly investigate the effect of NMDA on vascular tone of isolated aortic rings. Aorta rings were mounted onto a DMT 610 myograph system (DMT-USA International, Atlanta, GA). In a preliminary experiment, single additions of NMDA (10−6 to 10−3 M) to the tissue bath had no effect on vascular tone. It is noteworthy that a similar NMDA concentration range had no effect on isolated cerebral arteries (Simandle et al., 2005). Therefore, a higher concentration range (1.25–10 mM) of NMDA was cumulatively added to the tissue bath; this paradigm mimicked the continuous exposure of the vasculature to the infused NMDA in our aforementioned NMDA in vivo experiment. The functional integrity of the vascular rings was verified by constructing cumulative concentration-response curves with 0.001 to 3 μM phenylephrine (PE) according to established protocol (Harris et al., 2010) in all aortic rings that were used in the NMDA experiment. Contractile responses were recorded with PowerLab/8SP, and data were acquired and analyzed by LabChart (version 7) Pro software (ADInstruments) as reported previously (Harris et al., 2010).
Experiment 4: Effect of NOS Inhibition on Systemic NMDA-Evoked Pressor Responses.
Integrative, ex vivo, and in vitro studies were conducted to elucidate the role of NOS-derived NO in the pressor response elicited by peripheral NMDAR activation. Nine groups of conscious male rats (n = 4–6) were used in this experiment. In the first part of this experiment, two groups of rats received the nonselective NOS inhibitor Nω-nitro-l-arginine methyl ester (l-NAME) hydrochloride (10 mg/kg i.v) 90 min before NMDA (125, 250, 500, and 1000 μg/kg i.v.) or an equal volume of saline. Because l-NAME caused significant and sustained increase in BP that lasted until and during systemic NMDA injection, two additional groups of rats received phenylephrine (2.7 μg/kg/min), infused at a rate of 18 μl/min, before systemic NMDA or equal volumes of saline. The chosen phenylephrine infusion regimen (based on a preliminary experiment) and previous findings (El-Mas et al., 2006) elicited a similar rise in BP to that caused by l-NAME. At the conclusion of the experiment, the vascular tissues were collected for the measurement of vascular NO. To corroborate the impact of these treatments on vascular NO level in the absence of CNS involvement, a parallel experiment was conducted in which aortic sections from untreated animals were incubated with the following treatments for 30 min: 1) saline (control), 2) NMDA (100 μM), 3) l-arginine (10 mM), 4) l-NAME (100 μM), 5) PE (1 μM), 6) l-NAME + NMDA, or 7) PE + NMDA. Finally, because l-NAME pretreatment abrogated the NMDA-evoked pressor response in vivo, we elucidated the contribution of the constitutive isoforms of NOS, nNOS, and eNOS to this response by constructing the NMDA dose (125, 250, 500, and 1000 μg/kg i.v.) pressor response curve 30 min after selective inhibition of nNOS with Nω-propyl-l-arginine (NPLA) hydrochloride (150 μg/kg i.p.) or eNOS with N5-(1-Iminoethyl)-l-ornithine (l-NIO; 4 or 10 mg/kg i.v.). Control rats received saline (vehicle for nNOS or eNOS inhibitor) or the inhibitor alone. Pharmacologic iNOS inhibition was not attempted because the rapid onset (seconds) and short duration (<1 min) of the NMDA-evoked pressor response argued against the potential contribution of iNOS to the pressor response. Furthermore, preliminary Western blot studies on vasculature from vehicle- and NMDA-treated rats revealed no change in vascular iNOS (data not shown).
Drugs
N-methyl-d-aspartic acid, l-NAME, phenylephrine hydrochloride, hexamethonium chloride, 2′-7′-dichlorofluorescein, and dimethyl sulfoxide were purchased from Sigma-Aldrich. DAF-FM was purchased from Invitrogen. N5-(1-iminoethyl)-l-ornithine, Nω-propyl-l-arginine hydrochloride, dl-2-amino-5-phosphonopentanoic acid, and R(+)-3-amino-1-hydroxy-2-pyrrolidinone were purchased from Tocris Biosciences (Ellisville, MO). Sterile saline was purchased from B. Braun Medical (Irvine, CA).
Data Analysis and Statistics
Values were expressed as means ± S.E.M. Mean arterial pressure (MAP) was computed as 1/3 (systolic-diastolic) + diastolic. Statistical analyses were conducted by using a one-way or repeated-measures analysis of variance with Bonferroni post hoc test and Student's t test. Prism 5.0 software (GraphPad Software Inc., San Diego, CA) was used to perform statistical analyses. P < 0.05 was considered significant.
Results
Baseline blood pressure and heart rate values were similar in all groups of rats used in the study (Table 1).
TABLE 1.
Baseline values of MAP and HR
Values are means ± S.E.M. of four to six observations.
| Group | MAP | HR |
|---|---|---|
| mm Hg | beats/min | |
| Saline | 117 ± 8.1 | 389 ± 12.8 |
| NMDA | 120 ± 5.4 | 386 ± 10.5 |
| Saline infusion | 123 ± 5.9 | 373 ± 26.6 |
| NMDA infusion | 113 ± 2.5 | 365 ± 11.8 |
| AP-5 | 120 ± 6.5 | 390 ± 21.6 |
| AP-5 + NMDA infusion | 126 ± 4.9 | 386 ± 15.7 |
| Hexamethonium | 124 ± 1.8 | 306 ± 14 |
| Hexamethonium + NMDA | 130 ± 5.6 | 398 ± 18.1 |
| R-(+)-HA-966 | 129 ± 8.0 | 338 ± 12.7 |
| R-(+)-HA-966 + NMDA | 118 ± 3.7 | 344 ± 16.5 |
| l-NAME | 121 ± 5.5 | 330 ± 9.8 |
| l -NAME + NMDA | 132 ± 4.2 | 345 ± 19.4 |
| Phenylephrine | 133 ± 6.9 | 348 ± 12.7 |
| Phenylephrine + NMDA | 129 ± 4.6 | 377 ± 12.7 |
| l -NIO (4 mg/kg) + NMDA | 136 ± 8.2 | 395 ± 25.1 |
| l -NIO (10 mg/kg) + NMDA | 137 ± 5.5 | 379 ± 8.2 |
| NPLA (150 μg/kg) | 129 ± 4.7 | 370 ± 19.7 |
| NPLA (150 μg/kg) + NMDA | 114 ± 2.5 | 378 ± 16.2 |
NMDAR1 Protein Expression in the Cardiovascular System.
As illustrated in Fig. 1, the obligatory NMDAR subunit NMDAR1 (130 kDa) is expressed within the aorta, mesenteric arteries, and heart (Western blot).
Fig. 1.

Expression of the NMDAR obligatory subunit NMDAR1 (130 kDa) protein in aorta, mesenteric arteries (MA), and heart of male Sprague-Dawley rats.
Lack of Effect of Ganglion Blockade on NMDA-Evoked Cardiovascular Responses.
NMDA (250 and 500 μg/kg i.v.) elicited dose-related pressor responses consistent with those observed in the dose-pressor response curve in our preliminary studies (McGee and Abdel-Rahman, 2010) and in the experiments described below; heart rate also increased but the increases were not dose-related (Fig. 2). These NMDA-evoked pressor and tachycardic responses remained unaltered after hexamethonium (5 mg/kg i.v.) (Fig. 2). Statistical analysis revealed that NMDA-evoked MAP and HR responses at 250 μg/kg (P = 0.72 and 0.78, respectively) and 500 μg/kg (P = 0.25 and 0.54, respectively) in the absence or presence of hexamethonium were not significantly different (Fig. 2).
Fig. 2.

NMDA (250 or 500 μg/kg i.v.)-evoked changes in mean arterial pressure (top) and heart rate (bottom) before and after hexamethonium (5 mg/kg i.v.) in conscious male Sprague-Dawley rats. Values are means ± S.E.M. of four to six observations.
NMDAR Blockade Attenuates NMDA-Evoked Pressor Response.
In the first part of this experiment, NMDA infusion (180 μg/kg/min) produced significant (P < 0.05) increases in BP, which peaked at 10 min along with bradycardia (Fig. 3); despite its gradual decline, the BP of the NMDA-treated rats remained significantly (P < 0.05) higher than the corresponding control (saline) values (Fig. 3A). Pretreatment with the NMDAR antagonist AP-5, which had no significant effect on baseline blood pressure throughout the course of the experiment, abrogated the NMDA-evoked pressor response (Fig. 3A) but had no effect on the bradycardic response (Fig. 3B). The latter finding indicated that the bradycardic response was not baroreflex-mediated and seemed to have contributed to the gradual decline in the pressor response despite continued NMDA infusion (Fig. 3A). In the second experiment, pretreatment with the glycine/NMDAR antagonist R-(+)-HA-966 (10 mg/kg i.v.) significantly attenuated the dose-dependent pressor response (Fig. 4A) and the associated tachycardic response (Fig. 4B) elicited by systemic NMDA (125, 250, 500, and 1000 μg/kg i.v.). Compared with saline, R-(+)-HA-966 had no significant effect on blood pressure or heart rate throughout the course of the experiment (Fig. 4, C and D).
Fig. 3.
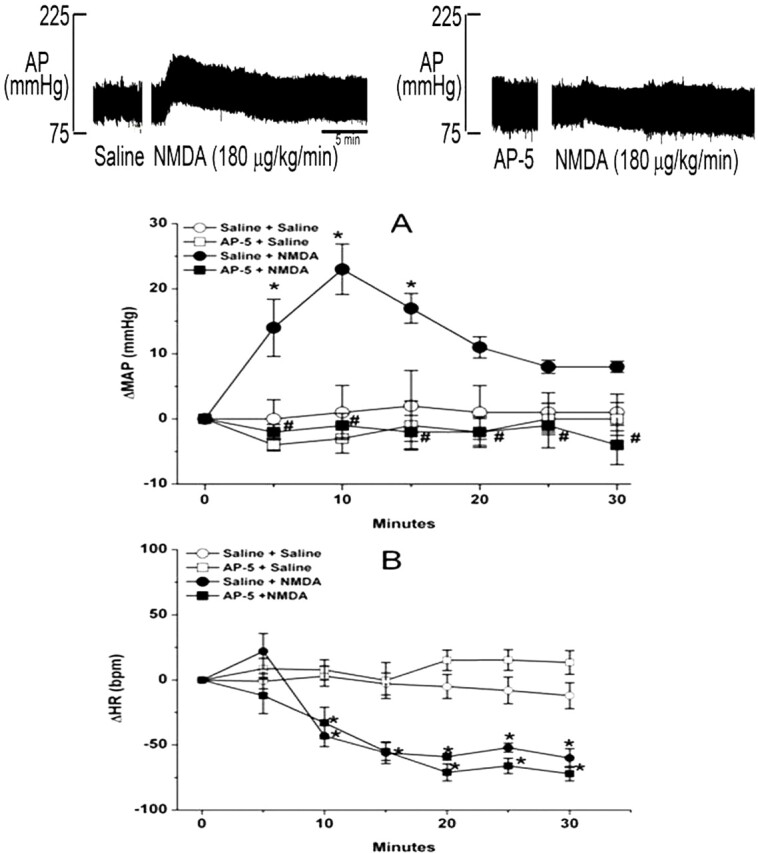
The NMDAR antagonist AP-5 abrogates the pressor (A) but not the bradycardic (B) response elicited by NMDA infusion (180 μg/kg/min; 9 μl/min) in conscious male Sprague-Dawley rats. NMDA or vehicle (saline) was infused for 30 min. Values are means ± S.E.M. of five observations. *, P < 0.05 versus saline (vehicle). #, P < 0.05 versus NMDA.
Fig. 4.

A and B, effect of the glycine/NMDAR antagonist R-(+)-HA-966 (10 mg/kg i.v.) or saline (vehicle) on intravenous NMDA-evoked changes in ΔMAP (A) and heart rate (ΔHR) (B) in conscious male Sprague-Dawley rats. C and D, effect of R-(+)-HA-966 or saline, given alone, on ΔMAP (C) and ΔHR (D). Values are means ± S.E.M. of four to six observations. #, P < 0.05 versus NMDA.
NMDA Increases Aortic Vascular Tone In Vitro.
Addition of cumulative concentrations of NMDA (1.25 and 10 mM) had little effect on the contractile force at lower concentrations (<2.5 mM) and resulted in increased contractile force at 5 mM (Fig. 5A). It is noteworthy that the response obtained with 5 mM NMDA was maximal because it did not increase further with 10 mM NMDA (Fig. 5A) or higher (data not shown). As a control for vascular integrity, phenylephrine produced concentration-dependent increases in contractile force of the same rings that were subjected to NMDA (Fig. 5B).
Fig. 5.

Contractile concentration-response curves constructed with the addition of NMDA (1.25–10 mM; A) or phenylephrine (0.001–3 μM; B) to aortic rings. Values are means ± S.E.M. of four observations.
NOS Inhibition Attenuates Systemic NMDA-Evoked Pressor Response.
Pretreatment with the nonselective NOS inhibitor l-NAME, which caused significant (P < 0.05) and sustained increase in BP, virtually abolished the pressor response elicited by NMDA (Fig. 6A). To control for the l-NAME-evoked pressor response, we infused phenylephrine to induce a pressor response of similar magnitude and duration. As illustrated in Fig. 6A, NMDA continued to produce a significant (P < 0.05) increase in BP in the presence of the phenylephrine-evoked pressor response. Saline (vehicle for NMDA) had no effect on BP in l-NAME- or phenylephrine-pretreated rats (Fig. 6B). Pretreatment with NPLA (150 μg/kg i.p.) significantly attenuated (Fig. 6C), whereas l-NIO (4 or 10 mg/kg i.v.) did not affect (Fig. 6D), the systemic NMDA-evoked pressor responses. Neither NPLA nor l-NIO influenced blood pressure.
Fig. 6.

A, effect of the nonselective NOS inhibitor l-NAME (10 mg/kg i.v.) or induction by PE infusion (2.7 μg/kg/min; 18 μl/min) of a comparable elevation in mean arterial pressure to that caused by l-NAME, on the pressor responses elicited by NMDA (125, 250, 500, or 1000 μg/kg i.v.). *, P < 0.05 versus l-NAME + NMDA. B, intravenous saline (vehicle for NMDA) was injected after l-NAME or phenylephrine administration. C and D, effect of pretreatment, 30 min earlier, with the selective nNOS inhibitor NPLA (150 μg/kg i.p.) (C) or the selective eNOS inhibitor l-NIO (4 or 10 mg/kg i.v.) (D) on the dose-pressor response curve elicited by intravenous NMDA. The vehicle for l-NIO and NPLA (saline) was administered as a control before NMDA. In C, *, p < 0.05 versus saline pretreatment group. Values are means ± S.E.M. of four to six observations.
Peripheral NMDAR Activation Enhances Vascular NO and ROS Generation.
Findings of the ex vivo studies in which vascular NOx levels (Fig. 7, A and B) were measured in aortic tissues collected at a time that coincided with the peak pressor response elicited by NMDA (1000 μg/kg) revealed that NMDA significantly (P < 0.05) increased aortic NOx levels, and such an increase was abrogated by pretreatment with l-NAME, NPLA, or R-(+)-HA-966 (Fig. 7, A and B) but remained in the presence of l-NIO or phenylephrine (Fig. 7, A and B). Finally, in vitro findings showed that the addition of NMDA to aortic rings caused a significant (P < 0.05) increase in vascular NO (DAF fluorescence), and such an increase was abrogated by l-NAME but not phenylephrine (Fig. 7, C and D). These in vitro findings support the involvement of vascular NMDAR in the NMDA-evoked increase in NO response and complement the in vivo and ex vivo findings.
Fig. 7.

A, effect of NMDA on aortic NOx content in the absence or presence of the nonselective NOS inhibitor l-NAME (10 mg/kg i.v.) or PE infusion (2.7 μg/kg/min; 18 μl/min). B, effect of NMDA in the absence or presence of the selective nNOS inhibitor NPLA (150 μg/kg i.p.), the selective eNOS inhibitor l-NIO (10 mg/kg i.v.), or the glycine/NMDAR antagonist R-(+)-HA-966 (10 mg/kg i.v.). Values are means ± S.E.M. of four to six observations in these ex vivo studies. C, NO generation in aortic sections measured by DAF-FM fluorescence in aortic sections from untreated rats (n = 10 for each treatment) in the presence of saline (control) (A), NMDA (100 μM) (B), the nonselective NOS inhibitor l-NAME (100 μM) (C), l-NAME + NMDA (D), PE (1 μM) (E), and PE + NMDA (F) incubated for 30 min before DAF (10 μM) addition. Sections were incubated at 37°C with DAF-FM. D, bar graphs showing DAF-FM fluorescence intensity; data were normalized to vehicle. *, P < 0.05 versus saline (vehicle). #, P < 0.05 versus NMDA.
As shown in Fig. 8, ROS levels were significantly (P < 0.05) higher in aortas collected at the peak of the pressor response elicited by NMDA (1000 μg/kg i.v.) and moderately elevated (P > 0.05) in the presence of l-NAME (Fig. 8B) or phenylephrine (Fig. 8C) compared with the levels in the aortas of vehicle-treated rats. The NMDA-evoked increase in vascular ROS was significantly (P < 0.05) attenuated in the presence of R-(+)-HA-966 (Fig. 8A) or l-NAME (Fig. 8B). By contrast, NMDA-evoked increase in vascular ROS was exacerbated (P < 0.05; compared with NMDA alone) in rats that received phenylephrine infusion (to control for l-NAME-evoked pressor response) (Fig. 8C). Finally, pretreatment with the selective nNOS inhibitor NPLA significantly (P < 0.05) attenuated the NMDA-evoked increase in vascular ROS (Fig. 8D). These ex vivo findings paralleled the blood pressure and vascular NO responses (Fig. 7) elicited by NMDAR activation in the presence of the aforementioned interventions.
Fig. 8.

ROS generation, measured by DCFH-DA, in aorta obtained from rats treated with saline (vehicle), NMDA, R-(+)-HA-966, or R-(+)-HA-966 + NMDA (A), saline (vehicle), NMDA, l-NAME, or l-NAME + NMDA (B), saline (vehicle), NMDA, PE, or PE + NMDA (C), and saline (vehicle), NMDA, NPLA, or NPLA + NMDA (D). Aorta homogenate was incubated at 37°C with DCF for 30 min. Kinetic readings were recorded at 5-min intervals. Relative fluorescence units (RFU) were normalized to protein and are expressed as RFU per microgram of protein per minute in the insets. Values are means ± S.E.M. of four to six observations. *, P < 0.05 versus saline (vehicle). #, P < 0.05 versus NMDA. V, vehicle; N, NMDA; R, R-(+)-HA-966; L, l-NAME; P, PE; NP, NPLA.
Discussion
The present study is the first to demonstrate a pressor response elicited by systemic NMDA in conscious rats along with the following important findings: 1) the obligatory subunit of the NMDAR, NMDAR1, is expressed within the vasculature and heart; 2) the NMDA-evoked pressor response is dose-dependent and mediated via the activation of peripheral NMDAR because it persisted after ganglion blockade (hexamethonium) and was attenuated by the NMDAR antagonist AP-5 or the glycine/NMDAR antagonist R-(+)-HA-966; 3) NMDA increased the contractile response of isolated aortic rings; and 4) prior nonselective NOS (l-NAME) or selective nNOS (NPLA) inhibition attenuated the peripheral NMDAR-mediated increases in BP and vascular NO and ROS. Collectively, the present integrative and molecular studies suggest a pivotal role for vascular nNOS-derived NO in ROS generation and the consequent peripheral NMDAR-mediated pressor response.
We are the first to demonstrate dose-dependent pressor response caused by systemic NMDA. This finding is in marked contrast to a modest depressor response in the only reported study, to our knowledge, on the effect of systemic NMDA on BP (Sitniewska et al., 2003). The NMDA doses and route of administration were similar in the two studies, but the use of anesthesia in the study by Sitniewska et al. may have confounded the BP response. Nonetheless, it was important to rule out the contribution of central NMDAR to the observed pressor response given NMDA's ability to influence BP in a complex manner that depends on the targeted area within the brain (Kubo et al., 1993; Rodrigues Dias et al., 2001) .
We adopted two approaches to ascertain the contribution of peripheral/vascular NMDAR to the pressor response. First, we showed that the obligatory NMDAR subunit NMDAR1 (Dingledine et al., 1999; Leung et al., 2002) is expressed in the vasculature (mesenteric arteries and aorta) and heart (Fig. 1), which agrees with previously reported findings (Leung et al., 2002). Second, prior ganglionic blockade (hexamethonium) had no effect on systemic NMDA-evoked pressor responses, which ruled out the contribution of a central/ganglionic component in the mediated pressor response. However, it was important to substantiate the involvement of the NMDAR in such a pressor response by demonstrating the ability of the selective NMDAR antagonist AP-5 to abrogate the systemic NMDA-evoked pressor response (Fig. 3). It is noteworthy that in the selected dose range AP-5 does not cross the blood-brain barrier (Dingledine et al., 1999) and had no effect on baseline BP in our study. Equally important, we demonstrated the involvement of the glycine modulatory site of the NMDAR in the peripheral NMDAR-mediated pressor response. This conclusion was based on the ability of the stereoselective blocker R-(+)-HA-966, which acts at the NMDAR strychnine-insensitive glycine site (Davies and Watkins, 1972; Vartanian and Taylor, 1991), to attenuate the systemic NMDA-evoked pressor response (Fig. 4). It is noteworthy that consistent with a previously reported study (Adam et al., 2001) the selected dose of R-(+)-HA-966 produced no change in basal BP. It is possible that R-(+)-HA-966's inability to fully abrogate the NMDA-evoked pressor response, as did AP-5, could be caused by its partial blockade of the NMDAR by targeting the glycine modulatory site (Foster and Kemp, 1989; Singh et al., 1990) or because we used a relatively low dose of the drug. It is noteworthy that higher R-(+)-HA-966 doses were not attempted because they are associated with central (behavioral) effects (McMillen et al., 2004). Furthermore, our in vitro findings provided the first direct evidence that NMDA caused contraction of isolated aortic rings (Fig. 5) when the responses were measured by a highly sensitive system and in accordance with a well established protocol (Harris et al., 2010). It is noteworthy that the contractile response caused by NMDA required high concentrations of the drug and abruptly reached a maximal response, which was substantially smaller than the maximal response caused by phenylephrine in the same aortic rings (Fig. 5). It is noteworthy that the inability of NMDA in concentrations up to 1 mM to influence aortic ring tone fully agrees with the inability of the same NMDA concentration to replicate the cerebral artery vasodilation in vivo when added to isolated cerebral arteries (Simandle et al., 2005). Nonetheless, despite the aforementioned limitations, these in vitro findings lend support to the in vivo and ex vivo findings, and collectively the data provide evidence for the involvement of the peripheral NMDAR and its glycine modulatory site in the systemic NMDA-evoked pressor response in conscious rats.
Subsequent studies focused on the molecular mechanisms implicated in the peripheral NMDAR-mediated pressor response. We hypothesized that an imbalance between the generation of vascular ROS and NO contributes to the peripheral NMDAR-mediated pressor response because these molecules produce vasoconstriction and vasodilation, respectively (Jin and Loscalzo, 2010; Schulz et al., 2011). Nonetheless, our experimental approaches also permitted testing the alternative hypothesis that enhanced generation of vascular NO might be implicated in the peripheral NMDAR-mediated pressor response. This premise is based on the ability of NMDAR-mediated NO generation to increase ROS production in neuronal tissue (Chianca et al., 2004). As a first step, we investigated the effect of nonselective NOS inhibition (l-NAME) on the peripheral NMDAR-mediated pressor response; abrogation of the latter by l-NAME implicated NOS-derived NO in such responses. However, we considered the possibility that the l-NAME-evoked substantial and sustained pressor response might have confounded data interpretation. To address this pitfall, we used a phenylephrine infusion paradigm to produce a pressor response similar in magnitude and duration to that caused by l-NAME. Under this experimental setting, systemic NMDA still caused pressor responses (Fig. 6). Together, these findings supported a role for NO in the peripheral NMDAR-mediated pressor response. However, additional studies were needed to identify the implicated NOS isoform and establish a causal role for NO generation in the NMDA-evoked pressor response, at least partly, via vascular ROS generation. Our ex vivo findings corroborated the integrative findings by demonstrating: 1) increased vascular NOx content in tissues isolated at the peak of the NMDA-evoked pressor response, and 2) abrogation of the increased vascular NOx by l-NAME but not phenylephrine (Fig. 7). These findings were confirmed in vitro when NMDA was added to aortic vascular sections in the absence or presence of l-NAME or phenylephrine (Fig. 7). The clear parallelism between the well controlled integrative findings (discussed above) and ex vivo and in vitro findings support a functional role for vascular NMDAR-mediated NO generation in controlling vascular tone. In the next series of studies, we investigated the potential contribution of eNOS and nNOS to the NMDA-evoked NO generation. Selective iNOS inhibition was not attempted because the short-lived (<1 min) pressor response argues against a role for iNOS, which requires minutes to hours to be activated (El-Mas et al., 2006; Smith et al., 2011).
We explored the possibility that eNOS-derived NO or eNOS uncoupling, which generates ROS (Förstermann and Li, 2011), contributes to the NMDAR-mediated pressor response. The inability of the selective eNOS inhibitor l-NIO to influence the pressor response (Fig. 6D) or the increase in vascular NOx (Fig. 7B), despite escalating its dose to 10 mg/kg, ruled out eNOS involvement in the NMDAR-mediated pressor response. Next, we investigated the role of nNOS because it is expressed in vascular smooth muscle cells and nitregic nerves within the vasculature (Forstermann and Sessa, 2012). Indeed, selective nNOS inhibition (NPLA) significantly attenuated the NMDAR-mediated pressor response (Fig. 6C). It is noteworthy that uncoupled nNOS-mediated ROS generation has been implicated in the paradoxical estrogen-induced contraction of coronary arteries and such response was attenuated by prior NOS inhibition (White et al., 2005). Therefore, we extended the ex vivo studies to investigate the impact of vascular NMDAR activation in the absence or presence of NPLA on vascular NO and ROS levels.
We showed that systemic NMDA enhanced ROS generation in vasculature, which fully agrees with a similar finding in primary cerebellar granule cells (Gunasekar et al., 1995). Furthermore, in agreement with findings that PE-evoked contraction is associated with increased vascular ROS (Hao et al., 2006; Tsai and Jiang, 2010), we showed that PE-evoked pressor response was associated with ex vivo vascular ROS generation. Nonetheless, it was imperative to affirm a causal role for the NO-ROS cascade in the peripheral NMDAR-mediated pressor response. First, l-NAME, but not PE, abrogated the NMDA-evoked ROS generation, and these findings mirrored the hemodynamic findings. It is noteworthy that the l-NAME enhancement of vascular ROS level, which agrees with findings in other preparations (Cao et al., 2010), might be related to its vasoconstrictor response; this notion is supported by the elevated vascular ROS in PE-treated animals (Fig. 8). It is imperative to note that nNOS inhibition (NPLA) or the glycine/NMDAR blockade with R-(+)-HA-966 had no effect on BP but attenuated the NMDAR-mediated increases in vascular NO and ROS and BP. Our findings are the first to implicate the strychnine-insensitive glycine site in peripheral NMDAR-mediated NO and ROS generation. Collectively, these findings support a causal role for vascular NO (nNOS)-dependent ROS generation in the peripheral NMDAR-mediated pressor response.
In conclusion, the present study provides the first evidence that peripheral NMDAR activation mediates dose-dependent pressor responses in conscious animals. The in vivo findings with selective pharmacological interventions, corroborated with complementary ex vivo and in vitro findings, have supported a causal role for nNOS-(NO)-dependent ROS generation in the NMDA-evoked pressor response. Furthermore, our findings support a role for the strychnine-insensitive glycine site in the peripheral NMDAR-mediated molecular and BP responses. Finally, the present studies pave the way for future studies on the role of peripheral NMDAR in BP control under physiological and pathophysiological (hypertension) conditions.
Acknowledgments
We thank Dr. Christopher Wingard and Dr. Erin Mann for assistance with aorta preparation and use of their DMT myograph for the vascular force generation experiments.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
- NMDA
- N-methyl-d-aspartate
- NMDAR
- NMDA receptor
- AP-5
- dl-2-amino-5-phosphonopentanoic acid
- BP
- blood pressure
- CNS
- central nervous system
- DAF-FM
- 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate
- DCF
- 2′,7′-dichlorofluorescein
- HR
- heart rate
- NO
- nitric oxide
- NOS
- NO synthase
- eNOS
- endothelial NOS
- iNOS
- inducible NOS
- nNOS
- neuronal NOS
- l-NAME
- Nω-nitro-l-arginine methyl ester
- l-NIO
- N5-(1-iminoethyl)-l-ornithine
- MAP
- mean arterial pressure
- Nox
- NADPH oxidase
- NOx
- nitrite/nitrate
- NPLA
- Nω-propyl-l-arginine
- PE
- phenylephrine
- R-(+)-HA-966
- R-(+)-3-amino-1-hydroxypyrrolid-2-one
- RFU
- relative fluorescence units
- ROS
- reactive oxygen species.
Authorship Contributions
Participated in research design: McGee and Abdel-Rahman.
Conducted experiments: McGee.
Performed data analysis: McGee.
Wrote or contributed to the writing of the manuscript: McGee and Abdel-Rahman.
References
- Adam F, Gairard AC, Chauvin M, Le Bars D, Guirimand F. (2001) Effects of sufentanil and NMDA antagonists on a C-fibre reflex in the rat. Br J Pharmacol 133:1013–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alderton WK, Cooper CE, Knowles RG. (2001) Nitric oxide synthases: structure, function and inhibition. Biochem J 357:593–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K, Krause KH. (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87:245–313. [DOI] [PubMed] [Google Scholar]
- Bonham AC, Chen CY. (2002) Glutamatergic neural transmission in the nucleus tractus solitarius: N-methyl-d-aspartate receptors. Clin Exp Pharmacol Physiol 29:497–502. [DOI] [PubMed] [Google Scholar]
- Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. (2009) NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci 12:857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C, Edwards A, Sendeski M, Lee-Kwon W, Cui L, Cai CY, Patzak A, Pallone TL. (2010) Intrinsic nitric oxide and superoxide production regulates descending vasa recta contraction. Am J Physiol Renal Physiol 299:F1056–F1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chianca DA, Jr, Lin LH, Dragon DN, Talman WT. (2004) NMDA receptors in nucleus tractus solitarii are linked to soluble guanylate cyclase. Am J Physiol Heart Circ Physiol 286:H1521–H1527. [DOI] [PubMed] [Google Scholar]
- Davies J, Watkins JC. (1972) Is 1-hydroxy-3-aminopyrrolidone-2 (HA-966) a selective excitatory amino-acid antagonist? Nat New Biol 238:61–63. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. (1999) The glutamate receptor ion channels. Pharmacol Rev 51:7–61. [PubMed] [Google Scholar]
- el-Mas MM, Abdel-Rahman AA. (1993) Role of NMDA and non-NMDA receptors in the nucleus tractus solitarius in the depressant effect of ethanol on baroreflexes. J Pharmacol Exp Ther 266:602–610. [PubMed] [Google Scholar]
- El-Mas MM, Fan M, Abdel-Rahman AA. (2009) Facilitation of myocardial PI3K/Akt/nNOS signaling contributes to ethanol-evoked hypotension in female rats. Alcohol Clin Exp Res 33:1158–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mas MM, Zhang J, Abdel-Rahman AA. (2006) Upregulation of vascular inducible nitric oxide synthase mediates the hypotensive effect of ethanol in conscious female rats. J Appl Physiol 100:1011–1018. [DOI] [PubMed] [Google Scholar]
- Förstermann U, Li H. (2011) Therapeutic effect of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. Br J Pharmacol 164:213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forstermann U, Sessa WC. (2012) Nitric oxide synthases: regulation and function. Eur Heart J 33:829–837, 837a–837d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster AC, Kemp JA. (1989) HA-966 antagonizes N-methyl-d-aspartate receptors through a selective interaction with the glycine modulatory site. J Neurosci 9:2191–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girouard H, Wang G, Gallo EF, Anrather J, Zhou P, Pickel VM, Iadecola C. (2009) NMDA receptor activation increases free radical production through nitric oxide and NOX2. J Neurosci 29:2545–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasekar PG, Kanthasamy AG, Borowitz JL, Isom GE. (1995) NMDA receptor activation produces concurrent generation of nitric oxide and reactive oxygen species: implication for cell death. J Neurochem 65:2016–2021. [DOI] [PubMed] [Google Scholar]
- Hao L, Nishimura T, Wo H, Fernandez-Patron C. (2006) Vascular responses to α1-adrenergic receptors in small rat mesenteric arteries depend on mitochondrial reactive oxygen species. Arterioscler Thromb Vasc Biol 26:819–825. [DOI] [PubMed] [Google Scholar]
- Harris GS, Lust RM, Katwa LC, Wingard CJ. (2010) Urotensin II alters vascular reactivity in animals subjected to volume overload. Peptides 31:2075–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Resources (1996) Guide for the Care and Use of Laboratory Animals 7th ed. Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council, Washington, DC. [Google Scholar]
- Jin RC, Loscalzo J. (2010) Vascular nitric oxide: formation and function. J Blood Med 2010:147–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katori T, Donzelli S, Tocchetti CG, Miranda KM, Cormaci G, Thomas DD, Ketner EA, Lee MJ, Mancardi D, Wink DA, et al. (2006) Peroxynitrite and myocardial contractility: in vivo versus in vitro effects. Free Radic Biol Med 41:1606–1618. [DOI] [PubMed] [Google Scholar]
- Kemp JA, Leeson PD. (1993) The glycine site of the NMDA receptor–five years on. Trends Pharmacol Sci 14:20–25. [DOI] [PubMed] [Google Scholar]
- Kubo T, Amano M, Asari T. (1993) N-methyl-d-aspartate receptors but not non-N-methyl-d-aspartate receptors mediate hypertension induced by carotid body chemoreceptor stimulation in the rostral ventrolateral medulla of the rat. Neurosci Lett 164:113–116. [DOI] [PubMed] [Google Scholar]
- Leung JC, Travis BR, Verlander JW, Sandhu SK, Yang SG, Zea AH, Weiner ID, Silverstein DM. (2002) Expression and developmental regulation of the NMDA receptor subunits in the kidney and cardiovascular system. Am J Physiol Regul Integr Comp Physiol 283:R964–R971. [DOI] [PubMed] [Google Scholar]
- McGee MA, Abdel-Rahman AA. (2010) Mechanism of peripheral NMDA receptor-mediated pressor response in conscious rats. FASEB J 24:lb523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillen BA, Joyner PW, Parmar CA, Tyer WE, Williams HL. (2004) Effects of NMDA glutamate receptor antagonist drugs on the volitional consumption of ethanol by a genetic drinking rat. Brain Res Bull 64:279–284. [DOI] [PubMed] [Google Scholar]
- Misko TP, Schilling RJ, Salvemini D, Moore WM, Currie MG. (1993) A fluorometric assay for the measurement of nitrite in biological samples. Anal Biochem 214:11–16. [DOI] [PubMed] [Google Scholar]
- Nassar NN, Li G, Strat AL, Abdel-Rahman AA. (2011) Enhanced hemeoxygenase activity in the rostral ventrolateral medulla mediates exaggerated hemin-evoked hypotension in the spontaneously hypertensive rat. J Pharmacol Exp Ther 339:267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. (2007) Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87:315–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues Dias AC, Talman WT, Colombari E. (2001) Hemodynamic effects elicited by microinjection of glutamatergic agonists into NTS of conscious rats. Am J Physiol Heart Circ Physiol 281:H1026–H1034. [DOI] [PubMed] [Google Scholar]
- Schulz E, Gori T, Münzel T. (2011) Oxidative stress and endothelial dysfunction in hypertension. Hypertens Res 34:665–673. [DOI] [PubMed] [Google Scholar]
- Simandle SA, Kerr BA, Lacza Z, Eckman DM, Busija DW, Bari F. (2005) Piglet pial arteries respond to N-methyl-d-aspartate in vivo but not in vitro. Microvasc Res 70:76–83. [DOI] [PubMed] [Google Scholar]
- Singh L, Donald AE, Foster AC, Hutson PH, Iversen LL, Iversen SD, Kemp JA, Leeson PD, Marshall GR, Oles RJ. (1990) Enantiomers of HA-966 (3-amino-1-hydroxypyrrolid-2-one) exhibit distinct central nervous system effects: (+)-HA-966 is a selective glycine/N-methyl-d-aspartate receptor antagonist, but (−)-HA-966 is a potent gamma-butyrolactone-like sedative. Proc Natl Acad Sci U S A 87:347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitniewska EM, Wiśniewska RJ, Wiśniewski K. (2003) The role of ionotropic receptors of glutaminic acid in cardiovascular system. A. The influence of ionotropic receptor NMDA agonist - 1R,3R-ACPD and antagonist - DL-AP7 on the systemic pressure in rats. Amino Acids 24:397–403. [DOI] [PubMed] [Google Scholar]
- Smith CJ, Santhanam L, Bruning RS, Stanhewicz A, Berkowitz DE, Holowatz LA. (2011) Upregulation of inducible nitric oxide synthase contributes to attenuated cutaneous vasodilation in essential hypertensive humans. Hypertension 58:935–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touyz RM. (2005) Reactive oxygen species as mediators of calcium signaling by angiotensin II: implications in vascular physiology and pathophysiology. Antioxid Redox Signal 7:1302–1314. [DOI] [PubMed] [Google Scholar]
- Tsai MH, Jiang MJ. (2010) Reactive oxygen species are involved in regulating α1-adrenoceptor-activated vascular smooth muscle contraction. J Biomed Sci 17:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartanian MG, Taylor CP. (1991) Different stereoselectivity of the enantiomers of HA-966 (3-amino-1-hydroxy-2-pyrrolidinone) for neuroprotective and anticonvulsant actions in vivo. Neurosci Lett 133:109–112. [DOI] [PubMed] [Google Scholar]
- White RE, Han G, Dimitropoulou C, Zhu S, Miyake K, Fulton D, Dave S, Barman SA. (2005) Estrogen-induced contraction of coronary arteries is mediated by superoxide generated in vascular smooth muscle. Am J Physiol Heart Circ Physiol 289:H1468–H1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Jung KJ, Kim JW, Yu BP, Chung HY. (2004) Alteration of soluble adhesion molecules during aging and their modulation by calorie restriction. FASEB J 18:320–322. [DOI] [PubMed] [Google Scholar]