

Abstract
Adenosine (Ado) is released in response to tissue injury, promotes hyperemia, and modulates inflammation. The proinflammatory effects of Ado, which are mediated by the A2B Ado receptor (AdoR), may exacerbate tissue damage. We hypothesized that selective blockade of the A2B AdoR with 3-ethyl-1-propyl-8-(1-(3-trifluoromethylbenzyl)-1H-pyrazol-4-yl)-3,7-dihydropurine-2,6-dione (GS-6201) during acute myocardial infarction (AMI) would reduce adverse cardiac remodeling. Male ICR mice underwent coronary artery ligation or sham surgery (n = 10–12 per group). The selective A2B AdoR antagonist GS-6201 (4 mg/kg) was given intraperitoneally twice daily starting immediately after surgery and continuing for 14 days. Transthoracic echocardiography was performed before surgery and after 7, 14, and 28 days. A subgroup of mice was killed 72 h after surgery, and the activity of caspase-1, a key proinflammatory mediator, was measured in the cardiac tissue. All sham-operated mice were alive at 4 weeks, whereas 50% of vehicle-treated mice and 75% of GS-6201-treated mice were alive at 4 weeks after surgery. Compared with vehicle, treatment with GS-6201 prevented caspase-1 activation in the heart at 72 h after AMI (P < 0.001) and significantly limited the increase in left ventricular (LV) end-diastolic diameter by 40% (P < 0.001), the decrease in LV ejection fraction by 18% (P < 0.01) and the changes in the myocardial performance index by 88% (P < 0.001) at 28 days after AMI. Selective blockade of A2B AdoR with GS-6201 reduces caspase-1 activity in the heart and leads to a more favorable cardiac remodeling after AMI in the mouse.
Introduction
Acute myocardial infarction (AMI) is characterized by ischemic necrosis of the myocardium followed by an intense inflammatory response (Frantz et al., 2009). The extent of the initial loss of viable myocardium and the intensity of the inflammatory response both are independent predictors of the ensuing cardiac remodeling characterized by cardiac enlargement and dysfunction (Abbate et al. 2003; Kelle et al., 2009).
Adenosine (Ado) is a ubiquitous small molecule released in response to tissue injury that promotes hyperemia and modulates inflammation through interaction with one of four types of cell surface membrane receptors (Gessi et al., 2011). The proinflammatory effects of Ado, which are mediated by the A2B Ado receptor (AdoR), may cause further tissue damage (Feoktistov and Biaggioni, 2011). In some models of inflammation blockade of the A2B AdoR has limited the intensity of the inflammatory response and improved healing, whereas in others A2B AdoR signaling seemed to inhibit inflammation (Mustafa et al., 2007; Eckle et al., 2008; Feoktistov and Biaggioni, 2011).
Caspase-1 is a key proinflammatory mediator representing the effector enzyme of the inflammasome, a macromolecular structure formed during tissue injury (Franchi et al., 2009). Caspase-1 is responsible for the amplification of the inflammatory responses by processing and releasing active interleukin (IL)-1β, which largely amplifies the inflammatory response by inducing the synthesis and release of numerous cytokines and adhesion molecules (Franchi et al., 2009; Dinarello, 2011). In mouse models of AMI or heart failure, caspase-1 acts as a contributor to further damage (Frantz et al., 2003; Merkle et al., 2007; Mezzaroma et al., 2011).
The current study was designed to investigate whether selective blockade of the A2B AdoR during acute myocardial infarction would reduce the inflammatory response induced by tissue injury, inhibit caspase-1, and reduce the adverse cardiac remodeling in a mouse model of severe ischemic cardiomyopathy caused by nonreperfused AMI. To validate A2B AdoR as the target for intervention, we used targeted silencing RNA given systemically.
Materials and Methods
Experimental AMI Model.
Adult out-bred male CD1 mice (8–12 weeks of age) were supplied by Harlan (Indianapolis, IN). The experiments were conducted under the guidelines for laboratory animals for biomedical research published by the National Institutes of Health (Institute of Laboratory Animal Resources, 1996). The study protocol was approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee. Experimental AMI was induced by permanent coronary artery ligation to induce a large nonreperfused infarct involving approximately 30% of the left ventricle and leading to an ischemic dilated cardiomyopathy (Abbate et al., 2008c; Mezzaroma et al., 2011). In brief, mice were orotracheally intubated under anesthesia (pentobarbital 50–70 mg/kg), placed in the right lateral decubitus position, then subjected to a left thoracotomy, pericardiectomy, and ligation of the proximal left coronary artery. The chest was closed, and the animals were allowed to recover. The mice surviving surgery were randomly assigned to the different groups of treatment (n = 6–15 per group). Sham operations were performed wherein animals underwent the same surgical procedure without coronary artery ligation (n = 4–8 per group). A timeline of the protocol of the study is shown in Fig. 1.
Fig. 1.

Timeline of the study protocol.
Treatment.
The A2B AdoR antagonist [3-ethyl-1-propyl-8-(1-(3-trifluoromethylbenzyl)-1H-pyrazol-4-yl)-3,7-dihydropurine-2,6-dione (GS-6201), also known as CVT-6883] (Sun et al., 2006; Karmouty-Quintana et al., 2012) was obtained from Gilead Sciences, Foster City, CA. GS-6201 is a high-affinity, selective A2B AdoR antagonist and a specific adenosine antagonist (Sun et al., 2006). GS-6201 has a calculated KB value (binding potency) of 2.2 ± 0.8 nM, a KI value (affinity) for the A2B AdoR of 8.3 ± 2.6 nM, and >500-fold higher affinity for A2B AdoR than for A1, A2A, and A3 AdoRs (Sun et al., 2006). GS-6201 (10 μM) had no significant effects on other common receptors, ion channels, transporters, and enzymes (Sun et al., 2006; Karmouty-Quintana et al., 2012). Mice were randomly assigned to treatment with GS-6201 (4 mg/kg) or a matching dose of vehicle administered intraperitoneally (final volume 0.13 ml) every 12 h for 14 days starting immediately after coronary artery ligation surgery. This dose was chosen to achieve similar exposure (1 nM plasma concentration of unbound GS-6201) as previous studies in other mouse models of inflammatory injury while taking into consideration differences in formulation and mouse strain (Sun et al., 2006; Karmouty-Quintana et al., 2012). An additional group of mice received a lower dose of GS-6201 (2 mg/kg) to explore a dose-response relationship. Two additional groups of mice received GS-6201 (4 mg/kg) starting 1 or 12 h after surgery to simulate a clinical scenario in which drug treatment may occur with some delay after AMI. An additional group of mice was treated with 0.13 ml of 0.9% NaCl as an additional control; however, because the data of vehicle treatment were not significantly different from those of NaCl treatment, only the results of vehicle treatment are reported here.
Caspase-1 Activation.
An additional subset of mice was sacrificed 72 h after surgery (n = 4–6 per treatment group). The heart was removed as a whole, and the pericardium and atria were removed. The remaining left and right ventricles were processed. The tissue activity of caspase-1 was determined by cleavage of a fluorogenic substrate (CaspACE; Promega, Madison, WI) (Abbate et al., 2008c). After homogenization using radioimmunoprecipitation assay buffer (Sigma-Aldrich, St. Louis, MO) containing a cocktail of protease inhibitors (Sigma-Aldrich) and centrifugation at 16,000 rpm for 20 min, 75 μg of protein from each sample were used for the assay according to the supplier's instructions. Fluorescence was measured after 60 min and expressed as arbitrary fluorescence units produced by one microgram of sample per minute (fluorescence/μg/min) and calculated as fold change compared with the caspase-1 activity in homogenates of the hearts of sham-operated mice.
Inflammatory Infiltrate.
To quantify the inflammatory infiltrate in the heart during AMI, we measured CD45 expression (a marker for leukocytes) in the heart by using Western blot. The hearts collected at 72 h after AMI were homogenized in radioimmunoprecipitation assay buffer (Sigma-Aldrich) supplemented with a protease inhibitor cocktail (Sigma-Aldrich) and centrifuged at 16,200g for 20 min. Thirty micrograms of each sample were diluted in Laemmli buffer, denatured for 10 min at 96°C, and resolved with SDS/polyacrylamide gel electrophoresis by using an 8% acrylamide gel to allow protein separation. The proteins were transferred onto a nitrocellulose membrane. After saturation with 5% milk in phosphate-buffered saline the membrane was incubated with a rat anti-mouse antibody raised against CD45 (R&D Systems, Minneapolis, MN). To normalize the protein loading a monoclonal antibody for β-actin (Sigma-Aldrich) was used. Enhanced chemiluminescence assay and autoradiography were used to detect the bands corresponding to CD45 and β-actin. The band intensity was determined by densitometric analysis using Scion Image software (Scion Corporation, Frederick, MD), and the results were expressed as percentage increase in intensity compared with the control sham samples.
Measurement of Circulating Levels of Cytokines and Soluble Adhesion Molecules.
The plasma concentrations of IL-1β, IL-6, tumor necrosis factor-α (TNF-α), and soluble adhesion molecules [E-selectin, intercellular adhesion molecule-1 (ICAM-1) and vascular cellular adhesion molecule (VCAM)] that are induced by IL-1β, were determined at day 28 after surgery by using Luminex kits obtained from Millipore Corporation (Billerica, MA) according to the manufacturer's instructions. A blood sample was obtained via a direct cardiac puncture immediately before killing the animals.
Echocardiography.
All mice underwent transthoracic echocardiography at baseline (before surgery) and at 7, 14, and 28 days after surgery (before sacrifice). Echocardiography was performed by using the Vevo770 imaging system (VisualSonics Inc., Toronto, Ontario, Canada) with a 30-MHz probe. The heart was visualized in bidimensional mode (B-mode) from parasternal short axis and apical views. We measured the left ventricular (LV) end-diastolic and end-systolic areas at B-mode and the LV end-diastolic diameter (LVEDD), LV end-systolic diameters (LVESD), LV anterior wall diastolic thickness (LVAWDT), and LV posterior wall diastolic thickness (LVPWDT) at monodimensional mode (M-mode), as described previously (Abbate et al., 2008c; Toldo et al., 2011) and according to the American Society of Echocardiography recommendations (Gardin et al., 2002). LV fractional shortening, LV ejection fraction (LVEF), and LV mass and eccentricity (LVEDD/LVPWDT ratio) were calculated (Gardin et al., 2002; Abbate et al., 2008c; Toldo et al., 2011). The transmitral and left ventricular outflow tract Doppler spectra were recorded from apical four-chamber views, and the myocardial performance index (MPI or Tei index) was calculated as the ratio of the isovolumetric contraction and relaxation time divided by the ejection time (Tei et al., 1995). LV stroke volume was calculated by using the velocity-time integral of the LV outflow tract flow multiplied by the LV outflow tract area, and cardiac output was calculated by multiplying LV stroke volume by the heart rate (HR) (Gardin et al., 2002; Abbate et al., 2008c; Toldo et al., 2011). Right ventricular (RV) enlargement was assessed by measuring the RV end-diastolic area in the parasternal short-axis view midventricular section, and RV systolic function was estimated by using M-mode and measuring the tricuspid annular plane systolic excursion (TAPSE) (Gardin et al., 2002; Toldo et al., 2011). The investigator performing and reading the echocardiogram was blinded to the treatment allocation.
Infarct Size Assessment.
After the 28-day echocardiogram, all mice were killed with a pentobarbital overdose and/or cervical dislocation. The hearts were explanted and fixed in formalin 10% for at least 48 h. A transverse section of the median third of the heart was dissected, included in paraffin, cut into 5-μm slides, and stained with Masson's trichrome (Sigma-Aldrich) (Abbate et al., 2008c). The areas of fibrosis and the whole left ventricle were determined by using computer morphometry with Image Pro Plus 6.0 software (Media Cybernetics, Inc., Bethesda, MD).
Hemodynamic Measurements.
In a subgroup of mice (n = 4 per each group) the LV apex was punctured 1 h after surgery, and a Millar catheter connected to a pressure transducer was inserted to measure LV peak systolic pressure and heart rate (Toldo et al., 2011).
A2B AdoR-Targeted Silencing RNA for Validation of the Target.
To confirm that the effects seen with GS-6201 were caused by inhibition of the A2B AdoR and not off-target effects, we used a previously validated approach of targeted silencing RNA in a subgroup of mice and measured its effects on caspase-1 activity in the heart and on cardiac enlargement and dysfunction (see Supplemental Methods) (Mezzaroma et al., 2011).
Statistical Analysis.
Data are presented as mean and S.E.M. for continuous variables and as number and percentage for the other variables. Differences between the three groups were analyzed by using one-way analysis of variance followed by Bonferroni-corrected Student's t test to compare two of the three groups. Changes in repeated measures of echocardiographic data were analyzed by using the random effects analysis of variance for repeated measures to determine the main effect of time, group, and time-by-group interaction. Survival analysis was performed by generating a Kaplan-Meyer survival curve and using logistic regression analysis. Calculations were completed by using the SPSS 15.0 package for Windows (SPSS Inc., Chicago, IL).
Results
GS-6201 Had No Hemodynamic Effects During Acute Myocardial Infarction.
Because Ado is a vasodilator, and to exclude that a difference in remodeling was caused by hemodynamic changes secondary to A2B AdoR antagonism using GS-6201, we measured left ventricular peak systolic pressure (LVPSP) and HR in mice treated with GS-6201 (4 m/kg) and those treated with vehicle. LVPSP was significantly reduced 1 h after coronary artery ligation, but was unaffected by treatment (Table 1).
TABLE 1.
Gross and hemodynamic data
Hemodynamic data were recorded 1 h after surgery.
| Group |
|||
|---|---|---|---|
| Sham | A2B AdoR Antagonist MI | Vehicle MI | |
| Age, weeks | 11 ± 1 | 12 ± 1 | 11 ± 1 |
| Weight, g | 32 ± 1 | 34 ± 1 | 32 ± 1 |
| LVPSP, mm Hg | 99 ± 3 | 57 ± 9* | 58 ± 8* |
| HR, min | 417 ± 13 | 428 ± 32 | 434 ± 18 |
, P < 0.001 vs. sham.
GS-6201 Inhibits Caspase-1 Activation and Inflammation.
Caspase-1 activation is part of a key proinflammatory mechanism in response to ischemic injury (Frantz et al., 2003; Merkle et al., 2007; Franchi et al., 2009; Dinarello, 2011; Mezzaroma et al., 2011). Treatment with GS-6201 (4 mg/kg) every 12 h prevented caspase-1 activation in the heart during AMI (Fig. 2). The intensity of the leukocyte (CD45+) infiltrate, measured as CD45 expression by Western blot, was also significantly reduced by treatment 72 h after AMI (Fig. 2). Caspase-1 activation results in the processing and release of active IL-1β that is usually present at very low tissue concentration and rapidly amplifies the inflammatory response by inducing the expression of secondary cytokines and adhesion molecules. IL-1β plasma levels were undetectable in all but two mice with AMI, whereas plasma levels of secondary cytokines (i.e., IL-6) were increased 28 days after AMI (Fig. 3). Treatment with GS-6201 (4 mg/kg) every 12 h for 14 days significantly reduced IL-6, TNF-α, E-selectin, ICAM-1, and VCAM plasma levels (Fig. 3).
Fig. 2.

Administration of GS-6201, given immediately after coronary ligation (4 mg/kg i.p. twice daily), prevented caspase-1 activation in the heart tissue measured 72 h after surgery and significantly inhibited leukocyte (CD45+) recruitment in the heart. *, P < 0.001 versus sham; #, P < 0.01 versus vehicle (n = 4–6 per group).
Fig. 3.

Treatment with GS-6201 (4 mg/kg) every 12 h for 14 days led to a significant reduction in plasma levels of inflammatory markers associated with AMI. Plasma concentrations of IL-6, TNF-α, soluble E-selectin (sE-selectin), soluble ICAM (sICAM), and soluble VCAM (sVCAM) were measured by using Luminex at day 28 after surgery. +, P < 0.05 versus vehicle; *, P < 0.001 versus sham.
Effects of A2B AdoR Antagonism with GS-6201 on Survival after Coronary Artery Ligation Surgery.
None of the sham-operated mice died. Half of the vehicle-treated mice (50%) survived to 28 days after coronary artery ligation surgery (P < 0.001 versus sham), whereas 75% of mice treated with GS-6201 were alive (P = 0.14) (Fig. 4).
Fig. 4.

Survival curves after AMI surgery: half of the vehicle-treated mice (50%) survived to 28 days after AMI surgery (P < 0.001 versus sham), whereas 75% of mice treated with GS-6201 were alive (P = 0.14).
Effects of GS-6201 on Cardiac Remodeling.
Cardiac remodeling was measured noninvasively by using transthoracic echocardiography. Examples of B-mode and M-mode recordings are shown in Fig. 5. Administration of GS-6201 (4 mg/kg) every 12 h starting at the time of surgery led to a significant attenuation of left and right ventricular enlargement and dysfunction at 7 days, which was maintained at 14 days and also at 28 days, 14 days after the last dose of drug was given (Fig. 6). At 28 days, LV enlargement after AMI was reduced by approximately 40% by GS-6201. LV systolic function was also significantly greater in the GS-6201 group (absolute difference in mean LVEF of 5%). The attenuation in cardiac remodeling was paralleled by preservation of myocardial diastolic/systolic performance (myocardial performance index) (Fig. 6). The hearts of mice treated with GS-6201 also showed less right ventricular enlargement and dysfunction (Fig. 6).
Fig. 5.

Examples of B-mode and M-mode echocardiographic images from a mouse treated with vehicle (top) and one treated with GS-6201 (4 mg/kg) every 12 h (bottom) 2 weeks after acute myocardial infarction surgery.
Fig. 6.
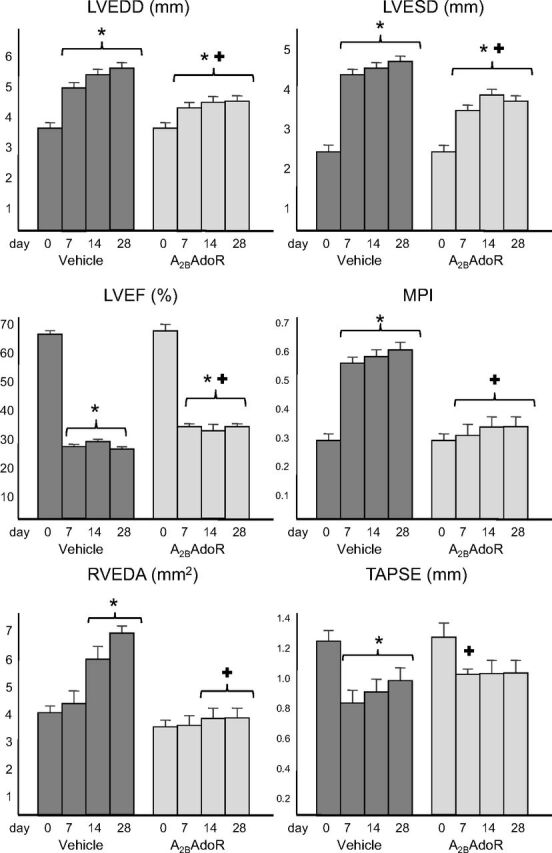
After coronary artery ligation surgery, vehicle-treated mice had a significant enlargement of the left and right ventricles [LVEDD, LVESD, and right ventricular end-diastolic area (RVEDA)] and a significant reduction in left and right ventricular function (LVEF, MPI, and TAPSE). Treatment with GS-6201 (4 mg i.p. twice daily for 14 days) significantly limited the cardiac enlargement and dysfunction. *, P < 0.001 versus baseline (or sham); +, P < 0.001 versus vehicle.
GS-6201 treatment led to a slightly thicker infarct scar (LVAWDT) thickness versus vehicle-treated mice and a similar degree of LV hypertrophy (LV mass) but no signs of eccentric dilatation (Fig. 7).
Fig. 7.

Compared with vehicle-treated mice, mice treated with GS-6201 (4 mg/kg every 12 h for 14 days) had a thicker infarct scar and a similar left ventricular mass but a significant difference in the degree of eccentricity (LVEDD/LVPWDT ratio) reflecting concentric versus eccentric hypertrophy, 4 weeks after acute myocardial infarction surgery. *, P < 0.05 versus sham; +, P < 0.05 versus vehicle.
Cardiac output was preserved in AMI mice compared with sham-operated mice, without significant differences between the two treatment groups (Table 2).
TABLE 2.
Doppler-derived left-ventricular stroke volume and cardiac output
Doppler echocardiography was completed 28 days after surgery.
| Group |
|||
|---|---|---|---|
| Sham | A2B AdoR Antagonist MI | Vehicle MI | |
| HR, min | 378 ± 23 | 370 ± 38 | 360 ± 32 |
| LV stroke volume, μl | 37 ± 3 | 27 ± 4* | 25 ± 4* |
| Cardiac output, ml/min | 14 ± 5 | 10 ± 4 | 9 ± 4 |
, P < 0.001 vs. sham.
The benefits of GS-6201 seemed to be dose-dependent, because a lower dose (2 mg/kg given every 12 h for 14 days) provided a smaller effect on cardiac enlargement (Fig. 8).
Fig. 8.

Treatment with GS-6201 at two different doses (2 or 4 mg i.p. twice daily) caused a dose-dependent reduction of the left and right ventricles (LVEDD) and preservation of left ventricular function (LVEF and MPI) 2 weeks after acute myocardial infarction surgery. +, P < 0.05 versus vehicle; *, P < 0.001 versus vehicle.
The Effects of A2B AdoR Antagonism with GS-6201 on Cardiac Remodeling Are Not Caused by a Reduction in Infarct Size.
At 28 days, the infarct scar measured at the midventricular section involved approximately 17% of the LV area and 30% of the LV circumference and was not significantly reduced by treatment with the A2B AdoR antagonist GS-6201 (4 mg/kg) every 12 h for 14 days (Fig. 9).
Fig. 9.
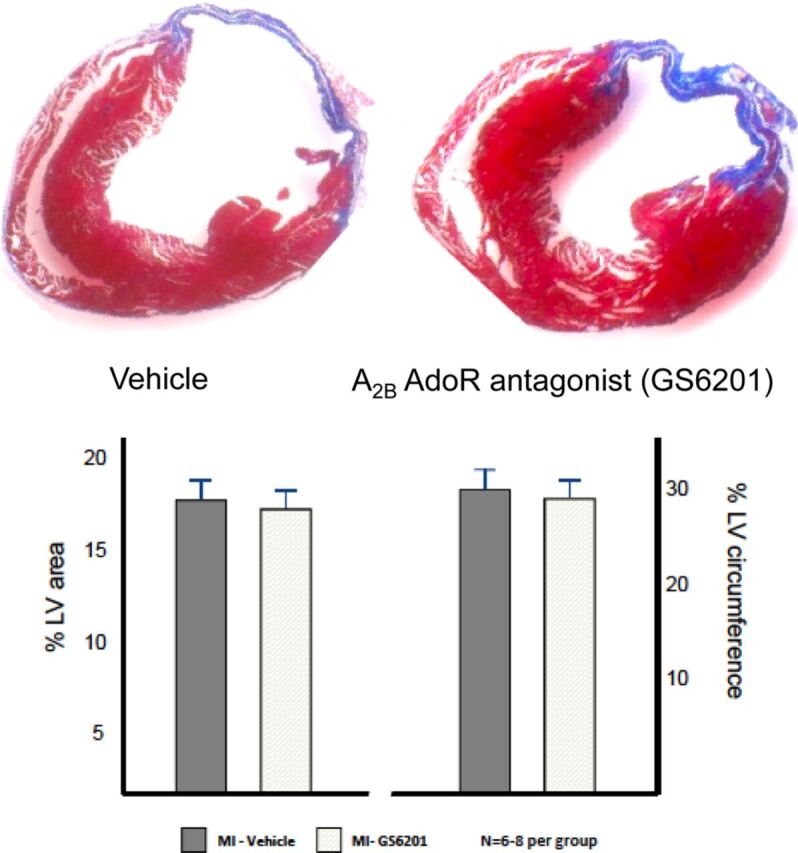
Masson trichrome-stained sections show infarct scar (blue) in vehicle-treated mice and mice treated with GS-6201 (4 mg/kg i.p. every 12 h for 14 days). The graph shows the mean values of the quantifications of the infarct size expressed as percentage of the area of the left ventricle section or as percentage of the circumference of the left ventricle.
Treatment Delay with GS-6201 and Effects on Cardiac Remodeling.
To simulate a clinically relevant scenario in which drug treatment may occur with some delay after AMI, we compared treatment with GS-6201 (4 mg/kg) every 12 h for 14 days, without delay with other groups in which GS-6201 was given with 1 or 12 h of delay. The treatment with a 1-h delay provided similar improvement in cardiac remodeling as no delay (Fig. 10). However, the treatment with a 12-h delay showed only minimal effects on adverse cardiac remodeling.
Fig. 10.

Treatment with GS-6201 (4 mg i.p. twice daily for 14 days) with a delay after AMI led to a time-dependent loss of effect on the reduction of the left and right ventricles (LVEDD and RVEDA) and on preservation of left ventricular function (LVEF and MPI) 4 weeks after AMI. *, P < 0.001 versus vehicle.
A2B AdoR-Targeted siRNA Inhibits Caspase-1 Activation and Limits Cardiac Dilatation and Dysfunction.
Treatment with A2B AdoR-targeted siRNA, but not scrambled siRNA, significantly inhibited caspase-1 activity in the heart during acute myocardial infarction and significantly limited cardiac enlargement and dysfunction measured 7 days after AMI (see Supplemental Results; Supplemental Fig. 1).
Discussion
Myocardial ischemia and the accompanying cellular injury are known to cause the release of cell contents, triggering a sterile inflammatory response promoting further dysfunction and heart failure (Frantz et al., 2009). This study shows for the first time that inhibition of Ado binding to the A2BAdoR with GS-6201 limits the inflammatory response and leads to a more favorable cardiac remodeling.
Ado is indeed rapidly released during tissue hypoxia and ischemia and binds rapidly to ubiquitous specific G protein-coupled receptors (AdoRs) (Gessi et al., 2011). There are four subtypes of AdoR: A1AdoR, primarily expressed in the heart, regulates electrical conduction; A3AdoR is expressed in rodent mast cell and regulates activation and degranulation in the mouse; and A2AAdoR and A2BAdoR have been linked to vascular tone and inflammation (Zhong et al., 2003; Feoktistov and Biaggioni, 2011; Gessi et al., 2011). A2AAdoRs are high-affinity AdoRs expressed on the membrane of various cell types including endothelial cells, leukocytes, and cardiomyocytes (Belardinelli et al., 1996; Zhong et al., 2003; Feoktistov and Biaggioni, 2011; Gessi et al., 2011). A2BAdoR are low-affinity receptors sometimes coexpressed in the same cells expressing A2AAdoR but in a low number and are therefore considered to be minimally relevant to Ado signaling in these cells, at least in unstressed conditions (Aherne et al., 2011; Feoktistov and Biaggioni, 2011). Whereas both A2AAdoR and A2BAdoR G protein-coupled receptors may signal through adenyl cyclase, A2BAdoR also signals through phospholipase C and the small GTP-binding protein p21ras, which is involved in inflammatory signaling involving the p38 mitogen-activated phosphokinase and the extracellular signal-regulated kinases (Feoktistov et al., 1999; Grant, et al. 2001; Schulte and Fredholm, 2003; Aherne et al., 2011; Feoktistov and Biaggioni, 2011). It is noteworthy that the expression of the A2BAdoR depends on stabilization of hypoxia inducible factor-1α and hence is highly sensitive to hypoxia and inflammation (Grant et al., 2001; Aherne et al., 2011; Feoktistov and Biaggioni, 2011; Koeppen et al., 2011).
Therefore, although in unstressed conditions Ado signaling through the A2BAdoR is probably not significant, in the context of tissue injury the A2BAdoR may play a significant role.
We hereby show that selective blockade of the A2BAdoR using GS-6201 blunted the inflammatory response during the infarction as reflected by a nearly complete reduction in caspase-1 activity, a significant reduction in infiltrating inflammatory cells early in the course of AMI, and a significant reduction in plasma cytokine and adhesion molecules 28 days after AMI. Caspase-1 is the enzymatically active component of the inflammasome, a macromolecular structure functioning as a “danger” sensor and involved in the processing of mature IL-1β and cell death (Franchi et al., 2009; Dinarello, 2011). Formation of the inflammasome and activation of caspase-1 in the heart leads to heart failure (Mezzaroma et al., 2011). The significant reduction in caspase-1 activity in the heart with selective blockade of A2BAdoR is in agreement with the proinflammatory signaling of the A2BAdoR (Feoktistov and Biaggioni, 2011).
Extracellular Ado, which functions as a signal for tissue injury, represents only one of the many potential triggers for sterile inflammation. Indeed, although we found a significant reduction in caspase-1 to near-normal levels, this determination was made at a single time point (72 h) and may not reflect complete inhibition of caspase-1 activity in the heart. It is noteworthy that an inflammatory response (measured as CD45 content) was evident in both groups, reflecting that A2BAdoR blockade with GS-6201 mitigates rather than eliminates the tissue inflammatory response during AMI, and suggests that other potential triggers contribute to the inflammatory response.
The role of A2BAdoR in myocardial ischemia is controversial (Aherne et al., 2011). In models of acute myocardial injury caused by ischemia/reperfusion, Ado consistently reproduces the beneficial effects of ischemic preconditioning (Feoktistov and Biaggioni, 2011; Gessi et al., 2011). The preconditioning-like effects of Ado are eliminated when signaling through the A2BAdoR is disrupted (Philipp et al., 2006; Wakeno et al., 2006). However, blockade of the A2BAdoR in the absence of ischemic preconditioning had no effects on the heart in such models. This suggests to us that although it may mediate some aspects of preconditioning A2BAdoR signaling is not inherently protective and unlikely to be the sole mediator of preconditioning.
In our mouse model of large nonreperfused myocardial infarction, A2BAdoR antagonism significantly limited left ventricular enlargement and systolic and diastolic dysfunction. The protective effects of the A2BAdoR were independent of an effect on infarct size. This is in agreement with the fact that in nonreperfused myocardial infarction in the rat adenosine has no effect on infarct size (Wakeno et al., 2006), but in apparent contrast with the report that the use of a A2BAdoR blocker [N-(4-cyanophenyl)-2-(4-(2,3,6,7-tetrahydro-2,6-dioxo-1,3-dipropyl-1H-purin-8-yl)-phenoxy) acetamide (MRS1754)] given with the adenosine agonist (2-chloro-adenosine) provided no benefit versus vehicle and eliminated the benefits of 2-chloro-adenosine alone (Wakeno et al., 2006). Unfortunately, there were no experiments in which the A2BAdoR blocker (MRS1754) was given in the absence of the adenosine agonist; therefore, the effects on A2BAdoR blockade on cardiac remodeling were not independently assessed (Wakeno et al., 2006). Moreover, although the intensity of the infiltrate seemed to be less, A2BAdoR blockade did not abolish recruitment of inflammatory cells in the infarct or prevent infarct scar formation, thus avoiding issues seen with other anti-inflammatory treatments.
A2BAdoR blockade limited left ventricular enlargement and dysfunction measured at M-mode echocardiography. A2BAdoR blockade also prevented changes in the MPI (Tei et al., 1995), reflecting preserved diastolic and systolic function, and in RV enlargement (Toldo et al., 2011), reflecting prevention of global remodeling measured at Doppler and B-mode echocardiography. Changes in cardiac structure and function assessed by using B-mode or Doppler in the mouse may, however, be affected by limited spatial and temporal resolution and an inability to fully correct for alignment.
Although most of the patients with AMI receive some form of intervention aimed at obtaining reperfusion, we chose the model of nonreperfused AMI, because incomplete myocardial reperfusion (no reflow) occurs in a large number of patients, which limits the benefit of reperfusion and is associated with a greater risk of subsequent heart failure (Abbate et al., 2008b).
In this model of nonreperfused myocardial infarction, mortality is high, and cardiac remodeling is global and involves the infarct and border zones as well as the unaffected remote left ventricle and the right ventricle (Abbate et al., 2008a; Toldo et al., 2011). Blockade of A2BAdoR seemed to have no effect on the infarct size, whereas A2BAdoR blockade may have prevented thinning and stretching of the infarct and border zones (infarct expansion) and protected the border zone and the remote myocardium, as evidenced by an improvement in both left and right ventricular dimensions and, importantly, LV function. This is in line with the benefits seen with numerous anti-inflammatory strategies that target caspase-1, the inflammasome, and IL-1β (Abbate et al., 2008c, 2010; Van Tassell et al., 2010a,b; Mezzaroma et al., 2011). The lack of detectable plasma IL-1β levels is not surprising because IL-1β is present in very low levels and has a very short half-life (Dinarello, 2011). Enhanced IL-1β activity is, however, reflected in the increased levels of IL-6, TNF-α, E-selectin, ICAM-1, and VCAM, which all are induced by IL-1β (Ikejima et al., 1990; Tosato and Jones, 1990; Tamaru et al., 1998; Dinarello, 2011).
In agreement with the fact that during myocardial ischemia and cellular injury adenosine is rapidly released and bound to cell surface receptors, we found that a delay of 12 h in the administration of the A2BAdoR antagonist significantly limited the benefits of A2BAdoR blockade on cardiac remodeling. Treatment with 12 h of delay, however, still showed protective effects in terms of preserved MPI, and we therefore cannot exclude that with longer treatment and/or follow-up this strategy may ultimately prove useful in ameliorating cardiac remodeling after AMI.
The finding of reduced caspase-1 activity in the heart and more favorable cardiac remodeling confirms the central role of caspase-1 in the myocardial response to myocardial ischemia. Overexpression of caspase-1 in the mouse heart leads to larger areas of ischemic damage, more severe cardiac enlargement, and a reduced survival after AMI; whereas caspase-1-deficient mice are protected after AMI (Frantz et al., 2003; Merkle et al., 2007; Mezzaroma et al., 2011).
GS-6201 was also associated with a numerically lower mortality compared with vehicle; however, this difference did not reach statistical difference probably because the study was not designed or powered to study the effects of GS-6201 on survival.
Our study is, however, limited because a direct cause-effect relationship between A2BAdoR antagonism and caspase-1 inhibition was not demonstrated. A2BAdoR antagonism could affect the expression of adhesion molecules on cardiac microvascular endothelial cells and hence reduce recruitment of inflammatory cells (Ryzhov et al., 2008). In our data, however, the effects of A2BAdoR antagonism on caspase-1 activity were out of proportion to the effects seen on leukocyte recruitment, suggesting that the inhibition of caspase-1 activity is not only an effect of reduced recruitment of leukocytes. However, because no data regarding the components of the leukocyte infiltrate were obtained, we cannot analyze whether A2BAdoR antagonism qualitatively affected the infiltrate. Moreover, A2BAdoR signaling may additionally affect cardiac remodeling independently of caspase-1: adenosine stimulates macrophages to produce osteopontin (Schneider et al., 2010) and release of interleukin-6, which induces differentiation of fibroblasts into myofibroblasts (Zhong et al., 2005), potentially affecting tissue remodeling and progression toward heart failure. In addition, although unlikely considering the selectivity of the GS-6201 (Sun et al., 2006), we cannot exclude that GS-6201 affected cardiac remodeling by blocking different AdoRs other than the A2BAdoR or even with an additional hitherto undetermined mechanism. This event is, however, even more unlikely, because the beneficial effects seen with GS-6201 were also seen with a different strategy aimed at reducing A2B AdoR expression using targeted siRNA (see Supplemental Information). Although not without technical limitations the siRNA approach offers complementary evidence for a proinflammatory and deleterious role of Ado and the A2BAdoR in infarct healing after myocardial ischemia. From a translational point of view, we used a clinically relevant treatment plan in which the animals were not pretreated and drug was administered after the onset of ischemia. Moreover, the effects of treatment were measured by using clinically relevant endpoints such as left ventricular enlargement and function. Finally, we treated the animals for a short period (2 weeks) and followed them for an additional period showing no evidence of rebound after withdrawal of treatment.
In conclusion, selective blockade of A2B AdoR with GS-6201 administered after the onset of ischemia reduces caspase-1 activity in the heart and leads to a more favorable cardiac remodeling after AMI in a mouse model of nonreperfused myocardial infarction.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
- AMI
- acute myocardial infarction
- Ado
- adenosine
- AdoR
- Ado receptor
- Ant
- antagonist
- B-mode
- bidimensional mode
- ECL
- enhanced chemiluminescence
- GS-6201
- 3-ethyl-1-propyl-8-(1-(3-trifluoromethylbenzyl)-1H-pyrazol-4-yl)-3,7-dihydropurine-2,6-dione
- HR
- heart rate
- ICAM-1
- intercellular adhesion molecule-1
- IL
- interleukin
- LV
- left ventricular
- LVAWDT
- LV anterior wall diastolic thickness
- LVEDD
- LV end diastolic diameter
- LVEF
- LV ejection fraction
- LVESD
- LV end systolic diameter
- LVPSP
- LV peak systolic pressure
- LVPWDT
- LV posterior wall diastolic thickness
- MI
- myocardial infarction
- M-mode
- monodimensional mode
- MPI
- myocardial performance index
- MRS1754
- N-(4-cyanophenyl)-2-(4-(2,3,6,7-tetrahydro-2,6-dioxo-1,3-dipropyl-1H-purin-8-yl)-phenoxy)acetamide
- RV
- right ventricular
- RVEDA
- RV end-diastolic area
- TAPSE
- tricuspid annular plane systolic excursion
- TNF-α
- tumor necrosis factor-α
- VCAM
- vascular cellular adhesion molecule.
Authorship Contributions
Participated in research design: Toldo, Zhong, Mezzaroma, Van Tassell, Zeng, and Abbate.
Conducted experiments: Toldo, Zhong, Mezzaroma, Kannan, and Abbate.
Wrote or contributed to the writing of the manuscript: Toldo, Zhong, Mezzaroma, Van Tassell, Kannan, Zeng, Belardinelli, Voelkel, and Abbate.
References
- Abbate A, Biondi-Zoccai GG, Brugaletta S, Liuzzo G, Biasucci LM. (2003) C-reactive protein and other inflammatory biomarkers as predictors of outcome following acute coronary syndromes. Semin Vasc Med 3:375–384. [DOI] [PubMed] [Google Scholar]
- Abbate A, Bussani R, Sinagra G, Barresi E, Pivetta A, Perkan A, Hoke NH, Salloum FN, Kontos MC, Biondi-Zoccai GG, et al. (2008a) Right ventricular cardiomyocyte apoptosis in patients with acute myocardial infarction of the left ventricular wall. Am J Cardiol 102:658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbate A, Kontos MC, Biondi-Zoccai GG. (2008b) No-reflow: the next challenge in treatment of ST-elevation acute myocardial infarction. Eur Heart J 29:1795–1797. [DOI] [PubMed] [Google Scholar]
- Abbate A, Salloum FN, Vecile E, Das A, Hoke NN, Straino S, Biondi-Zoccai GG, Houser JE, Qureshi IZ, Ownby ED, et al. (2008c) Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation 117:2670–2683. [DOI] [PubMed] [Google Scholar]
- Abbate A, Van Tassell BW, Seropian IM, Toldo S, Robati R, Varma A, Salloum FN, Smithson L, Dinarello CA. (2010) Interleukin-1β modulation using a genetically engineered antibody prevents adverse cardiac remodeling following acute myocardial infarction in the mouse. Eur J Heart Fail 12:319–322. [DOI] [PubMed] [Google Scholar]
- Aherne CM, Kewley EM, Eltzschig HK. (2011) The resurgence of A2B adenosine receptor signaling. Biochim Biophys Acta 1808:1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belardinelli L, Shryock JC, Ruble J, Monopoli A, Dionisotti S, Ongini E, Dennis DM, Baker SP. (1996) Binding of the novel nonxanthine A2A adenosine receptor antagonist [3H]SCH58261 to coronary artery membranes. Circ Res 79:1153–1160. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. (2011) Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117:3720–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckle T, Grenz A, Laucher S, Eltzschig HK. (2008) A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J Clin Invest 118:3301–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feoktistov I, Biaggioni I. (2011) Role of adenosine A2B receptors in inflammation. Adv Pharmacol 61:115–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feoktistov I, Goldstein AE, Biaggioni I. (1999) Role of p38 mitogen-activated protein kinase and extracellular signal-regulated protein kinase kinase in adenosine A2B receptor-mediated interleukin-8 production in human mast cells. Mol Pharmacol 55:726–734. [PubMed] [Google Scholar]
- Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. (2009) The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10:241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz S, Bauersachs J, Ertl G. (2009) Post-infarct remodeling: contribution of wound healing and inflammation. Cardiovasc Res 81:474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz S, Ducharme A, Sawyer D, Rohde LE, Kobzik L, Fukazawa R, Tracey D, Allen H, Lee RT, Kelly RA. (2003) Targeted deletion of caspase-1 reduces early mortality and left ventricular dilation following myocardial infarction. J Mol Cell Cardiol 35:685–694. [DOI] [PubMed] [Google Scholar]
- Gardin JM, Adams DB, Douglas PS, Feigenbaum H, Forst DH, Fraser AG, Grayburn PA, Katz AS, Keller AM, Kerber RE, et al. (2002) Recommendations for a standardized report for adult transthoracic echocardiography: a report from the American Society of Echocardiography's Nomenclature and Standards Committee and Task Force for a Standardized Echocardiography Report. J Am Soc Echocardiogr 15:275–290. [DOI] [PubMed] [Google Scholar]
- Gessi S, Merighi S, Varani K, Borea PA. (2011) Adenosine receptors in health and disease. Adv Pharmacol 61:41–75. [DOI] [PubMed] [Google Scholar]
- Grant MB, Davis MI, Caballero S, Feoktistov I, Biaggioni I, Belardinelli L. (2001) Proliferation, migration, and ERK activation in human retinal endothelial cells through A2B adenosine receptor stimulation. Invest Ophthalmol Vis Sci 42:2068–2073. [PubMed] [Google Scholar]
- Ikejima T, Okusawa S, Ghezzi P, van der Meer JW, Dinarello CA. (1990) Interleukin-1 induces tumor necrosis factor (TNF) in human peripheral blood mononuclear cells in vitro and a circulating TNF-like activity in rabbits. J Infect Dis 162:215–223. [DOI] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Resources (1996) Guide for the Care and Use of Laboratory Animals 7th ed. Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council, Washington, DC. [Google Scholar]
- Karmouty-Quintana H, Zhong H, Acero L, Weng T, Melicoff E, West JD, Hemnes A, Grenz A, Eltzschig HK, Blackwell TS, et al. (2012) The A2B adenosine receptor modulates pulmonary hypertension associated with interstitial lung disease. FASEB J 26:2546–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelle S, Roes SD, Klein C, Kokocinski T, de Roos A, Fleck E, Bax JJ, Nagel E. (2009) Prognostic value of myocardial infarct size and contractile reserve using magnetic resonance imaging. J Am Coll Cardiol 54:1770–1777. [DOI] [PubMed] [Google Scholar]
- Koeppen M, Eckle T, Eltzschig HK. (2011) Interplay of hypoxia and A2B adenosine receptors in tissue protection. Adv Pharmacol 61:145–186. [DOI] [PubMed] [Google Scholar]
- Merkle S, Frantz S, Schön MP, Bauersachs J, Buitrago M, Frost RJ, Schmitteckert EM, Lohse MJ, Engelhardt S. (2007) A role for caspase-1 in heart failure. Circ Res 100:645–653. [DOI] [PubMed] [Google Scholar]
- Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, Kannan HR, Menna AC, Voelkel NF, Abbate A. (2011) The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci U S A 108:19725–19730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa SJ, Nadeem A, Fan M, Zhong H, Belardinelli L, Zeng D. (2007) Effect of a specific and selective A2B adenosine receptor antagonist on adenosine agonist AMP and allergen-induced airway responsiveness and cellular influx in a mouse model of asthma. J Pharmacol Exp Ther 320:1246–1251. [DOI] [PubMed] [Google Scholar]
- Philipp S, Yang XM, Cui L, Davis AM, Downey JM, Cohen MV. (2006) Postconditioning protects rabbit hearts through a protein kinase C-adenosine A2b receptor cascade. Cardiovasc Res 70:308–314. [DOI] [PubMed] [Google Scholar]
- Ryzhov S, Solenkova NV, Goldstein AE, Lamparter M, Fleenor T, Young PP, Greelish JP, Byrne JG, Vaughan DE, Biaggioni I, et al. (2008) Adenosine receptor-mediated adhesion of endothelial progenitors to cardiac microvascular endothelial cells. Circ Res 102:356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider DJ, Lindsay JC, Zhou Y, Molina JG, Blackburn MR. (2010) Adenosine and osteopontin contribute to the development of chronic obstructive pulmonary disease. FASEB J 24:70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte G, Fredholm BB. (2003) The Gs-coupled adenosine A2B receptor recruits divergent pathways to regulate ERK1/2 and p38. Exp Cell Res 290:168–176. [DOI] [PubMed] [Google Scholar]
- Sun CX, Zhong H, Mohsenin A, Morschl E, Chunn JL, Molina JG, Belardinelli L, Zeng D, Blackburn MR. (2006) Role of A2B adenosine receptor signaling in adenosine-dependent pulmonary inflammation and injury. J Clin Invest 116:2173–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaru M, Tomura K, Sakamoto S, Tezuka K, Tamatani T, Narumi S. (1998) Interleukin-1β induces tissue- and cell type-specific expression of adhesion molecules in vivo. Arterioscler Thromb Vasc Biol 18:1292–1303. [DOI] [PubMed] [Google Scholar]
- Tei C, Ling LH, Hodge DO, Bailey KR, Oh JK, Rodeheffer RJ, Tajik AJ, Seward JB. (1995) New index of combined systolic and diastolic myocardial performance: a simple and reproducible measure of cardiac function–a study in normal and dilated cardiomyopathy. J Cardiol 26:357–366. [PubMed] [Google Scholar]
- Toldo S, Bogaard HJ, Van Tassell BW, Mezzaroma E, Seropian IM, Robati R, Salloum FN, Voelkel NF, Abbate A. (2011) Right ventricular dysfunction following acute myocardial infarction in the absence of pulmonary hypertension in the mouse. PloS One 6:e18102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosato G, Jones KD. (1990) Interleukin-1 induces interleukin-6 production in peripheral blood monocytes. Blood 75:1305–1310. [PubMed] [Google Scholar]
- Van Tassell BW, Seropian IM, Toldo S, Salloum FN, Smithson L, Varma A, Hoke NN, Gelwix C, Chau V, Abbate A. (2010a) Pharmacologic inhibition of myeloid differentiation factor 88 (MyD88) prevents left ventricular dilation and hypertrophy after experimental acute myocardial infarction in the mouse. J Cardiovasc Pharmacol 55:385–390. [DOI] [PubMed] [Google Scholar]
- Van Tassell BW, Varma A, Salloum FN, Das A, Seropian IM, Toldo S, Smithson L, Hoke NN, Chau VQ, Robati R, et al. (2010b) Interleukin-1 trap attenuates cardiac remodeling after experimental acute myocardial infarction in mice. J Cardiovasc Pharmacol 55:117–122. [DOI] [PubMed] [Google Scholar]
- Wakeno M, Minamino T, Seguchi O, Okazaki H, Tsukamoto O, Okada K, Hirata A, Fujita M, Asanuma H, Kim J, et al. (2006) Long-term stimulation of adenosine A2b receptors begun after myocardial infarction prevents cardiac remodeling in rats. Circulation 114:1923–1932. [DOI] [PubMed] [Google Scholar]
- Zhong H, Belardinelli L, Maa T, Zeng D. (2005) Synergy between A2B adenosine receptors and hypoxia in activating human lung fibroblasts. Am J Respir Cell Mol Biol 32:2–8. [DOI] [PubMed] [Google Scholar]
- Zhong H, Shlykov SG, Molina JG, Sanborn BM, Jacobson MA, Tilley SL, Blackburn MR. (2003) Activation of murine lung mast cells by the adenosine A3 receptor. J Immunol 171:338–345. [DOI] [PubMed] [Google Scholar]