

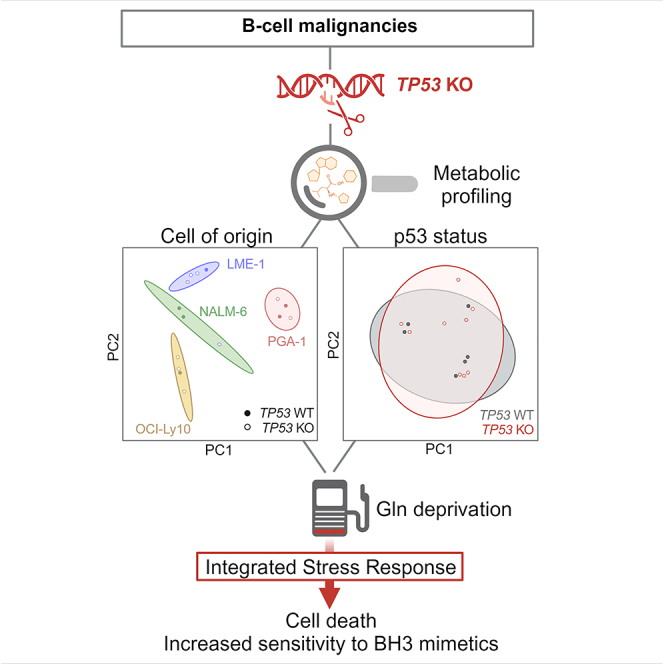
Summary
The tumor suppressor p53 has been described to control various aspects of metabolic reprogramming in solid tumors, but in B cell malignancies that role is as yet unknown. We generated pairs of p53 functional and knockout (KO) clones from distinct B cell malignancies (acute lymphoblastic leukemia, chronic lymphocytic leukemia, diffuse large B cell lymphoma, and multiple myeloma). Metabolomics and isotope tracing showed that p53 loss did not drive a common metabolic signature. Instead, cell lines segregated according to cell of origin. Next, we focused on glutamine as a crucial energy source in the B cell tumor microenvironment. In both TP53 wild-type and KO cells, glutamine deprivation induced cell death through the integrated stress response, via CHOP/ATF4. Lastly, combining BH3 mimetic drugs with glutamine starvation emerged as a possibility to target resistant clones. In conclusion, our analyses do not support a common metabolic signature of p53 deficiency in B cell malignancies and suggest therapeutic options for exploration based on glutamine dependency.
Subject areas: Microenvironment, Cell biology, Cancer, Metabolomics
Graphical abstract
Highlights
-
•
No common metabolic profile upon TP53 KO in distinct malignant B cell lines
-
•
Glutamine deprivation induces cell death and proliferation arrest regardless of p53
-
•
The integrated stress response drives cell death upon glutamine deprivation
-
•
Glutamine deprivation sensitizes malignant B cells to BH3 mimetics
Microenvironment; Cell biology; Cancer; Metabolomics
Introduction
p53 is the most mutated gene in human cancer, and the loss of its function is related to poor prognosis in most tumor types.1 Due to its role in regulating the response to DNA damage and apoptosis, cancer cells devoid of p53 function are frequently resistant to chemotherapy,2 which also holds true for hematological malignancies. In chronic lymphocytic leukemia (CLL), for example, TP53 defects highly affect outcome of chemoimmunotherapy and also alter response to Bruton’s tyrosine kinase inhibitors; therefore screening of p53 status is crucial.3,4,5 In diffuse large B cell lymphomas (DLBCLs) TP53 mutations are also associated with poor survival.6 In response to chemotherapeutic agents, p53 triggers apoptosis by upregulating NOXA and PUMA, which are BH3-only proteins that bind anti-apoptotic Bcl-2 family members and inhibit their interaction with the pro-apoptotic molecules Bax and Bak.1 BH3 mimetics are highly specific and potent drugs that induce apoptosis independently of p53 by mimicking the function of BH3-only proteins and thereby inhibiting the anti-apoptotic Bcl-2 family members.7 CLL patients with TP53 mutations respond to Bcl-2 inhibition therapy (venetoclax); however the emergence of resistance is more frequent within this group, therefore raising the question of whether sensitivity to BH3 mimetics is completely independent of p53 status in B cell malignancies.5
Besides its role in DNA damage response and apoptosis, it has become clear that p53 plays a crucial role in the regulation of cellular metabolism in both healthy and tumor cells.8,9 From a general point of view, p53 reduces glycolytic activity while boosting mitochondrial metabolism by increasing the expression of proteins related to the tricarboxylic acid (TCA) cycle and the electron transport chain (ETC).10,11 p53 has also been described to regulate amino acid and lipid metabolism, as well as the antioxidant response to reactive oxygen species (ROS).12 In turn, p53 is also involved in the response to nutrient starvation, by mobilizing alternative nutrient sources, increasing de novo biosynthesis pathways, or triggering compensatory catabolic routes to maintain cell survival, and also by driving cell-cycle arrest and ultimately elimination of cells if the stress is too strong or prolonged and cells cannot recover.13 The mechanisms by which cancer cells respond to glutamine deprivation, including p53 and others, and how glutamine deprivation can be exploited to trigger tumor cell death have been recently extensively reviewed by Jin et al.14 However, the metabolic signature driven by p53 and more specifically that driven by p53 loss appear to be rather tumor type-specific, and therefore findings cannot be easily extrapolated between different tumors.
This work investigates the effects of p53 loss in the metabolism and response to glutamine deprivation and BH3 mimetics in B cell malignancies from different origin. The primary goal of the study is to establish which common metabolic pathways (if any) are under the control of p53 in this group of tumors. Secondly, the specific involvement of p53 under conditions of glutamine deprivation and BH3 mimetics treatment has been addressed.
Results
Metabolic signatures in B cell malignancies are cell type-specific and independent of p53 status
In order to model malignancies originating at different states of B cell maturation, four cell lines were selected for this study: Nalm-6, PGA-1, OCI-Ly10, and LME-1, which originate from acute lymphoblastic leukemia, CLL, DLBCL, and multiple myeloma (MM), respectively (Table 1). All cell lines had TP53WT status and functional p53 activity. For each cell line, TP53-deficient clones (TP53KO) were generated with CRISPR-Cas9 technology by using a single guide RNA (sgRNA) targeting exon 2 of the gene, therefore disrupting full-length p53. CRISPR in malignant B cell lines was challenging, and efficiency was low in general and particularly low in Nalm-6, from which only one TP53KO clone could be generated. Loss of p53 expression was validated by western blotting, which showed absence of p53 protein levels and lack of p21 induction upon irradiation (Figure 1A). Parental and MOCK (Cas9+ sgRNA-) cells expressing wild-type (WT) TP53 were used as controls. TP53KO clones showed significantly reduced cell death upon treatment with fludarabine or irradiation as compared to their WT counterparts, confirming the effective suppression of p53 function (Figures S1A and S1B). To investigate the impact of p53 loss on the metabolism of malignant B cells, metabolomics analyses were performed in TP53WT and knockout (KO) cells from all four cell lines. Principal-component analysis (PCA) revealed that samples clustered based on cell of origin rather than TP53 status (Figure 1B), indicating that p53 loss did not drive a common metabolic signature in malignant B cell lines. Instead, cell line-intrinsic metabolic signatures defined by a predominant set of metabolites were identified (Figure 1C). Within these specific clusters of metabolites, no differences were observed according to p53 status, except in the Nalm-6 TP53KO clone, where metabolite abundance was higher compared to TP53WT cells. The most abundant metabolites from each cell type were subjected to pathway analysis. Most pathways identified were linked to amino acid and nucleotide metabolism, and no differences were observed according to p53 status (Figure 1C, Table S1). To further confirm that p53 did not drive a common metabolic signature in the cell lines studied, pathway analysis was repeated on the total metabolome pool by grouping cells according to TP53WT or KO status. Pathways involving metabolites of the upper glycolysis as well as the pentose phosphate pathway (PPP) were the most different between TP53WT and KO cells in all cell lines, yet differences were not statistically significant (Figure S1C). Specific analysis of the levels of PPP intermediates (G6P, 6PG, R5P, and PRPP) showed no global differences between TP53WT and KO clones across cell lines, only in PGA-1 and LME-1 the levels of PPP intermediates were consistently decreased in TP53KO clones compared to WT cells (Figure S1D). In accordance with metabolomics data, oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), indicators of mitochondrial and glycolytic metabolism, respectively, were not different between TP53WT and KO cells, as measured by extracellular flux analysis. In the single clone of Nalm-6 TP53KO, increased OCR and ECAR were detected (Figures 1D and S1E). Given that TP53 loss can confer genomic instability, comparative genomic hybridization (CGH) array analysis was performed in parental and TP53KO PGA-1 and Nalm-6 cells. A total of 8 and 9 genetic alterations were found in parental and TP53KO #1 PGA-1 cells, respectively, among which 7 were shared, indicating that p53 loss did not increase genomic instability in the conditions studied. It needs to be taken into account that the pressure for genomic rearrangements might be different in vitro than that in the tumor microenvironment. In parental Nalm-6 cells, 3 genetic alterations were found, which were also found in the TP53KO clone together with 4 new alterations (Table S2). Increased genomic alterations in the Nalm-6 TP53KO clone might explain the higher metabolic content and activity observed before. Nevertheless, loss of p53 had no effect on the growth of any of the cell lines (Figure S1F).
Table 1.
Phenotypical characterization of the cell lines used in this project
| Cell Line | Origin | Phenotype |
|---|---|---|
| Nalm-6 | ALL | CD3−, CD10+, CD19+, CD37−, cyCD79a+, CD80−, CD138+, HLA-DR+, sm/cyIgG-, cyIgM+, smIgM-, sm/cykappa-, sm/cylambda-; |
| PGA-1 | CLL (IGHV mutated) | CD3−, CD4−, CD5−, CD10−, CD13−, CD14−, CD19+, CD20+, CD34−, CD37+, CD80+, CD138, HLA-DR+ |
| OCI-Ly10 | DLBCL | ABC-subtype with chronic BCR signaling, CD79A and L265P MYD88 mutations |
| LME-1 | MM | CD38+, CD138+, CD45−, B2micro+, CD19−, CD56+, CD27− CD28−, CD117 dim |
Information for Nalm-6 and PGA-1 was obtained from https://www.dsmz.de, LME-1 and OCI-Ly10 were characterized in-house.
Figure 1.

Metabolic signatures in B cell malignancies are cell type-specific and independent of TP53 status
(A) TP53WT and TP53KO clones were generated from Nalm-6, PGA-1, OCI-Ly10, and LME-1 parental cell lines using CRISPR-Cas9 technology. Clones were subjected to 5 Gy irradiation (IR) and cultured for 16h. Representative western blots for p53 and p21 are shown.
(B) TP53WT and KO LME-1, Nalm-6, OCI-Ly10, and PGA-1 cells were cultured in complete medium for 48h and analyzed by LC-MS metabolomics. A total of 100 metabolites were identified. Metabolite abundance was further analyzed using MetaboAnalyst 5.0. Principal-component analysis (PCA) was performed grouping samples based on cell of origin or TP53 status.
(C) Heatmap depicting auto scaled metabolite abundance. p53 status is indicated by + (WT) and - (KO). Hierarchical clustering was applied to rows and columns. The most enriched clusters of metabolites in each cell line are indicated with black boxes. Pathway analysis was performed based on each cell line-specific group of metabolites. Pathways enriched with FDR<0.06 are depicted. Colored dots indicate the pathways enriched in each cell line.
(D) Mito stress test was performed on all cell lines in the same experimental conditions as described in B using a Seahorse XF96 analyzer. Representative oxygen consumption rate (OCR) profiles with experimental replicates are shown for each cell line as mean ± SD. See also Figure S1 and Tables S1.
Glutamine deprivation induces caspase-dependent cell death and proliferation arrest regardless of p53 status
The role of p53 as a nutrient sensor has been previously described in solid malignancies.15 Glucose deprivation and, more recently, glutamine deprivation have been shown to trigger starvation signals that, despite not being completely understood, ultimately lead to activation of p53.16,17 The consequences of this activation largely depend on fuel dependencies within each tumor and can lead to compensatory mechanisms to sustain metabolic rates, or to cell death. As recently reported by our group, primary CLL cells use glutamine as main fuel of the TCA cycle, and inhibition of glutamine uptake significantly increases their sensitivity to BH3 mimetics.18 Therefore, we reasoned that p53 status might be relevant in conditions of glutamine deprivation in B cell malignancies.
We first evaluated the effect of Gln deprivation (Gln-) on viability. Gln- induced cell death in all malignant B cell lines except for Nalm-6. When TP53KO cells were compared to their WT counterparts, a small but significant decrease in sensitivity upon Gln- was observed in PGA-1 and OCI-Ly10 TP53KO clones (Figure 2A). Cell death induced by Gln deprivation was completely or almost completely prevented when the caspase inhibitor Q-VD-OPh was added to the cultures in parallel to glutamine deprivation in LME-1 and PGA-1 cells, and was partially prevented in OCI-Ly10 (Figure 2B). This indicated that the main type of cell death triggered by glutamine deprivation is apoptosis, although the involvement of other types of cell death cannot be excluded, especially in OCI-Ly10 cells. To determine whether PGA-1 and OCI-Ly10 TP53KO clones had a survival advantage upon Gln-, competition assays were performed in PGA-1 cells. TP53KO clones had an advantage in Gln- and were the only viable cells left at day 16 (Figure 2C). However, it should be noted that the majority of cells were dead at this final time point. Assessment of absolute cell number along time showed that proliferation of TP53WT and KO PGA-1 cells was equally arrested in Gln- (Figure 2D), confirming that p53 loss conferred a small survival advantage but not a growth advantage in this cell line.
Figure 2.

Glutamine deprivation induces caspase-dependent cell death and proliferation arrest regardless of p53 status
(A) TP53WT and TP53KO cells were cultured in absence of glutamine (Gln-), and viability was assessed over time using flow cytometry after Mito-O/To-Pro-3 staining. (Nalm-6 N = 2; PGA-1 N = 4; OCI-Ly10 N = 3; LME-1 N = 4). Data are presented as mean ± SEM and differences were analyzed with two-way ANOVA with Tukey’s multiple comparison test (Grouped samples TP53WT vs. KO, ∗p < 0.05). For reference, viability was above 75% in all cell lines in complete media at all time points.
(B) TP53WT and TP53KO cells were cultured in the presence or absence of Gln and with Q-VD-OPh (5 μM) for 72 h (PGA-1 and LME-1) or 120 h (OCI-Ly10) and viability was assessed with Mito-O/To-Pro-3 staining (PGA-1, N = 2; OCI-Ly10, N = 1; LME-1, N = 1). Data are shown as mean ± SEM.
(C) Parental PGA-1 TP53WT cells (GFP-) were plated at equal quantity with either MOCK or p53KO (both GFP+) in control medium or Gln deprivation (Gln-). The percentage of GFP- and GFP+ cells within viable cells (Mito-O+/To-Pro3-) was assessed over time (N = 2). Data are shown as mean ± SEM.
(D) PGA-1 TP53WT and TP53KO cells were grown in complete media (Gln+) or Gln deprivation (Gln-) and cell density was assessed over time (N = 2). Data are shown as mean ± SEM.
(E) PGA-1 WT, MOCK, TP53KO #1 and #2 cells were cultured in complete media or glutamine deprivation (Gln-) for 24 h and subsequently incubated in the presence of 5 mM 13C6-Glucose. Mean enrichment indicates the 13C-labeled fraction of a given metabolite relative to the total amount of that metabolite within a sample. The figure shows the flux of glucose converted into nucleotides. WT cells grown in complete media had to be excluded from the analysis due to low quality of MS peaks. For complete overview of 13C tracing analysis, see also Figure S2.
To further investigate the proliferation arrest observed in PGA-1 cells upon Gln-, 13C-glucose isotope tracing was performed. In complete media conditions, 13C labeling from glucose was 50% and 60% in the purine nucleotides AMP and IMP, respectively, regardless of p53 status. However, in the absence of Gln, the fraction of 13C-labeled IMP or AMP was absent in TP53WT cells and was also decreased in TP53KO cells (to 40% in IMP and completely absent in AMP), indicating that nucleotides could no longer be synthesized (Figure 2E). Despite cells kept using glucose through the PPP, as evidenced by sustained 13C labeling in sedoheptulose 7-phosphate (S7P) (Figure S2), the lack of glutamine impeded the synthesis of nucleotides from Ribose-5-P. From a wider point of view, the distribution of 13C from glucose in TP53WT and KO cells was similar, confirming again that there were not major metabolic changes governed by p53 in these cells. The only two metabolites in which the labeling was different upon p53 loss were lactate and pyruvate, which were increased in TP53KO cells compared to WT. In contrast, metabolites of higher glycolysis did not differ between TP53WT and KO cells. The labeling in TCA cycle intermediates was low in complete media conditions, with the exception of citrate (first entrance point for glucose in the TCA cycle), and upon Gln- a shift in the entrance of glucose to the TCA cycle was observed in both TP53WT and KO cells. This shift consisted in increased synthesis of oxaloacetate (indirectly measured through aspartate) from pyruvate at the expenses of citrate, possibly to synthesize glutamine or Gln-derived products such as glutamate, in which we also found increased labeling from glucose upon Gln- (Figure S2). Overall, the most important changes upon Gln- were found at the level of nucleotide synthesis.
Glutamine deprivation induces cell death through the integrated stress response (ISR) pathway
To investigate the mechanisms underlying cell death upon Gln deprivation, we first assessed p53 activation in Gln-. Phosphorylation of p53 at serine 15 was robustly induced in PGA-1 and modestly induced in OCI-Ly10 cells, while p21 was upregulated only in PGA-1 (Figure 3A). Consistent with a role of p53 inducing cell death, a small survival advantage of TP53KO cells in both PGA-1 and OCI-Ly10 was observed before (Figure 2A). On the contrary, p53 was not activated in LME-1 or Nalm-6, where we previously did not observe differences in viability according to p53 status (Figure 2A). Of note, PGA-1 and OCI-Ly10 TP53KO cells, as well as LME-1 cells (both WT and KO), showed significantly decreased viability in Gln- (50% viability for PGA-1 and OCI-Ly10 and only 10%–20% viability for LME-1 at Day 3) (Figure 2A). Therefore, it was clear that other stress response pathways beyond p53 were involved in cell death induced by Gln- in these cell lines.
Figure 3.

Glutamine deprivation induces cell death through the integrated stress response pathway
(A) TP53WT cells were cultured in the presence or absence of glutamine (Gln-) for 48 h (Nalm-6, LME-1) or 72 h (PGA-1 and OCI-Ly10). Representative western blots showing total p53, P-p53 (Ser15) and p21 protein levels. A sample of PGA-1 cells irradiated (5 Gy) and cultured for 16 h was used as control for p53 induction.
(B and C) Cells were cultured in control media or glutamine deprivation (Gln-) for 72 h before RNA extraction. CHOP and ATF4 mRNA levels were measured by RT-qPCR in (B) parental cell lines (N = 3) and (C) PGA-1 TP53WT and KO clones (N = 4 CHOP, N = 3 ATF4).
(D) TP53WT or KO PGA-1 cells were cultured in glutamine deprivation in the presence of ISR inhibitor ISRIB (10 μM). Viability was assessed along time by flow cytometry analysis of Mito-O+/To-Pro-3- cells (N = 3). CHOP mRNA levels were measured by RT-qPCR (N = 2). Data are presented as mean ± SEM and differences were analyzed with t test (panel B) or two-way ANOVA with Tukey’s multiple comparison test (panels C and D) (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). Comparisons to Gln+ are indicated by ∗, while comparisons to Gln- are indicated by #. See also Figure S3.
Gln deficiency has been extensively shown to induce the ISR in healthy cells and solid malignancies, which results in phosphorylation of the eukaryotic translation initiation factor 2A (eIF2α), block of general protein synthesis, and translation of specific proteins such as the activating transcription factor 4 (ATF4), which engages the cell on a specific transcription program that, among others, increases the mRNA expression of ATF4 as a feedback loop. Sustained activation of the ISR and ATF4 translation induces the transcription of the pro-apoptotic transcription factor C/EBP Homologous Protein (CHOP), encoded by the gene DDIT3.19 Upon 72 h of Gln- in the presence of Q-VD-OPh, transcriptional induction of CHOP and ATF4 was observed in PGA-1, OCI-Ly10, and LME-1 cells. In line with the resistance to Gln deprivation-induced cell death previously observed, CHOP and ATF4 were not upregulated in Nalm-6 cells and the highest levels were observed in LME-1 cells, which were the most sensitive to glutamine deprivation (Figure 3B). Induction of ISR was already evident after 24 h of glutamine deprivation (Figure S3A). Importantly, induction of CHOP and ATF4 mRNA expression upon Gln- also occurred in TP53KO clones (Figures 3C and S3B). In order to assess the requirement of the ISR in cell death induction upon Gln-, the small molecule ISRIB was used. ISRIB binds to eIF2B promoting the assembly of the ternary complex for new protein synthesis and counteracting the inhibition of translation imposed by the phosphorylated form of eIF2α.19,20 ISRIB significantly protected cells from death upon Gln deprivation regardless of p53 status, and prevented the upregulation of CHOP upon Gln- in both TP53WT and TP53KO PGA-1 cells (Figure 3D). In fact, the rescue of viability was even more pronounced in TP53KO cells, where the ISR was the primary form of cell death in Gln- (Figure 3D). These results confirmed a role for ISR in glutamine deprivation-induced cell death in malignant B cells.
Glutamine deprivation sensitizes malignant B cells of different origin to BH3 mimetics
Two studies have suggested that p53 loss can confer a survival advantage in response to BH3 mimetic drugs in hematological malignancies.21,22 In order to address this relevant clinical question, viability of all four parental cell lines was assessed in complete media in the presence of venetoclax, S-63845, and A-1331852, which inhibit Bcl-2, Mcl-1, and Bcl-xL, respectively. All cell lines displayed substantial resistance to venetoclax and A-1331852 and different sensitivity to S-63845 (Figure 4A). As reference, the concentration of venetoclax in the blood of treated patients is 2.4 μΜ,23 and all cell lines studied except for OCI-Ly10 were resistant to even 10 μM venetoclax. Given that all cell lines except for PGA-1 were sensitive to S-63845, the response of TP53KO clones to this drug was further evaluated. Loss of p53 did not change the sensitivity to S-63845 in LME-1, OCI-Ly10, or PGA-1, whereas Nalm-6 TP53KO clone displayed a significant increase in resistance (Figure 4B). Accordingly, p53 loss in PGA-1 cells did not influence the mRNA expression of Bcl-2 family members, as assessed by reverse transcriptase multiplex ligation-dependent probe amplification (RT-MLPA).24,25 Analysis of the classical TP53 target genes CDKN1A (p21), BBC3 (PUMA), and PMAIP1 (NOXA) showed that p21 and PUMA were higher expressed in TP53WT cells than in the TP53KO counterparts in PGA-1 and LME-1, while the expression of NOXA remained constant between TP53WT and KO clones. Samples on RT-MLPA clustered based on cell of origin rather than TP53 status (Figure 4C).
Figure 4.

Most malignant cell lines are resistant to BH3 mimetics independently of p53 status
(A) TP53WT cell lines were treated with the indicated concentrations of venetoclax (Bcl-2 inhibitor, N = 2), A-1331852 (Bcl-XL inhibitor, N = 3) or S-63845 (Mcl-1 inhibitor, N = 3) for 24 h. Viability was measured by flow cytometry using Mito-O/To-Pro-3 staining.
(B) TP53WT and TP53KO clones deriving from Nalm-6 (N = 3), OCI-Ly10 (N = 2), LME-1 (N = 3), and PGA-1 (n = 1) were incubated with S-63845 for 24 h. Viability was assessed by flow cytometry using Mito-O/To-Pro-3 staining. Data are presented as mean ± SEM and differences were analyzed with two-way ANOVA with Tukey’s multiple comparison test (∗p < 0.05, ∗∗p < 0.01).
(C) PGA-1 TP53WT and TP53KO clones were cultured in complete media for 48 h, RNA was extracted and expression of TP53 transcriptional targets and apoptosis-related genes was investigated using RT-MLPA. Data were averaged and plotted as Z score using the ComplexHeatmap R package.
The next question was whether the expression of Bcl-2 family members changed upon Gln- and whether such analysis would provide a mechanism underlying the observed Gln deprivation-induced cell death. RT-MLPA analysis was performed in Gln- conditions in TP53WT and KO PGA-1 cells. Increased expression of BCL2L1 (Bcl-xL) was detected upon Gln- regardless of p53 status (Figure 5A) and was confirmed at the protein level (Figure 5B). A slight increase in BCL2 expression was observed only in TP53KO cells, possibly contributing to their survival advantage upon glutamine deprivation. Expression of CDKN1A (p21), BBC3 (PUMA), PMAIP1 (NOXA), and BCL2L11 (BIM), genes involved in induction of apoptosis or cell-cycle arrest, was upregulated upon Gln deprivation in both WT and TP53KO cells (Figure 5A).
Figure 5.

Glutamine deprivation sensitizes malignant B cells of different origin to BH3 mimetics
(A) PGA-1 TP53 WT and TP53 KO clones were cultured in compete or Gln- for 48 h, RNA was extracted and expression of TP53 transcriptional targets and apoptosis-related genes was investigated using RT-MLPA. Data were averaged and plotted as fold change to complete media in a heatmap using the ComplexHeatmap R package (N = 2). Crosses indicate missing data.
(B) Bcl-2 family members expression was analyzed by flow cytometry in PGA-1 TP53 WT and TP53 KO cells (N = 2).
(C) PGA-1 and OCI-Ly10 cells with different p53 status were incubated in Gln-for 16 h and subsequently treated with A-1331852 (Bcl-XL inhibition) (PGA-1 WT N = 6, the rest N = 3).
(D) Parental PGA-1 cells were incubated in Gln-for 16 h and subsequently treated with venetoclax (Bcl-2 inhibition, N = 2) or S-63845 (Mcl-1 inhibition, N = 2) for 24 h.
(E) PBMCs from TP53+/+ and TP53−/− CLL patients were cultured ex vivo in unstimulated conditions in complete media (Us) or on top of CD40L-expressing 3T3 fibroblasts to stimulate CD40 signaling in the presence or absence of Gln. Percentage of leukemic cells (CD5+CD19+) and CLL activation (CD19+CD95+) were measured 24 h post-stimulation. At that time, a fraction of CLL cells was harvested and treated with venetoclax for additional 24h. Viability was assessed by Mito-O/To-Pro-3 staining (TP53+/+ N = 7, TP53−/− N = 3). Data are presented as mean ± SEM and differences were analyzed with two-way ANOVA with Tukey’s multiple comparison test (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
The high expression of Bcl-xL detected upon Gln- suggested that cells might be dependent on this protein in order to survive under low-Gln conditions. To test this, we made use of the Bcl-xL inhibitor A-1331852, as all parental cells were resistant to it. PGA-1 and OCI-Ly10 cells were cultured in complete media or Gln- conditions in the presence of the drug. PGA-1 cells in Gln- showed significantly increased sensitivity to A-1331852, suggesting that Bcl-xL is relevant for at least part of the cell survival observed upon glutamine deprivation in these cell lines. Importantly, Gln- significantly increased the sensitivity of PGA-1 TP53KO cells to Bcl-xL inhibition, confirming again no differences with regards to p53 status and response to BH3 mimetics (Figure 5C). In OCI-Ly10, the sensitivity to the BH3 mimetic was not further increased upon Gln-, as the cell death observed was mainly due to the deprivation itself (Figure 5C). Gln deprivation also increased the sensitivity of PGA-1 cells to Bcl-2 inhibition by venetoclax, but not to Mcl-1 inhibition by S-63845 (Figure 5D).
To further investigate the translational role of our findings, we investigated the potential of glutamine deprivation in increasing the sensitivity of human primary samples from leukemic patients to BH3 mimetics. To do so, we cultured cells from chronic lymphocytic leukemia patients ex vivo either in control conditions (unstimulated) or on top of CD40L-expressing fibroblasts. As shown before by our group and others, in vitro CD40 stimulation mimics the lymph node microenvironment and renders cells resistant to the Bcl-2 inhibitor venetoclax.26 Patients were classified according to their p53 status. TP53−/− patients were selected based on mutated TP53 identified by sequencing and del(17p13) based on fluorescence in situ hybridization (FISH) analysis (Table 2). The percentage of leukemic cells (CD5+CD19+) in the PBMCs was similar between both groups. Both TP53+/+ and TP53−/− cells were activated upon CD40 stimulation, as observed by increased expression of the activation marker CD95, and became resistant to venetoclax upon CD40 stimulation. Remarkably, only cells from TP53+/+ patients were sensitized to venetoclax upon glutamine deprivation, while TP53−/− cells remained resistant (Figure 5E). While the data with TP53+/+ patients support our findings in cell lines, the fact that TP53−/− patients are not sensitized to BH3 mimetics indicates that in these patients, which harbor not only loss of p53 expression but also complete deletion of 17p13 locus, genomic instability rather than single deletion of p53 possibly determines their response to BH3 mimetics.
Table 2.
Patient table
| Patient ID | TP53 status | IGHV mutation status | Other relevant alterations | Treatment at sample acquisition |
|---|---|---|---|---|
| CLL01 | +/+ | mutated | – | untreated |
| CLL02 | +/+ | mutated | – | untreated |
| CLL03 | +/+ | unmutated | – | untreated |
| CLL04 | +/+ | unmutated | – | untreated |
| CLL05 | +/+ | n.a. | – | untreated |
| CLL06 | +/+ | unmutated | del(11q22), del(13q14) | untreated |
| CLL07 | +/+ | mutated | – | untreated |
| CLL08 | −/− | unmutated | – | untreated |
| CLL09 | −/− | unmutated | – | untreated |
| CLL10 | −/− | unmutated | del(11q22) and 9 other CGH aberrations = complex karyotype, pathogenic mutation in SF3B1 | Idelalisib |
Most relevant characteristics of the patients included in the study are summarized. TP53−/− was defined as both mutated TP53 and del(17p13).
Discussion
In this study we have addressed for the first time the question of whether p53 status confers a specific metabolic signature in malignant B cells, which we investigated by generating TP53KO cells from different B cell malignancies. Our results show that in B cell malignancies p53 does not drive a specific metabolic program, as opposed to the general assumption in the solid tumor metabolism field. In contrast, our study revealed clear differences in metabolic content as well as activity between cell lines at different stages of B cell maturation. Specifically, we did not find any significant differences in terms of flux from 13C-glucose in the PPP, which indicates that some of the roles that have been clearly stablished for p53 in solid tumor models9,27 are only partially applicable to B cell malignancies.
Only in tumor cells that are highly dependent on p53 loss, p53 status makes a significant difference at baseline, as it is the case of pancreatic ductal adenocarcinoma.28 In several tumor types, however, the role of p53 becomes especially relevant in case of genotoxic, oxidant, or nutrient stress. Several studies have shown that p53 senses nutritional stress resulting from glucose deprivation,13,15 but only few have explored the role of p53 in response to glutamine deprivation. Among the latter, three studies described a p53 pro-survival response in low-glutamine conditions, one through ROS sensing by B55α that involves p21 and glutaminase (GLS) expression,29 and the other two through upregulation of the amino acid transporters SLC1A3 and SLC7A3 as compensatory mechanisms.16,17
In our study, glutamine deprivation resulted in decreased viability in three out of the four cell lines tested regardless of p53 status, further confirming that p53 does not play a central role in the regulation of metabolism in B cell malignancies, also not in stress conditions. In two of the cell lines (PGA-1 and OCI-Ly10, from CLL and DLBCL, respectively), Gln- triggered p53 activation and viability was slightly decreased in TP53KO clones, indicating that p53 can have a minor role driving cell death in response to glutamine deprivation in certain cell types. However, p53 was not the main driver of Gln deprivation-induced cell death in the models studied. Instead, we show here that the ISR, which involves phosphorylation of eIF2α and selective translation of ATF4 and CHOP, is a common response pathway in all three cell lines sensitive to Gln starvation. The MM cell line LME-1 was the most sensitive to Gln deprivation and also the one where the highest induction of CHOP and ATF4 was observed. Inhibition of ISR rescued the viability in all cell lines, especially in the TP53 KO clones, confirming that p53 response has only a minor role upon amino acid deprivation in malignant B cells. The expression of NOXA, PUMA, and p21, classical targets of p53, was also induced in TP53KO cells, indicating that other pathways are involved in their transcriptional regulation. This is in accordance with a recent study which showed that ATF4 can take over p53 functions in tumors with mutated p53.30 In the publication mentioned earlier where p53 was described to induce the amino acid transporter SLC7A3 as a pro-survival mechanism upon glutamine deprivation, ATF4 was found to act upstream of p53 phosphorylation and to be essential for the transcriptional upregulation of SLC7A3. As the authors of the study also describe, sustained activation of the ISR can ultimately lead to cell death.17 It is therefore possible that ATF4, which we found increased in all cell lines susceptible to glutamine deprivation, also leads to the observed p53 phosphorylation in our experimental models.
Despite the four cell lines studied being metabolically distinct, three of them shared the fact that glutamine is an essential fuel for proliferation, with the exception of Nalm-6 (B-ALL). This means that our previously published findings with primary CLL cells, where we showed that glutamine is the most important fuel for the TCA cycle,18 are also possibly valid for DLBCL and MM. To what extent the TCA cycle in TP53−/− patients is also fueled by glutamine may be worthwhile to further explore, as we did find differences between TP53KO cell lines and TP53−/− primary CLL cells in terms of response to glutamine deprivation.
A second important question that we also aimed to answer with this study was whether p53 status can affect the sensitivity to BH3 mimetics in B cell malignancies. p53 induces apoptosis by upregulating BH3-only proteins,1 while BH3 mimetics act downstream of p53,7 and therefore they should be able to overcome p53 deficiency-related resistance. In fact, it has already been shown in clinical trials that CLL patients with TP53 mutations can benefit from Bcl-2 inhibition therapy (venetoclax).5,31,32 However, despite excellent initial responses, the emergence of resistance seems to be more likely in patients with poor genomic complexity, including 17p deletions and TP53 mutations.5,32,33 According to our in vitro data, single deletion of TP53 does not confer resistance to BH3 mimetics, which is in accordance with previous studies.34 In fact, most cell lines (TP53WT and KO) were resistant to Bcl-2 and Bcl-xL inhibition, while being sensitive to Mcl-1 inhibition. Only in Nalm-6, increased survival of TP53KO cells was observed upon Mcl-1 inhibition. We speculate that in vivo relapse of p53 dysfunctional clones upon BH3 mimetics treatment depends on the accumulation of de novo mutations giving a selective advantage to these clones and therefore cannot be recapitulated in vitro by CRISPR strategies. In line with this, we identified increased number of genetic alterations in Nalm-6 cells upon p53 loss, which correlated with higher metabolic activity and decreased sensitivity to Mcl-1 inhibition. We tested whether differences in metabolism upon p53 loss also occurred in a different B-ALL cell line (BV173), and that was not the case, indicating that those observed in Nalm-6 are probably due to increased genomic instability in this specific clone. Data from two previous studies found differences according to p53 status in response to the same drugs; however the doses used were lower than those used in our study and in the clinic.21,22 We think that the differences observed in these previous studies might be cell line-specific and potentially attributed to increased genomic instability in TP53KO clones.
Of relevance, when Gln deprivation was combined with BH3 mimetics, PGA-1 cells were more sensitive to Bcl-xL and Bcl-2 inhibition independently of p53 expression, while sensitivity to Mcl-1 inhibition remained the same. This might be attributed to the fact that Gln- increases the expression of Bcl-xL, which has been previously linked to venetoclax resistance in primary CLL samples.26,35 Increased Bcl-xL might counteract part of the pro-apoptotic effects of increased PUMA, NOXA and BIM observed at the mRNA level upon glutamine deprivation. In a similar manner, a previous publication showed that primary MM cells and cell lines are dependent on glucose and glutamine fueling and that glutamine deprivation increases the binding of the pro-apoptotic protein BIM to BCL-2, facilitating sensitization to venetoclax.36 Work performed on mouse hybridomas highly susceptible to glutamine deprivation also showed increased sensitivity to BH3 mimetics, in this case to the MCL-1 inhibitor obatoclax, upon removal of glutamine.37 Altogether, these data indicate that our recent findings about the potentiality of inhibiting Gln metabolism to sensitize primary CLL cells to BH3 mimetics18 are further supported by this and other studies and can possibly be extrapolated to other B cell malignancies and tumor types.
Importantly, our findings in cell lines were recapitulated in primary samples from TP53+/+ CLL patients, where we observed increased sensitivity to venetoclax upon Gln deprivation. However, this was not the case in three CLL patients with biallelic TP53 loss. This is most probably due to increased genomic instability,33 with high risk of acquiring other mutations over time, which can affect the response in the experimental setting. Since the available samples did not permit comparison with CLL cells before loss of TP53 in these patients, we could not make a direct comparison.
Functional p53 scores, such as the one recently developed by Durand et al. involving 13 genes specifically downregulated upon p53 silencing in MM, emerge as interesting strategies to identify patients with p53 dysfunction and determine whether therapeutic strategies such as the ones proposed by our study and others can be applied to this group of patients.38
In conclusion, with this work we provide clear answers to two clinically relevant questions in the field of B cell malignancies: i) p53 does not drive a common metabolic signature in these tumors and ii) p53 does not directly influence response to BH3 mimetics, neither when administered alone nor when administered in combination with glutamine starvation strategies. Besides, we broaden the scope of the potential use of inhibition of glutamine metabolism to overcome resistance to BH3 mimetics in several B cell malignancies.
Limitations of the study
This study was initiated based on a large body of literature that indicated that the tumor suppressor p53 has various but well-defined effects on cellular metabolism. We aimed to delineate common denominators of p53 on metabolism in several cell lines derived from B cell malignancies. We applied CRISPR-Cas9 to engineer p53 gene ablation to study such effects. In general, such an approach should ideally incorporate multiple gRNAs to target p53, and also non-targeting controls, in order to correct for off-target effects of single gRNAs. In addition, low efficiency of transfections in the B cell lines used, combined with low efficiency of p53 homozygous deletion, necessitated single-cell cloning, which could have enhanced clone-specific effects. In contrast to our initial hypothesis, we did not find shared p53-specific effects on metabolism in the distinct cell lines nor in the clones generated. Since almost all clones in fact metabolically resembled the parental cells, this in our opinion negated the necessity to include additional gRNAs, or to target non-essential gene loci. Nevertheless, we cannot definitely rule out that the minor distinctions that we subsequently observed upon additional studies among the cell lines and clones are clone-specific.
As mentioned earlier, knocking down TP53 in cell lines was challenging, as we observed cells with functional p53 being selected, which could be explained by increased p53-mediated DNA damage as a pro-survival response during genome editing by CRISPR-Cas9, as it has been reported before.39
It is important to note that the sgRNA used in this study targets exon 2 of TP53, where the classical translation start site of the gene is located, therefore suppressing the expression of full-length p53. The presence of a downstream translation start site leading to a truncated form of p53 (p53/47) has been described in some cell types, which lacks one of the two transcription-activation domains at the N-terminal side of p53 and therefore cannot transactivate classical p53 targets such as p21 but can increase transcription of other target genes such as MDM2.40 Whether p53/47 is expressed in the cell lines studied has not been investigated.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Bcl-2 BV421 | Biolegend | Cat no. 658709; RRID: AB_2563283 |
| anti-Bcl-xL APC | Cell Signaling Technology | Cat no. 12099S; RRID: AB_2797819 |
| anti-Mcl-1 PE | Cell Signaling Technology | Cat no. 65617S; RRID: AB_2799688 |
| anti-CD5 PE | eBioscience | Cat. no. 12-0059-42; RRID: AB_1548712 |
| anti-CD19 FITC | BD Biosciences | Cat. no. 555412; RRID: AB_395812 |
| anti-CD95 PE-Cy7 | Merck | Cat. no. 305621; RRID: AB_2100369 |
| anti-p53 | Calbiochem | Cat no. OP43; RRID: NA |
| anti-P-p53 (Ser15) | Cell Signaling | Cat no. 9284; RRID: AB_331464 |
| anti-p21 | Cell Signaling | Cat no. 2947; RRID: AB_823586 |
| anti-Beta-actin | SICGEN | Cat no. AB0145; RRID: AB_2895355 |
| Donkey αRabbit-IRDye680/e800 | LI-COR Biosciences | Cat no. 926-68023/926-32213; RRID: AB_10706167/RRID: AB_621848 |
| Donkey αMouse-IRDye680/e800 | LI-COR Biosciences | Cat no. 926-68072/926-32212; RRID: AB_10953628/RRID: AB_621847 |
| Chemicals, peptides, and recombinant proteins | ||
| Venetoclax | Sanbio | Cat no. A0776 |
| A-1331852 | ChemieTek | Cat no. CT-A133 |
| S-63845 | Chemgood | Cat no. C-1370 |
| ISRIB | Merck | Cat no. SML0843 |
| Oligonucleotides | ||
|
TP53 CRISPR gRNA: 5’-TCGACGCTAGGATCTGACTG-3’ TCGACGCTAGGATCTGACTG-3’ |
Designed in-house | N/A |
| RT-PCR CHOP_F: 5'-GGAGCATCAGTCCCCCACTT-3' | Designed in-house | N/A |
| RT-PCR CHOP_R: 5’-TGTGGGATTGAGGGTCACATC-3' | Designed in-house | N/A |
| RT-PCR ATF4_F: 5’-GGGACAGATTGGATGTTGGAGA-3’ | Designed in-house | N/A |
| RT-PCR ATF4_R: 5’-GGGACAGATTGGATGTTGGAGA-3’ | Designed in-house | N/A |
| RT-PCR TBP_F: 5'-CACATCACAGCTCCCCACCA-3’ | Designed in-house | N/A |
| RT-PCR TBP_R: 5'-TGCACAGGAGCCCAAGAGTG-3' | Designed in-house | N/A |
| Deposited data | ||
| Metabolomics and 13C isotope tracing data | MetaboLights | MTBLS9786 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Helga Simon-Molas (h.simonmolas@amsterdamumc.nl).
Materials availability
Cell lines generated in this study will be made available to interested researchers by the lead contact upon request.
Data and code availability
-
•
Metabolomics and 13C isotope tracing data are accessible in MetaboLights (EMBL-EBI)41: MTBLS9786.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyse the data reported in this work paper is available from the lead contact upon request.
Experimental model and study participant details
Cell lines and culture conditions
Nalm-6, PGA-1, OCI-Ly10 and LME-1 cells were cultured at 37°C and 5% CO2 in RPMI-1640 (ThermoFisher Scientific, Waltham, Massachusetts, US, #52400) supplemented with 10% foetal calf serum (FCS) (ThermoFisher Scientific, #10270-106), 2mM L-glutamine (Gln) (ThermoFisher Scientific, #25030024) and 100 U/mL Penicillin-Streptomycin (ThermoFisher Scientific, #15140-122). Parental cell lines were authenticated by short tandem repeat analysis in our laboratory and compared to https://www.cellosaurus.org/. LME-1 cells were generated at Amsterdam UMC location University of Amsterdam and were phenotyped in-house. More information about cell lines can be found in Table 1.
Patient material
After written informed consent, patient blood was obtained during diagnostic or follow-up procedures at the Departments of Hematology and Pathology of the Academic Medical Center Amsterdam. The studies were approved by the Academic Medical Center Ethical Review Board and conducted in agreement with the Declaration of Helsinki. Blood mononuclear cells of patients with CLL, obtained after Ficoll density gradient centrifugation (Pharmacia Biotech), were cryopreserved and stored as previously described.42 TP53+/+ and TP53-/- patient samples from our biobank were selected based on the results from TP53 sequencing and FISH analysis (Table 2). For CD40 activation, PBMCs were co-cultured with NIH3T3 fibroblasts stably transfected with human CD40L in the presence or absence of glutamine, or with negative control (NIH3T3 control fibroblasts) in complete media conditions, as described before.43 After 24h, percentage of leukemic cells and CLL activation were assessed. At that time, a fraction of CLL cells was harvested and treated with 0–10 μM venetoclax for additional 24h.
Method details
Generation of TP53 knock-out cell lines by CRISPR/Cas-9
TP53KO Nalm-6, PGA-1 and LME-1 cell lines were generated by lentiviral transduction as previously described.44 The following sgRNA was used to target exon 2 of TP53: 5’-TCGACGCTAGGATCTGACTG-3’. The sgRNA was cloned into the lentiCRISPRv2-puro plasmid (Addgene, #98290) and lentivirus particles were produced in HEK293T cells and used to transduce Nalm-6 and LME-1 parental cells. The sgRNA was cloned into pL-CRISPR.EFS.GFP plasmid (Addgene, #57818) and lentiviral particles were produced as previously mentioned and used to transduce PGA-1. Single-cell KO clones were obtained by performing limiting dilutions of the cell cultures in puromycin (Nalm-6, LME-1) or by single cell sorting of GFP+ cells (PGA-1). Parental OCI-Ly10 cells were transfected with the sgRNA:Cas9 RNP complex provided in the Gene Knockout Kit Human GKO_HS3_TP53 (Synthego) using the Amaxa 4D-nucleofector (Lonza, Basel, Switzerland), and single cell clones were obtained by limiting dilution. For all cell lines, MOCK cells were generated by using the aforementioned plasmids without the gRNAs and therefore constitute a control of Cas9-only expression.
Glutamine deprivation and treatments
For glutamine deprivation experiments, cells were washed with Gln-free RPMI-1640 (ThermoFisher Scientific, #31870) and cultured in Gln-free RPMI-1640 supplemented with 10% FCS and 100 U/mL Penicillin-Streptomycin. Final concentration of Gln in this media was 0,087mM, as determined with the Glutamine/Glutamate-Glo Assay (Promega Corporation, Madison, Wisconsin, US, #J8021). As control, complete media for these experiments was prepared by supplementing Gln-free media 10% FCS and 100 U/mL Penicillin-Streptomycin with 2mM Gln (final concentration). For experiments longer than 5 days, media was refreshed at day 5. Where indicated, cells were irradiated (IR, 5Gy) or treated with Venetoclax (Sanbio, #A0776; 0-100 μM), A-1331852 (ChemieTek, #CT-A133; 0 -100 μM), S-63845 (Chemgood, #C-1370; 0-100 μM) or ISRIB (Merck, #SML0843; 10 μM).
Flow cytometry
To assess protein levels of Bcl-2 family members, cells were stained with Fixable Viability Dye eFluor™ 780 (ThermoFisher Scientific, #65-0865-14) for 20min at 4°C, fixed and permeabilized with Cytofix/Cytoperm™ (BD Biosciences, #554714) following manufacturer’s instructions and subsequently stained with the following antibodies: anti-Bcl-2 BV421 (Biolegend, #658709), anti-Bcl-xL APC (Cell Signaling Technology, #12099S), anti-Mcl-1 PE (Cell Signaling Technology, #65617S) and anti-A1/Bfl-1 (kind gift of Prof. Dr. J. Borst, Leiden University Medical School, The Netherlands) for 20min at 4°C. Secondary goat anti-rabbit IgM+IgG-PE antibody (Southern Biotech, #4010-09S) was used to detect Bfl-1. Cells were analysed on a FACS Canto II cytometer (BD Biosciences, San Jose, CA, USA).
In primary CLL samples, percentage of leukemic cells and activation was measured by using the following antibodies: anti-CD5 PE (eBioscience, #12-0059-42), anti-CD19 FITC (BD Biosciences, #555412) and anti-CD95 PE-Cy7 (Merk, # 305621). Viable cells were gated using the Fixable Viability Dye eFluor™ 780.
Cell viability upon irradiation, glutamine deprivation or BH3 mimetics was assessed using MitoTracker™ Orange CMTMRos (Mito-O, Thermo Fisher Scientific, #M7510) and TO-PRO-3 (Thermo Fisher Scientific, #T3605). Cells were stained 25 min at 37°C with Mito-O, followed by incubation with TO-PRO-3 for 10 min and measured on FACS-Canto II cytometer (BD Biosciences). Viable cells were defined as Mito-O+ TOPRO-3-.
Cell growth was assessed in compete culture conditions or glutamine deprivation over time by counting cells using 123count™ eBeads Counting Beads (Thermo Fisher Scientific, # 01-1234-42).
All flow cytometry data were analysed using FlowJo v10 software (BD Biosciences).
Cell growth competition assays
Control parental PGA-1 (GFP-) cells were resuspended at a density of 200.000 cells/ml and plated at equal quantity with MOCK (GFP+) or TP53KO (GFP+) cells in complete medium or Gln deprivation (Gln-). GFP is co-expressed together with Cas9 via P2A cleavage site in pL-CRISPR.EFS.GFP, the plasmid used for CRISPR in PGA-1 cells. Cultures were maintained for 8 and 16 days (in experiments 1 and 2, respectively) and medium was refreshed at day 5. The relative proportion of viable GFP- or GFP+ cells was evaluated by Mito-O/To-Pro-3 staining and flow cytometry, gating on viable cells (MitoTracker™ Orange+ TO-PRO-3-) and GFP expression.
Comparative genomic hybridisation array
CGH array was performed on genomic DNA using the Affymetrix® Genome-Wide Human SNP Array 6.0 (CytoScan HD array, Affymetrix, Santa Clara, CA, USA). For data analysis, manufacturer’s protocols were followed and interpretation of copy number aberrations (CNAs) was carried out according to.45 Only aberrations present in at least 10% of the cells were reported. Genome coordinates were provided according to hg19/GRCh37 human reference genome and array profiles are described before.46
Extracellular flux analysis
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured by Mito Stress Test in a XF96 Extracellular Flux Analyzer (Agilent Technologies, Santa Clara, CA, USA), according to manufacturer’s instructions. Cells were resuspended in RPMI assay medium (Merck, #R1383) supplemented with 25 mM glucose, 1 mM pyruvate and 2 mM glutamine. 50,000 cells per well were plated in XF96 plates pre-coated with Poly-D-Lysine at 50μg/mL and centrifuged at 1700 rpm for 5 minutes with brake 0 to immobilize cells at the bottom of the wells. The correct formation of the cell monolayer was verified by microscope. XF plates were incubated in a non-CO2 incubator at 37°C for 1 hour to equilibrate the temperature. XF cartridges were hydrated overnight with 200 μl XF calibrant per well. Oxygen consumption was monitored under basal conditions and in response to Oligomycin (Merck, #O4876; 1 μM), FCCP (Merck, #C2920; 1.5 μM), Rotenone (Merck, #R8875; 100 nM) and Antimycin A (Merck, #A8674; 1 μM). Basal respiration rate was calculated as the difference between baseline OCR and OCR after inhibition of mitochondrial complexes I and III with rotenone and antimycin A, respectively. Results were analysed using Wave (Agilent Technologies).
Metabolomics analysis
Cells were cultured for 48 hours in complete medium and 5 million cells were pelleted from each condition. Absolute metabolite concentrations were measured by Human Metabolome Technologies (HMT) using capillary electrophoresis mass spectrometry (CE-TOFMS and CE-QqQMS) in the cation and anion analysis modes. Metabolites were extracted using 100% methanol supplemented with 550 μL of internal standard solution. Peaks detected in CE-TOFMS analysis were extracted using automatic integration software (MasterHands v.2.18.0.1 developed at Keio University)47 and those in CE-QqQMS analysis were extracted using automatic integration software (MassHunter Quantitative Analysis B.06.00, Agilent Technologies). All metabolite concentrations were calculated by normalizing the peak area of each metabolite with respect to the area of the internal standard and by using standard curves. Metabolomics data were analysed on MetaboAnalyst v5.0 by using the functions Statistical Analysis and Pathway Analysis with auto-scaling of the data.
13C6-glucose isotope tracing
PGA-1 cells, either WT (WT, MOCK) or TP53 KO (#1, #2) were cultured in complete media or Gln- media for 24h. Afterwards, cells were washed and cultured for 4h in fluxomics medium containing 5 mM D-Glucose-13C6 (Sigma, #389374), 2 mM glutamine and 50 μM Carnitine (Sigma, #C0283). Cells were pelleted, washed twice with ice-cold NaCl and analysed using a ZIC-cHILIC based full scan mode LC-MS platform following a liquid-liquid extraction in the presence of the internal standard Adenosine-15N5-monophosphate (5 nmol), as previously described.18 Data were analysed using Bruker TASQ software version 2.1.22.3. Relative isotope contribution was calculated using IsoCorrectoR Release 3.13.48
Western Blot
Protein lysates were obtained from 2-5 million cells (depending on experiments) and lysed using RIPA buffer. Protein concentration was measured by Pierce BCA assay (Thermo Fischer Scientific, #23227) and 50 μg of protein were loaded on Mini-PROTEAN TGX precast acrylamide gels (Biorad, #4569033), analysed by SDS-PAGE and transferred onto PVDF membranes. Membranes were blocked with 2% milk or 5% BSA and incubated with the following primary antibodies: anti-p53 (Calbiochem, #OP43), anti-p21 (Cell Signaling, #2947), anti-P-p53 (Ser15) (Cell Signaling, #9284), anti-Beta-actin (SICGEN, #AB0145-200) and secondary antibodies: Donkey αRabbit or αMouse-IRDye680/e800 (LI-COR Biosciences, Nebraska, United States). Membranes were developed using an Odyssey Imager (LI-COR).
Analysis of gene expression
Total RNA was isolated using the RNeasy kit (Qiagen, #74104) following manufacturer’s instructions. Gene expression of genes regulating apoptosis was analysed by reverse transcriptase multiplex ligation-dependent probe amplification (RT-MLPA) using SALSA MLPA KIT R011 Apoptosis mRNA (MRC-Holland, Amsterdam, The Netherlands), as previously described.24,25 Peaks were integrated with GeneMapper v5.0 and data were averaged and represented as Z-score or fold change, and plotted in heat maps using the R package ComplexHeatmap (https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html).
Gene expression of CHOP and ATF4 was analysed by RT-qPCR. RevertAid H Minus M-MuLV Reverse Transcriptase (Thermo Fisher scientific, #EP0451) and oligo(dT) primers were used for cDNA conversion. mRNA expression was quantified by RT-qPCR using SYBR green reaction Mix (Thermo Fisher scientific, #4385612). Primer sequences: ATF4_F: GGGACAGATTGGATGTTGGAGA, ATF4_R: GGGACAGATTGGATGTTGGAGA, CHOP_F: GGAGCATCAGTCCCCCACTT, CHOP_R: TGTGGGATTGAGGGTCACATC-3', TBP_F: CACATCACAGCTCCCCACCA, TBP_R: TGCACAGGAGCCCAAGAGTG. QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific) was used for analysis. Relative quantification (RQ) of mRNA expression was calculated with the equation RQ=2∧(−ΔΔCT) using TBP expression as housekeeping gene.
Quantification and statistical analysis
GraphPad Prism version 9.0.0 for Windows (GraphPad Software, Boston, Massachusetts USA) was used for the analysis. p values were calculated using t-test or two-way ANOVA with Tukey’s multiple comparison test. Differences were considered significant when p <0.05 (∗), p<0.01 (∗∗) or p<0.001 (∗∗∗). Comparisons performed in each case are detailed in figure legends. Importantly, TP53 WT (parental and MOCK) and KO (different TP53 clones) have been plotted separately in order to make the differences between the clones clearer and to facilitate the interpretation of the data. In some cases, statistical analyses have been performed between the two groups (TP53WT vs KO), these comparisons have been indicated in the corresponding figure legends.
Acknowledgments
This work was supported by the EU’s Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie grant agreement 766214 (META-CAN), Netherlands Organisation for Scientific Research/Netherlands Organization for Health Research and Development Vidi grant 91715337, ERC Consolidator: BOOTCAMP (864815), Lymph & Co.: 2018-LYCo-008, and Cancer Center Amsterdam grant 2022. The authors thank Dr. B. V. Schomakers and Dr. M. van Weeghel from the metabolomics core facility of Amsterdam UMC for help and advice with 13C isotope tracing experiments, Dr. Anne-Marie Van der Kevie-Kersemaekers and Dr. Clemens Mellink from the Genome Diagnostics Laboratory of Amsterdam UMC for support with CGH analyses, and Dr. M. Spaargaren, Dr. J. Guikema, and Dr. J. Borst for kindly providing cell lines and reagents used in this project.
Author contributions
Conceptualization: H.S.-M. and E.E.; methodology: C.M., H.S.-M., and E.E.; investigation and analysis: Z.C., C.M., I.A.M.D., G.C., E.A.K., and H.S.-M.; visualization: C.M. and H.S.-M.; writing – original draft: C.M., Z.C., H.S.-M., and E.E.; writing – review and editing: H.S.-M. and E.E.; funding acquisition and project administration: E.E.
Declaration of interests
The authors declare no competing interests.
Published: March 28, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109640.
Supplemental information
References
- 1.Vousden K.H., Lu X. Live or let die: The cell's response to p53. Nat. Rev. Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 2.Hollstein M., Sidransky D., Vogelstein B., Harris C.C. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 3.Stilgenbauer S., Schnaiter A., Paschka P., Zenz T., Rossi M., Döhner K., Bühler A., Böttcher S., Ritgen M., Kneba M., et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood. 2014;123:3247–3254. doi: 10.1182/blood-2014-01-546150. [DOI] [PubMed] [Google Scholar]
- 4.Landau D.A., Tausch E., Taylor-Weiner A.N., Stewart C., Reiter J.G., Bahlo J., Kluth S., Bozic I., Lawrence M., Böttcher S., et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525–530. doi: 10.1038/nature15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Te Raa G.D., Kater A.P. TP53 dysfunction in CLL: Implications for prognosis and treatment. Best Pract. Res. Clin. Haematol. 2016;29:90–99. doi: 10.1016/j.beha.2016.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Leroy K., Haioun C., Lepage E., Le Métayer N., Berger F., Labouyrie E., Meignin V., Petit B., Bastard C., Salles G., et al. p53 gene mutations are associated with poor survival in low and low-intermediate risk diffuse large B-cell lymphomas. Ann. Oncol. 2002;13:1108–1115. doi: 10.1093/annonc/mdf185. [DOI] [PubMed] [Google Scholar]
- 7.Diepstraten S.T., Anderson M.A., Czabotar P.E., Lessene G., Strasser A., Kelly G.L. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat. Rev. Cancer. 2022;22:45–64. doi: 10.1038/s41568-021-00407-4. [DOI] [PubMed] [Google Scholar]
- 8.Levine A.J., Oren M. The first 30 years of p53: growing ever more complex. Nat. Rev. Cancer. 2009;9:749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vousden K.H., Ryan K.M. P53 and metabolism. Nat. Rev. Cancer. 2009;9:691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- 10.Bensaad K., Tsuruta A., Selak M.A., Vidal M.N.C., Nakano K., Bartrons R., Gottlieb E., Vousden K.H. TIGAR, a p53-Inducible Regulator of Glycolysis and Apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 11.Berkers C.R., Maddocks O.D.K., Cheung E.C., Mor I., Vousden K.H. Metabolic regulation by p53 family members. Cell Metabol. 2013;18:617–633. doi: 10.1016/j.cmet.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simabuco F.M., Morale M.G., Pavan I.C.B., Morelli A.P., Silva F.R., Tamura R.E. p53 and metabolism: From mechanism to therapeutics. Oncotarget. 2018;9:23780–23823. doi: 10.18632/oncotarget.25267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Labuschagne C.F., Zani F., Vousden K.H. Control of metabolism by p53 – Cancer and beyond. Biochim. Biophys. Acta Rev. Cancer. 2018;1870:32–42. doi: 10.1016/j.bbcan.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin J., Byun J.K., Choi Y.K., Park K.G. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp. Mol. Med. 2023;55:706–715. doi: 10.1038/s12276-023-00971-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones R.G., Plas D.R., Kubek S., Buzzai M., Mu J., Xu Y., Birnbaum M.J., Thompson C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 16.Tajan M., Hock A.K., Blagih J., Robertson N.A., Labuschagne C.F., Kruiswijk F., Humpton T.J., Adams P.D., Vousden K.H. A Role for p53 in the Adaptation to Glutamine Starvation through the Expression of SLC1A3. Cell Metabol. 2018;28:721–736.e6. doi: 10.1016/j.cmet.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lowman X.H., Hanse E.A., Yang Y., Ishak Gabra M.B., Tran T.Q., Li H., Kong M. p53 Promotes Cancer Cell Adaptation to Glutamine Deprivation by Upregulating Slc7a3 to Increase Arginine Uptake. Cell Rep. 2019;26:3051–3060.e4. doi: 10.1016/j.celrep.2019.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Z., Simon-Molas H., Cretenet G., Valle-Argos B., Smith L.D., Forconi F., Schomakers B.V., van Weeghel M., Bryant D.J., van Bruggen J.A.C., et al. Characterization of metabolic alterations of chronic lymphocytic leukemia in the lymph node microenvironment. Blood. 2022;140:630–643. doi: 10.1182/blood.2021013990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Costa-Mattioli M., Walter P. The integrated stress response: From mechanism to disease. Science. 2020;368 doi: 10.1126/science.aat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sánchez-Vera I., Núñez-Vázquez S., Saura-Esteller J., Cosialls A.M., Heib J., Nadal Rodríguez P., Ghashghaei O., Lavilla R., Pons G., Gil J., Iglesias-Serret D. The Prohibitin-Binding Compound Fluorizoline Activates the Integrated Stress Response through the eIF2alpha Kinase HRI. Int. J. Mol. Sci. 2023;24 doi: 10.3390/ijms24098064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carter B.Z., Mak P.Y., Kornblau S.M., Tao W., Nishida Y., Ruvolo V., Cidado J., Drew L., Andreeff M. TP53 deficient/mutant AMLs are resistant to individual BH3 mimetics: high efficacy of combined inhibition of Bcl-2 and Mcl-1. Blood. 2019;134:1271. doi: 10.1182/blood-2019-124826. [DOI] [Google Scholar]
- 22.Thijssen R., Diepstraten S.T., Moujalled D., Chew E., Flensburg C., Shi M.X., Dengler M.A., Litalien V., MacRaild S., Chen M., et al. Intact TP-53 function is essential for sustaining durable responses to BH3-mimetic drugs in leukemias. Blood. 2021;137:2721–2735. doi: 10.1182/blood.2020010167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., Li Q., Shoemaker B.A., Thiessen P.A., Yu B., et al. PubChem 2023 update. Nucleic Acids Res. 2023;51:D1373–D1380. doi: 10.1093/nar/gkac956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eldering E., Spek C.A., Aberson H.L., Grummels A., Derks I.A., de Vos A.F., McElgunn C.J., Schouten J.P. Expression profiling via novel multiplex assay allows rapid assessment of gene regulation in defined signalling pathways. Nucleic Acids Res. 2003;31:e153. doi: 10.1093/nar/gng153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mackus W.J.M., Kater A.P., Grummels A., Evers L.M., Hooijbrink B., Kramer M.H.H., Castro J.E., Kipps T.J., van Lier R.A.W., van Oers M.H.J., Eldering E. Chronic lymphocytic leukemia cells display p53-dependent drug-induced Puma upregulation. Leukemia. 2005;19:427–434. doi: 10.1038/sj.leu.2403623. [DOI] [PubMed] [Google Scholar]
- 26.Thijssen R., Slinger E., Weller K., Geest C.R., Beaumont T., van Oers M.H.J., Kater A.P., Eldering E. Resistance to ABT-199 induced by microenvironmental signals in chronic lymphocytic leukemia can be counteracted by CD20 antibodies or kinase inhibitors. Haematologica. 2015;100:e302–e306. doi: 10.3324/haematol.2015.124560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang P., Du W., Wang X., Mancuso A., Gao X., Wu M., Yang X. P53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011;13:310–316. doi: 10.1038/ncb2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris J.P., 4th, Yashinskie J.J., Koche R., Chandwani R., Tian S., Chen C.C., Baslan T., Marinkovic Z.S., Sánchez-Rivera F.J., Leach S.D., et al. alpha-Ketoglutarate links p53 to cell fate during tumour suppression. Nature. 2019;573:595–599. doi: 10.1038/s41586-019-1577-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reid M.A., Wang W.I., Rosales K.R., Welliver M.X., Pan M., Kong M. The B55alpha subunit of PP2A drives a p53-dependent metabolic adaptation to glutamine deprivation. Mol. Cell. 2013;50:200–211. doi: 10.1016/j.molcel.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 30.Tian X., Ahsan N., Lulla A., Lev A., Abbosh P., Dicker D.T., Zhang S., El-Deiry W.S. P53-independent partial restoration of the p53 pathway in tumors with mutated p53 through ATF4 transcriptional modulation by ERK1/2 and CDK9. Neoplasia. 2021;23:304–325. doi: 10.1016/j.neo.2021.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seymour J.F., D. M.S., Pagel J.M., Kahl B.S., Wierda W.G. ABT-199 (GDC-0199) in relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL): High complete- response rate and durable disease control. J. Clin. Oncol. 2014;32:7015. doi: 10.1200/jco.2014.32.15_suppl.7015. [DOI] [Google Scholar]
- 32.Kater A.P., Wu J.Q., Kipps T., Eichhorst B., Hillmen P., D'Rozario J., Assouline S., Owen C., Robak T., de la Serna J., et al. Venetoclax Plus Rituximab in Relapsed Chronic Lymphocytic Leukemia: 4-Year Results and Evaluation of Impact of Genomic Complexity and Gene Mutations From the MURANO Phase III Study. J. Clin. Oncol. 2020;38:4042–4054. doi: 10.1200/JCO.20.00948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baliakas P., Jeromin S., Iskas M., Puiggros A., Plevova K., Nguyen-Khac F., Davis Z., Rigolin G.M., Visentin A., Xochelli A., et al. Cytogenetic complexity in chronic lymphocytic leukemia: definitions, associations, and clinical impact. Blood. 2019;133:1205–1216. doi: 10.1182/blood-2018-09-873083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anderson M.A., Deng J., Seymour J.F., Tam C., Kim S.Y., Fein J., Yu L., Brown J.R., Westerman D., Si E.G., et al. The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood. 2016;127:3215–3224. doi: 10.1182/blood-2016-01-688796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haselager M., Thijssen R., West C., Young L., Van Kampen R., Willmore E., Mackay S., Kater A., Eldering E. Regulation of Bcl-XL by non-canonical NF-kappaB in the context of CD40-induced drug resistance in CLL. Cell Death Differ. 2021;28:1658–1668. doi: 10.1038/s41418-020-00692-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bajpai R., Matulis S.M., Wei C., Nooka A.K., Von Hollen H.E., Lonial S., Boise L.H., Shanmugam M. Targeting glutamine metabolism in multiple myeloma enhances BIM binding to BCL-2 eliciting synthetic lethality to venetoclax. Oncogene. 2016;35:3955–3964. doi: 10.1038/onc.2015.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harnett C.C., Abusneina A., Clément J., Gauthier E.R. Inhibition of MCL-1 by obatoclax sensitizes Sp2/0-Ag14 hybridoma cells to glutamine deprivation-induced apoptosis. Cell Biochem. Funct. 2015;33:334–340. doi: 10.1002/cbf.3121. [DOI] [PubMed] [Google Scholar]
- 38.Durand R., Descamps G., Dousset C., Bellanger C., Maïga S., Alberge J.B., Derrien J., Cruard J., Minvielle S., Lilli N.L., et al. A p53 score derived from TP53 CRISPR/Cas9 HMCLs predicts survival and reveals major role of BAX in BH3 mimetics response. Blood. 2023;143 doi: 10.1182/blood.2023021581. [DOI] [PubMed] [Google Scholar]
- 39.Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018;24:927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 40.Yin Y., Stephen C.W., Luciani M.G., Fåhraeus R. p53 Stability and activity is regulated by Mdm2-mediated induction of alternative p53 translation products. Nat. Cell Biol. 2002;4:462–467. doi: 10.1038/ncb801. [DOI] [PubMed] [Google Scholar]
- 41.Yurekten O., Payne T., Tejera N., Amaladoss F.X., Martin C., Williams M., O'Donovan C. MetaboLights: open data repository for metabolomics. Nucleic Acids Res. 2024;52:D640–D646. doi: 10.1093/nar/gkad1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hallaert D.Y.H., Jaspers A., van Noesel C.J., van Oers M.H.J., Kater A.P., Eldering E. c-Abl kinase inhibitors overcome CD40-mediated drug resistance in CLL: implications for therapeutic targeting of chemoresistant niches. Blood. 2008;112:5141–5149. doi: 10.1182/blood-2008-03-146704. [DOI] [PubMed] [Google Scholar]
- 43.Kurtova A.V., Balakrishnan K., Chen R., Ding W., Schnabl S., Quiroga M.P., Sivina M., Wierda W.G., Estrov Z., Keating M.J., et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood. 2009;114:4441–4450. doi: 10.1182/blood-2009-07-233718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martens A.W.J., Janssen S.R., Derks I.A.M., Adams Iii H.C., Izhak L., van Kampen R., Tonino S.H., Eldering E., van der Windt G.J.W., Kater A.P. CD3xCD19 DART molecule treatment induces non-apoptotic killing and is efficient against high-risk chemotherapy and venetoclax-resistant chronic lymphocytic leukemia cells. J. Immunother. Cancer. 2020;8 doi: 10.1136/jitc-2019-000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schoumans J., Suela J., Hastings R., Muehlematter D., Rack K., van den Berg E., Berna Beverloo H., Stevens-Kroef M. Guidelines for genomic array analysis in acquired haematological neoplastic disorders. Genes Chromosomes Cancer. 2016;55:480–491. doi: 10.1002/gcc.22350. [DOI] [PubMed] [Google Scholar]
- 46.ISCN 2020: An International System for Human Cytogenomic Nomenclature (2020) (Cytogenetic and Genome Research). (2020). (S. Karger). [DOI] [PubMed]
- 47.Sugimoto M., Wong D.T., Hirayama A., Soga T., Tomita M. Capillary electrophoresis mass spectrometry-based saliva metabolomics identified oral, breast and pancreatic cancer-specific profiles. Metabolomics. 2010;6:78–95. doi: 10.1007/s11306-009-0178-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heinrich P., Kohler C., Ellmann L., Kuerner P., Spang R., Oefner P.J., Dettmer K. Correcting for natural isotope abundance and tracer impurity in MS-MS/MS- and high-resolution-multiple-tracer-data from stable isotope labeling experiments with IsoCorrectoR. Sci. Rep. 2018;8 doi: 10.1038/s41598-018-36293-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Metabolomics and 13C isotope tracing data are accessible in MetaboLights (EMBL-EBI)41: MTBLS9786.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyse the data reported in this work paper is available from the lead contact upon request.