

Abstract
Background
Antipsychotic drugs are the core treatment for schizophrenia. Treatment guidelines state that there is no difference in efficacy between the various first‐generation antipsychotics, however, low‐potency first‐generation antipsychotic drugs are sometimes perceived as less efficacious than high‐potency first‐generation compounds by clinicians, and they also seem to differ in their side effects.
Objectives
To review the effects of high‐potency, first‐generation perphenazine compared with low‐potency, first‐generation antipsychotic drugs for people with schizophrenia.
Search methods
We searched the Cochrane Schizophrenia Group Trials Register (October 2010).
Selection criteria
We included all randomised controlled trials (RCTs) comparing perphenazine with first‐generation, low‐potency antipsychotic drugs for people with schizophrenia or schizophrenia‐like psychoses.
Data collection and analysis
We extracted data independently. For dichotomous data we calculated risk ratios (RR) and their 95% confidence intervals (CI) on an intention‐to‐treat basis and using a random‐effects model.
Main results
The review currently includes four relevant randomised trials with 365 participants. The size of the included studies was between 42 and 158 participants with a study length between one and four months. Overall, the methods of sequence generation and allocation concealment were poorly reported. Most studies were rated as low risk of bias in terms of blinding. Overall, attrition bias in the studies was high.
The effects of perphenazine and low‐potency antipsychotic drugs seemed to be similar in terms of the primary outcome – response to treatment (perphenazine 58%, low‐potency antipsychotics 59%, 2 RCTs, n = 138, RR 0.97 CI 0.74 to 1.26 – moderate quality of evidence). There was also no clear evidence of a difference in acceptability of treatment with the number of participants leaving the studies early due to any reason, however results were imprecise (perphenazine 30%, low‐potency antipsychotics 28%, 3 RCTs, n = 323, RR 0.78 CI 0.35 to 1.76, very low quality of evidence).
There were low numbers of studies available for the outcomes experiencing at least one adverse effect (perphenazine 33%, low‐potency antipsychotics 47%, 2 RCTs, n = 165, RR 0.83 CI 0.36 to 1.95, low quality evidence) and experiencing at least one movement disorder (perphenazine 22%, low‐potency first‐generation antipsychotics 0%, 1 RCT, n = 69, RR 15.62 CI 0.94 to 260.49, low quality evidence), and the confidence intervals for the estimated effects did not exclude important differences. Akathisia was more frequent in the perphenazine group (perphenazine 25%, low‐potency antipsychotics 22%, 2 RCTs, n = 227, RR 9.45 CI 1.69 to 52.88), whereas severe toxicity was less so (perphenazine 42%, low‐potency antipsychotics 69%, 1 RCT, n = 96, RR 0.61 CI 0.41 to 0.89).
There were three deaths in the low‐potency group by four months but the difference between groups was not significant (perphenazine 0%, low‐potency antipsychotics 2%, 1 RCT, n = 96, RR 0.14 CI 0.01 to 2.69, moderate quality evidence). No data were available for our prespecified outcomes of interest sedation or quality of life. Data were not available for other outcomes such as relapse, service use, costs and satisfaction with care.
The event rates reported quote simple aggregates and are not based on the RRs.
Authors' conclusions
The results do not show a superiority in efficacy of high‐potency perphenazine compared with low‐potency first‐generation antipsychotics. There is some evidence that perphenazine is more likely to cause akathisia and less likely to cause severe toxicity, but most adverse effect results were equivocal. The number of studies as well as the quality of studies is low, with quality of evidence for the main outcomes ranging from moderate to very low, so more randomised evidence would be needed for conclusions to be made.
Keywords: Adult, Humans, Antipsychotic Agents, Antipsychotic Agents/therapeutic use, Perphenazine, Perphenazine/therapeutic use, Randomized Controlled Trials as Topic, Schizophrenia, Schizophrenia/drug therapy
Plain language summary
Perphenazine versus low‐potency first‐generation drugs for schizophrenia
Schizophrenia is a severe mental illness where people experience ‘positive symptoms’ (such as hearing voices, seeing things and having strange beliefs) and ‘negative symptoms’ (such as tiredness, apathy and loss of emotion).
Antipsychotic drugs are the main treatment for schizophrenia and can be grouped into older drugs (‘typical’ or first‐generation) and newer drugs (‘atypical’ or second‐generation), and within these groups you can have low strength (low‐potency) or high strength (high‐potency) antipsychotics. Perphenazine is a high‐potency first‐generation antipsychotic. Low‐potency antipsychotics are often seen by psychiatrists and health professionals as less effective in treating schizophrenia than high‐potency antipsychotic drugs; they also differ in side effects. Low‐potency antipsychotic drugs often cause sleepiness and low blood pressure whereas high‐potency antipsychotic drugs often produce movement disorders such as restlessness, shaking and tremors. Typical examples of low‐potency first‐generation antipsychotic drugs are chlorpromazine, chlorprothixene, thioridazine or levomepromazine.
The review aims to compare a high‐potency first‐generation antipsychotic, perphenazine with low‐potency first‐generation antipsychotics. A search for trials was run in 2010. Four trials that randomised a total of 365 people are included. The studies compared perphenazine with chlorpromazine, thioridazine and levomepromazine. Overall, the trials were of poor quality, poorly reported and small scale. Review authors also rated the quality of evidence for the main outcomes to range from moderate to very low quality.
It was found that perphenazine was not obviously clinically superior to low‐potency first‐generation antipsychotic drugs but was more likely cause the movement disorder akathisia (inner restlessness and the inability to sit still). Low‐potency first‐generation antipsychotics are thought more likely to cause side effects such as sedation and hypotension but evidence from this review showed people taking perphenazine were just as likely to experience hypotension as those taking first‐generation antipsychotics and no data were available for sedation. Other outcomes, such as re‐hospitalisation, costs, healthy days and quality of life were not addressed in the studies.
No firm conclusions can be made about perphenazine's superiority or inferiority over low‐potency first‐generation antipsychotics until newer and better conducted studies are completed.
This plain language summary has been written by a consumer, Benjamin Gray, from Rethink Mental Illness.
Summary of findings
Summary of findings for the main comparison. Comparison 1: PERPHENAZINE versus LOW‐POTENCY ANTIPSYCHOTIC DRUGS for schizophrenia.
| Comparison 1: PERPHENAZINE versus LOW‐POTENCY ANTIPSYCHOTIC DRUGS for schizophrenia | ||||||
| Patient or population: patients with schizophrenia Settings: hospital Intervention: Comparison 1: PERPHENAZINE versus LOW‐POTENCY ANTIPSYCHOTIC DRUGS | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Low‐potency first‐generation antipsychotic drugs | Perphenazine | |||||
| Response to treatment Follow‐up: 1‐4 months | Study population | RR 0.97 (0.74 to 1.26) | 138 (2 studies) | ⊕⊕⊕⊝ moderate1 | ||
| 594 per 1000 | 576 per 1000 (440 to 749) | |||||
| Moderate | ||||||
| 641 per 1000 | 622 per 1000 (474 to 808) | |||||
| Acceptability of treatment ‐ leaving the study early due to any reason Follow‐up: 1‐4 months | Study population | RR 0.78 (0.35 to 1.76) | 323 (3 studies) | ⊕⊝⊝⊝ very low1,2,3 | ||
| 285 per 1000 | 222 per 1000 (100 to 502) | |||||
| Moderate | ||||||
| 210 per 1000 | 164 per 1000 (73 to 370) | |||||
| Adverse effects ‐ at least one adverse effect Follow‐up: 2‐4 months | Study population | RR 0.83 (0.36 to 1.95) | 165 (2 studies) | ⊕⊕⊝⊝ low1,3 | ||
| 469 per 1000 | 389 per 1000 (169 to 915) | |||||
| Moderate | ||||||
| 420 per 1000 | 349 per 1000 (151 to 819) | |||||
| Adverse effects ‐ movement disorders ‐ at least one movement disorder Follow‐up: 6 weeks | Study population | RR 15.62 (0.94 to 260.49) | 69 (1 study) | ⊕⊕⊝⊝ low1,4 | ||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate | ||||||
| 219 per 1000 | 1000 per 1000 (206 to 1000) | |||||
| Death Follow‐up: mean 4 months | Study population | RR 0.14 (0.01 to 2.69) | 96 (1 study) | ⊕⊕⊕⊝ moderate3 | ||
| 62 per 1000 | 9 per 1000 (1 to 168) | |||||
| Moderate | ||||||
| 63 per 1000 | 9 per 1000 (1 to 169) | |||||
| Adverse effects ‐ other ‐ sedation | See comment | Not estimable | 0(0) | See comment | There were no data available for these important outcomes. | |
| Quality of life | ||||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Risk of bias: rated 'serious' ‐ many studies did not report the methods for sequence generation and/or allocation concealment, missing or unclear results for incomplete outcome data and selective reporting. 2 Inconsistency: rated 'serious' ‐ P value for heterogeneity was statistically significant (P 0.04) and the I2 = 70%. 3 Imprecision: rated 'serious' ‐ only few studies contribute data to this event and the CI was quite wide. 4 Imprecision: rated 'serious' ‐ only few studies contribute data to this event (event rate less than 300 and the CI was very wide.
Background
Description of the condition
Schizophrenia is often a chronic and disabling psychiatric disorder. It afflicts approximately one per cent of the population world wide with little gender differences (Berger 2003). The typical manifestations of schizophrenia are 'positive' symptoms such as fixed, false beliefs (delusions) and perceptions without cause (hallucinations), 'negative' symptoms such as apathy and lack of drive, disorganisation of behaviour and thought, and catatonic symptoms such as mannerisms and bizarre posturing (Carpenter 1994). The degree of suffering and disability is considerable, with 80% to 90% of people with schizophrenia not working (Marvaha 2004) and up to 10% dying by suicide (Tsuang 1978).
Description of the intervention
Antipsychotic drugs are the core treatment for schizophrenia. Both first‐ and second‐generation antipsychotic drugs block, to a greater or lesser extent, D2‐receptors in the brain. They can be classified according to their biochemical structure (e.g. butyrophenones, phenothiazines, thioxanthenes etc), their risk of producing movement disorders ('atypical' versus 'typical' antipsychotics) and the doses necessary for an antipsychotic effect (high‐potency versus low‐potency antipsychotics). The classification into high‐potency and low‐potency medication means that for low‐potency antipsychotic drugs, higher doses are necessary to obtain the same dopamine receptor occupancy and efficacy (Seeman 1975). In this context, perphenazine belongs to the high‐potency antipsychotic drug group. It is mostly indicated in schizophrenia, psychosis and the manic phases of bipolar disorder.
Low‐potency first‐generation antipsychotic drugs will be the comparator drugs in this review. Typical examples of low‐potency first‐generation antipsychotic drugs are chlorpromazine, chlorprothixene, thioridazine or levomepromazine. It is an old psychiatric dogma that can be found in textbooks and guidelines that ‐ with the exception of clozapine ‐ there is no difference in efficacy between any antipsychotic compounds (Gaebel 2006; Lehman 2004). Nevertheless, low‐potency antipsychotic drugs are often perceived as less efficacious than high‐potency compounds by clinicians, and high‐ and low‐potency antipsychotics also seem to differ in side effects. Low‐potency drugs have a high incidence of sedation or hypotonia, whereas high‐potency drugs produce more extrapyramidal side effects.
How the intervention might work
The theory is that schizophrenia is a chronic disorder caused by hyper‐dopaminergic states in the limbic system (Berger 2003). All antipsychotic drugs block dopamine receptors. Perphenazine (2‐[4‐[3‐(2‐chloro‐10H‐phenothiazin‐10‐yl) propyl]piperazin‐1‐yl]ethanol, Figure 1) is a phenothiazine which effectively treats the positive symptoms of schizophrenia, such as hallucinations and delusions (Hartung 2005). It is less potent than haloperidol but roughly five times more potent than chlorpromazine (Davis 1974). Therefore, it is sometimes also considered to be a medium‐potency antipsychotic and not a high‐potency antipsychotic. Perphenazine has a bioavailability of approximately 40% and a half‐life of eight to 12 hours (Berger 2003). It shares in general all the side effects of haloperidol, such as extrapyramidal side effects.
1.
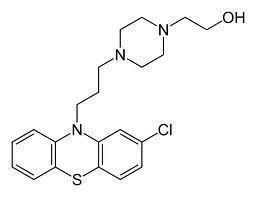
Perphenazine
Low‐potency medications have a lower affinity for dopamine receptors so that a higher dose is required to effectively treat symptoms of schizophrenia. They additionally block other receptors than dopamine, such as cholinergic or histaminergic receptors. This also explains the occurrence of adverse effects, which are less frequent with high‐potency drugs, such as sedation or hypotonia. The cutoff between high‐ and low‐potency drugs is not clear, but it has been tried to express their relationship in terms of dose equivalence. The most frequently applied concept is based on chlorpromazine equivalents according to Davis 1974 or Haase 1983, which provide data about comparable doses of various antipsychotic drugs to achieve an effect similar to 100 mg chlorpromazine.
Why it is important to do this review
Systematic reviews on the comparative effects of high‐potency versus low‐potency first‐ generation antipsychotic drugs are not available. Cochrane reviews on the effects of specific first‐generation antipsychotic drugs have been published, but they compared the effects of one antipsychotic drug versus any other antipsychotic drug (e.g. pimozide versus any other antipsychotic drug, Fenton 2007) and thus did not consider the important classification in high‐potency and low‐potency antipsychotics. Due to this lack of evidence treatment guidelines make statements such as “all conventional antipsychotics if adequately dosed have comparable efficacy” (German national schizophrenia guideline (Gaebel 2006); also see guideline of the World Federations of Societies of Biological Psychiatry (Falkai 2005)).
These guidelines contrast with the clinical impression that low‐potency first‐generation antipsychotic drugs are less efficacious than high‐potency conventional antipsychotic drugs. The clinical consequences to follow these guidelines are considerable, because high‐potency and low‐potency antipsychotics differ clearly in side effects. High‐potency antipsychotics often lead to strong extrapyramidal symptoms, low‐potency antipsychotics on the other hand have strong sedating properties and often also produce hypotension.
First‐generation antipsychotic drugs are still the mainstay of treatment in countries that cannot afford newer, expensive 'atypical' or 'second‐generation' antipsychotic drugs and even in some industrialised countries such as Germany, first‐generation antipsychotic medications still account for 50% of the market share (Lohse 2005). Recent studies on these more expensive second‐generation antipsychotics have also called into question their superiority (Jones 2006; Leucht 2009; Lieberman 2005). Therefore, research on older first‐generation agents is essential and has been asked for (Leucht 2009). This review will be part of a family of similar reviews (Table 2).
1. Series of similar reviews.
| Title | Reference |
| Haloperidol versus first‐generation antipsychotic drugs | Dold 2012 |
| Haloperidol versus low‐potency antipsychotic drugs | Tardy 2014b |
| Flupenthixol versus low‐potency antipsychotic drugs | Tardy 2014 |
| Fluphenazine versus low‐potency antipsychotic drugs | Tardy 2014a |
| Trifluoperazine versus low‐potency antipsychotic drugs | Tardy 2014c |
Objectives
To review the effects of the high‐potency first‐generation antipsychotic drug perphenazine versus low‐potency first‐generation antipsychotic drugs.
Methods
Criteria for considering studies for this review
Types of studies
We included only randomised studies with people suffering schizophrenia or related disorders in this review. We excluded quasi‐randomised trials, such as those where allocation is undertaken on surname. If a trial implied it was randomised without fully describing allocation, these were also included.
Types of participants
We included people with schizophrenia and schizophrenia‐like psychoses (schizophreniform and schizoaffective disorders) who had stabilised on antipsychotic medications. There is no clear evidence that the schizophrenia‐like psychoses are caused by fundamentally different disease processes or require different treatment approaches (Carpenter 1994). We also included studies that used diagnostic criteria other than ICD‐10 (International Statistical Classification of Diseases, tenth revision) or DSM‐IV ( Diagnostic and Statistical Manual of Mental disorders, fourth edition),. These diagnostic criteria are not meticulously used in clinical routine either, so broader inclusion criteria will enhance applicability.
We were interested in making sure that information is as relevant to the current care of people with schizophrenia as possible so proposed to clearly highlight the current clinical state (acute, early post‐acute, partial remission, remission) as well as the stage (prodromal, first episode, early illness, persistent) and as to whether the studies primarily focused on people with particular problems (for example, negative symptoms, treatment‐resistant illnesses).
Types of interventions
1. Perphenazine (a high‐potency first‐generation drug)
Any dose of oral mode of administration (no depots, no short‐acting parenteral forms of administration). We made an a priori decision that perphenazine will be the intervention because it is sometimes perceived to be more efficacious than low‐potency drugs by clinicians. Therefore, our hypothesis is that perphenazine is more effective, so that we have chosen it as the intervention.
2. Low‐potency first‐generation antipsychotic drugs
The control interventions were low‐potency first‐generation antipsychotic drugs, any oral form of administration and any oral dose. We used the dose equivalence tables presented by Davis 1974 and/or Haase 1983 and defined drugs as low potency which had equivalence doses roughly equal to or higher than chlorpromazine. The chlorpromazine equivalences of sulpiride are often estimated to be approximately 100. However, its properties are similar to those of amisulpride, which is an atypical antipsychotic and not within the scope of this review. Moreover, sulpiride does not cause a lot of sedation, which is another important characteristic of low‐potency antipsychotics. Therefore, we decided a priori to not consider sulpiride in this review.
Types of outcome measures
We analysed the outcomes for different lengths of follow‐up: up to three months (short term), six months (medium term) or more than six months (long term).
Primary outcomes
1. Response to treatment
Response to treatment as defined by the original studies
Secondary outcomes
1. Mental state: symptoms of schizophrenia
1.1 Overall symptoms ‐ average score/change in mental state 1.2 Positive symptoms ‐ average score/change in positive symptoms 1.3 Negative symptoms ‐ average score/change in negative symptoms
2. Global state: average score/change in global state
3. Relapse ‐ as defined by each of the studies
4. Leaving the study early
4.1 Acceptability of treatment ‐ leaving the study early due to any reason 4.2 Leaving the study early due to inefficacy of treatment 4.3 Leaving the study early due to side effects
5. Service use
5.1 Rehospitalisation
6. Adverse effects
6.1 At least one adverse effect 6.2 Extrapyramidal/movement disorders 6.3 Cardiac effects 6.4 Hypotension 6.5 Sedation 6.6 Weight gain 6.7 Other
6. Death
6.1 Death (all causes) 6.2 Suicide
8. Quality of life
9. Participant's/carer's satisfaction with care
10. Economic outcomes
11. 'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2008) and used GRADE profiler (GRADE 2004) to import data from Review Manager 5 (Review Manager 2008) to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes we have rated as important to patient care and decision making. We selected the following long‐term main outcomes for inclusion in the 'Summary of findings' table.
Response to treatment
Acceptability of treatment ‐ leaving the study early due to any reason
Adverse effects ‐ at least one adverse event
Adverse effects ‐ movement disorders ‐ at least one movement disorder
Adverse effects ‐ sedation
Death
Quality of life
Search methods for identification of studies
We applied no language restriction within the limitations of the search tools.
Electronic searches
We searched the ‘Cochrane Schizophrenia Group Trials Register’ for relevant studies (October 2010) using the phrase:
[(*perphenazine* in intervention of STUDY) OR (*perphenazine* in title, abstract and index terms of REFERENCE entered > = 1 May 2010.]
This register is compiled by systematic searches of major databases, handsearches and conference proceedings (see Group Module).
Searching other resources
1. Reference searching
We inspected the references of all identified included studies for more trials.
2. Previous reviews
We searched previous conventional reviews (Davis 1989; Klein 1969).
3. Personal contact
We contacted the first author of each included study for missing information and for the existence of further studies.
4. Drug companies
We contacted the original manufacturers of perphenazine and asked them for further relevant studies and for missing information on identified studies.
Data collection and analysis
Selection of studies
Two review authors (MT, MH) independently inspected all abstracts identified in the searches. We resolved disagreements by discussion; where doubt still remained, we acquired the full article for further inspection. Once we obtained the full articles, at least two review authors independently decided whether the studies met the review criteria. If disagreement could not be resolved by discussion, we resolved it with a third review author (SL) or sought further information from the study authors.
Data extraction and management
1. Extraction
Two review authors (MT, MH) independently extracted data from all selected trials. We decided post‐hoc to include all outcomes reported by a study, not only the predefined outcomes in the methods section. For the outcomes added post‐hoc only a random sample of 25% were independently extracted by a second review author (MH). When disagreement arose, we resolved it by discussion with a third review author (SL). Where this was not possible we contacted the study authors to resolve the dilemma.
2. Management
2.1 Forms
We extracted data onto simple, standard forms.
2.2 Scale‐derived data
We intended to include continuous data from rating scales only if: a. the psychometric properties of the measuring instrument have been described in a peer‐reviewed journal (Marshall 2000); and b. the measuring instrument was not written or modified by one of the trialists for that particular trial.
Ideally, the measuring instrument should either be i. a self‐report or ii. completed by an independent rater or relative (not the therapist), but we realise that this is not often reported clearly.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand, calculation of change needs two assessments (baseline and endpoint), which can be difficult in unstable and difficult to measure conditions such as schizophrenia. We decided to primarily to use endpoint data and only use change data if the former were not available. We planned to combine endpoint and change data in the analysis using mean differences (MD) rather than standardised mean differences (Higgins 2011, Chapter 9.4.5.2).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we aimed to apply the following standards to all data before inclusion: a) standard deviations (SDs ) and means are reported in the paper or obtainable from the authors; b) when a scale starts from the finite number zero, the SD, when multiplied by two, is less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution (Altman 1996)); c) if a scale started from a positive value (such as the Positive and Negative Syndrome Scale (PANSS,(Kay 1986), which can have values from 30 to 210), we planned to modify the calculation described above to take the scale starting point into account. In these cases, skew is present if 2 SD > (S‐S min), where S is the mean score and S min is the minimum score. Endpoint scores on scales often have a finite start and end point and these rules can be applied. We planned to enter skewed endpoint data from studies of fewer than 200 participants in additional tables rather than into an analysis. Skewed data pose less of a problem when looking at means if the sample size is large and we would have entered such data into syntheses. When continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not. We planned to enter change data into analysis.
2.5 Common measure
To facilitate comparison between trials, we intended to convert variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary
Where possible, we attempted to convert outcome measures to dichotomous data. This could be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. We generally assumed that if there had been a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the PANSS (Kay 1986), this could be considered as a clinically significant response (Leucht 2005a; Leucht 2005b). If data based on these thresholds were not available, we would have used the primary cut‐off presented by the original authors.
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicates a favourable outcome for perphenazine. Where keeping to this made it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'Not improved'), we reported data where the left of the line indicates an unfavourable outcome. We noted this in the relevant graphs.
Assessment of risk of bias in included studies
Again, review authors MT and MH worked independently to assess risk of bias by using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) to assess trial quality. This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting.
If the raters disagreed, we made the final rating by consensus with the involvement of another member of the review group (SL). Where inadequate details of randomisation and other characteristics of trials were provided, we contacted authors of the studies in order to obtain further information. We reported non‐concurrence in quality assessment, but if disputes arose as to which category a trial should be allocated, again, we sought resolution by discussion.
Measures of treatment effect
1. Dichotomous data
The review focused on binary data which are easy to interpret and can be intuitively understood. For binary outcomes, we calculated a standard estimation of the random‐effects risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios (OR) and that OR tend to be interpreted as RR by clinicians (Deeks 2000). This misinterpretation then leads to an overestimate of the impression of the effect.
2. Continuous data
For continuous outcomes, we estimated a mean difference (MD) between groups using the random‐effects model as this takes into account any differences between studies even if there is no statistically significant heterogeneity. We did not calculate standardised mean difference (SMD) measures. There was one exception to this rule, however: in the case of where scales were of such similarity to allow pooling, we calculated the SMD and, whenever possible, transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intraclass correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
If results from trials had not adjusted for clustering, we would have attempted to adjust the results for clustering, by multiplying the standard errors of the effect estimates (risk ratio or mean difference, ignoring clustering) by the square root of the design effect. The design effect is calculated as DEff = 1 + (M ‐ 1) ICC, where M is the average cluster size and ICC is the intra‐cluster coefficient (Higgins 2011). If an ICC was not available from the trial, other sources would have been used to impute ICCs (Campbell 2000)
If clustering had been incorporated into the analysis of primary studies, we would have presented these data as if from a non‐cluster randomised study, but adjusted for the clustering effect. If a cluster study had been appropriately analysed taking into account ICC and relevant data documented in the report, synthesis with parallel group randomised trials would have been possible using the generic inverse variance technique, where the natural logarithm of the effect estimate (and standard errors) for all included trials for that outcome would be calculated and entered into RevMan along with the log of the effect estimate (and standard errors) from the cluster randomised trial(s). We would have used methods described in section 7.7.7.2 and 7.7.7.3 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) to obtain standard errors.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in schizophrenia, randomised cross‐over studies could be eligible, but only data up to the point of first cross‐over.
3. Studies with multiple treatment groups
Where a study involved more than two treatment arms, if relevant, we would have presented the additional treatment arms in comparisons. If data were binary, we simply would have added these and combined within the two‐by‐two table. If data were continuous, we would have combined the data following the formula in section 7.7.3.8 (Combining groups) of the Handbook. Where the additional treatment arms were not relevant, we would not have reproduced these data. However, we did not include studies with multiple treatment groups.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). The loss to follow‐up in randomised schizophrenia trials is often considerable, calling the validity of the results into question. Nevertheless, it is unclear what degree of attrition leads to a high degree of bias. We did not exclude trials from outcomes on the basis of the percentage of participants completing them. We, however, used the 'Risk of bias' tool described above to indicate potential bias when more than 25% of the participants from the perphenazine group and low‐potency drug group left the studies prematurely (Xia 2009), when the reasons for attrition differed between the intervention and the control group, and when no appropriate imputation strategies were applied.
2. Dichotomous data
We presented data on a 'once‐randomised‐always‐analyse' basis, assuming an intention‐to‐treat (ITT) analysis. If the authors applied such a strategy, we used their results. If the original authors presented only the results of the per‐protocol or completer population, we assumed that those participants lost to follow‐up would have had the same percentage of events as those who remained in the study.
3. Continuous data
3.1 Attrition
We used ITT when available. We anticipated that in some studies, in order to do an ITT analysis, we would employ the method of last observation carried forward (LOCF) within the study report. As with all methods of imputation to deal with missing data, LOCF introduces uncertainty about the reliability of the results (Leon 2006). Therefore, where we used LOCF data in the analysis, we indicated this in the review.
3.2 Standard deviations
We first tried to obtain the missing values from the authors. If not available, where there were missing measures of variance for continuous data, but an exact standard error (SE) and confidence interval were available for group means, and either P value or T value were available for differences in mean, we calculated them according to the rules described in the Handbook (Higgins 2011). When only the SE was reported, we calculated standard deviations (SDs) by the formula SD = SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Handbook (Higgins 2011) present detailed formulae for estimating SDs from P, T or F values, confidence intervals, ranges or other statistics. If these formulae did not apply, we calculated the SDs according to a validated imputation method, which is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study's outcome and thus to lose information. We nevertheless examined the validity of the imputations in a sensitivity analysis excluding imputed values.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies without any comparison group to judge clinical heterogeneity.
We simply inspected all studies for clearly outlying situations or people which we had not predicted would arise. If such situations or participant groups arose, we fully discussed these.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods which we had not predicted would arise. If such methodological outliers arose, we fully discussed these.
3. Statistical
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2statistic
We investigated heterogeneity between studies by considering the I2 method alongside the Chi2 P value. The I2 provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2011). The importance of the observed value of I2 depends on i. magnitude and direction of effects and ii. strength of evidence for heterogeneity (e.g. P value from Chi2 test, or a confidence interval for I2).
An I2 estimate of 50% to 90%, accompanied by a statistically significant Chi2 statistic, may represent substantial heterogeneity (Section 9.5.2 ‐ Higgins 2011); we planned to explore reasons for heterogeneity. If the inconsistency was high and we found clear reasons, we presented data separately.
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Section 10 of the Handbook (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We had intended not to use funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar sizes.
Data synthesis
We employed a random‐effects model for analyses (Der‐Simonian 1986). We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This does seem true to us and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. Therefore, the random‐effects model is usually more conservative in terms of statistical significance, although as a disadvantage it puts added weight onto smaller studies, which can either inflate or deflate the effect size. We examined in a secondary analysis whether using a fixed‐effect model markedly changed the results of the primary outcome.
Subgroup analysis and investigation of heterogeneity
All subgroup analyses were performed only on the primary outcome response to treatment.
1. Subgroup analysis
1.1 Different low‐potency drugs
In one subgroup analysis we compared perphenazine with each low‐potency antipsychotic separately.
1.2 Clinical state, stage or problem
We proposed to undertake this review and provide an overview of the effects of perphenazine versus low‐potency antipsychotics for people with schizophrenia in general. In addition, however, we tried to report data on subgroups of people in the same clinical state, stage and with similar problems.
1.3 Investigation of heterogeneity
If inconsistency was high, we reported this. First, we investigated whether data had been entered correctly. Second, if data were correct, we visually inspected the graph and successively removed outlying studies to see if heterogeneity was restored. For this review, we decided that should this occur with data contributing to the summary finding of no more than around 10% of the total weighting, we would present the data. If not, we would not pool data but discuss the issues. We know of no supporting research for this 10% cut‐off but are investigating use of prediction intervals as an alternative to this unsatisfactory state.
When unanticipated clinical or methodological heterogeneity were obvious, we simply stated hypotheses regarding these for future reviews or versions of this review. We did not anticipate undertaking analyses relating to these.
Sensitivity analysis
1. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way as to imply randomisation. For the primary outcome, we included these studies and, if there was no substantive difference when we added the implied randomised studies to those with better description of randomisation, then we employed all data from these studies.
2. Implication of non double‐blind trials
We aimed to include trials in a sensitivity analysis if participants and treating psychiatrists were not blinded. For the primary outcome we included these studies and, if there was no substantive difference when we added the non double‐blind studies to the double‐blind studies, then we employed all data from these studies.
3. Assessment of dosage
We aimed to include trials in a sensitivity analysis if doses between perphenazine and low‐potency antipsychotics were clearly discrepant by our judgement based on the chlorpromazine equivalence tables in Davis 1974, Haase 1983 and Andreasen 2010. If, for the primary outcome, there was no substantive difference when we added studies with discrepant doses, then we employed all data from these studies.
Results
Description of studies
For substantive description of studies please see Characteristics of included studies and Characteristics of excluded studies tables.
Results of the search
The search strategy in the "Cochrane Schizophrenia Group Trials Register" generated 318 reports of which 17 studies were closely inspected (Figure 2).
2.
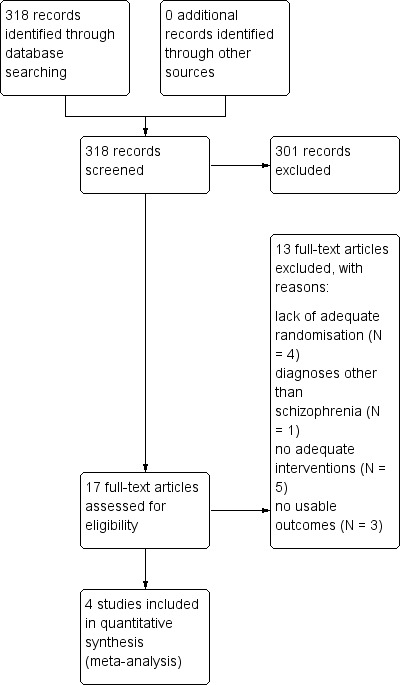
Study flow diagram.
Included studies
Four studies (365 participants) met the inclusion criteria.
1. Length of trials
Of the included studies, two studies (Hanlon 1965,;Shalev 1993) had a duration of four weeks, one of six weeks (Kurland 1961) and one study lasted up to four months (Adelson 1962). Long‐term studies were not available.
2. Participants
In three studies, participants were diagnosed according to clinical criteria, one study (Shalev 1993) diagnosed according to DSM‐III (APA 1980). The mean age was 36.1 years for those studies which indicated these data.
3. Setting
All studies were conducted in hospitals.
4. Study size
Hanlon 1965 was the largest study with 158 participants, followed by Adelson 1962 with 96 and by Kurland 1961 with 69 participants. Shalev 1993 randomised 42 participants.
5. Interventions
All studies compared perphenazine with low‐potency first‐generation antipsychotic drugs. In most studies flexible doses could be applied. The dose ranges were: 100 to 3000 mg/day for chlorpromazine (three studies) and perphenazine 8 to 240 mg/day. Two studies reported mean doses only, thioridazine 193 mg/day (Hanlon 1965) and levomepromazine 379 mg/day (Shalev 1993).
6. Outcomes
6.1 Response to treatment
Our primary outcome was response to treatment as defined by the original studies. Two studies (Adelson 1962; Shalev 1993) based response to treatment on clinical judgement by a psychiatrist and were the only studies that reported sufficient data for this outcome.
6.2 Mental state
None of the included studies reported scale derived data on mental state.
6.3 Leaving the study early
The number of participants leaving the study early were recorded for the categories any reason, adverse events and lack of efficacy. Three studies (Adelson 1962; Hanlon 1965; Kurland 1961) reported on this outcome.
6.4 Adverse effects
The following adverse effects: at least one adverse event, at least one movement disorder, akathisia, akinesia, dyskinesia, death, hypotension, leucopenia, neurologic symptoms (not otherwise specified), rash, severe toxicity and vasomotor episodes were reported in a dichotomous manner in terms of the number of participants with a given side effect.
6.5 Scale data
Although studies presented scale data, presentation was poor (see Characteristics of included studies) and we could not use any scale data for analyses.
6.6 Missing outcomes
None of the included studies reported on important outcomes such as relapse, service use, quality of life, participants´/carers´ satisfaction with care or economic outcomes.
Excluded studies
Thirteen studies were excluded. Four were not randomised (Hollister 1974; Schulsinger 1958; Smith 1959; Svestka 1972). One study was on a mixed population and it was not reported how many participants had schizophrenia (Nordic 1986). Five studies compared perphenazine with medications not of interest for this review. Of these, one study compared perphenazine with placebo (Akimoto 1966), two studies with second‐generation antipsychotics (Anon 2006; Loza 2001), one study examined the effects of perphenazine combined with chlorpromazine (Lapolla 1967) and one study compared perphenazine with oxypertine, which we could not easily classify as low‐potency or high‐potency (Svestka 1974). Moreover, Svestka 1974 was a cross‐over study without data for the first cross‐over phase. Three studies did not present any usable data for this review (Bennett 1961; Casey 1960; Vinar 1968).
Risk of bias in included studies
For graphical representations of our judgements of risk of bias please refer to Figure 3 and Figure 4. Full details of judgements are seen in the 'Risk of bias' tables.
3.
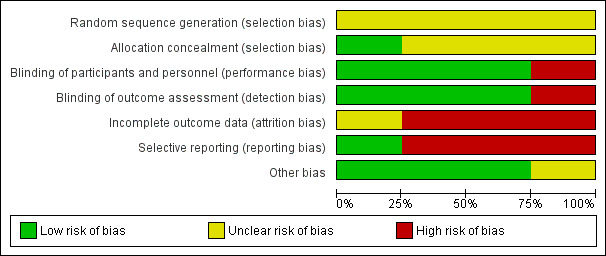
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
4.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Allocation
All studies were judged with a rating of unclear in terms of risk of bias concerning random sequence generation. All were described as randomised but none of them gave further information on sequence generation. Only one study provided some details on allocation concealment (Shalev 1993). All other trials were rated as unclear in this regard.
Blinding
All studies but one, which was an open RCT (Shalev 1993), were rated as low risk of bias in terms of blinding of both participants and personnel and of outcome assessment. Two studies used identical capsules for blinding (Adelson 1962; Kurland 1961). Adelson 1962 described that none of the treating staff were aware of the study design and the antipsychotics used. One study was also described as double‐blind, the treating physicians were only aware of the drugs generally involved in the study (Hanlon 1965).
Incomplete outcome data
One study was judged unclear in terms of incomplete outcome data (Adelson 1962). In the this study, two participants died and were replaced "in order to maintain the ANOVA design", but attrition was very low so that we are not sure whether this had an important impact on the results. We judged three studies to have a high risk due to incomplete outcome data. Of these, one study (Shalev 1993) did not report the number of participants leaving early separately for each drug, thus we could also not use this study in our analysis of 'leaving the study early'. Hanlon 1965 had an attrition rate of 15% (perphenazine) and 22% (low‐potency antipsychotics) and analysed only study completers. Kurland 1961 had a very high attrition rate of 89% (perphenazine) and 79% (low‐potency antipsychotics) and applied the last‐observation‐carried‐forward (LOCF) method.
Selective reporting
We judged one study to be free of selective reporting (Shalev 1993). Three studies were rated with a high risk of selective reporting. These studies did not (sufficiently) report on predefined outcomes (Adelson 1962, Hanlon 1965 and Kurland 1961).
Other potential sources of bias
We judged three studies to be free of other bias and one study with an unclear risk of bias. In Kurland 1961 participants received intramuscular medication on the first two days of the study.
Effects of interventions
See: Table 1
We calculated risk ratios (RR) with 95% confidence intervals (CIs) throughout..
1. Perphenazine versus low‐potency first‐generation antipsychotic drugs (FGA)
1.1 Response to treatment
There was no significant difference in response to treatment as defined by the original studies between perphenazine and low‐potency antipsychotics, neither in the short term (perphenazine 71%, low‐potency FGA 76%, 1 RCT, n = 42, RR 0.94 CI 0.65 to 1.35), nor in the medium term (perphenazine 52%, low‐potency FGA 52%, 1 RCT, n = 96, RR 1.00 CI 0.68 to 1.47), and also not overall (perphenazine 58%, low‐potency FGA 59%, 2 RCTs, n = 138, RR 0.97 CI 0.74 to 1.26).
1.2 Leaving the study
1.2.1 Due to any reason
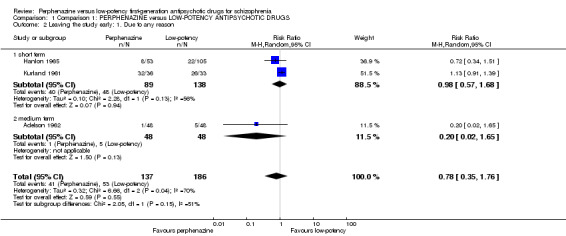
There was no significant difference between perphenazine and low‐potency FGA, neither in the short term (perphenazine 45%, low‐potency FGA 35%, 2 RCTs, n = 227, RR 0.98 CI 0.57 to 1.68), nor in the medium term (perphenazine 2%, low‐potency FGA 10%, 1 RCT, n = 96, RR 0.20 CI 0.02 to 1.65), and also not overall (perphenazine 30%, low‐potency FGA 28%, 3 RCTs, n = 323, RR 0.78 CI 0.35 to 1.76).
1.2.2 Due to adverse effects
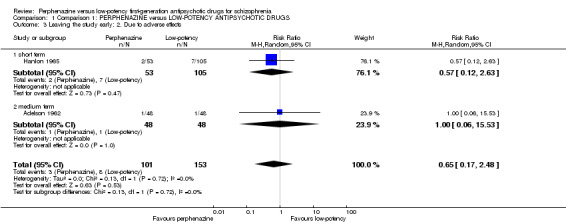
There was no significant difference between perphenazine and low‐potency FGA, neither in the short term (perphenazine 4%, low‐potency FGA 7%, 1 RCT, n = 158, RR 0.57 CI 0.12 to 2.63), nor in the medium term (perphenazine 2%, low‐potency FGA 2%, 1 RCT, n = 96, RR 1.00 CI 0.06 to 15.53), and also not overall (perphenazine 3%, low‐potency FGA 5%, 2 RCTs, n = 254, RR 0.65 CI 0.17 to 2.48).
1.2.3 Due to inefficacy
There was no significant difference between perphenazine and low‐potency FGA, neither in the short term (perphenazine 0%, low‐potency FGA 0%, 1 RCT, n = 158, RR not estimable), nor in the medium term (perphenazine 0%, low‐potency FGA 0%, 1 RCT, n = 96, RR not estimable), and also not overall (perphenazine 0%, low‐potency FGA 0%, 2 RCTs, n = 254, RR not estimable).
1.3 Adverse effects
1.3.1 General ‐ at least one adverse effect
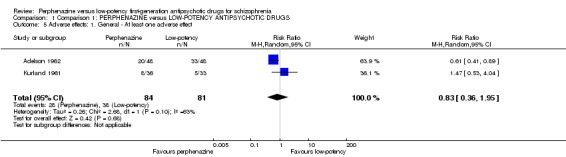
There was no significant difference between perphenazine and low‐potency FGA (perphenazine 33%, low‐potency FGA 47%, 2 RCTs, n = 165, RR 0.83 CI 0.36 to 1.95).
1.3.2 Specific
1.3.2.1 Movement disorders
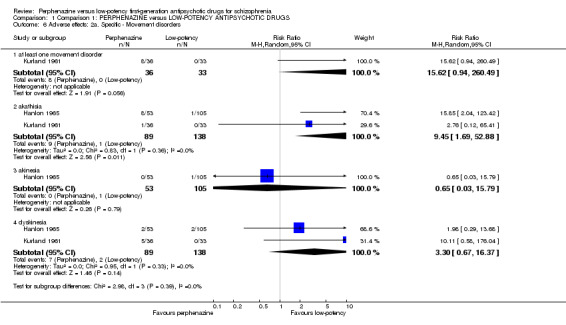
There was a trend in favour of low‐potency antipsychotics for the outcome of 'at least one movement disorder' but the difference was not statistically significant (perphenazine 22%, low‐potency FGA 0%, 1 RCT, n = 69, RR 15.62 CI 0.94 to 260.49). There was a significant difference in favour of low‐potency FGA for akathisia (perphenazine 25%, low‐potency FGA 22%, 2 RCTs, n = 227, RR 9.45 CI 1.69 to 52.88). There was no significant difference for akinesia (perphenazine 0%, low‐potency FGA 1%, 1 RCT, n = 158, RR 0.65 CI 0.03 to 15.79) and dyskinesia (perphenazine 9%, low‐potency FGA 1.5%, 2 RCTs, n = 227, RR 3.30 CI 0.67 to 16.37).
1.3.2.2 Other
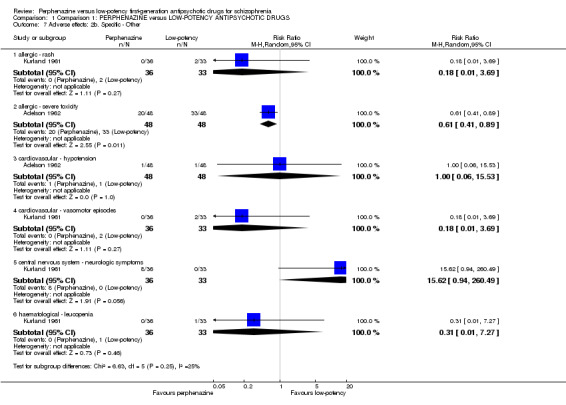
a. Allergic
There was no significant difference for the outcome of 'rash' (perphenazine 0%, low‐potency FGA 6%, 1 RCT, n = 69, RR 0.18 CI 0.01 to 3.69) but for 'severe toxicity' the finding was in favour of perphenazine (perphenazine 42%, low‐potency FGA 69%, 1 RCT, n = 96, RR 0.61 CI 0.41 to 0.89).
b. Cardiovascular
There was no significant difference for either 'hypotension' (perphenazine 2%, low‐potency FGA 2%, 1 RCT, n = 96, RR 1.00 CI 0.06 to 15.53) or 'vasomotor episodes' (perphenazine 0%, low‐potency FGA 6%, 1 RCT, n = 69, RR 0.18 CI 0.01 to 3.69).
c. Central nervous system
There was trend in favour of low‐potency antipsychotics for the outcome of 'neurological symptoms' but the difference was not statistically significant (perphenazine 22%, low‐potency FGA 0%, 1 RCT, n = 69, RR 15.62 CI 0.94 to 260.49).
d. Haematological
There was no significant difference in 'leucopenia' (perphenazine 0%, low‐potency FGA 3%, 1 RCT, n = 69, RR 0.31 CI 0.01 to 7.27).
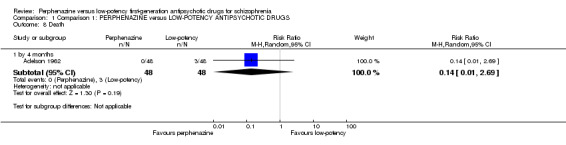
1.4 Death
There was no significant difference (perphenazine 0%, low‐potency FGA 2%, 1 RCT, n = 96, RR 0.14 CI 0.01 to 2.69).
1.5 Missing outcomes
There were no data on important other outcomes such as relapse, service use, quality of life or satisfaction with care.
2. Subgroup analyses
All subgroup analyses were conducted only on the primary outcome 'response to treatment' as defined by the original studies.
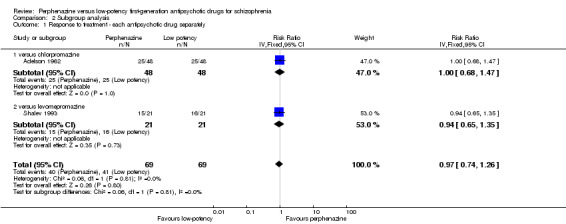
2.1 Single low‐potency FGA drugs
One study (Adelson 1962) compared perphenazine with chlorpromazine (perphenazine 52%, chlorpromazine 52%, 1 RCT, n = 96, RR 1.00 CI 0.68 to 1.47). Another study (Shalev 1993), compared perphenazine with levomepromazine (perphenazine 71%, levomepromazine 76%, 1 RCT, n = 42, RR 0.94 CI 0.65 to 1.35). Both comparisons were not significant. The other two studies did not report on response to treatment.
2.2 Clinical state, stage or problem
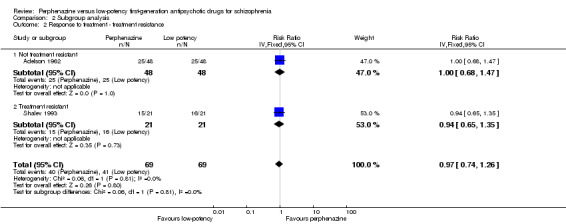
One study (Shalev 1993) included only participants who were treatment‐resistant and did not find a superiority of perphenazine compared to levomepromazine (perphenazine 71%, levomepromazine 76%, 1 RCT, n = 42, RR 0.94 CI 0.65 to 1.35). There was no significant difference compared to the other study (test for subgroup differences: Chi² = 0.06, df = 1 (P = 0.81), I² = 0%).
2.3 Investigation of heterogeneity
There was no heterogeneity in terms of the primary outcome clinically significant response to treatment' (P = 0.80, I2 = 0%).
3. Sensitivity analyses
All sensitivity analyses were conducted only on the primary outcome 'response to treatment' as defined by the original studies.
3.1 Exclusion of studies for which randomisation was implied because they were double‐blind
Two studies reported data on the primary outcome clinical response, both were described as randomised (Adelson 1962; Shalev 1993),
3.2 Exclusion of studies for which blinding was implied
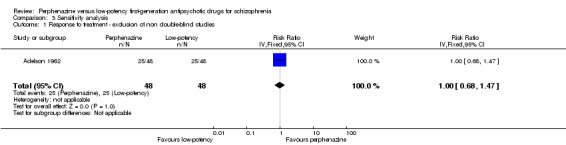
There was one study that was not double blinded (Shalev 1993). Excluding this study did not change the overall results (perphenazine 52%, low‐potency FGA 52%, 1 RCT, n = 96, RR 1.00 CI 0.68 to 1.47).
3.3 Assessment of dosage
The two included studies were not judged to have used unfair comparator doses.
3.4 Fixed‐effect model
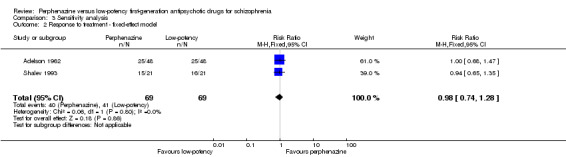
When a fixed‐effect model was applied, perphenazine was also not significantly different from low‐potency drugs (perphenazine 58%, low‐potency FGA 59%, 2 RCTs, n = 138, RR 0.98 CI 0.74 to 1.28).
4. Other results
4.2 Publication bias
Only two studies reported on the primary outcome clinical response, so that funnel plots were not meaningful.
4.3 'Summary of findings' table
The results of the outcomes response to treatment, acceptability of treatment (leaving the study early for any reason), at least one adverse effect, at least one movement disorder, sedation, death and quality of life were inspected more closely (see Table 1). Based on this tool we considered the results for the outcomes response to treatment and death to be moderate, for at least one adverse effect to be low and for leaving the study due to any reason and at least one movement disorder to be very low. Moreover, no data on the outcome sedation as well as quality of life were available. The judgements derived from this instrument were used for the discussion section of the review (see Discussion ‐ Summary of main results).
Discussion
Summary of main results
1. General
This review currently includes four trials with 365 participants that compared perphenazine with low‐potency first‐generation antipsychotics. The included studies were published from 1962 to 1993 in different countries and all were conducted in inpatients. The included studies were relatively small and all studies except one (Hanlon 1965) randomised less than 100 participants. Also, all studies were of short duration; two of the studies lasted four weeks, only one study lasted four months. Of the four studies that were included, only two studies presented data on the primary outcome response to treatment (Adelson 1962; Shalev 1993). The results did not show any difference of perphenazine compared to low‐potency first‐generation antipsychotics. This finding is line with the statements in treatment guidelines (Gaebel 2006; Lehman 2004) that low‐potency drugs are as efficacious as high‐potency antipsychotics such as perphenazine and contrasts with the clinical impression that low‐potency conventional antipsychotic drugs are less efficacious. This result was robust in the subgroup and sensitivity analyses, in which perphenazine was compared with each low‐potency drug separately and when a single open study was excluded. However, only two studies with a small number of participants reported data on the primary outcome addressed in these analyses (response to treatment) and the statistical power was very low, so no firm conclusions could be drawn. In summary, the obtained data are not ideal for making conclusions about the relative tolerability and efficacy of perphenazine compared to low‐potency first‐generation antipsychotics. Therefore, the results of this review are rather inconclusive.
2. Treatment effects
2.1 Response to treatment
The overall results of response to treatment reported by two studies do not suggest a difference in efficacy between perphenazine and low‐potency first‐generation antipsychotic drugs. This result supports early narrative reviews which were not based on meta‐analytic methods (Davis 1989; Klein 1969) and it does not confirm a clinical perception that low‐potency first‐generation antipsychotic drugs are less efficacious than higher‐potency ones such as perphenazine. However, only two studies reported on this outcome and the number of participants was low ‐ altogether 138 participants. Approximately 1000 participants need to be included in psychiatric meta‐analyses for the results to be robust (Trikalinos 2004), therefore this result was underpowered.
No usable data were presented by the included studies on ratings scales on global and mental state, thus it is not clear whether perphenazine or low‐potency antipsychotics are more effective in this regard.
2.2 Leaving the studies early
There was no significant difference between perphenazine and low‐potency first‐generation antipsychotics in the outcome leaving early due to any reason. As leaving early due to any reason comprises efficacy (leaving early due to inefficacy) and overall tolerability (leaving early due to adverse events), this finding suggests that perphenazine and low‐potency first‐generation antipsychotics are not different in their overall acceptability for people with schizophrenia. This is an indirect measure of acceptability, however. We also found no significant difference for leaving the study early due to adverse events. However, only three studies reported data on these outcomes, which is relatively little and more data would be needed for a clear interpretation here. No participants left the studies early due to inefficacy in the two studies that presented data on this outcome.
2.4 Adverse effects
In those studies which reported on adverse events, perphenazine produced more akathisia, a side effect which falls in the category of movement disorder. Low‐potency first‐generation antipsychotics caused more 'severe toxicity'. However, the study that reported on this outcome (Adelson 1962), included several symptoms such as convulsions, anorexia, weight loss, toxic confusion and death into this outcome which is thus a composite of various phenomena and difficult to interpret. Except for death, no data on those symptoms alone were reported. Altogether, only three of the four included studies reported adverse event data at all. Therefore no firm conclusions can be drawn.
2.4 Death
There were three deaths in the low‐potency group, but only one study reported data on this outcome and the results were not significant. More data on this important outcome would be needed for clear conclusions.
2.5. Missing outcomes
Missing outcomes on service use, quality of life, participants´/carers´ satisfaction with care or economic outcomes may be more important for afflicted people and policy makers than conventional measures of efficacy and tolerability. It is therefore disappointing that they are not available.
3. Publication bias
Due to the limited number of studies that reported on the primary outcome, the test for funnel plot asymmetry was not meaningful. But as efforts to make all data publicly available are quite recent while most of the studies were rather old, it is quite possible that publication bias exists.
4. Subgroup analyses and investigation of heterogeneity
The effects of perphenazine versus each single low‐potency first‐generation antipsychotic drug showed no significant superiority compared to chlorpromazine and levomepromazine. Also, there was no difference between the single comparisons. There was no significant difference between studies which included treatment resistant participants and the remaining studies. These two studies alone did also not show any significant difference between perphenazine and low‐potency first‐generation antipsychotics. Again, the database is very limited (two RCTs).
5. Sensitivity analyses
The exclusion of studies which were not described as double‐blind did not change the overall results in the primary outcome clinical response. The results of the primary outcome were not different when a fixed‐effect model instead of a random‐effects model was applied.
Overall completeness and applicability of evidence
1. Completeness
Several limitations, which are relevant for the conclusions of this meta‐analysis, must be considered. Of the four included studies, only two RCTs reported sufficient data for the primary outcome clinical response. Therefore, we believe that the evidence on the primary outcome is not complete. Similarly, often only one or two studies reported on secondary outcomes. The included studies only compared perphenazine with chlorpromazine, levomepromazine or thioridazine. Nothing can be said about the effects of perphenazine compared to other low‐potency first‐generation drugs such as chlorprothixene, mesoridazine or perazine so that again, there are evidence gaps. The evidence on predefined adverse events is particularly incomplete, as none of the included studies reported important side effects such as sedation, suicide or cardiac effects. There were also no data on quality of life, service use or satisfaction with care. New studies with better outcome reporting would be needed for stronger statements about the differences between fluphenazine and low‐potency first‐generation antipsychotic medication.
2. Applicability
Three studies were from the 1960s and one study from the 1990s, which could have led to limitations for applicability. Participants were diagnosed according to clinical diagnosis as operationalised diagnostic criteria such as DSM‐III or its more recent versions were not available. Thus, it is possible that those older studies included participants who nowadays would have another diagnosis than schizophrenia because at that time, the definition of schizophrenia was slightly different. Thus, applicability towards people diagnosed with schizophrenia nowadays must be made with caution. Furthermore, most of the included studies were characterised by small sample sizes (< 100 participants). Approximately 1000 participants need to be included in psychiatric meta‐analyses for the results to be robust (Trikalinos 2004). However, this meta‐analysis included altogether 365 participants, the primary outcome clinical response 138 participants, and was therefore underpowered.
Quality of the evidence
No study reported details on the randomisation method used. Some details about sequence generation and allocation concealment were only described in one study (Shalev 1993) and thus remained unclear. Three studies were described as double‐blind in that identical capsules were used. We therefore did not consider that there was an important source of bias in this regard. It might be argued that as antipsychotics differ in side effects, blinding might still not have worked. We felt that it is not clear that perphenazine and low‐potency first‐generation antipsychotics differ enough in this regard so that unblinding is likely and that the investigators have done what they could. Nevertheless, it would be desirable that future studies test the success of blinding. Predefined outcomes for the Table 1 such as sedation and quality of life were missing. There were also problems in terms of inconsistency and imprecise data. These issues led to relatively low quality ratings in the 'Summary of findings' table.
Potential biases in the review process
We pooled all low‐potency first‐generation antipsychotics in one group for all outcomes except for the primary outcome clinical response, for which we also carried out a subgroup analysis of perphenazine versus each single low‐potency first‐generation antipsychotic drug. As there were altogether only three different low‐potency antipsychotics, pooling the results of these antipsychotics should not have been a major problem. Also, the search is based on Cochrane Schizophrenia Trials Register, so it is possible that there are unpublished trials of which we are unaware. There is a possibility of publication bias, but due to the small number of trials, this could not be addressed. There is a risk of excluding studies that have merely failed to report relevant outcomes, rather than failing to measure them. Three of the excluded studies are now very old, and it has not been possible to ascertain whether outcome data were available at all. However, it is standard of the Cochrane Schizpohrenia Group, to exclude studies without relevant outcomes. As the search date is 2010, there is a lag time between search date and publication, which is a limitation to the review. However, it is very unlikely that there are new studies available.
Agreements and disagreements with other studies or reviews
We are not aware of other reviews on the efficacy of perphenazine versus low‐potency antipsychotic drugs.
Authors' conclusions
Implications for practice.
1. For clinicians
Clinicians should know that we did not find differences in the efficacy between perphenazine and low‐potency first‐generation antipsychotics. Low‐potency first‐generation antipsychotics appeared to produce fewer akathisia, but they were associated with other severe side effects, which the authors summarised as 'severe toxicity' in a single study. Due to the extremely limited number of studies and participants available, and due to their overall low quality, the results are very preliminary.
2. For people with schizophrenia
It might be important for people with schizophrenia to know that the side effect of akathisia, an unpleasant feeling of restlessness and the impossibility to sit still, was the only outcome that appeared more frequently under treatment with perphenazine than with low‐potency first‐generation antipsychotics, while 'serious toxicity' ‐ a composite of various adverse effects ‐ occurred more frequently in the low‐potency first‐generation group. It is likely that there are many more differences between perphenazine and low‐potency first‐generation antipsychotics, which we could simply not record due to the very limited amount of information available.
3. For managers/policy makers
There were no data on rehospitalisation, economic outcomes, healthy days or quality of life, which are very important outcomes for decision makers. Thus, it is not possible to make any recommendations apart from the fact that all of the examined drugs in this review have lost their patent protection and are therefore rather inexpensive.
Implications for research.
1. General
The reporting of outcomes in the included studies was generally insufficient. Few data were available, and long‐term effects were not reported at all. Strict adherence to the CONSORT statement (Moher 2010) would make such studies much more informative.
2. Specific
2.1 Reviews
Studies we have had to exclude because they were not directly relevant, however, do still show how this compound has been evaluated in many other ways. Some of these remain clinically relevant and may merit further systematic reviews (Table 3).
2. Comparisons for reviews suggested by excluded studies.
| Comparison | Excluded study tag |
| Perphenazine in combination with amitriptyline versus chlorpromazine alone for schizophrenia | Lapolla 1967 |
| Perphenazine versus atypical antipsychotics for schizophrenia | Anon 2006, Loza 2001 |
| Perphenazine versus mid‐potency antipsychotics for schizophrenia | Svestka 1974 (oxypertine) |
| Perphenazine versus placebo for schizophrenia | Akimoto 1966 |
2.2 Trials
The number of studies providing data on the primary outcome response to treatment, let alone on most adverse events was very low, and so was the overall quality of the included studies. New, better studies on the difference between perphenazine and low‐potency first‐generation antipsychotic drugs would be warranted, because first‐generation antipsychotic drugs are still frequently prescribed, not only in poorer countries but also in rich nations such as Germany. We make some suggestions for the design of a future study in Table 4.
3. Design of a future study.
| Methods | Allocation: randomised ‐ clearly described generation of sequence and concealment of allocation. Blinding: double ‐ described and tested. Duration: long term. |
| Participants | People with schizophrenia or schizophrenia‐like disorder. N = 500. Age: any. Sex: both. History: any. |
| Interventions | 1. Trifluoperazine (oral). 2. Any low‐potency antipsychotic (oral). |
| Outcomes | Response (primary outcome) Rehospitalisation Mental state (BPRS) Global state (CGI) Leaving the study early (including specific causes) Death (natural and unnatural causes) Side‐effects Quality of life Satisfaction with care Employment Economic/Costs |
BPRS ‐ Brief Psychiatric Rating Scale CGI ‐ Clinical Global Impression
Acknowledgements
The Cochrane Schizophrenia Group (CSzG) Editorial Base in Nottingham produces and maintains standard text for use in the methods sections of their reviews. We have used this text as the basis of what appears here and adapted it as required. We are indebted to the CSzG team for its assistance in the literature search and we thank the peer‐reviewers for their useful comments.
Data and analyses
Comparison 1. Comparison 1: PERPHENAZINE versus LOW‐POTENCY ANTIPSYCHOTIC DRUGS.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Response to treatment | 2 | 138 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.74, 1.26] |
| 1.1 short term | 1 | 42 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.65, 1.35] |
| 1.2 medium term | 1 | 96 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.68, 1.47] |
| 2 Leaving the study early: 1. Due to any reason | 3 | 323 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.35, 1.76] |
| 2.1 short term | 2 | 227 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.57, 1.68] |
| 2.2 medium term | 1 | 96 | Risk Ratio (M‐H, Random, 95% CI) | 0.20 [0.02, 1.65] |
| 3 Leaving the study early: 2. Due to adverse effects | 2 | 254 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.17, 2.48] |
| 3.1 short term | 1 | 158 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.12, 2.63] |
| 3.2 medium term | 1 | 96 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.06, 15.53] |
| 4 Leaving the study early: 3. Due to inefficacy | 2 | 254 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4.1 short term | 1 | 158 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4.2 medium term | 1 | 96 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5 Adverse effects: 1. General ‐ At least one adverse effect | 2 | 165 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.36, 1.95] |
| 6 Adverse effects: 2a. Specific ‐ Movement disorders | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 at least one movement disorder | 1 | 69 | Risk Ratio (M‐H, Random, 95% CI) | 15.62 [0.94, 260.49] |
| 6.2 akathisia | 2 | 227 | Risk Ratio (M‐H, Random, 95% CI) | 9.45 [1.69, 52.88] |
| 6.3 akinesia | 1 | 158 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.03, 15.79] |
| 6.4 dyskinesia | 2 | 227 | Risk Ratio (M‐H, Random, 95% CI) | 3.30 [0.67, 16.37] |
| 7 Adverse effects: 2b. Specific ‐ Other | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 allergic ‐ rash | 1 | 69 | Risk Ratio (M‐H, Random, 95% CI) | 0.18 [0.01, 3.69] |
| 7.2 allergic ‐ severe toxicity | 1 | 96 | Risk Ratio (M‐H, Random, 95% CI) | 0.61 [0.41, 0.89] |
| 7.3 cardiovascular ‐ hypotension | 1 | 96 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.06, 15.53] |
| 7.4 cardiovascular ‐ vasomotor episodes | 1 | 69 | Risk Ratio (M‐H, Random, 95% CI) | 0.18 [0.01, 3.69] |
| 7.5 central nervous system ‐ neurologic symptoms | 1 | 69 | Risk Ratio (M‐H, Random, 95% CI) | 15.62 [0.94, 260.49] |
| 7.6 haematological ‐ leucopenia | 1 | 69 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.01, 7.27] |
| 8 Death | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 by 4 months | 1 | 96 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.69] |
1.1. Analysis.
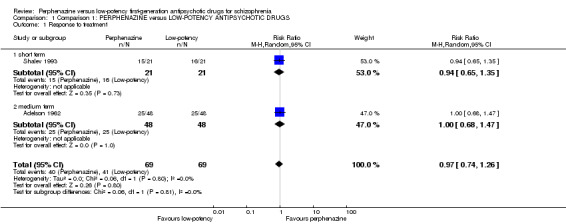
Comparison 1 Comparison 1: PERPHENAZINE versus LOW‐POTENCY ANTIPSYCHOTIC DRUGS, Outcome 1 Response to treatment.
1.2. Analysis.
Comparison 1 Comparison 1: PERPHENAZINE versus LOW‐POTENCY ANTIPSYCHOTIC DRUGS, Outcome 2 Leaving the study early: 1. Due to any reason.
1.3. Analysis.
Comparison 1 Comparison 1: PERPHENAZINE versus LOW‐POTENCY ANTIPSYCHOTIC DRUGS, Outcome 3 Leaving the study early: 2. Due to adverse effects.
1.4. Analysis.
Comparison 1 Comparison 1: PERPHENAZINE versus LOW‐POTENCY ANTIPSYCHOTIC DRUGS, Outcome 4 Leaving the study early: 3. Due to inefficacy.
1.5. Analysis.
Comparison 1 Comparison 1: PERPHENAZINE versus LOW‐POTENCY ANTIPSYCHOTIC DRUGS, Outcome 5 Adverse effects: 1. General ‐ At least one adverse effect.
1.6. Analysis.
Comparison 1 Comparison 1: PERPHENAZINE versus LOW‐POTENCY ANTIPSYCHOTIC DRUGS, Outcome 6 Adverse effects: 2a. Specific ‐ Movement disorders.
1.7. Analysis.
Comparison 1 Comparison 1: PERPHENAZINE versus LOW‐POTENCY ANTIPSYCHOTIC DRUGS, Outcome 7 Adverse effects: 2b. Specific ‐ Other.
1.8. Analysis.
Comparison 1 Comparison 1: PERPHENAZINE versus LOW‐POTENCY ANTIPSYCHOTIC DRUGS, Outcome 8 Death.
Comparison 2. Subgroup analysis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Response to treatment ‐ each antipsychotic drug separately | 2 | 138 | Risk Ratio (IV, Fixed, 95% CI) | 0.97 [0.74, 1.26] |
| 1.1 versus chlorpromazine | 1 | 96 | Risk Ratio (IV, Fixed, 95% CI) | 1.0 [0.68, 1.47] |
| 1.2 versus levomepromazine | 1 | 42 | Risk Ratio (IV, Fixed, 95% CI) | 0.94 [0.65, 1.35] |
| 2 Response to treatment ‐ treatment resistance | 2 | 138 | Risk Ratio (IV, Fixed, 95% CI) | 0.97 [0.74, 1.26] |
| 2.1 Not treatment resistant | 1 | 96 | Risk Ratio (IV, Fixed, 95% CI) | 1.0 [0.68, 1.47] |
| 2.2 Treatment resistant | 1 | 42 | Risk Ratio (IV, Fixed, 95% CI) | 0.94 [0.65, 1.35] |
2.1. Analysis.
Comparison 2 Subgroup analysis, Outcome 1 Response to treatment ‐ each antipsychotic drug separately.
2.2. Analysis.
Comparison 2 Subgroup analysis, Outcome 2 Response to treatment ‐ treatment resistance.
Comparison 3. Sensitivity analysis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Response to treatment ‐ exclusion of non double‐blind studies | 1 | 96 | Risk Ratio (IV, Fixed, 95% CI) | 1.0 [0.68, 1.47] |
| 2 Response to treatment ‐ fixed‐effect model | 2 | 138 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.74, 1.28] |
3.1. Analysis.
Comparison 3 Sensitivity analysis, Outcome 1 Response to treatment ‐ exclusion of non double‐blind studies.
3.2. Analysis.
Comparison 3 Sensitivity analysis, Outcome 2 Response to treatment ‐ fixed‐effect model.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Adelson 1962.
| Methods | Randomisation: randomly assigned to different treatments. Allocation: random assignment carried out by person having no other contact with the study and the entire study staff had no knowledge of the assignments made. Blinding: double ‐ all drugs supplied in two capsule sizes using Parke‐Davis standard pink capsules to appear identical. The manufactory laboratory of the school of pharmacy packaged the drugs in bottles which were labelled with only the patients´ name, cohort number, capsule size. None of the direct treating staff were aware of the design of the study, the drugs used, how many agents there were and whether or not there were any patients on placebo. Duration: 4 months. Design: parallel. Location: multicentre. Setting: inpatients. | |
| Participants | Diagnosis: hospitalised chronically ill schizophrenic participants (clinical diagnosis). N = 96. Gender: 48 M, 48 F. Age: n.i.. History: duration stable ‐ n.i., duration ill ‐ n.i., number of previous hospitalisations ‐ years: 96 = 2‐5 years, 96 = 6‐10 years, age at onset ‐ n.i., severity of illness ‐ n.i., baseline antipsychotic dose ‐ n.i.. |
|
| Interventions | 1. Perphenazine: flexible dose, allowed dose range 8 to 240 mg/day, mean dose 181.84 mg/day. N = 48. 2. Chlorpromazine: flexible dose, allowed dose range 100 to 3000 mg/day, mean dose 1800 mg/day N = 48. |
|
| Outcomes | Response to treatment: judgement of the treating psychiatrist. Leaving the study early. Adverse effects: at least one adverse effect, adverse effects ‐ other (e.g., hypotension, severe toxicity) Death Unable to use: Mental state: Multidimensional Scale for Rating Psychiatric Patients (MSRPP), Bender‐Gestalt Test and Minnesota Multiphasic Personality Inventory (MMPI) (all incomplete data, no SDs). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned to different treatments. |
| Allocation concealment (selection bias) | Unclear risk | Random assignment carried out by person having no other contact with the study and the entire study staff had no knowledge of the assignments made. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double ‐ all drugs supplied in two capsule sizes using Parke‐Davis standard pink capsules to appear identical. The manufactory laboratory of the school of pharmacy packaged the drugs in bottles which were labelled with only the patients´ name, cohort number, capsule size. None of the direct treating staff were aware of the design of the study, the drugs used, how many agents there were and whether or not there were any patients on placebo. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double ‐ all drugs supplied in two capsule sizes using Parke‐Davis standard pink capsules to appear identical. The manufactory laboratory of the school of pharmacy packaged the drugs in bottles which were labelled with only the patients´ name, cohort number, capsule size. None of the direct treating staff were aware of the design of the study, the drugs used, how many agents there were and whether or not there were any patients on placebo. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 2 chlorpromazine participants died and were replaced in order to maintain the analysis of variance design. 1 out of 48 perphenazine and 10 out of 144 participants from the low‐potency drug group left the study early, which is an acceptable rate. |
| Selective reporting (reporting bias) | High risk | MSRPP no SD, MMPI no data. |
| Other bias | Low risk | No clear other bias. |
Hanlon 1965.
| Methods | Randomisation: randomly assigned, no further details. Allocation: procedure not described. Blinding: double ‐ drugs dispensed in standard unmarked pink capsules no. 1 & 0 size, but treating ward physicians were aware of the various drugs and dosages involved in the study. Duration: 30 days. Design: parallel. Location: n.i.. Setting: inpatients. | |
| Participants | Diagnosis: psychotic (270) of which 232 were diagnosed as schizophrenic, 52 neurotics and/or personality disorder. N = 158. Gender: 160 M, 162 F. Age: mean 36.3 years. History: duration stable ‐ n.i., duration ill ‐ n.i., number of previous hospitalisations ‐ n.i., age at onset ‐ n.i., severity of illness ‐ MSRPP mean 41.4 (15.3), MACC mean 37.5 (10.4), baseline antipsychotic dose ‐ n.i.. |
|
| Interventions | 1. Perphenazine: flexible dose, mean dose 38.61 mg/day N = 53. 2. Chlorpromazine: flexible dose, mean dose 395.56 mg/day. N = 52. 3. Thioridazine: flexible dose, mean dose 193.46 mg/day. N = 53. Rescue medication: antiparkinson medication (biperiden), mild sedation (phenobarbital glutethimide). |
|
| Outcomes | Leaving the study early. Adverse effects: movement disorders (akathisia, dyskinesia, akinesia). Unable to use: Mental state: IMPS, MSRPP (all incomplete data, no means, no SDs). Behaviour: Behavioral Adjustment Scale incomplete data, no mean, no SD). Personality: Minnesota Multiphasic Personality Inventory (incomplete data, no mean, no SD). Ward observer measures (incomplete data, no mean, no SD). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned, no further details. |
| Allocation concealment (selection bias) | Unclear risk | Procedure not described. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double ‐ drugs dispensed in standard unmarked pink capsules no. 1 & 0 size, the treating ward physicians were only aware of the various drugs and dosages involved in the study. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double ‐ drugs dispensed in standard unmarked pink capsules no. 1 & 0 size, the treating ward physicians were only aware of the various drugs and dosages involved in the study. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 8 out of 53 (15%) participants from the perphenazine group and 46 out of 210 from the low‐potency group left the study early and not included in the final analysis (completers only). |
| Selective reporting (reporting bias) | High risk | MSRPP, MMPI, MACC (all no usable data, no means, no SDs). |
| Other bias | Low risk | No evidence for other bias. |
Kurland 1961.
| Methods | Randomisation: random selection, no further details. Allocation: procedure not described. Blinding: double. Dispensed in unmarked capsules of #0 and #1 size coloured pink to mask all identifying consistencies and colours of the drugs. Duration: 6 weeks. Design: parallel. Location: single centre. Setting: inpatients. |
|
| Participants | Diagnosis: predominantly schizophrenia (clinical diagnosis). N = 69. Gender: n.i.. Age: mean 39 years. History: duration stable – n.i., duration ill – n.i., number of previous hospitalisations – n.i., age at onset – n.i., severity of illness – n.i., baseline antipsychotic dose – n.i.. |
|
| Interventions | 1. Perphenazine: flexible dose, allowed dose range 24 to 96 mg/day, mean dose 30.83 mg/day. N = 36. 2. Chlorpromazine: flexible dose, allowed dose range 300 to 1200 mg/day, mean dose 401.35 mg/day. N = 33. Rescue medication: n.i.. |
|
| Outcomes | Leaving the study early. Adverse effects: at least one adverse effect, movement disorders (MD) (at least one MD, akathisia, dyskinesia), adverse effects ‐ other (leucopenia, neurologic symptoms, rash, vasomotor episodes). Unable to use: Mental state: MSRPP (no SD), PSTS (no SD) Behaviour: PRP (no usable data, no SD). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Random selection, no further details. |
| Allocation concealment (selection bias) | Unclear risk | Procedure not described. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double. Dispensed in unmarked capsules of #0 and #1 size coloured pink to mask all identifying consistencies and colours of the drugs. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double. Dispensed in unmarked capsules of #0 and #1 size coloured pink to mask all identifying consistencies and colours of the drugs. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 32 out of 36 from the perphenazine group left the study early (89%) of which 22% dropped out due to EPS. 26 out of 33 from the low‐potency group left early (79%). LOCF was applied. |
| Selective reporting (reporting bias) | High risk | MSRPP (no SD), PRP and PSTS (no usable data). |
| Other bias | Unclear risk | On the first two days of the study, participants received intramuscular medication, after that they were given oral mediations. |
Shalev 1993.
| Methods | Randomisation: randomly assigned, no further details. Allocation: treating psychiatrists were blind to the sequence in which the drugs were to be given to the patient. Blinding: not described, probably open. Duration: 4 weeks. Design: parallel (second part cross‐over). Location: single centre. Setting: inpatients. | |
| Participants | Diagnosis: chronic or subchronic schizophrenia (DSM‐III). N = 42. Gender: 35 F, 25 M. Age: mean 33 years. History: duration stable ‐ n.i., duration ill ‐ mean 4.8 years, number of previous hospitalisations ‐ mean 4.2, age at onset ‐ mean 28.2 years, severity of illness ‐ n.i., baseline antipsychotic dose ‐ n.i.. |
|
| Interventions | 1. Perphenazine: flexible dose, allowed dose range n.i., mean dose = 35.8 mg/day (SD 11.4). N = 21. 2. Levomepromazine: flexible dose, allowed dose range n.i., mean dose 379 mg/day (SD 128). N = 21. Rescue medication: antiparkinson medication. | |
| Outcomes | Response to treatment: clinical judgement by psychiatrist. Unable to use: Mental state: BPRS (incomplete data, analysed by responders and non‐responders, not by medication). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned, no further details. |
| Allocation concealment (selection bias) | Low risk | "Treating psychiatrists were blind to the sequence in which the drugs were to be given to the patient." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not described, probably open. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Not described, probably open. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 15 participants left the study early but it is not mentioned how many from which drug group. |
| Selective reporting (reporting bias) | Low risk | No evidence for selective outcome reporting. |
| Other bias | Low risk | No evidence for other bias. |
General Abbreviations n.i. ‐ not indicated mg ‐ milligram M ‐ male F ‐ female SD ‐ standard deviation LOCF ‐ Last‐observation‐carried‐forward DSM ‐ Diagnostic and Statistical Manual of Mental Disorders EPS ‐ extrapyramidal symptoms
Rating scales BPRS ‐ Brief Psychiatric Rating Scale IMPS ‐ Inpatient Multidimensional Psychiatric Rating Scale MACC ‐ Behavioral Adjustment Scale MMPI ‐ Minnesota Multiphasic Personality Inventory MSRPP ‐ Multidimensional Scale for Rating Psychiatric Patients PRP ‐ Psychotic Reaction Profile PSTS ‐ Psychiatric Scale of Target Symptoms
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Akimoto 1966 | Method: randomised. Participants: chronic schizophrenic participants. Intervention: perphenazine versus placebo. |
| Anon 2006 | Method: randomised. Participants: schizophrenic participants. Intervention: perphenazine versus atypical antipsychotics. |
| Bennett 1961 | Method: randomised. Participants: schizophrenia. Intervention: perphenazine, chlorpromazine. Outcome: no usable data. |
| Casey 1960 | Method: randomised. Participants: schizophrenia. Intervention: perphenazine, chlorpromazine. Outcome: no usable data. |
| Hollister 1974 | Method: not randomised. |
| Lapolla 1967 | Method: randomised. Participants: hospitalised schizophrenic participants. Intervention: perphenazine in combination with amitriptyline versus chlorpromazine alone. |
| Loza 2001 | Method: randomised. Participants: schizophrenic participants. Intervention: perphenazine versus atypical antipsychotics. |
| Nordic 1986 | Method: randomised. Participants: psychiatric participants with tardive dyskinesia, not exclusively people with schizophrenia, how many participants had schizophrenia or related disorders was not reported. |
| Schulsinger 1958 | Method: not randomised. |
| Smith 1959 | Method: not randomised. |
| Svestka 1972 | Method: not randomised. |
| Svestka 1974 | Method: not randomised, but double‐blind. Cross‐over study, results of first phase not reported separately. Participants: acute exacerbation of psychosis (mainly schizophrenia) Intervention: perphenazine versus oxypertine (difficult to classify, probably a mid‐potency antipsychotic). |
| Vinar 1968 | Method: randomised. Participants: schizophrenia. Intervention: perphenazine, chlorpromazine, levomepromazine, thioridazine. Outcome: no usable data, only total number of participants leaving early (no data for single drugs), no rating scale results (no mean, no SD). |
SD ‐ standard deviation
Differences between protocol and review
We decided post‐hoc to include all adverse effects reported by a study, not only the predefined adverse effects in the methods section. A randomised sample of 25% was extracted by the second review author (MH).
We have updated our methods section since the publication of the protocol to reflect new methodology used by The Cochrane Collaboration, and renamed some of our outcomes. These changes, we feel, do not affect the results or conclusions of the review.
Contributions of authors
Magdolna Tardy ‐ protocol development, trial selection, data extraction and report writing. Maximilian Huhn ‐ protocol development, trial selection, data extraction and report writing Stefan Leucht ‐ protocol development, checking of trial selection and data extraction, advice on report writing. Rolf Engel ‐ protocol development, advice.
Sources of support
Internal sources
Freistaat Bayern, Germany.
External sources
Bundesministerium für Bildung und Forschung Grant number 01KG09228816532, Germany.
Declarations of interest
Magdolna Tardy ‐ none to declare.
Maximilian Huhn ‐ none to declare.
Rolf Engel ‐ none to declare.
Stefan Leucht ‐ has received honoraria for lectures from Abbvie, Astra Zeneca, BristolMyersSquibb, ICON, EliLilly, Janssen, Johnson & Johnson, Roche, SanofiAventis, Lundbeck and Pfizer; honoraria for consulting/advisory boards from Roche, EliLilly, Medavante, BristolMyersSquibb, Alkermes, Janssen, Johnson & Johnson and Lundbeck. EliLilly has provided medication for a study with SL also primary investigator.
New
References
References to studies included in this review
Adelson 1962 {published data only}
- Adelson D, Epstein LJ. A study of phenothiazines with males and female chronically ill schizophrenic patients. Journal of Nervous and Mental Diseases 1962;134(1):543‐54. [DOI] [PubMed] [Google Scholar]
Hanlon 1965 {published data only}
- Hanlon TE, Michaux MH, Ota KY, Shaffer JW, Kurland AA. The comparative effectiveness of eight phenothiazines. Psychopharmacologia 1965;7:89‐106. [DOI] [PubMed] [Google Scholar]
Kurland 1961 {published data only}
- Kurland AA, Hanlon TE, Tatom MH, Ota KY, Simopoulos AM. The comparative effectiveness of six phenothiazine compounds, phenobarbital and inert placebo in the treatment of acutely ill patients: global measures of severity of illness. Journal of Nervous and Mental Disease 1961;133:1‐18. [DOI] [PubMed] [Google Scholar]
Shalev 1993 {published data only}
- Shalev A, Hermesh H, Rothberg J, Munitz H. Poor neuroleptic response in acutely exacerbated schizophrenic patients. Acta Psychiatrica Scandinavica 1993;87(2):86‐91. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Akimoto 1966 {published data only}
- Akimoto H, Shimazaki T, Shibata N, Sato Y, Takahashi R. Current status of pharmacotherapy in schizophrenia. Folia Psychiatrica et Neurologica Japonica 1966;20(1):1‐8. [DOI] [PubMed] [Google Scholar]
Anon 2006 {published data only}
- Anon. A new comparison of antipsychotic drugs. Harvard Mental Health Letter 2006;22(7):7. [PubMed] [Google Scholar]
Bennett 1961 {published data only}
- Bennett JL, Kooi KA. Five phenothiazine derivatives. Evaluation and toxicity studies. Archives of General Psychiatry 1961;4:413‐8. [Google Scholar]
Casey 1960 {published data only}
- Casey JF, Lasky JJ, Klett CJ, Hollister LE. Treatment of schizophrenic reactions with phenothiazine derivatives. American Journal of Psychiatry 1960;August 1960:97‐105. [DOI] [PubMed] [Google Scholar]
Hollister 1974 {published data only}
- Hollister LE, Overall JE, Kimbell IJ, Pokorny A. Specific indications for different classes of phenothiazines. Archives of General Psychiatry 1974;30(1):94‐9. [DOI] [PubMed] [Google Scholar]
Lapolla 1967 {published data only}
- Lapolla A. A double‐blind evaluation of chlorpromazine versus a combination of perphenazine and amitriptyline. International Journal of Neuropsychiatry 1967;3(5):403‐5. [Google Scholar]
Loza 2001 {published data only}
- Loza B, Kucharska‐Pietura K, Debowska G. Atypical versus typical antipsychotic treatment prognosis in first‐episode paranoid schizophrenia based on wcst and dichotic listening scores. European Neuropsychopharmacology 2001;11(3):285. [Google Scholar]
Nordic 1986 {published data only}
- Gerlach J. Tardive dyskinesia: pathophysiological mechanisms and clinical trials. L'Encephale 1988;14:227‐32. [PubMed] [Google Scholar]
- Nordic Dyskinesia Study Group. Effect of different neuroleptics in tardive dyskinesia and parkinsonism. A video‐controlled multicenter study with chlorprothixene, perphenazine, haloperidol and haloperidol + biperiden. Psychopharmacology 1986;90(4):423‐9. [PubMed] [Google Scholar]
- Povlsen UJ, Noring U, Meidahl B, Korsgaard S, Waehrens J, Gerlach J. The effects of neuroleptics on tardive dyskinesias. A video‐controlled, randomized study of chlorprothixene, perphenazine, haloperidol and haloperidol + biperiden [Neuroleptikas virkning pa tardive dyskinesier. En videokontrolleret, randomiseret undersogelse med klorprotixen, perfenazin, haloperidol og haloperidol + biperiden]. Ugeskrift for Laeger 1987;149(25):1682‐5. [PubMed] [Google Scholar]
Schulsinger 1958 {published data only}
- Schulsinger F, Jensen R. Trilafon (perphenazine) and largactil (chlorpromazine) in chronic schizophrenia. A comparison [Trilafon (perfenazin) og Largactil (klorpromazin) hos kronisk sindssyge]. Ugeskrift For Laeger 1958;120(12):366‐9. [PubMed] [Google Scholar]
Smith 1959 {published data only}
- Smith JA, Christian D, Mansfield E, Figaredo A. A graphic comparison of five phenothiazines. American Journal of Psychiatry 1959;116:392‐9. [DOI] [PubMed] [Google Scholar]
Svestka 1972 {published data only}
- Svestka J, Nahunek K. A comparison of pimozide with perphenazine in the treatment of acute schizophrenic psychoses. Activitas Nervosa Superior 1972;14(2):93‐4. [MEDLINE: ] [PubMed] [Google Scholar]
Svestka 1974 {published data only}
- Svestka J, Nahunek K, Rodova A, Ceskova E. A controlled comparison of oxypertine and perphenazine in schizophrenic psychoses. Activitas Nervosa Superior 1974;16(3):165‐6. [PubMed] [Google Scholar]
Vinar 1968 {published data only}
- Vinar O. Differences in therapeutic effects of phenothiazine drugs. Agressologie 1968;9(2):315‐7. [PubMed] [Google Scholar]
Additional references
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313:1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Andreasen 2010
- Andreasen NC, Pressler M, Nopoulos P, Miller D, Ho BC. Antipsychotic drug equivalents and dose‐years: a standardized method for comparing exposure to different drugs. Biological Psychiatry 2010;67:255‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
APA 1980
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Third Edition (DSM III). Washington DC: American Psychiatric Association, 1980. [Google Scholar]
Berger 2003
- Berger M. Psychische Erkrankungen. Klinik und Therapie. 2nd Edition. München: Urban & Fischer, 2003. [Google Scholar]
Bland 1997
- Bland JM, Kerry SM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use. Therapie 1999;54(4):405‐11. [PubMed] [Google Scholar]
Campbell 2000
- Campbell M, Grimshaw J, Steen N. Sample size calculations for cluster randomised trials. Changing Professional Practice in Europe Group (EU BIOMED II Concerted Action). Journal of Health Services Research and Policy 2000;5:12‐6. [DOI] [PubMed] [Google Scholar]
Carpenter 1994
- Carpenter WT, Buchanan RW. Schizophrenia. New England Journal of Medicine 1994;330:681‐90. [DOI] [PubMed] [Google Scholar]
Davis 1974
- Davis JM. Overview: maintenance therapy in psychiatry: I. Schizophrenia. American Journal of Psychiatry 1975;132(12):1237‐45. [DOI] [PubMed] [Google Scholar]
Davis 1989
- Davis JM, Barter JT, Kane JM. Antipsychotic drugs. Comprehensive Textbook of Psychiatry. Williams and Wilkins, 1989. [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Proceedings of the 8th International Cochrane Colloquium; 2000 Oct 25‐28th; Cape Town, South Africa. Cape Town, 2000.
Der‐Simonian 1986
- Der‐Simonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7:177‐88. [DOI] [PubMed] [Google Scholar]
Divine 1992
- Divine GW, Brown JT, Frazer LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7:623‐9. [DOI] [PubMed] [Google Scholar]
Dold 2012
- Dold M, Li C, Tardy M, Leucht S. Haloperidol versus first generation antipsychotics for schizophrenia. Cochrane Database of Systematic Reviews 2012, Issue 5. [DOI: 10.1002/14651858.CD009831] [DOI] [PMC free article] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey‐Smith G, Schneider M, Minder CSO. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;13:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31:140‐9. [DOI] [PubMed] [Google Scholar]
Falkai 2005
- Falkai P, Wobrock T, Lieberman J. World Federation of Societies of Biological Psychiatry (WFSBP) ‐ guidelines for biological treatment of schizophrenia, part 1: Acute treatment of schizophrenia. World Journal of Biological Psychiatry 2005;6:132‐91. [DOI] [PubMed] [Google Scholar]
Fenton 2007
- Fenton M, Rathbone J, Reilly J, Sultana A. Thioridazine for schizophrenia. Cochrane Database of Systematic Reviews 2007, Issue 3. [DOI: 10.1002/14651858.CD001944.pub2; PUBMED: 17636691] [DOI] [PMC free article] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(1):7‐10. [DOI] [PubMed] [Google Scholar]
Gaebel 2006
- Gaebel W, Falkai P, Weinmann S. Behandlungsleitlinie Schizophrenie. Darmstadt: Steinkopff, 2006. [Google Scholar]
GRADE 2004 [Computer program]
- GRADE Working Group. GRADE Profiler. Version 3.2. GRADE Working Group, 2004.
Gulliford 1999
- Gulliford MC, Ukoumunne OC, Chinn S. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Haase 1983
- Haase HJ. Dosierung der Neuroleptika. Ein Leitfaden für Klinik und Praxis unter besonderer Berücksichtigung psychotisch Kranker. Erlangen: Perimed Fachbuch‐Verlagsgesellschaft, 1983. [Google Scholar]
Hartung 2005
- Hartung B, Wada M, Laux G, Leucht S. Perphenazine for schizophrenia. Cochrane Database of Systematic Reviews 2005, Issue 1. [DOI: 10.1002/14651858.CD009369] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org 2011.
Jones 2006
- Jones PB, Barnes TRE, Davies L. Randomized controlled trial of the effect on quality of life of second‐ vs first‐generation antipsychotic drugs in schizophrenia ‐ cost utility of the latest antipsychotic drugs in schizophrenia study (CUtLASS 1). Archives of General Psychiatry 2006;63:1079‐86. [DOI] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda (NY): Multi‐Health Systems, 1986. [Google Scholar]
Klein 1969
- Klein DF, Davis JM. Diagnosis and Drug Treatment of Psychiatric Disorders. Baltimore, MD: Williams and Wilkins, 1969. [Google Scholar]
Lehman 2004
- Lehman AF, Lieberman JA, Dixon LB. Practice guideline for the treatment of patients with schizophrenia, second edition. American Journal of Psychiatry 2004;161:1‐56. [PubMed] [Google Scholar]
Leon 2006
- Leon AC, Mallinckrodt CH, Chuang‐Stein C, Archibald DG, Archer GE, Chartier K. Attrition in randomized controlled clinical trials: methodological issues in psychopharmacology. Biological Psychiatry 2006;59:1001‐5. [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. What does the PANSS mean?. Schizophrenia Research 2005;79:231‐8. [DOI] [PubMed] [Google Scholar]
Leucht 2005b
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of Brief Psychiatric Rating Scale Scores. British Journal of Psychiatry 2005;187:366‐71. [DOI] [PubMed] [Google Scholar]
Leucht 2009
- Leucht S, Corves C, Arbter D, Engel RR, Li C, Davis JM. Second‐generation versus first‐generation antipsychotic drugs for schizophrenia: a meta‐analysis. Lancet 2009;373(9657):31‐41. [PUBMED: 19058842] [DOI] [PubMed] [Google Scholar]
Lieberman 2005
- Lieberman JA, Stroup TS, McEvoy JP. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. New England Journal of Medicine 2005;353:1209‐23. [DOI] [PubMed] [Google Scholar]
Lohse 2005
- Lohse MJ, Lorenzen A, Müller‐Oerlinghausen B. Psychopharmaka. In: Schwabe U, Pfaffrath D editor(s). Arzneimittelverordnungsreport. Berlin: Springer, 2005:820‐64. [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Marvaha 2004
- Marwaha S, Johnson S. Schizophrenia and employment ‐ a review. Social Psychiatry and Psychiatric Epidemiology 2004;39:337‐49. [DOI] [PubMed] [Google Scholar]
Moher 2010
- Moher D, Hopewell S, Schulz KF, Montori V, Gøtzsche PC, Devereaux PJ, et al. CONSORT 2010 Explanation and Elaboration: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340:c869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychological Reports 1962;10:799‐12. [Google Scholar]
Review Manager 2008 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.0. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2008.
Schünemann 2008
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. The Cochrane Collaboration, 2008:359‐83. [Google Scholar]
Seeman 1975
- Seeman P, Lee T. Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science 1975;188(4194):1217‐9. [DOI] [PubMed] [Google Scholar]
Tardy 2014
- Tardy M, Leucht S, Potapov A, Engel R, Dold M, Kissling W. Flupenthixol versus low‐potency first‐generation antipsychotic drugs for schizophrenia. Cochrane Database of Systematic Reviews 2014, Issue 9. [DOI: 10.1002/14651858.CD009227; CD009227] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tardy 2014a
- Tardy M, Leucht S, Potapov A, Engel R, Huhn M, Kissling W. Fluphenazine versus low‐potency first‐generation antipsychotic drugs for schizophrenia. Cochrane Database of Systematic Reviews 2014, Issue 8. [DOI: 10.1002/14651858.CD009230; CD009230] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tardy 2014b
- Tardy M, Leucht S, Potapov A, Kissling W, Engel R, Huhn M, et al. Haloperidol versus low‐potency first‐generation antipsychotic drugs for schizophrenia. Cochrane Database of Systematic Reviews 2014, Issue 7. [DOI: 10.1002/14651858.CD009268; CD009268] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tardy 2014c
- Tardy M, Leucht S, Potapov A, Engel R, Dold M, Kissling W. Trifluoperazine versus low‐potency first‐generation antipsychotic drugs for schizophrenia. Cochrane Database of Systematic Reviews 2014, Issue 7. [DOI: 10.1002/14651858.CD009396; CD009396] [DOI] [PMC free article] [PubMed] [Google Scholar]
Trikalinos 2004
- Trikalinos TA, Churchill R, Ferri M, Leucht S, Tuunainen A, Wahlbeck K, et al. Effect sizes in cumulative meta‐analyses of mental health randomized trials evolved over time. Journal of Clinical Epidemiology 2004;57(11):1124‐30. [DOI] [PubMed] [Google Scholar]
Tsuang 1978
- Tsuang MT. Suicide in schizophrenics, manics, depressives, and surgical controls: a comparison with general population suicide mortality. Archives of General Psychiatry 1978;35:153‐5. [DOI] [PubMed] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]