

Abstract
Emerging infectious diseases of zoonotic origin are shaping today’s infectious disease field more than ever. In this article, we introduce and review three emerging zoonotic viruses. Novel hantaviruses emerged in the Americas in the mid-1990s as the cause of severe respiratory infections, designated hantavirus pulmonary syndrome, with case fatality rates of around 40%. Nipah virus emerged a few years later, causing respiratory infections and encephalitis in Southeast Asia, with case fatality rates ranging from 40% to more than 90%. A new coronavirus emerged in 2012 on the Arabian Peninsula with a clinical syndrome of acute respiratory infections, later designated as Middle East respiratory syndrome (MERS), and an initial case fatality rate of more than 40%. Our current state of knowledge on the pathogenicity of these three severe, emerging viral infections is discussed.
Keywords: emerging viruses, hantavirus pulmonary syndrome, Nipah virus disease, Middle East respiratory syndrome
INTRODUCTION
Emerging infectious diseases are an important part of medical history, exemplified by some of the most dramatic pandemics or larger epidemics throughout the centuries. The causative pathogens were often of zoonotic origin, and emergence/reemergence of zoonotic microbial pathogens is shaping today’s infectious disease field more than ever. Factors in emergence/reemergence are multifold and related to changes in human demographics and behavior, changes in land use and agricultural practices, development of technologies, changes in climate, alterations in genetics of microbial pathogens, and failing of health care systems and public health measures mostly in developing but to some degree also developed countries. In particular, the tremendous increase and speed in travel around the globe and to and from remote places provide a foundation for rapid distribution of microbial pathogens. Despite success over decades in the development of medical countermeasures largely against bacterial and fungal pathogens, worldwide public health remains particularly vulnerable against emerging viruses that cross the species barrier into humans. Among those, respiratory viruses are of great concern due to their general ease of transmission among humans. The fear of influenza epidemics/pandemics has dominated the emerging virus field over other, sometimes rogue, respiratory viruses that have not yet reached a similar level of attention, often due to still limited transmissibility. Some of these, however, pose a tremendous regional or global public health risk should the viruses gain enhanced transmissibility and pathogenicity through adaptation. Thus, work on emerging neglected or rogue respiratory viruses is essential for proper public health preparedness and response. This includes identifying the mechanisms of pathogenicity and enhanced transmissibility, a highly controversial topic in infectious disease research today.
In this article, we introduce and review three emerging zoonotic viruses that fulfill the criteria of a rogue respiratory pathogen. Novel hantaviruses, commonly known as causative agents of mild to severe kidney disease, designated nephropathia epidemica and hemorrhagic fever with renal syndrome (HFRS), emerged in the Americas in the mid-1990s, surprising public health officials as the cause of severe respiratory infections, later designated hantavirus pulmonary syndrome (HPS), with case fatality rates of around 40%. Only a few years later, Nipah virus emerged as a new paramyxovirus causing respiratory infections and encephalitis in Southeast Asia, with case fatality rates ranging from 40% to more than 90%. Most recently, a new coronavirus emerged on the Arabian Peninsula with a clinical syndrome of acute respiratory infections, later designated as Middle East respiratory syndrome coronavirus (MERS-CoV), and an initial case fatality rate of more than 40%. These three viral infections are currently not associated with efficient transmissibility among humans, but the viruses already display high pathogenicity for humans. Thus, these viral pathogens deserve serious attention as potential regional and/or global public health threats.
HANTAVIRUS
Hantavirus and Its Host
HantavirusesareauniquegroupofviruseswithinthefamilyBunyaviridaethatcanbeserologically, phylogenetically, and geographically divided into two general categories: Old and New World hantaviruses. Unlike other members of the Bunyaviridae, hantaviruses are not arthropod-borne but rather utilize small mammals as natural reservoirs and vectors. To date, hantaviruses that are pathogenic to humans are exclusively rodent-borne (1). Generally, hantaviruses occupy a specific and limited geographical niche based on the distribution of their natural host. Old World hantaviruses are associated with rodents of the Arvicolinae and Murinaesub families and are commonly found in Europe and Asia, whereas New World hantaviruses are associated with Sigmodontinae subfamily rodents and are found throughout the Americas (Figure 1) (2).
Figure 1.
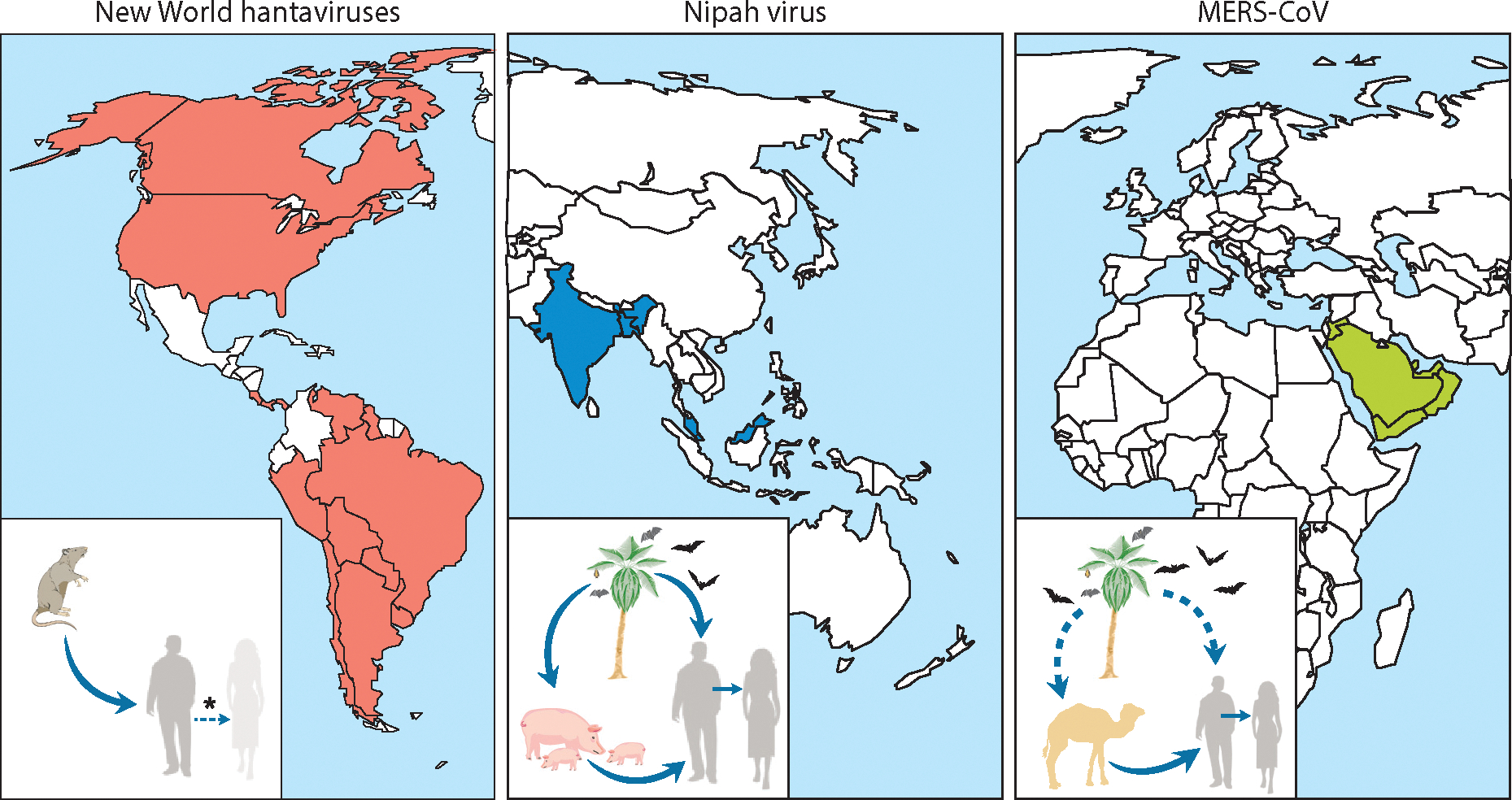
The geographic spread and zoonotic transmission of New World hantaviruses, Nipah virus, and Middle East respiratory syndrome coronavirus (MERS-CoV). Endemic regions where human cases have been identified are indicated on the map for each virus. Insets display the zoonotic transmission cycle of each virus. New World hantaviruses are transmitted directly from their rodent reservoir to humans; human-to-human transmission has occurred on rare occasion, but only for Andes virus (asterisk). Nipah virus is transmitted from its fruit bat reservoir directly or via an intermediate/amplifying host, the pig. Subsequent human-to-human transmission occurs regularly in Bangladesh. The transmission cycle of MERS-CoV is currently not completely resolved because the natural reservoir has not been definitively established. MERS-CoV is either transmitted directly from its reservoir in bats or dromedary camels, or is transmitted via dromedary camels as an intermediate/amplifying host; subsequent human-to-human and/or nosocomial transmission occurs regularly.
More than half of the 24 species of hantavirus currently recognized by the International Committee on Taxonomy of Viruses are associated with human disease, either HPS (caused by New World hantaviruses) or HFRS (caused by Old World hantaviruses) (1). In general, the clinical presentation of these two diseases is relatively different, with HPS primarily affecting the lungs and resulting in acute pulmonary manifestations, and HFRS targeting the kidneys and leading to acute renal insufficiencies. There is, however, growing evidence that the clinical distinction between HPS and HFRS is not as black and white as once thought, with renal complications and hemorrhage observed in cases of HPS and pulmonary manifestations noted with increasing frequency in cases of HFRS. Despite this, little is currently known about the pathophysiology of pulmonary disease associated with HFRS; therefore, the remainder of this section focuses exclusively on HPS, for which more information is available.
Hantavirus Disease Outbreaks and Incidence
In 1993, an outbreak of an unknown disease with characteristics of acute respiratory distress syndrome (ARDS) was reported in the Four Corners region of the southwestern United States (3). Between May and December 1993, 48 previously healthy people developed severe pulmonary illness, with more than 50% of these individuals rapidly succumbing to the infection of unknown etiology. The illness was quickly attributed to a novel hantavirus, the first pathogenic New World hantavirus to be described (4). The prototype virus was isolated and named Sin Nombre virus (SNV), and the disease was designated hantavirus pulmonary syndrome (HPS). Since 1993, cases of HPS have been documented throughout the Americas, with at least seven distinct viral species and related subspecies/genotypes, all associated with severe/lethal disease in humans (5). Although the severity of the initial Four Corners outbreak has never been matched in North America with respect to case numbers and fatality rate, multicase clusters have been observed, as was the case in Yosemite National Park in 2012, and serve as a reminder of the public health impact this rare but often fatal respiratory infection can have (5–8).
The true incidence of HPS across the Americas is difficult to determine due to differing diagnostic practices and nonstandardized reporting approaches in various regions. On average, fewer than 50 cases of HPS are diagnosed annually across North America. Between 1993 and 2011, 581 cases were confirmed in the United States (9). Disease in Canada is less frequent, with annual rates fluctuating between 2 and 8 cases (10). The incidence of HPS is more significant in South America, with more than 1,700 confirmed cases occurring in Argentina and Brazil between 1995 and 2011 (11, 12). In North America, the percentage of SNV infections that are associated with disease is believed to be nearly 100%. In contrast, South America may have a higher proportion of asymptomatic cases, with human seroprevalence rates between 2.2% in Chile and 14.3% in Argentina and Brazil (13–15). Like in North America, there is little hantavirus diversity in Chile, possibly due to isolation imparted by the Andes mountain range. The vast differences in asymptomatic cases observed between regions of North and South America seemingly correlate with the extent of genetically diverse viruses, many of which are suspected to be less virulent or potentially avirulent to humans, circulating in countries such as Argentina. Overall, HPS is fatal in approximately 30% of cases caused by SNV and 40–50% of cases caused by Andes virus (ANDV) (2).
Hantavirus Disease and Pathology
Clinically, HPS presents as a febrile disease characterized by bilateral interstitial pulmonary infiltrates and compromised respiratory function requiring supplemental oxygen and supportive care (16). The incubation period of HPS is generally between 9 and 33 days, with a median time to symptom onset of 14 to 17 days postexposure, although extended incubation periods of up to 51 days have been reported (17, 18). HPS is characterized by four phases of disease: febrile, cardiopulmonary, diuretic, and convalescent (Figure 2) (19). The febrile phase is characterized by nondescript symptoms, which often include lethargy, general weakness, and malaise. After 3 to 6 days of nondescript symptoms, patients enter the cardiopulmonary phase, which rapidly progresses from coughing and shortness of breath to severe respiratory distress requiring intubation and mechanical ventilation. This phase is characterized by vascular leakage, which occurs primarily in the lungs; hypoxemia; and cardiac complications. Death can occur within 36 to 48 h after the appearance of respiratory manifestations. In addition to respiratory failure, death is due to shock and myocardial dysfunction, which has led to the alternate designation of hantavirus cardiopulmonary syndrome. During the early stages of disease (i.e., around the time of symptom onset), virus is readily detectable in whole blood samples from patients, allowing for genotypic identification. The prognosis for patients who survive the respiratory insult and proceed to the diuretic phase is generally good, with rapid improvements and resolution of symptoms. The final or convalescent phase can last for months, and although long-term sequelae have not been identified, few studies have attempted to address this aspect of HPS.
Figure 2.
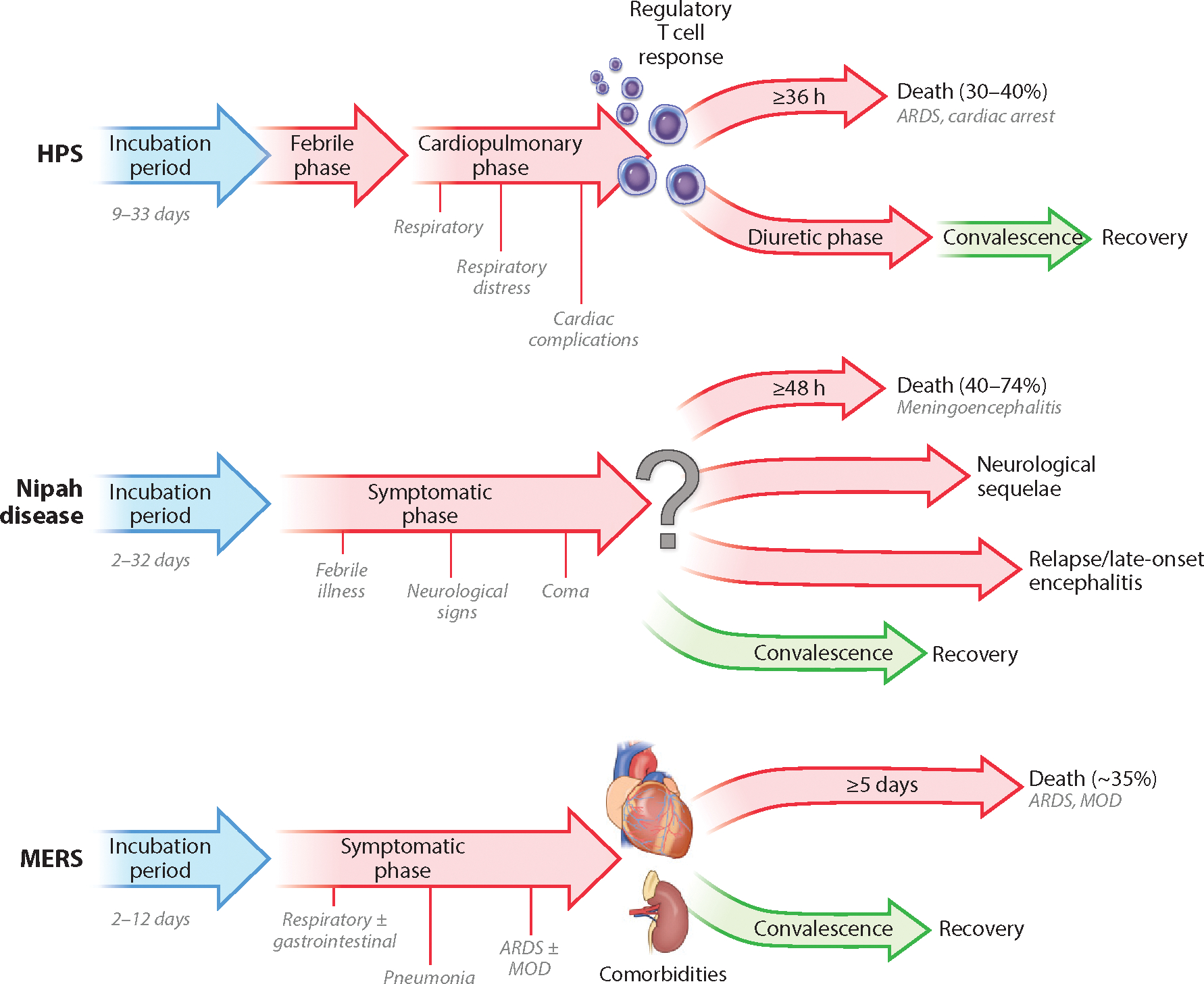
Disease progression in patients infected with New World hantaviruses, Nipah virus, or MERS-CoV. The different stages of disease are indicated, along with incubation time and minimum time from disease onset to death. Factors that are known to influence outcome of disease are also indicated. Abbreviations: ARDS, acute respiratory distress syndrome; HPS, hantavirus pulmonary syndrome; MERS, Middle East respiratory syndrome; MERS-CoV, Middle East respiratory syndrome coronavirus; MOD, multiorgan dysfunction.
Human infections most commonly occur by inhalation of virus-laden particles. Due to the extended incubation period, the early stages of infection and disease progression are not well established. In humans, hantaviruses primarily infect and replicate in endothelial cells, monocytes, and macrophages (Table 1) (3, 20, 21). Immunohistochemical staining conducted on tissue collected from fatal cases has demonstrated the presence of viral antigen in endothelial cells of capillaries and small vessels of several organs, including heart, kidney, spleen, bladder, pancreas, lymph node, skeletal muscle, intestine, adrenal, and adipose tissue (22, 23). Dendritic cells have also been shown to support hantavirus infection both in vivo and in vitro (21, 22). Despite the establishment of a systemic infection, pathological abnormalities associated with HPS are almost exclusive to the lungs and primarily consist of interstitial pneumonitis, mononuclear cell infiltrates, congestion, and alveolar edema (22).
Table 1.
Cell tropism of hantavirus, Nipah virus, and MERS-CoV
| Cell type | Virus | ||
|---|---|---|---|
| Hantavirus | Nipah virus | MERS-CoV | |
| Endothelial cells | + | + | + |
| Smooth muscle cells | + | + | − |
| Monocytes | + | − | − |
| Macrophages | + | + | + |
| Dendritic cells | + | − | + |
| Neurons | − | + | − |
| Upper respiratory tract epithelial cells | + | + | − |
| Alveolar pneumocytes | − | + | + |
A minus sign denotes that the indicated virus does not replicate in this cell type. A plus sign denotes that the indicated virus replicates in this cell type.
Animal Models to Study Hantavirus Disease
The study of hantaviruses and their associated diseases has been hampered by lack of a suitable animal model that reflects the disease progression observed in humans. Several small animal species have been evaluated as potential models for the study of HPS, with little success. Numerous laboratory rodents (mice, rats, guinea pigs, ferrets, gerbils, and hamsters) have been inoculated with various hantaviruses, and although they are susceptible to infection, much like the natural rodent hosts, they are generally asymptomatic (24). In 2001, the first authentic disease model for the study of hantaviruses was described (25). Hooper and colleagues reported that infection of Syrian hamsters (Mesocricetus auratus) with ANDV results in lethal illness that mirrors the cardiopulmonary phase of HPS. Following inoculation, hamsters undergo a 7–14-day prodrome, after which they abruptly transition into respiratory disease that is characterized by rapid and shallow breathing. Interestingly, although hamsters are highly susceptible to ANDV infection, with the 50% lethal dose for intramuscular and intraperitoneal injections calculated at less than 10 infectious units, other etiological agents of HPS (25, 26), including SNV, do not cause disease in this model (27, 28).
The pathogenesis of ANDV in hamsters has been intensely examined and provides important insight in to the pathophysiology of HPS in humans. Similar to the pathogenesis in humans, ANDV establishes a systemic infection in hamsters, with viral RNA and antigen readily detectable in the endothelium of essentially all organs examined (25, 29). Pathological abnormalities are generally limited to the lungs and include moderate to severe pulmonary edema and interstitial pneumonia (24). Red pulp congestion of the spleen and multifocal hepatitis, both of which have been described in humans, have also been reported in the hamster model, although the extent of each in hamsters appears to depend on the route of inoculation (24). In the days immediately preceding respiratory distress, hamsters become hypotensive and exhibit signs of cardiogenic shock as suggested by a rapidly increasing heart rate and heart rate variability (30). Hamsters also display an excessive and aberrant tissue-specific host immune response, which corresponds to disease onset and has been hypothesized to play a role in the development of HPS (29).
Over the past 15 years, the hamster model of HPS has been vital in elucidating the pathogenesis of HPS as well as characterizing potential medical countermeasures, including antiviral regimes, therapeutics targeting host-immune responses, and prophylactic and postexposure vaccine strategies. The importance of the hamster model cannot be understated; however, for illnesses that are potentially immune mediated, nonhuman primate (NHP) models are preferred due to the similarities in host responses between humans and NHPs (31). Several studies have shown that the majority of Old and New World NHPs are susceptible to infection with hantaviruses, although infection tends to be mild with little clinical relevance (24, 32). In 2014, the first NHP model for HPS was described in rhesus macaques (33). The key to development of the model appears to be the way inoculum was prepared: SNV propagated exclusively in deer mice as opposed to Vero cell culture. HPS in rhesus monkeys is strikingly similar to the human condition in many regards. Following inoculation, NHPs experience an incubation period of 15–22 days, after which respiratory manifestations appear (Figure 3), and the animals rapidly progress to a point where euthanasia is required. Similar to the disease in humans, the disease in NHPs is characterized by hematological abnormalities, including thrombocytopenia and leukocytosis, as well as pulmonary abnormalities, including edema characterized by moderate to severe interstitial pneumonia and thickening of the alveolar septae (Figure 4). Comparable to terminal disease in hamsters and humans, an excessive tissue-specific host immune response was noted in NHPs, the timing of which seemed to correspond with the hyperpermeability associated with the hallmark cardiopulmonary phase of HPS (29, 33). Although few studies have been conducted to date in the novel NHP model of HPS, the development of a gold-standard model for the study of HPS holds promise for future discoveries.
Figure 3.
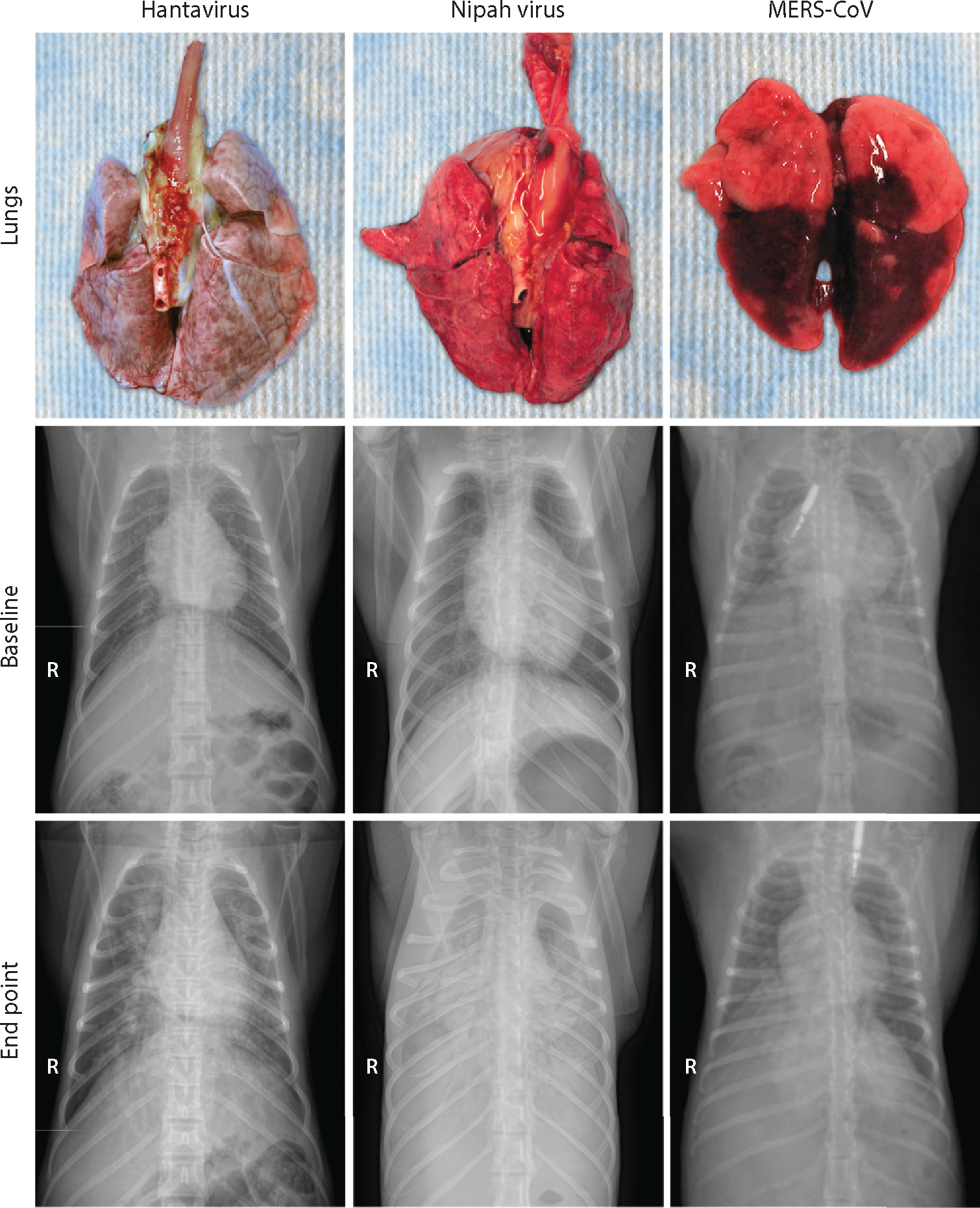
Gross pathological and radiographic changes in the lungs of nonhuman primates experimentally infected with a hantavirus, Nipah virus, or Middle East respiratory syndrome coronavirus (MERS-CoV). Lungs were collected from a Sin Nombre virus inoculated rhesus macaque at 15 days postinoculation (dpi); from a Nipah virus, strain Malaysia, inoculated African green monkey at 4 dpi; and from a MERS-CoV inoculated common marmoset at 6 dpi. Radiographs collected from these same animals are displayed. The top radiographs represent baseline; the bottom radiographs were taken at the endpoint of the experiment. The radiopaque objects in the two MERS-CoV radiographs are subcutaneously injected temperature transponders.
Figure 4.
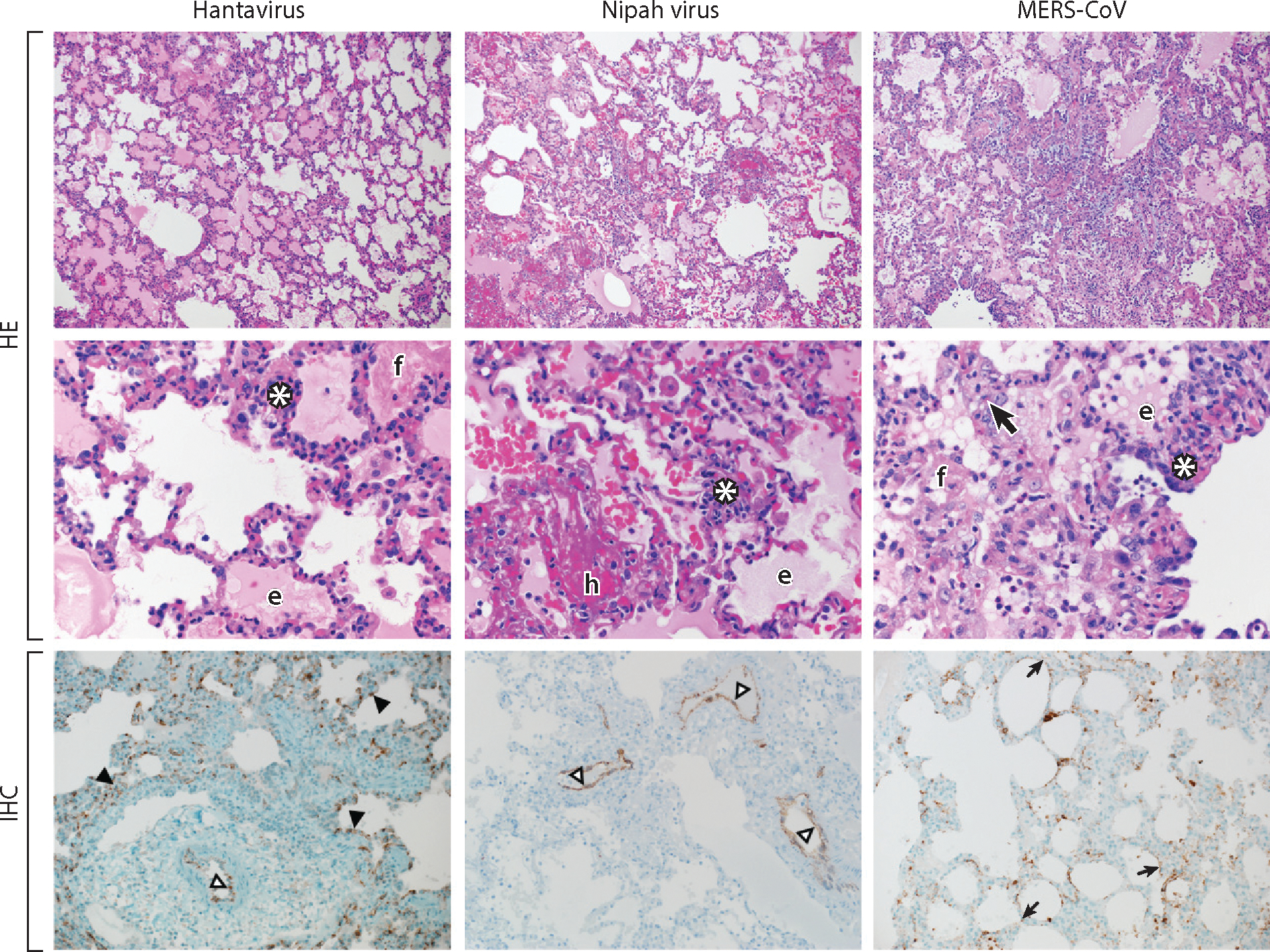
Histopathological changes in the lungs of nonhuman primates experimentally infected with a hantavirus, Nipah virus, or Middle East respiratory syndrome coronavirus (MERS-CoV). Lungs were collected from a Sin Nombre virus inoculated rhesus macaque; from a Nipah virus, strain Malaysia, inoculated African green monkey; and from a MERS-CoV inoculated common marmoset. Lung tissue was stained with hematoxylin eosin (HE) or a virus-specific antibody (IHC). Viral antigen is shown as brown-red staining.
Abbreviations and symbols: white asterisks, inflammatory cells; f, fibrin; e, edema; h, hemorrhage; bold black arrow, type II pneumocyte hyperplasia; black arrowheads, alveolar capillary endothelium; open arrowheads, arterial endothelium; small black arrows, alveolar pneumocytes. Magnification: top panels, 100×; middle panels, 400×; bottom panels, 200×.
Mechanisms of Hantavirus Disease
No single virulence factor has been identified to explain the differences between pathogenic and nonpathogenic hantaviruses, or why some cause HFRS and others cause HPS. Increased vascular permeability and leakage is a trait of both HPS and HFRS; however, the mechanisms responsible are largely unknown. It has been suggested that at least some hantaviruses may have direct effects on infected cells, which include apoptosis (34, 35) and cytopathic effect (36). However, other studies have found that pathogenic hantaviruses do not cause any apparent cytopathic effect in infected endothelial cells, and infection alone causes no disruption in the vascular endothelium, suggesting the pathology associated with hantaviral diseases, including HPS, is not exclusively or directly due to viral cytotoxicity (22, 37, 38). The prevailing theory on hantavirus pathogenesis is that disease is, at least in part, immune mediated. This hypothesis is based on clinical, epidemiological, and genetic analysis of human samples and is supported in part by in vitro and limited in vivo modeling studies.
Similar to other pathogenic RNA viruses, early regulation of the host cell responses appears to be critical for hantavirus replication. Several studies have demonstrated differences in the ability of pathogenic and nonpathogenic hantaviruses to suppress or not induce early innate responses in vitro, which may play a key role in establishment of infection (39–41). These findings are supported by studies in both hamsters and NHPs, which have demonstrated that early in infection, host responses are essentially acquiescent to viral infection with little or no immune activation; however, in the clinical stage of disease, intense and presumably deleterious responses are readily detectable, both circulating in serum as well as in specific tissues, as noted above (29, 33). Elevated cytokine levels (including TNF-α, IL-6, IL-10, and IFN-γ) as well as other soluble immunological mediators, including vascular endothelial growth factor (VEGF), have also been observed in hantaviral infections and may relate to symptoms (42–45). Further, increased numbers of cytokine-producing cells have been documented in tissue-specific samples of hantaviral diseases (kidney specimens for HFRS and lung specimens for HPS), as well as in animal models of HPS (29, 33, 46, 47). Comparisons of rodent reservoir and dead-end human host immune responses suggest the underlying mechanism may be differential activation of regulatory T cell responses (Figure 2). Although infected rodent reservoirs mount a regulatory T cell response (and do not develop disease) following infection, humans do not develop a robust regulatory T cell response, which may play an important role in the development of disease (48–50). Increased levels of activated CD8+ T cells have been documented in the acute stages of HPS; higher frequencies of circulating SNV-specific CD8+ T cells have been correlated with severe HPS (22, 51). Recent laboratory studies have demonstrated that hantavirus-specific cytotoxic T lymphocytes increased the permeability of infected endothelial cells following antigen recognition (52). Although the mechanisms responsible for the increased permeability were not addressed, a direct role of cellular immunity in the vascular leakage associated with HPS was suggested. Further support for the hypothesis of immune-related pathology is provided by the observation of a genetic predisposition associated with the severity of HPS (42, 51, 53).
Epidemiologically, the deductions of many of these findings are supported by the low incidence of HPS in children who by virtue of age have an immature immune system. While these observations could be dismissed by a possible reduction in exposure to hantaviruses in this age group, it is important to note that when hantaviral infection is confirmed in children and even adolescents, it is often atypical in presentation and could easily be misdiagnosed as a non-life-threatening respiratory infection (6). To date, few serological surveys have been conducted in young individuals, so an accurate determination of exposure/infection cannot be calculated.
As noted above, intense and potentially deleterious host immune responses following hantavirus infections have been documented in humans, hamsters, and NHPs; however, there is experimental evidence suggesting the pathophysiology of HPS may not be, at least entirely, immune mediated. Depletion studies in hamsters have shown that following inoculation with ANDV, animals specifically depleted for CD4+ or CD8+ T cells develop HPS, a result that is indistinguishable from that in immunologically normal, nondepleted hamsters (54). Further chemical destruction of the immune system renders hamsters susceptible to HPS-like disease following challenge with SNV, whereas immunologically normal hamsters are not susceptible to disease following challenge with SNV (55). The consequence of these findings for the pathophysiology of HPS in hamsters as well as NHPs and humans remains to be determined.
NIPAH VIRUS
Nipah Virus and Its Host
Nipah virus is one of three species in the genus Henipavirus of the Paramyxoviridae family, in addition to Hendra virus and Cedar virus (56, 57). Researchers proposed classifying the species Nipah virus into two genotypes: genotype M, containing strains from Malaysia and Cambodia, and genotype B, containing strains from Bangladesh and India (58). Unlike most paramyxoviruses, Nipah virus has a wide host range. Pteropus species fruit bats (also known as flying foxes) form the natural reservoir of Nipah virus (59–61). Experimental infection of flying foxes did not result in clinical disease, but animals shed virus and seroconverted (62). Spillover of Nipah virus from fruit bats into humans, pigs, dogs, and cats has been described (56). Moreover, Syrian hamsters, guinea pigs, ferrets, and African green monkeys can be infected with Nipah virus experimentally (63).
Nipah Virus Disease Outbreaks and Incidence
Nipah virus was discovered during a large outbreak of encephalitis that started in Malaysia in 1998 (56). During this outbreak, pigs formed an intermediate, amplifying reservoir that enabled Nipah virus transmission to humans on a large scale (Figure 1) and allowed spread of the outbreak to Singapore through transport of infected pigs (64). By the end of the outbreak in 1999, 276 people had become infected with Nipah virus, including 105 who suffered fatal encephalitis (56). Nipah virus reemerged in 2001 in India, where a large hospital-based outbreak resulted in 66 human cases with a case fatality rate of ~74% (65). A Nipah virus outbreak was first recognized in Bangladesh in 2001 (66, 67), and Nipah virus outbreaks with high case fatality rates have been identified in Bangladesh almost every year since then. Intermediate reservoirs do not seem to play a role in Nipah virus zoonoses in Bangladesh; rather, Nipah virus infections are thought to occur through the consumption of raw date palm sap that has been contaminated by Nipah virus–infected bats during collection (Figure 1) (68, 69). Respiratory disease is much more prevalent in Nipah virus patients in outbreaks in Bangladesh than in Malaysia (70). Probably as a result of this, human-to-human transmission occurs during Nipah virus outbreaks in Bangladesh; it was estimated that ~50% of Nipah virus cases in Bangladesh between 2001 and 2007 were the result of human-to-human transmission (71).
Nipah Virus Disease and Pathology
Nipah virus infection mainly results in encephalitis. Respiratory disease is also observed but not in all patients; respiratory disease is generally more prevalent in Nipah virus cases in Bangladesh (~69% of cases) than it was in cases in the Malaysia outbreak (~25% of cases) (70). Nipah virus–infected patients generally present with fever and altered mental status or reduced consciousness (72–76). Over time, neurological symptoms worsen and may result in coma and ultimately death (Figure 2). The case fatality rate of Nipah virus in the Malaysia and Singapore outbreak was ~40%; in Bangladesh the case fatality rate overall is ~70% but may reach 100% in some of the, generally small, outbreaks. Consistent with the neurological signs, magnetic resonance imaging revealed focal lesions disseminated throughout the central nervous system (CNS), mainly in the subcortical and deep white matter of the cerebral hemispheres (74, 76–78). In surviving patients, long-term neurological deficits are common (72). Moreover, late-onset or relapse encephalitis has been observed in survivors up to 11 years after the initial exposure to Nipah virus (Figure 2) (79–82).
Histopathological information on deceased patients is available only for patients from the Malaysian outbreak in 1998–1999. The main histopathological change observed in Nipah virus–infected patients was systemic vasculitis in small blood vessels and capillaries (83). This vasculitis was most prominent in the CNS, but was also detected in lungs, heart, and kidneys. Syncytia or multinucleated giant endothelial cells were observed in blood vessels (83).
Viral antigen was detected in the endothelium and neurons of the CNS, lungs, kidneys, heart, multinucleated giant cells in or lining the alveolar epithelium and macrophages, and multinucleated giant cells in spleen and lymph nodes (Table 1) (83).
Animal Models to Study Nipah Virus Disease
Several animal models have been developed for Nipah virus; Syrian hamsters, ferrets, and African green monkeys are currently the most commonly used models. In these three animal models, Nipah virus causes respiratory as well as neurological disease with lethal outcome (reviewed in 63). The animal models are mostly biased toward (fast) development of lethal respiratory disease (Figure 3); slower disease progression (i.e., through the use of a lower inoculum dose) allows the development of neurological signs. Nipah virus causes a systemic infection in Syrian hamsters, ferrets, and African green monkeys, with virus detected in lungs, CNS, liver, spleen, kidneys, and heart; virus can be detected in urine and in nose and throat swabs (84–88). Histological lesions are mainly observed in the respiratory tract and brain and consist of bronchointerstitial pneumonia (Figure 4) and widespread vasculitis; in parallel to observations in humans, endothelial cells and neurons in the CNS are infected by the virus (84–88). The African green monkey and ferret models have so far been used mainly for efficacy testing of vaccines and antiviral treatments; the hamster model is also used for efficacy testing, but has additionally been used to study Nipah virus pathogenicity and transmission.
Mechanisms of Nipah Virus Disease
In humans, Nipah virus pathology observed postmortem consists mainly of widespread vasculitis and encephalitis, with respiratory involvement in a subset of patients (83). From animal models, however, it is becoming clear that the respiratory tract plays an important part in the spread of Nipah virus to the CNS and/or systemically. Inoculation of pigs and Syrian hamsters has shown that Nipah virus can enter the CNS via olfactory neurons in the nasal cavity; in hamsters, this occurs within days of inoculation, before the onset of (respiratory) disease signs (89, 90). Viremia likely is another important component in the systemic spread of Nipah virus. Because Nipah virus replicates in endothelial cells, viremia may result in disruption of the blood-brain barrier and subsequent entry into the CNS (90). Disruption of the blood-brain barrier has indeed been observed in Nipah virus inoculated Syrian hamsters with neurological signs of disease (87); however, whether blood-brain barrier disruption was the cause or a result of CNS infection is currently not known. Nipah virus has been shown to attach to leukocytes without infecting them, thereby providing another mechanism for transport to different tissues (91).
Wild-type mice can be infected with Nipah virus but do not show clinical signs of disease upon infection. However, in IFNAR−/− mice lacking type I interferon (IFN) signaling, Nipah virus inoculation results in fatal encephalitis, indicating an important role for inhibition of innate immune signaling in the pathogenesis of Nipah virus (92).
Reverse genetics techniques became available for Nipah virus several years ago (93) but have so far only been used to elucidate the role of the nonstructural proteins V, W, and C in pathogenesis (94, 95). In vitro, these proteins inhibit IFN signaling (reviewed in 96). However, recombinant Nipah viruses lacking any one of these proteins were still able to suppress IFN signaling (95). In Syrian hamsters inoculated with recombinant viruses lacking V, W, or C, the virus lacking W did not show reduced virulence as compared with wild-type Nipah virus (95). However, the viruses lacking V or C were highly attenuated, with lack of clinical signs and 100% survival in inoculated animals. In Syrian hamsters inoculated with the virus lacking V, viral RNA could not be detected in tissues, although animals did seroconvert. In hamsters inoculated with a virus lacking C, viral RNA could be detected in several tissues, but at a level several orders of magnitude lower than in animals infected with wild-type Nipah virus (95). More detailed studies showed that in the lungs of Syrian hamsters inoculated with the recombinant Nipah virus lacking the C protein, the virus induced strong inflammation without edema or necrosis, whereas wild-type virus induced moderate inflammation with necrosis and edema (94). In primary human endothelial cells, the virus lacking C showed increased upregulation of genes encoding proinflammatory cytokines and chemokines. Taken together, these data indicate that protein C plays an important role in inhibiting the proinflammatory response upon Nipah virus infection, thereby preventing control of the infection by the immune system (94). However, other, currently undetermined, viral factors likely contribute to the development and severity of Nipah virus disease.
MIDDLE EAST RESPIRATORY SYNDROME CORONAVIRUS
Middle East Respiratory Syndrome Coronavirus and Its Host
MERS-CoV is classified as a lineage C betacoronavirus (97). All other known lineage C betacoronaviruses were found in bats, hence the natural reservoir of MERS-CoV was suspected to be bats (97, 98). Short sequence fragments detected in samples from bats had high sequence identity to MERS-CoV (99–102). A serosurvey of dromedary camels (Camelus dromedarius) in Oman revealed a very high prevalence of MERS-CoV neutralizing antibodies (103). Subsequent studies confirmed the presence of neutralizing antibodies in dromedary camels from Jordan, Qatar, Egypt, Dubai, and Saudi Arabia (104–107). Neutralizing antibodies were detected in sera from dromedary camels in Saudi Arabia going back as far as 1992 (104). MERS-CoV was also detected by RT-PCR in nasal swabs collected from dromedary camels from farms linked to human cases and from dromedary camels in Saudi Arabia (104, 106, 108, 109). Subsequently, MERS-CoV was isolated from nasal swabs taken from dromedary camels in Saudi Arabia and Qatar (104, 106). The detection of MERS-CoV in nasal swabs rather than rectal swabs suggested that MERS-CoV in dromedary camels is an upper respiratory tract infection. Although no formal proof exists that camels are infecting humans rather than humans infecting camels, the detection of MERS-CoV neutralizing antibodies in camel sera but not in human sera (Egypt) suggests that camels are the primary source of human infections. Whether dromedary camels are the natural reservoir of MERS-CoV or whether they are an intermediate or amplifying host is currently unclear (Figure 1), although the geographic range of the presence of neutralizing antibodies in these animals suggests that they are the natural reservoir.
Experimental inoculation of the potential reservoirs of MERS-CoV, bats and dromedary camels, has been performed (110). Inoculation of Jamaican fruit bats (Artibeus jamaicensis) did not result in clinical disease; however, viral RNA was detected in nose and throat swabs, blood, and several tissues, including lungs and nasal turbinates. Inoculation of dromedary camels with MERS-CoV resulted in mild clinical signs of disease. Dromedary camels shed virus mainly via the nose for a long period after inoculation. Moreover, MERS-CoV RNA was also detected in exhaled air (110).
Several studies have shown that the MERS-CoV receptor, dipeptidyl peptidase 4 (DPP4) (111), is a major determinant of host range (112–115). Although DPP4 is relatively conserved among species, variation exists in the region of DPP4 that binds to the MERS-CoV spike protein (116). Experimental studies, as well as modeling, have shown that DPP4 of mice, ferrets, and Syrian hamsters cannot effectively bind to the MERS-CoV spike protein (113, 114, 117, 118), whereas DPP4 from humans, rhesus macaques, common marmosets, dromedary camels, bats, horses, and goats can effectively bind (112, 115, 119–121).
Middle East Respiratory Syndrome Coronavirus Disease Outbreaks and Incidence
MERS-CoV was first isolated from a sputum sample from a man in Saudi Arabia with a fatal case of pneumonia and renal failure (122). Shortly after identification of this previously unknown virus, a cluster of undiagnosed respiratory disease in April 2012 in Jordan was retrospectively shown to have been caused by MERS-CoV, and the United Kingdom reported a case of MERS-CoV in a patient with severe respiratory disease transferred from Qatar (123). Sporadic cases continued to emerge until a hospital outbreak in April–May 2013 in Saudi Arabia caused a large accumulation of cases (124). In April 2014, cases again started to accumulate rapidly, most likely due to increased diagnostic testing resulting in the identification of many mild and asymptomatic cases. As of November 2014, 909 cases of MERS-CoV had been identified, including 331 fatal cases (125). The majority of cases occurred in Saudi Arabia, with additional cases in Qatar, Jordan, the United Arab Emirates, Yemen, and Kuwait (Figure 1), and imported cases occurred in the United Kingdom, Germany, France, Italy, Tunisia, Philippines, Malaysia, Greece, Egypt, the United States, and the Netherlands (125).
Although the MERS-CoV case fatality rate was initially very high at ~50%, more recently it has declined to around 35%, most likely due to the fact that more mild and asymptomatic cases are being detected during evaluation of patient contacts.
Middle East Respiratory Syndrome Coronavirus Disease and Pathology
MERS patients present with fever and respiratory symptoms such as coughing and shortness of breath; gastrointestinal symptoms also occur frequently (124, 126–133). Disease progresses to severe pneumonia requiring mechanical ventilation or extracorporeal membrane oxygenation (Figure 2). Fatal cases develop ARDS, often with multiorgan dysfunction and with acute renal failure as the main complication (127). One complicating factor in the pathogenesis of MERS-CoV is that almost all severe MERS-CoV cases have at least one comorbidity, such as diabetes, chronic kidney disease, chronic heart disease, hypertension, or chronic lung disease (Figure 2) (127). Unfortunately, these comorbidities are generally not replicated in animal models.
The pathology of MERS-CoV in humans is currently not well understood, mainly due to a lack of postmortem samples of fatal cases. Therefore, all data on the cell tropism of MERS-CoV are derived from ex vivo tissue culture experiments and animal models. Infection of human monocyte–derived macrophages in vitro resulted in upregulation of immune cell–recruiting cytokines and chemokines, of MHC class I and II, and of costimulation-related genes (134). In ex vivo human lung cultures, MERS-CoV replicated in ciliated and unciliated bronchiolar epithelium, type I and II pneumocytes, endothelial cells, and a very small percentage of alveolar macrophages (Table 1), a tropism that overlapped with the expression of DPP4 in the same cell types (134–136) and the MERS-CoV cell tropism observed in rhesus macaques and common marmosets (119, 120).
Animal Models to Study Middle East Respiratory Syndrome Coronavirus Disease
Although the search for an animal disease model started quickly after the discovery of MERS-CoV, it was not as straightforward as expected. Small animal models, such as mice and hamsters, were shown not to be susceptible to MERS-CoV (117,118). Neither were ferrets (114), a frequently used animal model for respiratory diseases. An NHP model was then established in rhesus macaques, thereby fulfilling Koch’s postulates for the causal relationship between the disease and MERS-CoV (121). In rhesus macaques, MERS-CoV causes a transient lower respiratory tract infection. Clinical signs develop within 24 h. Radiographically, varying degrees of localized infiltration and interstitial markings of the lungs are observed (119, 121, 137). Postmortem examinations at 3 days postinoculation (dpi) showed lesions throughout the lower respiratory tract, indicative of acute pneumonia. Lesions progressed into dark red-purple areas of pulmonary inflammation by 6 dpi. qRT-PCR analysis of tissues revealed widespread presence of hCoV-EMC/2012 in the nasal mucosa, trachea, mediastinal lymph nodes, and all six lung lobes at 3 dpi, with viral loads decreasing over time (119, 121). Virus was reisolated from lung tissue collected at 3 and 6 dpi. Histologically, lesions were characterized as multifocal, mild to marked, interstitial pneumonia. Virus replication occurred in type I and type II pneumocytes (119). Subsequently, a common marmoset model of MERS was developed (120). Clinical disease was more severe and of longer duration in common marmosets than in rhesus macaques. Some of the infected marmosets had to be euthanized due to the severity of the disease induced by MERS-CoV (120). In the lungs of infected common marmosets (Figure 3), up to ~1,000 times more viral RNA was detected than in the rhesus macaque lungs, and the bronchointerstitial pneumonia in marmosets was coalescing rather than multifocal (Figure 4). Moreover, MERS-CoV caused a systemic infection in common marmosets, with viremia starting at 3 dpi (120). Thus, the common marmoset model recapitulates severe human disease caused by MERS-CoV, whereas the rhesus macaque model recapitulates the milder cases.
A mouse model of MERS-CoV was developed by transducing mouse lungs with an adenovirus vector expressing human DPP4 (138). Although MERS-CoV does not cause severe disease in transduced, infected Balb/c mice, the virus replicates to high titers in the lungs and causes pneumonia. Moreover, development of the adenovirus vector allowed transduction and subsequent infection of several genetically modified mouse strains, thereby shedding some light on the role of different components of the immune system in MERS-CoV disease and clearance (138).
Mechanisms of Middle East Respiratory Syndrome Coronavirus Disease
In vitro studies have shown that MERS-CoV suppresses the induction of type I IFN after infection (134, 139, 140). Several viral proteins have been implicated in this suppression: ORF4a, ORF4b, ORF5, and M (141–143). ORF4a likely blocks the production of IFN through direct binding to double-stranded RNA (142, 144).
The use of an adenovirus 5 vector expressing human DPP4 to transduce different knockout mouse strains shed some light on the role of different components of the immune system in MERS-CoV disease (138). In MyD88−/− mice lacking Toll-like receptor signaling to induce IFN, MERS-CoV infection was more severe than in wild-type mice. In IFNAR−/− mice lacking IFN signaling, disease was as severe as in MyD88−/− mice; however, disease signs started two days earlier than in MyD88−/− mice (138). Clearance of virus was delayed in both mouse strains as compared with wild-type mice, suggesting that Toll-like receptor and IFN signaling play an important role in the development of MERS-CoV disease and clearance of the virus. Moreover, MERS-CoV did not cause more severe disease in TCRα−/− mice (lacking T cells), μMT mice (lacking B cells), or RAG1−/− SCID mice (lacking T and B cells); however, these experiments showed that T cells, but not B cells, are important for virus clearance because virus was not cleared in mice lacking T cells by the end of the experiment at 14 dpi (138).
In response to the observation of a high prevalence of comorbidities in severe MERS-CoV cases (127), the effect of immunosuppression on outcome of MERS-CoV infection was tested in the rhesus macaque model (145). Rhesus macaques were immunosuppressed through treatment with cyclophosphamide and dexamethasone starting at 2 weeks before MERS-CoV inoculation. Immunosuppressed rhesus macaques did not develop more severe disease than immunocompetent animals (145). However, immunosuppressed animals shed more virus, and viral loads in the lungs of these animals were significantly higher than in immunocompetent animals. Despite the increased virus replication in immunosuppressed animals, and in line with lack of increase in clinical disease, histological examination of the lungs showed a reduced inflammatory response in immunosuppressed rhesus macaques as compared with normal animals (145). These results suggested that the immune response to infection plays an important role in MERS-CoV pathogenesis.
Due to the very recent emergence of MERS-CoV and the initial lack of a small animal disease model, most questions surrounding MERS-CoV pathogenesis have not been addressed yet. Moreover, pathogenesis studies have been hampered by the absence of human postmortem data to compare with results in animal models. The availability of a small animal model in combination with reverse genetics techniques for MERS-CoV will be instrumental in increasing our understanding of the molecular determinants of MERS-CoV pathogenesis.
DISCUSSION
The mechanisms of disease underlying the severe infections caused by hantaviruses, Nipah virus, and MERS-CoV are poorly understood. Because MERS-CoV was discovered in 2012, it is perhaps not surprising that we are only starting to gain some knowledge on the pathogenesis of this virus. However, as is clear from the state of knowledge on hantavirus and Nipah virus pathogenesis, this lack of mechanistic data may not be resolved as quickly as is desirable. Hantavirus first caused an outbreak in North America in 1993 and has been causing fatal human cases every year since then, yet it is still poorly understood how this virus causes HPS and ultimately death. When we compare this with the amount of knowledge that is available on the pathogenesis of influenza A virus, where within months of the emergence of a new subtype in the human population the first scientific publications are available on pathogenicity markers, there is obviously a lot of room for improvement. Of course, new influenza A virus subtypes entering the human population are not considered rogue respiratory pathogens and are automatically perceived as a larger potential threat to public health than the viruses described in this review. However, human-to-human transmission already occurs with Nipah virus and MERS-CoV and with a selected portion of HPS-causing hantaviruses, and a small increase in transmission efficiency could potentially have a major impact on regional and/or global public health. More resources should thus be directed toward an understanding of the pathogenicity of the viruses described here and their mechanisms of transmission among humans.
One promising line of research in the hantavirus field is the comparison of infection in the natural reservoir with infection in humans and animal models of severe disease. In their rodent reservoirs, hantaviruses cause a persistent infection but no disease. Studying differences in how the infection is controlled by the immune system will likely lead to important insights into how hantaviruses cause severe disease in humans with respect to the immune system as well as other host factors. More detailed knowledge can be expected from the study of infection in the first NHP HPS model. Similar lines of research should be performed for Nipah virus and, potentially, MERS-CoV. Experimental infections of pteropid bats with Nipah virus have been performed, and studies as described above for hantaviruses are thus feasible at least for Nipah virus. Similar studies for MERS-CoV are lagging behind, as the reservoir has not definitively been identified and studies in camels, the natural or amplifying/interim host, are difficult to perform.
A lack of pathology from human autopsies is a huge limitation for understanding pathogenesis of the diseases caused by the three viruses discussed here. In this regard, some information exists for HPS infections, but Nipah virus and MERS-CoV cases are not commonly autopsied, as they occur in regions of the world that do not easily allow for such procedures for reasons of religion and tradition. Thus, our knowledge is largely based on animal models. A further limitation of most of the pathogenicity studies performed for all three viruses discussed in this review is that most research groups use a very limited number of strains for their pathogenicity experiments. For example, all Nipah virus pathogenicity studies have been performed with only two different strains, one representing the Malaysia genotype and one representing the Bangladesh genotype; almost all MERS-CoV studies have used a single strain. Surprisingly, only two disease models for HPS-causing hantaviruses have been established over two decades: the HPS hamster and rhesus macaque models utilizing ANDV and SNV, respectively. Although this allows for an easy comparison among studies performed by different laboratories, it is clear that much more genetic variation exists in nature. What is not clear, because of this limited strain use, is how this genetic variation affects pathogenicity. The discovery of less or more pathogenic strains of virus can lead to important discoveries on mechanisms of disease and should be pursued more actively.
ACKNOWLEDGMENTS
The authors thank Joe Prescott for providing Nipah virus figure panels and Ryan Kissinger and Anita Mora for help with figure preparation. Work on hantaviruses, henipaviruses, and coronaviruses in the Laboratory of Virology is supported by the Intramural Research Program of the NIAID.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
The Annual Review of Pathology: Mechanisms of Disease is online at pathol.annualreviews.org
This is a work of the US Government and is not subject to copyright protection in the United States.
LITERATURE CITED
- 1.Int. Comm. Taxon. Virol. (ICTV). 2013. Virus taxonomy: 2013 release. EC 45, Edinburgh, July 2013. http://aictvonline.org/virusTaxonomy.asp [Google Scholar]
- 2.Jonsson CB, Figueiredo LT, Vapalahti O. 2010. A global perspective on hantavirus ecology, epidemiology, and disease. Clin. Microbiol. Rev. 23:412–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duchin JS, Koster FT, Peters CJ, Simpson GL, Tempest B, et al. 1994. Hantavirus pulmonary syndrome: a clinical description of 17 patients with a newly recognized disease. N. Engl. J. Med. 330:949–55 [DOI] [PubMed] [Google Scholar]
- 4.Nichol ST, Spiropoulou CF, Morzunov S, Rollin PE, Ksiazek TG, et al. 1993. Genetic identification of a hantavirus associated with an outbreak of acute respiratory illness. Science 262:914–17 [DOI] [PubMed] [Google Scholar]
- 5.Macneil A, Nichol ST, Spiropoulou CF. 2011. Hantavirus pulmonary syndrome. Virus Res. 162:138–47 [DOI] [PubMed] [Google Scholar]
- 6.Webster D, Lee B, Joffe A, Sligl W, Dick D, et al. 2007. Cluster of cases of hantavirus pulmonary syndrome in Alberta, Canada. Am. J. Trop. Med. Hyg. 77:914–18 [PubMed] [Google Scholar]
- 7.Cent. Dis. Control Prev. 2012. Hantavirus pulmonary syndrome in visitors to a national park—Yosemite Valley, California, 2012. Morb. Mortal. Wkly. Rep. 61:952. [PubMed] [Google Scholar]
- 8.Roehr B 2012. US officials warn 39 countries about risk of hantavirus among travellers to Yosemite. BMJ 345:e6054. [DOI] [PubMed] [Google Scholar]
- 9.MacNeil A, Ksiazek TG, Rollin PE. 2011. Hantavirus pulmonary syndrome, United States, 1993–2009. Emerg. Infect. Dis. 17:1195–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drebot MA, Artsob H, Werker D. 2000. Hantavirus pulmonary syndrome in Canada, 1989–1999. Can. Commun. Dis. Rep. 26:65–69 [PubMed] [Google Scholar]
- 11.Figueiredo LT, Moreli ML, de-Sousa RL, Borges AA, de-Figueiredo GG, et al. 2009. Hantavirus pulmonary syndrome, central plateau, southeastern, and southern Brazil. Emerg. Infect. Dis. 15:561–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez VP, Bellomo CM, Cacace ML, Suarez P, Bogni L, Padula PJ. 2010. Hantavirus pulmonary syndrome in Argentina, 1995–2008. Emerg. Infect. Dis. 16:1853–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castillo C, Sanhueza L, Tager M, Munoz S, Ossa G, Vial P. 2002. Seroprevalence of antibodies against hantavirus in 10 communities of the IX Region of Chile where hantavirus infection were diagnosed. Rev. Med. Chile 130:251–58 [PubMed] [Google Scholar]
- 14.Campos GM, Moro de Sousa RL, Badra SJ, Pane C, Gomes UA, Figueiredo LT. 2003. Serological survey of hantavirus in Jardinopolis County, Brazil. J. Med. Virol. 71:417–22 [DOI] [PubMed] [Google Scholar]
- 15.Pini N, Levis S, Calderon G, Ramirez J, Bravo D, et al. 2003. Hantavirus infection in humans and rodents, northwestern Argentina. Emerg. Infect. Dis. 9:1070–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cent. Dis. Control Prev. 2010. Hantavirus pulmonary syndrome (hantavirus disease) (HPS) 2010 case definition. CSTE Pos. Statement 09-ID-17. http://wwwn.cdc.gov/nndss/script/casedef.aspx?CondYrID=697&DatePub=1/1/2010%2012:00:00%20AM [Google Scholar]
- 17.Young JC, Hansen GR, Graves TK, Deasy MP, Humphreys JG, et al. 2000. The incubation period of hantavirus pulmonary syndrome. Am. J. Trop. Med. Hyg. 62:714–17 [DOI] [PubMed] [Google Scholar]
- 18.Jonsson CB, Hooper J, Mertz G. 2008. Treatment of hantavirus pulmonary syndrome. Antivir. Res. 78:162–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Enria DA, Briggiler AM, Pini N, Levis S. 2001. Clinical manifestations of New World hantaviruses. Curr. Top. Microbiol. Immunol. 256:117–34 [DOI] [PubMed] [Google Scholar]
- 20.Mackow ER, Gavrilovskaya IN. 2001. Cellular receptors and hantavirus pathogenesis. Curr. Top. Microbiol. Immunol. 256:91–115 [DOI] [PubMed] [Google Scholar]
- 21.Raftery MJ, Kraus AA, Ulrich R, Kruger DH, Schonrich G. 2002. Hantavirus infection of dendritic cells. J. Virol. 76:10724–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zaki SR, Greer PW, Coffield LM, Goldsmith CS, Nolte KB, et al. 1995. Hantavirus pulmonary syndrome. Pathogenesis of an emerging infectious disease. Am. J. Pathol. 146:552–79 [PMC free article] [PubMed] [Google Scholar]
- 23.Borges AA, Campos GM, Moreli ML, Souza RL, Aquino VH, et al. 2006. Hantavirus cardiopulmonary syndrome: immune response and pathogenesis. Microbes Infect. 8:2324–30 [DOI] [PubMed] [Google Scholar]
- 24.Safronetz D, Ebihara H, Feldmann H, Hooper JW. 2012. The Syrian hamster model of hantavirus pulmonary syndrome. Antivir. Res. 95:282–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hooper JW, Larsen T, Custer DM, Schmaljohn CS. 2001. A lethal disease model for hantavirus pulmonary syndrome. Virology 289:6–14 [DOI] [PubMed] [Google Scholar]
- 26.Safronetz D, Hegde NR, Ebihara H, Denton M, Kobinger GP, et al. 2009. Adenovirus vectors expressing hantavirus proteins protect hamsters against lethal challenge with Andes virus. J. Virol. 83:7285–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wahl-Jensen V, Chapman J, Asher L, Fisher R, Zimmerman M, et al. 2007. Temporal analysis of Andes virus and Sin Nombre virus infections of Syrian hamsters. J. Virol. 81:7449–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Safronetz D, Prescott J, Haddock E, Scott DP, Feldmann H, Ebihara H. 2013. Hamster-adapted Sin Nombre virus causes disseminated infection and efficiently replicates in pulmonary endothelial cells without signs of disease. J. Virol. 87:4778–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Safronetz D, Zivcec M, Lacasse R, Feldmann F, Rosenke R, et al. 2011. Pathogenesis and host response in Syrian hamsters following intranasal infection with Andes virus. PLOS Pathog. 7:e1002426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campen MJ, Milazzo ML, Fulhorst CF, Obot Akata CJ, Koster F. 2006. Characterization of shock in a hamster model of hantavirus infection. Virology 356:45–49 [DOI] [PubMed] [Google Scholar]
- 31.Safronetz D, Geisbert TW, Feldmann H. 2013. Animal models for highly pathogenic emerging viruses. Curr. Opin. Virol. 3:205–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McElroy AK, Bray M, Reed DS, Schmaljohn CS. 2002. Andes virus infection of cynomolgus macaques. J. Infect. Dis. 186:1706–12 [DOI] [PubMed] [Google Scholar]
- 33.Safronetz D, Prescott J, Feldmann F, Haddock E, Rosenke R, et al. 2014. Pathophysiology of hantavirus pulmonary syndrome in rhesus macaques. PNAS 111:7114–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang JI, Park SH, Lee PW, Ahn BY. 1999. Apoptosis is induced by hantaviruses in cultured cells. Virology 264:99–105 [DOI] [PubMed] [Google Scholar]
- 35.Li XD, Lankinen H, Putkuri N, Vapalahti O, Vaheri A. 2005. Tula hantavirus triggers pro-apoptotic signals of ER stress in Vero E6 cells. Virology 333:180–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Markotic A, Hensley L, Geisbert T, Spik K, Schmaljohn C. 2003. Hantaviruses induce cytopathic effects and apoptosis in continuous human embryonic kidney cells. J. Gen. Virol. 84:2197–202 [DOI] [PubMed] [Google Scholar]
- 37.Sundstrom JB, McMullan LK, Spiropoulou CF, Hooper WC, Ansari AA, e tal. 2001. Hantavirus infection induces the expression of RANTES and IP-10 without causing increased permeability in human lung microvascular endothelial cells. J. Virol. 75:6070–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hardestam J, Klingstrom J, Mattsson K, Lundkvist A. 2005. HFRS causing hantaviruses do not induce apoptosis in confluent Vero E6 and A-549 cells. J. Med. Virol. 76:234–40 [DOI] [PubMed] [Google Scholar]
- 39.Levine JR, Prescott J, Brown KS, Best SM, Ebihara H, Feldmann H. 2010. Antagonism of type I interferon responses by New World hantaviruses. J. Virol. 84:11790–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spiropoulou CF, Albarino CG, Ksiazek TG, Rollin PE. 2007. Andes and Prospect Hill hantaviruses differ in early induction of interferon although both can downregulate interferon signaling. J. Virol. 81:2769–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alff PJ, Gavrilovskaya IN, Gorbunova E, Endriss K, Chong Y, et al. 2006. The pathogenic NY-1 hantavirus G1 cytoplasmic tail inhibits RIG-I- and TBK-1-directed interferon responses. J. Virol. 80:9676–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borges AA, Donadi EA, Campos GM, de Figueiredo GG, Saggioro FP, et al. 2013. Polymorphisms inhuman leukocyte antigens, human platelet antigens, and cytokine genes in hantavirus cardiopulmonary syndrome patients from Ribeirao Preto, Brazil. J. Med. Virol. 86(11):1962–70 [DOI] [PubMed] [Google Scholar]
- 43.Saggioro FP, Rossi MA, Duarte MI, Martin CC, Alves VA, et al. 2007. Hantavirus infection induces atypical myocarditis that may be responsible for myocardial depression and shock in hantavirus pulmonary syndrome. J. Infect. Dis. 195:1541–49 [DOI] [PubMed] [Google Scholar]
- 44.Linderholm M, Ahlm C, Settergren B, Waage A, Tarnvik A. 1996. Elevated plasma levels of tumor necrosis factor (TNF)-α, soluble TNF receptors, interleukin (IL)-6, and IL-10 in patients with hemorrhagic fever with renal syndrome. J. Infect. Dis. 173:38–43 [DOI] [PubMed] [Google Scholar]
- 45.Gavrilovskaya I, Gorbunova E, Koster F, Mackow E. 2012. Elevated VEGF levels in pulmonary edema fluid and PBMCs from patients with acute hantavirus pulmonary syndrome. Adv. Virol. 2012:674360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Temonen M, Mustonen J, Helin H, Pasternack A, Vaheri A, Holthofer H. 1996. Cytokines, adhesion molecules, and cellular infiltration in nephropathia epidemica kidneys: an immunohistochemical study. Clin. Immunol. Immunopathol. 78:47–55 [DOI] [PubMed] [Google Scholar]
- 47.Mori M, Rothman AL, Kurane I, Montoya JM, Nolte KB, et al. 1999. High levels of cytokine-producing cells in the lung tissues of patients with fatal hantavirus pulmonary syndrome. J. Infect. Dis. 179:295–302 [DOI] [PubMed] [Google Scholar]
- 48.Schountz T, Prescott J, Cogswell AC, Oko L, Mirowsky-Garcia K, et al. 2007. Regulatory T cell-like responses in deer mice persistently infected with Sin Nombre virus. PNAS 104:15496–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Easterbrook JD, Zink MC, Klein SL. 2007. Regulatory T cells enhance persistence of the zoonotic pathogen Seoul virus in its reservoir host. PNAS 104:15502–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Borges AA, Campos GM, Moreli ML, Moro Souza RL, Saggioro FP, et al. 2008. Role of mixed Th1 and Th2 serum cytokines on pathogenesis and prognosis of hantavirus pulmonary syndrome. Microbes Infect. 10:1150–57 [DOI] [PubMed] [Google Scholar]
- 51.Kilpatrick ED, Terajima M, Koster FT, Catalina MD, Cruz J, Ennis FA. 2004. Role of specific CD8+ T cells in the severity of a fulminant zoonotic viral hemorrhagic fever, hantavirus pulmonary syndrome. J. Immunol. 172:3297–304 [DOI] [PubMed] [Google Scholar]
- 52.Hayasaka D, Maeda K, Ennis FA, Terajima M. 2007. Increased permeability of human endothelial cell line EA.hy926 induced by hantavirus-specific cytotoxic T lymphocytes. Virus Res. 123:120–27 [DOI] [PubMed] [Google Scholar]
- 53.Borges AA, Donadi EA, Campos GM, Moreli ML, de Sousa RL, et al. 2010. Association of −308G/A polymorphism in the tumor necrosis factor-alpha gene promoter with susceptibility to development of hantavirus cardiopulmonary syndrome in the Ribeirao Preto region, Brazil. Arch. Virol. 155:971–75 [DOI] [PubMed] [Google Scholar]
- 54.Hammerbeck CD, Hooper JW. 2011. T cells are not required for pathogenesis in the Syrian hamster model of hantavirus pulmonary syndrome. J. Virol. 85:9929–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brocato RL, Hammerbeck CD, Bell TM, Wells JB, Queen LA, Hooper JW. 2014. A lethal disease model for hantavirus pulmonary syndrome in immunosuppressed Syrian hamsters infected with Sin Nombre virus. J. Virol. 88:811–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chua KB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, et al. 2000. Nipah virus: a recently emergent deadly paramyxovirus. Science 288:1432–35 [DOI] [PubMed] [Google Scholar]
- 57.Marsh GA, de Jong C, Barr JA, Tachedjian M, Smith C, et al. 2012. Cedar virus: a novel henipavirus isolated from Australian bats. PLOS Pathog. 8:e1002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lo MK, Lowe L, Hummel KB, Sazzad HM, Gurley ES, et al. 2012. Characterization of Nipah virus from outbreaks in Bangladesh, 2008–2010. Emerg. Infect. Dis. 18:248–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chua KB, Koh CL, Hooi PS, Wee KF, Khong JH, et al. 2002. Isolation of Nipah virus from Malaysian Island flying-foxes. Microbes Infect. 4:145–51 [DOI] [PubMed] [Google Scholar]
- 60.Rahman SA, Hassan SS, Olival KJ, Mohamed M, Chang LY, et al. 2010. Characterization of Nipah virus from naturally infected Pteropus vampyrus bats, Malaysia. Emerg. Infect. Dis. 16:1990–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sohayati AR, Hassan L, Sharifah SH, Lazarus K, Zaini CM, et al. 2011. Evidence for Nipah virus recrudescence and serological patterns of captive Pteropus vampyrus. Epidemiol. Infect. 139:1570–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Middleton DJ, Morrissy CJ, van der Heide BM, Russell GM, Braun MA, et al. 2007. Experimental Nipah virus infection in pteropid bats (Pteropus poliocephalus). J. Comp. Pathol. 136:266–72 [DOI] [PubMed] [Google Scholar]
- 63.Rockx B 2014. Recent developments in experimental animal models of Henipavirus infection. Pathog. Dis. 71:199–206 [DOI] [PubMed] [Google Scholar]
- 64.Mohd Nor MN, Gan CH, Ong BL. 2000. Nipah virus infection of pigs in peninsular Malaysia. Rev. Sci. Tech. 19:160–65 [DOI] [PubMed] [Google Scholar]
- 65.Chadha MS, Comer JA, Lowe L, Rota PA, Rollin PE, et al. 2006. Nipah virus-associated encephalitis outbreak, Siliguri, India. Emerg. Infect. Dis. 12:235–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hsu VP, Hossain MJ, Parashar UD, Ali MM, Ksiazek TG, et al. 2004. Nipah virus encephalitis reemergence, Bangladesh. Emerg. Infect. Dis. 10:2082–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Int. Cent. Diarrheal Dis. Res. Bangladesh. 2003. Outbreaks of encephalitis due to Nipah/Hendra-like viruses, Western Bangladesh. Health Sci. Bull. 1:1–6 [Google Scholar]
- 68.Luby SP, Rahman M, Hossain MJ, Blum LS, Husain MM, et al. 2006. Foodborne transmission of Nipah virus, Bangladesh. Emerg. Infect. Dis. 12:1888–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rahman MA, Hossain MJ, Sultana S, Homaira N, Khan SU, et al. 2011. Date palm sap linked to Nipah virus outbreak in Bangladesh, 2008. Vector Borne Zoonotic Dis. 12:65–72 [DOI] [PubMed] [Google Scholar]
- 70.Chong HT, Hossain J, Tan CT. 2008. Differences in epidemiologic and clinical features of Nipah virus encephalitis between the Malaysian and Bangladesh outbreaks. Neurol. Asia 13:23–26 [Google Scholar]
- 71.Luby SP, Hossain MJ, Gurley ES, Ahmed BN, Banu S, et al. 2009. Recurrent zoonotic transmission of Nipah virus into humans, Bangladesh, 2001–2007. Emerg. Infect. Dis. 15:1229–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chong HT, Kunjapan SR, Thayaparan T, Tong J, Petharunam V, et al. 2002. Nipah encephalitis outbreak in Malaysia, clinical features in patients from Seremban. Can. J. Neurol. Sci. 29:83–87 [DOI] [PubMed] [Google Scholar]
- 73.Chua KB, Goh KJ, Wong KT, Kamarulzaman A, Tan PS, et al. 1999. Fatal encephalitis due to Nipah virus among pig-farmers in Malaysia. Lancet 354:1257–59 [DOI] [PubMed] [Google Scholar]
- 74.Goh KJ, Tan CT, Chew NK, Tan PS, Kamarulzaman A, et al. 2000. Clinical features of Nipah virus encephalitis among pig farmers in Malaysia. N. Engl. J. Med. 342:1229–35 [DOI] [PubMed] [Google Scholar]
- 75.Hossain MJ, Gurley ES, Montgomery JM, Bell M, Carroll DS, et al. 2008. Clinical presentation of Nipah virus infection in Bangladesh. Clin. Infect. Dis. 46:977–84 [DOI] [PubMed] [Google Scholar]
- 76.Paton NI, Leo YS, Zaki SR, Auchus AP, Lee KE, et al. 1999. Outbreak of Nipah-virus infection among abattoir workers in Singapore. Lancet 354:1253–56 [DOI] [PubMed] [Google Scholar]
- 77.Lee KE, Umapathi T, Tan CB, Tjia HT, Chua TS, et al. 1999. The neurological manifestations of Nipah virus encephalitis, a novel paramyxovirus. Ann. Neurol. 46:428–32 [PubMed] [Google Scholar]
- 78.Sarji SA, Abdullah BJ, Goh KJ, Tan CT, Wong KT. 2000. MR imaging features of Nipah encephalitis. Am. J. Roentgenol. 175:437–42 [DOI] [PubMed] [Google Scholar]
- 79.Abdullah S, Chang LY, Rahmat K, Goh KJ, Tan CT. 2012. Late-onset Nipah virus encephalitis 11 years after the initial outbreak: a case report. Neurol. Asia 17:71–74 [Google Scholar]
- 80.Sejvar JJ, Hossain J, Saha SK, Gurley ES, Banu S, et al. 2007. Long-term neurological and functional outcome in Nipah virus infection. Ann. Neurol. 62:235–42 [DOI] [PubMed] [Google Scholar]
- 81.Tan CT, Goh KJ, Wong KT, Sarji SA, Chua KB, et al. 2002. Relapsed and late-onset Nipah encephalitis. Ann. Neurol. 51:703–8 [DOI] [PubMed] [Google Scholar]
- 82.Wong SC, Ooi MH, Wong MN, Tio PH, Solomon T, Cardosa MJ. 2001. Late presentation of Nipah virus encephalitis and kinetics of the humoral immune response. J. Neurol. Neurosurg. Psychiatry 71:552–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wong KT, Shieh WJ, Kumar S, Norain K, Abdullah W, et al. 2002. Nipah virus infection: pathology and pathogenesis of an emerging paramyxoviral zoonosis. Am. J. Pathol. 161:2153–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bossart KN, Zhu Z, Middleton D, Klippel J, Crameri G, et al. 2009. A neutralizing human monoclonal antibody protects against lethal disease in a new ferret model of acute Nipah virus infection. PLOS Pathog. 5:e1000642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.de Wit E, Bushmaker T, Scott D, Feldmann H, Munster VJ. 2011. Nipah virus transmission in a hamster model. PLOS Negl. Trop. Dis. 5:e1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Geisbert TW, Daddario-DiCaprio KM, Hickey AC, Smith MA, Chan YP, et al. 2010. Development of an acute and highly pathogenic nonhuman primate model of Nipah virus infection. PLOS ONE 5:e10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rockx B, Brining D, Kramer J, Callison J, Ebihara H, et al. 2011. Clinical outcome of henipavirus infection in hamsters is determined by the route and dose of infection. J. Virol. 85:7658–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wong KT, Grosjean I, Brisson C, Blanquier B, Fevre-Montange M, et al. 2003. A golden hamster model for human acute Nipah virus infection. Am. J. Pathol. 163:2127–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Munster VJ, Prescott JB, Bushmaker T, Long D, Rosenke R, et al. 2012. Rapid Nipah virus entry into the central nervous system of hamsters via the olfactory route. Sci. Rep. 2:736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Weingartl H, Czub S, Copps J, Berhane Y, Middleton D, et al. 2005. Invasion of the central nervous system in a porcine host by Nipah virus. J. Virol. 79:7528–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mathieu C, Pohl C, Szecsi J, Trajkovic-Bodennec S, Devergnas S, et al. 2011. Nipah virus uses leukocytes for efficient dissemination within a host. J. Virol. 85:7863–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dhondt KP, Mathieu C, Chalons M, Reynaud JM, Vallve A, et al. 2013. Type I interferon signaling protects mice from lethal henipavirus infection. J. Infect. Dis. 207:142–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yoneda M, Guillaume V, Ikeda F, Sakuma Y, Sato H, et al. 2006. Establishment of a Nipah virus rescue system. PNAS 103:16508–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mathieu C, Guillaume V, Volchkova VA, Pohl C, Jacquot F, et al. 2012. Nonstructural Nipah virus C protein regulates both the early host proinflammatory response and viral virulence. J. Virol. 86:10766–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yoneda M, Guillaume V, Sato H, Fujita K, Georges-Courbot MC, et al. 2010. The nonstructural proteins of Nipah virus play a key role in pathogenicity in experimentally infected animals. PLOS ONE 5:e12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Basler CF. 2012. Nipah and Hendra virus interactions with the innate immune system. Curr. Top. Microbiol. Immunol. 359:123–52 [DOI] [PubMed] [Google Scholar]
- 97.van Boheemen S, de Graaf M, Lauber C, Bestebroer TM, Raj VS, et al. 2012. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 3:e00473–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cotten M, Lam TT, Watson SJ, Palser AL, Petrova V, et al. 2013. Full-genome deep sequencing and phylogenetic analysis of novel human betacoronavirus. Emerg. Infect. Dis. 19:736–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Annan A, Baldwin HJ, Corman VM, Klose SM, Owusu M, et al. 2013. Human betacoronavirus 2c EMC/2012-related viruses in bats, Ghana and Europe. Emerg. Infect. Dis. 19:456–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Anthony SJ, Ojeda-Flores R, Rico-Chavez O, Navarette-Macias I, Zambrana-Torrelio CM, et al. 2013. Coronaviruses in bats from Mexico. J. Gen. Virol. 94:1028–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ithete NL, Stoffberg S, Corman VM, Cottontail VM, Richards LR, et al. 2013. Close relative of human Middle East respiratory syndrome coronavirus in bat, South Africa. Emerg. Infect. Dis. 19:1697–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Memish ZA, Mishra N, Olival KJ, Fagbo SF, Kapoor V, et al. 2013. Middle East respiratory syndrome coronavirus in bats, Saudi Arabia. Emerg. Infect. Dis. 19:1819–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Reusken C, Haagmans BL, Muller MA, Gutierrez C, Godeke GJ, et al. 2013. MERS-CoV neutralizing serum antibodies in dromedary camels: a comparative survey. Lancet Infect. Dis. 13:859–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Alagaili AN, Briese T, Mishra N, Kapoor V, Sameroff SC, et al. 2014. Middle East respiratory syndrome coronavirus infection in dromedary camels in Saudi Arabia. mBio 5:e00884–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Alexandersen S, Kobinger GP, Soule G, Wernery U. 2014. Middle East respiratory syndrome coronavirus antibody reactors among camels in Dubai, United Arab Emirates, in 2005. Transbound. Emerg. Dis. 61:105–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Haagmans BL, Al Dhahiry SH, Reusken CB, Raj VS, Galiano M, et al. 2014. Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect. Dis. 14:140–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Perera RA, Wang P, Gomaa MR, El-Shesheny R, Kandeil A, et al. 2013. Seroepidemiology for MERS coronavirus using microneutralisation and pseudoparticle virus neutralisation assays reveal a high prevalence of antibody in dromedary camels in Egypt, June 2013. Eurosurveillance 18:20574. [DOI] [PubMed] [Google Scholar]
- 108.Azhar EI, El-Kafrawy SA, Farraj SA, Hassan AM, Al-Saeed MS, et al. 2014. Evidence for camel-to-human transmission of MERS coronavirus. N. Engl. J. Med. 370:2499–505 [DOI] [PubMed] [Google Scholar]
- 109.Memish ZA, Cotten M, Meyer B, Watson SJ, Alsahafi AJ, et al. 2014. Human infection with MERS coronavirus after exposure to infected camels, Saudi Arabia, 2013. Emerg. Infect. Dis. 20:1012–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.van Doremalen N, de Wit E, Falzarano D, Scott D, Schountz T, et al. 2014. Modeling the host ecology of Middle East respiratory syndrome coronavirus (MERS-CoV): from host reservoir to disease. Annu. Meet. Am. Soc. Virol., 33rd, Fort Collins [Google Scholar]
- 111.Raj VS, Mou H, Smits SL, Dekkers DH, Muller MA, et al. 2013. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495:251–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Barlan A, Zhao J, Sarkar MK, Li K, McCray PB Jr, et al. 2014. Receptor variation and susceptibility to Middle East respiratory syndrome coronavirus infection. J. Virol. 88:4953–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cockrell AS, Peck KM, Yount BL, Agnihothram SS, Scobey T, et al. 2014. Mouse dipeptidyl peptidase4 is not a functional receptor for Middle East respiratory syndrome coronavirus infection. J. Virol. 88:5195–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Raj VS, Smits SL, Provacia LB, van den Brand JM, Wiersma L, et al. 2014. Adenosine deaminase acts as a natural antagonist for dipeptidyl peptidase 4-mediated entry of the Middle East respiratory syndrome coronavirus. J. Virol. 88:1834–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.van Doremalen N, Miazgowicz KL, Milne-Price S, Bushmaker T, Robertson S, et al. 2014. Host species restriction of Middle East respiratory syndrome coronavirus through its receptor dipeptidyl peptidase 4. J. Virol. 88:9220–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mou H, Raj VS, van Kuppeveld FJ, Rottier PJ, Haagmans BL, Bosch BJ. 2013. The receptor binding domain of the new Middle East respiratory syndrome coronavirus maps to a 231-residue region in the spike protein that efficiently elicits neutralizing antibodies. J. Virol. 87:9379–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Coleman CM, Matthews KL, Goicochea L, Frieman MB. 2014. Wild-type and innate immune-deficient mice are not susceptible to the Middle East respiratory syndrome coronavirus. J. Gen. Virol. 95:408–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.de Wit E, Prescott J, Baseler L, Bushmaker T, Thomas T, et al. 2013. The Middle East respiratory syndrome coronavirus (MERS-CoV) does not replicate in Syrian hamsters. PLOS ONE 8:e69127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.de Wit E, Rasmussen AL, Falzarano D, Bushmaker T, Feldmann F, et al. 2013. Middle East respiratory syndrome coronavirus (MERS-CoV) causes transient lower respiratory tract infection in rhesus macaques. PNAS 110:16598–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Falzarano D, de Wit E, Feldmann F, Rasmussen AL, Okumura A, et al. 2014. Infection with MERS-CoV causes lethal pneumonia in the common marmoset. PLOS Pathog. 10:e1004250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Munster VJ, de Wit E, Feldmann H. 2013. Pneumonia from human coronavirus in a macaque model. N. Engl. J. Med. 368:1560–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367:1814–20 [DOI] [PubMed] [Google Scholar]
- 123.Wise J 2013. Patient dies from novel coronavirus in UK. BMJ 346:f1133. [DOI] [PubMed] [Google Scholar]
- 124.Assiri A, McGeer A, Perl TM, Price CS, Al Rabeeah AA, et al. 2013. Hospital outbreak of Middle East respiratory syndrome coronavirus. N. Engl. J. Med. 369:407–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.World Health Organ. 2014. Global alert and response (GAR). Coronavirus infections. http://www.who.int/csr/don/archive/disease/coronavirus_infections/en/index.html [Google Scholar]
- 126.Albarrak AM, Stephens GM, Hewson R, Memish ZA. 2012. Recovery from severe novel coronavirus infection. Saudi Med. J. 33:1265–69 [PubMed] [Google Scholar]
- 127.Assiri A, Al-Tawfiq JA, Al-Rabeeah AA, Al-Rabiah FA, Al-Hajjar S, et al. 2013. Epidemiological, demographic, and clinical characteristics of 47 cases of Middle East respiratory syndrome coronavirus disease from Saudi Arabia: a descriptive study. Lancet Infect. Dis. 13:752–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Drosten C, Seilmaier M, Corman VM, Hartmann W, Scheible G, et al. 2013. Clinical features and virological analysis of a case of Middle East respiratory syndrome coronavirus infection. Lancet Infect. Dis. 13:745–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Guberina H, Witzke O, Timm J, Dittmer U, Muller MA, et al. 2014. A patient with severe respiratory failure caused by novel human coronavirus. Infection 42:203–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Guery B, Poissy J, El Mansouf L, Sejourne C, Ettahar N, et al. 2013. Clinical features and viral diagnosis of two cases of infection with Middle East respiratory syndrome coronavirus: a report of nosocomial transmission. Lancet 381:2265–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Memish ZA, Zumla AI, Al-Hakeem RF, Al-Rabeeah AA, Stephens GM. 2013. Family cluster of Middle East respiratory syndrome coronavirus infections. N. Engl. J. Med. 368:2487–94 [DOI] [PubMed] [Google Scholar]
- 132.Memish ZA, Zumla AI, AssiriA. 2013. Middle East respiratory syndrome coronavirus infections in healthcare workers. N. Engl. J. Med. 369:884–86 [DOI] [PubMed] [Google Scholar]
- 133.Puzelli S, Azzi A, Santini MG, Di Martino A, Facchini M, et al. 2013. Investigation of an imported case of Middle East respiratory syndrome coronavirus (MERS-CoV) infection in Florence, Italy, May to June 2013. Eurosurveillance 18:20564. [DOI] [PubMed] [Google Scholar]
- 134.Zhou J, Chu H, Li C, Wong BH, Cheng ZS, et al. 2013. Active replication of Middle East respiratory syndrome coronavirus replication and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J. Infect. Dis. 209:1331–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hocke AC, Becher A, Knepper J, Peter A, Holland G, et al. 2013. Emerging human Middle East respiratory syndrome coronavirus causes widespread infection and alveolar damage in human lungs. Am. J. Respir. Crit. Care Med. 188:882–86 [DOI] [PubMed] [Google Scholar]
- 136.Chan RW, Chan MC, Agnihothram S, Chan LL, Kuok DI, et al. 2013. Tropism and innate immune responses of the novel human betacoronavirus lineage C virus in human ex vivo respiratory organ cultures. J. Virol. 87:6604–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yao Y, Bao L, Deng W, Xu L, Li F, et al. 2014. An animal model of MERS produced by infection of rhesus macaques with MERS coronavirus. J. Infect. Dis. 209:236–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhao J, Li K, Wohlford-Lenane C, Agnihothram SS, Fett C, et al. 2014. Rapid generation of a mouse model for Middle East respiratory syndrome. PNAS 111:4970–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kindler E, Jonsdottir HR, Muth D, Hamming OJ, Hartmann R, et al. 2013. Efficient replication of the novel human betacoronavirus EMC on primary human epithelium highlights its zoonotic potential. mBio 4:e00611–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zielecki F, Weber M, Eickmann M, Spiegelberg L, Zaki AM, et al. 2013. Human cell tropism and innate immune system interactions of human respiratory coronavirus EMC compared to those of severe acute respiratory syndrome coronavirus. J. Virol. 87:5300–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Matthews KL, Coleman CM, van der Meer Y, Snijder EJ, Frieman MB. 2014. The ORF4b-encodedaccessory proteins of MERS-coronavirus and two related bat coronaviruses localize to the nucleus and inhibit innate immune signaling. J. Gen. Virol. 95(Pt. 4):874–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Niemeyer D, Zillinger T, Muth D, Zielecki F, Horvath G, et al. 2013. Middle East respiratory syndrome coronavirus accessory protein 4a is a type I interferon antagonist. J. Virol. 87:12489–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yang Y, Zhang L, Geng H, Deng Y, Huang B, et al. 2013. The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS-CoV) are potent interferon antagonists. Protein Cell 4:951–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Siu KL, Yeung ML, Kok KH, Yuen KS, Kew C, et al. 2014. Middle East respiratory syndrome coronavirus 4a protein is a double-stranded RNA-binding protein that suppresses PACT-induced activation of RIG-I and MDA5 in innate antiviral response. J. Virol. 88:4866–76 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 145.Prescott J, De Wit E, Falzarano D, Scott D, Feldmann H, Munster VJ. 2014. Defining the effects of immunosuppression in the rhesus model of Middle East respiratory syndrome (MERS). Annu. Meet. Am. Soc. Virol., 33rd, Fort Collins [Google Scholar]