

Abstract
To help provide a fundamental basis for use of microbial dissimilatory reduction processes in separating or immobilizing 99Tc in waste or groundwaters, the effects of electron donor and the presence of the bicarbonate ion on the rate and extent of pertechnetate ion [Tc(VII)O4−] enzymatic reduction by the subsurface metal-reducing bacterium Shewanella putrefaciens CN32 were determined, and the forms of aqueous and solid-phase reduction products were evaluated through a combination of high-resolution transmission electron microscopy, X-ray absorption spectroscopy, and thermodynamic calculations. When H2 served as the electron donor, dissolved Tc(VII) was rapidly reduced to amorphous Tc(IV) hydrous oxide, which was largely associated with the cell in unbuffered 0.85% NaCl and with extracellular particulates (0.2 to 0.001 μm) in bicarbonate buffer. Cell-associated Tc was present principally in the periplasm and outside the outer membrane. The reduction rate was much lower when lactate was the electron donor, with extracellular Tc(IV) hydrous oxide the dominant solid-phase reduction product, but in bicarbonate systems much less Tc(IV) was associated directly with the cell and solid-phase Tc(IV) carbonate may have been present. In the presence of carbonate, soluble (<0.001 μm) electronegative, Tc(IV) carbonate complexes were also formed that exceeded Tc(VII)O4− in electrophoretic mobility. Thermodynamic calculations indicate that the dominant reduced Tc species identified in the experiments would be stable over a range of Eh and pH conditions typical of natural waters. Thus, carbonate complexes may represent an important pathway for Tc transport in anaerobic subsurface environments, where it has generally been assumed that Tc mobility is controlled by low-solubility Tc(IV) hydrous oxide and adsorptive, aqueous Tc(IV) hydrolysis products.
Technetium (element 43) is present in the environment principally as a result of fallout from nuclear weapons testing, uranium enrichment, nuclear fuel processing, and disposal after pharmaceutical use (34). During nuclear fuel reprocessing, Tc is solubilized from spent fuels and is present in all waste streams (10) principally as the pertechnetate anion [Tc(VII)O4−]. Over the pH and Eh ranges typical of most groundwaters, Tc may exist in oxidation states VII, VI, V or IV, but in the absence of strong complexing agents Tc(VI) and Tc(V) may be expected to disproportionate to Tc(VII) and Tc(IV) (31). The dominant oxidation state under oxic conditions is Tc(VII), which is weakly sorbed by most soils and subsurface sediments at near neutral pH values (15, 35). Under anoxic conditions and in the absence of aqueous complexing agents other than OH−, Tc(IV) is largely immobile because it forms concentration-limiting solid phases and strong surface complexes with hydroxylated surface sites on Al and Fe oxides and clays (5, 9, 24, 31). Organisms capable of anaerobic energy metabolism, including the dissimilatory metal-reducing bacteria (DMRB) Shewanella putrefaciens, Shewanella alga, and Geobacter metallireducens (16, 36) and the sulfate-reducing bacterium Desulfovibrio desulfuricans (17, 18), have been shown to be capable of gaining energy for maintenance and growth by coupling the oxidation of organic C or H2 to the reduction of Tc(VII). The microbial reduction of Tc(VII) has been suggested as a potential mechanism for the removal of Tc from contaminated environments or waste streams (16, 19, 20).
In nonsulfidogenic, waste-contaminated groundwaters, the reduction of Tc(VII) may potentially occur directly, through enzymatic reduction, and/or indirectly via the microbial reduction of Fe(III) oxides and the formation of aqueous Fe(II) and/or Fe(II)-containing solids (6, 8, 12), which may in turn reduce Tc(VII) (4). The kinetics of Tc reduction and the aqueous chemistry of the environment are critical factors in determining which of these reactions dominates, as well as the nature of the hydrolysis and other complexation reactions that affect the form and stability of lower oxidation states of Tc. This information will be of key importance in developing waste and groundwater treatment strategies and assessing the mobility of lower oxidation states of Tc in groundwaters. However, the complex chemistry of Tc has provided formidable challenges to understanding the role of microorganisms in influencing its behavior under anaerobic environmental conditions. As a first step in assessing the combined effects of direct and indirect reduction processes on Tc speciation and mobility in waste and groundwaters, these studies examined the influence of electron donor and the presence of the inorganic complexing ligand bicarbonate on the rate of enzymatic Tc(VII) reduction by Shewanella putrefaciens CN32. The forms of solid- and aqueous-phase reduction products were determined by high-resolution transmission electron microscopy (TEM), X-ray absorption spectroscopy (XAS), and solution chemistry. Thermodynamic calculations were used to establish constraints on the solubility and chemical speciation of reduction products over a range of pH and Eh values commonly encountered in the subsurface.
MATERIALS AND METHODS
Cell preparation.
S. putrefaciens CN32, a facultative anaerobic bacterium, was originally isolated from an anaerobic sandstone at a depth of 250 m in the Morrison formation of northwestern New Mexico (8). CN32 was provided through the courtesy of D. Boone (Portland State University, Portland, Oreg.). Cells were cultured aerobically in tryptic soy broth (100 ml) and were incubated on a rotary shaker (100 rpm) at 30°C. The cultures were harvested after 16 h by centrifugation (5,000 × g, 15 min, 4°C).
Bacterial reduction.
The bacterial reduction of Tc(VII) was measured with time under anaerobic, nongrowth conditions. Bacterial cells were washed twice with a pH 7 solution of 30 mM NaHCO3 and 1.3 mM KCl (bicarbonate solution) or 0.85% NaCl (saline solution) that had been previously made anoxic by sparging it with a mixture of O2-free N2-CO2 (80:20; bicarbonate solution) or N2 (saline solution). Bacterial cells were resuspended in vials (20 ml) containing anoxic bicarbonate or saline solution (10 ml) and appropriate headspace gas to a density of ∼108 cells/ml. The vials were sealed with thick butyl rubber stoppers. All subsequent preparations and incubations were conducted at 30°C in an anaerobic glove box (Forma) containing an atmosphere of Ar (95%) and H2 (5%) and <10−4% O2. All reagents were added or samples removed by penetrating the stopper with a needle and syringe. Either Na lactate (10 mM) or H2 (4.5 × 10−4 M) was added as an electron donor. Technetium was added as pertechnetate [NH499Tc(VII)O4] (Amersham Life Sciences Products, Arlington Heights, Ill.) to achieve concentrations ranging from 1.86 × 105 to 2.23 × 107 dpm/ml and 5 μM to 6 mM, depending on the experiment. Enzymatic reduction was assumed as the loss of pertechnetate in filtered (see below) and unfiltered subsamples as a function of time. Pertechnetate was determined by direct extraction (33) and liquid scintillation counting of 99Tc (0.292 MEv beta). The pH and Eh were measured directly in the sealed vials by insertion of calibrated electrodes (Microelectrodes, Inc., Londonderry, N.H.). Because of potential interactions between H2 with the Pt electrode, Eh-sensitive dyes of known redox potential (2) were used as the standard measure of Eh at the end of the experiments and in aliquots of solutions with time. Three dyes (methylene blue, E0′, +11 mV; resazurin, E0′, −51 mV; and phenosafranin, E0′, −251 mV) were employed to bracket the Eh values in these systems, and therefore a range in Eh values is reported. Reduced Tc(IV) controls were prepared using SnCl2 (11) and hydrazine (23) as reductants.
Size distribution of Tc reduction products.
The relative size distribution (0.2, 0.01, and 0.001 μm) of Tc-containing solids formed upon reduction was determined by syringe filtration (Omega TM modified polyethersulfone; 13-mm Pall-Gelman Filtron) or centrifugation (Omega TM Filtron Nanosep; Pall-Gelman) of solution aliquots in combination with phosphorimaging or liquid scintillation counting to determine the concentration of 99Tc.
TEM.
Reduction products of >0.2 μm were imaged and analyzed using TEM. All samples were prepared in an anaerobic glove box to avoid oxidation of Tc(IV) solids. Whole mounts were prepared by placing a drop of cell suspension on a Formvar-coated Cu TEM grid. Grids were dried and stored in a sealed vial under an anaerobic atmosphere until they were transferred to the high vacuum of the TEM. Unstained whole mounts were analyzed by using a JEOL JEM 2000FX TEM apparatus, along with ancillary diagnostic equipment. Samples were observed at a 160-KeV accelerating voltage, and images were recorded on photographic film. Elemental analyses were accomplished using an Oxford-Link Isis energy-dispersive spectrometer (EDS) equipped with a SiLi detector. Electron diffraction patterns were obtained from representative solids shown to contain Tc by EDS in the saline-H2 systems. For solid-phase identification, diffraction patterns were digitized from photographic negatives using a flatbed scanner. Diffraction patterns were analyzed by search-match against the Powder Diffraction File (PDF) database (sets 1 to 48 and 70 to 85, year 1998) produced by the International Centre for Diffraction Data, Newtown Square, Pa. (Materials Data, Inc., Livermore, Calif.).
Samples of >0.2-μm solids for thin sectioning were fixed for 1 h by adding an anoxic solution of glutaraldehyde directly to the sample to yield a final fixative concentration of 2.5%. Fixed samples were gently mixed with an equal volume of 4% Noble agar. After solidifying them at room temperature, the samples were cut into 1-mm cubes and washed three times with an anoxic solution of 10 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)] buffer (pH 7) to remove the glutaraldehyde. The samples were slowly dehydrated using an anoxic graded ethanol series. Dehydrated samples were infiltrated with anoxic, low-viscosity LR-White embedding solution, placed in gelatin embedding capsules, and hardened by incubating them at 60°C for 1 h under an anoxic atmosphere. Embedded samples were sectioned to a 50-nm thickness inside an anaerobic glove box using a Leica Ultracut T microtome located in the glove box. Thin sections were placed on 200-mesh Cu TEM grids coated with a lacey carbon film. The unstained sections were examined using a JEOL 2010 TEM apparatus operating at a 200-kV accelerating voltage. Low-magnification and high-resolution images were digitally recorded using a 1,024 × 1,024 charge-coupled device camera (Gatan Instruments, Pleasantville, Calif.). Lattice fringe images were generated at high magnification to assist in the identification of solids and their crystallinity. The d-spacings were measured directly from the digital images and compared to those of standard Tc solids available from the PDF database.
Paper electrophoresis.
The charge and relative mobility of reduced Tc in filtrates were evaluated by paper electrophoresis (11) using cellulose polyacetate membrane paper (5.7 by 12.7 cm; Seprahpore; Gelman Sciences) and an electrophoresis chamber (Gelman) in the anaerobic glove box at 60 V for up to 1 h. Two solvent systems (30 mM NaHCO3–1.3M KCl and 10 mM HCl) were used to differentiate reduced Tc carbonate species. Tc(VII) and Tc reduction products were visualized and quantified by phosphorimaging (Bio-Rad) as described by Lloyd and Macaskie (16). Ammonium pertechnetate and the reduced Tc(IV) controls (above) served as standards for the calculation of retention factors.
X-ray absorption spectroscopy.
The oxidation state and the dimensionality of Tc electronic states and coordination were determined for selected solids and filtrates by using extended X-ray absorption fine structure (EXAFS) measurements conducted at the Stanford Synchrotron Radiation Laboratory under dedicated operating conditions (3.0 GeV and a current of 40 to 90 mA). Filtrates, precipitates, or slurries (0.1 to 0.2 ml) from enzymatic reduction experiments conducted at initial Tc concentrations of 3 to 6 mM were transferred into a Teflon tube and sealed under anaerobic conditions. The sealed tubes were placed in sample holders and secondary and tertiary containment cells with kapton windows, also under anaerobic conditions. The X-ray absorption analyses were conducted with the tertiary containers in a nitrogen glove bag to inhibit oxidation of the samples during the collection of absorption spectra. Spectra for Tc samples were collected at the Tc K-edge in the fluorescence and transmission geometries simultaneously, up to a photoelectron wave vector of 13 Å−1, using an energy-resolving 13-element Ge detector for the fluorescence measurement and nitrogen-filled ion chambers for the transmission measurement. The transmission signal of a Tc standard was measured simultaneously with each sample to provide energy calibration. Energy calibration was made by assigning the first inflection point in the absorption edge of the Tc standard to 21,044 eV. The absorption spectrum was normalized by fitting polynomials through the pre- and postedge regions. At E0, the value of the extrapolated preedge was set to zero and the difference between the extrapolations of the pre- and postedge polynomials was set to unity.
The EXAFS oscillations were extracted by fitting a polynomial spline function through the postedge region and normalizing the difference between this approximation of the solitary-atom EXAFS and the actual data with the absorption decrease calculated using the McMaster tables (22). Fourier transforms were taken over photoelectron wave vector ranges that varied on the basis of the signal-to-noise ratio for each sample. EXAFS nodes were selected as endpoints to the Fourier transform range, and a two-sigma-wide Gaussian window was used to dampen the EXAFS oscillations at the endpoints. The phase shift was not removed from the Fourier transforms, and, consequently, the peaks in the transform moduli appear 0.2 to 0.5 Å shorter than the actual distance from the absorber to the neighboring atoms. The phase and amplitude for the Tc-oxygen and Tc-Tc paths were calculated using the ab initio code FEFF7.02 (32, 37). These scattering paths were then parameterized and used to fit the experimentally measured EXAFS. The MoO2 structure was used to approximate the metal cation environment in TcO2 oxide. Citrate and diethylenetriamine pentaacetic acid complexes of reduced Tc were utilized to model the Tc-O-Tc and Tc-N scattering paths.
Thermodynamic calculations.
Equilibrium thermodynamic calculations were conducted iteratively with the enzymatic reduction experiments and analysis of Tc aqueous and solid phases to establish solubility, pH, and Eh limits on Tc reduction products and potential mobility under waste and groundwater conditions. The MINTEQA2 (version 3.11) geochemical reaction code (1) was used to calculate the aqueous speciation, saturation indices, and solubility of dissolved Tc for the solution compositions and conditions in the enzymatic reduction experiments. Eh-pH diagrams were calculated for Tc aqueous species using HSC Chemistry for Windows (version 2.03; Outokumpu Research Oy, Pori, Finland). The thermodynamic calculations were performed using the concentrations of dissolved Tc measured in the <0.001-μm (pore-size) filtrates at the end of the enzymatic reduction experiments. The thermodynamic databases in MINTEQA2 and HSC Chemistry were supplemented with Tc thermodynamic constants (Table 1) from Lemire and Jobe (14) to calculate aqueous speciation and solubility of Tc(IV), which was shown by XAS to be the dominant Tc reduction product in these systems. Thermodynamic constants for Tc(V) and Tc(VI) species and for Tc(IV) carbonate solid phases have not been published. Nordstrom and Munoz (29) and Langmuir (13) discuss geochemical modeling techniques and Eh-pH diagrams and their usefulness in understanding environmental behavior of metals in aqueous systems.
TABLE 1.
Thermodynamic constants from Lemire and Jobe (14) used for Tc aqueous speciation and solubility calculations
| Type and Tc oxidation state | Formula | ΔGf,298° (kJ/mol) |
|---|---|---|
| Aqueous species | ||
| III | Tc3+ | 88.4 |
| IV | TcO2+ | −118.0 |
| IV | TcO(OH)+ | −348.7 |
| IV | TcO(OH)20 (aq) | −567.5 |
| IV | [TcO(OH)20]2 (aq) | −1,172.2 |
| IV | TcO(OH)3− | −753.3 |
| IV | Tc(OH)(CO3)2− | −1,355 |
| IV | Tc(OH)2(CO3)23− | −1,483 |
| VII | TcO4− | −637.4 |
| Solidsa | ||
| IV | TcO2 (crystalline) | −404 |
| IV | TcO2 (electrodeposited solid)b | −379.3 |
Thermodynamic data for Tc(IV) carbonate solid phases have not been published.
Probably hydrated.
RESULTS
Effect of electron donor and solution composition on the rate and extent of Tc reduction.
S. putrefaciens CN32 was able to enzymatically reduce Tc(VII), but at very different rates depending upon the solution composition and the electron donor (Fig. 1). The Tc reduction rate [decrease in Tc(VII)] was most rapid with H2 as the electron donor, and reduction was complete in the saline and bicarbonate solutions after 5 and 21 h, respectively. Reduction rates were lower with lactate as the electron donor, with only 10 and 20% of the Tc(VII) reduced after 21 h in the saline and bicarbonate solutions, respectively. The results were highly reproducible over a broad Tc(VII) concentration range (5 μM to 6 mM). The Eh values of the unfiltered solutions after 21 h were −111 mV > Eh > −312 mV for the lactate electron donor and <−312 mV for the H2 electron donor. The pH values over the time of incubation were 7.5 ± 0.2, 7.2 ± 0.1, and 7.0 ± 0.0 for the bicarbonate-H2, bicarbonate-lactate, and saline-lactate treatments, respectively. The pH increased from 7.00 ± 0.1 to 7.90 ± 0.1 in the saline-H2 treatment, which was unbuffered and exhibited maximum Tc(VII) reduction. The increase in pH resulted from the consumption of H+ during the reduction of Tc(VII) as represented by the following equation:
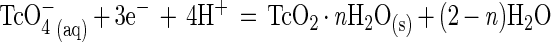 |
The results of thermodynamic calculations (discussed below) indicated that a change in pH over this range would not affect the chemical speciation of Tc in these systems. Controls with or without cells and electron donors maintained Eh values of >−49 mV and did not exhibit Tc reduction. The quantity of Tc(VII) associated with the cells (>0.2 μm) in the controls was <5% of the total. Residual (0.5 M HCl extractable) Fe(II,III) associated with the cells before initiation of enzymatic reduction amounted to 7.69E-19 mol per cell, or <0.006 μmol of total Fe per treatment, and was therefore insufficient to influence measured Tc(VII) reduction rates.
FIG. 1.

Reduction of Tc(VII) by S. putrefaciens CN32: effects of electron donor, solution composition, and reaction time on Tc distribution in filtrates. Symbols: ●, total Tc in the <0.2-μm filtrate; ■, TcO4− in the <0.2-μm filtrate; ○, Tc reduction products in the 0.2- to 0.001-μm fraction; □, Tc reduction products in the <0.001-μm filtrate. Data are means ± the standard deviation for duplicate assays. Controls with or without cells and electron donors did not exhibit Tc reduction.
Cell-associated Tc reduction products.
When H2 was used as the electron donor in saline solutions (Fig. 1), essentially all of the Tc(VII) was converted to a black solid phase that was completely retained by a 0.2-μm filter, a finding suggestive of the precipitation of Tc(IV) oxide (23). Unstained TEM images (Fig. 2A) of cell thin sections in this treatment (visualized by the presence of Tc) showed that Tc reduction products formed principally within the periplasmic region and on the outer membrane surface. Within the periplasmic region, Tc reduction products clearly formed invaginations that extended into the cytoplasm (Fig. 2A). It is not possible to determine from these observations, however, whether all Tc in the cytoplasm originated from this source. Cells suspended in saline solution with lactate as the electron donor exhibited similar properties (Fig. 2B). In bicarbonate solution with H2 as the electron donor, Tc precipitates were deposited in the periplasmic region and on the outer margins of the cells (Fig. 2C). The outer membrane was clearly visible as an electron-transparent region between the zones of Tc deposition. Invaginations of the cell periplasmic region were not observed in bicarbonate solutions. It is noteworthy that Tc precipitates did not form in or on the surfaces of cells in bicarbonate solutions when lactate was used as the electron donor (note the faint outline of cell in Fig. 2D). The extracellular particulates were fine grained when lactate was used as the electron donor compared to larger aggregates which were formed when H2 was the electron donor.
FIG. 2.

TEM images of S. putrefaciens CN32 after enzymatic reduction of Tc with different electron donors and solution compositions. Panels: A, saline-H2; B, saline-lactate; C, bicarbonate-H2; D, bicarbonate-lactate. The cells are unstained, and the images result from the presence of reduced Tc.
Characterization of the cell-associated Tc reduction products and results of the thermodynamic calculations provide strong evidence for the formation of sparingly soluble, amorphous Tc(IV) hydrous oxide. The precipitated Tc (Fig. 2A, B, and C) was amorphous or poorly crystalline, as determined by selected area electron diffraction and high-resolution TEM. The limited crystallinity of these solids did not result in definitive diffraction patterns that could be matched with solids in the PDF database. The EDS analyses showed that Tc precipitates contained Tc and O only. Microbially reduced Tc solids aged under anaerobic conditions for 6 months did not show evidence of increased crystallinity.
EXAFS analyses of the solids formed by bioreduction in the saline-H2 system indicated the presence of Tc(IV) oxide (Fig. 3). An Eh-pH diagram calculated for the saline-H2 system (Fig. 4) using the thermodynamic constants from Lemire and Jobe (14) and the detection limit concentration of total dissolved Tc (10−6.5 mol/liter) indicated that these solutions were oversaturated (shaded area in Fig. 4) with respect to hydrous TcO2 oxide at the Eh and pH of these experiments. Geochemical modeling (Fig. 5) using the same thermodynamic constants and concentration of dissolved Tc showed that the system was less than 2 orders of magnitude oversaturated with respect to hydrous TcO2 solid. Oversaturation would be consistent with experimental observations (Fig. 1), which indicated that Tc(VII) was reduced to Tc(IV) and removed by filtration (0.001 μm [pore size]). The modeling results also indicate that a neutral hydroxyl Tc(IV) complex is the predominate aqueous complex at the Eh (<−50 mV) and pH of these experiments. The calculated distribution of dissolved Tc complexes in the saline-H2 treatment was approximately 58% [Tc(IV)O(OH)2]20 (aq), 38% Tc(IV)O(OH)20 (aq), and 4% Tc(IV)O(OH)3−.
FIG. 3.

Fourier transform of the EXAFS of Tc products formed by microbial reduction of TcO4− in saline solution (>0.2-μm fraction) with H2 as the electron donor. A low-solubility Tc(IV) oxide is formed.
FIG. 4.

Eh-pH diagram for Tc in saline solution with H2 as the electron donor, indicating the dominant Tc aqueous species in solution and the Eh-pH regions (shaded area) theoretically oversaturated with respect to Tc(IV) hydrous oxide. The diagrams were calculated by using 5.3 × 10−7 mol/liter for total dissolved Tc and thermodynamic constants from Lemire and Jobe (14).
FIG. 5.

Calculated total concentration (solid line) of dissolved technetium (log moles/liter) in equilibrium with amorphous hydrous Tc(IV) oxide for different solution compositions as a function of Eh. The vertical dotted lines represent the limiting Eh values. The horizontal dashed lines represent the dissolved Tc measured in the <0.001-μm filtrate at the end of the incubation period as indicated in Fig. 1. The minimum detection limit for total dissolved Tc is 10−6.5 mol/liter.
Extracellular particulate Tc reduction products.
Total Tc in the <0.2-μm filtrates accounted for 70, 83, and 89% of the Tc in the bicarbonate-H2, saline-lactate, and bicarbonate-lactate systems, respectively, at the end of incubation. Thus, the major fraction of the reduction products in these treatments was not associated with the cells, which were removed by 0.2-μm filtration. A major portion of the reduced Tc in the <0.2-μm fraction was retained by 0.001-μm filters. A much smaller quantity (<11%) was in true solution (<0.001-μm filtrate) at the end of the incubation period. The EXAFS analyses of the <0.2-μm filtrate fraction of the saline-lactate, bicarbonate-H2, and bicarbonate-lactate systems indicated the presence of Tc(IV) oxide, as illustrated in Fig. 3. Solids from the bicarbonate-H2 system, collected on 0.01-μm and 0.001-μm filters, were black and were identified by EXAFS as Tc(IV) oxide as illustrated in Fig. 3.
The black color of the solids from the bicarbonate-H2 and saline-H2 systems contrasted with the amber and pink color of solids from the saline-lactate and bicarbonate-lactate systems, respectively. EXAFS analyses of these solids indicated that a mixture of Tc(IV) and Tc(VII) oxidation states was present, but there were insufficient quantities for identification of the Tc form. The saline-lactate filtrates (<0.001 μm) were calculated (Fig. 5) to be approximately 4 orders of magnitude oversaturated with respect to amorphous hydrous Tc(IV) oxide over the range of Eh values measured by the redox indicators (−111 to −312 mV). The measured dissolved Tc concentration was much higher than that predicted from equilibrium solubility, reflecting the lack of steady-state conditions, but the presence of solids (Fig. 1) is consistent with oversaturation. At the measured pH values and limiting Eh values of the experiments (determined colorimetrically), the concentrations of dissolved Tc in the <0.001-μm filtrates from the bicarbonate-hydrogen (Eh <−312 mV) and bicarbonate-lactate (−111 to −312 mV) systems were calculated to be slightly undersaturated (Fig. 6) with respect to hydrous Tc(IV) oxide. The geochemical modeling results (Fig. 5) indicated that these solutions were less than 0.5 orders of magnitude undersaturated with respect to hydrous Tc(IV) oxide, suggesting that this is the dominant solid phase controlling the dissolved Tc concentration. Thus, Tc(IV) oxide appears to be the dominant extracellular product common to all treatments, but the bicarbonate-lactate system may also contain (pink) Tc(IV) carbonate solids.
FIG. 6.

Eh-pH diagram for Tc in bicarbonate solution with H2 as the electron donor, indicating the dominant Tc aqueous species in solution, and Eh-pH regions (shaded areas) theoretically oversaturated with respect to Tc(IV) hydrous oxide. The diagrams were calculated by using 3.91 × 10−4 mol/liter for total dissolved Tc, 2.97 × 104 mol/liter for total dissolved carbonate, and thermodynamic constants from Lemire and Jobe (14).
Extracellular soluble Tc reduction products.
In the <0.001-μm filtrates, Tc(VII), which was determined by chemical extraction, constituted 98, 65, and 25% of the total Tc for the saline-lactate, bicarbonate-lactate, and bicarbonate-H2 systems, respectively. The rate of Tc(VII) reduction was much slower in these systems than in the saline-H2 system, and changes in Tc speciation were still taking place at the time of sampling. These factors, combined with the presence of Tc(VII) at low measured Eh values in these systems, suggest that the concentrations of Tc(VII) enzymatic reduction products had not reached steady-state conditions (Fig. 1). Assuming complete reduction of the initial Tc(VII) and the presence of carbonate from the oxidation of lactate, the final concentration of dissolved (<0.001-μm filtrate) Tc in the saline-lactate treatment was calculated to be comprised of approximately 72% [Tc(IV)O(OH)2]20 (aq), 26% Tc(IV)OH(CO3)2−, and 2% Tc(IV)O(OH)20 (aq).
The bicarbonate solutions (<0.2- and <0.001-μm filtrates) became progressively pink with time as Tc(VII) was reduced. Spectrophotometric analysis of these solutions indicated that the pink color was due to an absorption peak at 512 nm characteristic of Tc(IV) carbonates (30). Concentrations of Tc in the <0.001-μm filtrates were insufficient for unequivocal interpretation by EXAFS, but EXAFS verified the presence of Tc(VII) in the saline-lactate system and suggested the presence of Tc(IV) in the bicarbonate systems. Thermodynamic calculations for both bicarbonate systems indicated that >99.9% of the final concentrations of dissolved Tc over the pH and Eh range encompassing these experiments should be comprised of the negatively charged aqueous Tc(IV)OH(CO3)2− complex (Fig. 6). At the limiting Eh values of the experiments (determined colorimetrically), the calculated solubilities of hydrous Tc(IV) oxide (Fig. 5) in the bicarbonate-hydrogen (Eh <−312 mV) and bicarbonate-lactate (−111 to −312 mV) systems were several orders of magnitude higher than those for the other systems, reflecting the presence of soluble, reduced Tc carbonate species.
Further insight into the solid and aqueous forms of Tc in these systems was provided by paper electrophoresis, which documented changes in Tc speciation based on altered electrophoretic mobility in the <0.001-μm filtrates of bicarbonate and saline solutions (Fig. 7). There was essentially no Tc present in this fraction for the saline-H2 system. The saline-lactate system was predominately Tc(VII), as indicated by direct Tc(VII) measurements, but contained a small quantity of Tc that exceeded Tc(VII) in electrophoretic mobility as indicated by the gray area extending toward the cathode. The bicarbonate-lactate and bicarbonate-H2 systems also contained at least one component equivalent to or greater in electrophoretic mobility than Tc(VII). The Tc concentrations in these components, determined by phosphoimagery, approximated the difference between total Tc and Tc(VII) determined directly in the filtrates. Thus, it is likely that they comprise the reduced Tc in the soluble fraction (Fig. 1) and are the negatively charged Tc(IV) carbonate species indicated by spectrophotometric analysis and thermodynamic calculations (Fig. 6).
FIG. 7.

Paper electrophoretic separation of soluble (<0.001-μm filtrate) Tc reduction products in different solutions after microbial reduction of TcO4−. The intensity is proportional to the 99Tc concentration. The positive sign represents the position of the cathode.
DISCUSSION
S. putrefaciens CN32 was capable of using H2 and lactate as electron donors in the reduction of soluble Tc(VII) under nongrowth conditions, thus confirming previous observations that S. putrefaciens (ATCC 8071, Hammer strain) can reduce Tc(VII) to insoluble products of lower oxidation states (16). The current research demonstrated that the rate of Tc(VII) reduction by a subsurface strain of S. putrefaciens and the products of reduction were dependent upon solution composition and the electron donor. Bicarbonate was chosen as a naturally occurring inorganic ligand that forms soluble and insoluble complexes with some metals, while NaCl was used to represent dilute solutions with little or no complexation capacity but with sufficient ionic strengths to prevent bacterial lysis. H2 and lactate were chosen as the electron donors because they have previously been shown to be effective donors for metal reduction in S. putrefaciens (3) and because they are candidate electron donors for stimulating metal reduction in in situ and ex situ remediation processes. A summary of the conclusions of the Tc(VII) microbial enzymatic reduction experiments is given in Table 2.
TABLE 2.
Summary of the conclusions of microbial enzymatic reduction experiments
| Treatment | Extent of Tc(VII) reduction at 21 h (%) | Cellular association (% total) | Dominant solid-phase reduction product(s) | Dominant soluble reduction product(s) (% of total) |
|---|---|---|---|---|
| Saline-H2 | 100 | Periplasm, exterior of outer membrane—all cells (100) | Cell-associated amorphous Tc(IV) oxide | [TcO(OH)2]20 (aq) (0.1) |
| Saline-lactate | 29 | Periplasm—some cells; possibly cytoplasmic (7) | Extracellular Tc(IV) oxide | [TcO(OH)2]20 (aq); TcOH(CO3)2− (1.5) |
| Bicarbonate-H2 | 98 | Periplasm, exterior of outer membrane—all cells (23) | Extracellular Tc(IV) oxide | TcOH(CO3)2− (5.9) |
| Bicarbonate-lactate | 36 | Minor amounts in periplasm (1) | Extracellular Tc(IV) oxide, possible Tc carbonates | TcOH(CO3)2− (11) |
The rate and extent of Tc reduction by S. putrefaciens CN32 was much greater when H2 was provided as an electron donor, in both the saline and bicarbonate solutions, compared to lactate. This phenomenon was consistent with the results of related studies in which the rates of reduction of a variety of metals, including Cr(VI), U(VI), and both dissolved and solid forms of Fe(III), by S. putrefaciens strains CN32, MR-1, and BrY were consistently higher with H2 as the electron donor (Y. A. Gorby, unpublished data). The reasons for this are unclear, but there are several possible explanations. First, H2 is a relatively small, diffusible molecule that can readily cross the outer membrane of gram-negative bacteria and enter the periplasmic region, where it can be oxidized by soluble hydrogenases or hydrogenases associated with the cytoplasmic membrane. In contrast, lactate dehydrogenase is typically located in the cytoplasmic membrane or cytoplasm and transport of lactate to these sites may be rate limiting. Also, electrons from this process feed through intermediates such as NADH before entering the electron transport chain, adding additional steps that can slow the process. A second possible explanation is that electrons liberated during the oxidation of H2 enter a separate and perhaps less complex electron transport chain than is used when lactate is the electron donor. The enzymes and pathways involved in H2 and lactate oxidation in S. putrefaciens have not been thoroughly investigated. A soluble hydrogenase from Desulfovibrio desulfuricans has been shown to transfer electrons from H2 directly to a soluble c3 cytochrome that served as the terminal metal reductase (21). This two-component, in vitro system reduced a wide range of metals, including Fe(III), U(VI), and Cr(VI). Because D. desulfuricans cannot grow with metals as electron acceptors, the significance of this phenomenon is unknown. However, the results do demonstrate the potential for an abbreviated pathway for electrons during the reduction of metals with H2 as the electron donor. The rate of Tc(VII) reduction by D. desulfuricans also was found to be much greater when H2 was supplied as the electron donor in comparison to lactate (18), a result consistent with an abbreviated electron transport pathway. Lactate dehydrogenase is presumed to function similarly to formate dehydrogenase, which has been studied in the S. putrefaciens MR-1 (26). Electrons gained from the oxidation of formate by formate dehydrogenase enter an electron transport chain through a number of identified quinone compounds. The electron transport chain, which is composed of a complex series of Fe-S proteins and cytochromes, then serves to direct electrons toward outer-membrane cytochromes that may function as terminal metal reductases (20). Because of multiple enzyme steps in formate, and presumably lactate, oxidation it might be expected that the rate of reduction would be inherently slower than with H2, where a system composed of a limited number of electron transport components, possibly as few as two, catalyzes the oxidation of H2 and the reduction of Tc(VII).
S. putrefaciens MR-1 and four other strains of S. putrefaciens localize high concentrations of membrane-bound cytochromes (>80%) to the outer membrane when grown anaerobically with fumarate (28). At least four distinct c-type cytochromes that are readily reoxidized by Fe(III)-citrate have been identified in the outer membrane of MR-1 (27). Given the difference in standard potential, E0, values for the Tc(VII)O4−/TcO2 (solid) (E0 = 0.75 V) and Fe(III)-citrate/Fe(II)-citrate− (E0 = 0.31 V) redox couples, the outer membrane cytochromes in S. putrefaciens would be theoretically capable of reducing TcO4−. This, coupled with the known presence of c-type cytochromes in the periplasm of S. putrefaciens and the association of Fe reductase activity with the cell membrane (25), is consistent with the observation of Tc(IV) precipitates within the periplasm, in association with the outer cell surface, and possibly with the cell membrane. Due to the relative insolubility of Tc(IV) in this system, solids would be expected to precipitate at the site of reduction, and/or possibly bind to cell components, particularly when Tc(IV) is rapidly generated in the absence of complexing ligands such as in the saline-H2 treatment. With H2 as the electron donor in pH 7 MOPS (morpholinepropanesulfonic acid) buffer, Tc was shown to be associated with the periphery of the cells in D. desulfuricans (18). The participation of periplasmic hydrogenases in the reduction of Tc(VII) by S. putrefaciens cannot be ruled out. In fact, several putative genes coding for periplasmic hydrogenase genes, with high homology to Desulfovibrio periplasmic hydrogenases, have been identified from whole genome sequencing of S. putrefaciens MR-1 (M. F. Romine, personal communication). The enzymatic reduction of metals by S. putrefaciens is clearly a complex process involving multiple proteins that will require further investigations to elucidate the role of specific proteins in the reduction of metals and the localization of these activities within cells. Although Tc(VII) reduction was also rapid in bicarbonate solutions with H2 as the electron donor, and some Tc was again associated with the periplasm and outer cell surface, there was a much greater proportion (76%) of bioreduced Tc that was present in the fine-grained (0.2 to 0.001 μm) extracellular fraction, with lower concentrations of a soluble (<0.001-μm filtrate) species, likely TcIVOH(CO3)2− (Table 2). There is strong evidence from EXAFS and TEM analyses, supported by Eh and pH calculations and equilibrium modeling, that amorphous Tc(IV) hydrous oxide was formed inside and outside the cell (Fig. 1 and 2) in both saline and bicarbonate systems when H2 was the electron donor. This solid was oversaturated in the extracellular solution at the pH and Eh of the experiments and was calculated to be stable over a broad pH and Eh range (Fig. 4 and 6).
In the lactate-bicarbonate system, the formation of pink extracellular Tc(IV) solids was suggestive of carbonate complexes, but the concentrations of bioreduced Tc were insufficient to develop supporting evidence. The absence of thermodynamic data for Tc(IV) carbonate solids (which have not been previously identified) precluded calculating their equilibrium aqueous solubilities. In this regard, it must also be recognized that distinctly different chemical environments may exist within and outside the cell but they are likely closely interrelated. For example, soluble Tc(IV) species, such as carbonate complexes, that form in the periplasm may diffuse to the cell exterior, where precipitation of Tc solids may be more favorable due to differences in such factors as pH, Eh, and pCO2 derived from lactate oxidation. Microenvironmental differences between the cell surface-periplasm and the bulk solutions may have contributed to differences in Fe(II) biominerals generated as a result of reduction of hydrous ferric oxide by S. putrefaciens CN32 (7). The biologically mediated formation of reduced Tc solids during enzymatic reduction will clearly require further investigation if these processes are to be used in remediation of Tc dissolved in groundwaters.
The combined evidence also demonstrates that in conjunction with cell-associated and extracellular particulates (0.2 to 0.001 μm), soluble (<0.001-μm filtrates) Tc(IV) carbonate complexes were formed in the bicarbonate-H2 and bicarbonate-lactate systems. Much remains to be learned about the form and mobility of aqueous Tc(IV) carbonate complexes since there is uncertainty regarding the stoichiometries, and therefore the charges, for these species (5, 30). However, the results of paper electrophoresis suggested that these complexes were more negative than Tc(VII) as TcO4−. The results of thermodynamic calculations were consistent with their presence in the saline-lactate system as well. The effect of low concentrations of the soluble carbonates (pink) and of Tc(IV) hydrous oxides (black) likely resulted in the amber color of the saline-lactate system. Although the reduction of Tc(VII) to Tc(IV) must entail a transfer of three electrons which offers the opportunity for formation of the intermediate oxidation states Tc(VI) and Tc(V), there was no evidence of their presence in these systems.
Implications.
The results of this research have implications for the fate and transport of Tc in the subsurface, where anoxic conditions may develop naturally or as a result of in situ stimulation of microbial activity. In anoxic sediments, DMRB may play an important role in directly altering the form and mobility of Tc. However, the complex chemistry of Tc under reducing conditions requires careful consideration of both aqueous and solid-phase products resulting from dissimilatory reduction, which in turn will be strongly dependent upon the electron donor and solution composition. These studies suggest that direct bacterial dissimilatory reduction will result in rapid removal of Tc(VII) from groundwaters through the formation of low-solubility Tc(IV) solids, which will control Tc solubility unless Tc(IV) is stabilized in the aqueous phase by complexing ligands such as carbonate. The formation of highly electronegative, soluble Tc(IV) carbonate complexes indicates that it may be necessary to reassess current concepts of Tc transport in anaerobic, carbonate-enriched groundwaters, where Tc mobility has been considered to be controlled largely by the low solubility of Tc(IV) hydrous oxide. With apparent electronegativities greater than Tc(VII)O4−, which forms only weak surface complexes with hydroxylated surface sites on Al and Fe oxides and clays and is only minimally retarded in oxic groundwaters, the presence of these Tc species would have clear implications for bioremediation strategies and suggests that transport through anaerobic aquifers needs to be considered as a possible pathway in assessing the Tc radiation dose to humans. Ultimately, prediction of Tc behavior in anaerobic nonsulfidogenic groundwater systems will require a fundamental understanding of the mechanisms for direct (enzymatic) and indirect (e.g., biogenic Fe) Tc reduction and the factors controlling the combined effects of these process. The complex chemistry of Tc provides formidable challenges and the potential for misinterpretation in defining the chemical speciation and stability of Tc reduction products over a range of environmental conditions. A combination of rigorous analytical and modeling approaches will continue to be required to address these fundamental questions.
ACKNOWLEDGMENTS
This research was supported by the Natural and Accelerated Bioremediation Research Program (NABIR), Office of Biological and Environmental Research, U.S. Department of Energy (DOE). Pacific Northwest National Laboratory is operated for the DOE by Battelle Memorial Institute under contract DE-AC06-76RLO 1830.
The insights provided by T. R. Garland on the chemistry of Tc in these systems was invaluable. We appreciate the assistance of A. Dohnalkova with the high-resolution TEM analyses. The continued support of F. J. Wobber is greatly appreciated. We also thank David Boone of Portland State University for providing S. putrefaciens CN32 to us from the Subsurface Microbial Culture Collection, supported through Florida State University by DOE grant no. DE-FG05-90ER61039.
REFERENCES
- 1.Allison J D, Brown D S, Novo-Gradac K J. MINTEQA2/PRODEFA2, a geochemical assessment model for environmental systems, version 3.0 (user's manual). EPA/600/3-91/021. U.S. Athens, Ga: Environmental Protection Agency; 1991. [Google Scholar]
- 2.Breznak J A, Costilow R N. Physicochemical factors in growth. In: Gerhardt P, Murray R G E, Wood W A, Krieg N R, editors. Methods for general and molecular bacteriology. Washington, D.C.: American Society for Microbiology; 1994. pp. 137–154. [Google Scholar]
- 3.Caccavo F, Blankemore R P, Lovley D R. A hydrogen-oxidizing, Fe(III)-reducing microorganism from the Great Bay Estuary, New Hampshire. Appl Environ Microbiol. 1992;58:3211–3216. doi: 10.1128/aem.58.10.3211-3216.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cui D, Eriksen T E. Reduction of pertechnetate in solution by heterogeneous electron transfer from Fe(II) containing geologic material. Environ Sci Technol. 1996;30:2263–2269. [Google Scholar]
- 5.Eriksen T E, Ndalamba P, Bruno J, Caceci M. The solubility of TcO2 · nH2O in neutral to alkaline solutions under constant pCO2. Radiochim Acta. 1992;58/59:67–70. [Google Scholar]
- 6.Fredrickson J K, Gorby Y A. Environmental processes mediated by iron-reducing bacteria. Curr Opin Biotechnol. 1996;7:287–294. doi: 10.1016/s0958-1669(96)80032-2. [DOI] [PubMed] [Google Scholar]
- 7.Fredrickson J K, Zachara J M, Kennedy D W, Dong H, Onstott T C, Hinman N W, Li S W. Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium. Geochim Cosmochim Acta. 1998;62:3239–3257. [Google Scholar]
- 8.Fredrickson J K, Zachara J M, Kennedy D W, Dong H, Onstott T C, Hinman N W, Li S W. Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium. Geochim Cosmochim Acta. 1999;62:3239–3257. [Google Scholar]
- 9.Haines R I, Owen D G, Vandergraaf T T. Technetium-iron oxide reactions under anaerobic conditions: a Fourier transform infrared, FTIR study. Nuclear J Can. 1987;1:32–37. [Google Scholar]
- 10.Hartman M J, Dresel P E, editors. Hanford site groundwater monitoring for fiscal 1997. PNNL-11973. Richland, Wash: Pacific Northwest National Laboratory; 1988. [Google Scholar]
- 11.Jackson G E, Byrne M J, Hunter R, Woudenberg M. Technetium-99m labelling of bis-oxime ligands. Appl Radiat Isot. 1994;45:581–586. doi: 10.1016/0969-8043(94)90201-1. [DOI] [PubMed] [Google Scholar]
- 12.Kostka J E, Nealson K H. Dissolution and reduction of magnetite by bacteria. Environ Sci Technol. 1995;29:2535–2540. doi: 10.1021/es00010a012. [DOI] [PubMed] [Google Scholar]
- 13.Langmuir D. Aqueous environmental geochemistry. Upper Saddle River, N.J: Prentice Hall; 1997. [Google Scholar]
- 14.Lemire R J, Jobe D J. Predicted behaviour of technetium in a geological disposal vault for used nuclear fuel—ramifications of a recent determination of the enthalpy of formation of TcO2(cr) In: Murphy W M, Knecht D A, editors. Scientific basis for waste management. XIX. Materials research symposium proceedings. Material Res. Soc. 412:873. 1996. [Google Scholar]
- 15.Lieser K H, Bauscher Ch. Technetium in the hydrosphere and in the geosphere. I. Chemistry of technetium and iron in natural waters and influence of the redox potential on the sorption of technetium. Radiochim Acta. 1987;42:213. [Google Scholar]
- 16.Lloyd J R, Macaskie L E. A novel PhosphorImager-based technique for monitoring the microbial reduction of technetium. Appl Environ Microbiol. 1996;62:578–582. doi: 10.1128/aem.62.2.578-582.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lloyd J R, Nolting H-F, Sole V A, Bosecker K, Mascaskie L E. Technetium reduction and precipitation by sulfate-reducing bacteria. Geomicrobiol J. 1998;15:45–58. [Google Scholar]
- 18.Lloyd J R, Ridley J, Khizniak T, Lyalikova N N, Macaskie L E. Reduction of technetium by Desulfovibrio desulfuricans: biocatalyst characterization and use in a flowthrough bioreactor. Appl Environ Microbiol. 1999;65:2691–2696. doi: 10.1128/aem.65.6.2691-2696.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lovley D R. Dissimilatory metal reduction. Annu Rev Microbiol. 1993;47:263–290. doi: 10.1146/annurev.mi.47.100193.001403. [DOI] [PubMed] [Google Scholar]
- 20.Lovley D R. Bioremediation of organic and metal contaminants with dissimilatory metal reduction. J Ind Microbiol. 1995;14:85–93. doi: 10.1007/BF01569889. [DOI] [PubMed] [Google Scholar]
- 21.Lovley D R, Widman P K, Woodward J C, Phillips E J P. Reduction of uranium by cytochrome c3 of Desulfovibrio vulgaris. Appl Environ Microbiol. 1993;59:3572–3576. doi: 10.1128/aem.59.11.3572-3576.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McMaster W H, Del Grande N K, Mallett J H, Hubbell J H. Compilation of x-ray cross sections. UCRL-50174. Livermore, Calif: Lawrence Livermore National Laboratory; 1969. [Google Scholar]
- 23.Meyer R E, Arnold W D, Case F I. Valence effects on solubility of Tc(IV) oxides. ORNL-6199. Oak Ridge, Tenn: Oak Ridge National Laboratory; 1986. [Google Scholar]
- 24.Meyer R E, Arnold W D, Case F I, O'Kelley G D. Solubilities of Tc(IV) oxides. Radiochim Acta. 1991;55:11–18. [Google Scholar]
- 25.Myers C R, Myers J M. Ferric reductase is associated with the membranes of anaerobically grown Shewanella putrefaciens MR-1. FEMS Microbiol Lett. 1993;108:15–22. [Google Scholar]
- 26.Myers C R, Myers J M. Role of menaquinone in the reduction of fumarate, nitrate, iron(II) and manganese(IV) by Shewanella putrefaciens MR-1. FEMS Microbiol Lett. 1993;114:215–222. [Google Scholar]
- 27.Myers C R, Myers J M. Outer membrane cytochromes of Shewanella putrefaciens MR-1: spectral analysis, and purification of the 83-kDa c-type cytochrome. Biochim Biophys. 1997;1326:307–318. doi: 10.1016/s0005-2736(97)00034-5. [DOI] [PubMed] [Google Scholar]
- 28.Myers J M, Myers C R. Isolation and sequence of omcA, a gene encoding a decaheme outer membrane cytochrome c of Shewanella putrefaciens MR-1, and detection of omcA homologs in other strains of S. putrefaciens. Biochim Biophys. 1998;1373:237–251. doi: 10.1016/s0005-2736(98)00111-4. [DOI] [PubMed] [Google Scholar]
- 29.Nordstrom D K, Munoz J L. Geochemical thermodynamics. Menlo Park, Calif: Benjamin/Cummings Publishing Co., Inc.; 1985. [Google Scholar]
- 30.Pacquette J, Lawrence W E. A spectroelectrochemical study of the technetium(IV)/technetium(III) couple in bicarbonate solutions. Can J Chem. 1985;63:2639–2373. [Google Scholar]
- 31.Rard J A. Critical review of the chemistry and thermodynamics of technetium and some of its inorganic compounds and aqueous species. UCRL-53440. Livermore, Calif: Lawrence Livermore National Laboratory; 1983. [Google Scholar]
- 32.Rehr J J, Zabinsky S I, Albers R C. High-order multiple scattering calculations of x-ray-absorption fine structure. Phys Rev Lett. 1992;69:3397. doi: 10.1103/PhysRevLett.69.3397. [DOI] [PubMed] [Google Scholar]
- 33.Tribalat S, Beydon J. Isolement du technetium. Anal Chimi Acta. 1953;8:22–28. [Google Scholar]
- 34.Wildung R E, McFadden K M, Garland T R. Technetium sources and behavior in the environment. J Environ Qual. 1979;8:156–161. [Google Scholar]
- 35.Wildung R E, Garland T R, McFadden K M, Cowan C E. Technetium sorption in surface soils. In: Desmet G, Myttenaere C, editors. Technetium in the environment. London, England: Elsevier Applied Science Publishers; 1984. pp. 115–129. [Google Scholar]
- 36.Wildung R E, Gorby Y A, Fredrickson J K, Li S W, Plymale A E. Reduction of technetium by dissimilatory metal reducing Shewanella sp. Calgary, Alberta, Canada: First Annual International Biometals Symposium; 1997. [Google Scholar]
- 37.Zabinsky S I, Rehr J J, Ankudinov A, Albers R C, Eller M J. Multiple scattering calculations of x-ray absorption spectra. Phys Rev B. 1995;52:2995. doi: 10.1103/physrevb.52.2995. [DOI] [PubMed] [Google Scholar]