

Abstract
Background
Retained placenta affects 0.5% to 3% of women following delivery and it is a major cause of maternal death due to postpartum haemorrhage. Usually, retained placenta has been managed by manual removal or curettage under anaesthesia, which may be associated with haemorrhage, infection and uterine perforation. Medical management to facilitate the delivery of the retained placenta could be a safe alternative avoiding surgical intervention.
Objectives
To assess the effectiveness and safety of prostaglandins for the management of retained placenta.
Search methods
We searched the Cochrane Pregnancy and Childbirth Group's Trials Register (1 December 2013), LILACS (1982 to 1 December 2013), SciELO (1998 to 1 December 2013), Web of Science (2001 to 1 December 2013), openSIGLE (1997 to 1 December 2013), World Health Organization International Clinical Trials Registry Platform (ICTRP) (1 December 2013) and the metaRegister of Controlled Trials (mRCT) (1 December 2013). We also contacted authors of included studies and reviewed the reference lists of retrieved studies.
Selection criteria
Randomised controlled clinical trials comparing the use of prostaglandins (or prostaglandin analogues) with placebo, expectant management, tocolytic drugs, any other prostaglandins or surgical interventions for the management of retained placenta after vaginal delivery of singleton live infants of 20 or more weeks of gestation.
Data collection and analysis
Two review authors independently assessed trials for inclusion and assessed trial quality. Two review authors independently extracted data. Data were checked for accuracy. Any disagreements were resolved through consensus or consultation with a third review author when required. Authors of the included studies were contacted for additional information.
Main results
We included three trials, involving 244 women. The studies were considered to be at high risk of bias.
The prostaglandins used were PG E2 analogue (sulprostone) in 50 participants and PG E1 analogue (misoprostol) in 194 participants at a dose of 250 mcg and 800 mcg respectively. The prostaglandins compared with placebo, were not superior in reducing the rate of manual removal of placenta (average risk ratio (RR) 0.82; 95% confidence interval (CI) 0.54 to 1.27), severe postpartum haemorrhage (RR 0.80; 95% CI 0.55 to 1.15), need for blood transfusion (RR 0.72; 95% CI 0.43 to 1.22), mean blood loss (mean difference (MD) ‐205.26 mL; 95% CI ‐536.31 to 125.79, random‐effects) and the mean time from injection to placental removal (MD ‐7.00 minutes; 95% CI ‐21.20 to 7.20). Side‐effects were no different between groups (vomiting, headache, pain and nausea between injection and discharge from the labour ward), with the exception of shivering, which was more frequent in women receiving prostaglandins (RR 10.00; 95% CI 1.40 to 71.49). We did not obtain any data for the primary outcomes of maternal mortality and the need to add another therapeutic uterotonic.
Authors' conclusions
Currently there is limited, very low‐quality evidence relating to the effectiveness and the safety using prostaglandins for the management of retained placenta. Use of prostaglandins resulted in less need for manual removal of placenta, severe postpartum haemorrhage and blood transfusion but none of the differences reached statistical significance. Much larger, adequately powered studies are needed to confirm that these clinically important beneficial effects are not just chance findings.
Similarly, no differences were detected between prostaglandins and placebo in mean blood loss or the mean time from injection to placental removal (minutes) or side‐effects (vomiting, headache, pain and nausea between injection and discharge from the labour ward) except for 'shivering' which was more frequent in women who received prostaglandin. The included studies were of poor quality and there is little confidence in the effect estimates; the true effect is likely to be substantially different. We can not make any recommendations about changes to clinical practice. More high‐quality research in this area is needed.
Keywords: Female; Humans; Pregnancy; Abortifacient Agents, Nonsteroidal; Abortifacient Agents, Nonsteroidal/adverse effects; Abortifacient Agents, Nonsteroidal/therapeutic use; Dinoprostone; Dinoprostone/adverse effects; Dinoprostone/analogs & derivatives; Dinoprostone/therapeutic use; Labor Stage, Third; Misoprostol; Misoprostol/adverse effects; Misoprostol/therapeutic use; Oxytocics; Oxytocics/adverse effects; Oxytocics/therapeutic use; Placenta, Retained; Placenta, Retained/drug therapy; Prostaglandins; Prostaglandins/adverse effects; Prostaglandins/therapeutic use; Randomized Controlled Trials as Topic
Plain language summary
Prostaglandins for management of retained placenta
Medical research evidence is sparse and insufficient to support the routine use of the prostaglandins for the management of retained placenta.
Retained placenta affects 0.5% to 3% of women following delivery and is a major cause of maternal death caused by postpartum haemorrhage. A retained placenta is usually managed by manual removal or curettage under anaesthesia (which is not always immediately available). Surgical procedures themselves can be associated with haemorrhage and also infection and uterine perforation. Prostaglandins or their analogues, administered by any route, could be an alternative treatment especially in developing countries. Such medical management may facilitate the delivery of the retained placenta and be a safer alternative to surgery.
The review identified three randomised controlled studies (involving 244 women) that compared the use of prostaglandins with placebo. Currently there is limited, very low‐quality evidence relating to the effectiveness and the safety using prostaglandins for the management of retained placenta. Use of prostaglandins resulted in less need for manual removal of placenta, severe postpartum haemorrhage and need for blood transfusion but none of the differences reached statistical significance. Much larger, adequately powered studies are needed to confirm that these clinically important beneficial effects are not just chance findings. Similarly, no differences were detected between prostaglandins and placebo in mean blood loss or the mean time from injection to placental removal (minutes). The prostaglandin was administered by intravenous infusion (E2 analogue sulprostone) in one study including 50 women and was orally or sublingually administered (E1 analogue misoprostol) in the other two studies including 194 women.
Shivering was more frequent in women receiving the prostaglandin but there were no clear differences in vomiting, headache, maternal pain or nausea compared with placebo. The trials were small and of poor methodological quality. The quality of evidence is very low due to study limitations, inconsistency and imprecise results (few women and outcome events with wide confidence intervals). Two studies were stopped early due to an apparent benefit.
Summary of findings
Summary of findings for the main comparison. prostaglandins for retained placenta.
| Prostaglandins for retained placenta | ||||||
| Patient or population: women with retained placenta Settings: all care setting Intervention: prostaglandins | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Prostaglandins | |||||
| Manual removal of placenta Manual removal after intervention Follow‐up: 30‐45 minutes | Study population | RR 0.82 (0.54 to 1.27) | 244 (3 studies) | ⊕⊝⊝⊝ very low1,2,3 | ||
| 561 per 1000 | 460 per 1000 (303 to 712) | |||||
| Low | ||||||
| 10 per 1000 | 8 per 1000 (5 to 13) | |||||
| High | ||||||
| 30 per 1000 | 25 per 1000 (16 to 38) | |||||
| Severe postpartum haemorrhage Objectively or subjectively measured after intervention/Estimated blood loss in ML. Follow‐up: 30‐45 minutes | Study population | RR 0.80 (0.55 to 1.15) | 194 (2 studies) | ⊕⊝⊝⊝ very low3,4 | ||
| 432 per 1000 | 346 per 1000 (238 to 497) | |||||
| Low | ||||||
| 30 per 1000 | 24 per 1000 (17 to 34) | |||||
| High | ||||||
| 100 per 1000 | 80 per 1000 (55 to 115) | |||||
| Blood transfusion Blood transfusion during puerperium Follow‐up: 1 to 8 weeks | Study population | RR 0.72 (0.43 to 1.22) | 244 (3 studies) | ⊕⊝⊝⊝ very low3,4 | ||
| 224 per 1000 | 161 per 1000 (96 to 274) | |||||
| Low | ||||||
| 60 per 1000 | 43 per 1000 (26 to 73) | |||||
| High | ||||||
| 130 per 1000 | 94 per 1000 (56 to 159) | |||||
| Mean blood loss Objectively or subjectively measured after intervention/Mean blood loss in ML. Follow‐up: 30 to 45 minutes | The mean blood loss in the control groups was 0 millilitres | The mean blood loss in the intervention groups was 205.26 lower (536.31 lower to 125.79 higher) | 244 (3 studies) | ⊕⊝⊝⊝ very low1,5,6 | ||
| Mean time from injection to placenta removal Mean time from injection to placenta removal. Follow‐up: 30 to 45 minutes | The mean time from injection to placenta removal in the control groups was minutes | The mean time from injection to placenta removal in the intervention groups was 7.0 lower (21.2 lower to 7.2 higher) | 99 (1 study) | ⊕⊝⊝⊝ very low1,6 | ||
| Maternal pain between injection and discharge from labour ward Maternal pain between injection and discharge from labour ward Follow‐up: 1 to 24 hours | Study population | RR 0.91 (0.43 to 1.96) | 124 (2 studies) | ⊕⊝⊝⊝ very low1,3 | ||
| 172 per 1000 | 157 per 1000 (74 to 338) | |||||
| Low | ||||||
| 100 per 1000 | 91 per 1000 (43 to 196) | |||||
| High | ||||||
| 200 per 1000 | 182 per 1000 (86 to 392) | |||||
| Nausea between injection and discharge from labour ward Nausea between injection and discharge from labour ward Follow‐up: 1 to 24 hours | Study population | RR 1.72 (0.15 to 19.41) | 124 (2 studies) | ⊕⊝⊝⊝ very low1,2,6 | ||
| 34 per 1000 | 59 per 1000 (5 to 669) | |||||
| Low | ||||||
| 30 per 1000 | 52 per 1000 (5 to 582) | |||||
| High | ||||||
| 100 per 1000 | 172 per 1000 (15 to 1000) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Failure to adherence to intention‐to‐treat principle. Selective reporting. Stopping early for benefit. 2 Large variation in effect. Confidence intervals overlap.Substantial heterogeneity. 3 Few women and outcome events. Confidence interval include null effect, include appreciable harm or benefit. Not optimal information size. 4 Lack of blinding. Failure to adherence to intention‐to‐treat principle. Selective reporting. Stopping early for benefit. 5 Large variation in effect. Confidence intervals do not overlap. Substantial heterogeneity. 6 Few women. Confidence interval includes null effect, include appreciable harm or benefit. Not optimal information size.
Background
Description of the condition
Postpartum haemorrhage (PPH) is an important cause of maternal mortality (Sosa 2009) and it accounts for nearly one quarter of all maternal deaths world‐wide with an estimated 125,000 deaths per year (Carroli 2008). The overall incidence of PPH is about 6% with wide variations around the world; the highest rates occur in Africa with 10.5%; North America, Europe, Oceania and Latin America have intermediate rates and the lowest rates are found in Asia (2.6%) (Carroli 2008). PPH is also associated with serious morbidity including the need for blood transfusion, renal failure, coagulation deficiencies, anaemia and hysterectomy or other surgical procedures with subsequent consequences on fertility (Bodelon 2009).
One of the main causes of PPH is retained placenta, which affects 0.5% to 3.0% of women following delivery, and is a major cause of maternal death due to PPH. A further 15% to 20% of the PPH maternal deaths are due to retained placenta. After uterine atony, retained placenta is the second major indication for blood transfusion in the third stage of labour (Owolabi 2008). Retained placenta is a potentially preventable cause of PPH (Hoveyda 2001).
There is no consensus about the length of the third stage of labour after which a placenta should be called ‘retained’. In Europe, manual removal of placentae are advised at anytime between 20 minutes and over one hour after delivery (Weeks 2008). The choice of timing is a balance between the PPH risk of leaving the placenta in situ, the likelihood of spontaneous delivery and the knowledge that the manual removal itself causes haemorrhage (Rizwan 2009). Observational studies have demonstrated that a third stage longer than 30 minutes was associated with higher rates of PPH, higher rates of blood transfusions and dilatation and curettage, compared with a third stage of 30 minutes or less (Deneux‐Tharaux 2009). Because there was no increase in these medical complications until the third stage exceeded 30 minutes, it is suggested that above this time an intervention is mandatory (Combs 1991).
Description of the intervention
Prostaglandins (PG) are molecules responsible for physiologic reactions that act as intermediaries in several processes involved during pregnancy including term labour, postpartum involution, and placental–fetal vascular dynamics. Their biosynthesis is limited by the activity of the enzyme arachidonic acid cyclo‐oxygenase, which catalyses the transformation of arachidonic acid into prostaglandin (Miller 2006).
Prostaglandins receptors are present in both, pregnant and nonpregnant, uteri and their concentration in the myometrial tissue increases at the beginning of labour. Prostaglandins have effects on the myometrium and cervix, whereas the activity of oxytocin is limited to the uterine muscle and it is in fact, strictly dependent on calcium concentration (Arias 2000).
Prostaglandins E and F are the most important types of prostaglandins with uterotonic activity and have a relevant advantage compared with oxytocin in terms of biological activity. Prostaglandins E and F can be administered, and are absorbed by, any route including intravenous, oral, sublingual, vaginal or intracervical administration with variable incidence of side‐effects (Arias 2000). The knowledge that prostaglandins can be delivered to the retro placental myometrium by any route has stimulated a lot of interest.
How the intervention might work
Ultrasound studies have improved the understanding of the third stage of labour. One study (Herman 1993), using ultrasonography demonstrated that retro placental myometrial contraction is paramount to produce shearing forces on the interface between the placenta and myometrium, leading to its detachment. A prolonged third stage is due to contractile failure in the retro placental area (Weeks 2008), with difficulties for occluding the blood flow through the arcuate and radial arteries to the placental fragments with the consequences of retained placenta and PPH (Weeks 2001).
Prostaglandins have a potent uterotonic activity caused by their effect of increasing intracellular calcium and activating myosin light chain kinase. The influx of calcium caused by prostaglandins, however, is probably caused by interaction with calcium channels, which is different from the mechanism of oxytocin (Arias 2000).The role of prostaglandins in uterine muscle contractions during labour is well known.
The advantages mentioned above, allow prostaglandins to act specifically at the contractile failure area, stimulating contractions in the retro placental myometrium (Weeks 2008). Medical management to facilitate the delivery of the retained placenta with prostaglandins could potentially provide a safer alternative, involving early treatment and reducing the risk of complications.
Why it is important to do this review
Usually, the retained placenta has been managed by manual removal or curettage with general anaesthesia (which is not immediately available in the majority of cases), and which may be associated with haemorrhage, infection and uterine perforation. Medical management to facilitate the delivery of the retained placenta could be a safer alternative that avoids surgical intervention (Sundaram 2009). Nowadays, oxytocin umbilical vein injection and tocolysis have been the medical interventions systematically evaluated for treating retained placenta with no effect (Nardin 2011) and with limited evidence available for tocolysis (Abdel‐Aleem 2011). To date, no systematic review has examined the role of prostaglandins for the medical management of retained placenta. Prostaglandins or their analogues, administered by any route, could offer theoretical advances especially in developing countries. It is important to assess the effectiveness and safety of prostaglandins for the management of retained placenta. Medical management to facilitate the delivery of the retained placenta with prostaglandins could potentially provide a safer alternative involving an earlier treatment and reducing the morbidity and mortality associated with this condition.
Objectives
To assess the effectiveness and safety of prostaglandins for the management of retained placenta.
Methods
Criteria for considering studies for this review
Types of studies
All published and unpublished randomised controlled clinical trials comparing the use of prostaglandins (or prostaglandin analogues) with placebo, expectant management, tocolytic drugs, any other prostaglandin or surgical interventions for the management of retained placenta. Quasi‐randomised trials, cluster‐randomised trials and trials using a cross‐over design were not included.
Types of participants
All women having a vaginal delivery of singleton live infants of 20 or more weeks of gestational age, with a retained placenta, regardless of the management of the third stage of labour or the history of prior caesarean delivery. We defined retained placenta as a third stage exceeding 30 minutes after delivery of the infant. We excluded studies in which women have a clear diagnosis of placenta accreta.
Types of interventions
There are a number of existing or planned Cochrane reviews on the topic of management of retained placenta. The current list includes the following.
Surgical management of retained placenta.
Tocolysis for management of retained placenta (Abdel‐Aleem 2011).
Prostaglandins for management of retained placenta (this review).
Other pharmacological interventions for management of retained placenta.
Non‐pharmacological and non‐surgical interventions for managing retained placenta (Mockler 2012).
To avoid duplication, each individual Cochrane review on this topic will include comparisons only with the interventions listed above it in the list. Consequently, this review (which is number three in the list) included comparisons of prostaglandin (or prostaglandin analogue) (any type, dose, and route except umbilical vein injection (UVI)) versus any other prostaglandin (or prostaglandin analogue) (any type, dose, and route except UVI), tocolysis (2), surgical management (1) and placebo/expectant management.
This strategy will avoid the same comparisons being included in more than one of the original Cochrane reviews. The Cochrane reviews listed above will not include comparisons with UVI, which is covered in a separate review (Nardin 2011).
Types of outcome measures
In order to maximise consistency between reviews on the topic of managing retained placenta, a number of core outcomes have been identified and these were incorporated into all the reviews on this topic.
Primary outcomes
Maternal mortality.
Manual removal of the placenta.
Severe postpartum haemorrhage (defined as clinically estimated blood loss greater than or equal to 1000 mL).
Blood transfusion.
Need to add other therapeutic uterotonic.
Secondary outcomes
Serious maternal morbidity (hysterectomy, admission to intensive care, renal or respiratory failure, and other additional surgical procedures to treat PPH different to manual removal of placenta, related to the randomised interventions).
Postpartum haemorrhage (PPH) (defined as clinically estimated or measured blood loss greater than or equal to 500 mL).
Maternal postpartum anaemia (defined by the haemoglobin concentration according to local standards).
Mean blood loss (mL).
Mean time from injection to placental removal (minutes).
Perinatal fall in haemoglobin levels (defined as decrease in previous haemoglobin concentration levels by at least 10%).
Need for iron tablets during the puerperium.
Subsequent surgical evacuation of retained products of conception.
Diastolic blood pressure greater than 100 mmHg between injection and discharge from the labour ward.
Vomiting between injection and discharge from the labour ward.
Shivering between injection and discharge from the labour ward.
Nausea between injection and discharge from the labour ward.
Headache between injection and discharge from the labour ward.
Maternal pain between injection and discharge from the labour ward.
Maternal dissatisfaction with third stage management.
Secondary PPH (after 24 hours and before six weeks).
Bleeding needing readmission.
Need for treatment with antibiotics.
Maternal fatigue.
Breastfeeding at discharge from hospital.
Search methods for identification of studies
Electronic searches
We contacted the Trials Search Co‐ordinator to search the Cochrane Pregnancy and Childbirth Group’s Trials Register (1 December 2013).
The Cochrane Pregnancy and Childbirth Group’s Trials Register is maintained by the Trials Search Co‐ordinator and contains trials identified from:
monthly searches of the Cochrane Central Register of Controlled Trials (CENTRAL);
weekly searches of MEDLINE;
weekly searches of Embase;
handsearches of 30 journals and the proceedings of major conferences;
weekly current awareness alerts for a further 44 journals plus monthly BioMed Central email alerts.
Details of the search strategies for CENTRAL, MEDLINE and Embase, the list of handsearched journals and conference proceedings, and the list of journals reviewed via the current awareness service can be found in the ‘Specialized Register’ section within the editorial information about the Cochrane Pregnancy and Childbirth Group.
Trials identified through the searching activities described above are each assigned to a review topic (or topics). The Trials Search Co‐ordinator searches the register for each review using the topic list rather than keywords.
In addition, we searched regional databases because they could be an important source of additional studies from journals not indexed in other international databases Castro 2002. These were: LILACS (1982 to 1 December 2013) Castro 1999 (Appendix 1) and SciELO (1998 to 1 December 2013) (Appendix 2). We searched Web of Science (2001 to 1 December 2013) (Appendix 3) for conference proceedings, dissertations and theses and openSIGLE (1997 to 1 December 2013) (Appendix 4) for grey literature.
We searched the World Health Organization International Clinical Trials Registry Platform (ICTRP) (1 December 2013) (Appendix 5) and the metaRegister of Controlled Trials (mRCT) (1 December 2013) (Appendix 6) for ongoing studies.
Searching other resources
We contacted the first author of the included studies and sent a comprehensive list of relevant articles along with the inclusion criteria for the review, asking for any additional studies published or unpublished which might be relevant. Additionally, we searched the citation lists from reviewed articles and other relevant publications.
We did not apply any language restrictions.
Data collection and analysis
Selection of studies
Two review authors independently assessed for inclusion all the potential titles and abstracts of studies retrieved as a result of the search strategy to reduce the possibility that relevant reports were discarded. The review authors were masked to information of the article such as journal title, authors, institutions, magnitude and direction of the results. Any disagreement was resolved through consensus or, if required, by consulting a third review author.
We retrieved the full text of an article if there was any doubt as to whether the article should be included or excluded.
Data extraction and management
We designed a form to extract data. For eligible studies, two review authors extracted data independently using the agreed form; those review authors were thematic and methodological experts. Discrepancies were resolved through consensus or, if required, by consulting a third review author.
The data extracted included the following.
Methods
Location of the study. Setting.
Trial design.
Power calculation performed.
Method used to generate random allocation.
Methods used to maintain allocation concealment.
Number of women enrolled, randomised, excluded after randomisation, and analysed.
Use of any method of blinding of the researchers to the intervention in order to evaluate outcomes.
Number of participants lost to follow‐up in the groups.
Use of intention‐to‐treat analysis.
Funding sources, reported.
Ethical issues: use of signed informed consent and ethics approval.
Participants
Inclusion and exclusion criteria.
Baseline information on the participants in order to have comparable intervention and control groups at entry (management of the third stage of labour, retained placenta definition, exclusion of women with a clear diagnosis of placenta accreta).
Interventions
Total number of intervention groups.
Types of interventions: prostaglandins (or analogues) types, doses, route of administration a duration intervention.
Adherence to planned intervention and other interventions in the groups under evaluation.
Outcomes
Outcomes stated in methods versus outcomes reported in results.
How secondary outcomes were defined.
Differences between groups for outcome assessment.
Time of follow‐up of participants to measure outcomes.
How adverse event reports were validated.
This information was collated and presented in the tables Characteristics of included studies and Characteristics of excluded studies. Data were entered into Review Manager (RevMan 2012) and checked for accuracy. When information regarding any of the above was unclear, we contacted authors of the original reports to provide further details.
Assessment of risk of bias in included studies
Two review authors independently assessed risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). Any disagreement was resolved by consensus or by involving a third assessor when necessary. Review authors who assessed the risk of bias were blinded to the names of authors, institutions, journals and results of studies and were theme and methodology experts.
(1) Random sequence generation (checking for possible selection bias)
We described for each included study the method used to generate the allocation sequence in sufficient detail to allow an assessment of whether it should produce comparable groups.
The methodology was assessed as:
low risk of bias (any truly random process, e.g. random number table; computer random number generator);
high risk of bias (any non‐random process, e.g. odd or even date of birth; hospital or clinic record number);
unclear risk of bias (insufficient information about the sequence generation process to permit judgement of 'low' or 'high').
(2) Allocation concealment (checking for possible selection bias)
We described for each included study the method used to conceal allocation to interventions prior to assignment and we assessed whether intervention allocation could have been foreseen in advance of, or during recruitment, or changed after assignment.
We assessed the methods as:
low risk of bias (e.g. telephone or central randomisation; consecutively numbered sealed opaque envelopes);
high risk of bias (open random allocation; unsealed or non‐opaque envelopes, alternation; date of birth);
unclear risk of bias.
(3.1) Blinding of participants and personnel (checking for possible performance bias)
We described for each included study the methods used, if any, to blind study participants and personnel from knowledge of which intervention a participant received. We considered that studies were at low risk of bias if they were blinded, or if we judged that the lack of blinding would be unlikely to affect results. We assessed blinding separately for different outcomes or classes of outcomes.
We assessed the methods as:
low, high or unclear risk of bias for participants;
low, high or unclear risk of bias for personnel.
(3.2) Blinding of outcome assessment (checking for possible detection bias)
We described for each included study the methods used, if any, to blind outcome assessors from knowledge of which intervention a participant received. We assessed blinding separately for different outcomes or classes of outcomes.
We assessed the methods used to blind outcome assessment as:
low, high or unclear risk of bias.
(4) Incomplete outcome data (checking for possible attrition bias due to the amount, nature and handling of incomplete outcome data)
We described for each included study, and for each outcome or class of outcomes, the completeness of data including attrition and exclusions from the analysis. We stated whether attrition and exclusions were reported and the numbers included in the analysis at each stage (compared with the total randomised participants), if reasons for attrition or exclusion were reported, and whether missing data were balanced across groups or were related to outcomes. Where sufficient information was reported, or could be supplied by the trial authors, we planned to re‐included missing data in the analyses which we undertook.
We assessed methods as:
low risk of bias (e.g. no missing outcome data; missing outcome data balanced across groups);
high risk of bias (e.g. numbers or reasons for missing data imbalanced across groups; ‘as treated’ analysis done with substantial departure of intervention received from that assigned at randomisation);
unclear risk of bias.
We used a cut‐off point of 20% to consider that a study was at low or high risk of bias according to the level of missing data.
(5) Selective reporting (checking for reporting bias)
We described for each included study how the possibility of selective outcome reporting bias was investigated and what we found.
We assessed the methods as:
low risk of bias (where it was clear that all of the study’s pre‐specified outcomes and all expected outcomes of interest to the review had been reported);
high risk of bias (where not all the study’s pre‐specified outcomes had been reported; one or more reported primary outcomes were not pre‐specified; outcomes of interest were reported incompletely and so could not be used; study failed to include results of a key outcome that would have been expected to have been reported);
unclear risk of bias.
(6) Other bias (checking for bias due to problems not covered by (1) to (5) above)
We described for each included study any important concerns we had about other possible sources of bias.
We assessed whether each study was free of other problems that could cause risk of bias:
low risk of other bias;
high risk of other bias;
unclear whether there is risk of other bias.
(7) Overall risk of bias
We made explicit judgements about whether studies were at high risk of bias, according to the criteria given in the Handbook (Higgins 2011). With reference to (1) to (6) above, we assessed the magnitude and direction of the bias and whether we considered it was likely to impact on the findings. We explored the impact of the level of bias through undertaking sensitivity analyses ‐ see Sensitivity analysis.
We used the GRADE approach in order to produce a 'Summary of findings' table. We downgraded the quality of evidence depending on the presence of the following factors.
• Downgrade quality level for:
study limitations;
inconsistency of results;
indirectness of evidence;
imprecision;
publication bias.
Measures of treatment effect
Dichotomous data
For dichotomous data, results are presented as summary risk ratio (RR) with 95% confidence intervals (CI).The RR like the relative effect measure has consistency, works well with low or high rates of events and it is easier to interpret in clinical practice.
Continuous data
For continuous data, we used the mean difference (MD) as outcomes were measured in the same way between trials. If necessary, we would have used the standardised mean difference (SMD) to combine trials that measured the same outcome, but used different methods.
Unit of analysis issues
Other unit of analysis issues
In future updates of this review, if we identify a clinical trial in which participants are randomised to several intervention groups, we will determine which intervention groups are relevant and to avoid confusion for the reader, we will report all intervention groups of the study in the table of Characteristics of included studies in the notes cell, as well as we will provide a detailed description of the intervention groups relevant to the review and only use these groups in our analyses. In order to avoid a unit‐of‐analysis error for a study that could contribute multiple, correlated, comparisons, we will combine groups to create a single pair‐wise comparison.
Dealing with missing data
For included studies, we noted levels of attrition. We planned to investigate the impact of including studies with high levels of missing data in the overall assessment of treatment effect by using sensitivity analysis if high‐quality studies were included in the meta‐analysis. For all outcomes, we carried out analyses, as far as possible, on an intention‐to‐treat basis, i.e. we attempted to include all participants randomised to each group in the analyses, and all participants were analysed in the group to which they were allocated, regardless of whether or not they received the allocated intervention. The denominator for each outcome in each trial was the number randomised minus any participants whose outcomes were known to be missing.
Assessment of heterogeneity
We assessed statistical heterogeneity in each meta‐analysis using the T², I² and Chi² statistics. We regarded heterogeneity as substantial if the I² was greater than 30% and either the T² was greater than zero, or there was a low P value (less than 0.10) in the Chi² test for heterogeneity. Where we identified substantial heterogeneity, we explored it by pre‐specified subgroup analysis.
Assessment of reporting biases
In future updates of this review, if there are 10 or more studies in the meta‐analysis we will investigate reporting biases (such as publication bias) using funnel plots. We will assess funnel plot asymmetry visually. If asymmetry is suggested by a visual assessment, we will perform exploratory analyses to investigate it.
Data synthesis
We carried out statistical analysis using the Review Manager (RevMan 2012). We used fixed‐effect meta‐analysis for combining data when it was reasonable to assume that studies were estimating the same underlying treatment effect: i.e. where trials were examining the same intervention, and the trials’ populations and methods were judged sufficiently similar. We used random‐effects meta‐analysis if there were clinical heterogeneity sufficient to expect that the underlying treatment effects differed between trials, or if substantial statistical heterogeneity was detected, to produce an overall summary, if an average treatment effect across trials was considered clinically meaningful. The random‐effects summary was treated as the average range of possible treatment effects and we discussed the clinical implications of treatment effects differing between trials. If the average treatment effect was not clinically meaningful, we did not combine trials.
Subgroup analysis and investigation of heterogeneity
We planned to undertake subgroup analyses and sensitivity analyses if we identified substantial heterogeneity.
We planned the following subgroup analyses:
comparison of different prostaglandin type;
comparison of different prostaglandin administration route;
comparison of the time to intervention administration (less than 60 minutes; 60 minutes or more).
We planned to restricted subgroup analyses to the primary outcome: need for manual removal of placenta.
For fixed‐effect inverse variance meta‐analyses, we assessed differences between subgroups by interaction tests. For random‐effects and fixed‐effect meta‐analyses using methods other than inverse variance, we assessed differences between subgroups by inspection of the subgroups’ confidence intervals; non‐overlapping confidence intervals indicated a statistically significant difference in treatment effect between the subgroups.
Sensitivity analysis
We planned sensitivity analyses according to used definition of prolonged third stage (less than 60 minutes; 60 minutes or more) and to explore the effects of fixed‐effect or random‐effects analysis for outcomes with statistical heterogeneity. In future updates of this review, we will carry out sensitivity analyses for others aspects of the review that also might affect the results, such as the risk of bias associated with the quality of the included trials based on overall 'Risk of bias' assessment (low versus unclear and high risk of bias).
Results
Description of studies
Results of the search
A total of 1038 references were retrieved and reviewed after duplication. Of these, nine were initially screened as randomised controlled trials (RCTs).
Three studies that met the inclusion criteria were identified (SeeFigure 1). The included studies (van Beekhuizen 2006; van Stralen 2013; van Beekhuizen 2013) were published manuscripts. We excluded six studies (Habek 2007; Harara 2011; Notten 2012; Paavonen 2012; Rogers 2007; Sundaram 2007).
1.
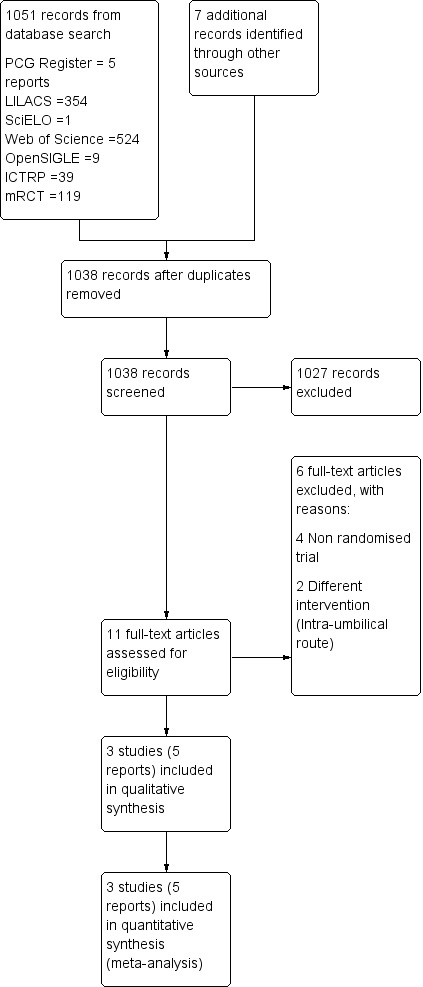
Study flow diagram.
Included studies
The three studies had small sample sizes and included a total of 244 women; the largest trial included 99 participants (van Stralen 2013). One study (van Beekhuizen 2006) was developed in two phases, the first one was a randomised phase comparing prostaglandin with placebo; and the second one had a non‐randomised assignation to the branch of prostaglandin. In order to carry out the analysis planned in our a priori protocol, we just included the randomised phase results with 50 participants. The studies were from The Netherlands and Tanzania and were published in English.
Population
The three studies included 244 participants (PG E2 analogue sulprostone with 50 participants and PG E1 analogue misoprostol with 194 participants), were multicentric and recruited women who were admitted for hospital delivery or who had home delivery. Two studies (van Beekhuizen 2006; van Beekhuizen 2013) included pregnancies with gestational age ≥ 28 weeks and the other one (van Stralen 2013) included pregnancies with at least 25 completed weeks of pregnancy. The studies included women having singleton pregnancy regardless of parity or previous history of manual removal of placenta or caesarean delivery. One study (van Beekhuizen 2006) included six women who received the intervention after operative vaginal delivery and 25 women with a previous history of curettage. van Stralen 2013 also included six women with history of postpartum haemorrhage (PPH). The third study (van Beekhuizen 2013) did not mention the characteristics of the included women. During delivery, all women received active management of labour with oxytocin in two studies (van Beekhuizen 2006; van Beekhuizen 2013), but this intervention was not mentioned in the other one (van Stralen 2013).
Interventions
One study (van Beekhuizen 2006) compared PG E2 analogue (sulprostone) at a single doses of 250 mcg by 30 minutes of intravenous infusion with placebo (saline solution) and included 50 women. The other two (van Beekhuizen 2013; van Stralen 2013) compared PG E1 analogue (misoprostol) with placebo at a single doses of 800 mcg administered orally dissolved in water or sublingually administrated and included 194 women.
Time to intervention administration and time limit for manual removal of the placenta
The time to intervention administration after delivery of the newborn was 30 minutes in one study (van Beekhuizen 2013) and 60 minutes the other two. The limit time for manual removal of the placenta was 30 minutes in two studies (van Beekhuizen 2006; van Beekhuizen 2013) and 45 minutes in the other one (van Stralen 2013).
Outcomes
Although the included studies reported at least one prespecified primary outcome of this review, there were some differences in reporting and definition of the outcomes. The trials recorded the need of manual removal of the placenta, blood transfusion, mean blood loss (mL), nausea and maternal pain between injection, discharge from the labour ward and severe postpartum haemorrhage. One study (van Stralen 2013) also registered mean time from injection to placental removal (minutes), vomiting, shivering and headache between injection and discharge from the labour ward.
Length of follow‐ up
The women were followed up for 12 to 24 hours (van Beekhuizen 2013) and from six to eight weeks postpartum (van Stralen 2013). In the third study (van Beekhuizen 2006), the timing of follow‐up was not specified.
Excluded studies
Six studies were excluded for the following reasons: four studies were not RCTs and two studies included a different intervention (Intra‐umbilical route). See Characteristics of excluded studies.
Risk of bias in included studies
Allocation
Two trials (van Beekhuizen 2006; van Beekhuizen 2013) adequately reported the method of random generation as using a computer‐generated randomisation list, and the concealment of allocation method by using sequentially numbered sealed envelopes, making selection bias at entry unlikely. The other trial (van Stralen 2013), did not report the method of random generation used but, based on the information provided during correspondence with the author, they implemented a computer‐generated randomisation list and sequentially numbered drugs containers, also making selection bias at entry unlikely. We judged all three studies to be at low risk of bias for selection bias.
Blinding
One study (van Beekhuizen 2006), did not adequately report the method implemented to blind study participants, outcome assessor and personnel from knowledge of which intervention a participant received. We contacted the principal author to obtain the necessary information for this judgment. Because the blinding method was reported as “…The physician in charge was blinded to the trial medication…” we assessed the risk of detection bias separately for each outcome.
Manual removal of placenta was objectively assessed and this outcome was measured during delivery. The blinding of participants and key study personnel was ensured and it was unlikely that there was detection bias. A time limit of 30 minutes for manual removal of placenta was established, in an effort to diminish the risk of performance bias. The outcomes pain and nausea were subjectively assessed but it was unclear when theses outcomes were measured. The method implemented in order to blind participants and key study personnel was not adequately reported, making an unclear the risk of bias.
Blood loss (measured in mL) was assessed objectively for women admitted for hospital delivery (determined by weight) and subjectively for women who had home delivery (estimated by the referring midwife). The method implemented in order to blind the participants and key study personnel was not adequately reported, which we considered resulted in an unclear the risk of bias. For blood transfusion and side‐effects such as pain and nausea, the author did not mention the method implemented to blind participants and key study personnel, making unclear the risk of bias.
In van Stralen 2013, the pills containing the interventions were identical and placed in identical containers in order to blind study participants and personnel from knowledge of which intervention a participant received, and a time limit of 45 minutes for manual removal of placenta was established, in an effort to diminish the risk of performance bias. Because blinding was broken after randomisation to some women for "breastfeeding reasons” during the postpartum stage, we assessed the risk of detection bias separately for each outcome.
Outcomes such as manual removal of placenta, severe postpartum haemorrhage, mean blood loss (measured in mL, estimated by collecting and weighing all blood, including in swabs and drapes) and mean time from injection to placental removal (minutes) were objectively assessed and these outcomes were measured during delivery. Blinding of participants and key study personnel was ensured and it was unlikely that blinding could have been broken, making detection bias improbable. Outcomes such as vomiting, shivering, nausea, headache and maternal pain between injection and discharge from the labour ward were subjectively assessed and these outcomes were measured during the first two hours after the intervention. Blinding of participants and key study personnel was ensured and it was unlikely that blinding could have been broken, thus making detection bias unlikely.
For need for blood transfusion, the author did not mention any criteria used to defined the outcome and it was measured during puerperium. Because blinding was broken after randomisation to some women according to "the obstetrician judgment" during the postpartum stage, some clinicians could have had knowledge about the women’s intervention. No blinding or incomplete blinding, and the way the outcome was measured makes detection bias likely.
Finally, the third study (van Beekhuizen 2013) implemented a technique of over encapsulation for both the 800 μg misoprostol tablets and the placebo tablets so that all tablets were the same size, and the placebo had a bitter taste and was dissolved sublingually similar to misoprostol. Because the method implemented to blind study participants and personnel from knowledge of which intervention a participant received, we assessed as unlikely the risk of performance and detection bias.
Incomplete outcome data
In one included trial (van Beekhuizen 2006), 15.2% of the women were excluded from the analysis for violations of the treatment protocol. One woman withdrew her consent; one women mentioned after inclusion that she had a cardiac condition that was a contraindication to participate in the study; in three women, the placenta was expelled within 60 minutes after delivery of the infant before trial medication, and in four women, blood loss exceeded 1000 mL before medication; the authors carried out an “as‐treated” analysis because they only included the results from those women who had both been assigned randomly and who had actually received the trial medication making attrition bias likely.
In another study (van Stralen 2013), follow‐up data were available for 38 (78%) women in the misoprostol group and 44 (86%) women in the placebo group. Follow‐up for the outcomes: manual removal of the placenta, blood transfusion and severe postpartum haemorrhage were completed with just one loss, but the follow‐up data for side‐effects such as vomiting, shivering, nausea, headache and pain were incomplete with more than 20% missing data. No explanation for these losses to follow‐up were provided, leading to a high risk of attrition bias for.
In the remaining trial (van Beekhuizen 2013), two women were excluded from the analysis because of follow‐up loss, one in each arm. Because the proportion of missing data compared with observed event risk was not enough to have a clinically relevant impact of the intervention effect estimate, we assessed this as being an unlikely the risk of attrition bias.
Selective reporting
In one study (van Beekhuizen 2006). the study protocol is not available and it is unclear if the published reports included all expected outcomes, including those that were prespecified. The report did not have sufficient information to permit judgment of “Yes” or “No” (rated as 'unclear' risk of bias). For the remaining trials, outcomes such as blood transfusion, PPH and haemoglobin at discharge from hospital were not pre specified in the protocol (ISRCTN16104753 (van Beekhuizen 2013) and ISRCTN45330307 (van Stralen 2013)), but were reported during the publication, thus raising the issue of reporting bias.
Other potential sources of bias
Two studies (van Beekhuizen 2006; van Beekhuizen 2013) had a potential source of bias related to a formal 'stopped early' rule due to apparent benefit; the other study (van Stralen 2013) appears to be free from other sources of bias.
In summary, according to the criteria applied for the sensitivity analysis, the trials were of poor methodological quality or there was insufficient information for inclusion into the high‐quality group (Figure 2; Figure 3).
2.
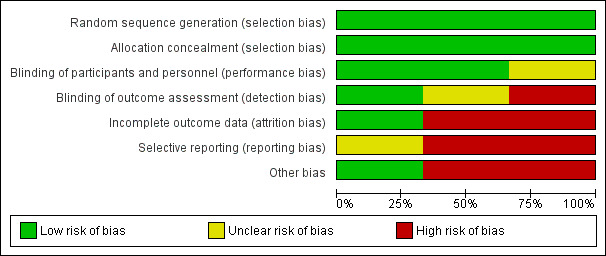
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Effects of interventions
See: Table 1
Overall, we included three trials, involving a total of 244 women. We carried out a total of six meta‐analysis (more than one trial analysed) and four comparisons correspond to a single‐trial analysis.
To display outcomes that were investigated only in single studies, we used a single forest plot in order to summarise the information (Higgins 2011).
Prostaglandins versus placebo (primary outcomes)
Manual removal of placenta
The following results correspond to the meta‐analysis of all three studies (van Beekhuizen 2006; van Beekhuizen 2013; van Stralen 2013). There was no significant difference between the prostaglandins and the placebo group in the rate of manual removal of placenta (average risk ratio (RR) 0.82, 95% confidence interval (CI) 0.54 to 1.27; three trials, 244 women, Tau² = 0.09, I² = 60%) (Analysis 1.1).
1.1. Analysis.
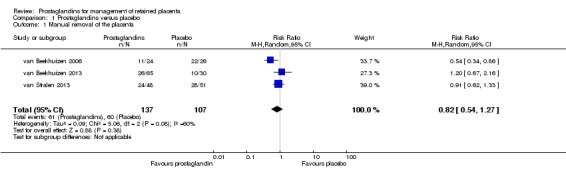
Comparison 1 Prostaglandins versus placebo, Outcome 1 Manual removal of the placenta.
Severe postpartum haemorrhage
We reported results corresponding to the meta‐analysis of two studies (van Beekhuizen 2013; van Stralen 2013). There was no significant difference in the rate of severe postpartum haemorrhage (RR 0.80, 95% CI 0.55 to 1.15; two trials, 194 participants; I2 = 0%) (Analysis 1.2).
1.2. Analysis.
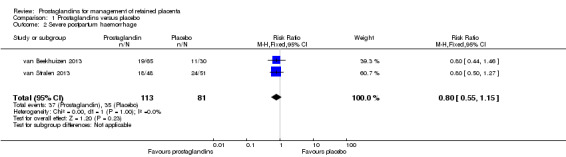
Comparison 1 Prostaglandins versus placebo, Outcome 2 Severe postpartum haemorrhage.
Blood transfusion
This following results correspond to the meta‐analysis of three studies (van Beekhuizen 2006; van Beekhuizen 2013; van Stralen 2013). There was no significant difference between the prostaglandin group and the placebo group in the rate of blood transfusion (RR 0.72, 95% CI 0.43 to 1.22; three trials, 244 women, I2 = 0%) (Analysis 1.3).
1.3. Analysis.
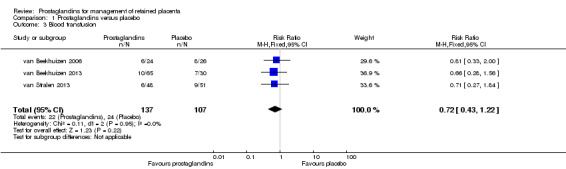
Comparison 1 Prostaglandins versus placebo, Outcome 3 Blood transfusion.
Prostaglandins versus placebo (secondary outcomes)
Mean blood loss (mL)
The following results correspond to meta‐analysis of three studies (van Beekhuizen 2006; van Beekhuizen 2013; van Stralen 2013). There was no significant difference between the prostaglandin group and the placebo group in the mean blood loss (mean difference (MD) ‐205.26 mL; 95% CI ‐536.31 to 125.79, three trials, 244 women, random‐effects, Tau² = 63417.09, I2 = 75%) (Analysis 1.4).
1.4. Analysis.
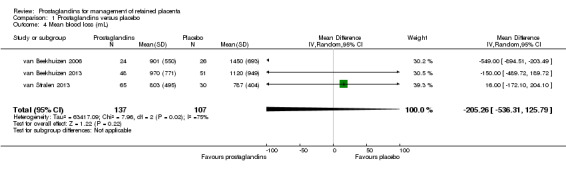
Comparison 1 Prostaglandins versus placebo, Outcome 4 Mean blood loss (mL).
Mean time from injection to placental removal (minutes)
We reported results corresponding to a single‐trial analysis (van Stralen 2013). There was no significant difference in the mean time from injection to placental removal (MD ‐7.00 minutes; 95% CI ‐21.20 to 7.20; one trial, 99 participants) (Analysis 1.5).
1.5. Analysis.
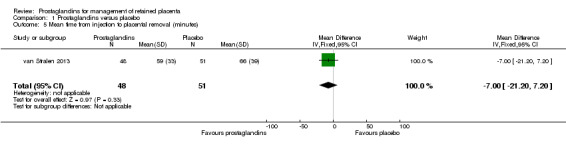
Comparison 1 Prostaglandins versus placebo, Outcome 5 Mean time from injection to placental removal (minutes).
Vomiting between injection and discharge from the labour ward
The following result corresponds to a single‐trial analysis (van Stralen 2013). There was no significant difference in the rate of vomiting between injection and discharge from the labour ward (RR 5.37, 95% CI 0.29 to 100.43; one trial, 74 participants) (Analysis 1.6).
1.6. Analysis.
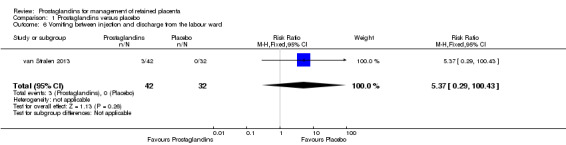
Comparison 1 Prostaglandins versus placebo, Outcome 6 Vomiting between injection and discharge from the labour ward.
Shivering between injection and discharge from the labour ward
We reported results corresponding to a single‐trial analysis (van Stralen 2013). There was a significant difference favouring placebo in the rate of shivering between injection and discharge from the labour ward (RR 10.00, 95% CI 1.40 to 71.49; one trial, 70 participants) (Analysis 1.7).
1.7. Analysis.
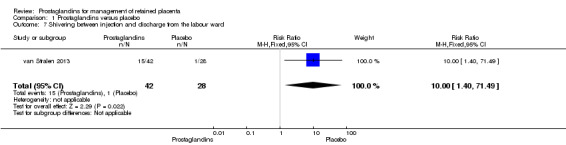
Comparison 1 Prostaglandins versus placebo, Outcome 7 Shivering between injection and discharge from the labour ward.
Nausea between injection and discharge from the labour ward
The following results correspond to meta‐analysis of two studies (van Beekhuizen 2006; van Stralen 2013). There was no significant difference between the prostaglandin group and the placebo group in the rate of nausea between injection and discharge from the labour ward (average RR 1.72, 95% CI 0.15 to 19.41; two trials, 124 women, Tau² = 1.38, I² = 43%) (Analysis 1.8).
1.8. Analysis.
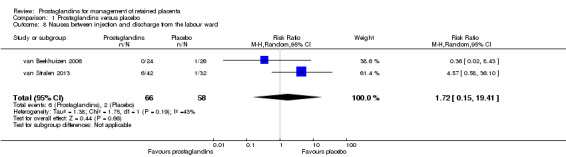
Comparison 1 Prostaglandins versus placebo, Outcome 8 Nausea between injection and discharge from the labour ward.
Headache between injection and discharge from the labour ward
We reported results corresponding to a single‐trial analysis (van Stralen 2013). There was no significant difference in the rate of headache between injection and discharge from the labour ward (RR 0.76, 95% CI 0.05 to 11.72; one trial, 74 participants) (Analysis 1.9).
1.9. Analysis.
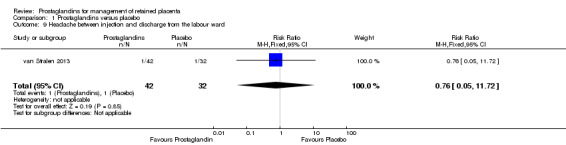
Comparison 1 Prostaglandins versus placebo, Outcome 9 Headache between injection and discharge from the labour ward.
Maternal pain between injection and discharge from the labour ward
The reported results corresponding to meta‐analysis of two studies (van Beekhuizen 2006; van Stralen 2013). There was no significant difference in the rate of maternal pain between injection and discharge from the labour ward (RR 0.91, 95% CI 0.43 to 1.96; two trials, 124 participants; I2 = 0%) (Analysis 1.10).
1.10. Analysis.
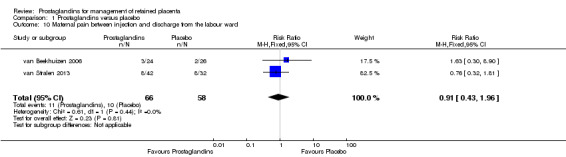
Comparison 1 Prostaglandins versus placebo, Outcome 10 Maternal pain between injection and discharge from the labour ward.
Subgroup analysis and investigation of heterogeneity
Subgroup analyses were carried out comparing the different prostaglandin type, the administration route and the time to intervention. The subgroup analyses were restricted to the primary outcome: need for manual removal of placenta according to the protocol.
When we explored the heterogeneity source by prostaglandin type (Analysis 2.1), the interaction test for subgroup differences was statistically significant (P = 0.04). For the subgroup relating to E2 prostaglandin (sulprostone), the rate of manual removal of placenta was significantly minor for the intervention group (RR 0.54, 95% CI 0.34 to 0.86; one trial, 50 women) contrasting with the E1 prostaglandin (misoprostol) subgroup, where the intervention did not show a significant difference (average RR 0.99, 95% CI 0.72 to 1.36; two trials, 194 participants, I2 = 0%) compared with placebo.
2.1. Analysis.
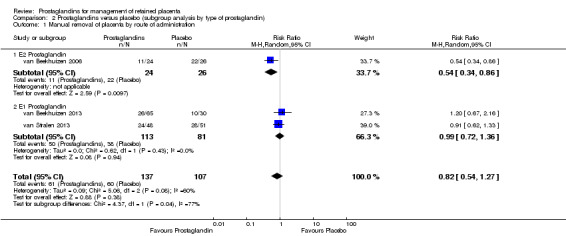
Comparison 2 Prostaglandins versus placebo (subgroup analysis by type of prostaglandin), Outcome 1 Manual removal of placenta by route of administration.
The tests for subgroup effect by administration route (intravenous infusion, oral and sublingual) (Analysis 3.1) and by the time to intervention administration (less than 60 minutes versus 60 minutes or more) (Analysis 4.1) were not statistically significant.
3.1. Analysis.
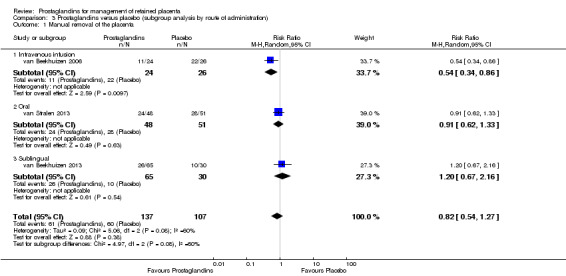
Comparison 3 Prostaglandins versus placebo (subgroup analysis by route of administration), Outcome 1 Manual removal of the placenta.
4.1. Analysis.
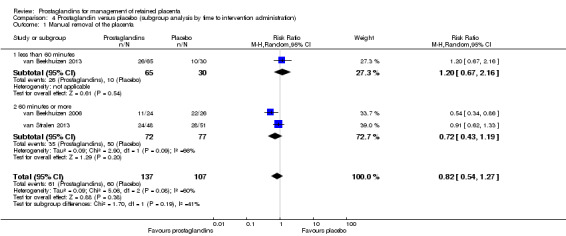
Comparison 4 Prostaglandin versus placebo (subgroup analysis by time to intervention administration), Outcome 1 Manual removal of the placenta.
Sensitivity analysis
We could not carry out the planned sensitivity analyses based on the quality of the included trials because all of the included studies were assessed as having a high risk of bias. Our sensitivity analyses based on the definition of prolonged third stage (less than 60 minutes; 60 minutes or more) showed the same result as that obtained for the time to intervention subgroup analysis.
Discussion
Summary of main results
We included three studies (involving 244 women) comparing the use of prostaglandins versus placebo for the management of retained placenta. Use of prostaglandins resulted in less need for manual removal of placenta, severe postpartum haemorrhage and blood transfusion but none of the differences reached statistical significance. Much larger, adequately powered studies are needed to confirm that these clinically important beneficial effects are not just chance findings.
Similarly, no differences were detected between prostaglandins and placebo in mean blood loss or the mean time from injection to placental removal (minutes) or side‐effects (vomiting, headache, pain and nausea between injection and discharge from the labour ward) except for 'shivering' which was more frequent in women who received prostaglandin. For the primary outcomes maternal mortality and need to add other therapeutic uterotonic, we did not obtain any data.
Overall completeness and applicability of evidence
Although comprehensive searches were conducted in order to retrieve all published and unpublished randomised clinical trials, this systematic review included only three trials of poor methodological quality and with small sample sizes. Additionally, the data are incomplete, and some of important clinical outcomes were not reported. For example, none of the included studies assessed the incidence of maternal mortality, need to add other therapeutic uterotonics, serious maternal morbidity, maternal postpartum anaemia or maternal satisfaction with treatment. There were also no comparisons between the medical management with prostaglandin and usual care. Consequently, the effectiveness and safety of medical management with prostaglandin compared with manual removal of retained placenta, is still unknown.
The applicability of the evidence outside the research setting is limited; however, these studies were all conducted in similar clinical settings and included a variety of clinical situations. Of the interventions analysed in the review, the PG E1 analogue (misoprostol) is the only prostaglandin that may be available in different clinical settings; contrasting with the PG E2 analogue (sulprostone), which is expensive, and requires refrigeration; as a result, it is not affordable in developing countries.
Quality of the evidence
The three included studies were judged to be at a high risk of bias and the evidence quality is considered to be very low (Table 1). There is little confidence in the effect estimates; the true effect is likely to be substantially different. The quality of evidence is very low due to study limitations (lack of blinding, failure to adhere to the intention‐to‐treat principle, selective reporting and stopping early due to apparent benefit), inconsistency (unexplained variability in some results) and imprecise results (few women and outcome events with wide confidence intervals).
We could not evaluate publication bias, because there were too few included studies and the trials were small. There remains some concern about publication bias.
Potential biases in the review process
Publication bias is a possibility in this review because of the limited number of studies and the small sample sizes. It is known that the risk of publication bias is probably higher for reviews that are based on small trials. Another important limitation of this systematic review is the measurement bias present in the available studies, especially when the outcomes were assessed subjectively. Finally, there was substantial heterogeneity for some outcomes and our investigation of heterogeneity sources (which was based on three studies) has limited value.
Authors' conclusions
Implications for practice.
Currently there is limited, low‐quality evidence relating to the effectiveness and safety of using prostaglandins for the management of retained placenta. Use of prostaglandins resulted in less need for manual removal of placenta, severe postpartum haemorrhage and blood transfusion but none of the differences reached statistical significance. Much larger, adequately powered studies are needed to confirm that these clinically important beneficial effects are not just chance findings.
Similarly, no differences were detected between prostaglandins and placebo in mean blood loss or the mean time from injection to placental removal (in minutes) or side‐effects (vomiting, headache, pain and nausea between injection and discharge from the labour ward) except for 'shivering', which was more frequent in women who received prostaglandin. The included studies were of poor quality and there is little confidence in the effect estimates; the true effect is likely to be substantially different. We can not make any recommendations about changes to clinical practice. More high‐quality research in this area is needed.
Implications for research.
There is an urgent need for high‐quality randomised controlled trials on treatments for women with retained placenta, particularly comparing manual removal and medical treatment including prostaglandins. Further research should focus on avoiding risk of bias such as, lack of blinding, failure to adhere to the intention‐to‐treat principle, selective reporting and stopping early for apparent benefit. Future studies should report important clinical outcomes such as: maternal mortality, the need to add another therapeutic uterotonic, the presence of serious maternal morbidity, the frequency of maternal postpartum anaemia or subsequent surgical evacuation of retained products of conception inter alia.
Acknowledgements
We would like to thank Dr MY Martínez‐Velásquez and Dr A Bautista‐Charry for their participation and clinical perspective during the production of this systematic review.
As part of the pre‐publication editorial process, this review has been commented on by four peers (an editor and three referees who are external to the editorial team), a member of the Pregnancy and Childbirth Group's international panel of consumers and the Group's Statistical Adviser.
Appendices
Appendix 1. LILACS search strategy
Keywords CONTAINS: ((Pt randomised controlled trial OR Pt controlled clinical trial OR Mh randomised controlled trials OR Mh random allocation OR Mh double‐blind method OR Mh single‐blind method) AND NOT (Ct animal AND NOT (Ct human and Ct animal)) OR (Pt clinical trial OR Ex E05.318.760.535$ OR (Tw clin$ AND (Tw trial$ OR Tw ensa$ OR Tw estud$ OR Tw experim$ OR Tw investiga$)) OR ((Tw singl$ OR Tw simple$ OR Tw doubl$ OR Tw doble$ OR Tw duplo$ OR Tw trebl$ OR Tw trip$) AND (Tw blind$ OR Tw cego$ OR Tw ciego$ OR Tw mask$ OR Tw mascar$)) OR Mh placebos OR Tw placebo$ OR (Tw random$ OR Tw randon$ OR Tw casual$ OR Tw acaso$ OR Tw azar OR Tw aleator$) OR Mh research design) AND NOT (Ct animal AND NOT (Ct human and Ct animal)) OR (Ct comparative study OR Ex E05.337$ OR Mh follow‐up studies OR Mh prospective studies OR Tw control$ OR Tw prospectiv$ OR Tw volunt$ OR Tw volunteer$) AND NOT (Ct animal AND NOT (Ct human and Ct animal))) AND Keywords CONTAINS: ((Labor stage, third) OR (third labor stage) OR (uterine hemorrhage) OR (puerperal disorders) OR (postpartum hemorrhage) OR (obstetric labor complications) OR (placenta, retained) OR (retained placenta) OR (placenta diseases))
Appendix 2. SciELO search strategy
Integrate CONTAINS: ((Labor stage, third) OR (third labor stage) OR (uterine hemorrhage) OR (puerperal disorders) OR (postpartum hemorrhage) OR (obstetric labor complications) OR (placenta, retained) OR (retained placenta) OR (placenta diseases)) AND ((prostaglandins) OR (dinoprostone) OR (pge2 alpha) OR (alpha, pge2) OR (prostaglandin e2 alpha) OR (prostaglandin e2) OR (prepidil gel) OR (prostenon) OR (prostaglandin f1) OR (prostaglandin e2 methyl ester) OR (dinoprost) OR (pgf2) OR (prostaglandin f2alpha) OR (oxytocics) OR (prostaglandins, synthetic) OR (pg analogs) OR (prostaglandin analogues) OR (prostaglandin analogs) OR (misoprostol) OR (cytotec) OR (prostaglandins f, synthetic) OR (prostaglandin f analogues) OR (prostaglandin f analogs) OR (carboprost) OR (abortifacient agents, nonsteroidal) OR (fenprostalene) OR (meteneprost) OR (sulprostone) OR (prostaglandins f) OR (prostaglandins e, synthetic) OR (prostaglandin e analogues) OR (prostaglandins e) OR (f2‐isoprostanes) OR (prostaglandin e1 methyl ester) OR (prostaglandin e2 ethanolamide) OR (gemeprost) OR (cervagem) OR (prostaglandin f2alpha ethanolamide) OR (2,3‐dinor‐8‐iso‐prostaglandin‐f(2alpha)) OR (16‐methyl prostaglandin e2) OR (dinoprost tromethamine) OR (prostaglandin f2alpha tromethamine) OR (lutalyse) OR (minprostin f2 alpha) OR (prostin f2) OR (carboprost tromethamine)) AND ((clinical trial) OR (randomised clinical trial) OR (controlled clinical trial))
Terms In Spanish. Integrada CONTENIENDO: ((tercer estadio de parto) (tercer estadio del parto) OR (hemorragia uterina) OR (desorden puerperal) OR (hemorragia postparto) OR (complicacion obstetrica) OR (complicacion del trabajo de parto) OR (placenta retenida) OR (retencion de placenta) (retencion de la placenta) OR (enfermedad placentaria)) AND ((prostaglandinas) OR (dinoprostone) OR (pge2 alpha) OR (alpha, pge2) OR (prostaglandina e2 alpha) OR (prostaglandina e2) OR (prepidil gel) OR (prostenon) OR (prostaglandina f1) OR (prostaglandina e2 methyl ester) OR (dinoprost) OR (pgf2) OR (prostaglandina f2 alpha) OR (oxitocicos) OR (prostaglandinas, sinteticas) OR (pg analogos) OR (prostaglandina analogos) OR (misoprostol) OR (cytotec) OR (prostaglandina f, sintetica) OR (prostaglandina f analogos) OR (carboprost) OR (agente abortivo) OR (agente abortivo no esteroideo) OR (fenprostalene) OR (meteneprost) OR (sulprostone) OR (prostaglandina f) OR (prostaglandina e, sintetica) OR (prostaglandina e analogos) OR (prostaglandina e) OR (f2‐isoprostanes) OR (prostaglandina e1 methyl ester) OR (prostaglandina e2 ethanolamide) OR (gemeprost) OR (cervagem) OR (prostaglandina f2alpha ethanolamide) OR (2,3‐dinor‐8‐iso‐prostaglandin‐f(2alpha)) OR (16‐methyl prostaglandina e2) OR (dinoprost tromethamine) OR (prostaglandina f2alpha tromethamine) OR (lutalyse) OR (minprostin f2 alpha) OR (prostin f2) OR (carboprost tromethamine)) AND ((expermiento clinico) OR (experimento clinico aleatorizado) OR (experimento clinico controlado) OR (experimental))
Appendix 3. Web of Science search strategy
Topic CONTAINS: ((Labor stage, third) OR (third labor stage) OR (uterine hemorrhage) OR (puerperal disorders) OR (postpartum hemorrhage) OR (obstetric labor complications) OR (placenta, retained) OR (retained placenta) OR (placenta diseases)) AND Topic CONTAINS: ((prostaglandins) OR (dinoprostone) OR (pge2 alpha) OR (alpha, pge2) OR (prostaglandin e2 alpha) OR (prostaglandin e2) OR (prepidil gel) OR (prostenon) OR (prostaglandin f1) OR (prostaglandin e2 methyl ester) OR (dinoprost) OR (pgf2) OR (prostaglandin f2alpha) OR (oxytocics) OR (prostaglandins, synthetic) OR (pg analogs) OR (prostaglandin analogues) OR (prostaglandin analogs) OR (misoprostol) OR (cytotec) OR (prostaglandins f, synthetic) OR (prostaglandin f analogues) OR (prostaglandin f analogs) OR (carboprost) OR (abortifacient agents, nonsteroidal) OR (fenprostalene) OR (meteneprost) OR (sulprostone) OR (prostaglandins f) OR (prostaglandins e, synthetic) OR (prostaglandin e analogues) OR (prostaglandins e) OR (f2‐isoprostanes) OR (prostaglandin e1 methyl ester) OR (prostaglandin e2 ethanolamide) OR (gemeprost) OR (cervagem) OR (prostaglandin f2alpha ethanolamide) OR (2,3‐dinor‐8‐iso‐prostaglandin‐f(2alpha)) OR (16‐methyl prostaglandin e2) OR (dinoprost tromethamine) OR (prostaglandin f2alpha tromethamine) OR (lutalyse) OR (minprostin f2 alpha) OR (prostin f2) OR (carboprost tromethamine))
Appendix 4. OpenSIGLE search strategy
Keywords CONTAINS: (Labor stage, third) OR (third labor stage) OR (uterine hemorrhage) OR (puerperal disorders) OR (postpartum hemorrhage) OR (obstetric labor complications) OR (placenta, retained) OR (retained placenta) OR (placenta diseases)
Appendix 5. International Clinical Trials Registry Platform search strategy
Search 1
postpartum haemorrhage OR postpartum hemorrhage OR post‐partum haemorrhage OR post‐partum hemorrhage OR post partum haemorrhage OR post partum hemorrhage OR retained placenta (Title)
Search 2
retained placenta ‐ (Condition)
Search 3
placenta (Title) AND prostaglandin OR prostaglandins OR misoprostol (Intervention)
Search 4
placenta (Condition) AND prostaglandin OR prostaglandins OR misoprostol (Intervention)
Appendix 6. metaRegister of Controlled Trials (mRCT) search strategy
postpartum hemorrhage
postpartum haemorrhage
post partum hemorrhage
post partum haemorrhage
retained placenta
Data and analyses
Comparison 1. Prostaglandins versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Manual removal of the placenta | 3 | 244 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.54, 1.27] |
| 2 Severe postpartum haemorrhage | 2 | 194 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.55, 1.15] |
| 3 Blood transfusion | 3 | 244 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.43, 1.22] |
| 4 Mean blood loss (mL) | 3 | 244 | Mean Difference (IV, Random, 95% CI) | ‐205.26 [‐536.31, 125.79] |
| 5 Mean time from injection to placental removal (minutes) | 1 | 99 | Mean Difference (IV, Fixed, 95% CI) | ‐7.0 [‐21.20, 7.20] |
| 6 Vomiting between injection and discharge from the labour ward | 1 | 74 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.37 [0.29, 100.43] |
| 7 Shivering between injection and discharge from the labour ward | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 10.0 [1.40, 71.49] |
| 8 Nausea between injection and discharge from the labour ward | 2 | 124 | Risk Ratio (M‐H, Random, 95% CI) | 1.72 [0.15, 19.41] |
| 9 Headache between injection and discharge from the labour ward | 1 | 74 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.05, 11.72] |
| 10 Maternal pain between injection and discharge from the labour ward | 2 | 124 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.43, 1.96] |
Comparison 2. Prostaglandins versus placebo (subgroup analysis by type of prostaglandin).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Manual removal of placenta by route of administration | 3 | 244 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.54, 1.27] |
| 1.1 E2 Prostaglandin | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 0.54 [0.34, 0.86] |
| 1.2 E1 Prostaglandin | 2 | 194 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.72, 1.36] |
Comparison 3. Prostaglandins versus placebo (subgroup analysis by route of administration).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Manual removal of the placenta | 3 | 244 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.54, 1.27] |
| 1.1 Intravenous infusion | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 0.54 [0.34, 0.86] |
| 1.2 Oral | 1 | 99 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.62, 1.33] |
| 1.3 Sublingual | 1 | 95 | Risk Ratio (M‐H, Random, 95% CI) | 1.2 [0.67, 2.16] |
Comparison 4. Prostaglandin versus placebo (subgroup analysis by time to intervention administration).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Manual removal of the placenta | 3 | 244 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.54, 1.27] |
| 1.1 less than 60 minutes | 1 | 95 | Risk Ratio (M‐H, Random, 95% CI) | 1.2 [0.67, 2.16] |
| 1.2 60 minutes or more | 2 | 149 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.43, 1.19] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
van Beekhuizen 2006.
| Methods | • Trial design: The Netherlands. Multicentric randomised clinical trial, parallel, 2 arms. • Funding sources reported: not mentioned. • Ethical issues: use of signed informed consent and ethics approval. |
|
| Participants | • Inclusion and exclusion criteria Exclusion criteria: Blood loss ≥ 1000 mL. Reduction in diastolic blood pressure ≥ 20 mmHg. Tachycardia ≥ 120 beat/minutes. Gynaecologic infection. General history. Age < 18 or > 40 years. Gestational age ≥ 28 weeks. Asthma, bronchitis. Epilepsy. Cardiac disease. Hypertension, pre‐eclampsia, HELLP syndrome. Liver failure, renal failure. Stomach ulcer, ulcerative colitis. Sickle cell anaemia, b‐thalassaemia. Glaucoma. • Study participants were recruited from women who were admitted for hospital delivery and from women who had been referred because of retained placenta after home delivery. • Women who were delivered in the hospital all received active management of labour with oxytocin 10 IU intramuscularly, and controlled cord traction. The women who were referred because of retained placenta after home delivery received the same treatment after they had arrived in the hospital. The administration of study medication started 60 minutes after the delivery of the Infant. |
|
| Interventions | • Total number of intervention groups: 2 groups. Intervention: sulprostone (synthetic prostaglandin‐E2 derivative) 250 mg by 30 minutes of intravenous infusion. Single doses. 24 participants. Control: placebo by 30 minutes of intravenous infusion. Single doses. 26 participants. |
|
| Outcomes | • The primary outcome was the presence or absence of manual removal of placenta. • The secondary outcome variable was the amount of blood loss (mL). The amount of in‐hospital blood loss was determined by weight; blood loss before entering the hospital was estimated by the referring midwife. Adverse event reports (shivering, headache, pain, vomiting and nausea, hypotension and hypertension) were recorded and measured subjectively. |
|
| Notes | Correspondence: yes. We send a letter asking to the principal author about the method implemented in the study in order to blind the study participants and personnel. We used an open question. The author answered: "The RCT was double blinded: After the patient was recruited a sealed enveloped was handed to an 'independent' midwife who was not involved in taking care of the patient. She opened the envelope and prepared the study medication and handed the blinded study medication to the midwife/nurse who took care of the patient. This was to ensure that both the patient, the doctor and the midwife were not aware whether the patient received placebo or study medication". Comment: unclear risk for performance and detection bias. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | “…Study medication was randomised in blocks of 4…” Comment: probably done. |
| Allocation concealment (selection bias) | Low risk | “…the allocation of sealed envelopes was in the sequence of enrolment…” Comment: probably done. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Did not adequately reported the method implemented to blind study participants and personnel from knowledge of which intervention a participant received “…The physician in charge was blinded to the trial medication…” Comment: unclear risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Did not adequately reported the method implemented to blind study participants and personnel from knowledge of which intervention a participant received “…The physician in charge was blinded to the trial medication…” Comment: unclear risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Authors did a “As‐treated” analysis because they only included the results from those women who had both been assigned randomly and who had actually received the trial medication. |
| Selective reporting (reporting bias) | Unclear risk | The report did not have sufficient information to permit judgment of “Yes” or “No”. |
| Other bias | High risk | Stopped early rule due to apparent benefit. |
van Beekhuizen 2013.
| Methods | • Trial design: Tanzania. Multicentric randomised clinical trial, parallel, 2 arms. • Funding sources, reported: yes. The trial was supported through the Stimuleringsfonds of the Dutch Society of Tropical Medicine and the German Gesellschaft für Internationale Zusammenarbeitung. • Ethical issues: use of signed informed consent and ethics approval. |
|
| Participants | • Inclusion and exclusion criteria Inclusion criteria: Delivered of a baby of 1 kg or more or at a gestational age of 28 weeks or more. Exclusion criteria: Haemoglobin concentration less than 100 g/L (6.2 mmol/l). Blood loss more than 750 mL. Pulse rate more than 120 beats per minute. Diastolic blood pressure reduction after delivery more than 20 mmHg. • Study participants were recruited from women who were admitted for hospital delivery. • Women who were delivered in the hospital received all active management of labour with oxytocin 5 IU and controlled cord traction. |
|
| Interventions | • Total number of intervention groups: 2 groups. Intervention: misoprostol 800 μg sublingually administered to the mother. Single doses. 65 participants. Control: placebo sublingually administered to the mother. Single doses. 30 participants. |
|
| Outcomes | • The primary outcome was the presence or absence of manual removal of placenta. • The secondary outcome variable was the amount of blood loss (mL), postpartum haemorrhage, blood transfusion and haemoglobin at discharge from hospital.The blood loss was calculated by weighing self‐absorbable mattresses. |
|
| Notes | Correspondence: no required. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Allocation was in accordance with the sequence of enrolment in each of the 7 hospitals." Comment: probably done. |
| Allocation concealment (selection bias) | Low risk | "The randomisation scheme used balanced variable blocks: in the labor ward were closed envelopes containing the registration papers in addition to the blinded study medication." Comment: probably done. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "A technique of over encapsulation was used for both the 800‐μg misoprostol tablets and the placebo tablets. All tablets were the same size, and the placebo had a bitter taste and dissolved sublingually similar to misoprostol." Comment: probably done. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "A technique of over encapsulation was used for both the 800‐μg misoprostol tablets and the placebo tablets. All tablets were the same size, and the placebo had a bitter taste and dissolved sublingually similar to misoprostol." Comment: probably done. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Follow‐up data were available for 65/66women in the misoprostol group and 30/31 women in the placebo group. |
| Selective reporting (reporting bias) | High risk | Postpartum haemorrhage (>1 L) and haemoglobin at discharge from hospital (g/dL) were not pre specified in the protocol stage (ISRCTN16104753) but were reported during the publication. |
| Other bias | High risk | Stopped early rule due to apparent benefit. |
van Stralen 2013.
| Methods | • Trial design: The Netherlands. Multicentric randomised clinical trial, parallel, 2 arms. • Funding sources, reported: yes. “…No funding was received for this trial…”. • Ethical issues: use of signed informed consent and ethics approval. |
|
| Participants | • Inclusion and exclusion criteria Inclusion criteria: Absence of excessive blood loss (as judged by the attending obstetrician). At least 25 completed pregnancy weeks. Minimum participation age was 18 years. Fluent in the Dutch language in word and script. Exclusion criteria: Postpartum haemorrhage, defined as more than 1000 mL within 60 minutes after birth of the newborn. • Study participants were recruited from women who were admitted for hospital delivery and from women who had been referred because of retained placenta after home delivery. • Active management of labour: not mentioned. |
|
| Interventions | • Total number of intervention groups: 2 groups. Intervention: misoprostol 800 μg dissolved in water administered orally to the mother. Single doses. 48 participants. Control: placebo dissolved in water administered orally to the mother. Single doses. 51 participants. |
|
| Outcomes | • Primary outcome was number of manual removals. •The secondary outcomes were amount of blood loss, blood transfusion, postpartum haemorrhage, interval between delivery of the baby and delivery of the placenta and side‐effects of the study medication. The blood loss was measured collecting and weighing all blood, including in swabs and drapes. |
|
| Notes | Correspondence: yes. We send a letter asking to the principal author about the method implemented into study in order to generate the random allocation and the method used to blind the study participants and personnel. We used an open question. The author answered: “our pharmacist provided us with coded blank pills, produced by the hospital pharmacy containing either placebo, or misoprostol. Pills were identical and placed in identical numbered containers (1,2,3,4 etc). After the inclusion period ended, the pharmacist gave us the key to the codes used for the medication. This ensured blinding of personnel and participants and proper allocation…” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | “…We conducted a double blinded, multicenter randomised trial…” Protocol registered ISRCTN45330307. Comment: probably done. |
| Allocation concealment (selection bias) | Low risk | “…After inclusion, a blank envelope was drawn containing study medication and a case record form. The Leiden University Medical Center pharmacist prepared the envelope and study medication. The study medication was provided in a single number coded container…” Comment: probably done. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The blinding was broken after randomisation to some women, according to "the obstetrician judgment" by “…Breastfeeding reasons...” during postpartum stage. Is unlikely to suffer performance bias because the physicians were not aware of the treatment given and a time limit of 45 minutes was established for manual removal the placenta. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blood transfusion: the author did not mention any used criteria. The blinding was broken after randomisation to some women, according to "the obstetrician judgment" during postpartum stage. Some clinicians could have knowledge about the women’s intervention. Comment: No blinding or incomplete blinding, and the outcome measurement is likely to be influenced by lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | “…Follow‐up data were available for 38 (78%) women in the misoprostol group and 44 (86%) women in the placebo group…” Follow‐up data for side‐effects were incomplete with a level of missing data more than 20%. |
| Selective reporting (reporting bias) | High risk | Blood transfusion and postpartum haemorrhage were not pre specified in the protocol stage (ISRCTN45330307) but were reported during the publication. |
| Other bias | Low risk | Study appear to be free of other sources of bias. |
HELLP: haemolysis elevated liver enzymes and low platelets syndrome IU: international unit
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Habek 2007 | Not a randomised controlled trial. |
| Harara 2011 | Different intervention. |
| Notten 2012 | Not a randomised controlled trial. |
| Paavonen 2012 | Not a randomised controlled trial. |
| Rogers 2007 | Randomised controlled trial, different intervention. |
| Sundaram 2007 | Not a randomised controlled trial. |
Contributions of authors
Carlos Fernando Grillo‐Ardila and Ariel Iván Ruiz‐Parra conducted the titles selection, assessed trial quality, extracted data, carried out data analysis and wrote the first draft of the systematic review and subsequent amendments. Hernando G Gaitán assessed trial quality, carried out data analysis and commented on and revised the first draft of the systematic review. Nelcy Rodriguez‐Malagón commented on and revised the first draft of the systematic review.
Sources of support
Internal sources
No sources of support supplied
External sources
Universidad Nacional de Colombia, Colombia.
Declarations of interest
None known.
New
References
References to studies included in this review
van Beekhuizen 2006 {published data only}
- Beekhuizen HJ, Groot ANJA, Boo T, Burger D, Jansen N, Lotgering FK. Sulprostone reduces the need for the manual removal of the placenta in patients with retained placenta: a randomized controlled trial. American Journal of Obstetrics and Gynecology 2006;194:446‐50. [DOI] [PubMed] [Google Scholar]
van Beekhuizen 2013 {published data only}
- Beekhuizen HJ, Pembe AB, Fauteck H, Lotgering FK. Treatment of retained placenta with misoprostol: a randomised controlled trial in a low‐resource setting (Tanzania). BMC Pregnancy and Childbirth 2009;9:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beekhuizen HJ, Tarimo V, Pembe AB, Fauteck H, Lotgering FK. A randomized controlled trial on the value of misoprostol for the treatment of retained placenta in a low‐resource setting. International Journal of Gynecology and Obstetrics 2013;122(3):234‐7. [DOI] [PubMed] [Google Scholar]
van Stralen 2013 {published data only}
- Stralen G. Misoprostol in the management of retained placenta ‐ a safe alternative for manual removal? A randomised controlled trial. Current Controlled Trials (www.controlled‐trials.com/) (accessed 30 October 2007).
- Stralen G, Veenhof M, Holleboom C, Roosmalen J. No reduction of manual removal after misoprostol for retained placenta: a double‐blind, randomized trial. Acta Obstetricia et Gynecologica Scandinavica 2013; Vol. 92, issue 4:398‐403. [DOI] [PubMed]
References to studies excluded from this review
Habek 2007 {published data only}
- Habek D, Franicevic D. Intraumbilical injection of uterotonics for retained placenta. International Journal of Gynecology & Obstetrics 2007;99(2):105‐9. [DOI] [PubMed] [Google Scholar]
Harara 2011 {published data only}
- Harara R. Intraumbilical injection of three different uterotonics in the management of retained placenta. Journal of Obstetrics and Gynaecology Research 2011;37(9):1203‐7. [DOI] [PubMed] [Google Scholar]
Notten 2012 {published data only}
- Notten F, Scheepers L. Consecutive intra‐umbilical vein injection of misoprostol and intravenous sulprostone in the management of retained placenta: a retrospective cohort study. American Journal of Obstetrics and Gynecology 2012;206(1):S139. [DOI] [PubMed] [Google Scholar]
Paavonen 2012 {published data only}
- Paavonen J. Intravenous sulprostone infusion in the management of retained placenta ‐ three‐year university hospital experience. Acta Obstetricia et Gynecologica Scandinavica 2012;91:117. [DOI] [PubMed] [Google Scholar]
Rogers 2007 {published data only}
- Rogers MS, Yuen PM, Wong S. Avoiding manual removal of placenta: evaluation of intra‐umbilical injection of uterotonics using the Pipingas technique for management of adherent placenta. Acta Obstetricia et Gynecologica Scandinavica 2007;86(1):48‐54. [DOI] [PubMed] [Google Scholar]
Sundaram 2007 {published data only}
- Sundaram S. Rectal misoprostol compared with 15‐methyl prostaglandin F2 alpha for retained placenta after second‐trimester delivery. Obstetrics and Gynecology 2007;109(4):S108. [DOI] [PubMed] [Google Scholar]
Additional references
Abdel‐Aleem 2011
- Abdel‐Aleem H, Abdel‐Aleem MA, Shaaban OM. Tocolysis for management of retained placenta. Cochrane Database of Systematic Reviews 2011, Issue 1. [DOI: 10.1002/14651858.CD007708] [DOI] [PubMed] [Google Scholar]
Arias 2000
- Arias F. Pharmacology of oxytocin and prostaglandins. Clinical Obstetrics and Gynecology 2000;43:455‐68. [DOI] [PubMed] [Google Scholar]
Bodelon 2009
- Bodelon C, Bernabe‐Ortiz A, Schiff MA, Reed SD. Factors associated with peripartum hysterectomy. Obstetrics & Gynecology 2009;114:115‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Carroli 2008
- Carroli G, Cuesta C, Abalos E, Gulmezoglu AM. Epidemiology of postpartum haemorrhage: a systematic review. Best Practice & Research Clinical Obstetrics & Gynaecology 2008;22:999‐1012. [DOI] [PubMed] [Google Scholar]
Castro 1999
- Castro AA, Clark OA, Atallah AN. Optimal search strategy for clinical trials in the Latin American and Caribbean Health Science Literature Database (LILACS database): Update [Letter]. Sao Paulo Medical Journal/ Revista Paulista de Medicina 1999;117(3):138‐9. [DOI] [PubMed] [Google Scholar]
Castro 2002
- Castro AA, Clark OA. Searching the Literatura Latino Americana e do Caribe em Ciencias da Saude (LILACS) database improves systematic reviews. International Journal of Epidemiology 2002;31(1):112‐4. [DOI] [PubMed] [Google Scholar]
Combs 1991
- Combs CA, Laros RK, Jr. Prolonged third stage of labor: morbidity and risk factors. Obstetrics & Gynecology 1991;77:863‐7. [PubMed] [Google Scholar]
Deneux‐Tharaux 2009
- Deneux‐Tharaux C, Macfarlane A, Winter C, Zhang WH, Alexander S, Bouvier‐Colle MH. Policies for manual removal of placenta at vaginal delivery: variations in timing within Europe. BJOG: an International Journal of Obstetrics and Gynaecology 2009;116:119‐24. [DOI] [PubMed] [Google Scholar]
Herman 1993
- Herman A, Weinraub Z, Bukovsky I, Arieli S, Zabow P, Caspi E, et al. Dynamic ultrasonographic imaging of the third stage of labor: new perspectives into third‐stage mechanisms. American Journal of Obstetrics and Gynecology 1993;168(5):1496‐9. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hoveyda 2001
- Hoveyda F, MacKenzie IZ. Secondary postpartum haemorrhage: incidence, morbidity and current management. BJOG: an International Journal of Obstetrics and Gynaecology 2001;108:927‐30. [DOI] [PubMed] [Google Scholar]
Miller 2006
- Miller SB. Prostaglandins in health and disease: an overview. Seminars in Arthritis and Rheumatism 2006;36:37‐49. [DOI] [PubMed] [Google Scholar]
Mockler 2012
- Mockler JC, East CE. Non‐pharmacological and non‐surgical interventions for managing retained placenta. Cochrane Database of Systematic Reviews 2012, Issue 5. [DOI: 10.1002/14651858.CD009854] [DOI] [Google Scholar]
Nardin 2011
- Nardin JN, Weeks A, Carroli G. Umbilical vein injection for management of retained placenta. Cochrane Database of Systematic Reviews 2011, Issue 5. [DOI: 10.1002/14651858.CD001337.pub2] [DOI] [PubMed] [Google Scholar]
Owolabi 2008
- Owolabi AT, Dare FO, Fasubaa OB, Ogunlola IO, Kuti O, Bisiriyu LA. Risk factors for retained placenta in southwestern Nigeria. Singapore Medical Journal 2008;49:532‐7. [PubMed] [Google Scholar]
RevMan 2012 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.2. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2011.
Rizwan 2009
- Rizwan N, Abbasi RM, Jatoi N. Retained placenta still a continuing cause of maternal morbidity and mortality. Journal of the Pakistan Medical Association 2009;59:812‐4. [PubMed] [Google Scholar]
Sosa 2009
- Sosa CG, Althabe F, Belizan JM, Buekens P. Risk factors for postpartum hemorrhage in vaginal deliveries in a Latin‐American population. Obstetrics & Gynecology 2009;113(6):1313‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sundaram 2009
- Sundaram S, Diaz JP, Gonzalez‐Quintero VH, Verma U. Rectal misoprostol vs 15‐methyl prostaglandin F2alpha for retained placenta after second‐trimester delivery. American Journal of Obstetrics and Gynecology 2009;200:e24‐6. [DOI] [PubMed] [Google Scholar]
Weeks 2001
- Weeks AD. The retained placenta. African Health Sciences 2001;1:36‐41. [PMC free article] [PubMed] [Google Scholar]
Weeks 2008
- Weeks AD. The retained placenta. Best Practice & Research Clinical Obstetrics & Gynaecology 2008;22:1103‐17. [DOI] [PubMed] [Google Scholar]