

Abstract
Tissue development and regeneration involve tightly coordinated and integrated processes: selective proliferation of resident stem and precursor cells, differentiation into target somatic cell type, and spatial morphological organization. The role of the mechanical environment in the coordination of these processes is poorly understood. We show that multipotent cells derived from native cardiac tissue continually monitored cell substratum rigidity and showed enhanced proliferation, endothelial differentiation, and morphogenesis when the cell substratum rigidity closely matched that of myocardium. Mechanoregulation of these diverse processes required p190RhoGAP, a guanosine triphosphatase–activating protein for RhoA, acting through RhoA-dependent and -independent mechanisms. Natural or induced decreases in the abundance of p190RhoGAP triggered a series of developmental events by coupling cell-cell and cell-substratum interactions to genetic circuits controlling differentiation.
INTRODUCTION
Mechanical cues influence various cellular phenotypes, including cell morphology, proliferation, differentiation, migration, and collective cell organization (1–4). These cues include the rigidity of the surrounding matrix or cell adhesion substratum (5–7). However, it is not clear whether narrowly controlled substratum rigidity is essential for multiple tightly integrated complex processes that mediate progressive cell and tissue proliferation, differentiation, and morphogenesis during tissue development and repair. Furthermore, the molecular networks underlying the influence of substratum rigidity on diverse cell and tissue types remain poorly characterized (8).
Understanding the mechanical control of tissue development and regeneration has important practical implications. For various putative adult stem cells, survival and integration into damaged tissue after transplantation has been reported to be very modest, which might limit their use in tissue regeneration applications. Cardiosphere-derived cells (CDCs) are an example of such multipotent cells, currently in clinical trials to induce cardiac regeneration after myocardial infarct (9). CDCs, obtained from heart tissue explants (or endomyocardial biopsies of patients), can decrease infarct size and pathological cardiac remodeling and improve hemodynamics when transplanted into rodent or porcine hearts after myocardial infarction (10, 11). However, the mechanisms by which CDCs provide these beneficial effects are not well understood. In particular, it is not clear what controls their differentiation into two distinct lineages, cardiomyogenic (9) and endothelial (12), both of which contribute to the restoration of functional heart tissue.
Here, we demonstrate that when the mechanical rigidity of the cell substratum matches the rigidity of heart tissue, it can act as a pleiotropic cue that coordinately influences CDC proliferation, endothelial differentiation, and subsequent collective morphogenesis. We furthermore demonstrate that these ostensibly independent processes involved in tissue repair are tightly integrated and temporally controlled by the substratum rigidity through the same signaling pathway. Our work implicates the abundance of p190RhoGAP, a guanosine triphosphatase (GTPase)–activating protein(GAP) for RhoA, as a key factor that controls the transduction of extracellular mechanical rigidity signals and determines the resulting cellular phenotypes by acting through both RhoA-dependent and -independent mechanisms.
RESULTS
Substratum rigidity influences single-cell and collective-cell organization of CDCs
To investigate whether mechanical properties of cell adhesion substratum influence the outcome of a long-term culture of multipotent rat CDCs, we cultured these cells for 12 days on polyacrylamide gels coated with fibronectin (10 μg/ml), which were designed to have a gradient of rigidity values (Young’s modulus) ranging from 8 to 21 kPa (Fig. 1, A and B, and fig. S1A). Over the duration of the culture, cells cultured on the narrow zone of intermediate rigidity values (12 to 16 kPa), which matches the rigidity of the myocardium (13, 14) (Fig. 1B) and is referred to hereafter as the myocardium rigidity–mimicking substratum (MRS), exhibited complex and dynamic morphogenic changes, ultimately forming well-organized cellular networks more than 90% of the time, with easily identifiable rounded gaps, or lacunae (Fig. 1B), resembling cell networks frequently formed by endothelial cells on softer substrata, such as Matrigel (5). Individual CDCs assumed markedly different cell sizes within a day of seeding on homogeneous polyacrylamide substrata of different rigidities (fig. S1, B to E), whereas they assumed fibroblast-like morphology when cultured on glass coated with fibronectin (fig. S1, F and G).
Fig. 1.
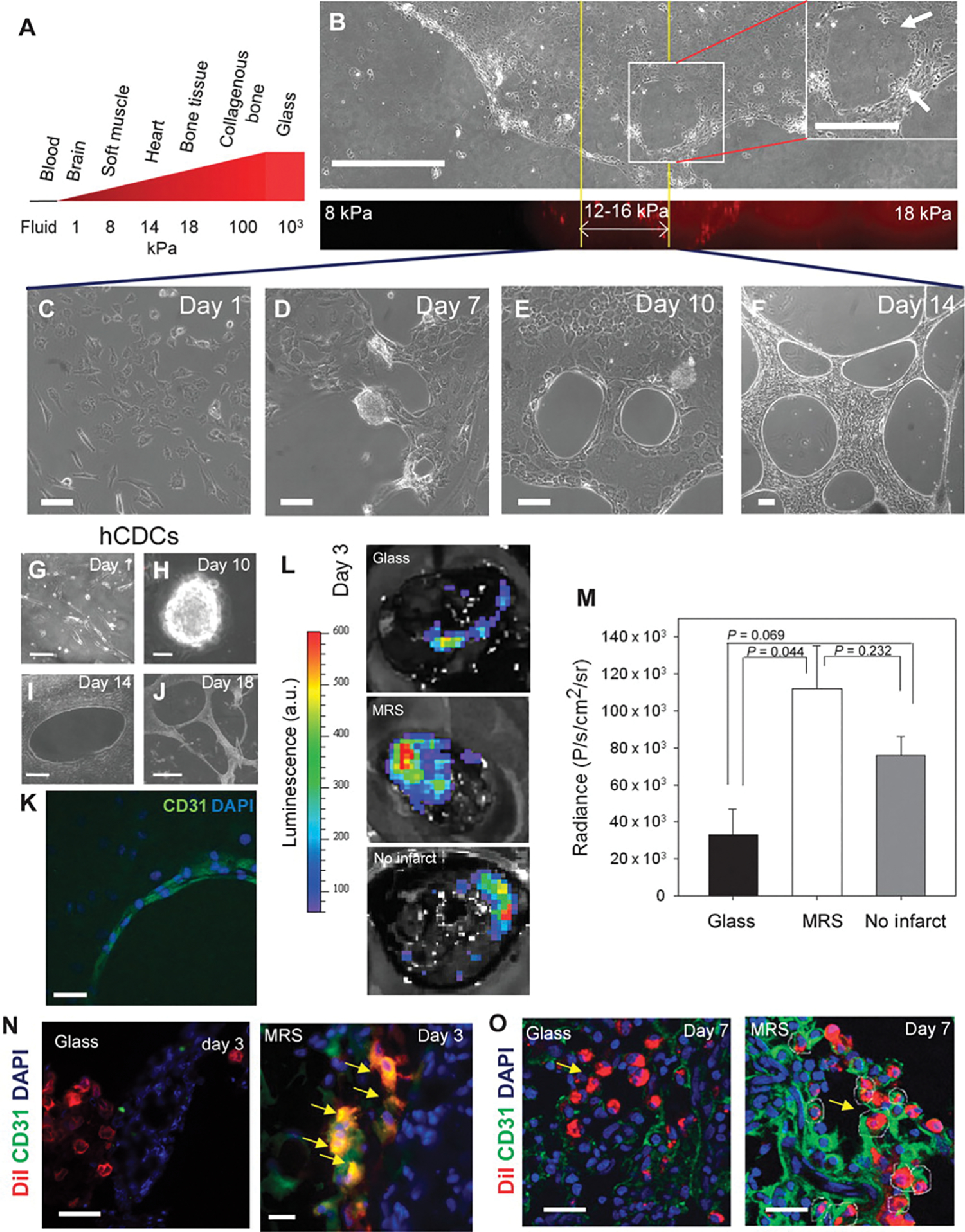
Rat CDCs undergo complex morphogenesis suggestive of endothelial differentiation when cultured on the substratum with rigidity matching that of the heart tissue. (A) Schematic showing approximate substratum rigidities of living tissues. (B) CDCs cultured for 10 days on polyacrylamide substratum with a spatial rigidity gradient form lacunae only in the rigidity range of 12 to 16 kPa. (C to F) Time-lapsed images of CDCs cultured on polyacrylamide gels of 14-kPa rigidity (MRS). (G to J) Time-lapsed images of human CDCs (hCDCs) cultured on MRS. (K) Confocal image of CD31-positive cells (green) lining lacunae. DAPI, 4′,6-diamidino-2-phenylindole. (L) Bioluminescence intensities of CDCs expressing lv-luciferase cultured on glass (left) and on MRS (center) for 7 days before injecting in infarcted rat and in healthy rat heart (right). a.u., arbitrary units. (M) Quantification of bioluminescence radiance in (I). Error bars, SEM; n = 3 rats. Statistical significance measured by paired t tests. (N and O) CDCs precultured on MRS integrate into blood vessels of rat heart. Representative images of DiI+ CDCs precultured on glass, or MRS for 7 days before injection at the site of myocardial infarct in rats. Images were acquired 3 days (N) and 7 days (O) after injection. DiI+CD31+ CDCs observed in 66% of sections analyzed for (N) (n = 12 from 3 rats) and in 25% of sections analyzed for (O) (n = 12 from 3 rats). Yellow arrowheads show DiI+CD31+ CDCs integrated in a lumen. Scale bars, 250 μm (A), 20 μm (C to F), 50 μm (G, K, N, and O), 100 μm (H), 250 μm (I), and 1 μm (J).
More dynamically resolved analysis revealed that CDCs attached to spatially homogeneous MRS, spread within a day (Fig. 1C), proliferated to create a monolayer within 3 days, forming multiple semi-adherent cell spheroid aggregates containing tightly packed and proliferating cells (movies S1 and S2). The spheroids ultimately dispersed, through outflow of streams of adherent cells by day 7 of culture (Fig. 1D), which was accompanied by formation of organized cellular networks (movies S1 to S4). The networks took a defined shape by day 10, with cells forming rounded gaps tightly juxtaposed with each other (Fig. 1E), but continued to spread and increase in density (Fig. 1F and fig. S1, H and I) over 18 days of culture (fig. S1I). Furthermore, CDCs cultured on MRS for 10 days formed three-dimensional (3D) cord networks when detached and reseeded in Matrigel (fig. S1J), whereas those cultured on glass did not (fig. S1K), suggesting that a large fraction of endothelial cells emerged within the population of CDCs cultured on MRS. The results were also recapitulated for human CDCs extracted from endomyocardial biopsies, albeit with delayed kinetics (Fig. 1, G to J). Individual human CDCs often curved to form looped structures, similar to those observed for endothelial cells (15) 7 days after seeding, further supporting endothelial characteristics of the emerging cell population (fig. S1L).
Rigidity cue enhances the abundance of endothelial markers in vitro and enhances cell survival and vascular integration in vivo
The emergence of endothelial cells from CDCs on MRS was further supported by the observation that cells lining the lacunae were positive for the endothelial markers CD31 [also known as PECAM-1 (platelet endothelial cell adhesion molecule-1) (16)] (Fig. 1K) and VEGFR2 (vascular endothelial growth factor receptor 2) (fig. S2A). Using a rat model of myocardial infarction, we explored the in vivo fate of CDCs lentivirally transduced with luciferase driven by the cytomegalovirus (CMV) promoter (CDC-lv-luciferase) and precultured on glass or on MRS for 7 days. A significantly higher number of CDCs cultured on MRS survived in the infarct 2 days after injection compared to those cultured on glass (Fig. 1, L and M). The ischemic microenvironment in the infarct was a major obstacle to cell survival, as also evidenced by the higher number of transplanted CDCs that survived after injection in a healthy heart (Fig. 1, L and M). Furthermore, cells precultured on MRS, but not on glass, gave rise to CD31+ cells by day 3 after injection (Fig. 1N) and occasionally displayed more pronounced integration into cardiac blood vessels by 7 days after injection (Fig. 1O).
Enrichment of CD31+ endothelial cells was also observed when human and rat CDCs were cultured on MRS, but not substrata on other rigidities (Fig. 2A and fig. S2, B and C). These cells also exhibited increased mRNA abundance of two transcription factors associated with endothelial differentiation, GATA2 (Fig. 2B and fig. S2D) and TFII-1 (Fig. 2B); increased abundance of the angiopoietin receptor Tie2 (fig. S2E); and increased binding to tomato lectin [which recognizes glycophorin, a glycoprotein abundant on endothelial cell walls (17)] (fig. S2F) after 7 days of culture on MRS, but not on glass. Silencing GATA2 (GATA2KD CDCs) disrupted the formation of cellular networks or lacunae on MRS in all samples tested (fig. S2G), suggesting that GATA2 is essential for the morphogenesis of endothelial cell–like lacunae. These data suggested that an appropriate rigidity cue enhances both cell survival and tissue integration of CDCs.
Fig. 2.
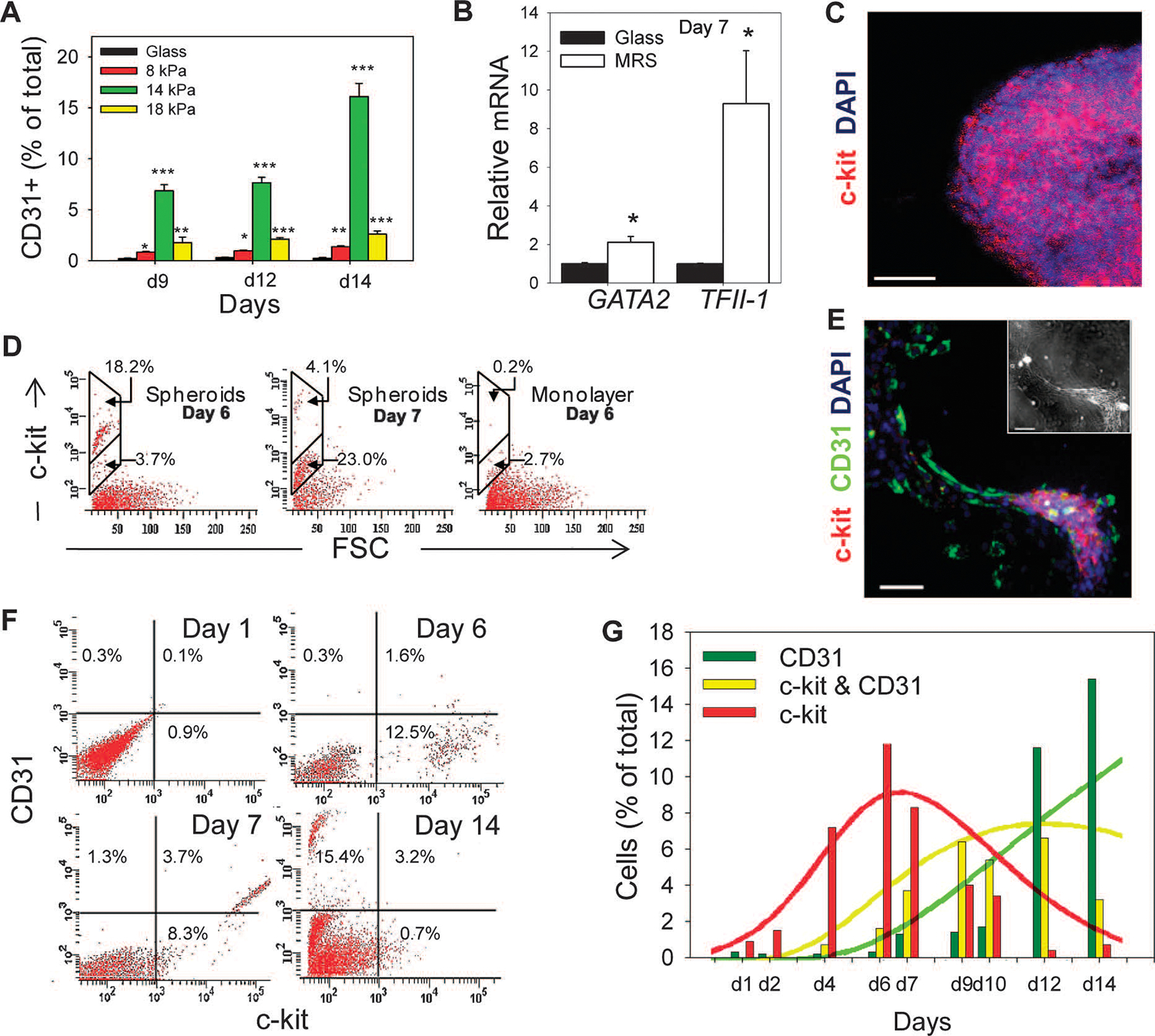
Substratum rigidity directs endothelial differentiation of CDCs. (A) Time course flow cytometric analysis of CDCs cultured on substrata of different rigidities. (B) Reverse transcription–PCR (RT-PCR) analysis of GATA2 and TFII-1 abundance in CDCs cultured on MRS and on glass. (C) Confocal microscopy shows high abundance of c-kit in spheroids. (D) Distribution of c-kit+ cells in CDCs cultured on MRS in mechanically isolated spheres 6 days (left) and 7 days (center) after cell seeding and from remaining adherent cells (right). (E) Immunostaining of CDCs cultured on MRS for 7 days for c-kit in cell aggregates and for CD31 in lacunae-forming cells (see inset for phase contrast). (F and G) Time course flow cytometric analysis of c-kit and CD31 abundance in CDCs cultured on MRS (F). Results quantified in (G) as percentages of c-kit+/CD31− (green), c-kit+/CD31+ (yellow), and c-kit−/CD31+ (red) cells in the total population (bars; based on the quadrant definition in (F). Results of the computational simulation of CDC proliferation and differentiation color-coded similarly (n = 2 cultures). Scale bars, 50 μm (C and E). *P < 0.05; **P < 0.01; ***P < 0.001. Error bars, SEM.
Cardiac tissue matching promotes endothelial differentiation
CDCs contain a small subpopulation of cells positive for c-kit (9) [also known as the stem cell factor receptor (SCFR) (18, 19)]. This cell subpopulation is a putative source of progenitors for endothelial and myocardial cell differentiation. We investigated whether the semi-adherent spheroids that formed on MRS, which were the apparent source of endothelial cells in our experiments, contained c-kit+ cells. Confocal microscopy of cytospinned spheroids and flow cytometry of mechanically isolated spheroids revealed that c-kit+ cells were enriched in these cell aggregates on day 6 (Fig. 2, C and D) and that c-kit abundance dropped by day 7 (Fig. 2D). In contrast, there were very few c-kit+ cells in the remaining cells in the monolayer (Fig. 2D).
We observed close apposition of c-kit+ spheroids and CD31+ cells within the cell streams emerging from these spheroids (Fig. 2E). Time course analysis of c-kit and CD31 abundance indicated that culture on MRS induced an increase in c-kit+ cells that peaked on day 6 (Fig. 2, Fand G, red bars), with this subpopulation then giving rise to a c-kit+ and CD31+ (c-kit+/CD31+) subpopulation, which peaked in percentage by day 9 (Fig. 2G, yellow bars). These cells eventually decreased in relative number coincident with emergence of c-kit−/CD31+ cells (Fig. 2G, green bars). The dynamics of the observed waves of sequential c-kit+, c-kit+/CD31+, and CD31+ cells were captured by a simple mathematical model (text S1), which accounted for both proliferation and differentiation of the initial progenitor population (Fig. 2G, solid lines) and was consistent with the hypothesis that MRS promoted endothelial differentiation of c-kit+ CDCs through an intermediate c-kit+/CD31+ state. EdU (5-ethynyl-2′-deoxyuridine) staining indicated that proliferative cells were largely confined to spheroid cell aggregates (Fig. 3A and fig. S2H). Formation of spheroid aggregates coincided with a sudden increase in proliferation after day 6, which was not observed when CDCs were cultured on softer or more rigid substrata, as indicated by the degree of extracellular reduction of NADH (reduced form of NAD+ nicotinamide adenine dinucleotide) produced by mitochondrial respiration (Fig. 3B). To further test whether c-kit+ cells differentiate into CD31+ endothelial cells, we sorted c-kit+ and c-kit− cells from CDCs cultured on MRS for 6 days and recultured them on MRS separately for 8 days. Flow cytometry revealed enhanced differentiation of c-kit+ cells into CD31+ cells (Fig. 3, C and D).
Fig. 3.
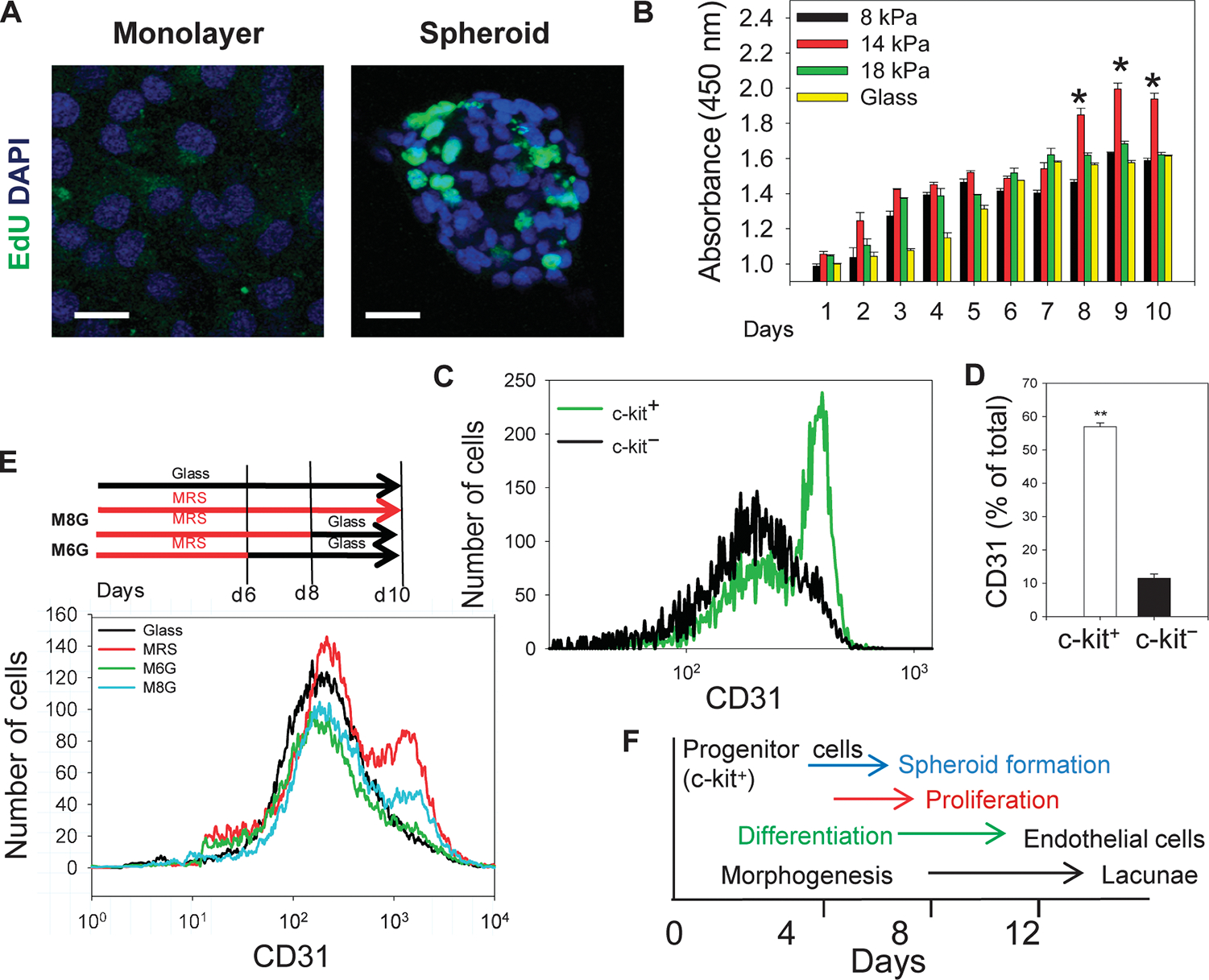
A persistent rigidity cue is required for proliferation and endothelial differentiation of CDCs. (A) Confocal imaging of EdU staining in CDCs in a monolayer and a spheroid. Scale bars, 20 μm. (B) CDCs cultured on MRS, but not substrata, of different rigidities show a sudden increase in proliferation as assessed by the WST-8 assay. (C) Flow cytometric analysis of CD31 abundance after 7 days of culture on MRS in c-kit+ and c-kit− sorted cells from CDCs cultured on MRS for 7 days. (D) Quantification of (C). (E) Persistent rigidity sensing is required for differentiation. Schematic of experimental procedure. (Top) CDCs were cultured on MRS or on glass for 10 days (Glass) and on MRS for 6 days (M6G) and 8 days (M8G) followed by transfer onto glass for the remainder of the 10-day period. (Bottom) Experimental results of CD31 flow cytometry. (F) Schematic summarizing the time distribution of spheroid formation, cell proliferation, differentiation, and cell network morphogenesis. *P < 0.05; **P < 0.01. Error bars, SEM.
To test whether substratum rigidity as an external cue was continuously required for endothelial differentiation and collective morphogenesis of CDCs (as opposed to rigidity sensing only at the time of the initial cell plating), we transiently cultured CDCs on MRS (for 0, 6, 8, and 10 days) and then transferred them by means of a nonenzymatic dissociation method onto glass for the remainder of the experiment (10, 4, 2, and 0 days, respectively) (Fig. 3E and fig. S2, I to L). We then reseeded the cells on Matrigel and determined their capacity to form 3D networks (fig. S2, I to L). As duration of the incubation on MRS decreased, progressively fewer CD31+ cells were formed (Fig. 3E). The degree of cell network formation on Matrigel, although not analyzed, also visually correlated with the duration of preculture on MRS, with cells cultured entirely on glass forming no detectable networks, whereas those precultured for 6, 8, or 10 days of MRS formed progressively more mature networks (fig. S2, I to L). These results suggest that physiologically relevant substratum rigidity could indeed control temporally overlapping processes in c-kit+ precursor cells, including the proliferation in spheroids, endothelial differentiation, and collective cell morphogenesis into lacunae, because these cells continuously monitored the mechanical microenvironment (Fig. 3F).
Substratum rigidity regulates dynamic changes in the abundance of p190RhoGAP, YAP, and p120-catenin
Together, the results described above indicate a strong influence of substratum rigidity on the emergence of both c-kit+ endothelial cell precursors and their endothelial differentiation through a complex set of morphogenetic events (Fig. 1, C to F). We sought to explore the molecular mechanisms underlying these processes. Substratum rigidity sensing is thought to both depend on and regulate integrin-nucleated focal adhesion complexes (20). As substrate rigidity increases, the abundance and recruitment of integrins to sites of adhesion also increase (21, 22). The abundance of β1 and β3 integrin (Fig. 4A and fig. S3A) was lower in CDCs cultured for 3 days on MRS compared to that in glass, consistent with the finding that cells assume a smaller surface area on MRS (fig. S1, D, F, and G). Notably, the distribution of integrin molecules showed higher variance in cells cultured on MRS than on glass, suggesting considerable “noise” in the degree of integrin regulation (Fig. 4A and fig. S3A). Actin stress fibers and focal adhesions appeared to be more mature in cells cultured on MRS (fig. S3B). The abundance and phosphorylation of focal adhesion kinase (FAK) decreased in cells cultured for 3 days on MRS as compared to glass (fig. S3, C and D). Downstream signaling through FAK can enhance cytoskeletal reorganization, affecting both the cell shape and cell-substratum interactions (23). Total abundance and phosphorylation of myosin light chain (MLC) increased more than threefold in CDCs cultured on MRS compared to those cultured on glass (fig. S3, C and E). Signaling through the GTPase RhoA results in phosphorylation and activation of MLC (24), and RhoA activity was higher in cells cultured on MRS (fig. S3F). Focal adhesion–mediated signaling has been suggested to enhance Rac activation (25, 26), which, in turn, can negatively crosstalk with RhoA by activating RhoA-specific GAPs, such as p190RhoGAP (27, 28). p190RhoGAP also interacts directly with phosphorylated FAK (29–31) and has been implicated in substratum rigidity sensing (31–33). Immunoprecipitation assays revealed that p190RhoGAP formed a complex with phosphorylated FAK (fig. S3G). Neutralizing antibody against β1 integrin, but not β3 integrin, decreased p190RhoGAP amounts in CDCs, indicating that the abundance of p190RhoGAP may be regulated by integrin-mediated signaling (Fig. 4B). Both the protein and the mRNA abundance of p190RhoGAP (Fig. 4, C and D, and fig. S3H) were significantly decreased in CDCs cultured for 7 days on MRS compared to glass. Immunostaining for p190RhoGAP in CDCs cultured for 7 days on glass showed diffuse p190RhoGAP localization in the cytosol, lamellipodia, and band formations at the cell membrane (Fig. 4E), suggestive of focal adhesion organization and enhanced FAK activity and FAK-p190RhoGAP complex formation on glass (fig. S3G). The membrane localization of p190RhoGAP decreased in cells cultured on MRS (Fig. 4E), suggesting locally high activity of RhoA at lamellipodia or other membrane structures, which might destabilize focal adhesions and decrease cell spreading leading to relatively smaller cell area (fig. S1G) (34). Time course analysis of p190RhoGAP abundance over 10 days suggested that it dropped after 4 to 6 days of CDC culture on MRS, which coincided with cell aggregate formation (Fig. 4F and fig. S3I).
Fig. 4.
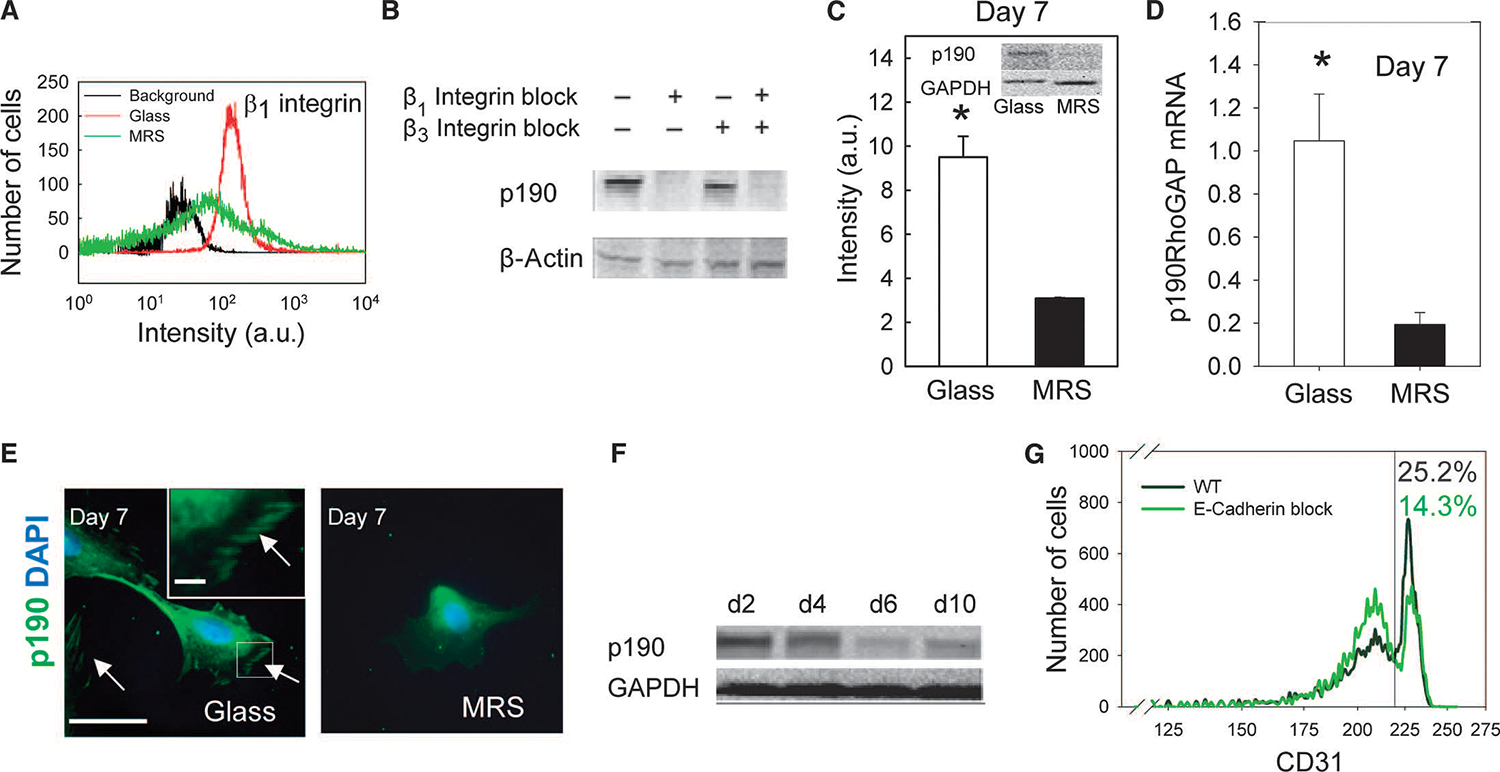
Substratum rigidity regulates focal adhesion–mediated RhoA and p190RhoGAP signaling. (A) Flow cytometric analysis of β1 integrin in CDCs after 3 days of culture. (B) Immunoblotting of p190RhoGAP in CDCs cultured on glass in the presence of blocking antibodies against β1 or β3 integrin for 3 days. β-Actin was used as the control. (C) Immunoblotting analysis of p190RhoGAP in CDCs cultured on MRS or glass for 7 days. Representative bands shown in inset. n = 3. (D) RT-PCR analysis of p190RhoGAP mRNA in CDCs. (E) Immunostaining of p190RhoGAP in CDCs on glass (left) or MRS (right) 7 days after seeding; localized staining indicated by arrows. n = 4 cultures for each condition. Scale bars, 50 μm. (F) Immunoblotting analysis of p190RhoGAP in CDCs cultured on MRS for different periods of time. Lower panel shows GAPDH control. (G) Flow cytometric analysis of CD31 abundance in CDCs cultured on MRS without (control, green) and with neutralizing E-cadherin antibody (black). Quantification of gated CD31+ cells shown in inset. n = 3 samples. *P < 0.05. Error bars, SEM.
That formation of cell spheroids preceded endothelial differentiation suggested the importance of cell-cell adhesion molecules, particularly E-cadherin. Indeed, a subpopulation of CDCs cultured on MRS showed a punctate distribution of E-cadherin, indicating cell-cell junction formation (35) (fig. S3, J to L). Neutralizing antibody against E-cadherin decreased cell differentiation, further suggesting the crucial importance of transient cell aggregation in regulation of CDC differentiation (Fig. 4G). The downstream effector of E-cadherin, p120-catenin, promotes cell-cell contact formation (36, 37), and the abundance of p120-catenin was transiently increased during the cell aggregation stage (Fig. 5A and fig. S4A). Knocking down p120-catenin inhibited spheroid formation and cell-network morphogenesis, and cells appeared to assume a polarized morphology (fig. S4, B and C). Consistent with findings in other systems (36), p120-catenin formed a complex with p190RhoGAP (fig. S4D) and was colocalized with p190RhoGAP (Fig. 5B and fig. S4E). Knockdown of p190RhoGAP increased the abundance of p120-catenin (Fig. 5C and fig. S4F), suggesting that p190RhoGAP inhibited p120-catenin by increasing its cytosolic localization and decreasing its protein abundance. The abundance of p120-catenin increased by day 6 and then decreased by day 9 (Fig. 5D and fig. S4G), with the latter coinciding with spheroid dispersal to form lacunae (Fig. 1, D and E). Thus, the decrease in p190RhoGAP abundance during spheroid formation can transiently increase p120-catenin amounts, further enhancing cell-cell contacts. Knocking down GATA2 led to modestly increased p120-catenin abundance in MRS-cultured cells (fig. S4H). This finding suggested that cell-cell contacts may ultimately weaken as a result of an increase in GATA2 abundance, which could antagonize p120-catenin, leading to a decrease in its abundance. The weakening of cell-cell contacts may account for dispersal of spheroids concurrently with endothelial differentiation.
Fig. 5.
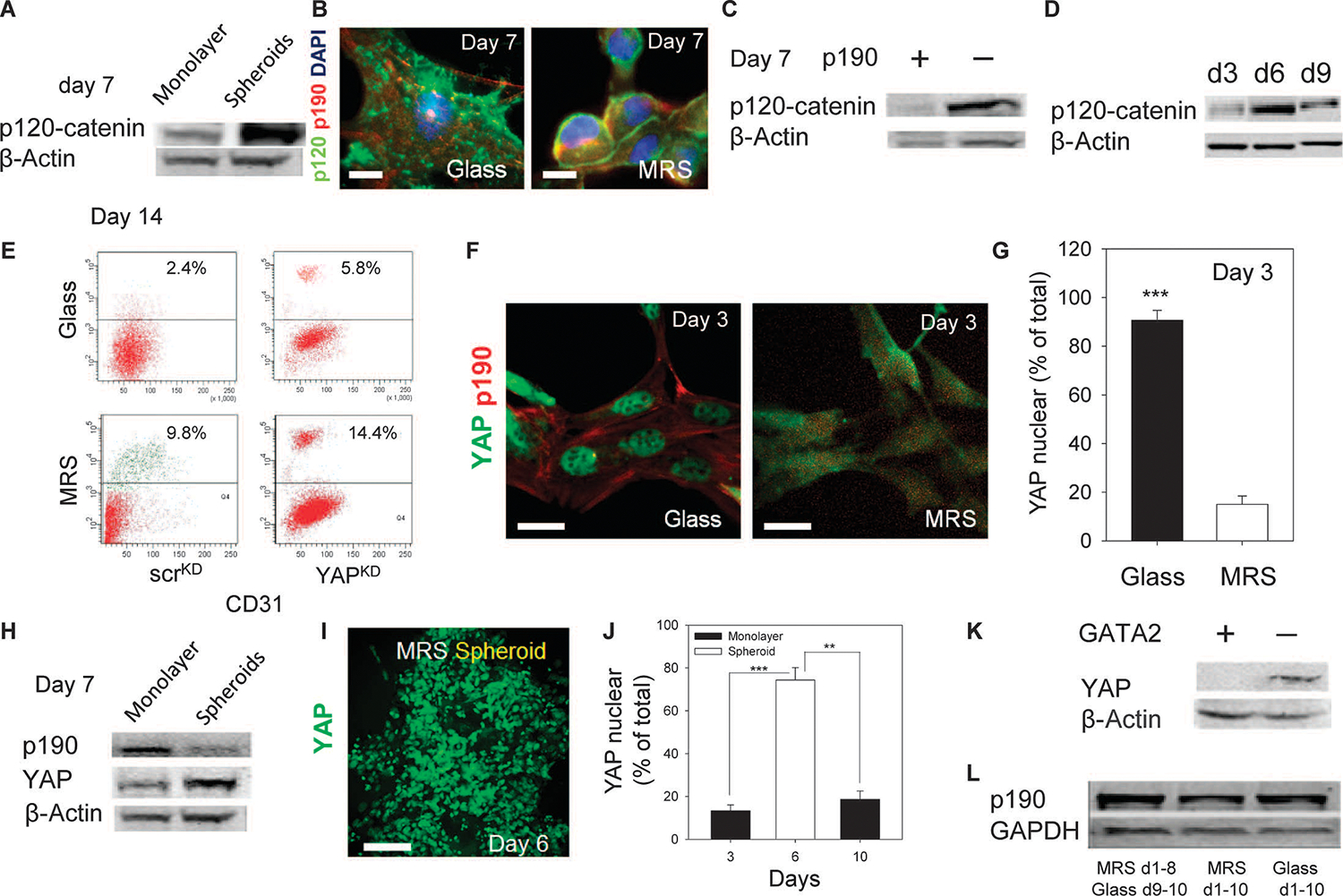
Substratum rigidity regulates the abundance and localization of p120-catenin and YAP. (A) p120-catenin abundance in CDCs cultured on MRS in spheroids, and remaining adherent cells. (B) Immunostaining for p120-catenin and p190RhoGAP in CDCs cultured on glass (left) or on MRS (right). Colocalization analysis is shown in fig. S4E. (C) p120-catenin abundance in CDCs transfected with control scrambled small interfering RNAs (siRNAs) (scrKD) or siRNAs directed against p190RhoGAP (p190KD). (D) Time course analysis of p120-catenin abundance in CDCs cultured on MRS. (E) Flow cytometric analysis of CD31 in scrKD and YAPKD CDCs. n = 2 cultures. (F) Immunostaining reveals that YAP has a predominantly nuclear localization in CDCs cultured on glass and a cytosolic distribution in CDCs cultured on MRS. (G) Quantification of nuclear localization in (F). n = 3 samples. (H) Analysis of p190RhoGAP, YAP, and p120-catenin abundance in cell monolayers and spheroid cell aggregates cultured on MRS. (I) Two-photon imaging of YAP in a CDC spheroid. (J) Quantification of nuclear localization of YAP. (K) Immunoblotting analysis of YAP in scrKD and GATA2KD CDCs. (L) p190RhoGAP abundance in CDCs cultured on MRS for 8 days followed by 2 days on glass, or cells cultured on MRS and glass for 10 days. Scale bars, 20 μm (B and F) and 100 μm (I). P values determined by paired t tests, **P < 0.01; ***P < 0.001. Error bars, SEM.
The transcriptional coactivator YAP (Yes-associated protein) is a member of the Hippo signaling pathway that has been implicated in both mechanosensing and regulation of cell survival and proliferation under various cell-cell and cell-substratum contact conditions (38). Knockdown of YAP (fig. S4I) augmented endothelial cell differentiation (Fig. 5E). In agreement with previous data, YAP localized to the nuclei in cells cultured on glass, but was cytosolic in cells cultured on MRS before spheroid formation (Fig. 5, F and G, and fig. S4J), suggesting a correlation between YAP localization and substrate rigidity. YAP abundance (Fig. 5H and fig. S4, J and K) and nuclear localization (Fig. 5, I and J, and fig. S4J) increased during spheroid formation, in agreement with its possible activation due to engagement of E-cadherin, as previously proposed (38, 39). The timing of this transient increase coincided with the increase in cell proliferation and the peak in c-kit+ cell numbers (Fig. 2, F and G), supporting an important role of YAP in progenitor cell proliferation (40). Further, p190RhoGAP knockdown modestly increased YAP abundance (fig. S4L). Although the reasons for subsequent decrease in YAP abundance were not clear, we found that GATA2 knockdown led to an increase in the amount of YAP (Fig. 5K), suggesting an antagonistic relationship between these two proteins. Thus, the increase in GATA2 expression at the point of emergence of CD31+ cells (Fig. 2B) may decrease YAP abundance when cells reengage contact with the substratum. As noted above, decreases in YAP abundance enhanced endothelial differentiation (Fig. 5E).
The findings described above suggested that the transient formation of spheroid cell aggregates was important for enhanced cell proliferation and differentiation, but it was unclear whether cells in these aggregates were continuously sensitive to the effects of substratum rigidity and whether cell-cell interactions other than those triggered by immediate cell-cell contact were important during this stage of the process. When CDCs were reseeded on glass after an initial culture on MRS, p190RhoGAP abundance increased and CD31 cell surface abundance decreased compared to CDCs continuously cultured on MRS (Fig. 5L and fig. S4M). These results suggest that continuous monitoring of the substratum rigidity can lead to gradual reversible decrease in p190RhoGAP abundance in a subpopulation of CDCs, which correlates with initiation of endothelial differentiation and morphogenesis. Further, CDCs cultured on MRS for 14 days and perfused every 8 hours with fresh medium did not show decreased differentiation into CD31+ cells, suggesting a limited role of paracrine signaling in cellular phenotypes (fig. S4, N and O).
p190RhoGAP regulates substratum rigidity–mediated endothelial differentiation of CDCs
Our findings suggested that decreased p190RhoGAP abundance was critical for endothelial differentiation. Indeed, p190RhoGAP silencing by short hairpin RNA (shRNA) in CDCs cultured on MRS (fig. S5, A and B) significantly increased the percentage of CD31+ cells (Fig. 6A), increased GATA2 protein abundance (Fig. 6B), and increased mRNA abundance for GATA2 (Fig. 6C) and TFII-1 (Fig. 6C) compared to the scrambled shRNA control cells cultured on MRS. Further, GATA2 mRNA abundance was higher in p190RhoGAP knockdown cells than in control cells after 10 days of culture both on MRS and on glass (Fig. 6D).
Fig. 6.
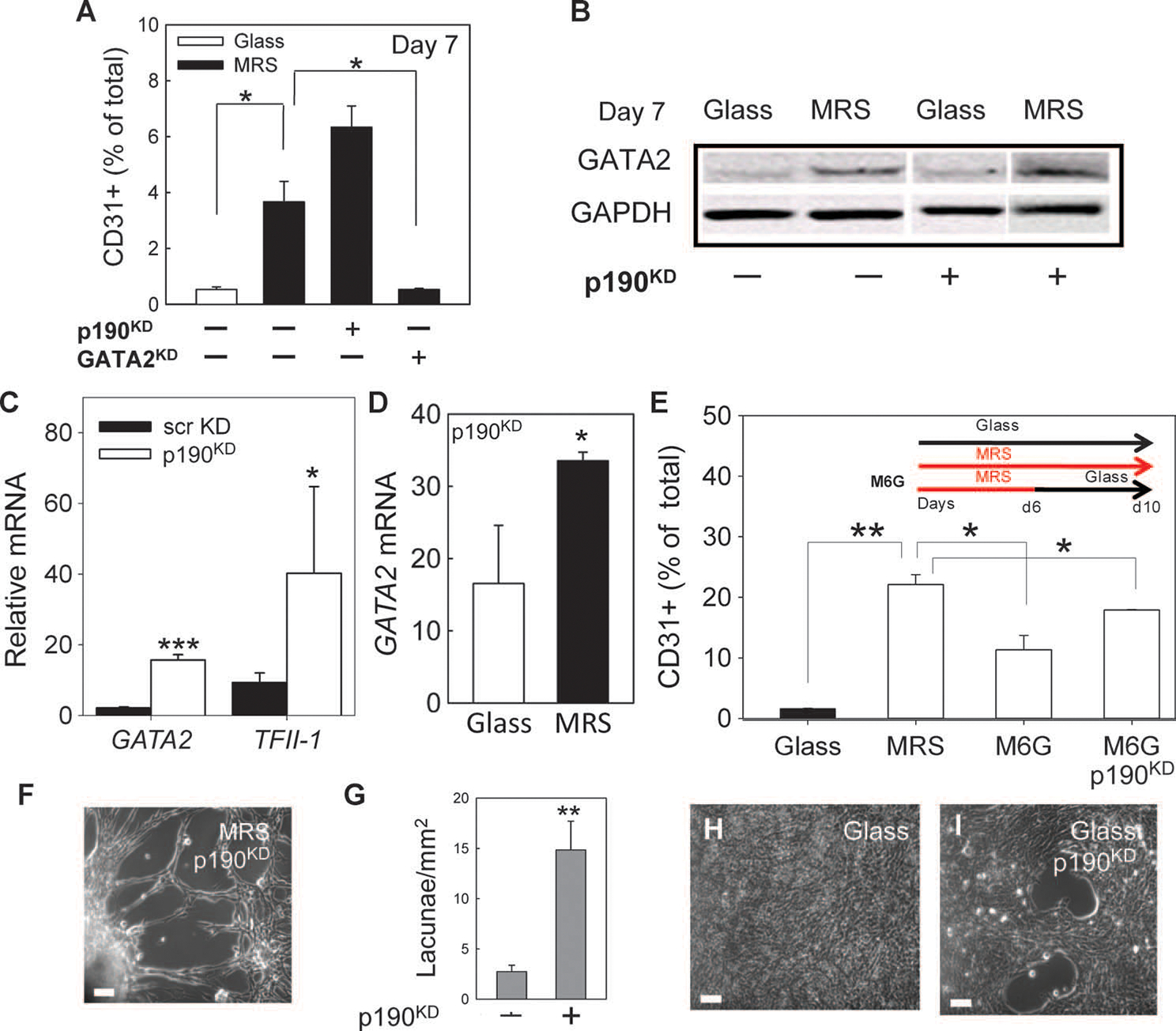
Decreased abundance of p190RhoGAP promotes endothelial differentiation and morphogenesis into cellular networks. (A) Flow cytometric analysis of CD31+ scrKD, p190KD, and GATA2KD CDCs. (B) Immunoblotting analysis of GATA2 in scrKD and p190KD CDCs. Noncontiguous lanes from a single experiment are indicated by white lines. (C) RT-PCR analysis of GATA2 and TFII-1 mRNA in p190KD CDCs relative to scrKD CDCs cultured on MRS. (D) RT-PCR analysis of GATA2 mRNA in p190KD CDCs cultured on glass relative to scrKD cultured on glass (left bar) and in p190KD CDCs cultured on MRS relative to scrKD CDCs cultured on glass (right bar). (E) Flow cytometric analysis of CD31 in scrKD CDCs cultured for 10 days on glass or on MRS and in scrKD and p190KD cells cultured for 6 days on MRS followed by 4 days on glass (M6G). (F) p190KD cells form lacunae network on MRS within 10 days. (G) Quantification of lacunae density formed by p190KD and scrKD cells on MRS. n = 3 samples. (H and I) Cell organization after 10 days of culture of scrKD (H) and p190KD (I) CDCs on glass. Scale bars, 20 mm (F, H, and I). *P < 0.5; **P < 0.01; ***P < 0.001. Error bars, SEM.
We further investigated whether p190RhoGAP silencing could also compensate for the loss of rigidity-sensing signaling. p190RhoGAP knockdown cells were cultured on MRS for 6 days (when peak formation of spheroids occurred) and transferred for further culture to glass for an additional 4 days (Fig. 6E). Flow cytometry revealed that cells continued to differentiate into CD31+ endothelial cells, in contrast to control cells that gave rise to a significantly smaller CD31+ subpopulation (Fig. 6E). Following transfer onto Matrigel after 6 days of culture on MRS and an additional 4 days of culture on glass, p190RhoGAP knockdown cells continued to form lacunae networks in all viable samples (fig. S5, C and D). p190RhoGAP silencing also led to the formation of denser and more pronounced cell networks on MRS after 10 days of culture (Fig. 6, F and G). In contrast to the control cells that formed unstructured cell monolayers in all observed cases (Fig. 6H), p190RhoGAP knockdown cells occasionally formed cell networks even on glass (Fig. 6I).
We next explored whether decreasing p190RhoGAP abundance enhances endothelial differentiation in vivo and promotes integration of CDCs into the host myocardium. Analysis of tissue 3 weeks after the injection of cells labeled with DiI, a lipophilic membrane stain retained in cells for many weeks, indicated that whereas the integration of DiI+CD31+ wild-type CDCs in blood vessels (fig. S6A) appeared to be limited, the integration of DiI+CD31+ p190RhoGAP knockdown CDCs appeared to occur more frequently (fig. S6B).
To investigate whether the kinetics of endothelial differentiation of CDCs could be modulated by p190RhoGAP abundance, we cultured p190RhoGAP knockdown cells on MRS for 14 days and investigated them by flow cytometry (Fig. 7, A to C, and fig. S7, A to D). In contrast to unperturbed cells (Fig. 2, F and G), p190RhoGAP knockdown cells displayed accelerated kinetics of differentiation, including an earlier onset of proliferation of c-kit+ cells (by day 4), earlier emergence of c-kit+/CD31+ cells, and earlier emergence of c-kit−/CD31+ cells (Fig. 7D and fig. S7E). To gain further insight into the dynamics of CDC subpopulations, we simulated the mathematical model described above over a wide range of parameters (fig. S7D). Although separately varying the rates of proliferation and differentiation of c-kit+ and c-kit+/CD31+ cells captured some of the observed changes in the subpopulation dynamics—for example, a higher peak for c-kit+ cells (Fig. 7A and fig. S7A) and an earlier emergence of c-kit+/CD31+ cells (Fig. 7B and fig. S7B)—only when both the proliferation and the differentiation rates were increased (Fig. 7C and fig. S7C) did the predictions closely match with the experimental data (Fig. 7D).These results suggest that MRS-induced decrease in p190RhoGAP abundance can increase both proliferation and differentiation of c-kit+ and c-kit+/CD31+ cells.
Fig. 7.
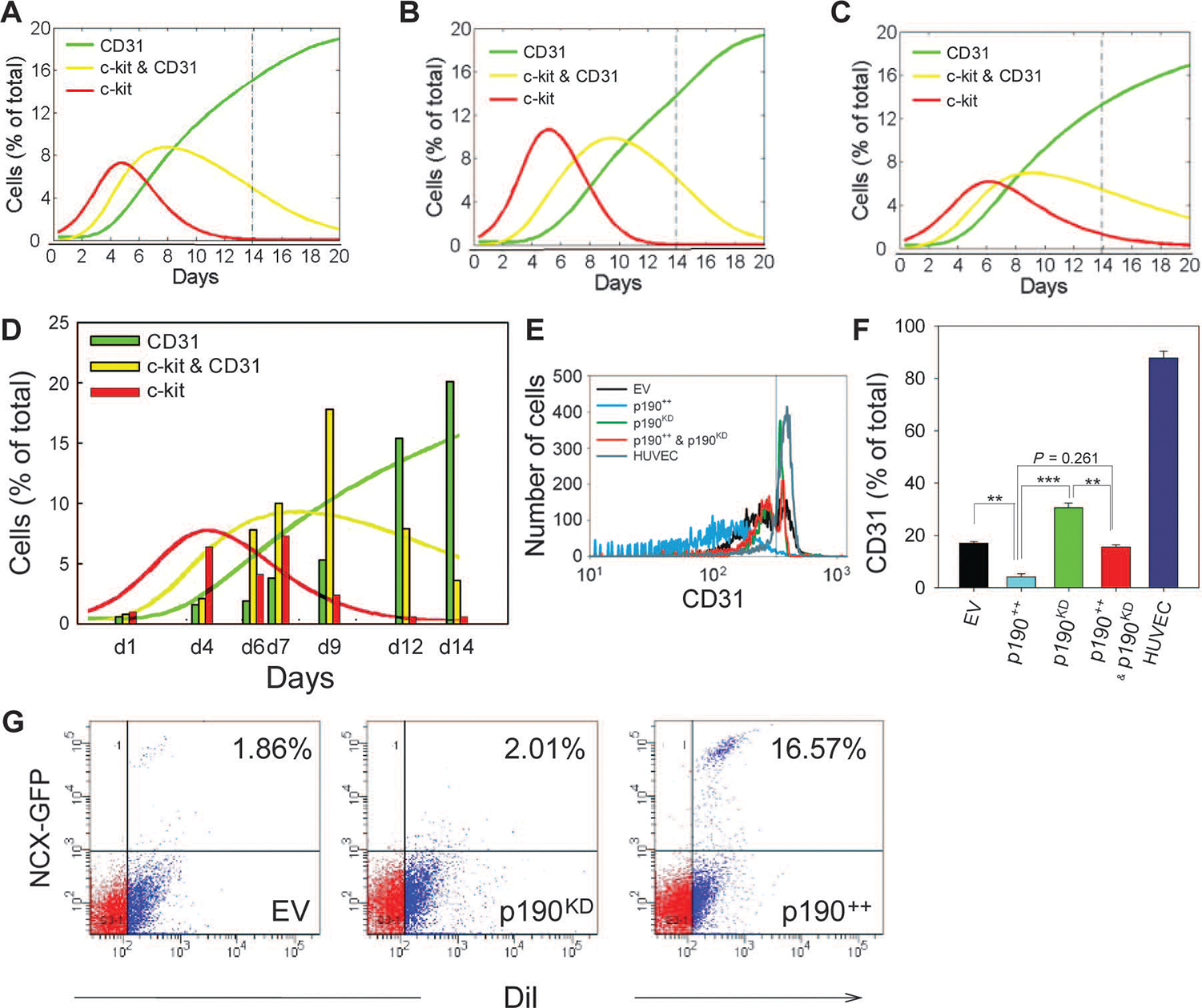
p190RhoGAP regulates MRS-induced CDC proliferation and endothelial differentiation. (A to D)Computational model and experimental analysis of CDC proliferation and differentiation. Representative simulation results showing effects of enhanced proliferation and differentiation (see also fig. S7), with increased proliferation rates for c-kit /CD31 and c-kit+/CD31+ cells (A), increased differentiation rates for c-kit+/CD31− and c-kit+/CD31+ cells (B), and increased rates for both proliferation and differentiation (C). Time course flow cytometry analysis of p190KD CDCs (vertical bars) (D). Overlaid solid lines show predictions of computational model calculated in (C). (E) Flow cytometric analysis of CD31 abundance of CDCs transduced with empty SV40 plasmid (EV, black), p190RhoGAP overexpression vector (p190++, blue), p190KD CDCs (green), p190++CDCs rescued with p190RhoGAP shRNA (p190++ and p190KD, red), and human umbilical cord endothelial cells (HUVEC) (cyan). (F) Quantification of percentage of CD31+ cells in (E). (G) Flow cytometric analysis of DiI-labeled CDC-lv-NCX-GFP cells transduced with empty SV40 expression vector (EV), p190RhoGAP overexpression vector (p190++), and p190RhoGAP shRNA (p190KD) in coculture with NRVMs for 10 days on MRS (n = 2 samples). Gates were created with negative control samples for DiI− and wild-type CDCs. In all panels: *P < 0.05; **P < 0.01. Error bars, SEM.
The crucial role of decreased p190RhoGAP abundance in the regulation of endothelial differentiation of CDCs was further supported by ectopic overexpression of p190RhoGAP through lentiviral transfection. Transduced cells cultured on MRS for 14 days displayed significantly reduced endothelial differentiation (Fig. 7, E and F). Surprisingly, cells overexpressing p190RhoGAP also displayed increased amounts of early cardiomyogenic protein markers including Nkx2.5 (fig. S8, A and D), cardiac troponin I (fig. S8B), and connexin-43 (fig. S8, C and D). When cells were cocultured on MRS in the presence of neonatal rat ventricular myocytes (NRVMs), p190RhoGAP abundance correlated with expression of green fluorescent protein (GFP) driven by the promoter of the NCX gene, a late cardiomyogenic marker (Fig. 7G). DiI-labeled CDCs overexpressing p190RhoGAP injected into a rat heart also appeared to show integration with the host myocardium and increased cardiac troponin I abundance (fig. S8E).
p190RhoGAP mediates its effects through both RhoA-dependent and -independent signaling
p190RhoGAP is a direct regulator of RhoA activity, and RhoA has previously been implicated in rigidity sensing (41–43). We therefore questioned whether the enhanced precursor cell proliferation and differentiation observed in CDCs when cultured on MRS were regulated through RhoA. Indeed, RhoA activity was higher in cells on MRS compared to that on glass (fig. S3F). Furthermore, enhancing RhoA activity with a pharmacological activator, CN01, resulted in a higher percentage of CD31+ cells after 10 days of culture (Fig. 8A), consistent with the potential role of RhoA in controlling endothelial differentiation. However, this effect of RhoA could be attributed to either enhanced proliferation of precursor cell subpopulations or enhanced differentiation, or both. Therefore, we assessed proliferation in the presence of RhoA activity modifiers with the WST-8 assay. We observed that CN01 treatment increased cell proliferation in control cells cultured for 6 days on either glass or MRS, indicating that RhoA can enhance the proliferation of these cells (Fig. 8B). Similarly, p190RhoGAP knockdown enhanced proliferation of cells cultured on glass, an effect that was further enhanced on MRS (Fig. 8B). To decouple the proliferative effect of p190RhoGAP silencing from RhoA activation, we analyzed the proliferation of p190RhoGAP knockdown cells cultured on MRS and treated with a RhoA inhibitor, CT04, starting 6 days after cell seeding, during formation of spheroid cell aggregates. Proliferation of CDCs was inhibited after CT04 treatment, indicating that inhibition of RhoA reduces cell proliferation even after p190RhoGAP was silenced (Fig. 8B).
Fig. 8.
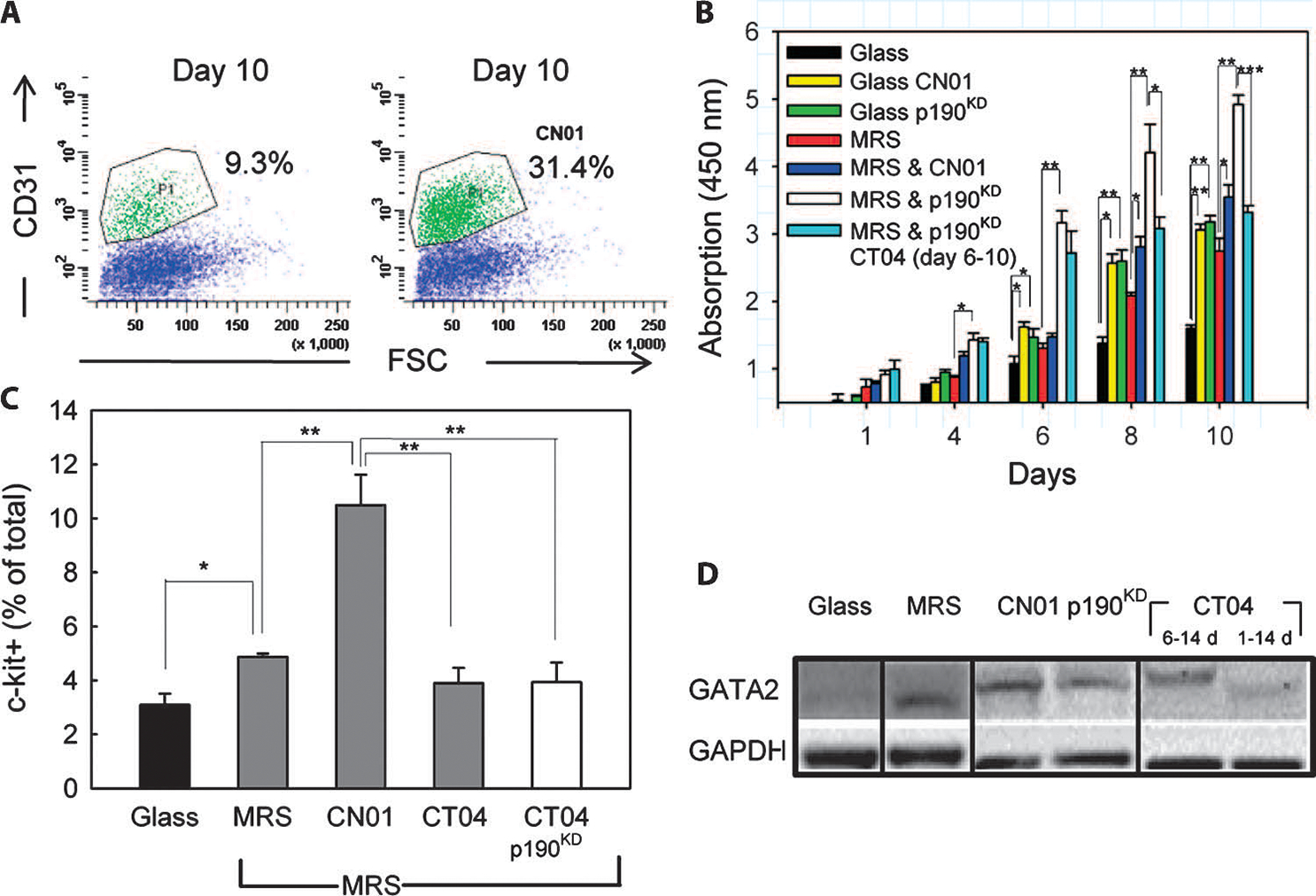
p190RhoGAP regulates MRS-induced CDC proliferation in a RhoA-dependent manner. (A) Flow cytometric analysis of CD31 in cells cultured on MRS without (left) and with CN01 treatment (right). n = 2 samples. (B) Time-resolved WST-8–based cell proliferation analysis of wild-type cells cultured on glass and MRS without or with CN01, and p190KD or scrKD control cells cultured on MRS. Rightmost bars correspond to p190KD treated with a RhoA inhibitor, CT04, during culture on MRS. (C) Flow cytometric analysis of c-kit+ cells cultured for 7 days on glass, or MRS with CN01, or CT04 treatment throughout experiment, or CT04 treatment after 6 days of culture. (D)GATA2 abundance in scrKD or p190KD CDCs cultured on the indicated substrata, with the indicated treatments, and for the indicated time periods. Noncontiguous lanes from a single experiment are indicated by black lines. Data are representative of 2 experiments. *P < 0.05; **P < 0.01; ***P < 0.001. Error bars, SEM.
We then studied whether RhoA activation directly promoted proliferation of the c-kit+ subpopulation of cells. Flow cytometry performed after 7 days of cell culture revealed that CN01 treatment significantly increased the fraction of c-kit+ cells compared to that of untreated cells cultured on glass or MRS (Fig. 8C). Conversely, CT04 treatment resulted in a reduction of c-kit+ cells cultured on MRS compared to those cultured on glass, whether p190RhoGAP was silenced or not. These data indicate that RhoA activation enhances cell proliferation on MRS by increasing the proliferation of c-kit+ cells.
We further investigated whether RhoA activity was essential for the increase in endothelial differentiation of CDCs cultured on MRS by analyzing GATA2 abundance in p190RhoGAP knockdown cells (Fig. 8D). CN01 treatment appeared to increase GATA2 abundance in cells cultured on MRS, consistent with the increase in CD31+ cells with CN01 treatment (Fig. 8A). Furthermore, CT04 treatment throughout the duration of cell culture appeared to reduce GATA2 protein abundance. However, when p190RhoGAP knockdown cells were treated with CT04 after the sixth day of culture (when proliferative c-kit+ aggregates had already formed), there appeared only a slight reduction in GATA2 abundance compared to that in untreated p190RhoGAP knockdown cells (Fig. 8D). Thus, p190RhoGAP knockdown or the reduction in p190RhoGAP protein abundance in cells cultured on MRS could enhance the differentiation of c-kit+ cells into endothelial cells independently of RhoA activation.
We then explored the mechanism of Rho-independent effects of p190RhoGAP. Previous studies suggested that p190RhoGAP directly interacts with GATA2 and thereby regulates GATA2 subcellular localization (5). Coimmunoprecipitation analysis revealed that GATA2 forms a complex with p190RhoGAP in CDCs (fig. S9A). We therefore examined whether p190RhoGAP perturbations could affect the amount and localization of GATA2 protein. GATA2 was not detectable in CDCs cultured on glass (fig. S9B), whereas it could be detected in cells cultured on MRS within 3 days, mostly in the perinuclear cytosol (Fig. 9A and fig. S9C).Within 10 days, GATA2 was predominantly found in the nuclei, coincident with decreased amount of p190RhoGAP (Fig. 9B and fig. S9D). Furthermore, in p190RhoGAP knockdown cells, the nuclear localization of GATA2 was enhanced on days 3 and 10 of culture (Fig. 9, C and D, and fig. S9, E and F) compared to that in the initial state (Fig. 9E). These data suggest that MRS regulates the amount of GATA2 protein, and increases nuclear localization of GATA2, at least partially through p190RhoGAP-dependent cytosolic targeting (Fig. 9, E and F).
Fig. 9.
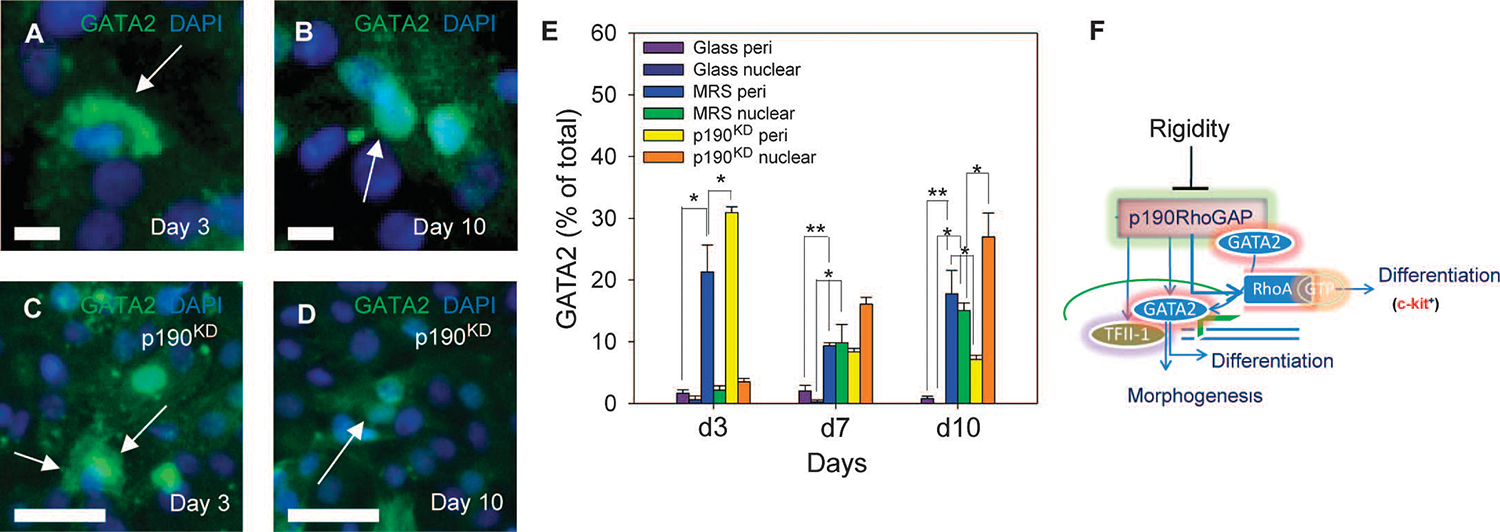
p190RhoGAP regulates substratum rigidity–induced endothelial differentiation through a RhoA-independent mechanism. (A to D) In scrKD cells cultured on MRS for 3 days (A) and 10 days. (B), GATA2 changes from a mostly perinuclear localization to a mostly nuclear localization (arrows). In p190KD cells cultured on MRS for 3 days (C) and 10 days (D), GATA changes from a nuclear and cytoplasmic localization to a predominantly nuclear localization by day 10 (arrows). (E) Analysis of perinuclear, and nuclear GATA2 localization in wild-type CDCs and p190KD CDCs cultured on glass and MRS for 3, 7, and 10 days. (F) Schematic showing the putative role of the rigidity-induced decrease in p190RhoGAP abundance in promoting cell proliferation, endothelial differentiation, and morphogenesis through regulation of RhoA activation and abundance and subcellular localization of GATA2. Scale bars, 20 μm (A to D). *P < 0.05; **P < 0.01. Error bars, SEM.
We finally investigated whether RhoA activation could directly influence the morphogenesis of the differentiating endothelial cells into lacunae-containing networks. Cells cultured on MRS in the presence of CN01 appeared to form structured cellular networks 14 days after plating (fig. S10A), similar to the behavior of control cells cultured on MRS (Fig. 1E). In contrast, treatment with CT04 over 2 weeks of culture appeared to abolish network formation (fig. S10B). Furthermore, even when CT04 was applied after 6 days of culture, complete lacunae failed to form (fig. S10C), indicating that increased RhoA activity in cells cultured on MRS can have a considerable effect over multiple stages of the morphogenic process.
DISCUSSION
Our results suggest that cell substratum rigidity, when matching the estimated physiological rigidity of the tissue, can coordinately control proliferation, differentiation, and morphogenesis of cells isolated from native heart tissue (Fig. 10, A and B). Mechanistically, the influence of the substratum rigidity on the distinct but interlinked processes leading to formation of structured cellular networks depends on the same molecular species, p190RhoGAP, acting both in the canonical, RhoA-dependent manner and in a RhoA-independent fashion (Fig. 10A). This molecule promotes VEGFR abundance and morphogenesis in differentiated human umbilical vein endothelial cells, suggesting that p190RhoGAP acts in a pleiotropic manner (5). Our analysis suggests that as multipotent endothelial and cardiac precursor cells undergo amplification and differentiation, followed by morphogenesis, the same molecular circuit is at play (Fig. 10B). The choice between the available fates can be directly controlled by endogenous regulation of p190RhoGAP concentration by mechanical and possibly other cues and can be directly controlled by exogenous perturbations of abundance of this molecule.
Fig. 10.
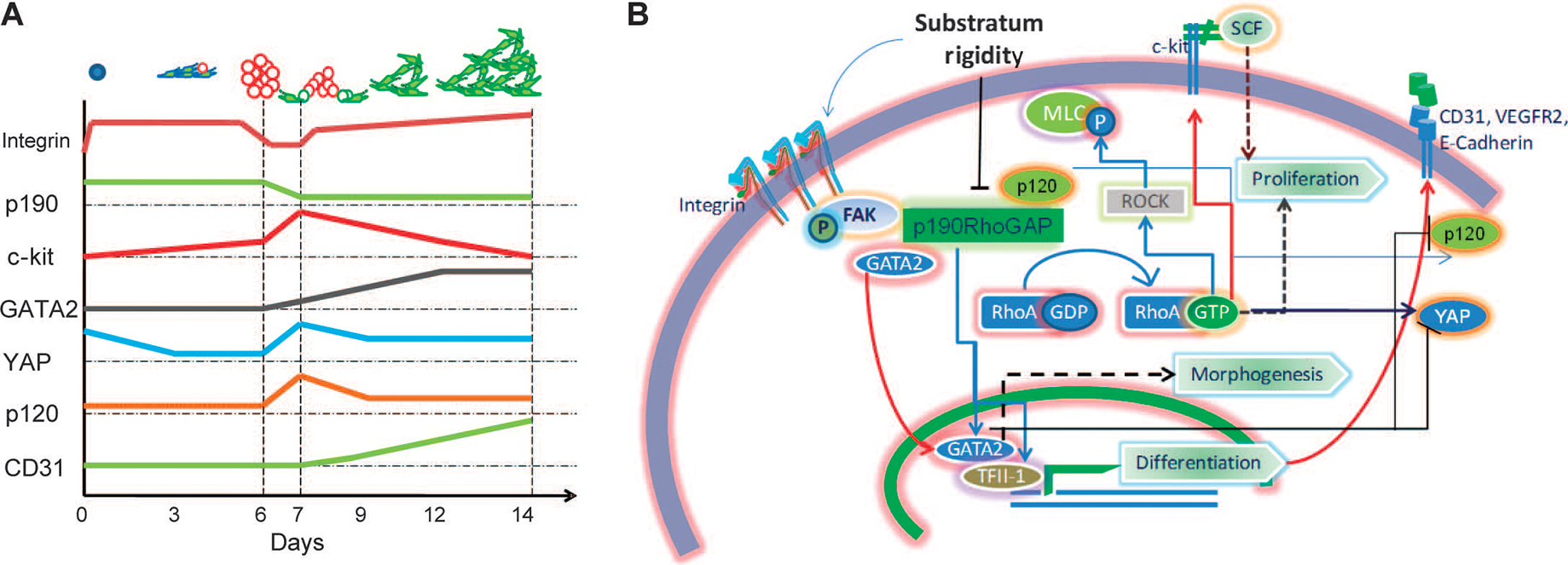
Substratum rigidity coordinates progenitor cell proliferation, endothelial differentiation, and lacunae morphogenesis through co-regulation of various signaling modules by p190RhoGAP. (A) Schematic representing the time course of abundance dynamics of various key players identified in mechanotransduction pathway involved in rigidity-mediated endothelial differentiation of CDCs. Vertical lines represent day 0 (when cells are seeded), day 6 (when spheroids form and mature), day 7 (when spheroids start dispersing), and day 14 (when cellular networks are formed). (B) Schematic summary of the proposed mechanism of substratum rigidity–induced phenotypic changes regulated by p190RhoGAP abundance. Decreased p190RhoGAP abundance increases RhoA activity, leading to enhanced proliferation of c-kit+ stem cells and morphogenetic changes through ROCK and MLC. p190RhoGAP silencing increases abundance and plasma membrane localization of p120-catenin, thereby enhancing cell-cell contact formation. p120-catenin abundance decreases during spheroid dispersal. Decreased p190RhoGAP abundance also increases the amounts of TFII-1 and GATA2 and localization of GATA2 to the nucleus, resulting in endothelial differentiation, and control of collective cell morphology. Substratum rigidity also controls YAP localization and abundance, with a transient rise during cell aggregation and a subsequent reduction during the ensuing increase in GATA2 abundance. GATA2 promotes the increased abundance of CD31 and VEGFR, events that accompany endothelial differentiation.
Decreased abundance of p190RhoGAP globally in the cell and locally in lamellipodia can lead to local de-inhibition of RhoA, thereby causing increased cell tension and more rounded cell shapes early in the culture process, which in combination with decreased integrin abundance can reduce cell contact with the underlying substratum and promote formation of the spheroid-like cell aggregates containing proliferating cells enriched in c-kit (43). Transient, E-cadherin and p120-catenin–dependent formation of these tight cell aggregates was essential for the differentiation process and depended on a transient increase in the abundance and nuclear localization of YAP, a molecule implicated in progenitor cell survival (40) that is sensitive to E-cadherin engagement and changes in mechanical cell environment (39). The p190RhoGAP-dependent increase in GATA2 abundance correlated with decreased abundance of YAPand p120-catenin. Thus, a gradual decrease in p190RhoGAP abundance in cells cultured on MRS can control the dispersal of cell aggregates, along with the onset and progression of endothelial differentiation through both canonical Rho-dependent changes in cell arrangement and noncanonical effects on GATA2 (Fig. 10A). Our results suggest that the persistent presence of the mechanical cue is essential for the decrease in p190RhoGAP abundance and the effects observed.
Dependence of CDC lineage selection on p190RhoGAP is interesting because p190RhoGAP abundance can be controlled by extracellular mechanical signals, which are relatively easier to control in a precise manner. Because a considerable hurdle faced in cardiac stem cell therapy is the lack of vascularization of the engineered tissue, these findings could have potentially important implications for tissue engineering and cardiovascular therapy.
Overall, p190RhoGAP may play a role as a central integrator of mechanical and chemical cues, although its function in controlling the differentiation of various progenitors is likely to be cell type– and context-dependent (5, 44). Because p190RhoGAP associates with various proteins, including p120-catenin (36), p120RasGAP (45, 46), FAK (45), c-Src (47), and cadherins (48), it is possible that the context of its action might in part be determined by the availability and activation of different binding partners of p190RhoGAP under diverse circumstances.
Because endothelial differentiation relies on the constant presence of a mechanical cue, it suggests more generally that the tissue regeneration and repair may depend on a constant monitoring of the mechanical environment. An abrupt change in mechanical signaling could adversely affect differentiation processes. Thus, the effectiveness of tissue regeneration dependent on CDCs and, likely, other types of adult stem and precursor cells could be hampered after formation of the scar tissue in the context of myocardial infarction as a result of the considerably increased rigidity of the scar compared to that of normal myocardium (49). Developing tissue can gradually change its rigidity, which may in turn influence differentiation pathways (50). On the other hand, the flexibility of p190RhoGAP regulation and its susceptibility to chemical as well as mechanical signaling inputs allow for the possibility that preconditioning of endothelial progenitors before their therapeutic use could change p190RhoGAP abundance and thus promote vascular regeneration. More generally, our results suggest the importance of the feedback between rigidity of a developing or regenerating tissue and the control of cell growth and differentiation, which may be critical for adaptive development and maintenance of structurally complex tissues and organs (Fig. 10B).
MATERIALS AND METHODS
Isolation and propagation of rat and human CDCs
CDCs were isolated and maintained with methods described by Smith et al. (9). WKY rats were heparinized and euthanized by sodium pentobarbital. Hearts were excised, blood was removed with ice-cold Tyrode’s solution (Sigma-Aldrich, T1788), and hearts were minced in small explants (<1 mm) after removal of gross connective tissue. Isolated fragments were partially digested with collagenase and placed on fibronectin (50 μg/ml; Sigma-Aldrich, F0895)–coated tissue culture plates (Corning, CLS330167) about 1 cm apart in complete explant medium (CEM) [Iscove’s modified Dulbecco’s medium (IMDM) (Gibco, 12440), 20% fetal bovine serum (FBS) (Gibco, 16000–044), 1% GlutaMAX (Gibco, 35050–061), 1% penicillin/streptomycin (Gibco, 15140122), and 0.5% 2-mercaptaethanol (Invitrogen, 21985023)]. Fibroblast-like cells migrated out of explants and formed a monolayer over which small, putative stem cells migrated and to which they attached. These cells were selectively harvested with Versene (Invitrogen, 15040–066) and seeded on a poly-l-lysine(BD Biosciences, 354210)–coated surface at a density of ~25,000 cells/ml in cardiosphere medium [35% IMDM and 65% Dulbecco’s modified Eagle’s medium (DMEM)/F-12 mix (Invitrogen, 11330032), 3.5% FBS, 1% penicillin/streptomycin, 1% GlutaMAX, 0.1 mM 2-mercaptoethanol, thrombin (Biopharm Laboratories, 91–050), B-27 (Invitrogen, A11576SA), basic fibroblast growth factor (bFGF) (80 ng/ml; PeproTech, 106096–92-8), epidermal growth factor (EGF) (25 ng/ml; PeproTech, AF-100–15), and cardiotrophin-1 (4 ng/ml; PeproTech, 300–32)]. Adherent cells were discarded after 2 weeks, and floating cardiospheres were cultured on a fibronectin (50 μg/ml)–coated surface in CEM and expanded as a monolayer until passage 2, frozen, and stored at −80°C. Cells were used between passages 3 and 7 and were passaged with 0.05% trypsin-EDTA (Invitrogen, 25300–054) at a split ratio of 1:3. For human CDCs, a similar procedure was used on biopsy fragments derived from endomyocardial biopsy.
Preparation of polyacrylamide gels with variable rigidity
Polyacrylamide gels with varying rigidities were prepared according to the protocol by Lo et al. (51), with slight variation. Polyacrylamide was crosslinked to glass by pretreating a glass coverslip (22 mm by 22 mm) for 3 min with 3-(trimethoxysilyl)propyl-methylacrylate (Sigma-Aldrich, M6514) solution, prepared as suggested by the vendor, and washed with distilled and deionized (DI) water. After polyacrylamide polymerization, gels were treated with 100 μl of sulfo-SANPAH (0.5 mg/ml; Pierce, 22589) and exposed to ultraviolet radiation (302 nm) for 20 min, washed with DI water, and coated with fibronectin (10 μg/ml) overnight. A gradient of rigidity was created by placing individual drops of 10 ml each of two solutions with low (0.08%) and high (0.4%) bisacrylamide concentrations 1.5 cm apart on the coverslip, which was then flattened with another coverslip, thereby resulting in mixing of two solutions and creating a gradient. Bisacrylamide solution (0.08%) was mixed with Texas red–conjugated 10,000 MW (molecular weight) dextran (Invitrogen, D1863) for visualization of the gradient. CDCs were seeded at a density of 100,000 per coverslip, and the medium was changed the next day to remove unattached cells (10 to 50% of the population, with varying consistency). CEM was changed on days 1, 4, 7, 10, and 13. When treating with CN01 (Cytoskeleton Inc.), 50% of the final volume of CEM containing CN01 (2 μg/ml) was added to the cell culture every 12 hours so that half of the old medium was retained. A similar protocol was adopted for CT04 (Cytoskeleton Inc.).
Plasmids
Plasmids containing human p190RhoGAP shRNA (clone ID: TRCN0000022184, TRCN0000022185), GATA2 shRNA (clone ID: TRCN0000085418), p120-catenin shRNA (clone ID: TRCN0000122984–86), YAP shRNA (clone ID: TRCN0000107266–67), and scrambled shRNA (Addgene, 1864) based on pLKO1 vector were purchased from ChemCore, Johns Hopkins Medical Institutions (JHMI) (52). The p190RhoGAP overexpression plasmid was prepared by cloning the human open reading frame(ORF)segment (OHS1770) (Open Biosystems) in a simian virus 40 (SV40) lentiviral expression vector.
Lentiviral shRNA transduction
Human embryonic kidney (HEK) cells were plated at 50% confluency and transfected with pLKO1-based shRNA expression plasmid, vesicular stomatitis virus glycoprotein (VSVG) (Addgene, 8454), and Δ8.2 dvpr (Addgene, 8455) with FuGENE 6 (Roche, 11814443001). The medium was changed after 12 hours. Supernatant was collected at 72 and 96 hours after transfection and transferred to CDC culture (at 70% confluency) with an equal amount of CEM and polybrene (Sigma-Aldrich, Al-118) at a final concentration of 10 μg/ml. The expression of the control plasmid (containing GFP in the pLKO1 vector) was assessed 4 days after transduction with flow cytometry, and more than 90% of the cells were transduced.
Flow cytometry
Cells were detached from the substrate with Hanks’-based enzyme-free cell dissociation buffer (Gibco, 13150) for 15 min at 37°C, quenched with excess medium, and washed twice with phosphate-buffered saline (PBS) by centrifugation at 1200 rpm for 5 min. Cells were blocked with 1% AlbuMAX (Gibco, 11020021), incubated with primary antibodies for 30 min on ice with repeated vortexing, washed with PBS, and incubated with Alexa Fluor 488 (Molecular Probes, A11001)– or Alexa Fluor 594 (Molecular Probes, A11012)–conjugated secondary antibodies for 30 min. Cells were washed with PBS and collected for flow cytometry with the relevant controls. Antibodies used were against CD31 (BD Biosciences, 555444) and c-kit (BD Biosciences, 555713). Flow cytometry was performed with FACSDiva (BD Biosciences), and analyses were performed with FACSDiva 6.0 (BD Biosciences) or MATLAB 2006. Statistical analyses were performed and graphs were plotted with SigmaPlot 9 (Systat Software Inc.).
Immunocytochemistry and immunohistology
For EdU staining, cells were pulse-treated with Click-iT EdU (Invitrogen, C10083) for 4 hours and fixed after 24 hours. For immunocytochemistry, samples were washed with ice-cold PBS, fixed with 4% paraformaldehyde (Sigma-Aldrich, F8775) for 20 min at room temperature, permeabilized with 0.05% Triton X-100 (Sigma-Aldrich) for 15 min, washed twice with PBS, and blocked with 10% goat serum and 1% AlbuMAX (Gibco, 11020021) for 1 hour. Samples were incubated with primary antibody for 1 hour followed by three washes and incubation with Alexa Fluor–conjugated secondary antibody (Molecular Probes) for 1 hour. For EdU staining, fixed and permeabilized samples were treated for 1 hour with EdU detection reagent. Samples were washed, mounted to microscopy slides with a drop of SlowFade (Invitrogen, S26938), and sealed with medical adhesive (Hollister, 7730). In immunohistology analysis, right ventricles in rat hearts were surgically excised and frozen with optimal cutting temperature compound (Ted Pella Inc., OCT 27050) and sectioned at the Johns Hopkins Histology center in 10-μm-thick sections, and the above protocol was used for staining.
For coimmunoprecipitation analysis, primary antibody or control immunoglobulin G (IgG) was incubated overnight at 4°C with cell lysates recovered from polyacrylamide-coated glass-bottom dishes (Live Assay, 35200001) followed by incubation with protein G–Sepharose beads (GE Healthcare) for 3 hours. Beads were washed four times in lysis buffer. Proteins were eluted in SDS sample buffer and separated by SDS–polyacrylamide gel electrophoresis (SDS-PAGE). Antibodies used in immunoblot and immunoprecipitation as says were p190RhoGAP (Bethyl Laboratories, A301–736A), GATA2 (Santa Cruz, sc-9008), and control IgG (Santa Cruz, sc-2025).
Proliferation analysis with WST-8 assay
Polyacrylamide gels of differing rigidity were created in a glass-bottom 96-well plate (Live Assay, 96W1501) with the procedure described above, and CDCs were cultured at 1000 cells per well. To estimate cell number, we added 10 μl of Cell Counting Kit-8 solution (Dojindo, CK04–11) containing WST-8 to the relevant wells, and we recorded absorbance at 450 nm in a multiwell plate luminometer (Lmax, Molecular Devices) after incubation for 4 hours. Background recorded from wells not containing any cells was subtracted. Statistical analysis and graph generation were performed with functions in MATLAB 2006.
Epifluorescence and confocal microscopy
Phase-contrast microscopy was performed with Nikon SPOT 2.0. Images were acquired with a SPOT Insight 2MP camera (Diagnostic Instruments Inc.) using SPOT Imaging software (Diagnostic Instruments Inc.) and processed with Adobe Photoshop (Adobe Systems Inc.). Epifluorescence microscopy was performed with a Zeiss Axiovert 2000 with 10×, 20×, and 40× apochromat objective lenses with a numerical aperture (NA) of 1.4. A Lambda LS Xenon arc lamp (Sutter Instrument) was used for excitation, and acquisition was performed with a Cascade 512BII camera (Roper Scientific Inc.). Images were acquired and processed with SlideBook 4.2.0 (Intelligent Imaging Innovations Inc.). Processing was performed with Adobe Photoshop (Adobe Systems Inc.).
Confocal microscopy was performed by means of a Zeiss LSM510 Meta CLSM microscope with 20× and 40× apochromat objective lenses (NA = 1.4). Pinholes were adjusted to 1 mm. For excitation, a blue diode (405 nm), green argon laser (488 nm), and red HeNe laser (568 nm) were used. Images were processed with Zeiss Meta software version 3.5. CD31+-lined blood vessels more than 5 mm in diameter containing DiI+-injected CDCs were counted in each examined section.
Immunoblot analysis
The cells were lysed in M-PER mammalian protein extraction reagent (Thermo Scientific, 78503) with 1% protease inhibitor cocktail (Sigma-Aldrich, S7830) and NuPAGE sample reducing agent (Invitrogen, NP0004) containing 10 mM dithiothreitol (DTT) (Sigma-Aldrich, D0632). Protein was quantified with the Coomassie Plus protein assay (Thermo Scientific, 23236). Immunoblot analysis was performed as described previously with modifications (53). Protein (15 to 20 μg) was fractionated in 4 to 12% or 12% NuPAGE gels (Invitrogen, NP0321BOX) and transferred onto nitrocellulose membranes (Bio-Rad, 162–0150). The antibodies used were FAK mouse monoclonal (#610087) from BD Biosciences; phospho-FAK (Tyr397) rabbit polyclonal (#3283), MLC2 rabbit polyclonal (#3672), phospho-MLC 2 (Ser19) mouse monoclonal (#3675S), mouse IgG–horseradish peroxidase (HRP) (#7076), and rabbit IgG-HRP (#7074) from Cell Signaling Technology Inc.;glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (I-19) goat polyclonal (#sc-48166) and goat IgG-HRP (#sc-2020) from Santa Cruz. The immune complexes were visualized with Western Lightning Plus-ECL Enhanced Chemiluminescence Substrate (PerkinElmer, NEL103E001EA). The nitrocellulose membranes analyzed were stripped with ReBlot Plus mild antibody stripping solution (Millipore, 2504) and reused for determining the abundance of GAPDH as a loading control. Unless otherwise indicated, representative data from three independent CDC culture experiments are shown.
Real-time reverse transcription–polymerase chain reaction
RNA was isolated from CDCs with TRIzol (Invitrogen, 15596–026) and precipitated in isopropanol with 2 μg of nuclease-free glycogen (Invitrogen, 10814–010) and treated with DNAse I solution (Ambion, AM2222). First-strand complementary DNA (cDNA) synthesis was performed with iScript cDNA Synthesis system (Bio-Rad, 170–8891) for5 μg of total RNA. cDNA obtained was diluted 1:10, and 2 μl was added to each polymerase chain reaction (PCR) with the primers listed below. Real-time PCR was conducted with iQ SYBR Green Supermix (Bio-Rad, 170–8880) in the iCycler Real-Time PCR Detection System (Bio-Rad). The abundance of each target mRNA relative to 18S ribosomal RNA (rRNA) was calculated with the threshold cycle as , where , as described by Rey et al. (54). The difference in relative mRNA abundance was found by dividing the values of the respective target mRNAs.
Matrigel assay for tube formation
Formation of cord and tube-like networks was observed with 96-well plates that contained BD Matrigel Matrix (BD BioCoat, 354150). Cells were trypsinized and washed twice, counted, and seeded. Cells (2500 and 5000) were plated with either 2.5 or 5% heat-inactivated FBS in IMDM and placed in a 5% CO2 incubator at 37°C for 5 hours. Tube formation was observed with phase-contrast microscopy at 5× magnification on a Zeiss Axiovert 2000.
Bioluminescence imaging
Bioluminescence imaging was performed 3 days after cell transplantation with an IVIS 200 optical imaging device (Caliper Life Sciences) equipped with a high-sensitivity charge-coupled device (CCD) detection system that is cryogenically cooled. Two million CDCs were lentivirally transduced with luciferase gene driven by CMV promoter, cultured on MRS or glass for 7 days, and injected at the site of infarct and in healthy hearts. Before imaging, each rat was anesthetized with 1 to 2% isoflurane, intraperitoneally injected with luciferase substrate luciferin (150 mg/kg) (Caliper Life Sciences), and kept immobilized for 20 min after luciferin injection. Animals were killed, and hearts were isolated and imaged. Signal processing was performed at the site of a manually chosen region of interest, and total flux (photons/s) was computed with Xenogen Living Image 2.5 software. The bioluminescence imaging signal was normalized with rat heart transduced with wild-type CDCs. Images were processed with Adobe Photoshop software.
Statistical analysis
P values were determined by Student’s t test applied only to data with normal distributions and were calculated with SigmaPlot 11 and 12 (Systat Software Inc.).
Supplementary Material
Text S1. ODE-based mathematical model.
Fig. S1. Substratum rigidity influences single-cell shape and multicell organization of CDCs.
Fig. S2. MRS promotes CDC proliferation and endothelial differentiation.
Fig. S3. MRS influences cell-substratum and cell-cell interaction dynamics in CDC cultures.
Fig. S4. p190RhoGAP mediates cell-cell interaction in aggregate formation by regulating p120-catenin and YAP abundance and localization.
Fig. S5. Characterization of p190RhoGAP-shRNA transduction.
Fig. S6. CDCs integrate in blood vessels in rat hearts.
Fig. S7. Model simulation of cell proliferation and differentiation.
Fig. S8. Increase in p190RhoGAP abundance promotes myocardial differentiation of CDCs.
Fig. S9. p190RhoGAP sequesters GATA2 in the cytosol.
Fig. S10. RhoA influences cell morphology.
Movies S1 to S4. Dynamics of cell network formation from CDC spheroid aggregates.
Acknowledgments:
We extend our gratitude to M. Delannoy and B. Smith (Johns Hopkins School of Medicine Microscopy Center) for their invaluable help in confocal microscopy and to S. Stäger (assistant professor, Department of Immunology, JHMI) for her help in flow cytometry.
Funding:
This work was supported by NIH grants 1R21EB008562–01A1 (to A.L.) and R01HL092985 (to R.M.A.) and Basic Science Research Program through the National Research Foundation of Korea (grant no. 2010–0010840) funded by the Ministry of Education, Science and Technology (to E.H.A.). A.L. is supported by an “Early Excellence Award” from the American Asthma Foundation. M.E.H. was funded by NIH hematology postdoctoral training grant T32HL007525. K. was supported by American Heart Association (AHA) Predoctoral Fellowship 10PRE4160120. D.-H.K. was supported by AHA Predoctoral Fellowship 0815104E, University of Washington Department of Bioengineering start-up fund, and Perkins Coie Award for Discovery.
Footnotes
Competing interests: K. has a controlling stake in Live Assay, which provided glass-bottomed tissue culture dishes for the studies.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Guo WH, Frey MT, Burnham NA, Wang YL, Substrate rigidity regulates the formation and maintenance of tissues. Biophys. J. 90, 2213–2220 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vogel V, Sheetz M, Local force and geometry sensing regulate cell functions. Nat. Rev. Mol. Cell Biol. 7, 265–275 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Montell DJ, Morphogenetic cell movements: Diversity from modular mechanical properties. Science 322, 1502–1505 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Kim DH, Lipke EA, Kim P, Cheong R, Thompson S, Delannoy M, Suh KY, Tung L, Levchenko A, Nanoscale cues regulate the structure and function of macroscopic cardiac tissue constructs. Proc. Natl. Acad. Sci. U.S.A. 107, 565–570 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mammoto A, Connor KM, Mammoto T, Yung CW, Huh D, Aderman CM, Mostoslavsky G, Smith LE, Ingber DE, A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature 457, 1103–1108 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim DH, Wong PK, Park J, Levchenko A, Sun Y, Microengineered platforms for cell mechanobiology. Annu. Rev. Biomed. Eng. 11, 203–233 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Kshitiz, Kim DH, Beebe DJ, Levchenko A, Micro- and nanoengineering for stem cell biology: The promise with a caution. Trends Biotechnol. 29, 399–408 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gupta K, Kim DH, Ellison D, Smith C, Kundu A, Tuan J, Suh KY, Levchenko A, Lab-on-a-chip devices as an emerging platform for stem cell biology. Lab Chip 10, 2019–2031 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Smith RR, Barile L, Cho HC, Leppo MK, Hare JM, Messina E, Giacomello A, Abraham MR, Marbán E, Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation 115, 896–908 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Johnston PV, Sasano T, Mills K, Evers R, Lee ST, Smith RR, Lardo AC, Lai S, Steenbergen C, Gerstenblith G, Lange R, Marbán E, Engraftment, differentiation, and functional benefits of autologous cardiosphere-derived cells in porcine ischemic cardiomyopathy. Circulation 120, 1075–1083 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Makkar RR, Smith RR, Cheng K, Malliaras K, Thomson LE, Berman D, Czer LS, Marbán L, Mendizabal A, Johnston PV, Russell SD, Schuleri KH, Lardo AC, Gerstenblith G, Marbán E, Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): A prospective, randomised phase 1 trial. Lancet 379, 895–904 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis DR, Zhang Y, Smith RR, Cheng K, Terrovitis J, Malliaras K, Li TS, White A, Makkar R, Marbán E, Validation of the cardiosphere method to culture cardiac progenitor cells from myocardial tissue. PLoS One 4, e7195 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore SW, Roca-Cusachs P, Sheetz MP, Stretchy proteins on stretchy substrates: The important elements of integrin-mediated rigidity sensing. Dev. Cell 19, 194–206 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engler AJ, Sen S, Sweeney HL, Discher DE, Matrix elasticity directs stem cell ineage specification. Cell 126, 677–689 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Folkman J, Haudenschild C, Angiogenesis in vitro. Nature 288, 551–556 (1980). [DOI] [PubMed] [Google Scholar]
- 16.Weil BR, Kushner EJ, Diehl KJ, Greiner JJ, Stauffer BL, DeSouza CA, CD31+ T cells, endothelial function and cardiovascular risk. Heart Lung Circ. 20, 659–662 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schubert SY, Benarroch A, Monter-Solans J, Edelman ER, Primary monocytes regulate endothelial cell survival through secretion of angiopoietin-1 and activation of endothelial Tie2. Arterioscler. Thromb. Vasc. Biol. 31, 870–875 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sandstedt J, Jonsson M, Lindahl A, Jeppsson A, Asp J, C-kit+ CD45–cells found in the adult human heart represent a population of endothelial progenitor cells. Basic Res. Cardiol. 105, 545–556 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Miyamoto S, Kawaguchi N, Ellison GM, Matsuoka R, Shin’oka T, Kurosawa H, Characterization of long-term cultured c-kit+ cardiac stem cells derived from adult rat hearts. Stem Cells Dev. 19, 105–116 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Puklin-Faucher E, Sheetz MP, The mechanical integrin cycle. J. Cell Sci. 122, 179–186 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang YK, Wang YH, Wang CZ, Sung JM, Chiu WT, Lin SH, Chang YH, Tang MJ, Rigidity of collagen fibrils controls collagen gel-induced down-regulation of focal adhesion complex proteins mediated by α2β1 integrin. J. Biol. Chem. 278, 21886–21892 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Katsumi A, Orr AW, Tzima E, Schwartz MA, Integrins in mechanotransduction. J. Biol. Chem. 279, 12001–12004 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Kallergi G, Agelaki S, Markomanolaki H, Georgoulias V, Stournaras C, Activation of FAK/PI3K/Rac1 signaling controls actin reorganization and inhibits cell motility in human cancer cells. Cell. Physiol. Biochem. 20, 977–986 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Mizutani T, Kawabata K, Koyama Y, Takahashi M, Haga H, Regulation of cellular contractile force in response to mechanical stretch by diphosphorylation of myosin regulatory light chain via RhoA signaling cascade. Cell Motil. Cytoskeleton 66, 389–397 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Hotchin NA, Kidd AG, Altroff H, Mardon HJ, Differential activation of focal adhesion kinase, Rho and Rac by the ninth and tenth FIII domains of fibronectin. J. Cell Sci. 112 (Pt. 17), 2937–2946 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Mackay DJ, Esch F, Furthmayr H, Hall A, Rho- and rac-dependent assembly of focal adhesion complexes and actin filaments in permeabilized fibroblasts: An essential role for ezrin/radixin/moesin proteins. J. Cell Biol. 138, 927–938 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nimnual AS, Taylor LJ, Bar-Sagi D, Redox-dependent downregulation of Rho by Rac. Nat. Cell Biol. 5, 236–241 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Burridge K, Wennerberg K, Rho and Rac take center stage. Cell 116, 167–179 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Lim ST, Chen XL, Tomar A, Miller NL, Yoo J, Schlaepfer DD, Knock-in mutation reveals an essential role for focal adhesion kinase activity in blood vessel morphogenesis and cell motility-polarity but not cell proliferation. J. Biol. Chem. 285, 21526–21536 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Playford MP, Vadali K, Cai X, Burridge K, Schaller MD, Focal adhesion kinase regulates cell-cell contact formation in epithelial cells via modulation of Rho. Exp. Cell Res. 314, 3187–3197 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mammoto A, Mammoto T, Ingber DE, Rho signaling and mechanical control of vascular development. Curr. Opin. Hematol. 15, 228–234 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Maddox AS, Burridge K, RhoA is required for cortical retraction and rigidity during mitotic cell rounding. J. Cell Biol. 160, 255–265 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bass MD, Morgan MR, Roach KA, Settleman J, Goryachev AB, Humphries MJ,p190RhoGAP is the convergence point of adhesion signals from α5β1 integrin and syndecan-4. J. Cell Biol. 181, 1013–1026 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Z, Tan JL, Cohen DM, Yang MT, Sniadecki NJ, Ruiz SA, Nelson CM, Chen CS, Mechanical tugging force regulates the size of cell–cell junctions. Proc. Natl. Acad. Sci. U.S.A. 107, 9944–9949 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams CL, Chen YT, Smith SJ, Nelson WJ, Mechanisms of epithelial cell–cell adhesion and cell compaction revealed by high-resolution tracking of E-cadherin–green fluorescent protein. J. Cell Biol. 142, 1105–1119 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wildenberg GA, Dohn MR, Carnahan RH, Davis MA, Lobdell NA, Settleman J, Reynolds AB, p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell 127, 1027–1039 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Molina-Ortiz I, Bartolomé RA, Hernández-Varas P, Colo GP, Teixidó J, Over expression of E-cadherin on melanoma cells inhibits chemokine-promoted invasion involving p190RhoGAP/p120ctn-dependent inactivation of RhoA. J. Biol. Chem. 284, 15147–15157 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S, Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Kim NG, Koh E, Chen X, Gumbiner BM, E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Natl. Acad. Sci. U.S.A. 108, 11930–11935 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao B, Tumaneng K, Guan KL, The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 13, 877–883 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen B, Gao H, Mechanical principle of enhancing cell-substrate adhesion via pretension in the cytoskeleton. Biophys. J. 98, 2154–2162 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS, Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev. Cell 6, 483–495 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Kim TJ, Seong J, Ouyang M, Sun J, Lu S, Hong JP, Wang N, Wang Y, Substrate rigidity regulates Ca2+ oscillation via RhoA pathway in stem cells. J. Cell. Physiol. 218, 285–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ingber DE, Folkman J, Mechanochemical switching between growth and differentiation during fibroblast growth factor–stimulated angiogenesis in vitro: Role of extracellular matrix. J. Cell Biol. 109, 317–330 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tomar A, Lim ST, Lim Y, Schlaepfer DD, A FAK-p120RasGAP-p190RhoGAP complex regulates polarity in migrating cells. J. Cell Sci. 122, 1852–1862 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bradley WD, Hernández SE, Settleman J, Koleske AJ, Integrin signaling through Arg activates p190RhoGAP by promoting its binding to p120RasGAP and recruitment to the membrane. Mol. Biol. Cell 17, 4827–4836 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller LD, Lee KC, Mochly-Rosen D, Cartwright CA, RACK1 regulates Src-mediated Sam68 and p190RhoGAP signaling. Oncogene 23, 5682–5686 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Noren NK, Arthur WT, Burridge K, Cadherin engagement inhibits RhoA via p190RhoGAP. J. Biol. Chem. 278, 13615–13618 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Engler AJ, Carag-Krieger C, Johnson CP, Raab M, Tang HY, Speicher DW, Sanger JW, Sanger JM, Discher DE, Embryonic cardiomyocytes beat best on a matrix with heart-like elasticity: Scar-like rigidity inhibits beating. J. Cell Sci. 121, 3794–3802 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Young JL, Engler AJ, Hydrogels with time-dependent material properties enhance cardiomyocyte differentiation in vitro. Biomaterials 32, 1002–1009 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lo CM, Wang HB, Dembo M, Wang YL, Cell movement is guided by the rigidity of the substrate. Biophys. J. 79, 144–152 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, Carpenter AE, Foo SY, Stewart SA, Stockwell BR, Hacohen N, Hahn WC, Lander ES, Sabatini DM, Root DE, A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Ahn EH, Chang CC, Talmage DA, Loss of anti-proliferative effect of all-trans retinoic acid in advanced stage of breast carcinogenesis. Anticancer Res. 29, 2899–2904 (2009). [PubMed] [Google Scholar]
- 54.Rey S, Lee K, Wang CJ, Gupta K, Chen S, McMillan A, Bhise N, Levchenko A, Semenza GL, Synergistic effect of HIF-1a gene therapy and HIF-1-activated bone marrow-derived angiogenic cells in a mouse model of limb ischemia. Proc. Natl. Acad. Sci. U.S.A. 106, 20399–20404 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Text S1. ODE-based mathematical model.
Fig. S1. Substratum rigidity influences single-cell shape and multicell organization of CDCs.
Fig. S2. MRS promotes CDC proliferation and endothelial differentiation.
Fig. S3. MRS influences cell-substratum and cell-cell interaction dynamics in CDC cultures.
Fig. S4. p190RhoGAP mediates cell-cell interaction in aggregate formation by regulating p120-catenin and YAP abundance and localization.
Fig. S5. Characterization of p190RhoGAP-shRNA transduction.
Fig. S6. CDCs integrate in blood vessels in rat hearts.
Fig. S7. Model simulation of cell proliferation and differentiation.
Fig. S8. Increase in p190RhoGAP abundance promotes myocardial differentiation of CDCs.
Fig. S9. p190RhoGAP sequesters GATA2 in the cytosol.
Fig. S10. RhoA influences cell morphology.
Movies S1 to S4. Dynamics of cell network formation from CDC spheroid aggregates.