

Abstract
Background
Supravalvar aortic stenosis (SVAS) is a characteristic feature of Williams–Beuren syndrome (WBS). Its severity varies: ~20% of people with Williams–Beuren syndrome have SVAS requiring surgical intervention, whereas ~35% have no appreciable SVAS. The remaining individuals have SVAS of intermediate severity. Little is known about genetic modifiers that contribute to this variability.
Methods and Results
We performed genome sequencing on 473 individuals with Williams–Beuren syndrome and developed strategies for modifier discovery in this rare disease population. Approaches include extreme phenotyping and nonsynonymous variant prioritization, followed by gene set enrichment and pathway‐level association tests. We next used GTEx v8 and proteomic data sets to verify expression of candidate modifiers in relevant tissues. Finally, we evaluated overlap between the genes/pathways identified here and those ascertained through larger aortic disease/trait genome‐wide association studies. We show that SVAS severity in Williams–Beuren syndrome is associated with increased frequency of common and rarer variants in matrisome and immune pathways. Two implicated matrisome genes (ACAN and LTBP4) were uniquely expressed in the aorta. Many genes in the identified pathways were previously reported in genome‐wide association studies for aneurysm, bicuspid aortic valve, or aortic size.
Conclusions
Smaller sample sizes in rare disease studies necessitate new approaches to detect modifiers. Our strategies identified variation in matrisome and immune pathways that are associated with SVAS severity. These findings suggest that, like other aortopathies, SVAS may be influenced by the balance of synthesis and degradation of matrisome proteins. Leveraging multiomic data and results from larger aorta‐focused genome‐wide association studies may accelerate modifier discovery for rare aortopathies like SVAS.
Keywords: adaptive/innate immune system, elastin (ELN), extreme phenotype, pathway analysis, supravalvar aortic stenosis, Williams–Beuren syndrome
Subject Categories: Aortic Dissection, Aneurysm, Stenosis, Vascular Disease, Coronary Artery Disease
Nonstandard Abbreviations and Acronyms
- SVAS
supravalvar aortic stenosis
- WBS
Williams–Beuren syndrome
Clinical Perspective.
What Is New?
Variation in genes in several pathways, including matrisome and adaptive/innate pathways, is associated with supravalvar aortic stenosis severity in people with Williams–Beuren syndrome.
Unbalanced expression of genes controlling extracellular matrix synthesis and degradation is common in aortopathies, including aneurysm and bicuspid aortic valve, suggesting overlapping mechanisms for supravalvar aortic stenosis and these conditions.
What Are the Clinical Implications?
New methodologies enabling identification of genetic modifiers in rare conditions may improve risk stratification for newly diagnosed individuals and identify novel pathway‐based targets for therapeutics.
In rare diseases, where sample size is small, fine phenotyping, extreme phenotype cohorting, and pathway‐based analyses are viable strategies for modifier discovery.
Leveraging multiomics data and accumulated knowledge from larger aortopathy genome‐wide association studies may accelerate discovery of targets and treatments for rare aortic diseases like supravalvar aortic stenosis.
Williams–Beuren syndrome (WBS, MIM # 194050), caused by deletion of 1.5 to 1.8 Mb on human 7q11.23, is a multisystem disorder characterized by distinctive facies, a typical neurodevelopmental profile, and cardiovascular disease. 1 It occurs in 1 of 7500 live births 2 and is de novo in almost all cases. The cardiovascular disease in WBS is primarily mediated by the deletion of elastin (ELN) from this region 3 , 4 , 5 , 6 , 7 and consists of large and medium artery stenosis in the setting of a more global decrease in arterial caliber. Supravalvar aortic stenosis (SVAS), which is the narrowing of the ascending aorta above the aortic valve, commonly complicates WBS. 8 , 9 , 10 It can be focal or may consist of a more gradual narrowing along a longer segment of the aortic arch. Although more than 95% of individuals with WBS share the same basic deletion on chromosome 7q11.23, their outcomes for focal SVAS vary: about 20% have clinically significant discrete SVAS requiring surgical intervention in infancy or childhood; in contrast, about 35% of individuals with WBS never develop significant discrete SVAS, although varying levels of long segment stenosis may be present. 9 , 11 , 12 It has been unclear what features (genetic or otherwise) predispose to these extreme outcomes.
The application of genome‐wide association studies (GWAS) to the identification of modifiers for rare conditions such as WBS has been challenging because most existing GWAS methods were developed for studies with thousands of participants. These numbers are unattainable for most rare disease studies, including WBS. Likewise, techniques to improve power, such as paired expression quantitative trait loci analysis 13 of affected tissues, are challenging in difficult‐to‐access tissues such as the aorta. As such, existing studies have primarily focused on correlation of phenotype with variants within the disease‐specific locus or region. 14 , 15 Therefore, alternative computational and analytic strategies are needed for the study of rare diseases using genome sequencing data.
To overcome this challenge and to identify modifiers contributing to SVAS severity, we devised a set of strategies centered on the question of whether those with extreme SVAS phenotypes exhibit a relative burden of nonsynonymous variants (hereafter variants) that are enriched in a small number of biological pathways. The concept of pathway enrichment, which has been widely used in mRNA expression studies, 16 has been recently incorporated into GWAS analysis. 11 , 17 , 18 For application in our smaller sample size WBS study, we aimed to increase power for discovery by prioritizing the most influential common and rarer variants—those with greater likelihood of a functional impact 19 —and variants with allele frequency (AF) differences between the extreme phenotype groups, thereby reducing the total number of variants to be considered for downstream pathway enrichment. 20 , 21 The pathways identified give us a bird's‐eye view of the molecules and processes that synergize to influence disease outcomes for SVAS.
Once pathways are identified, we sought additional evidence to confirm the relevance of the candidates to disease by examining tissue‐specific expression of the genes using public data sets. Then, by harnessing existing GWAS data on common aortic conditions like aneurysm, 22 , 23 , 24 , 25 , 26 , 27 , 28 bicuspid aortic valve, 29 , 30 calcific valve stenosis, 31 , 32 , 33 and aortic size, 34 , 35 , 36 , 37 we examine overlap between the modifier pathways discovered for SVAS and these more commonly studied conditions. Of particular interest is the notion of an imbalance between extracellular matrix accumulation and destruction at the hands of immune system players in a host of aortic conditions. 38 , 39 , 40 , 41 Such synergies should allow future investigators to drill down further into the pathways uncovered by our methods to determine how they affect the aorta.
Methods
Data Availability
Variant data are made available as part of the data supplement (Data S1‐S3).
Consent
All participants alive at the time of enrollment or their caregivers signed informed consent forms to participate in research that included genome evaluation. One hundred and eighty participants signed consent approved by the National Institutes of Health (NIH) Institutional Review Board (NCT02706639), 197 signed consent approved by the Reno Institutional Review Board of the University of Nevada, 20 signed consent approved by the University of Toronto Health Sciences Research Ethics Board (those 217 were shared under the umbrella of the Nevada‐Toronto collaboration), 10 signed consent approved by the Boston Children's Hospital Institutional Review Board, and 64 consented to participate in the Telethon Biobank in Italy and were approved by Fondazione IRCCS Casa Sollievo della Sofferenza Ethics Board. Two additional NIH samples were derived from tissue donated after death and were considered exempt. The data were analyzed under the NIH‐approved protocol.
Sequencing and Quality Assessment
See Supplemental Methods for sequencing and sample quality details. Briefly, samples were evaluated for relatedness and genomic sex was compared with family‐reported sex. Overall genomic variation within the cohort, with the genotyping matrix of 142 829 autosomal nonsynonymous single‐nucleotide variants (SNVs), was assessed with a principal component analysis and visualized in a plot of principal components 1 and 2 (PC1–2) using the “bigstatsR” package. 42 By using clustering information of the individuals, as shown in Figure S1, along with available self‐reported race and ethnicity data as a proxy for continental‐level ancestry, we imputed missing race and ethnicity data. The race and ethnicity‐linked clusters in Figure S1 are similar to those generated by the larger UK Biobank study, 43 suggesting appropriate representation of genotypes.
Extreme Phenotyping of Individuals With WBS
Participants with WBS were classified based on severity of their SVAS into 4 groups: (1) clinically significant/surgical as defined by a history of surgical intervention in the supravalvar aorta (“surgical SVAS”), (2) mild‐to‐moderate (defined as presence of any SVAS for which surgery was neither recommended nor performed), (3) no SVAS, meaning no appreciable “discrete” stenosis, and (4) unclassified. A combination of parental report and available medical records (cardiologist note, echo, cardiac catheterization report, or surgical reports) was used to assign phenotype. Because the degree of SVAS may increase over the first few years of life, we required that a participant be at least 3 years of age to be listed as “no SVAS.” Consequently, an additional category of unclassified participants who were either too young to classify as “no SVAS” or did not have adequate data for the clinician to confidently assign a phenotypic designation was created. In the classified surgical and no SVAS cases, records were determined to be adequate to justify the classification. Parental report was not used in isolation to assign the SVAS outcome.
Our modifier evaluation focused on comparisons of those with extreme phenotypes, that is, those with surgical SVAS (n=88) and those with no SVAS (n=137). We assessed these 225 individuals for differences in variant burden (defined as the sum of 0s, 1s, and 2s for genotypes 0/0, 0/1, and 1/1, respectively, for the set of variants of an individual, among the 100 744 autosomal nonsynonymous SNVs) based on research cohort membership (Boston, NIH, Nevada‐Toronto, Telethon), chromosomal sex (XX, XY), sequence batch (year 2017, year 2020), or sample type (blood, saliva, immortalized cells) with separate Wilcoxon tests. See Supplemental Methods for a detailed description of our 7‐point variant prioritization strategy.
The summary statistics of the variants that support the findings of this study are available in Data S1‐S3.
Statistical Analysis
The Wilcoxon test implemented in JMP16 software (SAS Institute Inc., Cary, NC) was used for all comparisons in the supplemental figures. R software (https://www.R‐project.org/) implemented through Rstudio (http://www.rstudio.com) was used for generating the principal component plot. The P values calculated from pathway enrichment and association tests were adjusted using the Benjamini and Hochberg method. A cutoff of adjusted P value (false discovery rate [FDR] value)<0.05 was used for selection of enriched pathways and associated pathways.
Results
Demographic Information
The 473 participants with WBS were classified into 4 categories: no SVAS (n=137), mild–moderate SVAS (n=189), surgical SVAS (n=88), and unclassified (n=59). Demographic information is presented in Table. The relative proportions of participants in the no SVAS, mild–moderate SVAS, and surgical SVAS categories are similar to those previously reported in the literature. 9 , 11 , 12 The median age at the last phenotyping event was 9 years, with an interquartile range from 4 to 18 years. Based on self‐report and PC1–2 based imputation for those missing self‐identified race and ethnicity, 421 are of European ancestry, 14 have African ancestry, 5 are of Asian ancestry, and the remaining 33 individuals represented in orange in the PC1–2 of Figure S1 are likely an admixture of European, Asian, and Latine/admixed American ancestry. The percentage of surgical SVAS in each of the 4 cohorts is Boston 20%, Telethon in Italy 25%, NIH 16%, and Nevada‐Toronto 20% (including unclassified participants).
Table 1.
Demographic Information for the 473 Participants With WBS in the Study
| Variable | All patients | No SVAS | Mild SVAS | Surgical SVAS | Unclassified SVAS |
|---|---|---|---|---|---|
| n=473 | n=137 | n=189 | n=88 | n=59 | |
| Sex | |||||
| Female | 236 | 73 | 98 | 35 | 30 |
| Male | 237 | 64 | 91 | 53 | 29 |
| Ancestry | |||||
| European | 421 | 119 | 170 | 80 | 52 |
| Asian | 5 | 3 | 0 | 2 | 0 |
| African | 14 | 7 | 5 | 1 | 1 |
| Mixed | 33 | 8 | 14 | 5 | 6 |
| Age | |||||
| Median | 9 | 13 | 7 | 10 | 2.6 |
| Age range, y | 0.01–62.6 | 3.73–62.6 | 0.01–46 | 0.1–45 | 0.12–60 |
SVAS indicates supravalvar aortic stenosis; and WBS, Williams–Beuren syndrome.
Consistent with previous reports, 44 , 45 the proportion of male participants to female participants was significantly higher in the surgical SVAS group than in the no SVAS group (P=0.048 by χ2 test). Each of the ancestry‐ and sex‐based subgroups had ratios of 1.2 to 2.1 individuals with no SVAS to each person with surgical SVAS. The only exception was the African ancestry subgroup in which 7 had “no SVAS” and 1 had “surgical SVAS,” leading to a 7:1 ratio.
For the extreme phenotype cohort (only those with surgical SVAS and no SVAS, n=225), variant burden did not vary by sample collection site (Figure S2A, P=0.36), chromosomal sex (Figure S2B, P=0.34), sequencing batch (Figure S2C, P=0.46), or sample type (Figure S2D, P=0.21). We noticed, however, that 8 individual samples in the Nevada‐Toronto and NIH collections exhibited increased variant numbers. Those 8 were mixed in terms of sample type, year of sequencing, and chromosomal sex but all belonged to the PC 1–2 cluster ascribed to those of African ancestry. Relatively increased variation is a well‐known feature of the genomic structure of this subgroup. 46 Because our analysis relies on differences in AF between members of the 2 extreme phenotype groups, skew in alleles related to ancestral background (in this case 7 in the no SVAS group and only 1 in the surgical SVAS group) could be conflated with disease outcome. As such, we performed our subsequent analyses using the 217 individuals (87 with surgical SVAS and 130 with no SVAS) without skew, as shown in Figure 1A. To show how our method performs in the presence of ancestry‐associated phenotype skewing, comparisons of these findings (n=217) to the findings when the 8 individuals with African ancestry (1 surgical SVAS, 7 no SVAS; n=225) are included are reported in Supplemental Analysis.
Figure 1. Flow chart of fine phenotyping of SVAS in 473 individuals with WBS and schematic flow chart outlining modifier identification and validation.
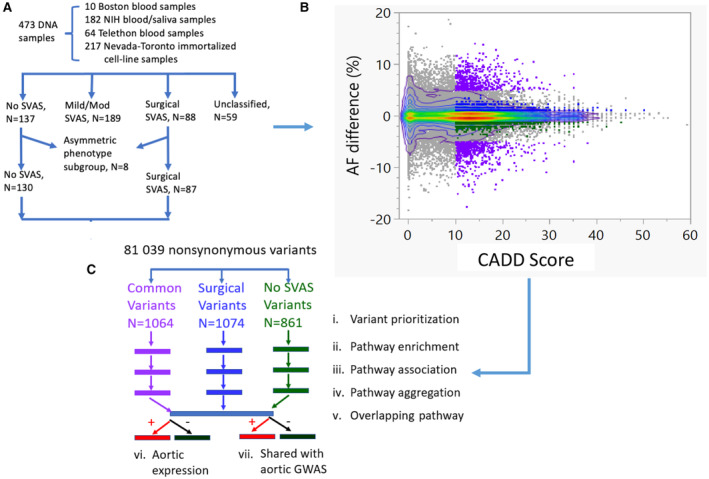
A, Phenotypes were assigned as described in the methods. The asymmetric phenotype subgroup includes 8 individuals with African ancestry: 7 with no SVAS and 1 with surgical SVAS. B, 81039 nonsynonymous variants were plotted based on putative pathogenicity (CADD score, x axis) and AF differences between the surgical minus the no SVAS groups (y axis). The color gradient depicts the density of the variants. Ninety percent of the 81 039 variants are inside the outer‐most bivariate smoothed contour (purple) in the 2‐dimensional plot. C, Steps i–iv are repeated 3 times for the subsets of variants with CADD score>10: (1) “common variants”: those with AF difference between surgical SVAS and no SVAS >5%, (2) “surgical variants” present only in the surgical group (AF>1% in the surgical group and AF=0% in the no SVAS group); (3) “no SVAS variants” present only in the no SVAS group (AF=0% in the surgical group and AF>1% in the no SVAS group). The prioritization process yields 1064 common variants (purple), 1074 “surgical SVAS variants” (blue), and 861 “no SVAS” variants (green). Subsequent pathway enrichment, association, and aggregation revealed 13 key pathways of interest. Validation of genes from key pathways was performed using publicly available data sets (vi and vii). AF indicates allele frequency; CADD, combined annotation dependent depletion; GWAS, genome‐wide association studies; NIH, National Institutes of Health; SVAS, supravalvar aortic stenosis; and WBS, Williams–Beuren syndrome.
Prioritization of Variants
Because our sample size is too small to power discovery of SNV/gene‐level association using established statistical packages, we instead focused on pathway‐level association with SVAS severity in the 217 extreme phenotype participants (Figure 1A). The scatter plot of combined annotation dependent depletion (CADD) score versus AF difference between the surgical and no SVAS groups for the 81 039 variants (after excluding 7671 SNVs with missing CADD scores) is shown in Figure 1B. The maximum difference in AF between the extreme phenotype groups is 18.7%. The variants with extremely high CADD score (CADD >30) were generally rare and showed little difference in AF between the extreme phenotype groups.
We applied our 7‐step strategy, shown in Figure 1C, to perform 3 separate analyses (for steps i–iv, see Supplemental Methods for further details) on variants with CADD_phred score (CADD score) >10 and a clear difference in AF between the 2 extreme phenotype groups. These analyses included (1) “common variants” that are present at a higher rate in 1 of the 2 groups (|AF(surgical)‐AF(no SVAS)|>5%); (2) “surgical variants” present only in the surgical group (AF>1% in the surgical and AF=0 in the no SVAS group); and (3) “no SVAS variants” present only in the no SVAS group (AF=0 in the surgical and AF>1% in the no SVAS group). This prioritization method yielded 1064 “common” variants in 914 genes, 1074 “surgical” variants in 995 genes, and 861 “no SVAS” variants in 816 genes.
Among the 1064 common variants (Data S1), 15 SNVs were stopgain, stoploss, and startloss (see Table S1). Of the 914 genes, 792 have 1 variant each; 104 genes carry 2; and ZAN, CDH23, and ZNF568 have 5 common variants each. No significant differences in the per‐individual burden of the 1064 variants were observed by collection location (P=0.95), chromosomal sex (P=0.07), sequencing batch (P=0.52), sample type (P=0.89), or SVAS status (P=0.70) (Figure S3A through S3E).
Pathway Enrichment and Association Tests of Common Variants in Pathways as a Function of SVAS Severity
We hypothesized that variants with larger AF differences between phenotype groups may be part of the same pathways and may work together to influence physiologic or cellular functions. To identify pathways with an increased burden of candidate variants, we performed pathway enrichment using the 914 common variant genes; this identified 44 pathways (Table S2; step ii in Figure 1C).
We then formally tested each of the 44 enriched pathways for association with SVAS severity (step iii in Figure 1C). The results from the 3 methods, RQT, sequence kernel association test (SKAT), and sequence kernel association test‐optimal (SKAT‐O), are shown in Table S3. The results from SKAT and SKAT‐O are similar. Thirty‐nine of the 44 pathways met the cutoff of FDR<0.05 on both the RQT test and the SKAT or SKAT‐O test (Figure S4). Some overlap exists across the 39 pathways. Based on this observation, we manually consolidated the 39 original pathways to 13 key pathways (Figure 2A) by grouping pathways with similar functions and overlapping genes, taking as the representative pathway the 1 with the greatest number of variant‐affected genes (step iv in Figure 1C). The 13 key pathways include extracellular matrix (ECM; here we maintained both NABA_CORE_MATRISOME 47 (core matrisome) to represent the structural ECM and NABA_MATRISOME 47 (matrisome), which includes both ECM and ECM‐associated proteins like proteases and growth factors), sensory/olfactory signaling, innate immune, developmental biology, polymerase II transcription, metabolism of lipids, transport of small molecule, ciliopathies, adaptive immune, PI3KAKT, disease of metabolism, and endocytosis.
Figure 2. Depiction of SVAS modifier pathways identified through gene set enrichment and association testing.
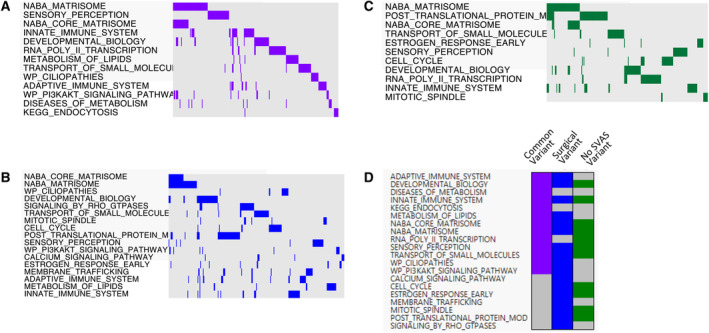
A, Thirteen aggregated common variant pathways. The most enriched pathways are presented in order of statistical significance. The colors represent the presence (purple) and absence (gray) of genes in that pathway (genes are represented by columns across the top of the image). B, Seventeen aggregated surgical SVAS variant pathways in blue. C, Eleven aggregated no SVAS variant pathways in dark green. D, Overlapping of 20 pathways in the 3 sets of pathway analyses. See Figures S4, S5, and S6 for the full (preaggregation) pathways. SVAS indicates supravalvar aortic stenosis.
Pathway Enrichment and Association Tests Using the “Surgical” Variants
As in the common variant analysis, we performed pathway enrichment using the 995 genes (1074 variants, Data S2) prioritized in the “surgical variant” analysis. Variants from 496 of the 995 gene were statistically enriched (FDR value <0.05) into 71 pathways (Table S4). The association tests by RQT and SKAT/SKAT‐O yielded significant results for 58 out of the 71 pathways with FDR value <0.05 (Table S5). In addition to enrichment in ECM pathways, as seen in the common variant analysis, we also observed enrichment for pathways including apoptotic cleavage of cellular protein/apoptotic execution phase, cell cycle/M phase, ciliopathies, developmental biology, mitotic spindle, RHO‐GTPASE, and PI3KAKT. The 58 pathways in Figure S5 were once again manually consolidated to 17 key pathways with overlapping gene content, as shown in Figure 2B. Interestingly, 28 of the 87 individuals with surgical SVAS possessed at least 1 gene with a less common variant among the 18 genes in the mitotic spindle pathway, and 47 exhibited at least 1 gene with a less frequent variant among the 36 genes in the cell cycle pathway. In total, 59 of the 87 individuals possessed a variant in one or both pathways.
Pathway Enrichment and Association Tests Using the “No SVAS” Variants
We identified 25 enriched pathways (with FDR value <0.05) from the 816 genes (861 variants, Data S3) identified in the “no SVAS” variant analysis. Twenty‐three (Figure S6) of the 25 were confirmed by RQT and SKAT/SKAT‐O (Table S6). The 23 pathways were aggregated to 11 key pathways: ECM, posttranslational protein modification, transport of small molecule, developmental biology, RNA polymerase II transcription, cell cycle, sensory, innate immune, estrogen response, and mitotic spindle, shown in Figure 2C.
Overlapping the Pathways Enriched by Common Variants, Surgical Variants, and No SVAS Variants
We then compared the 3 sets of key pathways (Figure 1C, step v, and Figure 2D). The developmental biology, ECM, innate immune, sensory, and transport of small molecule pathways were discovered in all 3 analyses, whereas the others were present in only 1 or 2 sets.
Influence of Ancestry and Phenotypic Skew on Pathway Selection
To determine the impact of skew in a genetic background subgroup, we repeated the 3 sequential allele frequency‐based analyses in the cohort of 225 individuals with WBS, including the additional 8 individuals with African ancestry. Inclusion of these 8 participants had a mild impact on the number of significant pathways from association tests for the common variants analysis and little impact on the number of pathways from the surgical SVAS analysis In contrast, we noted a dramatic increase in the number of pathways identified through the no SVAS analysis. The details of the comparisons are provided in Supplemental Analysis. Of note, the top pathways remained consistent in both analyses.
Core Matrisome Pathway Genes With Modifier Variants Are Expressed in Human Aorta
Because the pathways discovered by this strategy are key to SVAS outcomes, the genes should be expressed by tissues relevant to that pathology. Although some gene products (like those in immune‐mediated or endocrine pathways) are not predicted to be produced by native vascular cells, other products like ECM proteins are expected to be generated and deposited locally. As such, we assessed which of the 76 genes in the core matrisome pathway from our 3 analyses' mRNAs could be detected in human aorta. mRNA from 44 of the 74 genes for which data were available in GTEX (59%) were detected in large arteries (see Figure 3A for tiers of expression). Notably, ACAN (aggrecan), a previously described serum biomarker for detection of aorta dissection, 48 was uniquely expressed in adult human aorta.
Figure 3. mRNA and protein from core matrisome modifier genes found in vascular tissues.
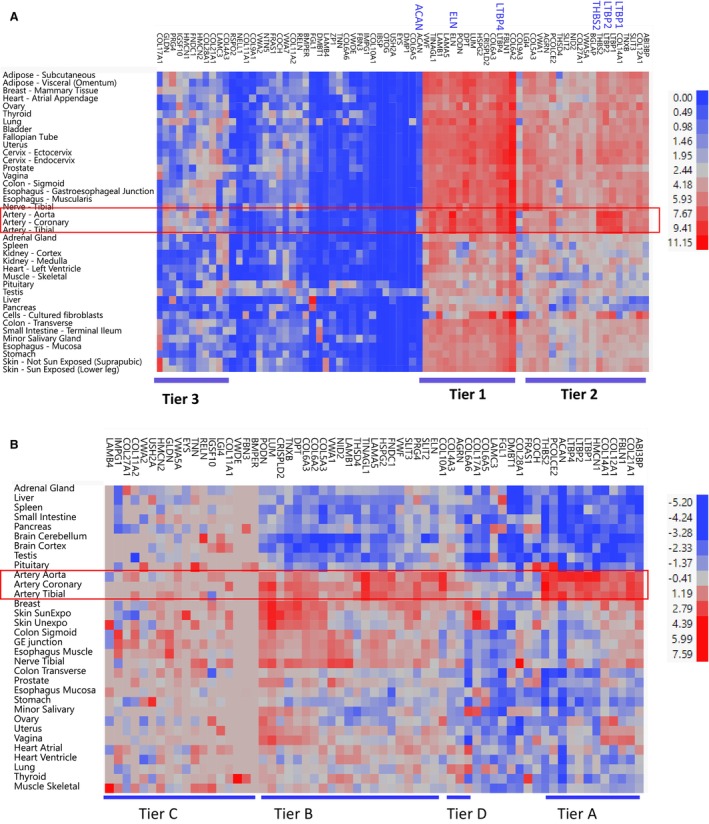
A, Clustering of log2 transformed expression of the 74 core matrisome genes in 37/54 human tissues (the expression in the 15 brain tissues, cell‐EBV, and whole blood are not shown to improve visibility of the remaining 37 tissue names) from GTEX v8 database reveals 3 tiers of expression: Tier 1 contains 15 genes, including ELN, LTBP4, and ACAN that are highly expressed in aorta; Tier 2 contains 19 genes with more moderate expression; and Tier 3 contains 10 genes with lower (but still positive) expression in aorta and other tissues. Of note, 2 of 76 genes identified in our modifier screen were not assessed in the GTEX mRNA database. B, Two‐way clustering of 63 protein levels present in 32 normal human tissues also reveals varied levels of expression: Tier A contains 12 genes, including ACAN, FBLN1, HMCN1, and LTBP4 uniquely expressed in aorta, coronary, and tibial tissues; Tier B contains 22 genes including ELN highly expressed in aorta and other tissues; Tier C contains 17 genes with moderate expression in aorta and other tissues; Tier D contains 2 genes with lowest expression in aorta and artery coronary and artery tibial. Of note, 2 of 76 genes identified in our modifier screen were not assessed in the GTEX mRNA database. Similarly, 13 of the 76 genes were not queried in the protein database. EBV indicates Epstein‐Barr virus.
We next investigated the protein levels of the core matrisome gene products in 32 human tissues. Overall, 84% (53/63) of the protein products present in the https://www.proteinatlas.org/ 49 database from the 76 core matrisome genes identified in our study were present in adult aortic tissue and thus able to have an impact on aortic outcomes. Twelve proteins, including ACAN, FBLN1, HMCN1, and LTBP4, were highly and uniquely expressed in adult aorta (Tier A, Figure 3B).
Innate Immune Pathway Genes With Modifier Variants in Human Aorta
We also looked at the expression of the 45 genes with common variants present in the innate immune pathway in the GTEx v8 database and the proteomics database. 49 As expected, the majority of these genes are expressed in immune tissues (white blood cells, spleen, appendix) as evidenced by mRNA (Figure S7A) and protein (Figure S7B). Interestingly, several of the 45 genes, including ICAM3, ITGAL, MMP9, MMP25, and TLR1, are highly expressed in immune cells with little to no expression elsewhere (including the aorta).
SVAS Modifiers Overlap With Genes Identified Through GWAS of Aortic Disease and Size
Variants in the ECM pathway predicted to modify phenotypic outcomes in WBS may perform similar functions in other aortopathies as well. To evaluate this possibility, we addressed 2 primary questions: (1) Is the matrisome/ECM pathway enriched in other GWAS on aortic diseases or aortic caliber? and (2) Do any aorta‐specific genes identified in those studies overlap with the specific matrisome/ECM pathway genes we identified in the present study?
First, we examined the 86 genes identified in the 13 aortic disease studies found in the National Human Genome Research Institute‐European Bioinformatics Institute GWAS catalog as of September 23, 2021 (see Table S7 for a description of those studies). We found enrichment for the NABA_MATRISOME pathway in those studies (FDR value=2.7E‐04) and additionally noted that 13 of the 86 genes found were included in 11 of our 20 SVAS modifier pathways (Figure 4A). The strongest overlap was seen with genes identified in aortic aneurysm studies (n=9), whereas 2 were ascertained in GWAS of aortic valve stenosis and 4 were noted in studies of aortic vessel or valve calcification. The 5 matrisome/ECM pathway genes: ACAN, COL6A6, CRISPLD2, HMCN2, and MMP9, were found in 4 aortic disorders. Three genes, IL6R, PCSK9, and SYMD2, are of particular interest due to existing clinical studies showing the potential for therapeutic intervention. 50 , 51 , 52 MMP9, found in the matrisome, innate immune, and developmental biology pathways in our study (Figure 4A), has been extensively studied in cancers, aging, and vascular diseases. 53 , 54 , 55
Figure 4. Overlap of genes identified in this study with published aortic disease/trait GWAS.
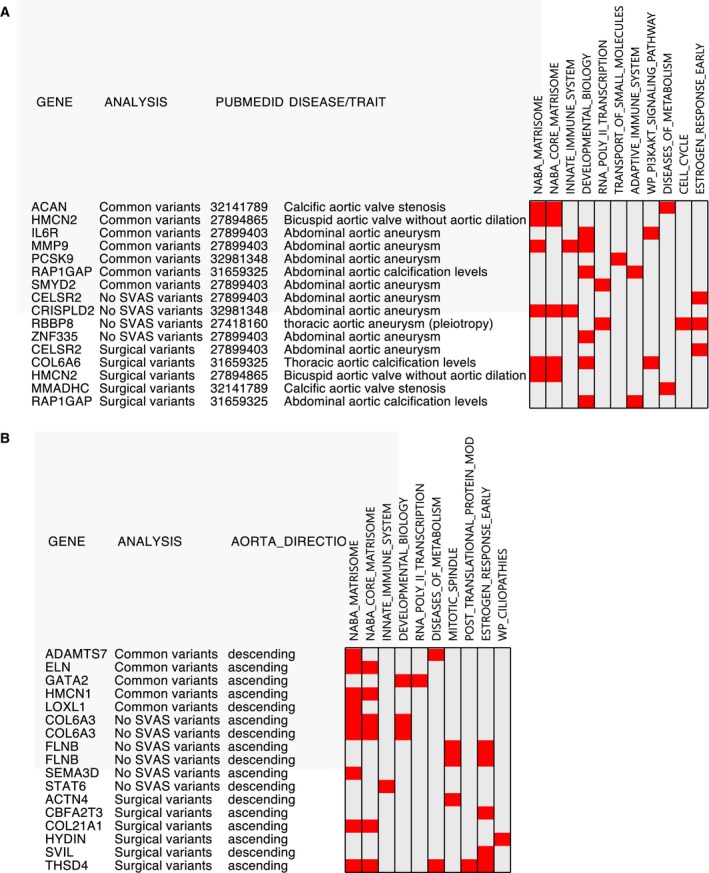
A, Comparison of variants identified in aortic disease GWAS (NHGRI‐EBI GWAS catalog as of September 23, 2021). Thirteen genes involved in 11 pathways in our study were identified in the 13 previously published studies. B, Fifteen genes overlapped between the list of 117 genes in Pirruccello et al's study 36 of aortic size and our SVAS study. Notably, both ELN and HMCN1, identified from ascending aorta, are human aorta specific. GWAS indicates genome‐wide association studies; NHGRI‐EBI, National Human Genome Research Institute‐European Bioinformatics Institute; and SVAS, supravalvar aortic stenosis.
We similarly applied these 2 questions to studies evaluating biomorphic traits of the aorta. Recently, Pirruccello et al 36 identified 117 genes associated with variation in ascending and descending thoracic aortic caliber in ≈40 000 adults enrolled in the UK Biobank (median age ≈64 years). As in the disease‐driven studies, we found enrichment of the NABA_MATRISOME pathway (FDR value=4.7E‐04) in this data set. Likewise, we found that 15 of their 117 genes overlapped with 10 of our 20 pathways (Figure 4B). Of these, 8 genes are part of the NABA_Matrisome pathway. Notably, both ELN and HMCN1, both human aorta specific genes/proteins on the matrisome pathway, were identified in the ascending thoracic aorta analysis. 36 Although not genetic markers per se, low‐density lipoprotein direct and apolipoprotein B are the top 2 clinical features inversely associated with ascending thoracic aorta diameter in Pirruccello et al's study, highlighting a potential role for lipids in affecting outcomes related to aortic dimensions. The summary of genes with common variants present in both our SVAS study and the previous large GWAS studies on aortic disorders is shown in Figure 5.
Figure 5. Summary of overlapping genes affected by common variants in our SVAS study and previous GWAS and depiction of interactions between key pathways inside the aortic wall.
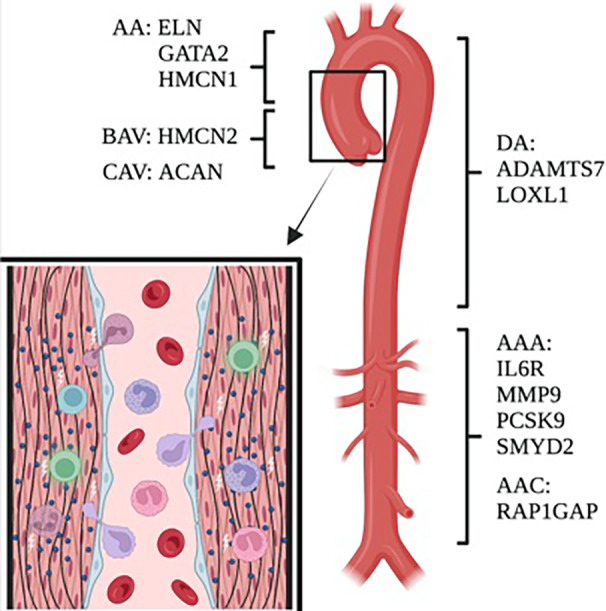
Immune cells, shown in a variety of colors, circulate in the blood and may enter the vessel wall to participate in vascular remodeling. Many of the protein products of the genes shown here are known to participate in this process. AA indicates ascending aorta; AAA, abdominal aortic aneurysm; AAC, abdominal aortic calcification; BAV, bicuspid aortic valve; CAV, calcific aortic valve; DA, descending aorta; GWAS, genome‐wide association studies; and SVAS, supravalvar aortic stenosis.
Discussion
WBS, like many diseases of haploinsufficiency, exhibits wide variability in outcomes. SVAS, a common vascular feature of the condition, varies from life‐threatening to not appreciable in people with the typical 7q11.23 deletion. Although previous mouse and human studies have shown the potential for background genetic variation 56 or environmental exposures 57 to influence vascular outcomes in the setting of elastin insufficiency, the only feature repeatedly shown to be associated with more severe vascular outcome is male sex, 44 , 45 a finding further replicated in the present study. By validating (and expanding) the findings of our earlier proof‐of‐concept exome study 11 in this, the largest WBS genome study to date, we now confirm the importance of background variation in matrisome, immune, and other pathways for influencing vascular outcomes in WBS.
Our approach included 3 major steps: (1) identification of pathways in which gene variation is associated with extreme outcomes, (2) interrogation of the identified genes for expression in tissues relevant to SVAS, and (3) assessment of overlap of the genes identified here with those ascertained in larger aortic GWAS. A similar approach can be undertaken to identify modifiers in other rare conditions.
Matrisome Pathway Variants Confirmed as Key Modifiers of SVAS Outcomes
As in Parrish et al, 11 we detected a strong association between variation in core matrisome and matrisome‐associated pathway genes and extreme SVAS outcomes. ELN, the gene within the WBS deletion that drives the vasculopathy, 6 encodes a smooth muscle cell‐produced extracellular matrix protein that imbues aortic tissue with elasticity. Elastin is deposited in the extracellular space following interactions with other ECM molecules 58 , 59 such as collagens, fibrillins, and fibulins. 60 , 61 , 62 , 63 Elastic fibers interact with the cell through integrin and proteoglycan interactions and are remodeled in response to changes in vascular mechanics and inflammatory processes by matrix metalloproteases 54 , 55 , 64 , 65 , 66 , 67 and other proteases in the extracellular space (Figure 4C). As such, the finding that variation in the genes that make up the matrisome may influence SVAS outcomes is not surprising.
Because components of the ECM are expressed in many tissues, we sought additional confirmation that the modifier genes we identified were relevant to aortic outcomes. Review of publicly available data from GTEX and https://www.proteinatlas.org/ 49 confirmed expression in the aorta (Figures 3A and 3B), with a subset (including ACAN, ELN, HMCN1, and LTBP4) being preferentially expressed there. Because these collections are limited to adult tissues, it is possible that inclusion of developing/pediatric tissues could further increase this percentage. Additionally, we also found significant enrichment in matrisome variants in genomes from individuals with other aortopathies (Figure 4A) and in studies evaluating aortic caliber (Figure 4B). Together, these findings support the role of the matrisome in a variety of aortic outcomes and highlight the validity of a pathway‐based approach in identifying relevant modifiers.
Immune Pathways Highlight the Potential Influence of Inflammation on Mediating SVAS Outcomes
Although it is reassuring that our methods identified expected modifier pathways like the matrisome, the identification of less obvious pathways may hold greater potential for advancement in the field. Review of the literature suggests a growing association between immune regulation and aortic disease. For example, TLR3, a gene on the innate/adaptive immune pathways identified in this study, was recently identified as a central regulator of calcification of the aortic valve. 68 Likewise, researchers recently showed that inhibition of the mTOR (mechanistic target of rapamycin) pathway (PI3KAK) alters tissue biomechanics and cell function in mouse and iPS models of elastin insufficiency. 63 , 69 Additionally, our group previously showed an increase in aortic diameter for Eln +/− ; Rag1 −/− mice that lack B and T cells, 11 and more recently Lin et al 70 showed an influx of monocytes to the area developing stenosis in a new model of elastin insufficiency, the TaglnCre; Eln Fl/Fl . Correspondingly, although healthy aorta exhibits relatively few inflammatory cells and secretion products, a review of published SVAS pathology images notes a neointima with immune cell accumulation and concomitant expression of MMPs (matrix metallopeptidases), including MMP2, 7, and 9, as well as their inhibitors, in some patient specimens. 65 , 71 MMP9 controls the access of monocytes and T cells to the vascular wall in large vessel vasculitis. 55 As such, it is thought that MMP9 contributes to the degradation of ECM proteins during the development of SVAS 65 and aneurysms. 72 These studies suggest complex interactions between ECM molecules and immune cell produced matrix modifiers in aortic media in patients with aneurysmal and stenotic aortopathies.
Pathways Underlying SVAS, Aneurysm, and Bicuspid Aortic Valve Disorders
The concept that modifiers of aortic outcomes may be shared across diseases was recently discussed in an editorial that posited that phenotypes like vascular stenosis and aneurysm may exist on a spectrum 73 and disorders on both ends of the spectrum may share common modifiers. Intriguingly, many of the matrisome genes identified as modifiers in our study have also been reported in aneurysm studies in humans and mice (reviewed in Jana et al 38 , 74 ). Likewise, immune actors are commonly implicated in the pathologic aortic remodeling phenomenon that precedes aneurysm development. 38 , 75 , 76 Our analysis of GWAS on aortic aneurysm 22 , 23 , 24 , 25 , 26 , 27 , 28 and bicuspid aortic valve 29 , 30 suggest that matrisome and innate immune pathways are key modifiers of multiple aortopathy types (Figure 5). MMP9 25 and CRISPLD2 26 were identified in studies of aortic aneurysm whereas ACAN, 32 an aortic‐specific gene, and HMCN2 30 were moderately associated with biscupid aortic valve in 2 separate studies. Although biscupid aortic valve is a valve disease, the aortas of such individuals often bear the stigmata of aneurysm, including elastic fiber fragmentation and increased MMPs, 77 as has been described for SVAS. Together, these findings suggest that health of a tissue is dependent on its ability to balance the rate (or total quantity) of ECM protein synthesis with matrix degradation. When this balance is disturbed, ineffective or destructive remodeling occurs. Although not specifically tested in this study, genetic variation that further perturbs this balance may, therefore, be reasonably expected to influence outcomes. Further ranking of variants based on weighted impact on SVAS outcomes as part of a polygenic score may inform future targeted models aimed at testing the relationship between the primary WBS deletion and background gene variation.
Additional Pathways Identified by “Surgical SVAS Only” and “No SVAS” Analyses
Enrichment analyses performed on recurrent variants unique to either the surgical or no SVAS subset revealed an association with cell cycle/mitotic spindle apparatus and estrogen responsiveness pathways, among others. The cell cycle pathway is intriguing, considering the known increase in smooth muscle cell proliferation seen in SVAS lesions. 63 , 71 Likewise, estrogen signaling pathways could underlie the reported increase in stenosis severity in men relative to women. 11 , 45 Sex hormone effects have also been shown to affect outcomes in other vascular diseases such as vascular Ehlers‐Danlos syndrome 78 and Marfan syndrome. 79 More than 67% of the 217 individuals in the surgical SVAS or no SVAS groups had at least 1 variant in genes in cell cycle pathway, and 34% of those with surgical SVAS had a rarer variant in the estrogen pathway, suggesting that even rarer variants within a pathway could cumulatively occur frequently enough to be considered viable modifiers. With growing information from phased haplotypes from long‐read sequencing, the net effect of rare and common variants of a gene on a haplotype can be studied and will likely be a driving factor in future genomic research.
Limitations of the Study
In this study, we found a difference in extreme outcome frequency in our African ancestry subgroup (7 no SVAS versus 1 surgical) that differed from the other cohorts (1.2 SVAS versus 1 surgical). The differences in the pathway sets are driven by variants that are common (AF >5%) in the individuals of African descent (whose representation in the extreme phenotypes is asymmetric) but rare in those of European, Asian, and Latine/admixed American backgrounds. Because our method is driven by differences in AF between extreme phenotype groups, attention to this limitation in future applications of this method should be considered. Currently, the literature contains no population genetic or cardiovascular study on people with WBS of African descent, and further efforts are needed to increase diversity in rare disease reports. 80 Broad representation is needed to employ robust statistical models that can incorporate samples of multiple ancestries. 81
In addition, we limited our focus to nonsynonymous variants with moderate or higher impact. In future work, we will extend our approaches to consider noncoding variants including those in 3' and 5' UTRs as well as more distant enhancer sites, and we will improve our SVAS classification methods for no SVAS versus mild SVAS and mild SVAS versus surgical SVAS. Multiple layers of statistical testing may increase the rate of false pathway discovery. Although we have used orthogonal methods to substantiate and replicate our top findings, lower tier pathways will need to be similarly validated in future research.
Future Studies
Our study raises new questions to be addressed in future work. First, mechanistic studies are needed to better understand how variation in the matrisome and inflammatory genes/pathways directly contributes to differences in SVAS outcomes. Animal models may be useful in this regard, but further prioritization of variants/pathways will be needed to make this technically tenable. Application of more recent methodologies, such as single cell RNAseq in affected tissues, can allow identification of key cell types within the vessel wall that are most relevant to stenosis severity. Likewise, focusing on the genes identified in the present study may accelerate hypothesis‐driven analyses aimed at the detection of relevant genetic (polygenic risk score calculation) and serum‐based biomarkers for identification of patients with the propensity for surgical SVAS. Key targets could include ACAN, 48 HMCN1, LTBP4, or macrophage/monocyte‐specific gene/protein MMP9. 64 , 66 Additionally, one of the most important aspects of modifier identification is the potential for implementation of novel therapeutics. Given the overlap we found between genes relevant to SVAS and other aortopathies, future efforts leveraging therapies under investigation for those conditions may allow rapid development of therapeutics for treatment of SVAS. For example, the contributions of MMP9 to aortic aneurysm have been studied 53 , 54 , 55 and a variety of MMP inhibitors have been developed for vascular diseases. 82 Regulating matrix proteases during critical periods for SVAS development in children with WBS may be one promising therapeutic strategy. Likewise, enhancing ECM proteins quantity or quality in the aortic wall of those with elastin insufficiency, as was done by regulating LTBP4, 83 may be another promising direction to pursue.
Conclusions
Taken together, our findings have enabled the discovery of new pathways in which the presence of gene variation is associated with more extreme outcomes. These same strategies can easily be implemented for other rare disease applications.
Sources of Funding
The NIH effort was supported by the National Heart, Lung, and Blood Institute Division of Intramural Research, NIH (B.A.K.). B.P.R. and C.A.M. were supported by grants from the Williams Syndrome Association (WSA) and L.R.O. received funding from the Canadian Institutes of Health Research (MOP77720. C.B.M. received support from the National Institute of Neurological Disorders and Stroke (NIH R01 NS35102) and the WSA (WSA 0104 and WSA 0111). C.A.M. and L.R.O. were also partially supported by subcontracts from NIH R01 NS35102. The Genomic Disorder Biobank of the Telethon Network of Genetic Biobanks was supported by Telethon Italy grant GTB12001G, G.M.).
Disclosures
None.
Supporting information
Acknowledgments
This work used the computational resources of the NIH High Performance Computing Biowulf cluster (http://hpc.nih.gov ). We would like to thank the individuals with WBS for contributing their DNA samples to this study and the parents who brought their family members to the clinics to be evaluated, provided the medical/surgical reports used for phenotyping, and answered follow‐up questions. Their participation has made the study possible. We also thank Dr Robert Hufnagel, Dr Dustin Baldridge, and members of the Kozel lab for discussion and comments. Data server websites: gnomAD database: https://gnomad.broadinstitute.org/; National Human Genome Research Institute‐European Bioinformatics Institute GWAS catalog database: https://www.ebi.ac.uk/gwas/; mysigDB data: http://www.gsea‐msigdb.org/gsea/msigdb/annotate.jsp; TOPMED imputation server: https://imputation.biodatacatalyst.nhlbi.nih.gov/; GTEX v8 database: https://www.gtexportal.org/home/.
Preprint posted on MedRxiv September 22, 2022. doi: https://doi.org/10.1101/2022.09.21.22280107.
This article was sent to Jacquelyn Y. Taylor, PhD, PNP‐BC, RN, FAHA, FAAN, Guest Editor, for review by expert referees, editorial decision, and final disposition.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.123.031377
For Sources of Funding and Disclosures, see page 13.
References
- 1. Kozel BA, Barak B, Kim CA, Mervis CB, Osborne LR, Porter M, Pober BR. Williams syndrome. Nat Rev Dis Primers. 2021;7:42. doi: 10.1038/s41572-021-00276-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stromme P, Bjornstad PG, Ramstad K. Prevalence estimation of Williams syndrome. J Child Neurol. 2002;17:269–271. doi: 10.1177/088307380201700406 [DOI] [PubMed] [Google Scholar]
- 3. Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, Spallone P, Stock AD, Leppert M, Keating MT. Hemizygosity at the elastin locus in a developmental disorder, Williams Syndrome. Nat Genet. 1993;5:11–16. doi: 10.1038/ng0993-11 [DOI] [PubMed] [Google Scholar]
- 4. Hillier LW, Fulton RS, Fulton LA, Graves TA, Pepin KH, Wagner‐McPherson C, Layman D, Maas J, Jaeger S, Walker R, et al. The DNA sequence of human chromosome 7. Nature. 2003;424:157–164. doi: 10.1038/nature01782 [DOI] [PubMed] [Google Scholar]
- 5. Jurado LAP, Peoples R, Kaplan P, Hamel BCJ, Francke U. Molecular definition of the chromosome 7 deletion in Williams syndrome and parent‐of‐origin effects on growth. Am J Hum Genet. 1996;59:781–792. [PMC free article] [PubMed] [Google Scholar]
- 6. Li DY, Toland AE, Boak BB, Atkinson DL, Ensing GJ, Morris CA, Keating MT. Elastin point mutations cause an obstructive vascular disease, supravalvular aortic stenosis. Hum Mol Genet. 1997;6:1021–1028. doi: 10.1093/hmg/6.7.1021 [DOI] [PubMed] [Google Scholar]
- 7. Valero MC, de Luis O, Cruces J, Jurado LAP. Fine‐scale comparative mapping of the human 7q11.23 region and the orthologous region on mouse chromosome 5G: the low‐copy repeats that flank the Williams‐Beuren syndrome deletion arose at breakpoint sites of an evolutionary inversion(s). Genomics. 2000;69:1–13. doi: 10.1006/geno.2000.6312 [DOI] [PubMed] [Google Scholar]
- 8. Beuren AJ, Apitz J, Harmjanz D. Supravalvular aortic stenosis in association with mental retardation and a certain facial appearance. Circulation. 1962;26:1235–1240. doi: 10.1161/01.cir.26.6.1235 [DOI] [PubMed] [Google Scholar]
- 9. Collins RT II. Cardiovascular disease in Williams syndrome. Circulation. 2013;127:2125–2134. doi: 10.1161/CIRCULATIONAHA.112.000064 [DOI] [PubMed] [Google Scholar]
- 10. Williams JC, Barratt‐Boyes BG, Lowe JB. Supravalvular aortic stenosis. Circulation. 1961;24:1311–1318. doi: 10.1161/01.cir.24.6.1311 [DOI] [PubMed] [Google Scholar]
- 11. Parrish PCR, Liu D, Knutsen RH, Billington CJ, Mecham RP, Fu YP, Kozel BA. Whole exome sequencing in patients with Williams‐Beuren syndrome followed by disease modeling in mice points to four novel pathways that may modify stenosis risk. Hum Mol Genet. 2020;29:2035–2050. doi: 10.1093/hmg/ddaa093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pober BR, Johnson M, Urban Z. Mechanisms and treatment of cardiovascular disease in Williams‐Beuren syndrome. J Clin Invest. 2008;118:1606–1615. doi: 10.1172/JCI35309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu B, Pjanic M, Wang T, Nguyen T, Gloudemans M, Rao A, Castano VG, Nurnberg S, Rader DJ, Elwyn S, et al. Genetic regulatory mechanisms of smooth muscle cells map to coronary artery disease risk loci. Am J Hum Genet. 2018;103:377–388. doi: 10.1016/j.ajhg.2018.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arnaud P, Milleron O, Hanna N, Ropers J, Ould Ouali N, Affoune A, Langeois M, Eliahou L, Arnoult F, Renard P, et al. Clinical relevance of genotype‐phenotype correlations beyond vascular events in a cohort study of 1500 Marfan syndrome patients with FBN1 pathogenic variants. Genet Med. 2021;23:1296–1304. doi: 10.1038/s41436-021-01132-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Meester JAN, Peeters S, Van Den Heuvel L, Vandeweyer G, Fransen E, Cappella E, Dietz HC, Forbus G, Gelb BD, Goldmuntz E, et al. Molecular characterization and investigation of the role of genetic variation in phenotypic variability and response to treatment in a large pediatric Marfan syndrome cohort. Genet Med. 2022;24:1045–1053. doi: 10.1016/j.gim.2021.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee J, Kim YJ, Lee J; Consortium TD‐G , Kim BJ, Lee S, Park T. Gene‐set association tests for next‐generation sequencing data. Bioinformatics. 2016;32:i611–i619. doi: 10.1093/bioinformatics/btw429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li X, Li Z, Zhou H, Gaynor SM, Liu Y, Chen H, Sun R, Dey R, Arnett DK, Aslibekyan S, et al. Dynamic incorporation of multiple in silico functional annotations empowers rare variant association analysis of large whole‐genome sequencing studies at scale. Nat Genet. 2020;52:969–983. doi: 10.1038/s41588-020-0676-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–D894. doi: 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hindy G, Dornbos P, Chaffin MD, Liu DJ, Wang M, Selvaraj MS, Zhang D, Park J, Aguilar‐Salinas CA, Antonacci‐Fulton L, et al. Rare coding variants in 35 genes associate with circulating lipid levels‐a multi‐ancestry analysis of 170,000 exomes. Am J Hum Genet. 2022;109:81–96. doi: 10.1016/j.ajhg.2021.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Rheenen W, Shatunov A, Dekker AM, McLaughlin RL, Diekstra FP, Pulit SL, van der Spek RA, Vosa U, de Jong S, Robinson MR, et al. Genome‐wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet. 2016;48:1043–1048. doi: 10.1038/ng.3622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bown MJ, Jones GT, Harrison SC, Wright BJ, Bumpstead S, Baas AF, Gretarsdottir S, Badger SA, Bradley DT, Burnand K, et al. Abdominal aortic aneurysm is associated with a variant in low‐density lipoprotein receptor‐related protein 1. Am J Hum Genet. 2011;89:619–627. doi: 10.1016/j.ajhg.2011.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bradley DT, Hughes AE, Badger SA, Jones GT, Harrison SC, Wright BJ, Bumpstead S, Baas AF, Gretarsdottir S, Burnand K, et al. A variant in LDLR is associated with abdominal aortic aneurysm. Circ Cardiovasc Genet. 2013;6:498–504. doi: 10.1161/CIRCGENETICS.113.000165 [DOI] [PubMed] [Google Scholar]
- 24. Gretarsdottir S, Baas AF, Thorleifsson G, Holm H, den Heijer M, de Vries JP, Kranendonk SE, Zeebregts CJ, van Sterkenburg SM, Geelkerken RH, et al. Genome‐wide association study identifies a sequence variant within the DAB2IP gene conferring susceptibility to abdominal aortic aneurysm. Nat Genet. 2010;42:692–697. doi: 10.1038/ng.622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jones GT, Tromp G, Kuivaniemi H, Gretarsdottir S, Baas AF, Giusti B, Strauss E, Van't Hof FN, Webb TR, Erdman R, et al. Meta‐analysis of genome‐wide association studies for abdominal aortic aneurysm identifies four new disease‐specific risk loci. Circ Res. 2017;120:341–353. doi: 10.1161/CIRCRESAHA.116.308765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klarin D, Verma SS, Judy R, Dikilitas O, Wolford BN, Paranjpe I, Levin MG, Pan C, Tcheandjieu C, Spin JM, et al. Genetic architecture of abdominal aortic aneurysm in the million veteran program. Circulation. 2020;142:1633–1646. doi: 10.1161/CIRCULATIONAHA.120.047544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. LeMaire SA, McDonald ML, Guo DC, Russell L, Miller CC III, Johnson RJ, Bekheirnia MR, Franco LM, Nguyen M, Pyeritz RE, et al. Genome‐wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat Genet. 2011;43:996–1000. doi: 10.1038/ng.934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van't Hof FN, Ruigrok YM, Lee CH, Ripke S, Anderson G, de Andrade M, Baas AF, Blankensteijn JD, Bottinger EP, Bown MJ, et al. Shared genetic risk factors of intracranial, abdominal, and thoracic aneurysms. J Am Heart Assoc. 2016;5:e002603. doi: 10.1161/JAHA.115.002603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fulmer D, Toomer K, Guo L, Moore K, Glover J, Moore R, Stairley R, Lobo G, Zuo X, Dang Y, et al. Defects in the exocyst‐cilia machinery cause bicuspid aortic valve disease and aortic stenosis. Circulation. 2019;140:1331–1341. doi: 10.1161/CIRCULATIONAHA.119.038376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gago‐Diaz M, Brion M, Gallego P, Calvo F, Robledo‐Carmona J, Saura D, Sanchez V, Bermejo J, Sevilla T, Newton‐Cheh C, et al. The genetic component of bicuspid aortic valve and aortic dilation. An exome‐wide association study. J Mol Cell Cardiol. 2017;102:3–9. doi: 10.1016/j.yjmcc.2016.11.012 [DOI] [PubMed] [Google Scholar]
- 31. Malhotra R, Mauer AC, Lino Cardenas CL, Guo X, Yao J, Zhang X, Wunderer F, Smith AV, Wong Q, Pechlivanis S, et al. HDAC9 is implicated in atherosclerotic aortic calcification and affects vascular smooth muscle cell phenotype. Nat Genet. 2019;51:1580–1587. doi: 10.1038/s41588-019-0514-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Theriault S, Dina C, Messika‐Zeitoun D, Le Scouarnec S, Capoulade R, Gaudreault N, Rigade S, Li Z, Simonet F, Lamontagne M, et al. Genetic association analyses highlight IL6, ALPL, and NAV1 As 3 new susceptibility genes underlying calcific aortic valve stenosis. Circ Genom Precis Med. 2019;12:e002617. doi: 10.1161/CIRCGEN.119.002617 [DOI] [PubMed] [Google Scholar]
- 33. Theriault S, Gaudreault N, Lamontagne M, Rosa M, Boulanger MC, Messika‐Zeitoun D, Clavel MA, Capoulade R, Dagenais F, Pibarot P, et al. A transcriptome‐wide association study identifies PALMD as a susceptibility gene for calcific aortic valve stenosis. Nat Commun. 2018;9:988. doi: 10.1038/s41467-018-03260-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Benjamins JW, Yeung MW, van de Vegte YJ, Said MA, van der Linden T, Ties D, Juarez‐Orozco LE, Verweij N, van der Harst P. Genomic insights in ascending aortic size and distensibility. EBioMedicine. 2022;75:103783. doi: 10.1016/j.ebiom.2021.103783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Francis CM, Futschik ME, Huang J, Bai W, Sargurupremraj M, Teumer A, Breteler MMB, Petretto E, Ho ASR, Amouyel P, et al. Genome‐wide associations of aortic distensibility suggest causality for aortic aneurysms and brain white matter hyperintensities. Nat Commun. 2022;13:4505. doi: 10.1038/s41467-022-32219-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pirruccello JP, Chaffin MD, Chou EL, Fleming SJ, Lin H, Nekoui M, Khurshid S, Friedman SF, Bick AG, Arduini A, et al. Deep learning enables genetic analysis of the human thoracic aorta. Nat Genet. 2022;54:40–51. doi: 10.1038/s41588-021-00962-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tcheandjieu C, Xiao K, Tejeda H, Lynch JA, Ruotsalainen S, Bellomo T, Palnati M, Judy R, Klarin D, Kember RL, et al. High heritability of ascending aortic diameter and trans‐ancestry prediction of thoracic aortic disease. Nat Genet. 2022;54:772–782. doi: 10.1038/s41588-022-01070-7 [DOI] [PubMed] [Google Scholar]
- 38. Jana S, Hu M, Shen M, Kassiri Z. Extracellular matrix, regional heterogeneity of the aorta, and aortic aneurysm. Exp Mol Med. 2019;51:1–15. doi: 10.1038/s12276-019-0286-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van Varik BJ, Rennenberg RJ, Reutelingsperger CP, Kroon AA, de Leeuw PW, Schurgers LJ. Mechanisms of arterial remodeling: lessons from genetic diseases. Front Genet. 2012;3:290. doi: 10.3389/fgene.2012.00290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lindeman JH, Abdul‐Hussien H, van Bockel JH, Wolterbeek R, Kleemann R. Clinical trial of doxycycline for matrix metalloproteinase‐9 inhibition in patients with an abdominal aneurysm: doxycycline selectively depletes aortic wall neutrophils and cytotoxic T cells. Circulation. 2009;119:2209–2216. doi: 10.1161/CIRCULATIONAHA.108.806505 [DOI] [PubMed] [Google Scholar]
- 41. Thompson RW, Geraghty PJ, Lee JK. Abdominal aortic aneurysms: basic mechanisms and clinical implications. Curr Probl Surg. 2002;39:110–230. doi: 10.1067/msg.2002.121421 [DOI] [PubMed] [Google Scholar]
- 42. Prive F, Aschard H, Ziyatdinov A, Blum MGB. Efficient analysis of large‐scale genome‐wide data with two R packages: bigstatsr and bigsnpr. Bioinformatics. 2018;34:2781–2787. doi: 10.1093/bioinformatics/bty185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O'Connell J, et al. The UK biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li FF, Chen WJ, Yao D, Xu L, Shen JY, Zeng Y, Shi Z, Ye XW, Kang DH, Xu B, et al. Clinical phenotypes study of 231 children with Williams syndrome in China: a single‐center retrospective study. Mol Genet Genomic Med. 2022;10:e2069. doi: 10.1002/mgg3.2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sadler LS, Pober BR, Grandinetti A, Scheiber D, Fekete G, Sharma AN, Urban Z. Differences by sex in cardiovascular disease in Williams syndrome. J Pediatr. 2001;139:849–853. doi: 10.1067/mpd.2001.118889 [DOI] [PubMed] [Google Scholar]
- 46. Genomes Project Consortium , Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics. 2012;11:M111.014647. doi: 10.1074/mcp.M111.014647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Konig KC, Lahm H, Dressen M, Doppler SA, Eichhorn S, Beck N, Kraehschuetz K, Doll S, Holdenrieder S, Kastrati A, et al. Aggrecan: a new biomarker for acute type A aortic dissection. Sci Rep. 2021;11:10371. doi: 10.1038/s41598-021-89653-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jiang L, Wang M, Lin S, Jian R, Li X, Chan J, Dong G, Fang H, Robinson AE, Consortium GT, et al. A quantitative proteome map of the human body. Cell. 2020;183:269–283 e219. doi: 10.1016/j.cell.2020.08.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cai T, Zhang Y, Ho YL, Link N, Sun J, Huang J, Cai TA, Damrauer S, Ahuja Y, Honerlaw J, et al. Association of Interleukin 6 receptor variant with cardiovascular disease effects of interleukin 6 receptor blocking therapy: a phenome‐wide association study. JAMA Cardiol. 2018;3:849–857. doi: 10.1001/jamacardio.2018.2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Interleukin‐6 Receptor Mendelian Randomisation Analysis C , Swerdlow DI, Holmes MV, Kuchenbaecker KB, Engmann JE, Shah T, Sofat R, Guo Y, Chung C, Peasey A, et al. The interleukin‐6 receptor as a target for prevention of coronary heart disease: a Mendelian randomisation analysis. Lancet. 2012;379:1214–1224. doi: 10.1016/S0140-6736(12)60110-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–1722. doi: 10.1056/NEJMoa1615664 [DOI] [PubMed] [Google Scholar]
- 53. Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110:625–632. doi: 10.1172/JCI15334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pyo R, Lee JK, Shipley JM, Curci JA, Mao D, Ziporin SJ, Ennis TL, Shapiro SD, Senior RM, Thompson RW. Targeted gene disruption of matrix metalloproteinase‐9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J Clin Invest. 2000;105:1641–1649. doi: 10.1172/JCI8931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Watanabe R, Maeda T, Zhang H, Berry GJ, Zeisbrich M, Brockett R, Greenstein AE, Tian L, Goronzy JJ, Weyand CM. MMP (matrix metalloprotease)‐9‐producing monocytes enable T cells to invade the vessel wall and cause vasculitis. Circ Res. 2018;123:700–715. doi: 10.1161/CIRCRESAHA.118.313206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kozel BA, Knutsen RH, Ye L, Ciliberto CH, Broekelmann TJ, Mecham RP. Genetic modifiers of cardiovascular phenotype caused by elastin haploinsufficiency act by extrinsic noncomplementation. J Biol Chem. 2011;286:44926–44936. doi: 10.1074/jbc.M111.274779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Addissie YA, Troia A, Wong ZC, Everson JL, Kozel BA, Muenke M, Lipinski RJ, Malecki KMC, Kruszka P. Identifying environmental risk factors and gene‐environment interactions in holoprosencephaly. Birth Defects Res. 2021;113:63–76. doi: 10.1002/bdr2.1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kielty CM. Elastic fibres in health and disease. Expert Rev Mol Med. 2006;8:1–23. doi: 10.1017/erm.2013.9 [DOI] [PubMed] [Google Scholar]
- 59. Kozel BA, Mecham RP. Elastic fiber ultrastructure and assembly. Matrix Biol. 2019;84:31–40. doi: 10.1016/j.matbio.2019.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bax DV, Rodgers UR, Bilek MM, Weiss AS. Cell adhesion to tropoelastin is mediated via the C‐terminal GRKRK motif and integrin alphaVbeta3. J Biol Chem. 2009;284:28616–28623. doi: 10.1074/jbc.M109.017525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kozel BA, Ciliberto CH, Mecham RP. Deposition of tropoelastin into the extracellular matrix requires a competent elastic fiber scaffold but not live cells. Matrix Biol. 2004;23:23–34. doi: 10.1016/j.matbio.2004.02.004 [DOI] [PubMed] [Google Scholar]
- 62. Krishnamurthy VK, Evans AN, Wansapura JP, Osinska H, Maddy KE, Biechler SV, Narmoneva DA, Goodwin RL, Hinton RB. Asymmetric cell‐matrix and biomechanical abnormalities in elastin insufficiency induced aortopathy. Ann Biomed Eng. 2014;42:2014–2028. doi: 10.1007/s10439-014-1072-y [DOI] [PubMed] [Google Scholar]
- 63. Misra A, Sheikh AQ, Kumar A, Luo J, Zhang J, Hinton RB, Smoot L, Kaplan P, Urban Z, Qyang Y, et al. Integrin beta3 inhibition is a therapeutic strategy for supravalvular aortic stenosis. J Exp Med. 2016;213:451–463. doi: 10.1084/jem.20150688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chiao YA, Dai Q, Zhang J, Lin J, Lopez EF, Ahuja SS, Chou YM, Lindsey ML, Jin YF. Multi‐analyte profiling reveals matrix metalloproteinase‐9 and monocyte chemotactic protein‐1 as plasma biomarkers of cardiac aging. Circ Cardiovasc Genet. 2011;4:455–462. doi: 10.1161/CIRCGENETICS.111.959981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dridi SM, Foucault Bertaud A, Igondjo Tchen S, Senni K, Ejeil AL, Pellat B, Lyonnet S, Bonnet D, Charpiot P, Godeau G. Vascular wall remodeling in patients with supravalvular aortic stenosis and Williams Beuren syndrome. J Vasc Res. 2005;42:190–201. doi: 10.1159/000085141 [DOI] [PubMed] [Google Scholar]
- 66. Li T, Jing JJ, Yang J, Sun LP, Gong YH, Xin SJ, Yuan Y. Serum levels of matrix metalloproteinase 9 and toll‐like receptor 4 in acute aortic dissection: a case‐control study. BMC Cardiovasc Disord. 2018;18:219. doi: 10.1186/s12872-018-0958-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mecham RP, Broekelmann TJ, Fliszar CJ, Shapiro SD, Welgus HG, Senior RM. Elastin degradation by matrix metalloproteinases. Cleavage site specificity and mechanisms of elastolysis. J Biol Chem. 1997;272:18071–18076. doi: 10.1074/jbc.272.29.18071 [DOI] [PubMed] [Google Scholar]
- 68. Gollmann‐Tepekoylu C, Graber M, Hirsch J, Mair S, Naschberger A, Polzl L, Nagele F, Kirchmair E, Degenhart G, Demetz E, et al. Toll‐like receptor 3 mediates aortic stenosis through a conserved mechanism of calcification. Circulation. 2023;147:1518–1533. doi: 10.1161/CIRCULATIONAHA.122.063481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jiao Y, Li G, Li Q, Ali R, Qin L, Li W, Qyang Y, Greif DM, Geirsson A, Humphrey JD, et al. mTOR (mechanistic target of rapamycin) inhibition decreases mechanosignaling, collagen accumulation, and stiffening of the thoracic aorta in elastin‐deficient mice. Arterioscler Thromb Vasc Biol. 2017;37:1657–1666. doi: 10.1161/ATVBAHA.117.309653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lin CJ, Hunkins BM, Roth RA, Lin CY, Wagenseil JE, Mecham RP. Vascular smooth muscle cell subpopulations and neointimal formation in mouse models of elastin insufficiency. Arterioscler Thromb Vasc Biol. 2021;41:2890–2905. doi: 10.1161/ATVBAHA.120.315681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Urban Z, Riazi S, Seidl TL, Katahira J, Smoot LB, Chitayat D, Boyd CD, Hinek A. Connection between elastin haploinsufficiency and increased cell proliferation in patients with supravalvular aortic stenosis and Williams‐Beuren syndrome. Am J Hum Genet. 2002;71:30–44. doi: 10.1086/341035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sakalihasan N, Delvenne P, Nusgens BV, Limet R, Lapiere CM. Activated forms of MMP2 and MMP9 in abdominal aortic aneurysms. J Vasc Surg. 1996;24:127–133. doi: 10.1016/s0741-5214(96)70153-2 [DOI] [PubMed] [Google Scholar]
- 73. Luperchio TR, Kozel BA. Extending the spectrum in aortopathy: stenosis to aneurysm. Curr Opin Genet Dev. 2022;76:101962. doi: 10.1016/j.gde.2022.101962 [DOI] [PubMed] [Google Scholar]
- 74. Pinard A, Jones GT, Milewicz DM. Genetics of thoracic and abdominal aortic diseases. Circ Res. 2019;124:588–606. doi: 10.1161/CIRCRESAHA.118.312436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kuivaniemi H, Platsoucas CD, Tilson MD III. Aortic aneurysms: an immune disease with a strong genetic component. Circulation. 2008;117:242–252. doi: 10.1161/CIRCULATIONAHA.107.690982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sakalihasan N, Michel JB, Katsargyris A, Kuivaniemi H, Defraigne JO, Nchimi A, Powell JT, Yoshimura K, Hultgren R. Abdominal aortic aneurysms. Nat Rev Dis Primers. 2018;4:34. doi: 10.1038/s41572-018-0030-7 [DOI] [PubMed] [Google Scholar]
- 77. Verma S, Siu SC. Aortic dilatation in patients with bicuspid aortic valve. N Engl J Med. 2014;370:1920–1929. doi: 10.1056/NEJMra1207059 [DOI] [PubMed] [Google Scholar]
- 78. Bowen CJ, Calderon Giadrosic JF, Burger Z, Rykiel G, Davis EC, Helmers MR, Benke K, Gallo MacFarlane E, Dietz HC. Targetable cellular signaling events mediate vascular pathology in vascular Ehlers‐Danlos syndrome. J Clin Invest. 2020;130:686–698. doi: 10.1172/JCI130730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nucera M, Heinisch PP, Langhammer B, Jungi S, Mihalj M, Schober P, Luedi MM, Yildiz M, Schoenhoff FS. The impact of sex and gender on aortic events in patients with Marfan syndrome. Eur J Cardiothorac Surg. 2022;62:62. doi: 10.1093/ejcts/ezac305 [DOI] [PubMed] [Google Scholar]
- 80. Graham G. Disparities in cardiovascular disease risk in the United States. Curr Cardiol Rev. 2015;11:238–245. doi: 10.2174/1573403X11666141122220003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Chen H, Huffman JE, Brody JA, Wang C, Lee S, Li Z, Gogarten SM, Sofer T, Bielak LF, Bis JC, et al. Efficient variant set mixed model association tests for continuous and binary traits in large‐scale whole‐genome sequencing studies. Am J Hum Genet. 2019;104:260–274. doi: 10.1016/j.ajhg.2018.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Raffetto JD, Khalil RA. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol. 2008;75:346–359. doi: 10.1016/j.bcp.2007.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Demonbreun AR, Fallon KS, Oosterbaan CC, Vaught LA, Reiser NL, Bogdanovic E, Velez MP, Salamone IM, Page PGT, Hadhazy M, et al. Anti‐latent TGFbeta binding protein 4 antibody improves muscle function and reduces muscle fibrosis in muscular dystrophy. Sci Transl Med. 2021;13:eabf0376. doi: 10.1126/scitranslmed.abf0376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Li H, Durbin R. Fast and accurate long‐read alignment with burrows‐wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single‐cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhbannikov IY, Arbeev KG, Yashin AI. Rqt: an R package for gene‐level meta‐analysis. Bioinformatics. 2017;33:3129–3130. doi: 10.1093/bioinformatics/btx395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhan X, Hu Y, Li B, Abecasis GR, Liu DJ. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics. 2016;32:1423–1426. doi: 10.1093/bioinformatics/btw079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gogarten SM, Sofer T, Chen H, Yu C, Brody JA, Thornton TA, Rice KM, Conomos MP. Genetic association testing using the GENESIS R/Bioconductor package. Bioinformatics. 2019;35:5346–5348. doi: 10.1093/bioinformatics/btz567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Jiang L, Zheng Z, Fang H, Yang J. A generalized linear mixed model association tool for biobank‐scale data. Nat Genet. 2021;53:1616–1621. doi: 10.1038/s41588-021-00954-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Variant data are made available as part of the data supplement (Data S1‐S3).