

Abstract
Direct intercellular transfer of cellular components is a recently described general mechanism of cell–cell communication. It is a more non-specific mode of intercellular communication that is not actively controlled by the participating cells. Though membrane bound proteins and small non-protein cytosolic components have been shown to be transferred between cells, the possibility of transfer of cytosolic proteins has not been clearly established, and its mechanism remains unexplained. Using a cell–cell pair of metastatic melanoma and endothelial cells, known to interact at various stages during cancer progression, we show that cytosolic proteins can indeed be transferred between heterotypic cells. Using precise relative cell patterning we provide evidence that this transfer depends on extent of the interface between heterotypic cell populations. This result is further supported by a mathematical model capturing various experimental conditions. We further demonstrate that cytosolic protein transfer can have important functional consequences for the tumor–stroma interactions, e.g., in heterotypic transfer of constitutively activated BRAF, a common melanoma associated mutation, leading to an enhanced activation of the downstream MAPK pathway. Our results suggest that cytosolic protein transfer can have important consequences for regulation of processes involving physical co-location of heterotypic cell types, particularly in invasive cancer growth.
Introduction
Cell–cell communication plays an important role in various physiological or pathological contexts. For example, interaction of leukocytes with the endothelium can guide the wound healing process, while communication of cancer cells with endothelial or connective tissues regulates metastasis.1–3 These cell–cell interactions include autocrine,4 paracrine,5 and juxtacrine intermolecular signaling,6,7 or indirect cell interaction through force application onto the extracellular matrix by fibroblasts and other cell types.8 Another, less understood mechanism that cells employ to communicate with each other is the direct intercellular transfer of cellular components between adjacent cells.9–14 This mechanism essentially differs from other forms of cell–cell communication because it is a considerably less specific and controlled form of communication, wherein the induced phenotypic changes depend on the expression of corresponding molecular components in just one (donor) of the interacting cell types.14
Previous studies reported on the functional transfer of various membrane proteins and small non-protein cytosolic components between homotypic and heterotypic cell types.14–18 Since proteins constitute the bulk of functional machinery in the cell, and are primarily responsible for most of the cellular phenotypes, intercellular protein transfer can be of considerable physiological significance. However, although transfer of membrane components has been known to occur for quite some time, the evidence for intercellular transfer of cytosolic proteins has remained weak or inconsistent.9,15,19 The existence of such transfer can be of particular importance in the context of tumor–stroma interactions, many of which are thought to govern invasive cancer spread, e.g., in advanced stages of melanoma. Cytosolic or membrane associated proteins like beta-catenin have been shown to be transferred intercellularly via exosomes,20 while the existence of tunneling nanotubes (TNTs) has been documented in malignant cancer cells in culture, as well as in vivo.21 Although a definitive demonstration of intercellular protein transfer between cells has been lacking, these studies highlight that intercellular transfer of cytosolic proteins from cancer cells to other cell types is possible, including those that could elicit a phenotypic response in the recipient cells. Here, we demonstrate that cytosolic protein transfer indeed occurs upon heterotypic interaction between aggressive melanoma cells and endothelial cells, whose interactions occur during metastatic events for example, during angiogenesis22 and extravasation.23 Since protein transfer can occur through a range of mechanisms that are frequently difficult to establish,9,15,21 we suggest a new strategy to unravel the mechanistic details through controlled patterning of cells during co-culture. Using this technique, we suggest that intercellular cytosolic protein transfer occurs via direct cell–cell contact, through formation of transient cell–cell fusions.
Finally we argue that cytosolic protein transfer can have important functional consequences. In particular, using co-culture of two cancer cell types, with one containing a constitutively active BRAF, present in a large percentage of invasive melanomas,24 we show that prolonged heterotypic co-cultures can result in transfer of signaling molecules, activating the downstream mitogen-activated kinase (MAPK) pathway.
Results
Cancer cells acquire the capability to physically interact with the endothelium as they become metastatic.1,2 While cancer in its radial growth stage is localized at the site of its origin, as it becomes more metastatic, it acquires the capability to interact with the endothelium and excavate to newer sites of colonization. Therefore, as a physiologically relevant model of heterotypic cellular interaction pair, we chose a metastatic melanoma cell type (1205Lu cells), and human umbilical cord endothelial cells (HUVECs).25 1205Lu cells were lentivirally transduced with a green fluorescent protein (GFP) expressing plasmid, clonally selected, and sorted using fluorescence assisted cell sorting (FACS) to ensure clonal GFP expressing population with a small population wide variability. Prior analysis of intercellular protein transfer suggested that it can depend strongly on the duration of co-culture and the ratio of the donor and recipient cells. To explore whether GFP can be transferred and to test the dependence of the transfer efficiency on the co-culture parameters, 1205Lu-GFP cells were co-cultured with HUVEC cells in varying ratios, and varying initial seeding densities, and maintained in 1% serum to minimize proliferation (Fig. 1A). Flow cytometry was performed 1, 3, 5, and 7 days after cell seeding and GFP levels in recipient HUVECs were measured vs. cells cultured without co-culture (Fig. 1B). The GFP fluorescence intensity values in donor (1205Lu-GFP) and recipient (HUVEC) cells varied by several orders of magnitude, permitting unambiguous designation of the cell type in co-culture without the need for additional labeling of the acceptor cells. We found that co-culture of 1205Lu-GFP cells and HUVECs resulted in an increase in GFP levels in HUVECs in a co-culture duration and cell seeding density dependent manner. When cells were seeded in a 1 : 1 ratio with a low initial density (ρ = 10 000 cm−2), average GFP levels of HUVECs increased slowly (Fig. 1C), as compared to when the seeding ratio of donor cells was 3 times higher than recipient cells (Fig. 1D). These data indicated that observable GFP transfer does increasingly occur when cells are co-cultured for over 1 week, and is dependent on the donor to recipient cell ratio. Indeed, increasing the density of initial cell seeding enhanced both the kinetics, and the extent of GFP transfer from donor to recipient cells, as observed for ρ = 50 000 cells per cm2 (Fig. 1E and Fig. S1, ESI†). The trend of dependence on both the donor/recipient ratio and cell seeding density continued for higher values of these parameters (Fig. 1F–G and Fig. S1, ESI†).
Fig. 1.

Heterotypic cytosolic protein transfer. (A) Schematic of the experimental design. HUVECs and 1205Lu-GFP cells are randomly mixed in different ratios and initial seeding densities, and time course analysis of protein transfer from 1205Lu-GFP cells to HUVECs is observed by flow cytometry. The right panel shows an example of flow cytometry showing GFP intensity of HUVECs and 1205Lu-GFP cells randomly mixed and co-cultured with an initial ratio of 1 : 1 and seeding density of 100 000 cm−2. (B–G) Flow cytometry analysis of average GFP intensity of the recipient HUVECs after co-culture with 1205Lu-GFP cells shows an increase in average GFP intensity over time in co-culture with different kinetics. Average GFP intensity of HUVECs after 1, 3, 5 and 7 days of co-culture with 1205Lu-GFP cells with initial seeding cell density of 10 000 cm−2 with the ratio of HUVECs and 1205Lu-GFP cells being 1 : 1 (B), 1 : 3 (C), with initial seeding cell density of 50 000 cm−2 with the ratio of HUVECs and 1205Lu-GFPcells being 1 : 1 (D), 1 : 3 (E), with initial cell seeding density of 100 000 cm−2 with the ratio of HUVECs and 1205Lu-GFP cells being 1 : 1 (F), and 1 : 3 (G). In all panels above, *, P < 0.05, **, P < 0.01, ***, P < 0.001, between conditions connected with horizontal bars. Error bars represent the standard error of mean (s.e.m), n = 3 for independent co-culture experiments.
Various mechanisms proposed to account for intercellular protein transfer fall into two general groups: those postulating release/re-incorporation of small membrane vesicles and transient cell–cell fusion. For many reported instances of transfer, the available data do not permit one to unequivocally distinguish between these mechanisms.9,13,26 E.g., the dependence of the transfer efficiency on the cell seeding density (Fig. 1) is consistent with both possibilities, as an increased number of interacting cells can lead to both enhanced cell–cell contact and elevated number of secreted episomal membrane vesicles. To distinguish between these putative mechanisms, we developed a new assay allowing controlled variation of the interface between donor and recipient cells, while preserving the same overall number of cells (or seeding density) in the co-culture. This was achieved using stencil masks micro-fabricated from PDMS (poly dimethoxy silane), a common biocompatible material used for fabrication of microfluidic devices. Using this methodology, donor and recipient cells could be co-cultured to create a heterotypic cell interface in the form of a straight line (Fig. 2A inset, 2D), or a circle with a diameter of 2000 μm (Fig. 2B inset, 2E). A random mixture of cell types maximizing the effective interface between the cells was also used as a control (Fig. 2C inset, 2F). Overall, cell micropatterning allowed us to vary the extent of initial heterotypic cell interactions from low (1205Lu-GFP cells and HUVECs separated by a line), medium (1205Lu-GFP cells in circular islands surrounded by HUVECs), high (random co-culture), and no interaction (cells cultured separately). We estimated the interaction length per unit area (ILA) as the parameterized line where heterotypic interaction occurred normalized by the total area covered by the cells (Fig. 2A–C, ESI†). The ILA was 11 times higher in circular border co-cultures vs. the straight line ones (Fig. 2A and B). By contrast, in the case of random mixing of cells, if we assumed HUVECs and 1205Lu cells to be squares of sides 10 μm and 5 μm respectively, organized in a checkerboard fashion, the length of heterotypic interaction was 2186 times higher compared to the straight line (Fig. 2A and C), representing the upper limit of the interaction interface.
Fig. 2.
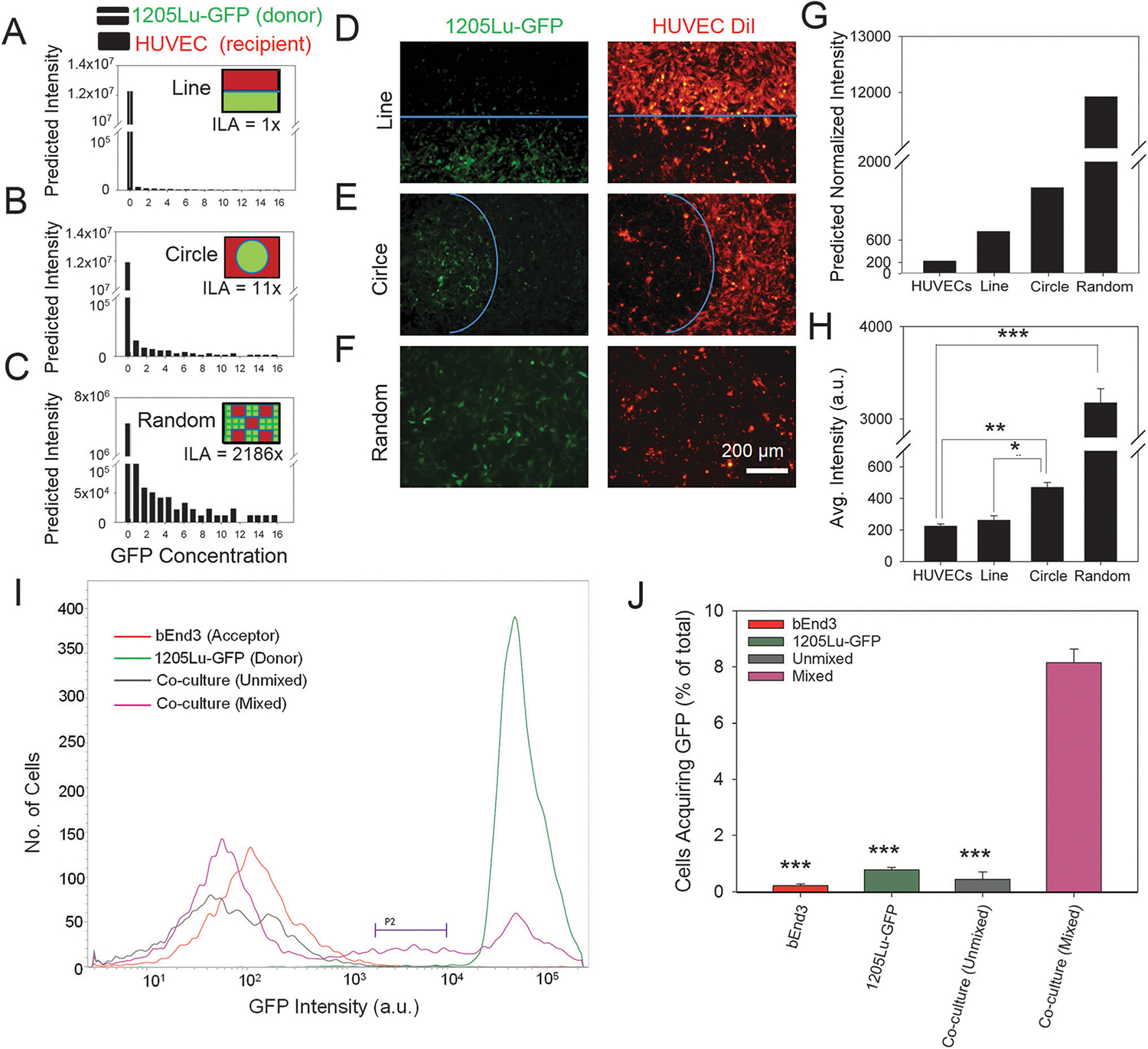
Cytosolic intercellular protein transfer occurs via direct cell–cell contact. The computational model showing distribution of GFP concentration in recipient HUVECs with the ILA (heterotypic interactive length per unit area) being 1× (A), 11× (B), and 2186× (C); striped bars in A–C refer to donor GFP +ve cells, while black bars refer to GFP levels in recipient cells; schematic showing the design of computational model and experimental design of patterning DiI-HUVECs (red) and 1205Lu-GFP cells (green) in a line, circle, or randomly to control the ILA (shown in solid blue line) shown in the inset of A–C. Microfabricated stencil based patterning of DiI-labeled HUVECs and 1205Lu-GFP cells with ILA topology being a line (D), circle (E), and randomly distributed cells (F); in A–F the initial seeded area for each cell type was kept equal, while changing the ILA. Computationally calculated GFP intensity of HUVECs at an interface with 1205Lu-GFP cells in the form of a line, circle, or in a random mixture (G); intensity is normalized to the initial experimental value of unlabeled HUVECs. Average GFP intensity of HUVECs after 7 days of co-culture with 1205Lu-GFP cells interacting with the HUVECs at an interface designed as a line, circle, or in a random mixture (H). (I and J) Cytosolic intercellular protein transfer requires cells to be physically in proximity; (H) representative flow cytometry profiles showing GFP expression in a population of bEnd3 endothelial cells and 1205Lu-GFP cells cultured separately (red, and green respectively), and of bEnd3 endothelial cells co-cultured with 1205Lu-GFP cells separated by a porous membrane but sharing the medium (gray), and co-cultured with direct cell–cell contacts (pink). P2 shows the subpopulation of bEnd3 endothelial cells that have received GFP from 1205Lu-GFP cells; (J) quantified analysis of percentage of cells in the region defined by P2 in I showing that physical contacts of cells is a requirement for intercellular cytosolic protein transfer. In all the above panels, *, n = 3 for independent co-culture experiments, P < 0.05, **, P < 0.01, ***, P < 0.001, between conditions connected with horizontal bars; error bars represent the standard error of mean (s.e.m).
To set expectations for the experimental data provided by this new technique, we implemented the geometric arrangements described above within a previously reported mathematical model, postulating cell contact mediated intercellular protein transfer.13 The model predicted that in the contact mediated but not vesicle based transfer mechanism, random mixing of donor and recipient cells would result in the maximum transfer of cellular components from donor to recipient cells (Fig. 2C and G), followed by a lower efficiency of transfer in a circle of donor surrounded by recipient cells (Fig. 2B and G), with the lowest transfer extent achieved with ILA limited to a straight line (Fig. 2A and G). These predictions were in sharp contrast to the approximately equal values of the transfer efficiency expected for the vesicle mediated transfer mechanism when the medium was thoroughly mixed, as was the case in our subsequent experiments. Indeed, in this case, the efficiency is only dependent on the relative number of donor cells, which was approximately equal under all conditions examined.
To test these predictions experimentally, HUVECs were labeled with DiI, and patterned with 1205Lu-GFP cells as described above in glass-bottomed dishes coated with 10 μg ml−1 of fibronectin (Fig. 2D–F). Flow cytometry after 7 days of co-culture revealed a high correlation between ILA and GFP transfer (Fig. 2H). With a small ILA for donor and recipient cell types (heterotypic interaction over a straight line), the GFP transfer was not observed (Fig. 2D and H), while increasing the ILA by 11 times (heterotypic interactions via circular islands of donor surrounded by recipient cells) showing a significant increase in average GFP intensity in the recipient cells (Fig. 2E and H). Random mixing of cells, maximizing ILA, further significantly increased the extent of GFP transfer (Fig. 2F and H). The modeling and experimental results were in close agreement (cf. Fig. 2G and H), strongly favoring the contact mediated mechanism of cytosolic protein transfer.
To further confirm whether heterotypic cell–cell contact is necessary for intercellular protein transfer, and if this transfer is more general in nature, we co-cultured 1205Lu-GFP cells with bEnd3 endothelial cells either by mixing them together, or by physically separating them with a porous membrane with a pore size of 0.4–1 μm (Fig. 2I). We found that co-culture of physically separated cells accompanied by mixing of the medium did not result in transfer of GFP from 1205Lu-GFP cells to bEnd3 endothelial cells. In contrast cells that were mixed together in the same chamber showed the highest percentage of cells that acquired GFP (Fig. 2J). These data indicate that cell–cell contact is an essential requirement for intercellular transfer of GFP, and likely other cytosolic proteins.
Cytoskeleton and membrane properties can have a considerable effect on the efficiency of intercellular protein transfer. For instance, functional cytoskeletons can both increase the rate of cell migration and thus the probability of encounter between donor and recipient cells, and also enhance the stability of intercellular contacts, as has been shown e.g., for intercellular TNTs.26 We repeatedly found evidence of thin membranous connections between HUVECs and 1205Lu cells resembling TNTs (Fig. 3A), which suggested that transient cell–cell fusion might have occurred. Indeed, Cytochalasin B (cytoB), a blocker of contractile actin microfilament formation also known to de-stabilize TNTs, significantly decreased GFP transfer, further providing evidence for contact mediated transfer of cytosolic proteins (Fig. 3B).27 We also tested whether altering the stability of actin microfilaments, and thereby the average length of TNT-like nanotubes, could influence intercellular protein transfer. To understand the effect of nanotube stability on intercellular protein transfer across distance, we simulated the transfer by assuming that the donor cells were patterned as circular islands, surrounded by recipient cells, similar to our patterned co-culture experiments (Fig. 2B and E). This model predicted that a decreased stability of nanotube formation can result in a decreased transfer of proteins across the intercellular distance (Fig. 3C), consistent with the experimental results. Membrane fluidity can also alter the efficiency of intercellular transfer of various cellular components,13 although its relevance to transfer cytosolic rather than membrane proteins is not obvious. Treatment with cyclodextrin, known to manipulate the cellular content of cholesterol,28 did not affect the extent of GFP transfer, though increasing membrane fluidity by linoleic acid treatment facilitated higher protein transfer (Fig. 3D). Increased membrane fluidity may possibly influence cytosolic protein transfer by either increasing the effective contact area between cells, or increased propensity of material exchange when the cells come in contact via actin based structures, e.g. TNTs. These data indicate that the characteristics of structural cellular components, e.g., the cytoskeleton and the membrane, can strongly influence the efficiency of transfer, in a manner consistent with the contact mediated transfer mechanism.
Fig. 3.

Intercellular protein transfer is regulated by membrane properties. (A) The representative phase-contrast image showing tunneling nanotubes connecting a HUVEC and a 1205Lu cell; scale bar = 5 μm. (B) Average GFP intensity in HUVECs when co-cultured with 1205Lu-GFP cells in the presence of cytochalasin B for 7 days with an initial seeding. (C) Computational model simulation of a patterned co-culture set-up without, and with decreased nanotube stability (or the average nanotube length decreased by 50%), causing high diffusion of cytosolic proteins due to enhanced formation of transient heterotypic cell–cell bonds. Time lapse acquirement of GFP levels is shown in HUVECs at various distances from the 1205Lu-GFP cells; the distance is shown in units of HUVEC cell lengths. (D) Flow cytometry analysis of co-culture of HUVECs and 1205Lu-GFP cells with a 1 : 3 ratio at an initial total density of 100 000 cm−2 for 7 days in the presence of DMSO (Cntrl), Cyclodextrin (CycloD), and Linoleic Acid (LA). In the panels above, *, P < 0.05, ***, P < 0.001; error bars represent the standard error of mean (s.e.m.).
We then explored if transfer of cytosolic proteins may have functional consequences, particularly in the context of interaction between cancer cells, and between cancer cells and stroma. Of particular consequence may be the transfer of signaling proteins, both due to frequently activating mutations found in signal transduction networks of cancer cells and due to amplifying effects of signaling cascades. We focused on constitutively activated BRAF mutation (valine replaced at locus 600 by glutamic acid, BRAF-V600E), present in a majority of invasive melanomas, and responsible for constitutive activation of the pro-proliferative mitogen activated kinase (MAPK) pathway.29,30 As donor cells we chose human malignant melanoma A375 cells, while as a recipient partner we selected human embryonic kidney 293T cells. 293T cells are known to have properties characteristic of both cancer and epithelial phenotypes, and their co-culture with A375 cells could serve as a dual model for heterotypic cancer cell interaction, as well as cancer–stroma interaction. HEK cells were labeled with a very high molecular weight fluorescently labeled dextran to preserve the identity of cells during co-culture, since cytosolic molecules have been shown to be transferred in a molecular weight dependent manner.13,26 Cells were co-cultured for 12 days at a high density (100 000 cm−2) in the presence of linoleic acid to maximize potential protein transfer, and with 2% serum to minimize cell proliferation while maintaining survival. Dextran labeled 293T cells were sorted and probed for BRAF-V600E and downstream signaling molecules in the MAPK pathway (Fig. 4A). A recently available highly specific antibody against BRAF-V600E allowed us to confirm the high abundance of BRAF-V600E in A375 cancer cells,24 while control 293T cells did not contain the mutated BRAF protein (Fig. 4B), and also had reduced phosphorylation of Erk1/2 (Fig. 4B). We probed for BRAF-V600E in 293T cells sorted post co-culture and found a readily detectable transfer of BRAF-V600E (Fig. 4C and D, Fig. S2A, ESI†). Importantly, we also found a corresponding increase in phosphorylation of Erk1/2 in sorted 293T cells indicating that it is possible for intercellular transfer of signaling molecules to influence downstream signal transduction pathways (Fig. 4E and F, Fig. S2B, ESI†).
Fig. 4.
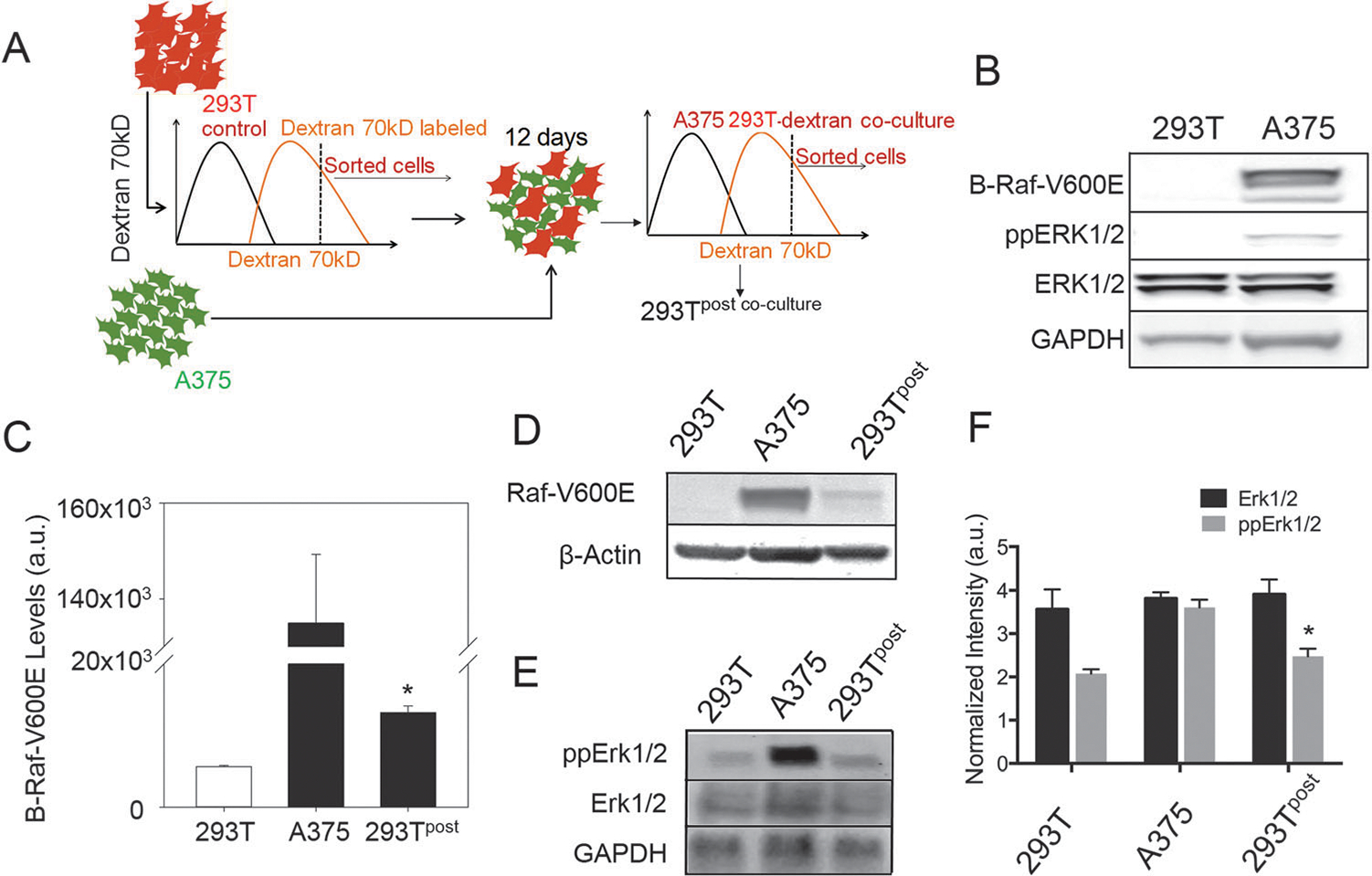
Signaling molecules can transfer between cells influencing cell phenotypes. (A) Schematic showing the experimental plan. A cell type is labeled with a high molecular weight dextran (that does not get transferred in significant amount to preserve cell type-specific labeling), and mixed with the donor cell type. Cells are co-cultured for 12 days, and sorted by dextran labeling using fluorescence assisted cell sorting (FACS), and probed for the transfer of proteins from the donor cells. (B) Immunoblot images showing that A375 cells were positive for BRAF point mutated at locus 600 with valine substituted for glutamic acid, and had increased Erk1/2 phosphorylation; the lower lane shows abundance of GAPDH as the control. (C) Quantitative immunoblot analysis of control 293T cells and sorted 293T cells after co-culture shows that a very small, but significant amount of constitutively active BRAF (V600E) gets transferred from A375 to 293T cells; *, P < 0.05 shows the standard error of mean between the 293T cells before and after co-culture, n = 3 for independent co-culture experiments. (D) The immunoblot image of 293T, A375, and FACS sorted 293T cells after co-culture with A375 shows transfer of BRAF-V600E in 293T cells; the lower lane shows abundance of β-actin as the control. (E) The immunoblot image of control 293T cells and sorted 293T cells after co-culture shows an increase in Erk1/2 phosphorylation in sorted cells; the lower lane shows abundance of GAPDH as the control. (F) Immunoblot quantification for E showing relative abundance of Erk1/2 and phospho-Erk1/2 in A375, 293T, and sorted 293T cells after co-culture with A375; *, P < 0.05 shows standard error of mean between the 293T cells before and after co-culture, n = 3 for independent co-culture experiments.
Discussion
Cell–cell communication plays an important role in maintaining tissue homeostasis as well as in regulating tissue response to stimuli. Cell–cell communication by direct transfer of cellular components can provide a non-genetic means to acquire new phenotypes by the recipient cells, allowing them to transiently acquire novel characteristics. This, still not well appreciated form of cellular communication can affect our understanding of a variety of physiological and pathological states. Previous studies,31 including ours,13 reported lack of evidence for cytosolic protein transfer, though other reports have indicated that such a phenomenon does exist.21,32,33 Here we address this paradoxical inconsistency by a more controlled co-culture methodology. We show that cytosolic proteins can be transferred between cells and that this transfer occurs via direct cell–cell interaction. However, we also found that cytosolic protein transfer occurs with slow kinetics, requiring a long time frame for accumulation of transferred proteins in target cells for detection by conventional methods. Further, difficulty in detecting cytosolic protein transfer may also be due to the dependence of the kinetics of protein transfer on cell density, heterotypic cell ratio, and membrane characteristics.
How does cytosolic protein transfer occur mechanistically is a question with significant bearing on cellular and tissue physiology. Our study shows that this transfer can occur via direct cell–cell interaction. To distinguish this mechanism of protein transfer from the frequently studied alternative: transfer by formation of membrane vesicles, or exosomes, we established a new method based on microfabrication to control the length of the interface between heterotypic cells in co-culture. Controlled patterning of cells in heterotypic co-cultures has already been used in the setting of melanoma-endothelial paracrine communication, yielding new insights into the mechanisms of interaction between these cell types.25,34 In our system, if only the interface but not the numbers of donor and recipient cells varies, it would be expected that a different efficiency of transfer would occur when transfer occurs via cell–cell contact, but not by vesicles. Indeed, we found that protein transfer depended on cell–cell interaction, exhibiting higher evidence of transfer with increased effective heterotypic cell interaction length. The topology of cell–cell interactions is highly organized and results in specific patterns of heterotypic cell–cell interaction, for example stem cells are closely surrounded by stromal cells in their niches,35 presenting opportunities for intercellular protein transfer. Since, lithography can allow for arbitrary patterning contours, our assay could serve as a useful platform to study the phenomenon of intercellular protein transfer with different physiologically relevant topologies of tissue interaction.
Evidence for cell fusion and nanotube transfer of cytosolic proteins provides an immediate explanation for somewhat inconclusive evidence of its existence found in diverse experimental systems. As suggested by the recently proposed mathematical model,27 transfer of membrane components though TNTs is expected to be much faster than transfer of cytosolic components. Specifically membrane bound proteins may get transferred by exchange of membrane components between juxtaposed cells, while cytosolic components may get transferred by diffusion through TNTs transiently connecting the cells. We had postulated that transfer of cytoplasmic components by TNTs would result in a characteristic pattern of intercellular protein transfer where membrane bound proteins will get transferred in a size independent manner, while those in the cytoplasm will transfer in a size dependent manner.27 Thus one can predict that the transfer can be dramatically decreased for larger proteins or for cases when TNTs are short lived. For instance, if cells move with respect to each other sufficiently fast, transfer of larger, heavier proteins or protein complexes can be precluded. On the other hand, if one can stabilize TNTs, transfer of such proteins can be enhanced, as shown in this study. By contrast, the efficiency of transfer of cytosolic compounds by vesicle transport would not depend on the physical parameters of the cargo, leading to a potentially high degree of transfer for any cytosolic protein. Overall, if the cell-fusion mediated transfer mechanism is indeed general and widespread, important limitations on the transferred components may exist and lead to variable observation results.
Here we also show that critically important cytosolic proteins can be shared between the donor melanoma cells and other recipient cell types. In particular, we found that a commonly mutated protein, BRAF, could be transferred in its constitutively active form and supply a considerable increase in the activity of the downstream mitogen-activated protein kinase, Erk1/2. Invasive melanoma growth can create complex interfaces between the tumor and stromal regions, including finger-like projections of invading cells into the underlying dermis.25,36 This complex interface can enhance the probability of protein transfer and endow the stromal cells with new functionality. For instance, enhanced activity of Erk1/2 is thought to underlay the response to various growth factors controlling angiogenic growth, potentially leading to enhanced vasculature dependent metastatic processes.
In summary, our results strongly suggest that cytosolic proteins can be functionally transferred between juxtaposed cells leading to non-genetic spreading of the corresponding phenotypic traits. This finding adds to a growing list of types of transferred cellular components further redefining our concepts of cell identity and relationship between cellular genotype and phenotype.
Methods
Cell culture
Melanoma cell line 1205Lu (American Type Culture Collection, Manassas, VA) stably transfected with GFP was cultured at 37 °C and 5% CO2 in a Dulbecco’s modified Eagle’s medium (DMEM) containing 2 mM L-glutamine, 50 U ml−1 of penicillin, and 50 μg ml−1 of streptomycin with 10% fetal bovine serum (Invitrogen). Human umbilical cord endothelial cells (HUVECs) were cultured in endothelial growth medium (EGM-2, Lonza), and the medium was changed every other day. Under all co-culture conditions, cells were cultured in a 50 : 50 mixture of 1205Lu medium and EGM-2, and only 50% of the medium was changed each day, retaining the older remaining medium within the culture. This method allowed for a more gradual change in the cellular microenvironment than a complete change of medium would ensue, while also ensuring at least a daily mixing of medium. A375 cells and 293T cells were cultured in the medium in which 1205Lu were cultured.
Reagents
Linoleic acid (Sigma, L5900) was added to the medium at a concentration of 2.5 μM one day after cell attachment, and medium was replenished each day with the addition of linoleic acid. Methyl-β-cyclodextrin (Sigma, 4555) was added to the medium at a concentration of 1 mM one day after cell attachment, and medium replenished each day during the course of the experiment. Cytochalasin B (Sigma, C2743) was added to the medium at a concentration of 50 μM one day after cell attachment, and medium replenished each day during the course of the experiment. Dextran with a molecular weight of 70 kD conjugated with Texas Red (Life Technologies, D1864) was used to label cell types with minimal expectation of lateral intercellular transfer. Dextran was loaded into cells in the presence of a pinocytic influx reagent (Life Technologies, I14402) at a concentration of >100 μM. Cells were FACS sorted to ensure a narrow range of highly fluorescent cells to ensure dextran availability after cell divisions.
Cell patterning
Cells were patterned using stereolithography based PDMS mold incorporated tissueware (LiveAssay) coated with fibronectin.25 1205Lu cells were cultured in the holes, and left to adhere for 6 hours. Stencils were gently removed using a pair of forceps, unadhered cells were washed off, and HUVECs were cultured thereafter. After 6 hours, unadhered cells were washed off. Co-cultures were maintained for the duration of the experiments in a mixed medium.
Flow cytometry
Cells were detached from the substrate using Hanks’-based enzyme free cell dissociation buffer (GIBCO, 13150) for 15 min at 37 °C, quenched using excess media, washed twice with PBS using centrifugation at 1200 rpm for 5 min. Cells were analyzed using BD Facsaria II, with a minimum of 10 000 cells collected under each condition. For experiments where cells needed to be sorted, conditions were gated with appropriate negative and positive controls. Statistical analyses were performed and the graph plotted using Sigma Plot 12 (Systat Software Inc.).
Immunoblotting
Cells were lysed using RIPA buffer (Thermo Scientific) with a 1% protease inhibitor cocktail (Sigma-Aldrich, S7830) and the NuPAGe sample reducing agent (Invitrogen, NP0004) containing 10 mM dithiothreitol (DTT) (Sigma-Aldrich, D0632). Lysates were centrifuged at 13 000 × g at 4 °C for 15 min and the supernatant was collected and quantified using the BCA assay kit (Thermo Scientific, 23227). Protein lysates were appropriately diluted with sample buffer, heated at 70 °C for 10 min, cooled and separated on a 4–20% w/v SDS PAGE gel (BioRad), and subsequently transferred to the nitrocellulose membrane (BioRad). The membrane was blocked for 1 hour in blocking solution consisting of TBST (10 mM Tris, pH 8.0, 1% w/v Tween 20), and 5% BSA (BioRad). Thereafter the membrane was incubated in 1 : 500 diluted solution of anti-BRAF antibody (Abcam, 33899), 1 : 250 diluted solution of anti-BRAF-V600E antibody (Ventana), 1 : 500 diluted solution of Erk1/2 (Cell Signaling, 9102), 1 : 500 diluted solution of phosphor-Erk1/2 (Cell Signaling, 9101), 1 : 1000 diluted solution of GAPDH (Cell Signaling, 2118), and 1 : 1000 diluted solution of β-actin antibody (Cell Signaling, 4967) in blocking solution at 4 °C overnight. Membranes were then washed for 1 hour with TBST and blocked again for 1 hour before incubating in HRP conjugated secondary antibodies (Pierce, anti-mouse, 31160, and anti-rabbit, 31188) diluted 1 : 5000 for 1 hour. Membranes were washed repeated with TBST and analyzed using the Biorad gel imaging system and ChemiDoc XRS+ with the ECL Western blotting substrate (Pierce, 32106). When required, membranes were stripped of antibodies using restore stripping buffer (Thermo Scientific).
Supplementary Material
Insight, innovation, integration.
Cells are conventionally considered as separate, autonomous entities that maintain their own identities while communicating with their environment and neighboring cells. However, a recently identified phenomenon of intercellular transfer of cellular components indicates that the cellular identity is vaguer than previously thought. However, though evidence for transfer of membrane proteins has been significant, our study suggests that even cytosolic proteins can be transferred between similar or dissimilar cells. We also unravel some of the mysteries regarding the mechanism of this transfer and suggest that this transfer occurs via direct cell–cell contact. We also show that invasive cancer cells can transfer signaling molecules to their neighbors presenting a way for cancer cells to transiently pass on novel characteristics to their neighbors.
Acknowledgements
We would like to extend our gratitude to Dr Jun O. Liu, Department of Pharmacology, The Johns Hopkins Medical Institutions, and to Dr Rhoda M. Alani, Department of Dermatology, Boston University for providing us with a steady supply of HUVECs and 1205Lu cells during the course of the experiments.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ib00209a
References
- 1.Orr FW, Wang HH, Lafrenie RM, Scherbarth S and Nance DM, J. Pathol, 2000, 190, 310–329. [DOI] [PubMed] [Google Scholar]
- 2.Kramer RH and Nicolson GL, Proc. Natl. Acad. Sci. U. S. A, 1979, 76, 5704–5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Somers WS, Tang J, Shaw GD and Camphausen RT, Cell, 2000, 103, 467–479. [DOI] [PubMed] [Google Scholar]
- 4.Lichtenberger BM, Tan PK, Niederleithner H, Ferrara N, Petzelbauer P and Sibilia M, Cell, 2010, 140, 268–279. [DOI] [PubMed] [Google Scholar]
- 5.Abou-Khalil R, Le Grand F, Pallafacchina G, Valable S, Authier FJ, Rudnicki MA, Gherardi RK, Germain S, Chretien F, Sotiropoulos A, Lafuste P, Montarras D and Chazaud B, Cell Stem Cell, 2009, 5, 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bosenberg MW and Massague J, Curr. Opin. Cell Biol, 1993, 5, 832–838. [DOI] [PubMed] [Google Scholar]
- 7.Singh AB and Harris RC, Cell. Signalling, 2005, 17, 1183–1193. [DOI] [PubMed] [Google Scholar]
- 8.Etienne-Manneville S and Hall A, Cell, 2001, 106, 489–498. [DOI] [PubMed] [Google Scholar]
- 9.Ahmed KA and Xiang J, J. Cell. Mol. Med, 2011, 15, 1458–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li M, Aliotta JM, Asara JM, Wu Q, Dooner MS, Tucker LD, Wells A, Quesenberry PJ and Ramratnam B, J. Biol. Chem, 2010, 285, 6285–6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pap E, Pallinger E, Pasztoi M and Falus A, Inflammation Res, 2009, 58, 1–8. [DOI] [PubMed] [Google Scholar]
- 12.Prochiantz A, Methods Mol. Biol, 2011, 683, 249–257. [DOI] [PubMed] [Google Scholar]
- 13.Niu X, Gupta K, Yang JT, Shamblott MJ and Levchenko A, J. Cell Sci, 2009, 122, 600–610. [DOI] [PubMed] [Google Scholar]
- 14.Levchenko A, Mehta BM, Niu X, Kang G, Villafania L, Way D, Polycarpe D, Sadelain M and Larson SM, Proc. Natl. Acad. Sci. U. S. A, 2005, 102, 1933–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guescini M, Leo G, Genedani S, Carone C, Pederzoli F, Ciruela F, Guidolin D, Stocchi V, Mantuano M, Borroto-Escuela DO, Fuxe K and Agnati LF, Exp. Cell Res, 2012, 318, 603–613. [DOI] [PubMed] [Google Scholar]
- 16.Agnati LF, Guidolin D, Leo G, Guescini M, Pizzi M, Stocchi V, Spano PF, Ghidoni R, Ciruela F, Genedani S and Fuxe K, J. Recept. Signal Transduction Res, 2011, 31, 315–331. [DOI] [PubMed] [Google Scholar]
- 17.Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A and Rak J, Nat. Cell Biol, 2008, 10, 619–624. [DOI] [PubMed] [Google Scholar]
- 18.Davis DM, Nat. Rev. Immunol, 2007, 7, 238–243. [DOI] [PubMed] [Google Scholar]
- 19.Liu T, Li R, Pan T, Liu D, Petersen RB, Wong BS, Gambetti P and Sy MS, J. Biol. Chem, 2002, 277, 47671–47678. [DOI] [PubMed] [Google Scholar]
- 20.Chairoungdua A, Smith DL, Pochard P, Hull M and Caplan MJ, J. Cell Biol, 2010, 190, 1079–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lou E, Fujisawa S, Morozov A, Barlas A, Romin Y, Dogan Y, Gholami S, Moreira AL, Manova-Todorova K and Moore MA, PLoS One, 2012, 7, e33093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carmeliet P and Jain RK, Nature, 2000, 407, 249–257. [DOI] [PubMed] [Google Scholar]
- 23.Wendel C, Hemping-Bovenkerk A, Krasnyanska J, Mees ST, Kochetkova M, Stoeppeler S and Haier J, PLoS One, 2012, 7, e30046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chien AJ, Haydu LE, Biechele TL, Kulikauskas RM, Rizos H, Kefford RF, Scolyer RA, Moon RT and Long GV, PLoS One, 2014, 9, e94748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howard JD, Moriarty WF, Park J, Riedy K, Panova IP, Chung CH, Suh KY, Levchenko A and Alani RM, Pigm. Cell Melanoma Res, 2013, 26, 697–707. [DOI] [PubMed] [Google Scholar]
- 26.Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ and Lotvall JO, Nat. Cell Biol, 2007, 9, 654–659. [DOI] [PubMed] [Google Scholar]
- 27.Suhail Y, Kshitiz J. Lee, Walker M, Kim DH, Brennan MD, Bader JS and Levchenko A, Bull. Math. Biol, 2013, 75, 1400–1416. [DOI] [PubMed] [Google Scholar]
- 28.Wu SN, Yeh CC, Huang HC and Yang WH, Cell. Physiol. Biochem, 2011, 28, 959–968. [DOI] [PubMed] [Google Scholar]
- 29.Chien AJ, Moore EC, Lonsdorf AS, Kulikauskas RM, Rothberg BG, Berger AJ, Major MB, Hwang ST, Rimm DL and Moon RT, Proc. Natl. Acad. Sci. U. S. A, 2009, 106, 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anastas JN, Kulikauskas RM, Tamir T, Rizos H, Long GV, von Euw EM, Yang PT, Chen HW, Haydu L, Toroni RA, Lucero OM, Chien AJ and Moon RT, J. Clin. Invest, 2014, 124, 2877–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rustom A, Saffrich R, Markovic I, Walther P and Gerdes HH, Science, 2004, 303, 1007–1010. [DOI] [PubMed] [Google Scholar]
- 32.Hofmann JP, Denner P, Nussbaum-Krammer C, Kuhn PH, Suhre MH, Scheibel T, Lichtenthaler SF, Schatzl HM, Bano D and Vorberg IM, Proc. Natl. Acad. Sci. U. S. A, 2013, 110, 5951–5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chu PP, Bari S, Fan X, Gay FP, Ang JM, Chiu GN, Lim SK and Hwang WY, Cytotherapy, 2012, 14, 1064–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stine MJ, Wang CJ, Moriarty WF, Ryu B, Cheong R, Westra WH, Levchenko A and Alani RM, Cancer Res, 2011, 71, 2433–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rompolas P, Mesa KR and Greco V, Nature, 2013, 502, 513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rigel DS, Robinson JK, Ross M, Friedman RJ, Cockerell CJ, Lim HW, Stockfleth E and Kirkwood JM, Cancer of the Skin, Elsevier, 2011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.