

SUMMARY
Regulatory T (Treg) cells integrate diverse environmental signals to modulate their function for optimal suppression. Translational regulation represents a favorable mechanism for Treg cell environmental sensing and adaptation. In this study, we carry out an unbiased screen of the Treg cell translatome and identify serum/glucocorticoid-regulated kinase 1 (SGK1), a known salt sensor in T cells, as being preferentially translated in activated Treg cells. We show that high salt (HS) drives thymic Treg cells to adopt a T helper type 17 (Th17)-like phenotype and enhances generation of Th17-like induced Treg cells in a SGK1-dependent manner, all the while maintaining suppressive function. Salt-mediated Th17-like differentiation of Treg cells was evident in mice fed with HS diet or injected with HS-preconditioned T cells. Overall, SGK1 enables Treg cells to adapt their function in response to environmental cues. By understanding these environmental-sensing mechanisms, we envision targeted approaches to fine-tune Treg cell function for better control of inflammation.
In Brief
Yang et al. demonstrate that high salt promotes Th17-like functional adaptation of both thymic and induced Foxp3+ Treg cell subsets through a salt-sensor protein, SGK1. The translationally regulated SGK1 pathway enables Treg cells to fine-tune their effector function in response to environmental cues during the dynamic course of inflammation.
Graphical Abstract
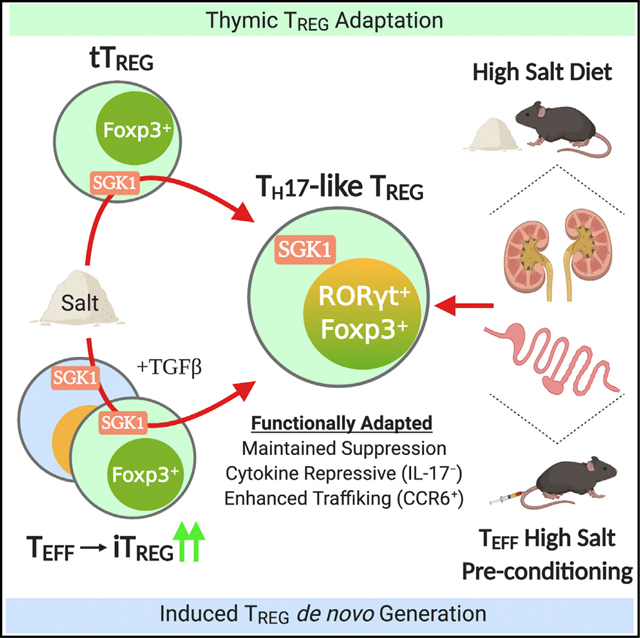
INTRODUCTION
While the immune system is capable of mounting rapid, efficient, and adapted responses to danger, it must ensure a subsequent return to tissue homeostasis and peripheral tolerance. Critical to this function are Foxp3+ regulatory T (Treg) cells, a subset of CD4+ T cells with potent immunosuppressive and tolerogenic functions (Sakaguchi et al., 2008; Shevach, 2009; Rudensky 2011). Treg cells are defined by the expression of Foxp3, a master transcriptional regulator that orchestrates the core transcriptional and epigenetic program that endows Treg cells with their suppressive abilities. Genetic defects in Foxp3 provoke uncontrolled pro-inflammatory effector T (Teff) cell activation and cause severe multi-organ autoimmunity in humans and mice (Brunkow et al., 2001; Wildin et al., 2001). In order to optimally control pro-inflammatory Teff cell responses, Treg cells recognize and localize to the inflammatory environment of their target cells (Wohlfert and Belkaid, 2010; Hori, 2014). Thus, Treg cells must employ diverse mechanisms to sense and integrate environmental cues and adjust their gene expression to ensure proper function in a particular inflammatory milieu (Piccirillo, 2013; Istomine et al., 2016).
Recent evidence indicates that Treg cells can manifest variable degrees of functional adaptation in order to ensure a timely and context-dependent modulation of immune responses in inflammatory environments. Treg cells can adapt by upregulating the necessary homing and cytokine-sensing machinery to localize to sites of Teff-cell-mediated inflammation and promote tissue homeostasis. This functional adaptation is often characterized by Foxp3+ Treg cells co-expressing the lineage-defining transcription factor of their target Teff cells, which include T-bet (T helper type 1 [Th1]), GATA-3 (Th2), and RORγt (Th17), in response to polarizing inflammatory signals (e.g., interleukin-12 [IL-12], IL-4, and IL-6, respectively) (Koch et al., 2009; Yu et al., 2015; Yang et al., 2016). For example, Treg cells expressing transcription factors controlling the Th17 transcriptional program like RORγt and STAT3 were previously shown to upregulate the chemokine receptor CCR6, crucial for the homing and suppressive ability of Treg cells in the gut and kidney during Th17-cell-driven inflammation (Kim et al., 2017; Yang et al., 2016; Chaudhry et al., 2009). In addition, recent studies provide evidence that adapted Treg cells may actually contribute to the onset and maintenance of inflammation and pathology under conditions of acute or chronic inflammation (Kluger et al., 2016; Alvarez et al., 2019). In such settings, inflammatory mediators may destabilize Foxp3 expression, in turn forcing the generation of inflammatory IL-17A and/or interferon-γ (IFN-γ)-producing exTreg cells which promote local inflammatory responses (Zhou et al., 2008; Voo et al., 2009; Yurchenko et al., 2012). While the mechanisms driving exTreg cell generation are largely unknown, the co-expression of Th-cell-defining transcription factors, like RORγt, in Foxp3+ Treg cells may represent a distinct functional fate or an intermediate state before conversion into exFoxp3 Teff cells in autoimmune or infectious diseases. Cytokine factors like IL-1β and IL-6 are well described to promote Treg cell differentiation into a Th17-like phenotype (Yang et al., 2008; Li et al., 2010; Alvarez et al., 2019). However, the environmental cues, often non-immune in nature, controlling Treg cell functional adaptation and potential lineage conversion into pro-inflammatory Teff cells remain poorly understood.
Regulation of mRNA translation is a mechanism by which Treg cells acquire the ability to rapidly respond to environmental cues. Unlike gene transcription, translational regulation provides a rapid and energetically favorable mechanism of shaping the proteome of a given cell and tailoring cell function to the extracellular contexts (Mohr and Sonenberg, 2012; Piccirillo, 2013). To this end, we previously carried out the first unbiased assessment of global translation in primary murine Treg and Teff cells and successfully identified distinct translational signatures distinguishing Treg and Teff cells (Bjur et al., 2013). Using the same approach, we identify that the serum/glucocorticoid-regulated kinase 1 (SGK1) is preferentially translated in activated Treg cells compared to activated Teff cells. While SGK1 has pleiotropic functions (Lang et al., 2014), it is an important signal transducer of sodium chloride (salt)-sensing in various cells downstream of the p38/NFAT5 pathway (Bell et al., 2000; Chen et al., 2009; Kleinewietfeld et al., 2013). In conventional CD4+ T cells, high-salt (HS) conditions were shown to promote Th17 cell differentiation of CD4+ T cells in autoimmunity in a SGK1-dependent manner (Kleinewietfeld et al., 2013; Wu et al., 2013). Similarly, it was reported that Foxp3+ T cells can sense HS conditions (Hernandez et al., 2015; Luo et al., 2019), although the links among HS, SGK1, and Treg cell differentiation remain ill defined. Given the prevalence of salt at important sites of Th17 cell inflammation like the gut and kidneys, we predicted that salt sensing may be an important mechanism by which Treg cells can adapt to a Th17-like phenotype and thus provide context-dependent regulatory function.
In this study, we explored the environmental sensing role of SGK1 on the modulation of Treg cell development, differentiation, and function in response to salt. We show that the mRNA encoding SGK1 is preferentially translated in activated Foxp3+ Treg cells compared to activated conventional Teff cells and constitutes part of the Treg cell translatome. In vitro HS conditions force the expression of the Th17 master transcriptional regulator RORγt in Helios+ and Helios− thymic Treg (tTreg) cells and enhance the generation of Th17-like de novo induced Treg (iTreg) cells in a SGK1- and NaCl-dependent manner and without affecting Treg cell suppressive function. Salt-mediated Th17-like differentiation in Treg cells was evident in mice fed a HS diet (HSD) or injected with HS-preconditioned T cells, conditions that are associated with increased Th17-cell-driven inflammation in gut and kidney tissues. Thus, salt-mediated SGK1 activation represents a mechanism for Treg cells to integrate environmental signals and adapt functionally to local inflammatory responses.
RESULTS
Salt-Sensing Kinase SGK1 mRNA Is Preferentially Translated in Foxp3+ Treg Cells
Translational control of gene expression enables immune cells to rapidly adapt and deliver appropriate responses through integration of diverse environmental signals (Mohr and Sonenberg, 2012; Piccirillo, 2013). Ribosome assembly is the most immediate step prior to mRNA translation and protein synthesis, and differences observed by comparing cytosolic and polyribosomal (ribosome-rich and actively translated) mRNA can reliably identify changes in gene-specific, translational activity that contribute to the proteome of the cell (King and Gerber, 2016; Piccirillo et al., 2014). Recently, we reported the first genome-wide study of translational control in primary CD4+ Teff and Treg cells (Bjur et al., 2013). In our study, we obtained parallel measurements of cytosolic and polysome-associated mRNA levels in ex vivo and in vitro T cell receptor (TCR)-activated Teff and Foxp3+ Treg cells and discovered distinct mRNA translational signatures distinguishing Teff and Treg cells (Figure 1A; Bjur et al., 2013). Here, we undertook an unbiased screen of the ~700 differentially translated mRNAs in activated CD4+ T cell subsets in order to identify candidate genes whose expression could selectively impact Treg cell development or function.
Figure 1. Salt-Sensing Kinase SGK1 mRNA Is Preferentially Translated in Foxp3+ Treg Cells.

(A) Schematic diagram showing the workflow of translatome profiling in CD4+ T cell subsets. FACS-purified Teff and Treg cells that were naive (directly ex vivo) or TCR activated (in vitro) were treated with cycloheximide to immobilize ribosomes on the mRNA. Polysome-associated (≥3 ribosomes) mRNAs were fractionated from total cytosolic mRNA by sedimentation through a sucrose gradient. Total cytosolic mRNA and polysome-associated mRNA were probed with microarrays to quantify mRNA level. Polysome-associated mRNA pools describe the translatomes of CD4+ T cell subsets.
(B) Quantitative representation of SGK1 translational activity (after correction for cytosolic mRNA levels) in CD4+ Teff and Treg cells ex vivo or after in vitro activation. Data are shown as log2 fold change.
(C) Representative flow cytometric histograms and quantitative analysis of SGK1 protein expression in ex vivo CD4+ T cell subsets by intracellular staining of SGK1 and the assessment of geometric mean fluorescent intensity (gMFI).
(D) Representative flow cytometric histograms and quantitative analysis of SGK1 protein expression in in vitro TCR-activated CD4+ T cell subsets by intracellular staining of SGK1 and the assessment of geometric mean fluorescent intensity (gMFI).
(E) Representative flow cytometric histograms and quantitative analysis of SGK1 protein expression in CD4+ T cells treated with increasing concentration of salt through intracellular staining of SGK1 and the assessment of geometric mean fluorescent intensity (gMFI).
Polysomal profiling experiments (A and B) were performed in biological duplicates. Data shown in (D) and (E) are representative of three independent experiments with triplicates of each condition. Error bars represent mean ± standard deviation.
Our analysis revealed that the mRNA encoding SGK1 was one of the highest ranked transcripts preferentially translated in Treg cells compared to Teff cells in TCR-activated conditions (log2 fold increase = 4.414; false discovery rate [FDR] <15%; Figure 1B). SGK1 is a critical mediator of various cell stress responses through its sensing of several environmental signals such as hypertonicity, cell shrinkage, water/ion imbalance, and hormonal signals (Lang et al., 2014). Using flow cytometry, we show that ex vivo sorted murine Treg and Teff cells displayed a similar basal level of SGK1 protein expression (Figure 1C), recapitulating the polysomal profiling data where the translational activity of SGK1 mRNA levels are similar in ex vivo Treg and Teff cells (Figure 1B). However, SGK1 protein expression was expressed at a significantly higher level in activated Treg cells compared to Teff cells (Figure 1D), confirming the polysomal profile that Treg cells exhibit significant higher translation of SGK1 mRNA in TCR-activated conditions (Figure 1B). Several immune cell populations have been shown to respond to higher levels of extracellular salt (NaCl) with increased SGK1 expression and activity (Kleinewietfeld et al., 2013; Wu et al., 2013; Machnik et al., 2009). However, previous studies assessed SGK1 transcript abundance by qPCR or microarray and inferred SGK1 protein expression in T cells upon HS stimulation. Here, we assessed SGK1 protein levels directly through flow cytometry in primary CD4+ T cells and showed that HS conditions prominently increased SGK1 protein expression in CD4+ T cells in a dose-dependent manner (Figure 1E). Overall, the preferential mRNA translation of SGK1 in Treg cells, coupled with the key salt-sensing role of SGK1, strongly suggest a putative role of the salt-SGK1 signaling axis in the development and/or suppressive function of Treg cells.
High Salt Promotes Reprogramming of Thymic Helios+ and Helios− Treg Cells toward a Th17-like Phenotype via a SGK1-Dependent Mechanism
Salt-mediated signaling via SGK1 in T cells has been previously shown to enhance pathogenic Th17 cell differentiation (Kleinewietfeld et al., 2013). However, the role of salt and SGK1 in modulating Treg cell development, fate or function remains poorly defined. In this regard, we first sought to investigate if preferential translation of SGK1 predisposes Treg cells to reprogram into a Th17-like lineage upon HS stimulation. Th17-like RORγt+ Treg cells are a novel Treg cell subset possessing pleiotropic functions depending on the inflammatory milieu, and have been previously identified in the gut, lungs, and kidneys during Th17-mediated inflammation (Zhou et al., 2008; Kluger et al., 2016; Yang et al., 2016; Alvarez et al., 2019). To trace the ontogeny of Foxp3+ Treg cells in vitro, a mixed population consisting of fluorescence-activated cell sorting (FACS)-isolated Foxp3+ (GFP+) thymic Treg (tTreg) cells and CellTrace Violet (CTV) proliferation dye-labeled Foxp3− (GFP−) Teff cells from B6.Foxp3GFP reporter mice, was used to perform in vitro assays where cells of tTreg cell-origin were identified as CTV− (Figure 2A). Increased extracellular NaCl concentration (+40 Mm or +60 mM) significantly promoted the co-expression of the Th17 master transcription factor RORγt in tTreg cells (Figures 2B and 2C) while having no impact on T cell viability, survival, and proliferative potential (Figures S1A–S1C). Besides the increased frequency of RORγt+ tTreg cells, HS also induced a dose-dependent increase in the median fluorescent intensity (MFI) of RORγt protein expression and a concomitant reduction in Foxp3 protein expression in tTreg cells (Figures 2B, 2D, and 2E). To further validate the effect of salt on tTreg cells, we performed the same experiment using Foxp3+ tTreg (GFP+) cells isolated from the thymus of Foxp3-reporter mice and confirmed the same effects as those observed with splenic Foxp3+ tTreg cells (Figures S1A–S2C). Interestingly, tTreg cells did not produce pro-inflammatory cytokines such as IL-17A despite the significant expression of RORγt (Figure 2F), suggesting that the Th17-like phenotype induced by HS stimulation is not sufficient to completely abrogate the anti-inflammatory nature of Treg cells. Indeed, HS-pretreated tTreg cells exhibit potent and comparable suppression capacity to tTreg cells pretreated in control medium (Figures 2G and 2H), suggesting that HS-induced RORγt+ tTreg cells represent functional and adapted Treg cells.
Figure 2. High Salt Promotes Reprogramming of Thymic Treg Cells toward a Th17-like Phenotype via a SGK1-Dependent Mechanism.

(A) Schematic diagram showing the in vitro co-culture system that enables the tracking of cell ontogeny (top panel). FACS-purified CD4+ GFP− Teff cells from B6.Foxp3GFP mice were labeled with CellTrace Violet (CTV) proliferation dye and then combined with GFP+ thymic Treg (tTreg) cells at a ratio of 6:1. After co-culture, cells of tTreg cell origin were identified as CTV−. Similarly, cells of original Teff cell origin were identified as CTV+. Representative flow cytometry plots showing the gating strategy to differentiate the cells of either tTreg or Teff cell origins (bottom panel).
(B) tTreg and Teff cells were co-cultured (A) under increasing NaCl concentrations in the presence of Th17-polarizing cytokines for 72 h and in the presence or absence of a SGK1 inhibitor. Representative flow cytometry plots showing the expression of transcription factors RORγt and Helios in CTV− tTreg cells at 72 h.
(C) Quantitation of the frequency of RORγt+ cells in Foxp3+ CTV− tTreg cells.
(D and E) Quantitative analyses of RORγt (D) and Foxp3 (E) protein expressions in tTreg cells by the assessment of median fluorescent intensity (MFI).
(F) Representative flow cytometry plots showing the intracellular staining of IL-17A and RORγt in Teff cells or tTreg cells.
(G) Assessment of the suppressive function of tTreg cells cultured in normal salt (NS) or high salt (HS) medium in a Th17-polarizing condition, as described in (A), in an in vitro suppression assay using CD4+ GFP− Teff cells as responders. Representative flow cytometry histograms of the proliferation of Teff by the dilution of the CTV proliferation dye.
(H) Quantification of percent suppression at different Treg/Teff cell ratios.
(I) Quantitative analysis of the frequency of RORγt+ cells in Helios+ and Helios− subsets of tTreg cells.
Data are representative of three independent experiments with triplicates of each condition. Error bars represent mean ± standard deviation.
To demonstrate whether the effect of HS is mediated through the kinase activity of SGK1, we activated Teff and Foxp3+ tTreg cells in the presence of salt and in the presence or absence of a SGK1-specific pharmacological inhibitor (SGK1i; GSK650394) at a concentration at which no toxic effect is observed, all the while abrogating SGK1 kinase activity (Sherk et al., 2008; Kleinewietfeld et al., 2013). SGK1i completely abolished the effect of salt, as we observed a significant reduction in the frequency of RORγt+ tTreg as well as the level of RORγt protein expression (Figures 2B–2D) while maintaining Foxp3 expression in tTreg cells in HS conditions in the presence of SGK1i (Figure 2E).
Finally, we assessed the expression of the transcription factor Helios in these conditions, as it was recently described to identify a lineage of enhanced and stable suppressive Treg cells (Bin Dhuban et al., 2015; Thornton et al., 2019). Although most Foxp3+ Treg cells from thymus of adult mice are Helios+, we and others show that thymic Foxp3+ Treg cells include up to 20% of Helios− cells (Figures S2D and 2E; Thornton et al., 2010). Using total Foxp3+ tTreg cells, we observed that Helios− Treg cells were more susceptible to HS-induced Th17 reprogramming than their Helios+ counterparts (Figure 2I). To further examine whether salt is capable of influencing the differentiation of Helios+ tTreg cells, we performed the same assay using Helios+ Treg cells (purity >99.7%) isolated from Foxp3-Helios dual reporter mice (Figure S2F). Interestingly, HS significantly increased the frequency of exHelios and RORγt+ Treg cells from Helios+ sorted tTreg cells (Figures S2G–S2J). In addition, HS exposure leads to increased level of RORγt expression and a concomitant reduction in Helios and Foxp3 expression as measured by MFI (Figures S2K–S2M). The effects of HS on Helios+ tTreg cells were SGK1 dependent, as SGK1i abolished the effect of salt (Figures S2G–S2M). Therefore, HS drives both Helios+ and Helios− tTreg cells to adopt a Th17-like phenotype, although Helios− Treg cells displayed increased susceptibility to these salt-mediated effects. In summary, these results indicate that HS favors the reduction of Foxp3 and Helios expression and acquisition of a Th17-like phenotype in tTreg cells in a SGK1-dependent manner.
HSD Promotes the Differentiation of RORγt+ Th17-like Treg Cells in the Intestine and Kidneys in Mice
Since HS promoted the expression of RORγt in Foxp3+ Treg cells in vitro, we assessed the effect of salt on the generation of RORγt+ Treg cells in vivo. To this end, we used an HSD model consisting of an 8%-NaCl-enriched diet and a 1% NaCl in water given ad libitum to mice and examined T cell differentiation and cytokine production 2 weeks after the start of the diet. We isolated T cells infiltrating intestines and kidneys, since these organs are directly involved in salt absorption and excretion and are targets of strong Th17-cell-driven inflammatory disorders such as inflammatory bowel disease and crescentic glomerulonephritis, respectively (Maloy and Powrie, 2011; Krebs et al., 2017). In contrast to mice receiving a normal-salt diet (NSD), the frequencies and numbers of RORγt+ Teff cells were significantly higher in the ileum and kidneys of mice receiving HSD (Figures 3A, 3B, 3D, and 3E). While Treg cells in the NSD-fed mice are mostly RORγt− Foxp3+ Treg cells, HSD-fed mice had a significantly higher frequency and absolute number of RORγt+ Treg cells in the ileum and kidneys (Figures 3C and 3F). Indeed, the HSD-mediated differentiation of RORγt+ Treg cells was not observed in peripheral lymph nodes (pLNs), sites that did not encounter direct HS exposure or had not established Th17 cell inflammation (data not shown). In line with increased frequency of RORγt+ Teff cells, mice under HSD displayed significantly elevated frequencies of IL-17A-producing Teff cells in the ileum lamina propria (Figures 3G and 3H). In contrast, the proportions of IFN-γ-producing Teff cells in the ileum were not significantly different between mice receiving NSD or HSD (Figure 3I). Since RORγt+ Treg cells have been shown to identify Treg cells with improved trafficking to Th17 cell inflammatory sites through CCR6 upregulation and a fine-tuned suppressive function in situ (Kim et al., 2017; Yang et al., 2016; Chaudhry et al., 2009), we examined whether HS-promoted RORγt+ Treg cells exhibited similar functional properties. Indeed, HS-promoted RORγt+ Treg cells expressed high levels of CCR6, while the cellular frequency of RORγt+ Treg cells strongly correlates with the number of IL-17A+ T cells in the ileum (Figures 3J–3L), suggesting an improved ability of these cells to migrate to Th17-cell-driven inflammatory sites. Finally, consistent with our in vitro findings, the RORγt+ Treg cells found in HSD-treated mice were largely Helios−, whereas RORγt− Treg cells were Helios+ (Figures 3K and 3M). Our data show that HS promotes the emergence of Th17-like RORγt+ Treg cells that accumulate to sites susceptible to Th17 cell inflammation in vivo.
Figure 3. HSD Promotes the Differentiation of RORγt+ Th17-like Treg Cells in the Intestine and Kidneys in Mice.

(A and D) Flow cytometry analysis of the expression of Foxp3 and RORγt in CD4+ T cells isolated from the ileum lamina propria (A) and kidneys (D) of the mice fed with NS diet (NSD) or HS diet (HSD).
(B–F) Quantitative analyses of the frequency and the absolute cell counts of RORγt+ Teff cells (B and E) and RORγt+ Treg cells (C and F) isolated from the ileum lamina propria (B and C; red) and kidneys (E and F; orange) in mice fed with NSD or HSD.
(G) Flow cytometry analysis of IFN-γ and IL-17A production by intracellular staining of cytokines in CD4+ T cells isolated from the ileum lamina propria of the mice fed with NSD or HSD.
(H and I) Quantitative analyses of the frequency of IL-17A-producing cells (H) and IFN-γ-producing cells (I) in CD4+ T cells isolated from the ileum lamina propria of mice fed with NSD or HSD.
(J) Correlative analysis of absolute numbers of RORγt+ Treg and IL-17+ T cells in the lamina propria of mice fed with NSD or HSD.
(K) Flow cytometry analysis of the expression of CCR6 (left), Helios (right), and RORγt of Foxp3+ Treg cells isolated from the ileum of the mice fed with HSD.
(L) Quantitative analyses of the frequency of CCR6+ cells and the median fluorescent intensity (MFI) of CCR6 in RORγt+ or RORγt− Treg cells isolated from the ileum of the mice fed with HSD.
(M) Quantitative analyses of the frequency of Helios− median fluorescent intensity (MFI) of Helios in RORγt+ or RORγt− Treg cells isolated from the ileum of the mice fed with HSD.
Data are representative of three independent experiments with n = 6 mice of each treatment group. Error bars represent mean ± standard deviation.
HS Promotes the De Novo Generation of RORγt+ iTreg Cells in Th17-Polarizing Conditions through a SGK1-Dependent Mechanism
Since Helios−, in contrast to Helios+, tTreg cells showed a greater propensity to be reprogrammed toward a RORγt+ Foxp3+ T cell phenotype, we next investigated the effect of HS and the role of the SGK1 pathway on the development of iTreg cells, a subset of de novo–generated Foxp3+ Treg cells that emerge from CD4+ Foxp3− (conventional) T cells (Chen et al., 2003) and typically do not express Helios (Atarashi et al., 2011; Thornton et al., 2019). Th17 and iTreg cell development is counter-regulated with RORγt+ Th17 cells arising from conventional T cells activated in the presence of transforming growth factor β (TGF-β) and IL-6, while Foxp3+ iTreg cells require TGF-β and IL-2 (Bettelli etal., 2006). While HS (+40 mM and +60 mM) amplified RORγt+ Th17 cell differentiation under Th17-polarizing conditions, HS, but not normal salt (NS), Th17-polarizing condition also promoted the differentiation of Foxp3− (GFP−) conventional T cells into RORγt+ Foxp3+ iTreg cells (Figures 4A and 4B), which accounted for almost all of the HS-induced iTreg cells. In contrast, normal media had a minimal effect on Foxp3 induction (<0.5%), and Foxp3+ T cells induced from normal media in Th17-polarizing conditions did not co-express RORγt (Figures 4A and 4B). The number of RORγt+ iTreg cells generated in vitro was also significantly higher in +60 mM of salt when compared to +40 mM of salt or normal medium (Figure 4C). The expression of Foxp3, measured by MFI, was also significantly increased in RORγt+ iTreg cells in a salt-concentration-dependent manner (Figures 4A and 4D) Importantly, the effect of salt depended on the kinase activity of SGK1, as SGK1 pharmacological inhibition with SGK1i almost completely abrogated the induction of RORγt+ iTreg cells (Figures 4A–4C). Similar to the RORγt+ tTreg cells described in Figure 2, salt-induced RORγt+ iTreg cells are highly refractory to IL-17A production (Figure S3B), a phenotypic feature of functional, suppressive Treg cells. These observations suggest that salt-induced SGK1 activation enables the differentiation of Th17-like Foxp3+ iTreg cells from conventional Teff cells despite the counter-regulatory nature of Th17/iTreg cell differentiation.
Figure 4. HS Promotes the De Novo Generation of RORγt+ iTreg Cells in Th17-Polarizing Conditions through a SGK1-Dependent Mechanism.

(A) FACS-purified CD4+ GFP− Teff cells were cultured under the indicated increased NaCl concentrations in the presence of Th17-polarizing cytokines for 72 h, with or without SGK1 inhibitor. Representative flow cytometry plots showing the expression of transcription factors Foxp3 and RORγt in CD4+ T cells.
(B–D) Quantitative analyses of the frequency (B), absolute cell counts (C), and Foxp3 protein level (D) of RORγt+ iTreg cells after 72 h in culture.
(E) Representative flow cytometry plots showing the expression of transcription factors Foxp3 and RORγt in CD4+ T cells cultured in NS or HS conditions in Th17-polarizing conditions in the presence of SGK1i or furosemide.
(F) Quantification of the frequency of RORγt+ iTreg cells in the experiment described in (E).
Data are representative of three independent experiments with triplicates of each condition. Error bars represent mean ± standard deviation.
It is unknown whether T cells respond directly to NaCl or do so in response to general hyperosmotic and hypertonic stress. In order to assess a direct involvement of NaCl, we examined the effect of furosemide, a specific inhibitor of the Na+ channel, NKCC1, which was shown to mediate the SGK1-dependent effect on IL-23R expression and Th17 cell development (Norlander et al., 2017). Indeed, furosemide treatment completely abolished the effect of HS in promoting RORγt+ iTreg cell differentiation, mimicking the effect of SGK1 inhibition (Figures 4E and 4F). We then investigated how hyperosmolarity from increased concentration of biologically inert molecules (urea and mannitol) compared to HS (NaCl) conditions. While urea (hyperosmotic and isotonic) had no effect, mannitol (hyperosmotic and hypertonic) promoted RORγt+ iTreg cell differentiation, but to a much lesser extent than equivalent osmolarity of NaCl (Figure S4). The use of SGK1i was able to completely abrogate the effect of mannitol (Figure S4), confirming that both HS and hyperosmotic stress activates the SGK1 pathway to influence Treg cell phenotype. Overall, these results show that Na+ is central to the effect of salt on SGK1 activation and subsequently RORγt+ Treg cell differentiation.
In the Absence of the Th17-Polarizing Condition, High Salt Enhances TGF-β-Mediated Treg Cell Induction and Facilitates Co-expression of RORγt
Given the strong induction of iTreg cells in response to salt under Th17-polarizing conditions, we hypothesized that salt may act by potentiating TGF-β signaling. To investigate how HS and SGK1 signaling act on the TGF-β pathway, we first assessed the effect of HS conditions on Th1, Th2, Th17, and iTreg cell differentiation in vitro. We observed that polarizing conditions that involve TGF-β signaling, such as Treg and Th17 cell differentiation, but not Th1 or Th2 cell differentiation, were influenced by HS (Figures S1D and S1E). In TGF-β-mediated iTreg cell induction, HS (+60 mM) potentiated TGF-β-induced Foxp3 expression in CD4+ Teff cells across a wide range of TGF-β concentrations (0.2–5.0 ng/mL) (Figures 5A and 5B). Moreover, in addition to increasing the frequency of Foxp3+ iTreg cells, HS also enhanced the level of Foxp3 expression (MFI) in these cells (Figure 5C), suggesting that HS potentiated iTreg cell differentiation by enhancing the effect of TGF-β. Indeed, iTreg cells induced in the HS condition displayed superior suppressive function in vitro compared to iTreg cells induced in the NS condition (Figures S3B and S3C). Moreover, we confirmed that HS operated in a SGK1-dependent manner, as SGK1i significantly attenuated the effect of HS on the proportion of Foxp3+ Treg cells as well as the level Foxp3 expression (Figures 5F–5H).
Figure 5. In the Absence of Th17-Polarizing Conditions, HS Enhances TGF-β-Mediated Treg Cell Induction and Facilitates Co-expression of RORγt.

(A) FACS-purified CD4+ GFP− Teff cells were cultured in NS media or +60 mM NaCl and in the presence of various concentrations of TGF-β for 72 h. Representative flow cytometry plots showing the expression of transcription factors Foxp3 and RORγt in CD4+ T cells.
(B) Quantitative analyses of the efficiency of Foxp3 induction at indicated concentrations of TGF-β in CD4+ T cells cultured in NS (black) or HS (+60 mM; red) media.
(C) Quantitative analyses of the median fluorescent intensity (MFI) of Foxp3 in Foxp3+ iTreg cells.
(D) Quantitative analyses of the frequency of RORγt+ iTreg cells and the MFI of RORγt at indicated concentrations of TGF-β in CD4+ T cells cultured in NS (black) or HS (+60 mM; blue) media.
(E) Quantitative analyses of the median fluorescent intensity (MFI) of RORγt in Foxp3+ iTreg cells.
(F) The TGF-β-mediated iTreg cell induction assay with additional salt (A) was performed in the presence of SGK1 inhibitor. Representative flow cytometry plots showing the expression and frequency of Foxp3 in CD4+ T cells.
(G and H) Quantitative analyses of the frequency of Foxp3+ iTreg cells (G) and the median fluorescent intensity (MFI) of Foxp3 of iTreg cells (H).
(I) Representative flow cytometry plots showing the expression of Tbet, GATA3, and RORγt in the Foxp3+ iTreg cells described in (A).
(J) Quantitative analyses of the frequencies of Tbet or GATA3 or RORγt-positive cells in Foxp3+ iTreg cells.
Data are representative of three independent experiments with triplicates of each condition. Error bars represent mean ± standard deviation.
Strikingly, in addition to promoting Foxp3 expression, HS also facilitated co-expression of RORγt in iTreg cells in the absence of Th17-polarizing cytokines. We observed a substantial increase in the frequency of RORγt+ iTreg cells (~30%) when TGF-β induction is supplemented with HS compared to normal media (Figures 5A and 5D). Moreover, RORγt+ iTreg cells from the HS condition expressed significantly higher levels of RORγt than those from normal media (Figure 5E) but were not significant producers of IL-17A (Figure S3A). These observations suggest that while enhancing TGF-β-mediated Foxp3 induction, HS is also capable of promoting a RORγt+ phenotype in developing Treg cells in the absence of essential exogenous RORγt-inducing signals (e.g., IL-6 and IL-23) (Ivanov et al., 2006; Bettelli et al., 2007). Further analysis revealed that iTreg cells induced in HS conditions did not co-express other major lineage-defining transcription factors such as Tbet and GATA3, indicating that the HS-mediated iTreg cell differentiation is highly specific to the Th17 cell program (Figures 5I and 5J).
The differentiation of CD4+ T cell subsets is reciprocally regulated (Zhu et al., 2010). It is well recognized that Th1-, Th2-, and Th17-polarizing cytokines (i.e., IL-12, IL-4, and IL-6, respectively) have inhibitory effects on TGF-β-mediated Treg induction and vice versa (Prochazkova etal., 2012; Dardalhon etal., 2008; Bettelli et al., 2006). Thus, we assessed whether HS conditions influence IL-12- or IL-4-mediated Th1 and Th2 cell differentiation, respectively, in the presence of TGF-β (Bright and Sriram, 1998; Heath et al., 2000). In line with our previous results, HS significantly potentiated the effect of TGF-β, as we observed an increase in the frequency of Foxp3+ and a concomitant decrease in Tbet+ or GATA3+ T cells in vitro (Figures S5A–S5C and 5E–5G). Interestingly, the reduction in Tbet or GATA3 expression correlated with a substantial upregulation of RORγt expression in iTreg cells generated with exogenous TGF-β and in the presence of HS. At low TGF-β concentrations, HS was sufficient to give rise to more than 10% of RORγt+ iTreg cells, even in the presence of strong Th1- (high levels of IL-12) or Th2-cell-polarizing (high levels of IL-4) conditions, in contrast to normal media (Figures S5A, S5D, S5E, and S5H). Overall, these results suggest that HS potentiates TGF-β-mediated signaling in T cells, leading to a skewed polarization of T cell subsets toward Treg and Th17 cell lineages.
Pre-exposure of CD4+ Teff Cells in HS Conditions Promotes the Generation of Inflammatory RORγt+ Th17-like Treg Cells In Vivo
Lastly, we sought to determine whether the Treg- and Th17-cell-promoting effects observed were stable or required continuous exposure of T cells to HS in vivo. To address this key question, we used a model of T-cell-transfer-induced intestinal inflammation where pathogenesis is dictated by the balance of Th17 and Treg cells in the gut and determined by the local availability of TGF-β and IL-6 cytokines in situ (Mottet et al., 2003; Ostanin et al., 2009). Using this model, we are able to track the development of Th17 or iTreg cells in the gut, as well as the functional consequences of any changes in T cell phenotypes following transfer of CD4+ Teff cells that were pre-exposed to NS or HS in vitro. Here, FACS-purified (GFP−) Teff cells were activated in NS or HS in vitro for 3 days, followed by another FACS purification to ensure equal viabilities (>99%) of donor cells between the two treatment groups. These viable preconditioned GFP− Teff cells were then transferred intravenously (i.v.) into Tcrb−/− recipient mice, and the donor T cells were examined in the gut 12 days post-transfer (Figure 6A). Interestingly, in mesenteric lymph nodes (mLNs) and colons of mice that received HS-preconditioned Teff cells, we observed significantly higher frequencies of RORγt+ Th17 cells compared to mLN and colons of mice that received NS-preconditioned T cells (Figures 6B and 6C). There was also significantly higher induction of total iTreg cells and RORγt+ iTreg cells in mice that received HS-preconditioned Teff cells, confirming our in vitro findings (Figures 6B, 6D, and 6E). Compared to iTreg cells derived from NS-preconditioned Teff cells, HS-preconditioned Teff cells gave rise to significantly greater proportions of IL-17A+ RORγt+ Treg cells (Figures 6F and 6G). Moreover, the proportion and accumulation of RORγt+ Treg cells strongly correlated with both the numbers of total CD4+ T cells and IL-17-producing T cells in the mLNs (Figures 6H–6K). In line with this result, the absolute number of RORγt+ Treg also correlated with the number of colonic IL-17A-producing T cells and the degree of intestinal inflammation assessed by histopathology (Figure S6). In addition, RORγt+ iTreg cells were not readily detected in pLNs, sites that did not experience Th17-cell-driven inflammation (Figures 6H–6K). Indeed, no correlation between the frequency or number of RORγt+ Treg cells and CD4+ or IL-17A-producing T cells was observed in the pLN (Figures 6H–6K). These results suggest that RORγt+ iTreg cells developed from HS-preconditioned T cells accumulate and expand in Th17-cell-driven inflammatory sites. These Treg cells may influence inflammatory outcomes depending on additional inflammatory signals available. Overall, HS preconditioning is sufficient to promote Treg cell differentiation into a Th17-like phenotype in vivo.
Figure 6. Pre-exposure of CD4+ Teff Cells in HS Conditions Promotes the Generation of Inflammatory RORγt+ Th17-like Treg Cells In Vivo.

(A) Schematic diagram showing the workflow of the adoptive transfer of salt-preconditioned T cells. FACS-purified Ly5.1+ Teff cells were preconditioned in vitro in NS or HS (+60 mM) media for 3 days. After a secondary FACS purification on viable CD4+ GFP− Teff cells, the same number of NS-preconditioned or HS-preconditioned Teff cells was intravenously transferred into Ly5.2+ TCRβ−/− lymphopenic mice. Mice were sacrificed, and transferred cells were isolated and analyzed at day 12 post-transfer.
(B) Flow cytometry analysis of the expression of Foxp3 and RORγt in the transferred (ly5.1+) CD4+ T cells isolated from mesenteric lymph nodes (mLNs) and colon lamina propria.
(C–E) Quantitative analyses of the frequency of RORγt+ Teff cells (C), Foxp3+ Treg cells (D), and RORγt+ Treg cells (E) in CD4+ T cells isolated from mLNs and colon lamina propria.
(F) Representative flow cytometry plots showing Foxp3+ Treg cells isolated from mLNs and colon lamina propria.
(G) Quantitative analysis of IL-17A-producing cells of Foxp3+ Treg cells isolated from mLNs and colon lamina propria.
(H and I) Correlative analyses of total infiltration of CD4+ T cells with the proportion of RORγt+ Treg cells among Foxp3+ Treg cells (H), and the absolute number of RORγt+ Treg cells (I) in the mLN and peripheral lymph node (pLN).
(J and K) Correlative analyses of total infiltration of IL-17A+ T cells with the proportion of RORγt+ Treg cells among Foxp3+ Treg cells (J), and the absolute number of RORγt+ Treg cells (K) in the mLN and pLN.
Data shown in (A)–(G) are pooled from three independent experiments with n > 3 mice for each group. Data shown in (H)–(K) are representative of the three independent experiments. Error bars represent mean ± standard deviation.
DISCUSSION
The capacity of CD4+ Treg cells to sense environmental cues and adapt their functions to a particular inflammatory environment is crucial for the maintenance of immune homeostasis. However, the nature of the signals and underlying mechanisms controlling this functional adaptation remain unknown. Given the key role of mRNA translational regulation for rapid changes in gene expression and constitution of the cell proteome, we carried out a genome-wide polysomal profiling of CD4+ T cells and identified the SGK1 mRNA as being preferentially translated in activated Treg cells. Interestingly, recent studies proposed a role for SGK1 as a salt sensor in CD4+ T cells, which promoted the differentiation of pathogenic Th17 cells and exacerbated experimental autoimmune encephalomyelitis (Kleinewietfeld et al., 2013; Wu et al., 2013). Hence, we postulated that SGK1 may play a role in Treg cell functional adaptation in HS conditions. Here, we show that the salt-SGK1 signaling axis enables Treg cells to acquire a Th17-like phenotype in vitro and in vivo, thus establishing salt as a novel, non-immune factor that promotes Treg cell functional adaptation.
In our study, HS was sufficient to induce RORγt expression in both tTreg and iTreg cell subsets but did not induce the production of pro-inflammatory cytokines like IL-17A in vitro. Similarly, the observation of a moderate decrease in Foxp3 expression in thymic HS-promoted RORγt+ Treg cells does not suggest a loss of Treg identity and function, as these cells still express significant levels of Foxp3. This phenotypic alteration may represent a beneficial mechanism for Treg cells to co-express RORγt and all the while maintain the potential to undergo subsequent functional specialization. Importantly, RORγt+ Foxp3+ Treg cells have been readily detected in various models of inflammation and disease, and some groups have confirmed that these cells fully retain their suppressive activity in vitro and in vivo (Kim et al., 2017; Yang et al., 2016). Indeed, we show that HS-pretreated thymic and iTreg cell subsets, despite expression of RORγt, retain potent suppressive function. Moreover, RORγt+ Treg cells express high CCR6 levels and accumulate in Th17-cell-driven inflammatory sites, but not in non-inflammatory sites (pLN), confirming their adapted phenotype and function for Th1 cell inflammation in vivo. Taken together, our study shows that RORγt+ Treg cells represent functionally specialized Treg cells that adapt to HS conditions while maintaining suppressive functions.
A previous study showed that salt decreased the suppressive function and augmented IFN-γ production in human Foxp3-expressing T cells, all the while upregulating rorc, il17a, and il17f gene expression (Hernandez et al., 2015). In our study, salt did not increase IFN-γ expression but rather increased Th17-like differentiation in murine Treg cells. While we did not observe IL-17A production in vitro, IL-17A+ RORγt+ Foxp3+ iTreg cells were readily induced in vivo, suggesting that additional inflammatory signals, like IL-23 (Wu et al., 2018) or IL-1β (Alvarez et al., 2019), may promote the development of inflammatory Th17-like Treg cells. However, it remains to be determined if RORγt+ Foxp3+ Treg cells accumulate in tissues to more efficiently control evolving Th17 inflammation in situ or whether they are compromised in their functional stability and actively contribute to the onset and magnitude of inflammatory responses.
The majority of HS-induced RORγt+ Foxp3+ Treg cells do not express Helios, a transcription factor associated with stable and robust Treg suppressive function (Sebastian et al., 2016; Thornton et al., 2019). This observation is consistent with the evidence that Helios− Treg cells have the propensity for functional plasticity (Bin Dhuban et al., 2015; Thornton et al., 2019). However, Helios+ Treg cells isolated from Foxp3-Helios dual reporter mice are also susceptible to the effect of salt, but to a lesser extent than their Helios− counterpart. These results are in agreement with the notion that Treg cells with differential Helios expression may represent Treg cell subsets with different functions or fates but collectively ensure immune regulation during the dynamic course of inflammation. Whether salt is a stress factor that may influence Helios stability and subsequently contributes to Treg cell plasticity remains an important topic for future studies.
iTreg cell subsets are key players in maintaining immune homeostasis at mucosal surfaces. Our results are in opposition to a recent study that suggested that iTreg cells were unaffected by HS (Luo et al., 2019). We show that salt strongly potentiates TGF-β-mediated iTreg induction through a SGK1-dependent pathway and provide the first evidence that SGK1 modulates TGF-β signaling in CD4+ T cells, as specific SGK1 inhibition significantly attenuates HS-promoted, TGF-β-mediated iTreg cell induction. In fact, SGK1 is known to potentiate TGF-β signaling in other cells by promoting the sustained expression of pSMAD2/3 through phosphorylation and subsequent degradation of NEDD-4L, a ubiquitin ligase that otherwise labels SMAD2/3 for proteasomal degradation (Gao et al., 2009). Our results suggest that a similar mechanism may be involved in T cells.
In addition to potentiating iTreg generation, almost all iTreg cells generated in HS Th17-polarizing conditions co-express RORγt in a SGK1-dependent manner. This is in contrast to Th17 cell polarization in NS conditions, where CD4+ T cells differentiate exclusively to RORγt+ Th17 cells. Interestingly, the salt-induced RORγt+ iTreg cells stem from RORγt+, but not RORγt−, Teff cells in such conditions. These observations suggest that SGK1 activation potentiates the TGF-β pathway and enables RORγt+ T cells to further upregulate Foxp3 expression, although this effect may represent a compensatory mechanism by Foxp3 to reestablish transcriptional dominance in newly generated RORγt+ T cells. Strikingly, HS promotes RORγt+ iTreg cell induction in the absence of IL-6, even in the presence of strong Th1 and Th2 polarizing conditions, which inhibit RORγt expression. These results suggest that the effect of salt is TGF-β dependent but independent of other polarizing signals.
Previous work on CD4+ Teff cells described a mechanism by which HS-induced activation of SGK1 supports functional maturation of pathogenic Th17 cell by promoting IL-23R expression (Wu et al., 2013). However, the mechanism by which SGK1 promoted Th17 cell maturation was suggested to be secondary to previous RORγt induction, as IL-23R transcription is driven by RORγt, and thus occurs only in a later stage of Th17 cell differentiation. In contrast, we propose a mechanism by which SGK1 signaling induced by HS can directly promote RORγt expression in Foxp3+ Treg cells, thus acting upstream in Th17 polarization. Since no exogenous IL-23 was supplemented and T cells are not prominent producers of IL-23, we have limited indication that IL-23 is involved in the observed effect of salt, though a direct causative relationship was not tested. Future studies will aim to dissect the pathways downstream of SGK1 that enable RORγt transcriptional activation in Treg cells and understand the functional outcomes linked to SGK1 expression by these cells.
Our study highlights that translational regulation of gene expression is an efficient way for Treg cells to sense and integrate environmental signals like salt with the goal of adapting their function to the local inflammatory milieu. In this regard, we identified an essential role of the salt-sensing kinase SGK1 in modulating Treg cell differentiation and functional adaptation driven by salt. Specifically, we describe that HS conditions instruct a RORγt-mediated Th17-like phenotype in both tTreg and iTreg cells without compromising suppressive capacity, thus representing functionally specialized subsets of Treg cells. This study provides a potential basis to explain epidemiological observations of increased incidence of autoimmunity and chronic inflammatory diseases among populations consuming HSD. Further validation of the involvement of the SGK1 pathway in immune dysregulation will shed light into the design of novel targeted therapy for the control of Th17 cell responses or potentiation of Treg cell function in various autoimmune disorders.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
This study did not generate new unique reagents. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ciriaco A. Piccirillo (ciro.piccirillo@mcgill.ca).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6-Foxp3GFP knock-in mice were kindly provided by Dr. Alexander Rudensky (Memorial Sloan Kettering Cancer Centre, NY) and bred into CD45.1 (Ly5.1) congenic background in house. C57BL/6 WT mice and C57BL/6 TCRβ−/− mice were purchased from Taconic Laboratory and Jackson Laboratory, respectively. All mice were housed and bred in a single room under specific pathogen-free conditions at the Research Institute of McGill University Health Centre (RI-MUHC), except the C57BL/6 WT mice used for HSD experiments were housed and bred under specific pathogen-free conditions at McGill University and Genome Quebec Innovation Centre. All in vivo experiments used sex-matched littermates at 8–12 weeks of age, unless otherwise stated. All experiments and procedures were carried out in accordance with guidelines prescribed by McGill University and the RI-MUHC.
Primary Murine Cells
Fresh total splenocytes from C57BL/6-Foxp3RFP-HeliosGFP knock-in dual reporter mice were kindly provided by Dr. Ethan Shevach (National Institute of Allergy and Infectious Disease, National Institute of Health, Bethesda, MD).
METHOD DETAILS
Isolation and purification of T cells
For in vitro assays or pre-treatments, primary murine CD4+ T cells were isolated from spleen and peripheral lymph nodes from Foxp3GFP mice. Cells were stained with mouse CD4 microbeads (Miltenyi Biotec) as per manufacturer’s protocol. CD4+ cells were then positively selected using the Miltenyi AutoMACS (purity > 95%). If needed, MACS-enriched CD4+ cells were stained with CD4-AF700 and further purified into CD4+ GFP− TEFF cell and CD4+ GFP+ TREG cell subsets using a BD FACSAria Fusion cell sorter (purity > 99%). For isolation and purification of TREG cells from thymus, CD4+ CD8− GFP+ TREG cells were sorted on a BD FACSAria Fusion cell sorter after depleting CD8+ cells using the Miltenyi AutoMACS (purity > 99%). For isolation and purification of TREG cells from Foxp3GFP-HeliosGFP dual reporter mice, Helios+ TREG subset was sorted as CD4+ RFP+ GFP+ and Helios− TREG subset was sorted as CD4+ RFP+ GFP− (purity > 99.7%). For isolation of lymphocytes from spleens, peripheral lymph nodes and mesenteric lymph nodes, simple mechanical homogenization using frosted glass slides were used to liberate the cells from the organs. For isolation of lymphocytes from intestinal lamina propria, using a previously described protocol involving EDTM and Collagenase IV for the liberation of tissue resident leukocytes (Alvarez et al., 2019). The processed cell suspensions were filtered through a 70 μm cell strainer prior to staining or PMA/ionomycin stimulation. Kidneys were minced and incubated in HBSS containing 1 mg/ml Collagenase D (Roche) and 0.1 mg/ml DNase I for 45 minutes at 37°C. The kidneys were then washed with HBSS, and cells were dissociated using an enzyme-free cell dissociation buffer for 10 minutes at 37°C (GIBCO). The cell-containing supernatant was filtered through a 100 μm and then a 70 μm mesh before washing with PBS for FACS staining.
Polysome profiling
Cells isolated from peripheral lymph nodes and spleen of Foxp3GFP mice were autoMACS (Miltenyi Biotec) purified for CD4+ T cells. High purity (> 99%) populations of CD4+ TEFF cells and Foxp3+ TREG cells were further sorted based on Foxp3-GFP fusion protein expression by FACSAria Fusion cell sorter (BD Bioscience). For naive groups, cells were incubated with cycloheximide (100 μg/ml) immediately post-cell sorting to immobilize ribosomes on the mRNA. For activated groups, sorted cells were activated for 36h with plate bound α-CD3 and α-CD28 (5 μg/ml and 2 μg/ml, respectively; BD Bioscience) in the presence of recombinant hIL-2 (100U/ml; a kind gift from the Surgery Branch, NCI/NIH). Cycloheximide (100 μg/ml) was added to the medium at the end of the culture. Cytosolic and polysome-associated RNA directly ex vivo and post-activation in vitro were isolated using a sucrose gradient as described previously (Bjur et al., 2013). Data analysis were performed using our previously published methods and algorithms.
In vitro assays
For in vitro assays, FACS-sorted CD4+ Foxp3− (GFP−) TEFF cells were activated alone (1 × 105 cells), or co-cultured with various FACS-sorted TREG cells in 96-well flat-bottom plates previously coated with α-CD3 and α-CD28 (5 μg/ml and 2 μg/ml, respectively; BD Bioscience) in RPMI1640 medium (Wisent) supplemented with 10% FBS. In co-culture experiments, TREG and TEFF cells were combined at a 1:6 ratio (1.5 × 104 TREG: 9 × 104 TEFF) to recapitulate physiological balance of T cell activation and suppression while ensuring adequate number of TREG cells for reliable analysis. Original TEFF cells were labeled with Cell Trace Violet (CTV) Proliferation Dye (Invitrogen) prior to co-culture with unlabelled original tTREG cells for post-co-culture identification of the ontogeny of any resultant cells, regardless of their Foxp3 expression. For various T cell differentiation assays, polarizing cytokines were always added at the time of activation and no further cytokine was replenished. Recombinant mIL-6 (10 ng/ml or otherwise indicated; BioLegend) and mTGFβ1 (1 ng/ml or otherwise indicated; Novoprotein) were used for TH17 differentiation. iTREG induction was carried out in presence of recombinant mTGFβ1 (1 ng/ml, or otherwise indicated; Novoprotein) alone. Recombinant mIL-12 (10 ng/ml) and mIL-4 (10 ng/ml) were used for TH1 differentiation and TH2 differentiation, respectively. All in vitro assays were stopped and analyzed 72 hr post-activation unless otherwise described. Increase in NaCl concentration was achieved by supplementing negligible volume (5–7.5 μL per 250 μl) of sterile NaCl aqueous solution in culture medium. The additional 40 mM or 60 mM of NaCl were empirically tested to have no significant effect on cell survival and growth. SGK1 inhibitor (GSK650394, GlaxoSmithKline) was used at 0.75 μM, a concentration with no significant effect on cell survival and growth. Furosemide (Sigma Aldrich) was used at 100 μM, a concentration with no significant effect on cell survival and growth.
Suppression assay
FACS-sorted CD4+ CD45.1+Foxp3− (GFP−) TEFF cells were labeled with Cell Trace Violet (CTV) Proliferation Dye (Invitrogen) and used as responder cells for the suppression assay. 5 × 104 TEFF were plated in 96-well flat-bottomed plates together with 2 × 105 mitomycin-treated CD4− feeder cells in the presence of 1 μg/ml of soluble α-CD3ε (BD Bioscience). tTREG cells were pre-treated with control or high salt (+60 mM) medium in previously described TH17 polarizing conditions for 3 days, then FACS-purified again for CD4+ CD45.2+ GFP+ tTREG cells. iTREG cells were induced by TGFβ in control medium or high salt (+60 mM) medium for 3 days, then FACS-purified again for CD4+ CD45.2+ GFP+ iTREG cells. Purified TREG cells were plated with responder cells at 1:4, 1:8, and 1:16 ratios. Proliferation of CD45.1+ TEFF was assessed by flow cytometry at day 3. Suppression was calculated as based on the division index of TEFF cells cultured in the absence of TREG cells using the following formula: (1-(Division index of TEFF cultured in the presence of TREG cells / Division index of TEFF cultured in the presence of TREG cells))*100.
Flow cytometry
Single cell suspensions were first stained with fixable viability dye eFluor780 or eFluor506 (Invitrogen) in PBS following manufacturer’s instruction. Extracellular markers were stained with monoclonal antibodies with directly-conjugated fluorochromes in PBS. Subsequently, cells were fixed and permeabilized using the eBioscience Foxp3/transcription factor staining buffer kit (Invitrogen) according to manufacturer’s instruction. Intracellular and intranuclear markers were stained with monoclonal antibodies with directly-conjugated fluorochrome in 1 × permeabilization buffer (Invitrogen). Detailed extracellular and intracellular antibody information is listed in the Key Resource Table. For intracellular cytokine detection, cells were stimulated with PMA and ionomycin in the presence of monensin-based Golgi Stop (BD) for 3hrs at 37°C in 5% CO2 incubator prior to staining. Flow cytometry data were acquired on a BD LSRFortessa X-20 flow cytometer (BD Bioscience) and analyzed using FlowJo version 10 software (FlowJo, LLC).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Anti-mouse CD4-Alexa Fluor 700 | Thermo Fisher | Cat #56-0041-82; RRID:AB_493999 |
| Anti-mouse CD4-APC-eFluor780 | Thermo Fisher | Cat #47-0042-82; RRID:AB_1272183 |
| Anti-mouse 45.1-PerCP-Cy5.5 | Thermo Fisher | Cat #45-0453-82; RRID:AB_1107003 |
| Anti-mouse 45.1-APC-Cy7 | BD PharMingen | Cat #560579; RRID:AB_1727487 |
| Anti-mouse Foxp3-FITC | Thermo Fisher | Cat #11-5773-82; RRID:AB_465243 |
| Anti-mouse RORγt-PE | Thermo Fisher | Cat #12-6988-82; RRID:AB_1834470 |
| Anti-mouse RORγt-BV786 | BD Horizon | Cat #564723; RRID:AB_2738916 |
| Anti-mouse Helios-Pacific Blue | BioLegend | Cat #137220; RRID:AB_10690535 |
| Anti-mouse Helios-PE-Cy7 | Thermo Fisher | Cat #25-9883-42; RRID:AB_2637136 |
| Anti-mouse Helios-APC | BioLegend | Cat #137222; RRID:AB_10662900 |
| Anti-mouse Tbet-PE-Cy7 | Thermo Fisher | Cat #25-5825-82; RRID:AB_11042699 |
| Anti-mouse GATA3-BUV395 | BD Horizon | Cat #565448; RRID:AB_2739241 |
| Anti-mouse IFN-γ-PE-Cy7 | Thermo Fisher | Cat #25-7311-82; RRID:AB_469680 |
| Anti-mouse IFN-γ-PerCP-Cy5.5 | Thermo Fisher | Cat #45-7311-82; RRID:AB_1107020 |
| Anti-mouse IL-4-PE | Thermo Fisher | Cat #12-7041-41; RRID:AB_10854281 |
| Anti-mouse IL-17A-APC | Thermo Fisher | Cat #17-7177-81; RRID:AB_763580 |
| Anti-mouse IL-17A-PE | BD PharMingen | Cat #559502; RRID:AB_397256 |
| Anti-SGK1[Y238]-PE | Abcam | Cat #ab212093 |
| Anti-mouse CD3ε | Thermo Fisher | Cat #16-0031-86; RRID:AB_468849 |
| Anti-mouse CD28 | Thermo Fisher | Cat #16-0281-86; RRID:AB_468923 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Recombinant mouse IL-6 | BioLegend | Cat #575704 |
| Recombinant mouse TGF-β | Novoprotein | Cat #CK33 |
| Recombinant mouse IL-12p70 | Peprotech | Cat #210-12 |
| Recombinant mouse IL-4 | BioLegend | Cat #574302 |
| Sodium Chloride | Fisher Chemical | Cat #S671-3 |
| SGK1 inhibitor-GSK650394 | Tocris | Cat #3572; CAS: 890842-28-1 |
| Furosemide | Sigma-Aldrich | Cat #F4381, CAS:54-31-9 |
| Mitomycin C | Sigma-Aldrich | Cat #M4287, CAS: 50-07-7 |
| Phorbol 12-myristate 13-acetate | Sigma-Aldrich | Cat #P1585; CAS: 16561-29-8 |
| Ionomycin calcium salt | Sigma-Aldrich | Cat #I0634; CAS: 56092-82-1 |
| BD Golgi Stop Protein Transport Inhibitor | BD Biosciences | Cat #554724 |
| EDTA | Thermo Fisher | Cat #15575-038 |
| Collagenase IV | GIBCO | Cat #17104-019; CAS: 9001-12-1 |
| Collagenase D | Sigma-Aldrich | Cat #11088866001; CAS: 9001-12-1 |
| Cell Dissociation Buffer | GIBCO | Cat #13151-014 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| Fixable Viability Dye eFluor 780 | Thermo Fisher | Cat #65-0865-14 |
| Fixable Viability Dye eFluor 506 | Thermo Fisher | Cat #65-0866-18 |
| Foxp3/Transcription Factor Fixation/Permeabilization Kit | Thermo Fisher | Cat #00-5521-00 |
| Permeabilization Buffer 10X | Thermo Fisher | Cat #00-8333-56 |
| Cell Trace Violet Cell Proliferation Kit | Thermo Fisher | Cat #C34571 |
| CD4 (L3T4) Microbeads | Miltenyi Biotec | Cat #130-117-043 |
|
| ||
| Deposited Data | ||
|
| ||
| Polysomal Profiling of CD4+ T cells | Bjur et al., 2013 | GEO: GSE45401 |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| Mouse: C57BL/6 | Taconic Laboratory | B6NTac |
| Mouse: C57BL/6 Foxp3GFP | Laboratory of Alexander Rudensky | N/A |
| Mouse: C57BL/6 Ly5.1+ TCRβ−/− | Jackson Laboratory | B6.129P2-Tcrbtm1Mom/J Stock No.002118 |
| Mouse Cells: C57BL/6 Foxp3RFP HeliosGFP | Laboratory of Ethan Shevach | N/A |
|
| ||
| Software and Algorithms | ||
|
| ||
| FlowJo | FlowJo, LLC | https://www.flowjo.com/ |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| BioRender | Biorender.com | https://www.biorender.com/ |
| Anota | Larsson et al., 2011; Bjur et al., 2013 | https://www.bioconductor.org/packages/release/bioc/html/anota.html |
High salt diet feeding
Age-/sex-matched C57BL/6 littermates (8–10 g) were randomly assigned to receive a high-salt diet (HSD) or a normal salt diet (NSD) at the time of weaning (21 days of age). Mice fed with NSD received regular laboratory chow (0.3% NaCl, from Teklad irradiated laboratory animal diet, Envigo) and distilled water. The HSD regimen consisted of 8% NaCl in chow (custom made) and 1% NaCl in water. Mice were sacrificed after two weeks of HSD/NSD feeding and organs/tissues were collected for analysis.
Adoptive T cell transfer
FACS-sorted Ly5.1+ CD4+ TEFF cells were in vitro activated in presence of high salt for 72 hr. High salt pre-conditioned TEFF cells were FACS-sorted again to purify viable CD4+ GFP− TEFF cells (purity > 99%). 7.5 × 105 cells were intravenously transferred into Ly5.2+ TCRβ−/− lymphopenic recipient mice. Age-/sex-matched control group mice received the same number cells which underwent the same process but were pre-conditioned in standard medium. Experiment group and control group mice were co-housed for the entire duration of the experiment. Mice were monitored for signals of colitis and weight loss throughout the experiment. At day 12 post-transfer, mice were sacrificed, and organs/tissues were collected for analysis.
Intestinal inflammation score
Mean pathology score of the colon of each mouse was assessed by 2 distinct blinded observers, each score twice with re-randomization following the guidelines from Erben et al. (2014).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism, version 7 (GraphPad Software). Data are displayed as mean ± standard deviation. For comparison with two groups, unpaired Student’s t test was used to determine significance. For comparison with more than two groups, one-way ANOVA was used followed by multiple comparisons with Tukey’s post hoc test. For multivariable comparisons involving multiple groups, two-way ANOVA was performed followed by Sidak’s multiple comparisons test. Correlation coefficient was determined by two-tailed Pearson correlation analysis. P value of ≤ 0.05 was considered significant. P values were indicated on graphs as * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
DATA AND CODE AVAILABILITY
This study did not generate any unique datasets or code.
ADDITIONAL RESOURCES
This study did not generate any additional resources.
Supplementary Material
Highlights.
The mRNA encoding salt-sensing SGK1 is preferentially translated in Foxp3+ Treg cells
HS promotes Th17-like adaptation in both tTreg and iTreg cell subsets via SGK1
HS-induced RORγt+ Treg cells are functionally adapted and suppressive
Helios− Treg cells are more susceptible than Helios+ Treg cells to the effects of salt
ACKNOWLEDGMENTS
We thank Helen Mason for technical assistance on various aspects of this research. We thank the Immunophenotyping Platform and the Histopathology Platform of the Research Institute of McGill University Health Centre for excellent cell-sorting services and histology service, respectively. Financial support for this study came from Canadian Institutes of Health Research (CIHR) operating grant PJT-148821 (C.A.P.); the Canada Research Chair program (C.A.P.); Natural Sciences and Engineering Research Council of Canada (NSERC) discovery grant RGPIN/05541-2017 (J.Z.); and in part supported by the Intramural Research Program of NIAID, NIH (A.M.T. and E.M.S.). The graphical abstract was generated using the BioRender application (Biorender.com).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.01.002.
REFERENCES
- Alvarez F, Istomine R, Shourian M, Pavey N, Al-Aubodah TA, Qureshi S, Fritz JH, and Piccirillo CA (2019). The alarmins IL-1 and IL-33 differentially regulate the functional specialisation of Foxp3+ regulatory T cells during mucosal inflammation. Mucosal Immunol. 12, 746–760. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, et al. (2011). Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell LM, Leong ML, Kim B, Wang E, Park J, Hemmings BA, and Firestone GL (2000). Hyperosmotic stress stimulates promoter activity and regulates cellular utilization of the serum- and glucocorticoid-inducible protein kinase (Sgk) by a p38 MAPK-dependent pathway. J. Biol. Chem. 275, 25262–25272. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, and Kuchroo VK (2006). Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Oukka M, and Kuchroo VK (2007). T(H)-17 cells in the circle of immunity and autoimmunity. Nat. Immunol. 8, 345–350. [DOI] [PubMed] [Google Scholar]
- Bin Dhuban K, d’Hennezel E, Nashi E, Bar-Or A, Rieder S, Shevach EM, Nagata S, and Piccirillo CA (2015). Coexpression of TIGIT and FCRL3 identifies Helios+ human memory regulatory T cells. J. Immunol. 194, 3687–3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjur E, Larsson O, Yurchenko E, Zheng L, Gandin V, Topisirovic I, Li S, Wagner CR, Sonenberg N, and Piccirillo CA (2013). Distinct translational control in CD4+ T cell subsets. PLoS Genet. 9, e1003494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright JJ, and Sriram S. (1998). TGF-beta inhibits IL-12-induced activation of Jak-STAT pathway in T lymphocytes. J. Immunol. 161, 1772–1777. [PubMed] [Google Scholar]
- Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, and Ramsdell F. (2001). Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 27, 68–73. [DOI] [PubMed] [Google Scholar]
- Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, and Rudensky AY (2009). CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 326, 986–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, and Wahl SM (2003). Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 198, 1875–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Grigsby CL, Law CS, Ni X, Nekrep N, Olsen K, Humphreys MH, and Gardner DG (2009). Tonicity-dependent induction of Sgk1 expression has a potential role in dehydration-induced natriuresis in rodents. J. Clin. Invest. 119, 1647–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, Mitsdoerffer M, Strom TB, Elyaman W, Ho IC, et al. (2008). IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(−) effector T cells. Nat. Immunol. 9, 1347–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erben U, Loddenkemper C, Doerfel K, Spieckermann S, Haller D, Heimesaat MM, Zeitz M, Siegmund B, and Kühl AA (2014). A guide to histomorphological evaluation of intestinal inflammation in mouse models. Int. J. Clin. Exp. Pathol. 7, 4557–4576. [PMC free article] [PubMed] [Google Scholar]
- Gao S, Alarcón C, Sapkota G, Rahman S, Chen PY, Goerner N, Macias MJ, Erdjument-Bromage H, Tempst P, and Massagué J. (2009). Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-beta signaling. Mol. Cell 36, 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath VL, Murphy EE, Crain C, Tomlinson MG, and O’Garra A. (2000). TGF-beta1 down-regulates Th2 development and results in decreased IL-4-induced STAT6 activation and GATA-3 expression. Eur. J. Immunol. 30, 2639–2649. [DOI] [PubMed] [Google Scholar]
- Hernandez AL, Kitz A, Wu C, Lowther DE, Rodriguez DM, Vudattu N, Deng S, Herold KC, Kuchroo VK, Kleinewietfeld M, and Hafler DA (2015). Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J. Clin. Invest. 125, 4212–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori S. (2014). Lineage stability and phenotypic plasticity of Foxp3+ regulatory T cells. Immunol. Rev. 259, 159–172. [DOI] [PubMed] [Google Scholar]
- Istomine R, Pavey N, and Piccirillo CA (2016). Posttranscriptional and translational control of gene regulation in CD4+ T cell subsets. J. Immunol. 196, 533–540. [DOI] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, and Littman DR (2006). The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133. [DOI] [PubMed] [Google Scholar]
- Kim BS, Lu H, Ichiyama K, Chen X, Zhang YB, Mistry NA, Tanaka K, Lee YH, Nurieva R, Zhang L, et al. (2017). Generation of RORγt+ antigen-specific T regulatory 17 cells from Foxp3+ precursors in autoimmunity. Cell Rep. 21, 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King HA, and Gerber AP (2016). Translatome profiling: methods for genome-scale analysis of mRNA translation. Brief. Funct. Genomics 15, 22–31. [DOI] [PubMed] [Google Scholar]
- Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, and Hafler DA (2013). Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluger MA, Meyer MC, Nosko A, Goerke B, Luig M, Wegscheid C, Tiegs G, Stahl RA, Panzer U, and Steinmetz OM (2016). RORγt(+) Foxp3(+) cells are an independent bifunctional regulatory T cell lineage and mediate crescentic GN. J. Am. Soc. Nephrol. 27, 454–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, and Campbell DJ (2009). The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 10, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs CF, Schmidt T, Riedel JH, and Panzer U. (2017). T helper type 17 cells in immune-mediated glomerular disease. Nat. Rev. Nephrol. 13, 647–659. [DOI] [PubMed] [Google Scholar]
- Lang F, Stournaras C, and Alesutan I. (2014). Regulation of transport across cell membranes by the serum- and glucocorticoid-inducible kinase SGK1. Mol. Membr. Biol. 31, 29–36. [DOI] [PubMed] [Google Scholar]
- Larsson O, Sonenberg N, and Nadon R. (2011). anota: Analysis of differential translation in genome-wide studies. Bioinformatics 27, 1440–1441. [DOI] [PubMed] [Google Scholar]
- Li L, Kim J, and Boussiotis VA (2010). IL-1β-mediated signals preferentially drive conversion of regulatory T cells but not conventional T cells into IL-17-producing cells. J. Immunol. 185, 4148–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Xue Y, Wang J, Dang J, Fang Q, Huang G, Olsen N, and Zheng SG (2019). Negligible effect of sodium chloride on the development and function of TGF-beta-induced CD4(+) Foxp3(+) regulatory T cells. Cell Rep. 26, 1869–1879.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, Park JK, Beck FX, Müller DN, Derer W, et al. (2009). Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat. Med. 15, 545–552. [DOI] [PubMed] [Google Scholar]
- Maloy KJ, and Powrie F. (2011). Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474, 298–306. [DOI] [PubMed] [Google Scholar]
- Mohr I, and Sonenberg N. (2012). Host translation at the nexus of infection and immunity. Cell Host Microbe 12, 470–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottet C, Uhlig HH, and Powrie F. (2003). Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J. Immunol. 170, 3939–3943. [DOI] [PubMed] [Google Scholar]
- Norlander AE, Saleh MA, Pandey AK, Itani HA, Wu J, Xiao L, Kang J, Dale BL, Goleva SB, Laroumanie F, et al. (2017). A salt-sensing kinase in T lymphocytes, SGK1, drives hypertension and hypertensive end-organ damage. JCI Insight 2, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostanin DV, Bao J, Koboziev I, Gray L, Robinson-Jackson SA, Kosloski-Davidson M, Price VH, and Grisham MB (2009). T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G135–G146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccirillo CA (2013). Environmental sensing and regulation of gene expression in CD4+ T cell subsets. Curr. Opin. Immunol. 25, 564–570. [DOI] [PubMed] [Google Scholar]
- Piccirillo CA, Bjur E, Topisirovic I, Sonenberg N, and Larsson O. (2014). Translational control of immune responses: from transcripts to translatomes. Nat. Immunol. 15, 503–511. [DOI] [PubMed] [Google Scholar]
- Prochazkova J, Pokorna K, and Holan V. (2012). IL-12 inhibits the TGF-β-dependent T cell developmental programs and skews the TGF-β-induced differentiation into a Th1-like direction. Immunobiology 217, 74–82. [DOI] [PubMed] [Google Scholar]
- Rudensky AY (2011). Regulatory T cells and Foxp3. Immunol. Rev. 241, 260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Yamaguchi T, Nomura T, and Ono M. (2008). Regulatory T cells and immune tolerance. Cell 133, 775–787. [DOI] [PubMed] [Google Scholar]
- Sebastian M, Lopez-Ocasio M, Metidji A, Rieder SA, Shevach EM, and Thornton AM (2016). Helios controls a limited subset of regulatory T cell functions. J. Immunol. 196, 144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherk AB, Frigo DE, Schnackenberg CG, Bray JD, Laping NJ, Trizna W, Hammond M, Patterson JR, Thompson SK, Kazmin D, et al. (2008). Development of a small-molecule serum- and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res. 68, 7475–7483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevach EM (2009). Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 30, 636–645. [DOI] [PubMed] [Google Scholar]
- Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, and Shevach EM (2010). Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J. Immunol. 184, 3433–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton AM, Lu J, Korty PE, Kim YC, Martens C, Sun PD, and Shevach EM (2019). Helios+ and Helios− Treg subpopulations are phenotypically and functionally distinct and express dissimilar TCR repertoires. Eur. J. Immunol. 49, 398–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voo KS, Wang YH, Santori FR, Boggiano C, Wang YH, Arima K, Bover L, Hanabuchi S, Khalili J, Marinova E, et al. (2009). Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc. Natl. Acad. Sci. USA 106, 4793–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, et al. (2001). X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 27, 18–20. [DOI] [PubMed] [Google Scholar]
- Wohlfert E, and Belkaid Y. (2010). Plasticity of T reg at infected sites. Mucosal Immunol. 3, 213–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, Regev A, and Kuchroo VK (2013). Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 496, 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Chen Z, Xiao S, Thalhamer T, Madi A, Han T, and Kuchroo V. (2018). SGK1 governs the reciprocal development of Th17 and regulatory T cells. Cell Rep. 22, 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, Shah B, Chang SH, Schluns KS, Watowich SS, et al. (2008). Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 29, 44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang BH, Hagemann S, Mamareli P, Lauer U, Hoffmann U, Beckstette M, Föhse L, Prinz I, Pezoldt J, Suerbaum S, et al. (2016). Foxp3(+) T cells expressing RORγt represent a stable regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal inflammation. Mucosal Immunol. 9, 444–457. [DOI] [PubMed] [Google Scholar]
- Yu F, Sharma S, Edwards J, Feigenbaum L, and Zhu J. (2015). Dynamic expression of transcription factors T-bet and GATA-3 by regulatory T cells maintains immunotolerance. Nat. Immunol. 16, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurchenko E, Shio MT, Huang TC, Da Silva Martins M, Szyf M, Levings MK, Olivier M, and Piccirillo CA (2012). Inflammation-driven reprogramming of CD4+ Foxp3+ regulatory T cells into pathogenic Th1/Th17 T effectors is abrogated by mTOR inhibition in vivo. PLoS ONE 7, e35572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, et al. (2008). TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 453, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Yamane H, and Paul WE (2010). Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 28, 445–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.