

Abstract
The epigenome is multi-dimensional, with individual molecular components operating on different levels to control transcriptional output. Techniques that combine measurements of these features can reveal their precise correspondence in genomic space, or temporal connectivity, to better understand how they jointly regulate genes. ATAC-Me is an integrated method to probe DNA methylation and chromatin accessibility from a single DNA fragment library. Intact nuclei undergo Tn5 transposition to isolate DNA fragments within nucleosome-free regions. Isolated fragments are exposed to sodium bisulfite before library amplification and sequencing. A typical ATAC-Me experiment detects ~60–75k peak regions (p-value <0.05), covering approximately 3–4 million CpG sites with at least 5X coverage. These sites display a range of methylation values depending on the cellular and genomic context. The approach is well-suited for time course studies that aim to capture chromatin and DNA methylation dynamics in tandem during cellular differentiation. The protocol is completed in 2 days with standard molecular biology equipment and expertise. Analysis of resulting data uses publicly available software requiring basic bioinformatics skills to interpret results.
INTRODUCTION
Development of the protocol
Epigenetic mechanisms are fundamental to the gene regulatory processes that shape cellular development. The epigenome is multi-faceted with different biochemical features acting in both complementary and opposing ways to influence gene expression at a given locus. The individual gene regulatory contributions of these features, including chromatin accessibility, post-translational histone modifications, and DNA methylation (DNAme), has been highlighted by studies probing their locations genome-wide1–7. Steady-state comparisons of these profiles reveal that certain features often colocalize while others appear to be mutually exclusive8,9. Multi-omic analysis of data from cell-types at different developmental stages further shows, albeit indirectly, that these relationships change over time, particularly at gene enhancer elements, as cells become more differentiated10. It is well-established that a significant level of interplay exists between epigenetic modifications11,12; so, an integrative view of their dynamics, and how they may influence each other, is particularly relevant to understanding tightly regulated processes such as cellular differentiation.
DNAme is prevalent in vertebrate genomes and is dynamic during cellular differentiation. The ability to observe DNAme dynamics has been greatly augmented by the application of sodium bisulfite conversion methods to capture methylation at base pair resolution13–15. Numerous genome-wide studies have catalogued DNAme patterns across functionally specialized cell types and developmental states, finding that the most differential DNAme patterns often coincide with enhancers while a majority of promoters are stably hypomethylated14–16. DNAme is typically associated with stable repression of gene promoters and enhancers, while hypomethylation is considered permissive to transcription17,18. Additionally, the functional consequence of differential DNAme patterns in these regions appears to be affected by other epigenetic factors11. For example, in certain developmental contexts a subset of promoters are marked by a combination H3K27me3 (histone H3 tri-methylated lysine 27) and low DNAme, which may be indicative of a poised transcriptional state4. Low DNAme has also been associated with increased DNA accessibility and H3K27ac (histone H3 acetylated lysine 27) at putative enhancers7,19,20. Other studies have revealed unexpected “bivalent” permissive/repressive states, where high DNAme, H3k27ac and chromatin accessibility co-exist21,22. The cumulative effect of these distinct modifications can dictate the regulatory potential of the genomic region, thus influencing the resulting transcriptional program23. Given that gene regulation is an ordered process, it is possible that these bivalent states are short-lived during cellular transitions. To identify a more direct causal relationship between different facets of the epigenome requires observation of multiple features simultaneously. Joint profiling approaches are therefore uniquely positioned to measure multiple features while also providing information on their spatial and temporal relationships.
We developed ATAC-Me to directly assess chromatin accessibility and DNAme from a single DNA fragment library24,25. This method works by coupling assay for transposase accessible chromatin (ATAC) with bisulfite sequencing26–28. Initial enrichment of the chromatin accessible DNA is achieved by exposing nuclei to hyperactive Tn5 transposase assembled with methylated adaptors. The accessible fragment library is then treated with sodium bisulfite resulting in the deamination of non-methylated cytosines. By comparing sequenced reads to a reference genome, converted and unconverted cytosines are quantified at individual CpG sites to determine methylation levels. Nuclei can be sampled at a series of timepoints in response to a cellular stimulus. Capturing these two data types simultaneously throughout a period of dynamic gene regulation and within a changing chromatin environment provides valuable information on the spatiotemporal relationship between these two features.
Applications of the method
ATAC-Me is broadly applicable to research involving epigenetics and gene regulation. This method can be applied to understand the regulatory role of DNAme as it relates to chromatin accessibility in any cellular induction, differentiation or steady-state system. ATAC-Me is particularly well-suited for applications involving a timeseries, where multiple timepoints can result in high sample numbers and sequencing costs. Broadly, ATAC-Me can be used as a cost-effective and straightforward method for performing targeted methylation sequencing - similar to reduced representation bisulfite sequencing (RRBS) or sequence capture-based methylation profiling, but with a specific focus on open chromatin regions.
Comparison with other methods
Joint profiling of chromatin features and DNAme has been described previously using approaches such as NOME-seq (nucleosome occupancy and methylome sequencing), scNMT-seq (single-cell nucleosome, methylation and transcription sequencing) by extension, and ChIP-BS (chromatin immunoprecipitation coupled with bisulfite sequencing)19,29,30. In contrast with some approaches, ATAC-Me does not depend on cytosine frequency in specific dinucleotide contexts, so enhancers and other open chromatin regions representing a wide range of CpG densities are evaluated24. ATAC-Me is a straightforward approach and requires minimal molecular adaptation of existing Tn5-assisted methods including ATAC-seq and tagmentation-based whole genome bisulfite sequencing (WGBS). As the initial tagmentation step shares many similarities with the standard ATAC-seq method, publications are available for a variety of cell types to further assist in optimization31–33. To limit opportunities for input loss, our method uses a transposome assembly consisting of methylated adaptors which enables direct cloning of accessible fragments and prevents unwanted bisulfite conversion of sequencing-compatible fragment ends. Like standard ATAC-seq, fewer cells (50–200k) and fewer sequence reads (50–75 million) are necessary to obtain high, focused coverage (less expensive) of accessible DNA compared to some orthogonal methods (e.g., NOME-seq requires ≥200 million reads34). In addition, the cost effectiveness of the approach permits processing of numerous samples and time points. Notably, our method can interrogate transcription factor (TF) footprints despite loss of larger DNA fragments upon bisulfite treatment; however, similar to standard ATAC-seq, footprinting quality is predicated on sequencing depth and library complexity. In addition to identifying putative TF footprint sites, the DNAme state of these regions can be calculated, a feature that distinguishes ATAC-Me from other approaches.
Experimental design
Overview:
The ATAC-me method can be applied to a variety of cell types and differentiation schemes. Completion of the protocol from cell collection to library preparation can be accomplished in two days (Fig. 1). We have successfully applied the ATAC-Me protocol to a variety of cell types, including the B cell lymphoma line, BCBL1, the monocytic cell line, THP1, and H9 human embryonic stem cells (ESCs)35. Other cell lines, as well as primary cell populations, can be used as input to the ATAC-Me protocol, though each cell type may require slight optimization at the collection and tagmentation steps. We have found the below protocol to be an effective starting point for this optimization. Additionally, previously published standard ATAC-seq papers may provide further insight into beneficial modifications for other cell types36–38. Aside from cell type differences, the tagmentation step is greatly influenced by the activity of the Tn5 transposase. We purify the Tn5 in-house; however, regardless of enzyme source, the activity of adaptor-assembled enzyme may require optimization. Prior to use, we test the activity of enzyme aliquots on genomic DNA (gDNA), as outlined in Box 1. To initiate the ATAC-Me protocol, cells are harvested, and a single cell suspension must be achieved – a step that is likely to be unique to different systems and cell types. Following collection, 200,000 cells are lysed, and nuclei are collected. Cell number may be a limitation for the application of ATAC-Me in some systems. Generally, we have found that increased cell number is helpful for producing high complexity fragment libraries. Through subsampling of the accessible genome, ATAC approaches intentionally result in reduced complexity libraries compared to protocols that sample the entire genome. Within the sampled regions, all efforts are made to represent endogenous complexity in the final libraries (see Fig. 2a, b).
Figure 1. General workflow for the ATAC-Me protocol.
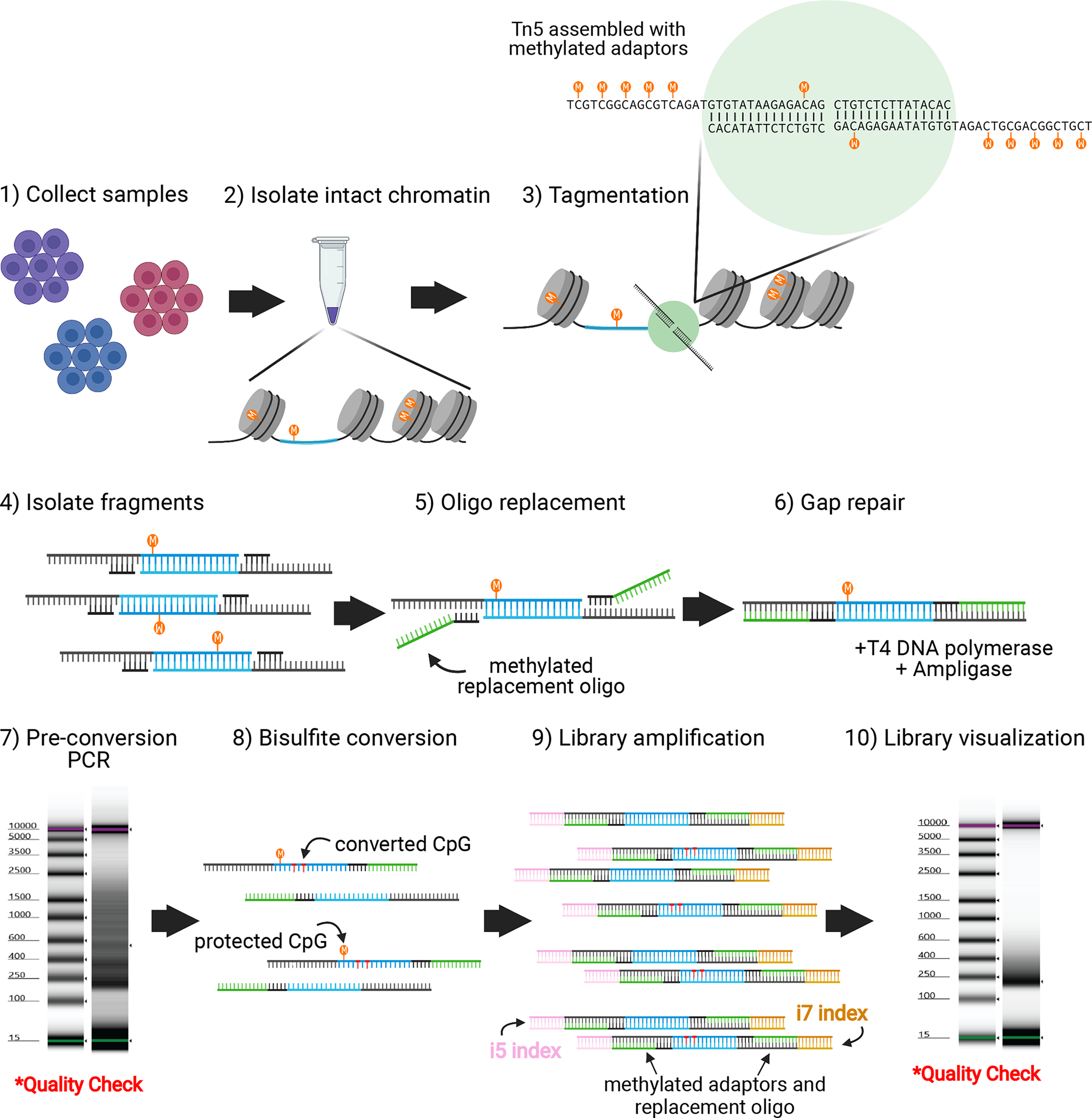
ATAC-Me is appropriate for use on a variety of cell types and treatment conditions, including flow sorted samples and in vitro differentiation systems. 200,000 cells are recommended as a starting point (compared to 50,000 in standard ATAC-seq protocols) to compensate for DNA input loss resulting from added experimental steps and sodium bisulfite degradation. Cells are pelleted by centrifugation and resuspended in a gentle lysis buffer to isolate nuclei with intact chromatin (procedure steps 7–14). Nuclei are incubated in Tn5 transposition reaction buffer with Tn5 assembled with methylated adaptors, displayed here as a green circle (procedure step 15–20). Tn5 accessible DNA fragments (blue) undergo column-based purification (procedure steps 21–28) followed by oligo replacement (procedure step 29) and gap repair steps to incorporate asymmetric, sequencing-compatible adaptor ends and complete ligation of DNA bottom strands (procedure steps 30–34). Due to the subsequent bisulfite conversion, adaptor sequences and replacement oligonucleotides (green) are methylated to preserve the sequence of the Illumina compatible adaptor ends (sequences are included in Table 1). A small aliquot of the resulting fragment pool is reserved for a quality check PCR amplification prior to bisulfite conversion (procedure steps 35–37). Fragment distribution of high-quality libraries should reflect standard ATAC libraries with nucleosomal banding patterns (procedure step 38). Fragments undergo heat denaturation and sodium bisulfite conversion (procedure steps 39–58) followed by limited PCR amplification and indexing for next generation sequencing (procedure steps 59–68). Indexed primers are depicted as yellow and pink lines. Final libraries should display a fragment distribution between 200–600bp. Figure created with Biorender.com. This schematic is labeled with the corresponding procedure steps.
Box 1. Tn5 transposase purification.
Transform pTXB1-Tn5 into E. coli strain C3013 (NEB) according to the manufacturer supplied protocol and plate onto ampicillin selection plates. Grow overnight at 37°C.
Inoculate colonies into 5mL LB + 100μg/mL ampicillin and incubate at 37°C, shaking at 220RPM until OD600 is approximately 1.0. Inoculate 5mL cultures into 1L LB +100μg/mL ampicillin and grow at 37°C, shaking at 220RPM in baffled flasks until OD600 is approximately 0.70.
Transfer cultures to an ice water bath and cool to 15°C. Add IPTG to a final concentration of 250μM followed by incubation at 23°C with shaking at 120RPM for approximately 4 hrs or until OD600 is approximately 2.1.
Harvest bacteria cultures by centrifugation at 5,000xg (F9–4×1000y rotor), 4°C for 10 min in 1L centrifuge bottles. Decant supernatant and store pellets at −80°C overnight.
Resuspend pellets in 60mL HEGX buffer (20 mM HEPES-KOH at pH 7.2, 0.8 M NaCl, 1 mM EDTA, 10% (vol/vol) glycerol, 0.2% (vol/vol) Triton X-100) supplemented with Complete EDTA-free protease inhibitor tablet.
Cool bacterial suspension on salt-ice to −1°C and subsequently lyse with 8 rounds of sonication using a 5mm tip (Branson) for 40 seconds at 70% duty cycle. Pellet lysate at 27,000xg (SS-34 rotor), 4°C for 30 min.
Prepare 10% (vol/vol) Poly(ethyleneimine) (PEI) solution by diluting 1:5 in sterile H2O. Clear lysate of contaminating E. coli genomic DNA by drop wise addition of 1.6mL 10% (vol/vol) neutralized (PEI) solution with slow stirring. The PEI lysate solution should begin to turn milky white as the DNA precipitate forms.
Pellet the PEI lysate mixture at 12,000 RPM (SS-34 rotor), 4°C for 10 min. Prepare purification column with 5mL chitin resin. Pre-wash column with 200mL HEGX buffer. Discard the pellet and load supernatant onto a preparedchitin column at 0.4mL/min flow rate, 4°C. Wash column with 300 mL HEGX buffer at a flow rate of 0.4mL/min.
Elute Tn5 transposase by the addition of 14mL HEGX + 100mM DTT to the column followed by a 48hr incubation at 4°C to induce cleavage from the column. Collect transposase in 1mL fractions from the column and subsequently analyze for protein concentration using a Bradford assay.
Pool fractions with the highest protein concentration and dialyze twice with 1L of 2x dialysis buffer (100mM HEPES, pH 7.2, 0.2M NaCl, 0.2mM EDTA, 2mM DTT, 0.2% (vol/vol) Triton-X 100, 20% (vol/vol) Glycerol). Dialyzed protein may be concentrated with Amicon Ultracel-30 centrifugal filters (Millipore) to a target range of 3–5 mg/mL.
Add 1 volume of 100% glycerol, prepare 200μl aliquots purified Tn5 transposase and store at −20°C.
Enzyme activity can be tested by incubating Tn5 with purified genomic DNA. Assemble transposomes as described in Steps 1–6 of the main Procedure. To test Tn5 activity, combine 50ng of genomic DNA, 5μl 5x Tris-DMF, 2.5μl assembled Tn5 and nuclease-free water up to 50μl and incubate at 55°C for 8 min. Stop the reaction by adding 125μl of DNA Binding Buffer from a DNA Clean & Concentrate-5 (Zymo, D4004) kit. The resulting fragment collection can be run on a 1% agarose gel at 100V for 50 minutes for visualization. Target fragment distributions will be between 200–1000bp.
Figure 2. Fragment distribution at quality check steps.
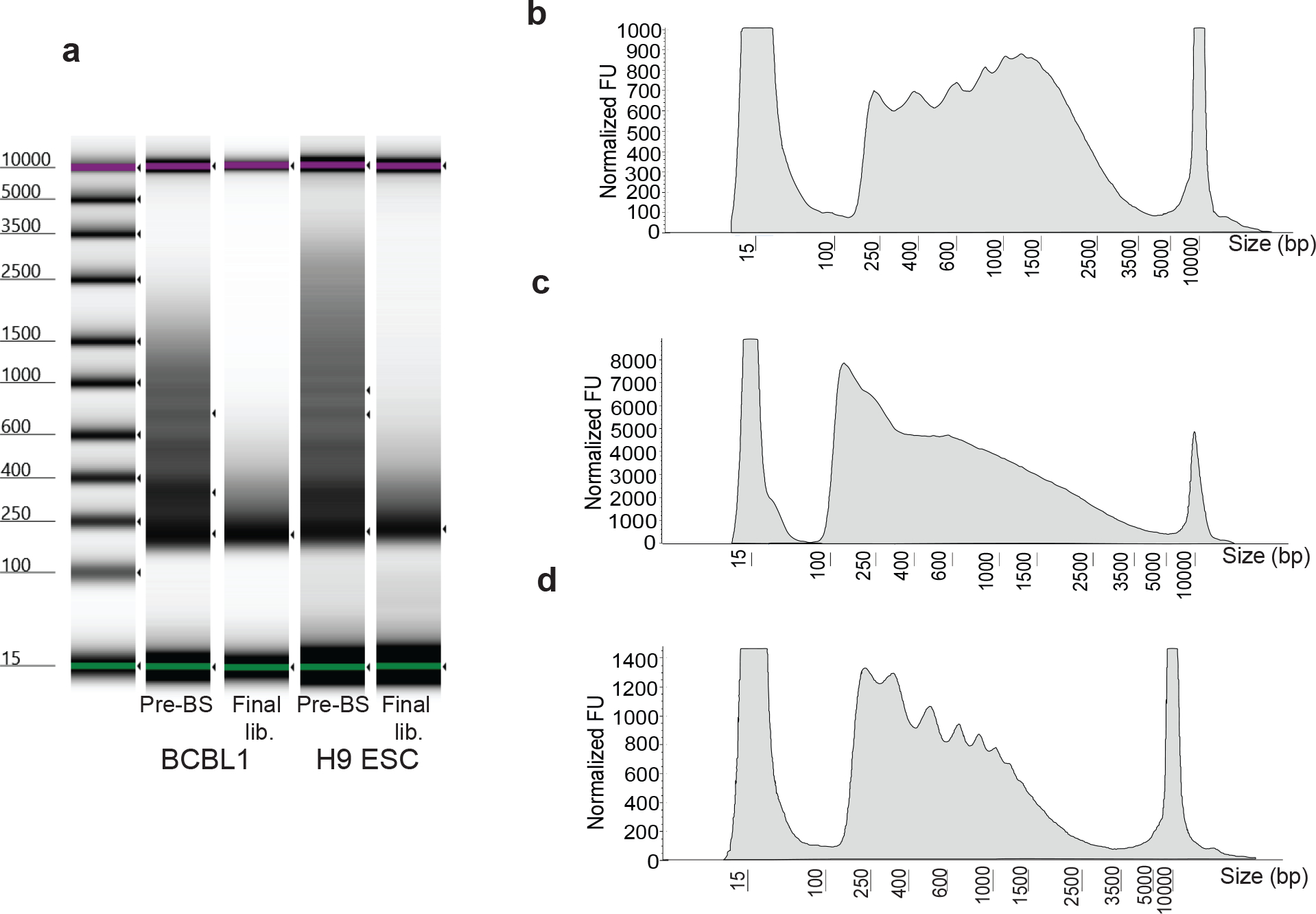
Representative fragment distribution of amplified libraries following gap repair (steps 35–38; before bisulfite treatment, pre-BS) and bisulfite conversion (steps 59–69; final lib.) of two different cell types including H9 embryonic stem cells grown as a monolayer on Matrigel and non-adherent BCBL1 cells grown in suspension (a). Gel-like images are captured from Agilent Tape Station results files. This step is a key quality control checkpoint to assess tagmentation quality. Prior to bisulfite conversion, high quality libraries should display distinct banding patterns resembling the characteristic nucleosome ladder (pre-BS), whereas lower quality libraries display a more diffuse fragment distribution. The banding pattern is diminished, and larger fragments are lost following bisulfite treatment and PCR (final lib). Over- and under-tagmentation of chromatin accessible fragments are common issues which may require optimization on a per cell type or per condition basis. Traces of electropherogram profiles of example pre-BS amplifications are shown (b-d). y-axis displays normalized fluorescence units (FU). Fragment distributions resulting from under-tagmentation are characterized by an accumulation of large fragments (b), whereas over-tagmentation is characterized by a loss of nucleosomal banding (c). An ideal tagmentation reaction preserves nucleosomal banding while also reducing the accumulation of large fragments (d).
We have maximized complexity within the “accessible” fragment library in a couple of ways: 1) by maintaining high yield at each stage of the ATAC-Me protocol, and 2) by minimizing PCR cycles during library amplification. As our analysis method removes PCR duplicates, excess amplification can reduce final library complexity. While we recommend 200,000 cells as input, we have been able to scale down the reaction to 50,000 cells for more difficult to obtain samples. Lowering cell numbers is likely to result in a reduction in library complexity. Fewer input cells will require a proportionate decrease in Tn5 and transposition reaction buffer (2.5μl and 47.5μl respectively) for the tagmentation steps. Isolated nuclei are then incubated with transposition reaction buffer and Tn5 transposase assembled with methylated adaptors (steps 7–20, see Table 1 for sequences). Due to the adaptor assembly structure, fragments must undergo a gap repair step prior to bisulfite conversion (steps 21–26). As a quality control, a small portion of gap repaired fragments can be amplified and visualized to evaluate the distribution of fragments (Fig. 2a, b). At this step, samples should display nucleosomal banding similar to standard ATAC-seq libraries. Cell type specific requirements for modification to the protocol can be evaluated at this stage. Improper tagmentation can been identified through loss of nucleosomal banding or an accumulation of large fragments (Fig. 2c, d). Gap repaired fragments are then exposed to sodium bisulfite, converting unmethylated cytosine to uracil (steps 31–50).
Table 1.
Oligonucleotide sequences.
| Tn5mC-Apt1 | T/iMe-dC/GT/iMe-dC/GG/iMe-dC/AG/iMe-dC/GT/iMe-dC/AGATGTGTATAAGAGA/iMe-dC/AG |
| Tn5mC1.1-A1block | /5Phos/CTGTCTCTTATACA/3ddC/ |
| Tn5mC-ReplO1 | /5Phos//iMe-dC/TGT/iMe-dC/T/iMe-dC/TTATA/iMe-dC/A/iMe-dC/AT/iMe-dC/T/iMe-dC//iMe-dC/GAG/iMe-dC//iMe-dC//iMe-dC/A/iMe-dC/GAGA/iMe-dC//3InvdT/ |
| Index N701 | CAAGCAGAAGACGGCATACGAGATTCGCCTTAGTCTCGTGGGCTCGG |
| Index N702 | CAAGCAGAAGACGGCATACGAGATCTAGTACGGTCTCGTGGGCTCGG |
| Index N703 | CAAGCAGAAGACGGCATACGAGATTTCTGCCTGTCTCGTGGGCTCGG |
| Index N704 | CAAGCAGAAGACGGCATACGAGATGCTCAGGAGTCTCGTGGGCTCGG |
| Index N705 | CAAGCAGAAGACGGCATACGAGATAGGAGTCCGTCTCGTGGGCTCGG |
| Index N706 | CAAGCAGAAGACGGCATACGAGATCATGCCTAGTCTCGTGGGCTCGG |
| Index N501 | AATGATACGGCGACCACCGAGATCTACACTAGATCGCTCGTCGGCAGCGTC |
| Index N502 | AATGATACGGCGACCACCGAGATCTACACCTCTCTATTCGTCGGCAGCGTC |
| Index N503 | AATGATACGGCGACCACCGAGATCTACACTATCCTCTTCGTCGGCAGCGTC |
| Index N504 | AATGATACGGCGACCACCGAGATCTACACAGAGTAGATCGTCGGCAGCGTC |
| Index N505 | AATGATACGGCGACCACCGAGATCTACACGTAAGGAGTCGTCGGCAGCGTC |
| Index N506 | AATGATACGGCGACCACCGAGATCTACACACTGCATATCGTCGGCAGCGTC |
Sequences are listed 5’ to 3’. Modification code is according to Integrated DNA Technology ordering. iMe-dC: 5-Methyl deoxycytidine, 5Phos: 5’ Phosphorylation, 3ddc: 3’Dideoxycytidine, 3InvdT: 3’ Inverted dT
Bisulfite conversion is a damaging process. Earlier versions of our protocol employed the EZ DNA Methylation-Lightning Kit (Zymo, D5030), which uses ammonium bisulfite to perform rapid deamination reactions. However, sodium bisulfite-based reactions have been shown to produce larger fragment sizes and higher yields compared to ammonium bisulfite39. Comparison between the Lightning and EZ DNA Methylation-Gold (Zymo, D5005) kits shows higher yield and a slightly larger fragment distribution when using the Gold kit (Fig. 3a). For this reason, we recommend using the EZ DNA Methylation-Gold Kit (or other commercially available sodium bisulfite-based kit) for conversion of ATAC-Me libraries. Amplification of purified, converted fragments incorporates dual barcoded adaptors for high throughput sequencing (steps 51–60, Figure 2). Resulting libraries are analyzed to evaluate regions of accessibility and quantify their methylation state at each collected time point.
Figure 3. Assessing library complexity versus data quality.
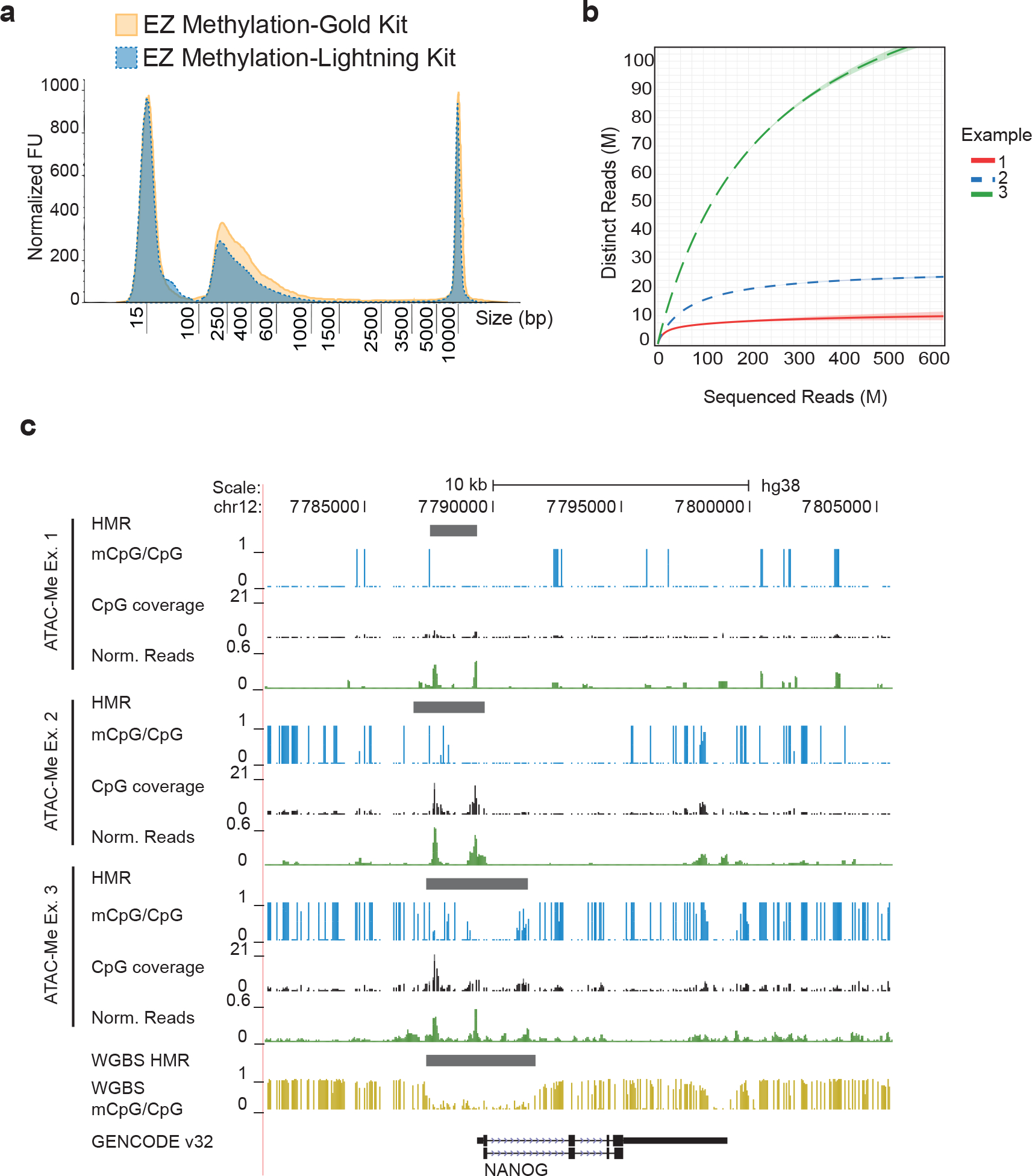
(a) Fragment distribution of libraries converted using the EZ Methylation-Gold Kit (gold solid line) and EZ Methylation-Lightning Kit (blue dotted line). Fragment distributions are representative of electropherograms visualized on Agilent Tape Station results files. y-axis displays normalized fluorescence units (FU). (b) Sequence complexity was calculated using Pre-seq57 for three example ATAC-Me libraries of H9 ESCs representing a range of sequenced DNA fragment complexities. (c) Genome Browser tracks surrounding the Nanog locus for three ATAC-Me examples of H9 ESCs are shown alongside tracks of whole genome methylation data from Xie et al. 201316 (gold track). Single CpG methylation levels are shown as vertical blue bars with height corresponding to fraction methylation (calculated as mCpG reads/total CpG reads). The read coverage of each CpG is represented as the height of the black bars. Reads normalized to library depth are shown in green to represent accessibility signal calculated for each sample. Hypomethylated regions (HMRs) were determined using the MethPipe software package and correspond to peaks called by Genrich software (available at https://github.com/jsh58/Genrich)48.
Controls:
During method development, genomic DNA sourced from Drosophila S2 cells was used to evaluate false positive methylation signal introduced during library preparation as Drosophila genomes are devoid of DNAme24. S2 cells may serve as a spike-in control for cross-library normalization, in which case they can be added at a ratio of 100:1 mammalian:Drosophila cells, or 1–2 % of total cell number prior to cell lysis. We have typically performed PCR amplification of pre-bisulfite converted samples to visualize fragment distribution and confirm nucleosomal banding. However, the conversion process is damaging, and these profiles cannot be seen in the final sequencing libraries. Therefore, in some cases, supplementing ATAC-Me datasets with standard ATAC-seq may be necessary to verify preservation of nucleosomal profiles. If preparing Tn5 in-house, one should test and optimize enzyme concentration and efficiency on genomic DNA prior to use in the ATAC-Me protocol.
Limitations
When considering ATAC-Me as a potential subsampling approach to profiling DNAme, it is important to note that ATAC-Me focuses primarily on nucleosome-free regions, so methods capable of subsampling both accessible and inaccessible loci may be preferred (RRBS). ATAC-Me relies on the assumption that inaccessible chromatin is methylated. The promiscuity of Tn5 does create some background level of reads, which permits estimation of DNAme levels of inaccessible loci. However, to verify that DNAme patterns are maintained outside of open chromatin, a complementary WGBS dataset may be necessary. Contamination by mitochondrial DNA is another challenge shared with other ATAC-inspired methodologies. Modified lysis protocols such as those that include digitonin or depletion by Cas9 paired with a library of mitochondrial guide RNAs can be incorporated to limit mitochondrial DNA presence in the libraries36,40. As is the case for all sodium bisulfite approaches, ATAC-Me is unable to distinguish between methylcytosine and hydroxymethylcytosine. Therefore, additional profiling of hydroxymethylcytosine (5hmC) may provide useful information about methylation states observed in ATAC-Me profiles, particularly in cell types displaying high levels of 5hmC41–44.
Expertise needed
ATAC-Me is streamlined for ease of use and almost all biochemistry and molecular biology graduate students should be able to perform this protocol. Analysis of resulting data does not require extensive bioinformatics experience as we have taken advantage of publicly available packages for trimming, mapping, peak calling, and methylation analysis.
MATERIALS
BIOLOGICAL MATERIALS
H9 human embryonic stem cells (ESCs) (gift from Gama Lab, Vanderbilt University; WiCell, cat. no. WA09; https://scicrunch.org/resolver/RRID:CVCL_9773)
BCBL1 cell line (gift from Karijolich lab, Vanderbilt University; DSMZ Cat. no. ACC-683, https://scicrunch.org/resolver/RRID:CVCL_0165)
CAUTION Cell lines used in this protocol should undergo regular authentication and mycoplasma testing.
REAGENTS
Tn5 (produced in-house, using pTXB1-Tn5 (https://scicrunch.org/resolver/RRID:Addgene_60240)), or purchased from Diagenode, cat. no. C01070010). See Reagent Setup and Picelli et al. for detailed purification.
*Critical* Tn5 activity should be tested on gDNA and chromatin prior to use in ATAC-Me for each batch used. This critical step applies primarily to Tn5 produced in-house, rather than commercially available Tn5.
DNA Clean & Concentrate-5 kit (Zymo Research, cat. no. D4004)
EZ DNA Methylation-Gold Kit (Zymo Research, cat. no. D5005)
*Critical* There are a variety of commercially available kits for sodium bisulfite conversion. Some kits are based on different chemistries that can result in lower DNA fragment yields and size distributions.
Matrigel (Corning, cat. no. CB-40234)
MTeSR™1 (Stem Cell Technologies, cat. no. 5850)
StemPro™ Accutase Cell Dissociation Reagent(Gibco, cat. no. A1110501)
Trypan Blue solution (Sigma-Aldrich, cat. no. T8154)
NEBNext Ultra II Q5 Master Mix (NEB, cat. no. M0544S)
T4 DNA Polymerase (NEB, cat. no. M0203S)
Ampligase® Enzyme and Buffer (Lucigen, cat. no. A3202K)
2x KAPA HiFi HotStart Uracil+ ReadyMix (Roche, cat. no. 07959052001)
IGEPAL® CA-630 (Sigma-Aldrich, cat. no. I8896)
Trizma® hydrochloride solution (Sigma-Aldrich, cat. no. T2319)
Dimethylformamide (Thermo Fisher Scientific, cat. no. D119–1)
D5000 ScreenTape (Agilent Technologies, cat. no. 5067–5588)
D5000 Reagents (Agilent Technologies, cat. no. 5067–5589)
100% Glycerol
100% Ethanol, molecular biology grade
Nuclease free water
Tn5 purification reagents
T7 Express lysY/Iq Competent E. coli (NEB, cat. no. C3013l)
LB Broth (Teknova, cat. no. L9140)
Ampicillin, Sodium Salt (Research Product International, cat. no. 69–52-3)
Isopropyl-β-D-thiogalactopyranoside (IPTG), Dioxane-free) (Thermo Fisher Scientific, cat. no. BP1755–1)
HEPES (Sigma-Aldrich, cat. no. 7365–45-9)
Sodium Chloride (Research Product International, cat. no. 7647–14-5)
Ethylenediaminetetraacetic acid (EDTA) (Research Product International, cat. no. 60–00-4)
Triton X-100 (Sigma-Aldrich, T cat. no. 8787)
cOmplete™, EDTA-free Protease Inhibitor Cocktail (Roche, cat. no. 4693132001)
DL-Dithiothreitol (DTT) (Research Product International, cat. no. 3483–12-3)
Poly(ethyleneimine) solution, ~50% (wt/vol) in H2O (Sigma-Aldrich, cat. no. 03880)
Chitin Resin (NEB, cat. no. S6651S)
Pierce™ Coomassie Plus (Bradford) Assay Kit (Thermo Fisher Scientific, 23236)
EQUIPMENT
Freezers: −20°C, −80°C
Thermocycler (Bio-Rad, model no. C1000)
Microcentrifuge (Benchmark Scientific, C1012)
Refrigerated centrifuge (Eppendorf, model no. 5425R)
TapeStation System (Agilent Technologies, model no. 4150)
Branson 540 Sonifer with a 5mm Tapered Microtip
Centrifuge (NuWind, NU-C200V)
Automated cell counter (Bio-Rad TC20)
Software
SAMtools 1.6 (https://github.com/samtools/samtools)
trim-galore 0.6.5 (https://github.com/FelixKrueger/TrimGalore)
cutadapt 1.18 (https://github.com/marcelm/cutadapt)
fastqc 0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
WALT v1.0 (https://github.com/smithlabcode/methpipe)
deeptools 3.3 (https://deeptools.readthedocs.io/en/develop/index.html)
R version 3.6.0 (https://www.r-project.org/)
pheatmap 1.0.12 (https://cran.r-project.org/web/packages/pheatmap/pheatmap.pdf)
TCseq 1.8.0 (https://rdrr.io/bioc/TCseq/f/inst/doc/TCseq.pdf)
tidyverse 1.3.0 (https://www.tidyverse.org/)
picard 2.18.27 (https://github.com/broadinstitute/picard)
preseq 2.0.0 (http://smithlabresearch.org/software/preseq/)
1.5mL Low Bind or siliconized Eppendorf Tubes (Eppendorf, cat. no. 022431021)
1.7mL microcentrifuge tubes (Thomas Scientific, cat. no. 1159M35)
Eppendorf 8 Tube Strip of 0.2ml PCR tubes (Fisher Scientific, cat. no. E0030124286)
Amicon Ultracel-30 Centrifugal filters (Millipore Sigma, UFC9030)
REAGENT SETUP
Cell Culture
H9 human embryonic stem cells (ESCs) are maintained on Matrigel coated 6-well plates in mTeSR1. Cells are inspected for morphological characteristics of ESCs, and expression of characteristic ESC markers (i.e., Oct4) is confirmed by flow cytometry or qPCR. When 80% confluent, cells are washed with sterile DPBS without calcium or magnesium prior to incubation with StemPro™ Accutase for 8 minutes at 37°C. Following incubation, Accutase is pipetted up and down to generate a single cell suspension.
Illumina compatible adaptor and PCR oligonucleotides
All oligonucleotides (see Table 1 for sequences) are synthesized by Integrated DNA Technology and suspended in STE Buffer (10 mM Tris-HCl pH 8.0, 50 mM NaCl, 1mM EDTA, pH 8.0) to a final concentration of 100 μM. Following resuspension, oligos can be stored at −20°C indefinitely.
ATAC lysis buffer
Prepare the lysis buffer containing 10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl and 0.1% (vol/vol) IGEPAL CA-630 vol/vol in nuclease free water. Aliquots can be stored at −20°C for up to one year.
Tagmentation reaction buffer (5x Tris-DMF)
Prepare the tagmentation reaction buffer containing 50 mM Tris-HCl pH 7.5, 25 mM MgCl2, and 50% vol/vol Dimethylformamide. Aliquots can be stored at −20°C for up to one year and diluted to 1x with nuclease-free water prior to use, as noted in step 15.
Tn5 transposase purification
Tn5 transposase can be prepared in-house as detailed previously24,45 and outlined in Box 1.
HEGX buffer
Prepare HEGX buffer containing 20 mM HEPES-KOH at pH 7.2, 0.8 M NaCl, 1 mM EDTA, 10% (vol/vol) glycerol , 0.2% (vol/vol) Triton X-100 in H2O.
PROCEDURE
Cell Collection, Lysis, and Transposition Reaction (Day 1)
Transposome Preparation Timing 2 h
** Critical: Magnesium is sometimes recommended to help stabilize two annealing oligos. This is not recommended for the Tn5 adapter oligo annealing. Magnesium ions are the catalytic ions required for Tn5 transposase activity. Any magnesium ion content during transposome assembly can result in concatemers of fully methylated adapter sequences.
- 1
-
2Incubate oligo mixture in a PCR thermocycler to anneal adapters as follows:
95°C, 3 min 65°C, 3 min Ramp down to 24°C, −1°C/cycle, 30 sec/cycle Hold at 24°C -
3
While oligo mixture is annealing, heat approximately 500 μL of 100% glycerol to 98°C for 2 min. While glycerol is still hot pipette 100 μL into a clean Eppendorf tube and cool on ice for 5 min.
-
4
After incubation add 100 μL oligo mixture from Step 2 into the cooled 100 μL of 100% glycerol. Mix well by vortexing and stirring with a micropipette tip. The resulting mixture is the final 5 μM adapter mixture in 50% glycerol.
*PAUSE POINT* Adapter mixture can be stored at −20°C for several months prior to assembly.
-
5Mix as follows:
Component Amount (μl) Final concentration 5 μM Methylated Adapters (Step 4) 10 0.5μM Tn5 (see Box 1, step 11) 10 Total 20 -
6
Let mixture stand at room temperature (20–27°C) for 60 min, then immediately place on ice. Any remaining assembled Tn5 can be stored at −20°C.
*PAUSE POINT* Assembled Tn5 can be stored at −20°C for several weeks.
Cell Lysis Timing 45 min
** Critical: It is recommended to process fewer than 15 samples per experiment to reduce sample processing times. It is also highly recommended to use low-bind or siliconized Eppendorf tubes as pellet formation of lysed cells is frequently diffuse and streaked with normal Eppendorf tubes (and may even be diffuse to the point of invisibility).
-
7
Pellet cells at 500xg for 5 min at 4°C and resuspend in 500 μL PBS, chilled on ice.
-
8
Count PBS cell suspension with a manual or automated cell counter. Recommended: mix cell suspension with an equal volume of Trypan blue solution before counting to assess cell viability.
-
9
According to the cell count, pipette a volume of PBS cell suspension corresponding to 2×105 cells into a pre-chilled 1.5mL Eppendorf tube.
-
10
Centrifuge cells in Eppendorf tube at 500g for 5 min at 4°C.
-
11
Remove and discard the supernatant and add 200 μL ice-cold ATAC lysis buffer.
-
12
Resuspend the cell pellet by gently pipetting 10 times with a 200 μL micropipette tip.
** Critical step: Cells should not be incubated with lysis buffer for longer than 10 minutes before centrifugation.
-
13
Immediately centrifuge cells in Eppendorf tube at 500g for 30 min at 4°C.
-
14
Remove and discard supernatant, being careful not to disturb pelleted nuclei and continue immediately to transposition reaction.
** Critical step: When removing supernatant from the nuclei pellet it is best to avoid removing the entire volume to avoid accidentally aspirating away nuclei. Generally leaving ≤ 5μL of supernatant is compatible with this protocol.
Transposition Reaction Timing 1.5–2 h
-
15Prepare transposition master mix by combining:
Component Amount (μl) Final concentration 5x Tris-DMF 40 1x Nuclease free water 150 Total 190 -
16
Resuspend the nuclei pellet in 190μL of transposition reaction master mix by gently pipetting up and down with a 200 μL micropipette tip 10 times.
-
17
Lastly, add 10μL assembled transposome (from Step 6) and flick gently to mix.
-
18
Divide resuspension into 4–1.5 mL tubes (50 μL / tube).
-
19
Incubate the transposition reaction at 37°C for 1 hr at 1200RPM in an Eppendorf Thermomixer.
-
20
After incubation, stop the reaction by pooling the four reactions together into a 1.5mL Eppendorf tube containing 1 mL Zymo DNA Binding Buffer from the Zymo DNA clean & concentrator-5 kit and vortexing briefly.
*PAUSE POINT* Reaction with DNA Binding Buffer can be stored overnight at −20°C. Samples must be allowed to warm to room temperature again when processing further as DNA binding buffer components usually precipitate at −20°C.
-
21
Pipette 600 μL transposition reaction and DNA binding buffer mixture into a Zymo DNA Clean and Concentrator-5 spin column.
-
22
Centrifuge at 12,500g for 30 sec at room temp. Discard flow through.
-
23
Repeat steps 21–22 with the same spin column to load the remaining volume of transposition reaction and DNA binding buffer transposition mixture. Discard flow through.
-
24
Wash spin column by adding 200 μL Zymo DNA wash buffer that has been previously supplemented with molecular grade 100% ethanol according to manufacturer instructions and centrifuging at 12,500g for 30 sec at room temperature. Discard flow through.
** Critical step: Old or improperly stored DNA wash buffer containing ethanol can result in inefficient DNA recovery during purification. It is recommended to always properly seal the wash buffer storage container or prepare aliquots of fresh wash buffer as needed.
-
25
Repeat wash step 24.
-
26
Transfer spin column to a clean 1.5 mL Eppendorf and add 15 μL nuclease free water directly to the column matrix.
-
27
Incubate spin column at room temperature for 2 min.
-
28
Centrifuge column at 12,500g for 1 min to elute DNA. Store the eluted DNA on ice or at −20°C.
*PAUSE POINT* Eluted DNA can be stored at −20°C for several weeks.
Gap Repair Timing 2 h
-
29Prepare the following gap repair master mix on ice:
Component Amount (μl) Final concentration 10 μM Tn5mC-Repl01 oligo (Table 1) 2 1 μM 10x Ampligase buffer 2 1x 50 mM NAD+ 0.5 1.25 mM 10 mM dNTPs (2.5 mM of each of the 4 dNTPs) 2 1 mM Total 6.5 ** Critical step: The catalytic activity of ampligase requires the cofactor NAD+ which is included in the 10x Ampligase buffer. However, NAD+ is unstable in aqueous solutions at alkaline pH. Thus, it may be beneficial for a more efficient ligase reaction to include a spike-in of NAD+ (prepared from single use aliquots stored at −80°C), as shown above, for a final additional NAD+ concentration of 1 mM.
-
30Mix the following on ice in a 0.2 ml PCR tube:
Component Amount (μl) Eluted DNA from transposition reaction (Step 28) 11 Gap repair master mix (Step 29) 6.5 Total 17.5 -
31Incubate the mixture in a thermocycler as follows:
50°C for 1 min 45°C for 10 min Ramp down to 37°C at −0.1°C/second Hold at 37°C -
32
Add 1 μL T4 DNA Polymerase and 2.5 μL Ampligase to the incubated sample without removing the tube from the thermocycler. Do not make a master mix of these two enzymes, as their respective storage buffers are likely incompatible. Add them in two separate additions instead. Mix well by pipetting up/down 10 times with a 20 μL micropipette or a multi-channel micropipette.
-
33Incubate as follows:
37°C for 30 min Hold at 4°C -
34
Purify the gap-repaired DNA fragments using the Zymo Clean and Concentrator-5 kit according to the manufacturer’s instructions. Elute in 22 μL nuclease-free water.
-
35To determine library distribution pre-bisulfite conversion, use 2 μL of eluted DNA fragments in a test amplification.
**Critical Step: Indices used for post-gap repair PCR are not used for sequencing and do not need to be unique per sample.**
-
36Run post-gap repair PCR with the following parameters:
Temperature Time No. of cycles 98°C 30 sec 1 98°C 5 sec 10 62°C 15 sec 72°C 30 sec 72°C 2 min 1 -
37
Purify PCR product using Zymo Clean and Concentrator kit according to the manufacturer’s instructions, eluting in 10 μL water.
*PAUSE POINT* Gap repaired DNA can be stored at −20°C overnight prior to performing the bisulfite conversion.
-
38
Analyze distribution of fragments on Agilent Tape Station using a D5000 screentape.
?Troubleshooting
Conversion and Library Preparation (Day 2)
Bisulfite conversion Timing 4 h
-
39
Prepare the Zymo EZ Methylation-Gold Kit CT Conversion Reagent according to manufacturer’s instructions. CT Conversion Reagent can be stored at −20°C for up to one month.
-
40Mix the following in a 1.5mL Eppendorf and vortex briefly:
Reagent Amount (μl) gap repaired DNA fragments (Step 34) 20 CT conversion reagent (Zymo EZ DNA Methylation-Gold Kit) 130 Total 150 -
41
Spin down the Eppendorf tube to ensure no droplets are left on the sides or lid.
-
42
Distribute the 150 μL of DNA and conversion reagent mixture equally across PCR tubes according to the volume limit specifications of your thermocycler.
-
43
Spin down the PCR tubes to ensure no droplets are left on the sides or lid.
** Critical step: Droplets on the sides of the tube are likely to be inefficiently denatured/bisulfite converted.
-
44Incubate the conversion reagent mixture as follows:
98°C for 10 min 64°C for 2.5 hours Hold at 4°C -
45
Add 600 μl of M-Binding Buffer from the Zymo EZ DNA Methylation-Gold Kit to the supplied spin column.
-
46
Recombine the 150μL of bisulfite converted DNA spread across multiple PCR tubes and add it to the spin column with the M-Binding buffer.
-
47
Invert the capped spin column 5–7 times to mix the M-Binding Buffer with the bisulfite converted DNA.
-
48
Centrifuge at 12,500g for 30 sec at room temperature. Discard the flow through.
-
49
Add 100 μl M-Wash Buffer that has been previously supplemented with molecular grade 100% ethanol according to the manufacturer instructions to the spin column.
** Critical step: Old or improperly stored DNA wash buffer containing ethanol can result in inefficient DNA recovery during purification. It is recommended to always properly seal the wash buffer storage container or prepare aliquots of fresh wash buffer as needed.
-
50
Centrifuge at 12,500g for 30 sec at room temperature.
-
51
Add 200 μl M-Desulphonation Buffer and let the column stand at room temperature for 20 min.
-
52
Centrifuge at 12,500g for 30 sec at room temperature. Discard the flow through.
-
53
Add 200 μl M-Wash buffer and centrifuge the column at 12,500g for 30 sec.
-
54
Repeat wash step 53.
-
55
Transfer the column to a clean 1.5mL Eppendorf tube.
-
56
Add 25 μL M-Elution Buffer directly to the column matrix.
-
57
Incubate spin column at room temperature for 2 min.
-
58
Centrifuge the column at 12,500g for 1 min at room temperature to elute DNA.
*PAUSE POINT* Eluted DNA can be stored at −20°C for several weeks.
PCR Amplification and Barcoding Timing 1 h
-
59Assemble the following PCR reaction:
-
60Incubate the PCR reaction as follows:
Temperature Time No. of cycles 98°C 45 sec 1 98°C 15 sec 10 62°C 30 sec 72°C 30 sec 72°C 2 min 1 -
61
Mix 250 μL of Zymo DNA Binding Buffer with the 50 μL PCR reaction in a 1.5mL Eppendorf tube.
-
62
Pipette the mixture into a Zymo DNA Clean and Concentrator-5 spin column.
-
63
Centrifuge at 12,500g for 30 sec at room temperature. Discard flow through.
-
64
Wash the spin column by adding 200 μL Zymo DNA wash buffer that has been previously supplemented with molecular grade 100% ethanol according to manufacturer instructions and centrifuging at 12,500g for 30 sec at room temperature. Discard the flow through.
-
65
Repeat wash step 64.
-
66
Transfer the spin column to a clean 1.5 mL Eppendorf tube and add 22 μL nuclease free water directly to the column matrix.
-
67
Incubate spin column at room temperature for 2 min.
-
68
Centrifuge the column at 12,500g for 1 min at room temperature to elute DNA. Store the eluted DNA on ice or at −20°C.
*PAUSE POINT* Eluted final libraries can be stored at −20°C for several months.
?Troubleshooting
Library Analysis and sequencing Timing 30 min
-
69
Measure concentration and fragment distribution of DNA libraries on an Agilent Tape Station using a D5000 screentape. Alternatively, a 5% bisacrylamide gel can be used.
-
70
If using an Illumina NovaSeq6000, sequence libraries using 2×150bp paired-end sequencing. We have typically found that requesting 75 million reads provides sufficient sequencing depth when libraries are appropriately complex. An ideal fragment distribution post conversion is shown in Fig. 2b.
Data Processing and Analysis (Day 3)
-
71
Trim adaptor sequences from the fastq reads using the TrimGalore46 wrapper for Cutadapt47 and FastQC. A nine base region must be additionally trimmed to avoid artificial methylation level calculation using the arguments, --clip_R1 9 –-clip_R2 9, as these bases were filled-in during library prep and will be unmethylated as a result. The methylatied CpGs contained in adaptors preserves the original sequence throughout the process of bisulfite conversion. As a result, standard trimming programs can recognize adaptor sequences in the final libraries.
-
72
Build reference genome index using WALT.48
-
73
Align reads using WALT49. Alignment tools must be able to accommodate C-T mismatches. This mapping tool requires the reference genome index build in step 72.
-
74
Calculate initial methylation rates for individual cytosines using methcounts from the Methpipe package48. Statistics for methylation levels can be calculated for cytosines regardless of context using the levels command. For most downstream analysis we focus on symmetric CpG context cystosines which can be selected for using the symmetric-cpgs command.
-
75
Perform further methylation analyses using the Methpipe pipeline, such as: calculating bisulfite conversion rate for libraries (bsrate), scanning for hypomethylated regions (hmr), and evaluating differential methylated regions between samples (methdiff,dmr).
-
76
Call peaks for accessibility analysis using a peak calling program such as MACS250 or Genrich (https://github.com/HodgesGenomicsLab/NatProtocols_ATACme). We currently use Genrich with options: -r -e chrX,chrY,chrM -E hg38.blacklist.bed -j -p 0.005 -q 0.01. Alternatively, we use MACS2 with options: -f BAMPE -g hs -q 0.05 -B --keep-dup all --broad --broad-cutoff 0.05. For comparison, we have also displayed MACS2 narrowPeak files generated using the options: -f BAMPE -g hs -q 0.05 -B --call-summits. Additional details can be found at https://github.com/HodgesGenomicsLab/NatProtocols_ATACme.
-
77
If applying ATAC-Me throughout a time course, identify temporal accessibility patterns using the R package TCseq51.
-
78
Calculate methylation levels of possible regions of interest identified from accessibility analyses using roimethstat from Methpipe48.
TIMING
Day 1: Cell collection to gap repair
Steps 1–4, anneal methylated adaptors: 1 hour
Steps 5 and 6, assemble transposome: 1 hour
Steps 7–14, cell lysis to pellet nuclei: 1 hour
Steps 15–20, transposition reaction: 1.5 hours
Steps 21–28, purify fragments: 15 minutes
Steps 29–34, gap repair and purification: 45 minutes
Steps 35–38, PCR amplification and fragment visualization: 45 minutes
Day 2: Bisulfite Conversion, library preparation, and sequencing
Steps 39–58, bisulfite conversion and purification: 4 hours
Steps 59–68, PCR amplification and barcoding: 1 hours
Step 69, fragment size distribution analysis: 30 minutes
Step 70, Sequencing: 1day to 2 weeks, dependent on sequencing facility
Steps 71–78, data analysis: 1 week to 1 month, dependent on user
TROUBLESHOOTING
Troubleshooting advice can be found in Table 2.
Table 2.
Troubleshooting table.
| Step | Problem | Possible reasons | Solutions |
|---|---|---|---|
| 14 | No nuclei pellet | Spin conditions | Extend spin time or speed |
| 38 | Loss of nucleosomal banding | Chromatin integrity disrupted during cell collection | Try alternative methods of cell dissociation during collection to maintain high viability |
| Over-tagmentation | Scale down reaction size relative to cell number or increase the starting number of cells | ||
| Decrease transposition incubation time | |||
| Accumulation of large size fragments | Under-tagmentation | Reduce cell numbers or scale up reaction size relative to cell number | |
| Increase transposition incubation time | |||
| 69 | Low yield | Insufficient lysis | Additional pipetting following addition of lysis buffer |
| Inaccurate cell count | Repeat cell counting, confirming single cell suspension has been achieved and viability is high (>80%) |
ANTICIPATED RESULTS
Assembled libraries should display accessibility profiles consistent with standard ATAC-seq libraries following the gap repair step. This can be assessed by PCR amplification and visualization of fragment distribution prior to bisulfite conversion (Fig. 2). Accordingly, as this fragment population is the input into bisulfite conversion, accessibility peaks called on final sequenced libraries should correlate with standard ATAC peaks when available for comparison24. The gap repair PCR quality check (Steps 35–38) is critical to evaluating the quality of the final library. The presence of nucleosomal banding following amplification can be used to confirm the subsampling of the accessible genome for methylation analysis. Following bisulfite conversion, nucleosomal banding is typically lost; therefore, we rely on the post gap repair fragment distribution to assess transposition quality (Fig. 2).
Library complexity refers to the number of distinct fragments sequenced at increasing levels of sequencing depth. High library complexity indicates low PCR duplicate rates and can be used to determine whether increased sequencing would be beneficial. This protocol has been optimized to reduce sample loss, as we have found it is important to retain as much of the original input DNA going into bisulfite conversion as possible to maintain some library complexity. For this reason, a final library concentration above 2.5 μM is recommended. We suggest starting with at least 200,000 cells when feasible, though using fewer cells may be possible. Inadequate sequencing depth and low complexity results in few high confidence peaks being called, making downstream analysis difficult. Fig. 3b and c display illustrative examples of libraries with varying degrees of quality and complexity. Statistics of library yield and key data processing steps for three ATAC-Me examples are included in Table 3. Low complexity libraries are characterized by higher duplicate rates and sparse CpG coverage and may show higher background read levels relative to reads stemming from accessible sites (see example #1, Table 3). High complexity libraries will have greater CpG coverage, thus producing dense methylation data mirroring whole genome methylation levels (see example #3, Table 3). However, ATAC-Me selectively samples the accessible genome and resulting fragment populations are intentionally less complex than whole genome libraries. High complexity may have the unintended result of high background reads, which can negatively impact the number of peaks called, as well as the fraction of reads in called peaks (FRiP score52). To maximize both the methylation and accessibility data produced by an ATAC-Me experiment, a balance must be struck between high complexity and low background (see example #2, Table 3).
Table 3.
Mapping and Peak Processing Statistics.
| Example 1 Low complexity/Sparse read coverage | Example 2 Intermediate complexity/Low background | Example 3 High complexity/High background | Merged Replicates** | ||
|---|---|---|---|---|---|
| Final library concentration (μM) | 1.3 | 6.6 | 33.9 | N/A | |
| Total reads | 59,241,310 | 87,868,338 | 81,734,480 | ||
| % Uniquely mapped | 62.8716 | 69.0193 | 64.6791 | ||
| non duplicate fraction | 0.0757579 | 0.133552 | 0.463592 | ||
| CpGs (coverage ≥ 5) | 270,381 | 1,302,954 | 2,089,067 | 3,379,747 | |
| Genrich | p-value (< 0.05) | q-value (< 0.05) | |||
| Peaks called | 456,542 | 169,122 | 80,894 | 69,886 | |
| FRiP* | 0.476 | 0.428 | 0.205 | 0.217 | |
| p-value (<0.01) | q-value (< 0.01) | ||||
| Peaks called | 34,718 | 60,870 | 29,990 | 50,199 | |
| FRiP* | 0.201 | 0.346 | 0.137 | 0.185 | |
| p-value (<0.005) | q-value (< 0.005) | ||||
| Peaks called | 57,944 | 73,068 | 31,091 | 43,792 | |
| FRiP* | 0.174 | 0.315 | 0.119 | 0.173 | |
| MACS2 *** | q-value (<0.05) narrowPeak | N/A | |||
| Peaks called | 15,322 | 64,127 | 76,310 | ||
| FRiP* | 0.08 | 0.277 | 0.131 | ||
| q-value (<0.05) broadPeak | |||||
| Peaks called | 15,294 | 58,423 | 64,903 | ||
| FRiP* | 0.085 | 0.232 | 0.138 | ||
| q-value (<0.005) narrowPeak | |||||
| Peaks called | 9,780 | 63,903 | 57,829 | ||
| FRiP* | 0.063 | 0.243 | 0.115 | ||
| q-value (<0.05) broadPeak | |||||
| Peaks called | 9,615 | 55,782 | 48,217 | ||
| FRiP* | 0.066 | 0.267 | 0.12 | ||
Fraction of reads in called peak regions. All reads mapping to non-mitochondrial chromosomes (non-chrM) in peak regions are divided by the total number of total non-chrM reads to evaluate the signal to noise ratio in ATAC-seq data. Background descriptions are based on this ratio. Generally, FRiP scores correlate with the number of regions called, and scores ≥ 0.2 are considered acceptable for standard ATAC-seq52, although ATAC-Me FRiP scores may differ. We have filtered out mitochondrial reads for this calculation as we do not call peaks on mitochondrial fragments.
Genrich can incorporate replicates into peak calling analysis and the resulting merged data is produced using the arguments described in “Data Processing”. MACS2 does not have this option. As a consequence, merged replicate data for MACS2 peaks will depend on the approach chosen for determining consensus peaks.
MACS2 has the option of calling “broad” or “narrow” peaks. We provide both here.
In the example data obtained from H9 ESCs, sequence reads were mapped using the mapping command contained in the MethPipe package48. In our analysis pipeline (Fig. 4a), methylation values are calculated from the merged data of all replicates using the MethPipe package48. This increases CpG coverage and confidence in calculated methylation values. Established algorithms for calling ATAC-seq peaks include MACS250 and Genrich (available at vhttps://github.com/jsh58/Genrich), which are referenced in the ENCODE uniform ATAC-seq data processing pipeline (https://www.encodeproject.org/atac-seq/) and the ATAC-seq best practices guidelines from Harvard Informatics (https://informatics.fas.harvard.edu/atac-seq-guidelines.html), respectively. We provide peak calling statistics from both MACS2 and Genrich for the three individual H9 example libraries to display a range of potential outcomes from libraries with different levels of quality (Table 3). A variety of p-value cutoffs are reported to illustrate how thresholds differ between the examples (Table 3). A comparison of peaks and their size distributions for individual libraries and peak calling conditions is displayed in Fig. 4b–e. For illustrative purposes, we provide results for individual libraries; however, a typical ATAC-Me experiment should be performed with at least two biological replicates per sample. While both peak calling methods performed reasonably well with ATAC-Me data, we recommend Genrich due to its ATAC-seq mode with built-in read-shift correction and its streamlined handling of biological replicates. It is common practice to shift mapped reads +4 bp and −5 bp on the positive and negative strands respectively when working with ATAC-seq data 53. This practice is beneficial prior to single nucleotide resolution analyses, such as transcription factor footprinting. We incorporate this shift at the peak calling stage of analysis. With two biological replicates, ATAC-Me detects 60–75k high confidence peak regions (p-value <0.05), covering approximately 3–4 million CpG sites with at least 5X coverage (see Merged Replicates column in Table 3).
Figure 4. Data analysis pipeline and peak caller characteristics.
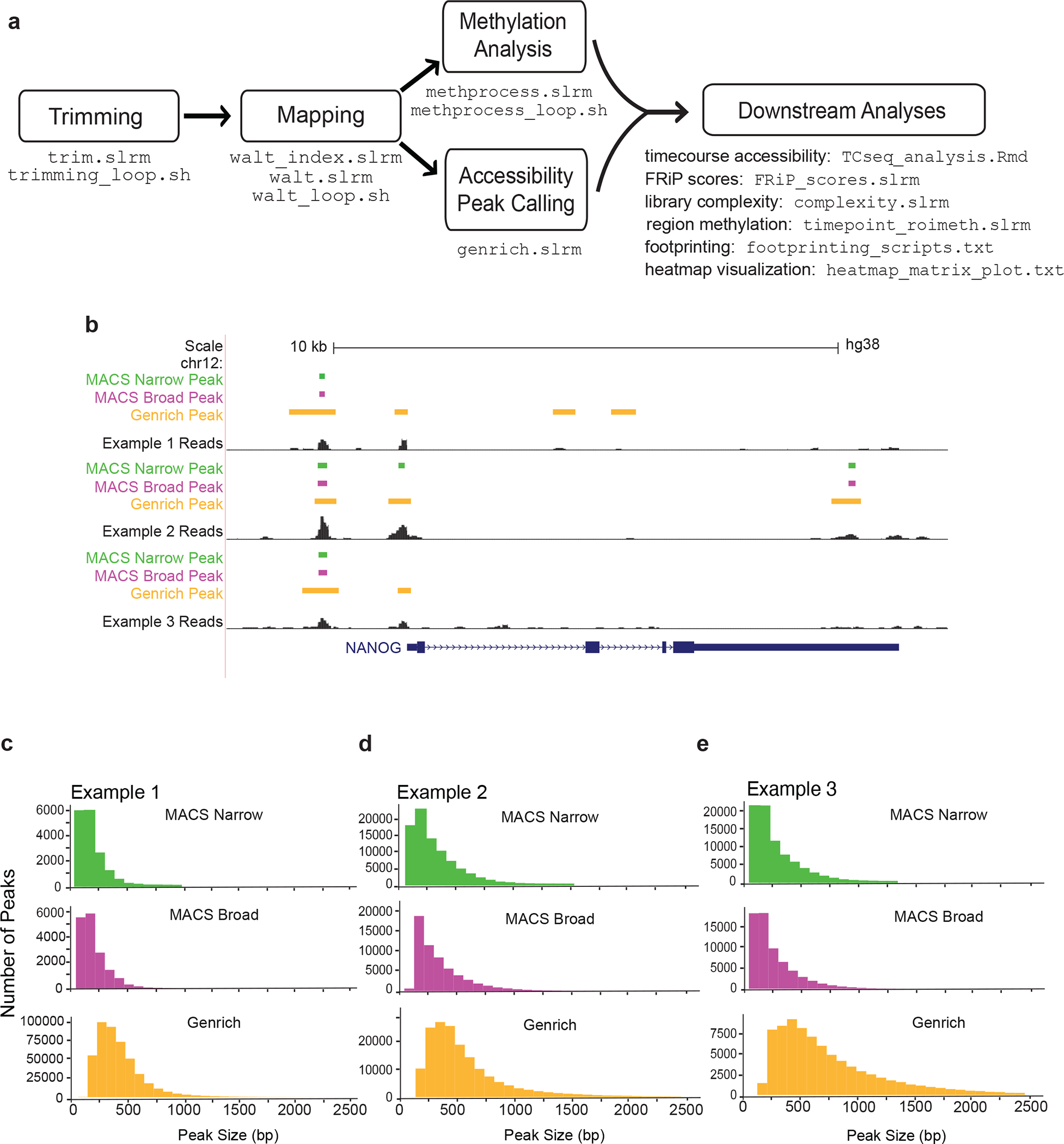
ATAC-Me data can be analyzed to determine both accessibility and methylation simultaneously. (a) Initial analysis steps are similar to other sequencing pipelines which begin with trimming of adaptors. These trimmed reads are then aligned to a reference index. As the fragments in ATAC-Me have been converted using sodium bisulfite they are aligned using a program that can accommodate T-rich reads. WALT49 is used to both create a reference genome index and map reads to this index. Following alignment, analysis branches to peak calling for accessibility using Genrich. Regions of increased chromatin accessibility have traditionally been identified through peak calling algorithms that determine genomic regions that contain a statistically significant enrichment of reads relative to background reads. Genrich incorporates biological replicates to create a set of consensus peaks. We provide additional details for MACS2, which can be used as an alternative to Genrich for calling peaks (See also Table 3). Parallel methylation analysis is performed using the MethPipe software48. A variety of downstream analyses are possible such as TF footprinting, differential accessibility, and regional methylation calculation. Scripts from this pipeline including example analyses are available at https://github.com/HodgesGenomicsLab/NatProtocols_ATACme56. (b) ATAC-Me signal tracks are displayed for MACS2 and Genrich peak calling methods, using a cutoff of p <0.05, to illustrate the differences between these two algorithms. We do not typically recommend calling peaks on samples individually, as there will be increased statistical strength and stringency when peak calling is performed on replicates and a consensus peak set is identified. (c-e) Histograms display the size distribution of regions called by Genrich and MACS2 (called using callpeak with and without the --broad argument, which composites broad regions from nearby highly enriched regions. At the lowest stringency p-value threshold (0.5), Genrich calls a larger number of peaks with wider size distributions.
Called peaks can then be integrated with parallel methylation quantification to more thoroughly understand how these two features interact at specific genomic loci (Fig. 5a–c). To highlight regions displaying a range of methylation levels we focused on the ~30,000 peaks found in non-coding genomic regions, excluding promoters (Fig. 5b). While most accessible regions are hypomethylated, a subset of regions display methylation levels above 75% (Fig. 5c,d). This highlights the capability of ATAC-Me to detect a continuum of methylation states across the accessible genome.
Figure 5. Anticipated results and ATAC-Me data visualization.
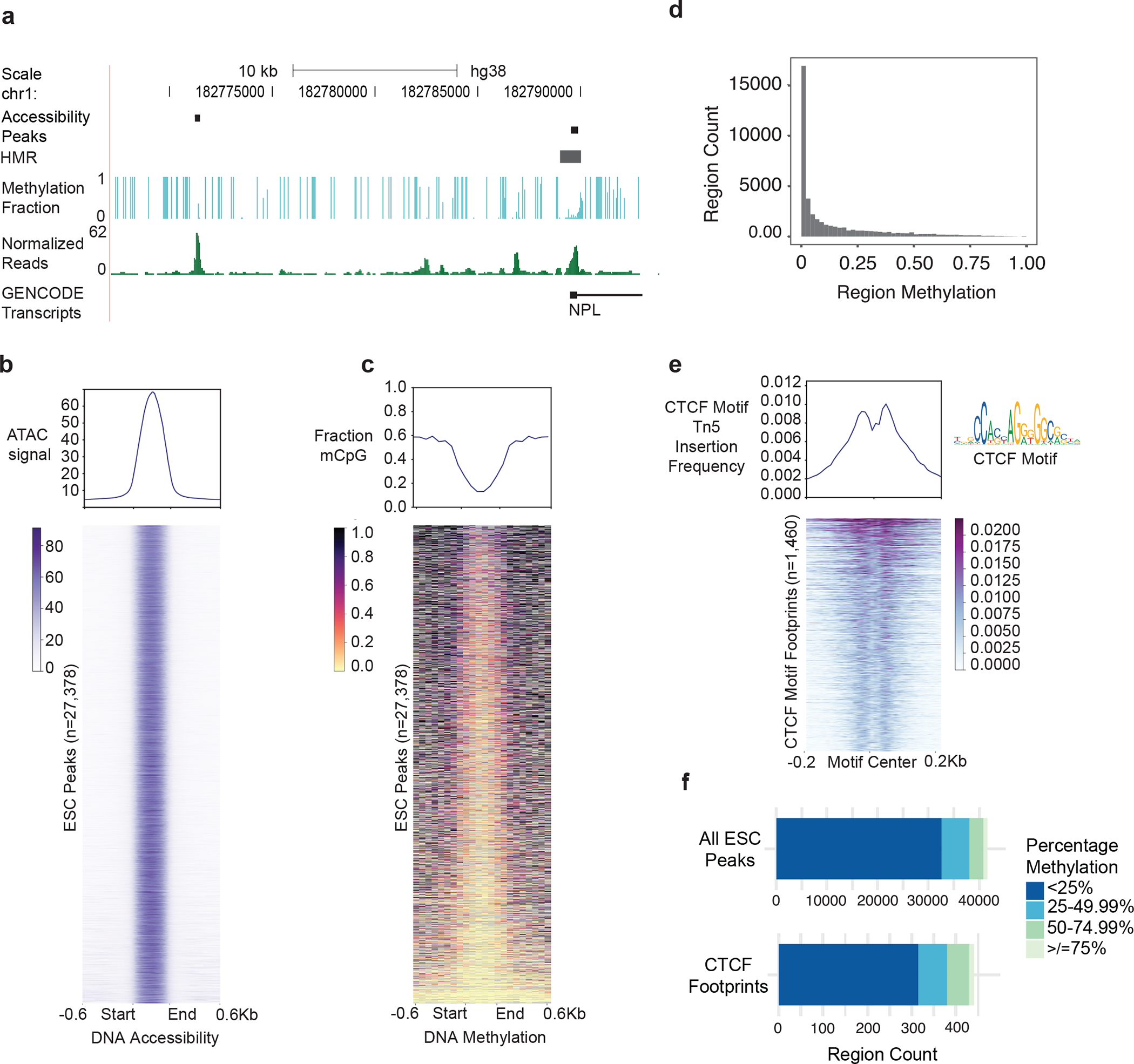
(a) A representative UCSC genome browser track showing peak regions called using Genrich, HMRs determined by MethPipe, methylation levels, and normalized read coverage in the hESC line, H9. (b) Heatmap of accessibility signal from ATAC-Me read count data for all non-promoter ESC peaks with <0.005 q-value according to Genrich (y-axis). The heatmap was generated using deepTools computeMatrix and plotHeatmap functions58. Count data is calculated in 10bp bins centered on peaks +/− 600bp (x-axis). Included regions are scaled to 450bp using the “scale-regions” function of computeMatrix. Regions are ordered along the vertical axis according to decreasing average methylation levels (shown in (c)). (c) Heatmap of methylation fractions of CpG sites averaged across 75bp bins demonstrates a range of methylation levels within peak regions. A larger bin size is used to calculate methylation levels compared to read count data due to low CpG frequencies in the genome. (d) Histogram summarizing data displayed in (c). (e) ATAC-Me retains the ability to map transcription factor footprints by calculating Tn5 insertion frequencies. The displayed heatmap shows H9 hESC open chromatin regions containing the CTCF motifs identified by TOBIAS software55. Regions are ordered by decreasing Tn5 insertion frequencies or “cut counts” centered on CTCF motifs +/−200bp. Frequencies are calculated for each 10bp bin along the x-axis using computeMatrix “reference-point” in deepTools. (f) The quartile distribution of methylation levels of accessible peak regions and CTCF footprints provides the numbers of peaks associated with each quartile. The profile plots displayed above heatmaps in (b), (c), and (d) summarize average values in each bin along the x-axis calculated for all displayed regions along the y-axis.
We previously showed that ATAC-Me peaks are differentially enriched for transcription factor motifs throughout a THP1 monocyte-to-macrophage time course24. Computational footprinting methods identify Tn5 cleavage events or “cut sites” from ATAC-seq data54,55. These approaches scan chromatin accessibility profiles to identify depletion of cleavage events. When combined with motif analysis, these methods are capable of identifying TF binding sites with high accuracy. Despite DNA fragment degradation from sodium bisulfite exposure, ATAC-Me retains the ability to map TF footprints (Fig. 5e). TF footprints can be specifically interrogated for methylation, which can provide more insight into the relationship between accessibility, DNAme, and gene regulation (Fig. 5f). In addition to footprinting, RNA-seq data can be paired with ATAC-Me to evaluate the consequence of observed regulatory dynamics on gene expression24.
ATAC-Me is a streamlined method making simultaneous capture of DNAme and chromatin accessibility technically simple. As these two features have been causally implicated in gene regulation during processes of dynamic gene expression changes, such as cellular differentiation, capturing these data will inform an integrated understanding of the regulatory landscape in a variety of systems. Results from the application of ATAC-Me can be generalized to increase understanding of the functional role of DNAme in different genomic and biological contexts.
AKNOWLEDGEMENTS
We thank VANTAGE for their advice in sequencing the libraries. We thank members of the Hodges lab for helpful discussions. We thank the Kariolich and Gama labs for cell lines. This work was supported by National Institutes of Health (K22 CA184308 to E.H., T32HD007502 to K.B).
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
CODE AVAILABILITY
Bioinformatic pipelines described in this protocol have been made available in GitHub (https://github.com/HodgesGenomicsLab/NatProtocols_ATACme)56.
DATA AVAILABILITY
Data used in this protocol have been deposited in the Gene Expression Omnibus database with accession number GSE166267. Fig. 5 is derived from the raw data.
REFERENCES
- 1.Albert I et al. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature 446, 572–576, doi: 10.1038/nature05632 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Barski A et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837, doi: 10.1016/j.cell.2007.05.009 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Johnson DS, Mortazavi A, Myers RM & Wold B Genome-wide mapping of in vivo protein-DNA interactions. Science 316, 1497–1502, doi: 10.1126/science.1141319 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Mikkelsen TS et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560, doi: 10.1038/nature06008 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cokus SJ et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452, 215–219, doi: 10.1038/nature06745 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boyle AP et al. High-resolution mapping and characterization of open chromatin across the genome. Cell 132, 311–322, doi: 10.1016/j.cell.2007.12.014 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thurman RE et al. The accessible chromatin landscape of the human genome. Nature 489, 75–82, doi: 10.1038/nature11232 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rada-Iglesias A et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283, doi: 10.1038/nature09692 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taberlay PC et al. Polycomb-repressed genes have permissive enhancers that initiate reprogramming. Cell 147, 1283–1294, doi: 10.1016/j.cell.2011.10.040 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Creyghton MP et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences of the United States of America 107, 21931–21936, doi: 10.1073/pnas.1016071107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atlasi Y & Stunnenberg HG The interplay of epigenetic marks during stem cell differentiation and development. Nature Publishing Group; 18, 643–658, doi: 10.1038/nrg.2017.57 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Kelsey G, Stegle O & Reik W Single-cell epigenomics: Recording the past and predicting the future. Science 358, 69–75, doi: 10.1126/science.aan6826 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Lister R et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322, doi: 10.1038/nature08514 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hodges E et al. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Molecular Cell 44, 17–28, doi: 10.1016/j.molcel.2011.08.026 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stadler MB et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480, 490–495, doi: 10.1038/nature10716 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Xie W et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 153, 1134–1148, doi: 10.1016/j.cell.2013.04.022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schubeler D Function and information content of DNA methylation. Nature 517, 321–326, doi: 10.1038/nature14192 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Jones PA Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13, 484–492, doi: 10.1038/nrg3230 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Brinkman AB et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res 22, 1128–1138, doi: 10.1101/gr.133728.111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.King Andrew D. et al. Reversible Regulation of Promoter and Enhancer Histone Landscape by DNA Methylation in Mouse Embryonic Stem Cells. Cell Reports 17, 289–302, doi: 10.1016/j.celrep.2016.08.083 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Charlet J et al. Bivalent Regions of Cytosine Methylation and H3K27 Acetylation Suggest an Active Role for DNA Methylation at Enhancers. Mol Cell 62, 422–431, doi: 10.1016/j.molcel.2016.03.033 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schlesinger F, Smith AD, Gingeras TR, Hannon GJ & Hodges E De novo DNA demethylation and noncoding transcription define active intergenic regulatory elements. Genome Res 23, 1601–1614, doi: 10.1101/gr.157271.113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmidl C et al. Lineage-specific DNA methylation in T cells correlates with histone methylation and enhancer activity. Genome Research 19, 1165–1174, doi: 10.1101/gr.091470.109 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barnett KR et al. ATAC-Me Captures Prolonged DNA Methylation of Dynamic Chromatin Accessibility Loci during Cell Fate Transitions. Mol Cell 77, 1350–1364 e1356, doi: 10.1016/j.molcel.2020.01.004 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kriaucionis S & Klose RJ ATACing DNA Methylation during Differentiation. Mol Cell 77, 1159–1161, doi: 10.1016/j.molcel.2020.02.026 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods 10, 1213–1218, doi: 10.1038/nmeth.2688 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adey A & Shendure J Ultra-low-input, tagmentation-based whole-genome bisulfite sequencing. Genome Res 22, 1139–1143, doi: 10.1101/gr.136242.111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Q et al. Tagmentation-based whole-genome bisulfite sequencing. Nature Protocols 8, 2022–2032, doi: 10.1038/nprot.2013.118 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Clark SJ et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nature Communications 9, 781, doi: 10.1038/s41467-018-03149-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelly TK et al. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res 22, 2497–2506, doi: 10.1101/gr.143008.112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fujiwara S, Baek S, Varticovski L, Kim S & Hager GL High Quality ATAC-Seq Data Recovered from Cryopreserved Breast Cell Lines and Tissue. Scientific Reports 9, 516, doi: 10.1038/s41598-018-36927-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scharer CD et al. ATAC-seq on biobanked specimens defines a unique chromatin accessibility structure in naïve SLE B cells. Scientific Reports 6, 27030, doi: 10.1038/srep27030 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lara-Astiaso D et al. Chromatin state dynamics during blood formation. Science 345, 943, doi: 10.1126/science.1256271 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rhie SK, Schreiner S & Farnham PJ Defining Regulatory Elements in the Human Genome Using Nucleosome Occupancy and Methylome Sequencing (NOMe-Seq). Methods Mol Biol 1766, 209–229, doi: 10.1007/978-1-4939-7768-0_12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barnett KR et al. ATAC-Me Captures Prolonged DNA Methylation of Dynamic Chromatin Accessibility Loci during Cell Fate Transitions. Molecular Cell 0, doi: 10.1016/j.molcel.2020.01.004 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Corces MR et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nature Methods 14, 959–962, doi: 10.1038/nmeth.4396 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trevino AE et al. Chromatin accessibility dynamics in a model of human forebrain development. Science 367, eaay1645, doi: 10.1126/science.aay1645 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bysani M et al. ATAC-seq reveals alterations in open chromatin in pancreatic islets from subjects with type 2 diabetes. Scientific Reports 9, 7785, doi: 10.1038/s41598-019-44076-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tierling S, Schmitt B & Walter J Comprehensive Evaluation of Commercial Bisulfite-Based DNA Methylation Kits and Development of an Alternative Protocol With Improved Conversion Performance. Genet Epigenet 10, 1179237x18766097, doi: 10.1177/1179237x18766097 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Montefiori L et al. Reducing mitochondrial reads in ATAC-seq using CRISPR/Cas9. Scientific Reports 7, 2451, doi: 10.1038/s41598-017-02547-w (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schutsky EK et al. Nondestructive, base-resolution sequencing of 5-hydroxymethylcytosine using a DNA deaminase. Nature Biotechnology 36, 1083–1090, doi: 10.1038/nbt.4204 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu M et al. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat Protoc 7, 2159–2170, doi: 10.1038/nprot.2012.137 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song C-X et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nature Biotechnology 29, 68–72, doi: 10.1038/nbt.1732 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu M et al. Base-Resolution Analysis of 5-Hydroxymethylcytosine in the Mammalian Genome. Cell 149, 1368–1380, doi: 10.1016/j.cell.2012.04.027 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Picelli S et al. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res 24, 2033–2040, doi: 10.1101/gr.177881.114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krueger F Trim galore. A wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files 516, 517 (2015). [Google Scholar]
- 47.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal 17, 10–12 (2011). [Google Scholar]
- 48.Song Q et al. A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PloS one 8, e81148–e81148, doi: 10.1371/journal.pone.0081148 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen H, Smith AD & Chen T WALT: fast and accurate read mapping for bisulfite sequencing. Bioinformatics 32, 3507–3509 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biology 9, R137–R137, doi: 10.1186/gb-2008-9-9-r137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu M & Gu L TCseq: Time course sequencing data analysis. R package version 1.12.1 (2020). [Google Scholar]
- 52.Landt SG et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 22, 1813–1831, doi: 10.1101/gr.136184.111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yan F, Powell DR, Curtis DJ & Wong NC From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis. Genome Biology 21, 22, doi: 10.1186/s13059-020-1929-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li Z et al. Identification of transcription factor binding sites using ATAC-seq. Genome Biology 20, 45, doi: 10.1186/s13059-019-1642-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bentsen M et al. ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nature Communications 11, 4267, doi: 10.1038/s41467-020-18035-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guerin L, Barnett KR & Hodges E Dual detection of chromatin accessibility and DNA methylation using ATAC-Me. HodgesGenomicsLab/NatProtocols_ATACme, doi: 10.5281/zenodo.5062153 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Daley T & Smith AD Predicting the molecular complexity of sequencing libraries. Nature Methods 10, 325–327, doi: 10.1038/nmeth.2375 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramírez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research 44, W160–W165, doi: 10.1093/nar/gkw257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data used in this protocol have been deposited in the Gene Expression Omnibus database with accession number GSE166267. Fig. 5 is derived from the raw data.