

Abstract
Objectives
To report novel biallelic PI4KA variants in a family presenting with pure hereditary spastic paraparesis.
Methods
Two affected sisters presented with unsolved hereditary spastic paraparesis and underwent clinical and imaging assessments. This was followed by short-read next-generation sequencing.
Results
Analysis of next-generation sequencing data uncovered compound heterozygous variants in PI4KA (NM_058004.4: c.[3883C>A];[5785A>C]; p.[(His1295Asn);(Thr1929Pro)]. Using ACMG guidelines, both variants were classified as likely pathogenic.
Discussion
Here, next-generation sequencing revealed 2 novel compound heterozygous variants in the phosphatidylinositol 4-kinase alpha gene (PI4KA) in 2 sisters presenting with progressive pure hereditary spastic paraparesis. Pathogenic variants in PI4KA have previously been associated with a spectrum of disorders including autosomal recessive perisylvian polymicrogyria, with cerebellar hypoplasia, arthrogryposis, and pure spastic paraplegia. The cases presented in this study expand the phenotypic spectrum associated with PI4KA variants and contribute new likely pathogenic variants for testing in patients with otherwise unsolved hereditary spastic paraparesis.
Introduction
PI4KA encodes phosphatidylinositol 4-kinase alpha (PI4KIIIα), a highly conserved enzyme that catalyzes phosphatidylinositol-4-phosphate (PI4P) production at the plasma membrane after recruitment by the EFR3 homolog A/B (EFR3A/B) and tetratricopeptide repeat domain 7 A/B (TTC7A/B) proteins.1,2 Biallelic pathogenic variants in PI4KA have previously been associated with autosomal recessive perisylvian polymicrogyria, with cerebellar hypoplasia and arthrogryposis [MIM: 616531],3 in addition to other less severe clinical presentations.4,5 Two patients presenting with autosomal recessive hereditary spastic paraparesis (HSP) harboring PI4KA variants (SPG84; MIM: 619621) have also been reported in the literature.5
HSPs encompass a group of disorders with core clinical features of bilateral lower limb spasticity, extensor plantar responses, and hyperreflexia.6 Over 70 genes have been associated with HSP, but there is considerable phenotypic overlap with other neurologic and metabolic disorders, including ataxias and leukodystrophies.6 Despite the use of next-generation sequencing technologies, many patients with HSP remain without a genetic diagnosis, complicated by the clinical and genetic heterogeneity of HSPs.
Here, we report 2 novel compound heterozygous PI4KA variants in 2 sisters with hereditary spastic paraparesis, providing novel genetic findings and highlighting the phenotypic heterogeneity of PI4KA-associated disorders.
Methods
Ethics and Informed Consent
The study was approved by the Human Research Ethics Committee of the University of Western Australia, and informed consent was obtained for study participants (Approval numbers: 2019/PID14033 and 2019/RA/4/20/1008). The proband and affected sister were recruited independently for this study by 2 groups from Perth, Australia, and Sydney, Australia, respectively.
Next-Generation Sequencing and Analysis
Proband DNA underwent testing on the nerve disease-targeted gene panel at Diagnostic Genomics, Pathwest (Perth), as previously described.7 Short-read whole-genome sequencing (WGS) was also performed on the proband and both parents by the Australian Genomics Research Facility (Melbourne), following GATK4 best-practices.8 Paired-end sequencing reads (150 base pairs) were generated using Illumina NovaSeq 6000 sequencing, with 30-fold average read depth per sample. Parent-proband trio WGS data were analyzed on the GENESIS platform.9
Short-read exome sequencing (WES) was independently performed on DNA from the affected sister by the New South Wales Health Pathology statewide sequencing service at Randwick Genomics Laboratory using the Twist Alliance VCGS exome capture kit on the lllumina NovaSeq 6000 sequencing system. Secondary and tertiary analyses were performed on the SOPHiA Data Driven Medicine platform (v5.10.30) using a hereditary spastic paraplegia gene panel (257 genes).
Variant Validation
Variants were confirmed by bidirectional Sanger sequencing of DNA from both affected sisters and other unaffected family members.
Data Availability
Data are available from the corresponding author on request.
Results
Case Presentation
The proband (II.3) and her affected sister (II.1) were of European background and born to healthy, nonconsanguineous parents (Figure 1A). Onset of symptoms in the proband started at age 12 years, with occasional leg spasms. Currently, the proband (aged 51 years) uses elbow crutches for walking and a wheelchair for longer distances. Neurologic examination showed slight weakness in the upper limbs, with normal tone. The proband scored 32/52 on the Spastic Paraplegia Rating Scale (SPRS). Hyperreflexia of both the upper and lower limbs, with bilateral upgoing plantar responses were present. Lower limbs show severe stiffness and spasticity, prohibiting assessment of lower limb strength. The patient shows no indication of dysarthria, dysphagia, or cognitive impairment. The proband also reports urinary urgency in the mornings. There was no involvement of the sensory nervous system. Brain MRI results show subtle periventricular hyperintensity predominantly in the occipital and parietal lobes extending into the posterior limb of the internal capsules (Figure 2A). There is also diffuse atrophy of the cervical and upper thoracic spinal cord (Figure 2B).
Figure 1. Investigation of Biallelic PI4KA Variants in a Family With Hereditary Spastic Paraparesis.

(A) Cosegregation of PI4KA variants with disease in the family. Proband indicated with a red arrow. Individuals whose DNA underwent genome sequencing is marked with an asterisk (*). (B) Sequence chromatograms of the c.3883C>A and c.5785A>C variants for all members in the family with available DNA. (C) Multiple sequence alignment of PI4KA orthologs across multiple species. Identical amino acid residues at His1295 and Thr1929 residue positions are highlighted in blue. Amino acids that are different to the human amino acid but with similar chemical properties are highlighted in yellow. UniProt accession codes for PI4KA orthologs provided in eTable 1. (D) Linearized protein schematic of PI4KA (N-terminus to C-terminus), annotated with known protein domains, variants identified in this study (red) and previously identified variants (black, missense; blue, nonsense or frameshift variants).3-5 a.a. = amino acid.
Figure 2. Proband MRI Images.
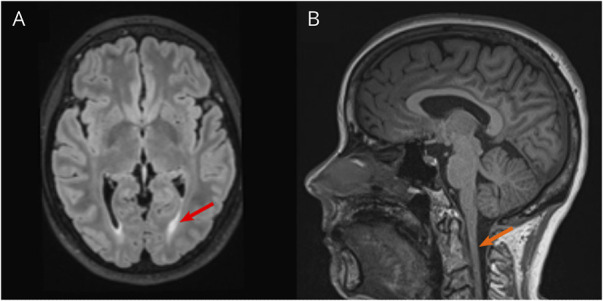
(A) Transverse brain MRI indicating posterior periventricular white matter changes (red arrow). (B) Sagittal MRI showing diffuse atrophy of the cervical spinal cord (orange arrow).
The affected sister (II.1) started presenting symptoms during her late teenage years. She would trip and fall over, with the problem gradually worsening over time. She presently (aged 56 years) reports falls every few months and requires 2 walking sticks for support. She has severe back spasms and pain, with increasing stiffness in the legs. The affected sister notes that lower back pains travel into the right leg and not the left. The patient shows no symptoms of dysarthria, dysphagia, or cognitive impairment. Neurologic examination revealed a severe spastic gait with bilateral footdrop. The SPRS score was 31/52. While upper limbs have preserved tone, strength, and proprioception, lower limbs show severe spasticity. The patient shows hyperreflexic knee jerks and ankle jerks with clonus, and bilaterally upgoing plantar responses. Brain MRI results were normal while MRI of the lumbar spine showed L3 nerve root impingement (not shown).
The clinical presentation of both affected sisters, compared with reported HSP cases with PI4KA variants,5 is summarized in Table 1.
Table 1.
Comparison of Clinical Details for Patients II.3 and II.1 With Other Cases in the Literature With Predominant Spastic Paraparesis Phenotype
| Verdura et al.5 | This study | |||
| Patient 9 | Patient 10 | II.3 | II.1 | |
| Sex | Male | Male | Female | Female |
| Ethnicity | European | Latin American | European | European |
| Age at onset | 17 y | 2 y | 12 y | Late teenage years |
| Age at last examination | 40 y | 18 y | 51 y | 56 y |
| Variant | c.5459_5461delAAG c.6156_6159delGACA |
c.4666G>A c.5159C>T |
c.3883C>A c.5785A>C |
c.3883C>A c.5785A>C |
| p.(Glu1820del) p.(Thr2053SerfsTer4) |
p.(Val1556Met) p.(Thr1720Ile) |
p.(His1295Asn) p.(Thr1929Pro) |
p.(His1295Asn) p.(Thr1929Pro) |
|
| Spasticity | ✓ | ✓ | ✓ | ✓ |
| Ataxia | — | — | — | ✓ Mild, upper limb |
| Upgoing plantar response | + Bilateral |
N/A | + Bilateral |
+ Bilateral |
| Epilepsy | — | — | — | — |
| Gross motor development | No delay | Delayed | No delay | No delay |
| Intellectual disability | Normal | Mild | Normal | Normal |
| Polymicrogyria | — | — | — | — |
| Cerebellar and/or brainstem abnormalities | Arachnoid cyst of posterior fossa | — | — | — |
| Leukodystrophy | — | — | — | — |
| Spinal cord abnormalities | Cervical spinal cord atrophy | Cervical spinal cord atrophy | Diffuse cervical spinal cord atrophy | L3 nerve root impingement |
| Other clinical information | Moderate mid-lumbar scoliosis | |||
Abbreviation: N/A = not available.
Genetic Findings
Previous genetic testing on a hereditary spastic paraplegia diagnostic targeted gene panel was negative for both affected sisters. Interrogation of parent-proband trio WGS data through GENESIS8 uncovered biallelic variants in PI4KA (NM_058004.4: c.[3883C>A];[5785A>C]; p.[(His1295Asn);(Thr1929Pro)]). Both variants were independently identified in the proband's sister through analysis of clinical WES data. No variants in other clinically relevant genes or candidate disease genes were identified. Variants were subsequently confirmed by Sanger sequencing and showed cosegregation with disease in the family following autosomal recessive inheritance (Figure 1, A and B).
The His1295 amino acid residue is conserved to zebrafish (Figure 1C, eTable 1). By contrast, the Thr1929 residue is only moderately conserved (Figure 1C, eTable 1). Both variants are present in gnomAD at very low allele frequencies (allele frequency <0.00001) and are predicted to have a damaging effect on the protein by multiple in silico tools. Both variants have a CADD score of 27. In addition, both variants are located in important PI4KA functional domains (Figure 1D). Of note, the p.(His1295Asn) variant occurs at the same amino acid position as a variant p.(His1295Arg) reported in a patient with developmental encephalopathy with hypomyelinating leukodystrophy and structural brain anomalies (Figure 1D).5 This patient additionally presented with spastic paraparesis. Thus, both variants were classified as likely pathogenic according to the American College of Medical Genetics and Genomics (ACMG) guidelines (c.3883C>A ACMG criteria: PM2, PM5, PP1, PP2, PP3; c.5785A>C ACMG criteria: PM1, PM2, PM3, PP1, PP2, PP3).
Discussion
Here, we present compound heterozygous variants in PI4KA (c.[3883C>A];[5785A>C]; p.[(His1295Asn);(Thr1929Pro)]) for 2 patients presenting with HSP. This is the second study reporting PI4KA as causing a HSP phenotype and therefore represents an important confirmatory study for the literature.
PI4KA variants were initially established as the cause of severe in utero autosomal recessive perisylvian polymicrogyria, with cerebellar hypoplasia and arthrogryposis [MIM: 616531].3 The recently proposed ‘PI4KA-spectrum’ has since expanded to include various immunologic, intestinal, neurodevelopmental, and neurologic presentations.4,5,10 Of note, patients with spastic paraplegia have been reported in the literature, one of which harbored a p.(His1295Arg) substitution.5 The reported phenotype matched that of the affected sisters in this family, who harbor a different missense variant in the same position (p.(His1295Asn)). In particular, the patient harboring the p.(His1295Arg) substitution presented with a complex manifestation of disease (including, hypomyelinating leukodystrophy, moderate intellectual disability, spastic paraparesis, ataxia, epilepsy, nystagmus, and severely delayed development).5 By contrast, our patients harboring the p.(His1295Asn) substitution presented with a predominant HSP phenotype. The highly conserved nature of this amino acid position (Figure 1C) suggests that the p.(His1295Asn) substitution is disease-causing.
Hypomorphic missense variants can result in milder clinical presentations, and this is a recognized phenomenon in neurogenetic diseases.11 Functional examination by Verdura et al.5 showed significant downregulation, but not absence, of PI4KIIIα and PI4P expression in patient fibroblasts. The discovery of these 2 additional missense variants offers a unique avenue to functionally investigate the contribution of hypomorphic PI4KA variants to a less severe phenotypic spectrum of disease.
PI4KIIIα is involved in lipid phosphoinositide metabolism.1,2 More broadly, these molecules play a crucial role in several pathways that govern cellular identity, growth, and development.1,2 The PI4P metabolite phosphatidylinositol-4,5-bisphosphate is particularly important for signal transduction, plasma membrane regulation, and myelination,1,2,5 suggesting that changes to PI4K catalytic function or complex formation will have lethal downstream effects. Indeed, mouse models with loss-of-function PI4KA variants result in embryonic lethality,12,13 while widespread developmental abnormalities are a consequence of pi4ka knockdown in zebrafish.14 Taken together, PI4KA and downstream metabolites play an important function evolutionarily, and any perturbations to the system will likely lead to disease.
Notably, PI4K2α, another member of the PI4K enzyme family, catalyzes PI4P formation.15 Mice with Pi4k2a knockouts initially presented normally, but progressively developed neurologic features (for example, tremor, limb weakness, and axonal degeneration of the spinal cord) reminiscent of late-onset human hereditary spastic paraplegia.15
Given the importance of PI4KIIIα in various cellular pathways, it may not seem surprising that pathogenic variants in the gene will result in a wide range of disease phenotypes. Some trends have emerged linking PI4KA variants to more severe phenotypes3-5,10 or variants restricted to patients of Amish ancestry.4,10 However, future studies may validate whether there are definitive genotype-phenotype correlations.
In conclusion, the study highlights hereditary spastic paraparesis as a milder phenotype within the ‘PI4KA-spectrum’. The findings have diagnostic implications: variants in PI4KA should be considered in patients with presentations beyond arthrogryposis.
Appendix. Authors
| Name | Location | Contribution |
| Jevin M. Parmar, BBiomedSc (Hons) | Rare Disease Genetics and Functional Genomics Group, Centre for Medical Research, University of Western Australia; Harry Perkins Institute of Medical Research, Nedlands, Australia | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
| Elyshia L. McNamara, BSc (Hons) | Rare Disease Genetics and Functional Genomics Group, Centre for Medical Research, University of Western Australia; Harry Perkins Institute of Medical Research, Nedlands, Australia | Major role in the acquisition of data; analysis or interpretation of data |
| Phillipa J. Lamont, MBBS, PhD, FRACP | Royal Perth Hospital, Australia | Major role in the acquisition of data |
| Kishore R. Kumar, MBBS, PhD, FRACP | Sydney Medical School, Faculty of Medicine and Health, University of Sydney, Camperdown; Garvan Institute of Medical Research, Darlinghurst; Molecular Medicine Laboratory, Concord Repatriation General Hospital, NSW Health Pathology; Department of Neurology, Concord Repatriation General Hospital, Australia | Drafting/revision of the manuscript for content, including medical writing for content |
| Audrey Rick, BSc | Rare Disease Genetics and Functional Genomics Group, Centre for Medical Research, University of Western Australia; Harry Perkins Institute of Medical Research, Nedlands, Australia | Major role in the acquisition of data |
| Marion Stoll, PhD | Molecular Medicine Laboratory, Concord Repatriation General Hospital, NSW Health Pathology; School of Medical Sciences, University of Sydney, Camperdown, Australia | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Pak Leng Cheong, MB ChB, BMedSc (Hons), MPhil, DPhil, FRCPA | Sydney Medical School, Faculty of Medicine and Health, University of Sydney, Camperdown; Molecular Medicine Laboratory, Concord Repatriation General Hospital, NSW Health Pathology, Australia | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data |
| Gianina Ravenscroft, PhD | Rare Disease Genetics and Functional Genomics Group, Centre for Medical Research, University of Western Australia; Harry Perkins Institute of Medical Research, Nedlands, Australia | Drafting/revision of the manuscript for content, including medical writing for content; study concept or design |
Study Funding
This project was supported by the Australian MRFF (APP2007681).
Disclosure
J.M. Parmar is supported by the Australian Government Research Training Program. G. Ravenscroft is supported by an Australian NHMRC EL2 Fellowship (APP2007769). All other authors report no disclosures relevant to the manuscript. Go to Neurology.org/NG for full disclosures.
References
- 1.Boura E, Nencka R. Phosphatidylinositol 4-kinases: function, structure, and inhibition. Exp Cell Res. 2015;337(2):136-145. doi: 10.1016/j.yexcr.2015.03.028 [DOI] [PubMed] [Google Scholar]
- 2.Raghu P, Joseph A, Krishnan H, Singh P, Saha S. Phosphoinositides: regulators of nervous system function in health and disease. Front Mol Neurosci. 2019;12:208. doi: 10.3389/fnmol.2019.00208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pagnamenta AT, Howard MF, Wisniewski E, et al. Germline recessive mutations in PI4KA are associated with perisylvian polymicrogyria, cerebellar hypoplasia and arthrogryposis. Hum Mol Genet. 2015;24(13):3732-3741. doi: 10.1093/hmg/ddv117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salter CG, Cai Y, Lo B, et al. Biallelic PI4KA variants cause neurological, intestinal and immunological disease. Brain. 2021;144(12):3597-3610. doi: 10.1093/brain/awab313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verdura E, Rodríguez-Palmero A, Vélez-Santamaria V, et al. Biallelic PI4KA variants cause a novel neurodevelopmental syndrome with hypomyelinating leukodystrophy. Brain. 2021;144(9):2659-2669. doi: 10.1093/brain/awab124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shribman S, Reid E, Crosby AH, Houlden H, Warner TT. Hereditary spastic paraplegia: from diagnosis to emerging therapeutic approaches. Lancet Neurol. 2019;18(12):1136-1146. doi: 10.1016/S1474-4422(19)30235-2 [DOI] [PubMed] [Google Scholar]
- 7.Beecroft SJ, Yau KS, Allcock RJN, et al. Targeted gene panel use in 2249 neuromuscular patients: the Australasian referral center experience. Ann Clin Transl Neurol. 2020;7(3):353-362. doi: 10.1002/acn3.51002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poplin R, Ruano-Rubio V, Depristo MA, et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv. Preprint posted online July 24, 2018. doi: 10.1101/201178 [DOI]
- 9.Gonzalez M, Falk MJ, Gai X, Postrel R, Schüle R, Zuchner S. Innovative genomic collaboration using the GENESIS (GEM.app) platform. Hum Mutat. 2015;36(10):950-956. doi: 10.1002/humu.22836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baple EL, Salter C, Uhlig H, Wolf NI, Crosby AH. PI4KA-related disorder. In: Adam MP, Feldman J, Mirzaa GM, et al., eds. GeneReviews®. University of Washington, Seattle; 2022. Accessed February 7, 2024. pubmed.ncbi.nlm.nih.gov/35951779/ [PubMed] [Google Scholar]
- 11.Minnerop M, Kurzwelly D, Wagner H, et al. Hypomorphic mutations in POLR3A are a frequent cause of sporadic and recessive spastic ataxia. Brain. 2017;140(6):1561-1578. doi: 10.1093/brain/awx095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bojjireddy N, Botyanszki J, Hammond G, et al. Pharmacological and genetic targeting of the PI4KA enzyme reveals its important role in maintaining plasma membrane phosphatidylinositol 4-phosphate and phosphatidylinositol 4,5-bisphosphate levels. J Biol Chem. 2014;289(9):6120-6132. doi: 10.1074/jbc.M113.531426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaillancourt FH, Brault M, Pilote L, et al. Evaluation of phosphatidylinositol-4-kinase IIIα as a hepatitis C virus drug target. J Virol. 2012;86(21):11595-11607. doi: 10.1128/JVI.01320-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma H, Blake T, Chitnis A, Liu P, Balla T. Crucial role of phosphatidylinositol 4-kinase IIIalpha in development of zebrafish pectoral fin is linked to phosphoinositide 3-kinase and FGF signaling. J Cell Sci. 2009;122(Pt 23):4303-4310. doi: 10.1242/jcs.057646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simons JP, Al-Shawi R, Minogue S, et al. Loss of phosphatidylinositol 4-kinase 2alpha activity causes late onset degeneration of spinal cord axons. Proc Natl Acad Sci U S A. 2009;106(28):11535-11539. doi: 10.1073/pnas.0903011106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available from the corresponding author on request.