

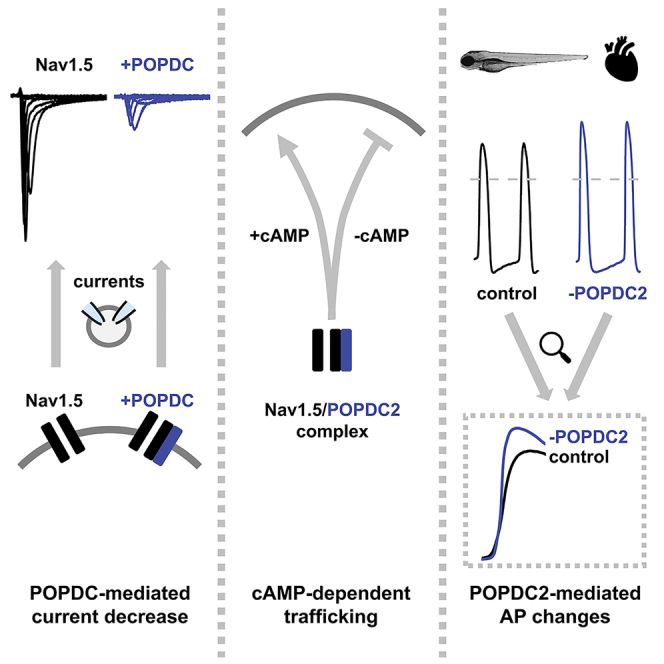
Summary
Popeye domain containing (POPDC) proteins are predominantly expressed in the heart and skeletal muscle, modulating the K2P potassium channel TREK-1 in a cAMP-dependent manner. POPDC1 and POPDC2 variants cause cardiac conduction disorders with or without muscular dystrophy. Searching for POPDC2-modulated ion channels using a functional co-expression screen in Xenopus oocytes, we found POPDC proteins to modulate the cardiac sodium channel Nav1.5. POPDC proteins downregulate Nav1.5 currents in a cAMP-dependent manner by reducing the surface expression of the channel. POPDC2 and Nav1.5 are both expressed in different regions of the murine heart and consistently POPDC2 co-immunoprecipitates with Nav1.5 from native cardiac tissue. Strikingly, the knock-down of popdc2 in embryonic zebrafish caused an increased upstroke velocity and overshoot of cardiac action potentials. The POPDC modulation of Nav1.5 provides a new mechanism to regulate cardiac sodium channel densities under sympathetic stimulation, which is likely to have a functional impact on cardiac physiology and inherited arrhythmias.
Subject areas: Biochemistry, Molecular biology, Physiology
Graphical abstract
Highlights
-
•
Popeye-domain-containing proteins (POPDC) modulate the cardiac sodium channel Nav1.5
-
•
POPDC2 modulates Nav1.5 surface expression and currents in a cAMP-dependent manner
-
•
Knock-down of popdc2 in embryonic zebrafish indicate altered sodium channel densities
-
•
Nav1.5 modulation might be relevant for sympathetic stimulation and arrhythmias
Biochemistry; Molecular biology; Physiology
Introduction
Popeye domain containing (POPDC) proteins are localized to the plasma membrane of striated muscle cells.1 They consist of a short extracellular domain followed by three transmembrane segments. The cytoplasmic part of the protein contains the conserved Popeye domain, which is a divergent cAMP-binding domain that binds cAMP with high affinity and specificity. This novel class of cAMP-binding proteins comprises three family members, POPDC1, also known as BVES, POPDC2 and POPDC3.2,3 POPDC1 and POPDC2 knock-out mice display a strong stress-induced sinoatrial node dysfunction.4 In POPDC1/2 double knock-out mice, a wide range of arrhythmias including atrial fibrillation, polymorphic ventricular tachycardia, extrasystoles, and atrio-ventricular block (AVB) was prevalent5 and in zebrafish, morpholino-mediated knock-down of popdc2 led to AVB.6 Recently, we and others described patients with disease-causing mutations in POPDC1, POPDC2 or POPDC3, leading in the case of POPDC1 variants to a combined phenotype of cardiac conduction abnormalities with autosomal recessive limb-girdle muscular dystrophy (LGMD), in the case of the POPDC2W188∗ variant to an isolated AVB without any extra-cardiac manifestation and for POPDC3 missense-variants to a novel form of LGMD lacking a cardiac manifestation, respectively.7,8,9,10 We found that variants in the different POPDC genes lead to a dysregulation of the two-pore domain potassium (K2P) channel TREK-1,7,8,9 yet the disease-causing mechanisms are still not completely understood.
POPDC proteins were described to interact with several proteins, including zonula occludens 1 protein (ZO-1),11 atypical protein kinase C (aPKC),12 dystrophin,13 dysferlin,13 guanine nucleotide exchange factor T (GEFT),14 vesicle-associated membrane protein 2 (Vamp2) and 3 (Vamp3),15 caveolin-3,16 PDE417 and recently, we described POPDC1 as a novel adenylyl cyclase 9 scaffold.18 However, besides TREK-1, no other ion channel is known to be functionally modulated by POPDC proteins. Since cardiac excitability and the pathogenesis of arrhythmias are primarily linked to ion channel (dys)function, the aim of our study was to identify additional cardiac ion channels modulated by POPDC proteins.
In a functional co-expression screen using Xenopus oocytes, we identified the voltage-gated cardiac sodium channel Nav1.5, encoded by the SCN5A gene, to be modulated by POPDC proteins. Nav1.5 is primarily found in cardiac muscle, where it mediates the fast influx of sodium ions across the cell membrane. In addition, Nav1.5 plays a major role for the impulse propagation throughout the heart. SCN5A variants can lead to a large variety of different cardiac arrhythmias including Long QT Syndrome 3 (LQT3),19,20 Brugada Syndrome (BrS),21 idiopathic ventricular fibrillation,22 atrial fibrillation,23 Progressive Cardiac Conduction Disorder (PCCD),24 Sick Sinus Syndrome (SSS)25 or overlap syndromes.26,27 The newly identified cAMP-dependent modulation of Nav1.5 by POPDC proteins describes a novel mechanism to control cardiac excitability and is therefore likely to play roles in the pathogenesis of different arrhythmias primarily affecting the cardiac conduction system.
Results
Functional screen of cardiac ion channels for a modulation by Popeye domain containing protein 2
POPDC variants cause cardiac arrhythmias mechanistically involving a dysregulation of TREK-1 channels. However, little is known about putative other ion channels that are regulated by POPDC proteins and specifically about the role of POPDC2, which is, besides POPDC1, the major isoform expressed in the heart. To probe for cardiac ion channels modulated by POPDC proteins, we screened a large set of cardiac ion channels for a potential modulation by POPDC2, using functional co-expression studies in Xenopus laevis oocytes and voltage-clamp recordings. We found that POPDC2 does not modulate the current amplitudes of the pacemaker channels HCN2 and HCN4 or the K2P channels TASK-1, TASK-4, and TRAAK (Figures 1A and 1E). Similarly, POPDC2 did not affect the voltage-dependent A-type potassium channels Kv1.4 (Figures 1B and 1E) and Kv4.3 (Figures 1C and 1E) encoding components of cardiac Ito currents. In addition, KCNQ1 encoding the α-subunit of the cardiac IKs current was not affected by the co-expression of POPDC2 (Figure 1E). A mutation of the Na+/K+-ATPase in the hiphop (hip) zebrafish line causes AVB,28 indicating a role of this pump in the cardiac conduction system. However, the K+-induced pump currents of the Na+/K+-ATPase were also not altered by the co-expression of POPDC2 (Figure 1E) in Xenopus laevis oocytes. In contrast, POPDC2 was found to reduce the current amplitude of the sodium channel Nav1.5 which mediates the cardiac INa current (Figures 1D and 1E), the key current for the cardiac action potential upstroke in the working myocardium and the cardiac conduction system, propagating the electrical signal to the working myocardium.
Figure 1.

Functional screen for cardiac ion channels modulated by POPDC2
(A–D) Representative current traces of (A) HCN4, applying a voltage step from −30 mV to −120 mV within 6 s, from a holding potential of −30 mV, (B) Kv1.4, applying a voltage step from −80 mV to +40 mV within 2 s, from a holding potential of −80 mV, (C) Kv4.3, applying a voltage step from −80 mV to +40 mV within 2 s, from a holding potential of −80 mV or (D) Nav1.5, applying a voltage step from −80 mV to −20 mV within 50 ms, from a holding potential of −80 mV, expressed alone (black) or together with POPDC2 (blue) in Xenopus laevis oocytes.
(E) Indicated cardiac channels or the Na+/K+ ATPase (Na/K) were expressed alone or together with POPDC2 in Xenopus oocytes and relative changes in current amplitudes by POPDC2 were plotted. Oocytes were stored in a storage solution lacking theophylline. The number of technical replicates (n) is indicated in parentheses. Data are presented as mean ± s.e.m. Significance was probed using a two-sided Student’s t-test. ∗∗∗, p < 0.001.
Co-immunoprecipitation of Nav1.5 and Popeye domain containing protein 2 and modulation of voltage-gated sodium currents by Popeye domain containing protein isoforms
Co-immunoprecipitation experiments in transiently transfected COS-7 cells, as well as in murine hearts, confirmed the protein-protein interaction of POPDC2 with Nav1.5 (Figures 2A and 2B).
Figure 2.

Interaction of POPDC proteins with the cardiac sodium channel Nav1.5 or the skeletal muscle sodium channel Nav1.4
(A) Co-immunoprecipitation of POPDC2-FLAG with Nav1.5-HA was performed in COS-7 cells using anti-HA antibodies. An empty vector served as control. The co-immunoprecipitation was subsequently detected with an anti-FLAG antibody (IB: α-Flag).
(B) Co-immunoprecipitation of POPDC2 and Nav1.5 in murine heart lysates (mheart) using anti Nav1.5 antibodies (IP: α-Nav1.5). The immunoprecipitation was subsequently detected with an anti-Nav1.5 (IB: α-Nav1.5) and the co-immunoprecipitation by an anti-POPDC2 antibody (IB: α-POPDC2). HeLa cells were used as a control.
(C) Oocytes were injected with cRNA for human Nav1.5 or co-injected with different murine POPDC isoforms and subsequently stored in a storage solution lacking theophylline. Currents were recorded with a P/N (N = 4) voltage-step protocol (holding potential −80 mV) and voltage-steps of 50 ms ranging from −70 to +80 mV in 10 mV increments (see inset).
(D and E) (D) Relative current amplitudes analyzed at −20 mV and normalized to Nav1.5. (E) Voltage-dependence of Nav1.5 activation in the absence or presence of POPDC1, POPDC2 or POPDC3. The respective voltages of half-maximal activation (V1/2) and the Boltzmann constants (k) are provided as inset. The number of technical replicates (n) is indicated in parentheses or bar graphs. Data are presented as mean ± s.e.m. Significance was probed using a two-sided Student’s t-test. ∗∗∗, p < 0.001.
(F and G) (F) POPDC2 does not modulate current amplitudes of Nav1.4, analyzed at −10 mV and normalized to Nav1.4 or (G) the shape of the voltage-dependence of Nav1.4 activation. The respective voltages of half-maximal activation (V1/2) and the Boltzmann constants (k) are provided as inset. The number of technical replicates (n) is indicated in parentheses or bar graphs. Oocytes were stored in a storage solution lacking theophylline. Data are presented as mean ± s.e.m. Significance was probed using a two-sided Student’s t-test. See also Figures S1 and S2.
In voltage-clamp recordings, the effect on Nav1.5-mediated current amplitudes was the same for all three murine POPDC isoforms (Figures 2C and 2D). All three POPDC isoforms caused a pronounced reduction of the Nav1.5 current amplitude of about 80% (Figures 2C and 2D; Figure S1A), while the voltage-dependence of activation was not affected (Figure 2E; Figures S1C and S1D). However, the current amplitudes, as well as the voltage-dependence of activation, of the Nav1.4 sodium channel, which is found in skeletal muscle, were not altered upon the co-expression of POPDC2 (Figures 2F and 2G; Figure S2).
Popeye domain containing protein 2 does not affect Nav1.5 current kinetics
Since we initially screened with POPDC2 for novel interaction partners, we continued our mechanistic studies of the Nav1.5 modulation with this isoform. As the observed current reduction could either result from an altered channel gating or trafficking, we first probed for changes in Nav1.5 current kinetics by POPDC2 (Figure 3A). However, voltage dependence of activation and inactivation, as well as the resulting window current, were not significantly altered by the co-expression of POPDC2 (Figure 3B; Figure S3). Likewise, the kinetics of inactivation (Figure 3C) and recovery from inactivation (Figures 3D–3F) were not changed by POPDC2. Thus, the overall kinetics of Nav1.5 currents were unchanged by POPDC2 and also the late or remaining current, evident at the end of the test pulse in Figure 3C remained unaltered. As channel gating is not affected and an isolated effect on Nav1.5 current amplitudes was observed, POPDC proteins most likely regulate the transport of Nav1.5 channels to the plasma membrane.
Figure 3.

POPDC2 does not alter the current kinetics of Nav1.5
(A) Current traces of human Nav1.5 expressed alone or co-expressed with human POPDC2. Representative traces are depicted at −30 mV, −20 mV and −10 mV.
(B) Voltage-dependence of activation and inactivation curves fitted to a Boltzmann equation. The respective voltages of half-maximal activation (V1/2) together with the Boltzmann constants (k), as well as the window currents, are provided as inset. For the V1/2 of activation measurements, the currents were recorded with a P/N (N = 4) voltage-step protocol (holding potential −80 mV) and voltage-steps of 50 ms ranging from −70 to +80 mV in 10 mV increments (see inset activation protocol). For the V1/2 of inactivation measurements, the currents were recorded with a P/N (N = 4) voltage-step protocol (holding potential −100 mV) and voltage-steps of 50 ms ranging from −120 to −10 mV in 10 mV increments with a final step to −30 mV for 50 ms (see inset inactivation protocol). The number of technical replicates (n) is indicated in parentheses. Data are presented as mean ± s.e.m.
(C) Representative current traces of the kinetics of inactivation recorded with a single-step protocol from −100 mV to −30 mV for 65 ms (inset illustrates voltage protocol).
(D) Representative recordings of the recovery from inactivation (inset illustrates voltage protocol).
(E) Time-course of the recovery from inactivation recorded with a two-pulse protocol, first step to −10 mV for 50 ms and the second test pulse stepped to −10 mV with increasing interpulse intervals. The number of technical replicates (n) is indicated in parentheses. Data are presented as mean ± s.e.m.
(F) Time constants of the fast and slow component of the recovery from inactivation (lower panel) and the relative contribution of the fast and slow component (upper panel). Values were obtained by a bi-exponential fit from the data in (E). Oocytes were stored in a storage solution lacking theophylline. The number of technical replicates (n) is indicated in parentheses. Data are presented as mean ± s.e.m. See also Figure S3.
Nav1.5 is modulated by Popeye domain containing protein 2 in a cyclic adenosine monophosphate-dependent manner
The K2P channel TREK-1 is modulated by POPDC proteins in a cAMP-dependent manner and the application of theophylline, a nonspecific phosphodiesterase inhibitor to increase intracellular cAMP levels, completely removes TREK-1 modulation by POPDC proteins.4,13 To investigate whether Nav1.5 modulation by POPDC2 is also cAMP-dependent, an effect that might provide an explanation for the previously described cAMP-dependent regulation of native INa in cardiomyocytes,29 we performed voltage-clamp experiments in oocytes injected with Nav1.5 cRNA alone or together with POPDC2 cRNA in the presence or absence of theophylline (Figure 4; Figure S4). Interestingly, the Nav1.5 modulation by co-expressing POPDC2 (Figures 4A and 4B) was completely antagonized under conditions of increased cAMP levels (Figures 4A and 4C). Thus, the modulation of Nav1.5 by POPDC2 appears to be cAMP-dependent, which might result in increased sodium currents under conditions of sympathetic stimulation.
Figure 4.

POPDC2 reduces Nav1.5 current amplitudes in a cAMP-dependent manner
(A) POPDC2 modulation of Nav1.5 is cAMP-dependent. Representative current traces recorded by voltage steps to −20 mV. Oocytes were injected with 10 ng Nav1.5 alone or 10 ng Nav1.5 together with 5 ng POPDC2 (50%) or 10 ng POPDC2 (100%), and stored in ND96 solution with or without theophylline before voltage-clamp measurements in oocytes. Inset illustrates the applied voltage protocol.
(B) Analyses of peak current amplitudes from experiments performed in (A), analyzed at −20 mV and normalized to Nav1.5. The number of technical replicates (n) is indicated in bar graphs. Data are presented as mean ± s.e.m. Significance was probed using a two-sided Student’s t-test. ∗, p < 0.05; ∗∗∗, p < 0.001.
(C) Oocytes were injected as in (A-B) but incubated in a storage solution containing theophylline. Relative current amplitudes were analyzed at −20 mV and normalized to Nav1.5. The number of technical replicates is indicated in bar graphs. Data are presented as mean ± s.e.m. See also Figure S4.
Popeye domain containing protein 2 co-localizes with Nav1.5 at the plasma membrane and intracellular compartments, regulating the surface expression of Nav1.5 in a cyclic adenosine monophosphate-dependent manner
To study the membrane and subcellular localization of Nav1.5 by fluorescence microscopy, HeLa cells were transfected with extracellularly HA-tagged Nav1.5, fixed and processed for immunofluorescence analysis using anti-HA antibodies (in magenta) (Figures 5A–5D). In non-permeabilized cells Nav1.5-HA was nicely stained at the plasma membrane (Figure 5A). In cells co-transfected with POPDC2-YFP (in cyan) the Nav1.5-mediated fluorescence overlapped (in white) with that of POPDC2-YFP, highlighting that Nav1.5 and POPDC2 are co-localized at the plasma membrane (Figure 5B). See also very evident co-staining of membrane protrusions indicated by arrows (Figure 5B). If the cells were permeabilized with 0.1% Triton X-100, intracellular membranous networks and vesicular compartments were also labeled by antibodies targeting the HA-tagged Nav1.5 channels. However, no obvious alterations in this subcellular distribution of Nav1.5-HA were observed following the co-transfection of POPDC2-YFP (Figure 5C vs; Figure 5D). Yet, some intracellular Nav1.5-HA-positive structures were co-stained by POPDC2-YFP (Figure 5D). See also clear co-stainings in the extensions of the cells indicated by arrowheads (Figure 5D). Overall, these labeling patterns suggest that Nav1.5 channels and POPDC2 subunits share at least in part identical subcellular compartments and co-localize at the plasma membrane.
Figure 5.

POPDC2 co-localizes with Nav1.5 at the plasma membrane and intracellular compartments and regulates the surface expression of Nav1.5 in a cAMP-dependent manner
(A and C) HeLa cells were transfected exclusively with Nav1.5-HA or (B and D) in combination with POPDC2-YFP. 24 h after transfection the cells were fixed, non-permeabilized (A-B) or permeabilized (C-D) and immunostained with anti HA antibody and Alexa 647 conjugated secondary antibody. The cells were then analyzed by confocal microscopy. Nuclear counterstaining with Hoechst 33342 is depicted in blue and nuclei of non-transfected cells are indicated by asterisk. Arrows point to either (A) Nav1.5- or (B) Nav1.5- plus POPDC2-YFP-stained membrane protrusions; PM, plasma membrane. Arrowheads in (D) depict Nav1.5-positive intracellular structures co-labelled by POPDC2-YFP, which are also designated by arrows in the single fluorescence images shown on the left. Scale bars, 10 μm.
(E) Cartoon of the chemiluminescence-based ELISA-assay performed in HeLa cells transiently transfected with Nav1.5HA to quantify the changes in Nav1.5 surface expression by co-transfection with POPDC2. In fixed cells, the primary antibody detects extracellularly HA-tagged Nav1.5 channels (Nav1.5HA) at the surface membrane while the peroxidase-enzyme (POD) which is linked to the secondary antibody catalyzes the oxidation of a luminol-substrate which is accompanied by the emission of light. Subsequently, the light emission can be quantified as relative light units (RLUs) as a measure of the surface expression of the channel.
(F) Chemiluminescence assay of extracellularly HA-tagged Nav1.5 to determine the relative changes in surface expression by co-expressed POPDC2. The number of biological replicates (N) is indicated in the bar graphs. Data are expressed as relative light units (RLUs). Data are presented as mean ± s.e.m. Significance was probed using a two-sided Student’s t-test. ∗, p < 0.05.
(G) Chemiluminescence assay of extracellularly HA-tagged Nav1.5 to determine the relative changes in surface expression by co-expressed POPDC2 in the presence of theophylline. The number of biological replicates (N) is indicated in the bar graphs. Data are expressed as relative light units (RLUs). Data are presented as mean ± s.e.m.
POPDC proteins caused an isolated reduction in current amplitudes without any changes to the gating of the channel (Figures 2 and 3), suggesting an effect of POPDC proteins on Nav1.5 channel transport to the plasma membrane. However, given that the surface membrane is very thin and that there is a naturally large cell-to-cell variability in membrane fluorescence imaging experiments, a reduction in surface expression is difficult to quantity by single-cell fluorescence microscopy. Therefore, we quantified Nav1.5 surface expression using an extracellular HA-tagged channel construct and a chemiluminescence-based ELISA assay in transiently transfected HeLa cells (Figures 5E–5G). Here, human POPDC2 co-transfection reduced Nav1.5 surface expression (Figure 5F) to a similar extent as observed for the current amplitudes in the voltage clamp experiments, supporting the hypothesis that the Nav1.5 current reduction is exclusively caused by a reduced number of channels at the plasma membrane. Strikingly, similar to the reduction in Nav1.5 current amplitudes (Figures 4B and 4C), the reduction in surface expression was antagonized by theophylline (Figure 5G), indicating as a primary mechanism of action that Nav1.5 is in the presence of cAMP more efficiently transported to the plasma membrane.
Knock-down of Popeye domain containing protein 2 leads to increased upstroke velocity and overshoot of cardiac action potentials in embryonic zebrafish hearts
Popdc2 knock-out mice, as well as the knock-in mice carrying a POPDC2W188∗ mutation, suffer from a degeneration of the sinoatrial node.4,9 Here, we elucidated the effect of a knock-down of popdc2 on cardiac action potentials in the zebrafish embryonic heart by patch-clamp experiments, as the action potentials resemble those of transitional sinoatrial node cells with a fast Nav1.5-mediated action potential upstroke. To this end, we knocked down the zebrafish popdc2 mRNA using morpholino-antisense oligonucleotide injections (POPDC2 MO) into the early zebrafish embryo as previously reported.6 Note, that we did not observe other alterations in cardiac development/structure than those that we have previously reported for morpholino oligonucleotide-mediated knock-down of popdc2.6 Exclusively focusing on the electrophysiological changes, we separated the intact and beating embryonic heart 48 h after fertilization (hpf), and measured the spontaneous action potentials using the whole-cell patch-clamp technique (Figure 6A). Neither the beating frequency nor the length of the action potential (APD50 and APD90) were altered (Figures 6B–6D). While we could not precisely quantify INa currents of the spontaneously beating embryonic zebrafish hearts due to space clamp problems, strikingly, the upstroke velocity was significantly increased and the action potential overshoot was more pronounced, which is in perfect agreement with increased Nav1.5 currents due to the knock-down of popdc2 by the morpholino injections (Figures 6E–6G). Taken together, these data suggest that POPDC2 might modulate the Nav1.5 current densities in vivo and support a functional role in cardiac physiology and pathophysiology.
Figure 6.

Morpholino-mediated knock-down of popdc2 modifies embryonic cardiac action potential in the zebrafish heart
(A) Representative voltage traces of cardiac action potentials in zebrafish embryos. POPDC2 MO: Morpholino-mediated knock-down of popdc2. hpf: hours postfertilization.
(B–D) Analysis of (B) action potential frequency, (C) APD50, and (D) APD90. The number of biological replicates is indicated in bar graphs. Data are presented individually but also as mean ± s.e.m.
(E) Representative voltage traces of cardiac action potentials in embryonic zebrafish focusing on the fast depolarization phase.
(F) Analysis of the action potential overshoot. The number of biological replicates is indicated in bar graphs. Data are presented individually but also as mean ± s.e.m. Significance was probed using a two-sided Student’s t test. ∗∗, p < 0.01.
(G) Analysis of the action potential upstroke velocity. The number of biological replicates is indicated in the individual bar graphs. Data are presented individually but also as mean ± s.e.m. Significance was probed using a two-sided Student’s t-test. ∗, p < 0.05.
Discussion
POPDC family members are cAMP-binding proteins involved in regulating cardiac excitability. Consistently, POPDC1 and POPDC2 deficient mice and mutant/morphant zebrafish suffer from severe cardiac arrhythmias.4,6,13 In addition, recently described POPDC variants led to a combined cardiac and skeletal muscle phenotype in the case of POPDC1,10,13 AVB for a POPDC2 variant9 or an isolated skeletal muscle phenotype for a set of different POPDC3 variants.8 Still, the mechanism of how POPDC variants lead to the arrhythmic phenotypes remained to a large fraction unclear and the only cardiac ion channel so far known to be modulated by POPDC proteins is the K2P channel TREK-1.4 The newly identified POPDC interaction partner Nav1.5, is associated with a variety of different arrhythmias, including sudden infant death syndrome (SIDS),30 LQTS3,19,20 BrS,21 idiopathic ventricular fibrillation,22 atrial fibrillation,23,31 PCCD,24 SSS25 or overlap syndromes.26,27 Thus, the modulation of Nav1.5 by POPDC proteins might be also relevant for the pathophysiology of different arrhythmias. As POPDC proteins also interact with many other proteins like ZO-1, atypical PKC, dystrophin, dysferlin, GEFT, MYC, PR61a, LRP6, Vamp2, Vamp3, NDRG4, and Caveolin-3,32 but also with XIRP1,33 PDE4,17 and AC9,18 it is difficult to extrapolate to which extent the cAMP-dependent modulation of Nav1.5 described here contributes to the phenotypes observed in the different transgenic animals and patients carrying POPDC variants. Although the interaction partners described above might be part of a complex modulating protein network that may be also species- and/or tissue-specific, these proteins are more likely to only indirectly affect the cardiac electrophysiology while Nav1.5 is clearly a major player regulating cardiac excitability.
As mentioned above, SCN5A mutations were associated with a large variety of different arrhythmias.19,20,21,22,23,24,25,26,27,30,34 Given these variations, it is hard to predict what disorder will be triggered by a specific SCN5A mutation. From studies on SSS and Lenègre Lev disease24,25 or Brugada syndrome21 one would rather expect that a loss-of-function is associated with the sinoatrial dysfunction or slowing of conductivity. Consistently, the genetic impairment of SCN5A in the porcine heart results in AVB35 and overexpression of SCN5A in the mouse heart causes shortened PR intervals.36 However, LQT3 patients with gain-of-function mutations in SCN5A often show sinoatrial bradycardia,27,37,38 mice carrying LQT3 mutations have decreased heart rates and slowed AV conduction39,40 and for LQT3 patients with the ΔKPQ SCN5A gain-of-function mutation an altered atrial and AV conduction was observed.41 Thus, similar as for sodium channel mutations in epilepsy, it appears that both, a gain or a loss-of-function, can cause AVB.42,43 In epilepsy, this scenario can be explained by the fact that a hyperpolarization can lead to decreased excitability, which is however also observed for depolarization, due to increased refractoriness of sodium channels. Moreover, other factors contribute to the manifestation of the phenotype i.e., for the SCN5A haploinsufficiency aging and fibrosis seem to play a major role in the development of the Lenègre-Lev conduction disorder.24 In addition, the diversity of cardiac sodium channel disorders might also result from the fact that depending on the nature or localization of the mutation, the modulation by different sodium channel subunits might be affected. Conversely, a mutation in a sodium channel subunit might cause a specific or novel phenotype, like in the case of POPDC1S201F or POPDC2W188∗, where a mixed dysregulation of TREK-1 and/or sodium channels might primarily affect the conduction system.
Despite that our study clearly indicates an effect of POPDC proteins on sodium channels, there are several limitations to our study and the current understanding of POPDC-mediated channel regulation and trafficking. POPDC proteins do not contain a classical conserved retention or retrieval signal and changes in ion channel trafficking appear to depend on the close interaction with the ion channel, especially as POPDC proteins increase TREK-1 surface expression while at the same time they decrease Nav1.5 membrane expression, both in a cAMP-dependent manner. In addition, we do not know the mechanism of how under high cAMP levels Nav1.5 is less efficiently transported to the plasma membrane or where the “non-trafficked” Nav1.5 channels primarily reside. One putative explanation might be that only POPDC proteins in a cAMP unbound state can assist in the last steps of Nav1.5 trafficking to the plasma membrane. As only a minor fraction of Nav1.5 channels are localized at the surface membrane, the large cytosolic pool of Nav1.5 channels appears apparently unchanged. Yet another point is that our screen for POPDC-regulated ion channels identified Nav1.5, while it might have missed other channels or those channel modulations that cannot be identified with a single voltage step protocol that we used for the initial screening. In addition, voltage-clamp recordings of oocytes are not as fast in clamp speed as patch-clamp recordings of transfected cells and oocytes do not fully reflect physiological conditions, for instance the lower temperatures compared to that in humans may influence protein stability and protein trafficking. As POPDCs strongly reduce the amplitudes of the Nav1.5 currents, wild-type channels have to be expressed at a high level, which cannot be fully clamped, so that one can also record currents from the strongly suppressed Nav1.5/POPDC complex. The insufficient clamping of the large Nav1.5 current in controls might cause that the suppressive effect of POPDCs on Nav1.5 is not fully captured and might be even underestimated. These clamping artifacts can be bypassed for the studies of the kinetics by analyzing only small currents and here, given the same limitations in clamp speed, we did not observe any significant differences between Nav1.5 alone or co-expressed with POPDCs. Note, that the oocyte expression system provided very important advantages for our study. For instance, one can express precise amounts and ratios of the channel and its subunits in each given oocyte. In contrast, transfected cells would uptake a variable amount of cDNA and not always in a fixed Nav1.5 to POPDC stoichiometry. In the oocyte system, each cell receives a precise amount of injected cRNA for all constructs, and there are no problems arising from variations in the series resistance, as in patch-clamp recordings. Therefore, voltage-clamp experiments in oocytes can detect even minor changes in current amplitudes, which is relatively difficult or not possible in patch-clamp recordings of mammalian cells. Despite the methodological limitations, we think that our data clearly indicates that POPDCs primarily reduce current amplitudes of Nav1.5 by a trafficking effect without changing the kinetics of the channels. Future studies are necessary to address, to which extent a putative sodium channel dysregulation contributes to the phenotypes observed in the POPDC1 and POPDC2 knock-out animals or the human diseases associated with POPDC1S201F and POPDC2W188∗, especially as POPDC proteins might be involved with other scaffolding proteins in a multi-channel regulation.
Similarly, as for TREK-1, Nav1.5 modulation by POPDC proteins is cAMP-dependent,13 since the reduction of Nav1.5 currents and surface expression was not observed when theophylline was applied to increase cAMP levels. These results are in line with previous findings, where increased cAMP levels, by direct cAMP application, phosphodiesterase inhibition and stimulation of adenylate cyclases resulted in a 30% increase of INa in canine cardiomyocytes.29 Mechanistically it was discussed, that the INa increase may be due to increased channel trafficking29,44 and our data in oocytes, showing that POPDC2 reduces the expression of Nav1.5 at the plasma membrane, strengthen this suggested mechanism. Thus, the cAMP-dependent modulation of sodium channels by POPDC proteins might contribute to this effect and result in increased sodium currents under conditions of sympathetic stimulation.
Summarizing, we propose that POPDC proteins are cAMP-dependent modulators of cardiac sodium channels, which provides a novel mechanism to regulate sodium channel densities and thus cardiac excitability under physiological conditions and in arrhythmias. Moreover, POPDC proteins are also promising to be studied in the pathogenesis of other types of congenital disorders related to sodium channel dysfunction.
Limitations of the study
This study was performed utilizing heterologous expression systems and an in situ embryonic zebrafish heart model and may therefore not fully resemble the situation in the human heart.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Nav1.5 (SCN5A) antibody (493-511) | Alomone | Cat#ASC-005; RRID: AB_2040001 |
| Anti-POPDC2 antibody | Sigma | Cat#HPA024255; RRID: AB_1855545 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, HRP | ThermoFisher Scientific | Cat#31460 |
| Chemicals, peptides, and recombinant proteins | ||
| Lipofectamine2000 | Invitrogen | Cat#11668027 |
| Dynabeads protein A magnetic beads | Thermo Scientific | Cat#10001D |
| ProLongDiamond | Thermo Scientific | Cat#P36961 |
| jetPRIME | Peqlab | Cat#101000027 |
| SuperSignal ELISA femto substrate | Thermo Scientific | Cat#37075 |
| Critical commercial assays | ||
| mMessage mMACHINE T7 transcription kit | Invitrogen | Cat#AM1344 |
| Pierce HA-Tag IP/Co-IP Kit | Thermo Scientific | Cat#26180 |
| Experimental models: Cell lines | ||
| COS-7 | DSMZ | Cat#ACC60; RRID: CVCL_0224 |
| HeLa | DSMZ | Cat#ACC57 |
| Experimental models: Organisms/strains | ||
| Xenopus laevis toads | Nasco | N/A |
| C57BL/6NCrl mouse | Charles River | N/A |
| AB zebrafish embryos | In house breeding | N/A |
| Oligonucleotides | ||
| 5′-CTTAATCTGGAATTAAACAGGAGAA-3′ | GeneTools | N/A |
| 5’-CCTCTTACCTCAGTTACAATTTATA-3’ | GeneTools | N/A |
| Software and algorithms | ||
| pClamp 10 | Molecular Devices | https://support.moleculardevices.com/s/article/Axon-pCLAMP-10-Electrophysiology-Data-Acquisition-Analysis-Software-Download-Page |
| OriginPro 2016 | OriginLab | https://www.originlab.com/2016 |
| Excel 2019 | Microsoft | N/A |
| Other | ||
| NanoDrop 2000 UV-Vis spectrophotometer | Thermo Scientific | N/A |
| TurboTec 10CD amplifier | npi | N/A |
| DMZ-Universal puller | Zeitz | N/A |
| STELLARIS confocal microscope | Leica Microsystems | N/A |
| GloMax 20/20 luminometer | Promega | N/A |
| Zeiss Examiner.D1 microscope | Carl Zeiss Microscopy | N/A |
| Multiclamp 700B amplifier | Molecular Devices | N/A |
| Digidata 1440A digitizer | Molecular Devices | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Niels Decher (decher@staff.uni-marburg.de).
Materials availability
Materials are available upon request.
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
No original code was reported in this study.
-
•
Any additional information related to this study will be fulfilled by the lead contact upon request.
Experimental model and study participant details
Cell lines
Authenticated COS-7 and HeLa cells were commercially purchased from the Leibniz Institute (“Deutsche Sammlung für Mikroorganismen und Zellkulturen” (DSMZ)). COS-7 cells were cultivated at 37°C and 5% CO2 in DMEM medium (Invitrogen) supplemented with 10% (v/v) FCS and 1% (v/v) Penicillin/Streptomycin solution (Invitrogen). One day before transfection, cells were grown on 60 mm plastic petri dishes (BD). HeLa cells were cultivated at 37°C and 5% CO2 in DMEM medium (Invitrogen) supplemented with 10% FCS (v/v) and 1% Penicillin/Streptomycin solution (v/v) (Invitrogen). One day before transfection, cells were grown on 35 mm plastic petri dishes (NUNC). The cell lines were tested for mycoplasma contamination routinely. COS-7 cells are derived from a male African green monkey, whereas HeLa cells are derived from a female human. These standard cell lines are used throughout the scientific community and therefore provide highly comparable data. Most importantly, the resulting data of these standard immortalized cell lines are transferrable to both sexes, in particular as the function of the expressed ion channels and their direct pharmacological modulation are also not sex specific.
Xenopus laevis toads
Oocytes were taken from ovarian lobes of anesthetized female Xenopus laevis toads (Nasco, Wisconsin, USA), anaesthetized with 2 g/l tricaine-methansulfonate (SIGMA, Taufkirchen, Germany). Xenopus laevis oocytes were utilized as a heterologous expression system in this study, and the isolation of oocytes is by nature only possible from female frogs. Nevertheless, the resulting data is transferrable to both sexes, considering that the function of the expressed ion channels and their direct pharmacological modulation are not sex specific.
Zebrafish embryos
Embryos were staged by hours postfertilization (hpf) at 28.5°C.45 Morpholinos were obtained from Gene Tools, resuspended in distilled H2O and independent of the sex of the embryo around 2 nl were injected into 1-cell stage embryos. The following morpholino was used: popdc2 MO1 (splice-acceptor site blocking of exon 2) 5′-CTTAATCTGGAATTAAACAGGAGAA-3′) at 1 ng/embryo.6 An equal amount of the standard control MO: 5’-CCTCTTACCTCAGTTACAATTTATA-3’ was used in each experiment serving as a control.
Ethics approval and consent to participate
The studies did not involve human participants, human data or human tissue. Xenopus laevis experiments were approved by the local ethics commission of the Regierungspräsidium Gießen (V54-19c 20 15 h 02 MR 20/28 Nr.A 4/2013). All animal housing and husbandry were performed under standard conditions in accordance with institutional (Philipps University of Marburg) and national ethical and animal welfare guidelines approved by the ethics committee for animal experiments at the Regierungspräsidium Gießen, Germany. All animal procedures performed conformed to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes and the current NIH guidelines.
Method details
Oocyte preparation, cRNA synthesis and injection
Oocytes were obtained from anesthetized Xenopus laevis toads and incubated in an OR2 solution containing in mM: NaCl 82.5, KCl 2, MgCl2 1, HEPES 5 (pH 7.5) substituted with 2 mg/ml collagenase II (Sigma) to remove residual connective tissue. Subsequently, oocytes were stored at 18°C in ND96 solution supplemented with 50 mg/l gentamycine, 274 mg/l sodium pyruvate and as indicated with or without 88 mg/l theophylline. cRNA was synthesized with the mMESSAGE mMACHINE-Kit (Ambion) following the manufacturer’s instructions. Quality of the cRNA was tested using gel electrophoresis and cRNA was quantified with the NanoDrop 2000 UV-Vis spectrophotometer (Thermo Scientific). Oocytes were each injected with 50 nl of cRNA. Oocytes injected with Na+/K+ ATPase were incubated for 4 h before recording in a K+ free solution containing (in mM) NaCl 96, CaCl2 1.8, MgCl2 1, HEPES 5 and sucrose 25, pH 7.4 (NaOH). To screen for cardiac ion channels modulated by POPDC2 the following cRNA amounts were injected and current amplitudes were analyzed at the respective voltages:
-3 ng TASK-1 versus 3 ng TASK-1 plus 2.5 ng POPDC2 (+40 mV),
-2.5 ng TRAAK versus 2.5 ng TRAAK plus 2.5 ng POPDC2 (+40 mV),
-20 ng TASK-4 versus 20 ng TASK-4 plus 2.5 ng POPDC2 (+40 mV),
-10 ng HCN2 versus 10 ng HCN2 plus 2.5 ng POPDC2 (-120 mV),
-25 ng HCN4 versus 25 ng HCN4 plus 2.5 ng POPDC2 (-120 mV),
-0.5 ng Kv4.3 versus 0.5 ng Kv4.3 plus 0.5 ng POPDC2 (+40 mV),
-0.05 ng Kv1.4 versus 0.05 ng Kv1.4 and 0.05 ng POPDC2 (+40 mV),
-15 ng KCNQ1 versus 15 ng KCNQ1 plus 2.5 ng POPDC2 (+40 mV),
-endogenous Na+/K+ ATPase versus endogenous Na+/K+ ATPase plus 5 ng POPDC2 (potassium induced current at -30 mV),
-10 ng Nav1.5 versus 10 ng Nav1.5 plus 2.5 ng POPDC2 (-20 mV).
Two-electrode voltage-clamp recordings in oocytes
The voltage-clamp recordings were performed with a TurboTEC 10CD (npi) amplifier and a Digidata 1200 Series (Axon Instruments) as A/D converter at room temperature (20 - 22°C). Micropipettes were made from borosilicate glass capillaries GB 150TF-8P (Science Products) and pulled with a DMZ-Universal Puller (Zeitz). The resistance of the recording pipettes was 0.5-1.5 MΩ when pipettes were filled with 3 M KCl solution. The recording solution (ND96) contained in mM: NaCl 96, KCl 2, CaCl2 1.8, MgCl2 1, HEPES 5 (pH 7.5). Oocytes injected with Na+/K+ ATPase were recorded with K+ free solution containing (in mM) NaCl 96, CaCl2 1.8, MgCl2 1, HEPES 5, sucrose 10 and BaCl2 5, pH 7.4 (NaOH) for inhibition of K+ channels. Subsequently 10 mM sucrose was replaced by KCl in the continuous presence of BaCl2. Data were acquired with Clampex 10 (Molecular Devices) and analyzed with Clampfit 10 (Molecular Devices) and OriginPro 2016 (OriginLab Corporation). All electrophysiological experiments were performed from at least three independent injections. For all Nav1.5 and Nav1.4 measurements the currents were recorded with a P/N (N=4) voltage-step protocol, with a negative pre-pulse executed form the holding potential before the test pulse.
Co-immunoprecipitation and Western blot
For CoIP analysis, COS-7 cells were transiently transfected with FLAG-tagged wild-type POPDC2 (POPDC2-WT) or the empty pcDNA3.1 (+) vector together with HA-tagged Nav1.5 using Lipofectamine2000 reagent (Invitrogen). Cells were harvested 24 h after transfection using a buffer containing 50 mM Tris (pH 8.0), 150 mM NaCl, 2 mM EDTA, 1% (v/v) Triton X-100, 0.25% gelatin, and protease inhibitors (Roche). Cell lysates were briefly sonicated and cleared twice by centrifugation at >18,000 g at 4°C for 30 min. A sample was taken as input control and the rest of the lysates was subjected to CoIP using the Pierce HA Tag IP/Co-IP Kit (Thermo Scientific) following manufacturer’s instructions. Proteins were size-separated and transferred onto nitrocellulose membrane (BioTrace NT, Pall). The membrane was washed with TBST (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.1% Tween-20) and blocked in 5% (w/v) low-fat milk in TBST for 1 h at room temperature and subsequently incubated with anti-FLAG antibody (Sigma-Aldrich, 1:1000 dilution) over night at 4°C. After three washes with TBST, the blots were incubated for 1 h at room temperature with horseradish peroxidase-coupled anti-mouse antibody (Vector Laboratories 1:1000 dilution). After washing, signals were detected using an enhanced chemiluminescence protein detection method (GE Healthcare). Exposed films were digitalized.
For co-immunoprecipitation experiments in mouse heart, euthanasia of the mouse was performed by isoflurane anesthesia followed by cervical dislocation. Cardiac tissue was isolated and subsequently washed and cut into small pieces in ice-cold PBS containing protease inhibitor cocktail. Tissue was pestled in liquid nitrogen, homogenized with a hollow needle in RIPA buffer containing in mM Tris-Base 50, NaCl 150 pH 7.4 with NaOH, 1% NP40 and 0.25% sodium deoxycholate supplemented with protease inhibitor cocktail and incubated on ice for 30 min. Finally, lysate was cleared by centrifugation for 30 min at 13,000 rpm at 4°C. For CoIP Dynabeads protein A magnetic beads (Thermo Fisher Scientific) were used following manufacturer’s instructions. Anti-Nav1.5 (SCN5A) antibody (493-511, Alomone) was used for precipitation and Western Blots were stained with anti-Nav1.5 (SCN5A) (493-511, Alomone, 1:1000) or anti-POPDC2 (Sigma, HPA 024255, 1:500) antibodies and peroxidase-conjugated anti-rabbit secondary antibody (Thermo, 31460, 1:8000).
Confocal fluorescence microscopy
HeLa cells grown on cover slips were fixed in 4% paraformaldehyde/4% sucrose in PBS++ for 15 min. Afterwards, cells were permeabilized with 0.1% Triton-X-100 for 10 min or not permeabilized followed by blocking in 5% BSA/ PBS++ for 1 h. Immunostaining was performed with the indicated primary antibodies in blocking reagent for 2 h or overnight. Secondary antibodies labelled with the indicated Alexa Fluor dyes were applied in PBS++ for 1 h. Nuclei were stained with Hoechst 33342. The cells were washed with PBS++ and mounted with ProLongDiamond (Thermo Fisher). Confocal images were acquired on a Leica STELLARIS equipped with a 93x glycerol planapochromat objective (Leica Microsystems).
Chemiluminescence assay in HeLa cells – quantification of surface expression
For the quantification of Nav1.5 expression at the plasma membrane, three copies of the hemagglutinin (HA) epitope were inserted at amino acid position 304 in the extracellular S5-S6 loop of domain I of Nav1.5. HeLa cells were transfected with the indicated constructs using jetPRIME (Peqlab). After 48 h, cells were fixed with 4% PFA (w/v) (in PBS), washed three times with PBS and blocked with 10% normal goat serum (v/v) (in PBS). Cells were stained with a monoclonal anti-HA primary antibody (HA-probe (F-7), Santa Cruz, dilution 1:100) and washed 4 times for 15 minutes with PBS. As a secondary antibody a horseradish-peroxidase (HRP)-conjugated antibody (goat anti-mouse IgG-HRP, Santa Cruz, dilution 1:5000) was used. After several washing steps with PBS (6 times for 20 minutes), surface expression was measured as relative light units (RLUs) in a luminometer (GloMax 20/20, Promega) using a luminogenic substrate (SuperSignal ELISA Femto, Thermo Scientific).
Whole cell patch clamp experiments in embryonic zebrafish hearts
Embryonic zebrafish hearts were dissected and measured by whole cell patch clamp technique as previously described.46 Briefly summarized, zebrafish embryos (48 hpf) were anesthetized with 0.02% tricaine for 1 to 2 min and dechorionated. Subsequently, the whole heart was separated from the thorax using fine forceps. Only spontaneously beating whole hearts were transferred to the recording chamber and studied at 22–24°C. The recording chamber was mounted on a Zeiss Examiner.D1 microscope (Carl Zeiss Microscopy, LLC, United States) with a 5 × /0.12 objective and a Zeiss AxioCam MRm camera (Carl Zeiss Microscopy, LLC, United States) and was perfused with solution containing 140 mM NaCl, 4 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 10 mM HEPES (pH 7.5). Patch pipettes were prepared from borosilicate glass capillaries GB 150TF- 8P (Science Products, Hofheim, Germany) using DMZ-Universal Puller (Zeitz, Martinsried, Germany) with tip resistances between 3 and 5 MΩ when filled with 120 mM KCl, 5 mM EGTA, 5 mM K2ATP, 10 mM HEPES, and 5 mM MgCl2 (pH 7.2). Membrane potential (Vm) was measured using a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale CA, USA) and Clampex 10.0 software (pClamp10, Molecular Devices, Sunnyvale CA, USA). Data were digitized at 10 kHz with a Digidata 1440A digitizer (Molecular Devices, Sunnyvale CA, USA), filtered at 10 kHz and electrode capacitance was compensated. After the positioning of the pipette adjacent to the atrium, a seal was reached by application of suction. Spontaneously generated atrial APs were recorded. AP parameters were analyzed manually from a series of five APs using Clampfit 10.0 software (pClamp10, Molecular Devices, Sunnyvale CA, USA), whereas the beating frequencies were analyzed over a time period of 30 s.
Quantification and statistical analysis
Sample or group sizes were not predetermined and the numbers of necessary experiments were estimated on the basis of previous experiments/literature in the field. In addition, no exclusion criteria were pre-established, and no data were excluded from the subsequent analysis. For the experiments, no randomization or blinding was performed and normal distribution was assumed. An unpaired two-sided Student's t-test was used to probe the significance. All data are presented as mean ± s.e.m.. Statistical analysis were performed using Microsoft Excel. The number of technical replicates (n) or biological replicates (N) is illustrated in the respective graphs. Significances are indicated with ∗, p<0.05; ∗∗, p<0.01; ∗∗∗, p<0.001 in the Figures.
Acknowledgments
This work was supported by a grant of the Universitätsklinikum Gießen und Marburg to S.R. and A.K.K. Funding: European Union (grant agreement No 965286 [MAESTRIA] to LF). Work in the Brand lab was funded by the British Heart Foundation (PG/14/46/30911, PG/14/83/31128). N.D. is supported by Deutsche Forschungsgemeinschaft grant DE1482-9/1. We thank Ursula Herbort-Brand and Simone Preis for expert technical assistance.
Author contributions
S.R., B.O.B. and T.C. carried out electrophysiological experiments. R.F.R.S, S.R. and M.K.-H.S performed the co-immunoprecipitation experiments. S.R. quantified the surface expression of Nav1.5, worked on the data analysis and figures. A.K.K. performed patch-clamp experiments of embryonic zebrafish hearts and worked on the data analyses and figures. L.F. helped in the discussion and writing of the paper. R.J. designed immunohistochemistry experiments, supervised M.K. and E.D.M. and helped writing the article. T.B. designed experiments, supervised R.F.R.S. and helped writing the article. C.H. helped designing the morpholino knock-down experiments in zebrafish. S.F. performed morpholino knock-down experiments in zebrafish, N.D. designed and coordinated the study. S.R. and N.D. drafted the article and all authors read and approved the final article.
Declaration of interests
L.F. has received institutional research grants and non-financial support from European Union, DFG, British Heart Foundation, Medical Research Council (UK), NIHR, and several biomedical companies. L.F. is listed as inventor on two patents held by the academic employer (Atrial Fibrillation Therapy WO 2015140571, Markers for Atrial Fibrillation WO 2016012783).
Published: April 9, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109696.
Supplemental information
References
- 1.Knight R.F., Bader D.M., Backstrom J.R. Membrane topology of Bves/Pop1A, a cell adhesion molecule that displays dynamic changes in cellular distribution during development. J. Biol. Chem. 2003;278:32872–32879. doi: 10.1074/jbc.M301961200. [DOI] [PubMed] [Google Scholar]
- 2.Andrée B., Hillemann T., Kessler-Icekson G., Schmitt-John T., Jockusch H., Arnold H.H., Brand T. Isolation and characterization of the novel popeye gene family expressed in skeletal muscle and heart. Dev. Biol. 2000;223:371–382. doi: 10.1006/dbio.2000.9751. [DOI] [PubMed] [Google Scholar]
- 3.Reese D.E., Zavaljevski M., Streiff N.L., Bader D. bves: A novel gene expressed during coronary blood vessel development. Dev. Biol. 1999;209:159–171. doi: 10.1006/dbio.1999.9246. [DOI] [PubMed] [Google Scholar]
- 4.Froese A., Breher S.S., Waldeyer C., Schindler R.F.R., Nikolaev V.O., Rinné S., Wischmeyer E., Schlueter J., Becher J., Simrick S., et al. Popeye domain containing proteins are essential for stress-mediated modulation of cardiac pacemaking in mice. J. Clin. Invest. 2012;122:1119–1130. doi: 10.1172/JCI59410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simrick S., Schindler R.F., Poon K.L., Brand T. Popeye domain-containing proteins and stress-mediated modulation of cardiac pacemaking. Trends Cardiovasc. Med. 2013;23:257–263. doi: 10.1016/j.tcm.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirchmaier B.C., Poon K.L., Schwerte T., Huisken J., Winkler C., Jungblut B., Stainier D.Y., Brand T. The Popeye domain containing 2 (popdc2) gene in zebrafish is required for heart and skeletal muscle development. Dev. Biol. 2012;363:438–450. doi: 10.1016/j.ydbio.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schindler R.F., Scotton C., French V., Ferlini A., Brand T. The Popeye Domain Containing Genes and their Function in Striated Muscle. J. Cardiovasc. Dev. Dis. 2016;3:22. doi: 10.3390/jcdd3020022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vissing J., Johnson K., Töpf A., Nafissi S., Díaz-Manera J., French V.M., Schindler R.F., Sarathchandra P., Løkken N., Rinné S., et al. POPDC3 Gene Variants Associate with a New Form of Limb Girdle Muscular Dystrophy. Ann. Neurol. 2019;86:832–843. doi: 10.1002/ana.25620. [DOI] [PubMed] [Google Scholar]
- 9.Rinné S., Ortiz-Bonnin B., Stallmeyer B., Kiper A.K., Fortmüller L., Schindler R.F.R., Herbort-Brand U., Kabir N.S., Dittmann S., Friedrich C., et al. POPDC2 a novel susceptibility gene for conduction disorders. J. Mol. Cell. Cardiol. 2020;145:74–83. doi: 10.1016/j.yjmcc.2020.06.005. [DOI] [PubMed] [Google Scholar]
- 10.De Ridder W., Nelson I., Asselbergh B., De Paepe B., Beuvin M., Ben Yaou R., Masson C., Boland A., Deleuze J.F., Maisonobe T., et al. Muscular dystrophy with arrhythmia caused by loss-of-function mutations in BVES. Neurol. Genet. 2019;5 doi: 10.1212/NXG.0000000000000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osler M.E., Chang M.S., Bader D.M. Bves modulates epithelial integrity through an interaction at the tight junction. J. Cell Sci. 2005;118:4667–4678. doi: 10.1242/jcs.02588. [DOI] [PubMed] [Google Scholar]
- 12.Wu Y.C., Liu C.Y., Chen Y.H., Chen R.F., Huang C.J., Wang I.J. Blood vessel epicardial substance (Bves) regulates epidermal tight junction integrity through atypical protein kinase C. J. Biol. Chem. 2012;287:39887–39897. doi: 10.1074/jbc.M112.372078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schindler R.F.R., Scotton C., Zhang J., Passarelli C., Ortiz-Bonnin B., Simrick S., Schwerte T., Poon K.L., Fang M., Rinné S., et al. POPDC1S201F causes muscular dystrophy and arrhythmia by affecting protein trafficking. J. Clin. Invest. 2016;126:239–253. doi: 10.1172/JCI79562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith T.K., Hager H.A., Francis R., Kilkenny D.M., Lo C.W., Bader D.M. Bves directly interacts with GEFT, and controls cell shape and movement through regulation of Rac1/Cdc42 activity. Proc. Natl. Acad. Sci. USA. 2008;105:8298–8303. doi: 10.1073/pnas.0802345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hager H.A., Roberts R.J., Cross E.E., Proux-Gillardeaux V., Bader D.M. Identification of a novel Bves function: regulation of vesicular transport. EMBO J. 2010;29:532–545. doi: 10.1038/emboj.2009.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alcalay Y., Hochhauser E., Kliminski V., Dick J., Zahalka M.A., Parnes D., Schlesinger H., Abassi Z., Shainberg A., Schindler R.F.R., et al. Popeye domain containing 1 (Popdc1/Bves) is a caveolae-associated protein involved in ischemia tolerance. PLoS One. 2013;8 doi: 10.1371/journal.pone.0071100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tibbo A.J., Mika D., Dobi S., Ling J., McFall A., Tejeda G.S., Blair C., MacLeod R., MacQuaide N., Gök C., et al. Phosphodiesterase type 4 anchoring regulates cAMP signaling to Popeye domain-containing proteins. J. Mol. Cell. Cardiol. 2022;165:86–102. doi: 10.1016/j.yjmcc.2022.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baldwin T.A., Li Y., Marsden A.N., Rinné S., Garza-Carbajal A., Schindler R.F.R., Zhang M., Garcia M.A., Venna V.R., Decher N., et al. POPDC1 scaffolds a complex of adenylyl cyclase 9 and the potassium channel TREK-1 in heart. EMBO Rep. 2022;23 doi: 10.15252/embr.202255208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennett P.B., Yazawa K., Makita N., George A.L., Jr. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- 20.Wang Q., Shen J., Splawski I., Atkinson D., Li Z., Robinson J.L., Moss A.J., Towbin J.A., Keating M.T. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 21.Chen Q., Kirsch G.E., Zhang D., Brugada R., Brugada J., Brugada P., Potenza D., Moya A., Borggrefe M., Breithardt G., et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 22.Akai J., Makita N., Sakurada H., Shirai N., Ueda K., Kitabatake A., Nakazawa K., Kimura A., Hiraoka M. A novel SCN5A mutation associated with idiopathic ventricular fibrillation without typical ECG findings of Brugada syndrome. FEBS Lett. 2000;479:29–34. doi: 10.1016/s0014-5793(00)01875-5. [DOI] [PubMed] [Google Scholar]
- 23.Olson T.M., Michels V.V., Ballew J.D., Reyna S.P., Karst M.L., Herron K.J., Horton S.C., Rodeheffer R.J., Anderson J.L. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293:447–454. doi: 10.1001/jama.293.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Probst V., Kyndt F., Potet F., Trochu J.N., Mialet G., Demolombe S., Schott J.J., Baró I., Escande D., Le Marec H. Haploinsufficiency in combination with aging causes SCN5A-linked hereditary Lenegre disease. J. Am. Coll. Cardiol. 2003;41:643–652. doi: 10.1016/s0735-1097(02)02864-4. [DOI] [PubMed] [Google Scholar]
- 25.Benson D.W., Wang D.W., Dyment M., Knilans T.K., Fish F.A., Strieper M.J., Rhodes T.H., George A.L., Jr. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A) J. Clin. Invest. 2003;112:1019–1028. doi: 10.1172/JCI18062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bezzina C., Veldkamp M.W., van Den Berg M.P., Postma A.V., Rook M.B., Viersma J.W., van Langen I.M., Tan-Sindhunata G., Bink-Boelkens M.T., van Der Hout A.H., et al. A single Na+ channel mutation causing both long-QT and Brugada syndromes. Circ. Res. 1999;85:1206–1213. doi: 10.1161/01.res.85.12.1206. [DOI] [PubMed] [Google Scholar]
- 27.Veldkamp M.W., Wilders R., Baartscheer A., Zegers J.G., Bezzina C.R., Wilde A.A.M. Contribution of sodium channel mutations to bradycardia and sinus node dysfunction in LQT3 families. Circ. Res. 2003;92:976–983. doi: 10.1161/01.RES.0000069689.09869.A8. [DOI] [PubMed] [Google Scholar]
- 28.Pott A., Bock S., Berger I.M., Frese K., Dahme T., Keßler M., Rinné S., Decher N., Just S., Rottbauer W. Mutation of the Na+/K+-ATPase Atp1a1a.1 causes QT interval prolongation and bradycardia in zebrafish. J. Mol. Cell. Cardiol. 2018;120:42–52. doi: 10.1016/j.yjmcc.2018.05.005. [DOI] [PubMed] [Google Scholar]
- 29.Baba S., Dun W., Boyden P.A. Can PKA activators rescue Na+ channel function in epicardial border zone cells that survive in the infarcted canine heart? Cardiovasc. Res. 2004;64:260–267. doi: 10.1016/j.cardiores.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 30.Wang D.W., Desai R.R., Crotti L., Arnestad M., Insolia R., Pedrazzini M., Ferrandi C., Vege A., Rognum T., Schwartz P.J., George A.L., Jr. Cardiac sodium channel dysfunction in sudden infant death syndrome. Circulation. 2007;115:368–376. doi: 10.1161/CIRCULATIONAHA.106.646513. [DOI] [PubMed] [Google Scholar]
- 31.Olesen M.S., Yuan L., Liang B., Holst A.G., Nielsen N., Nielsen J.B., Hedley P.L., Christiansen M., Olesen S.P., Haunsø S., et al. High prevalence of long QT syndrome-associated SCN5A variants in patients with early-onset lone atrial fibrillation. Circ. Cardiovasc. Genet. 2012;5:450–459. doi: 10.1161/CIRCGENETICS.111.962597. [DOI] [PubMed] [Google Scholar]
- 32.Brand T., Schindler R. New kids on the block: The Popeye domain containing (POPDC) protein family acting as a novel class of cAMP effector proteins in striated muscle. Cell. Signal. 2017;40:156–165. doi: 10.1016/j.cellsig.2017.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holt I., Fuller H.R., Schindler R.F.R., Shirran S.L., Brand T., Morris G.E. An interaction of heart disease-associated proteins POPDC1/2 with XIRP1 in transverse tubules and intercalated discs. BMC Mol. Cell Biol. 2020;21:88. doi: 10.1186/s12860-020-00329-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marangoni S., Di Resta C., Rocchetti M., Barile L., Rizzetto R., Summa A., Severi S., Sommariva E., Pappone C., Ferrari M., et al. A Brugada syndrome mutation (p.S216L) and its modulation by p.H558R polymorphism: standard and dynamic characterization. Cardiovasc. Res. 2011;91:606–616. doi: 10.1093/cvr/cvr142. [DOI] [PubMed] [Google Scholar]
- 35.Park D.S., Cerrone M., Morley G., Vasquez C., Fowler S., Liu N., Bernstein S.A., Liu F.Y., Zhang J., Rogers C.S., et al. Genetically engineered SCN5A mutant pig hearts exhibit conduction defects and arrhythmias. J. Clin. Invest. 2015;125:403–412. doi: 10.1172/JCI76919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu G.X., Remme C.A., Boukens B.J., Belardinelli L., Rajamani S. Overexpression of SCN5A in mouse heart mimics human syndrome of enhanced atrioventricular nodal conduction. Heart Rhythm. 2015;12:1036–1045. doi: 10.1016/j.hrthm.2015.01.029. [DOI] [PubMed] [Google Scholar]
- 37.Moss A.J., Zareba W., Benhorin J., Locati E.H., Hall W.J., Robinson J.L., Schwartz P.J., Towbin J.A., Vincent G.M., Lehmann M.H. ECG T-wave patterns in genetically distinct forms of the hereditary long QT syndrome. Circulation. 1995;92:2929–2934. doi: 10.1161/01.cir.92.10.2929. [DOI] [PubMed] [Google Scholar]
- 38.Bezzina C.R., Rook M.B., Wilde A.A. Cardiac sodium channel and inherited arrhythmia syndromes. Cardiovasc. Res. 2001;49:257–271. doi: 10.1016/s0008-6363(00)00272-8. [DOI] [PubMed] [Google Scholar]
- 39.Blana A., Kaese S., Fortmüller L., Laakmann S., Damke D., van Bragt K., Eckstein J., Piccini I., Kirchhefer U., Nattel S., et al. Knock-in gain-of-function sodium channel mutation prolongs atrial action potentials and alters atrial vulnerability. Heart Rhythm. 2010;7:1862–1869. doi: 10.1016/j.hrthm.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 40.Remme C.A., Verkerk A.O., Nuyens D., van Ginneken A.C.G., van Brunschot S., Belterman C.N.W., Wilders R., van Roon M.A., Tan H.L., Wilde A.A.M., et al. Overlap syndrome of cardiac sodium channel disease in mice carrying the equivalent mutation of human SCN5A-1795insD. Circulation. 2006;114:2584–2594. doi: 10.1161/CIRCULATIONAHA.106.653949. [DOI] [PubMed] [Google Scholar]
- 41.Zareba W., Sattari M.N., Rosero S., Couderc J.P., Moss A.J. Altered atrial, atrioventricular, and ventricular conduction in patients with the long QT syndrome caused by the DeltaKPQ SCN5A sodium channel gene mutation. Am. J. Cardiol. 2001;88:1311–1314. doi: 10.1016/s0002-9149(01)02097-5. [DOI] [PubMed] [Google Scholar]
- 42.George A.L., Jr. Inherited disorders of voltage-gated sodium channels. J. Clin. Invest. 2005;115:1990–1999. doi: 10.1172/JCI25505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamakawa K. Epilepsy and sodium channel gene mutations: gain or loss of function? Neuroreport. 2005;16:1–3. doi: 10.1097/00001756-200501190-00001. [DOI] [PubMed] [Google Scholar]
- 44.Hallaq H., Yang Z., Viswanathan P.C., Fukuda K., Shen W., Wang D.W., Wells K.S., Zhou J., Yi J., Murray K.T. Quantitation of protein kinase A-mediated trafficking of cardiac sodium channels in living cells. Cardiovasc. Res. 2006;72:250–261. doi: 10.1016/j.cardiores.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 45.Kimmel C.B., Ballard W.W., Kimmel S.R., Ullmann B., Schilling T.F. Stages of embryonic development of the zebrafish. Dev. Dynam. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 46.Arnaout R., Ferrer T., Huisken J., Spitzer K., Stainier D.Y.R., Tristani-Firouzi M., Chi N.C. Zebrafish model for human long QT syndrome. Proc. Natl. Acad. Sci. USA. 2007;104:11316–11321. doi: 10.1073/pnas.0702724104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
No original code was reported in this study.
-
•
Any additional information related to this study will be fulfilled by the lead contact upon request.