

Abstract
Background:
The human heart primarily metabolizes fatty acids and this decreases as alternative fuel use rises in heart failure with reduced ejection fraction (HFrEF). Patients with severe obesity and diabetes are thought to have increased myocardial fatty acid metabolism, but whether this is found in those who also have HF with preserved EF (HFpEF) is unknown.
Methods:
Plasma and endomyocardial biopsies were randomly selected from a two-center derived biobank of HFpEF (n=38), HFrEF (n=30) and non-failing donor control (n=20) tissue. Quantitative targeted metabolomics measured organic acids, amino acids, and acylcarnitines in myocardium (72 metabolites) and plasma (69 metabolites). The results were integrated with reported RNAseq data. Metabolomics were analyzed using agnostic clustering tools, Kruskal-Wallis test with Dunn’s test, and machine learning.
Results:
Agnostic clustering of myocardial but not plasma metabolites separated disease groups. Despite more obesity and diabetes in HFpEF vs. HFrEF (BMI 39.8 kg/m2 vs. 26.1 kg/m2, diabetes 70% vs. 30%, both p<0.0001), medium and long-chain acylcarnitines (mostly metabolites of fatty acid oxidation) were markedly lower in myocardium from both HF groups vs. control. In contrast, plasma levels were no different or higher than control. Gene expression linked to fatty acid metabolism was generally lower in HFpEF vs. control. Myocardial pyruvate was higher in HFpEF while the tricarboxylic acid cycle (TCA) intermediates succinate and fumarate were lower, as were several genes controlling glucose metabolism. Non-branched-chain and branched-chain amino acids (BCAA) were highest in HFpEF myocardium, yet downstream BCAA metabolites and genes controlling BCAA metabolism were lower. Ketone levels were higher in myocardium and plasma of HFrEF but not HFpEF patients. HFpEF metabolomics-derived subgroups showed few differences in BCAA metabolites but little else.
Conclusions:
Despite marked obesity and diabetes, HFpEF myocardium exhibited lower fatty acid metabolites compare HFrEF. Ketones and metabolites of the TCA cycle and BCAA were also lower in HFpEF, suggesting insufficient utilization of alternative fuels. These differences were not detectable in plasma and challenge conventional views of myocardial fuel utilization in HFpEF with marked diabetes and obesity and suggest substantial fuel inflexibility in this syndrome.
Keywords: HFpEF, human, metabolism, metabolomics, fatty acid oxidation, branched chain amino acid
Introduction
Heart failure with preserved ejection fraction (HFpEF) affects up to half the estimated 65 million heart failure patients worldwide, 1 with over 3 million people with HFpEF in the United States alone. 2 Clinical, hemodynamic, epidemiological, and blood-component features of this syndrome have been well described. However, much less is known about the pathobiology transpiring in the tissues of major organs involved. Furthermore, what is known stems largely from patients with HFpEF having severe hypertension, ventricular hypertrophy, and diastolic dysfunction. 3–6 While these co-morbidities remain relevant, they have become overshadowed by severe obesity, diabetes (DM) and metabolic syndrome. 7 The impact of this shift at the biological level remains little studied, although two recent studies found a significant impact of obesity on myocardial transcriptomics (notably on ATP synthesis pathways), 8 as well as on reducing calcium-activated myofilament function. 9
To date, most treatments tested for HFpEF have not been beneficial. The very recent exception are inhibitors of sodium glucose co-transporter 2 (SGLT2), 10, 11 drugs first developed for diabetes. The normal adult heart exhibits substantial metabolic substrate flexibility, primarily utilizing fatty acids (FA) but also capable of consuming glucose, ketones, lactate, and amino acids to generate ATP. 12 In HF with reduced EF (HFrEF), myocardial fuel utilization shifts away from FA to ketones and lactate, whereas glucose use may or may not increase. 12, 13 Direct myocardial data in humans with obesity/DM are lacking, but animal models have shown higher FA uptake and oxidation related in part to insulin resistance that lowers glucose metabolism.14 Whether this is also found in HFpEF patients with similarly severe obesity/DM is unknown. Metabolomics of peripheral blood has reported higher medium and long-chain FA (MLFA) in HFpEF, 15, 16, as is also found in HFrEF 15, 17, but as revealed by HFrEF, this does not guarantee elevation of myocardial levels. To address critical knowledge gaps, this study performed metabolomics of myocardial tissue and blood metabolomics obtained from patients with HFpEF, HFrEF, and non-failing controls.
Methods
Data availability
The majority of the data presented in this paper are provided in the Figures and Tables. The methods used for the metabolomic analysis are available in detail from the University of Pennsylvania Metabolomics Core. The raw metabolomics data coupled to de-identified metadata and the R script used to perform the analyses will be provided upon reasonable request.
HFpEF Study Population
Patients with HFpEF referred to the Johns Hopkins University HFpEF Clinic from 7/2016–10/2019 were screened for inclusion in this single center, cross-sectional study. Each patient provided informed consent and underwent research endomyocardial biopsy under a protocol approved by the Johns Hopkins Institutional Review Board. HFpEF diagnosis was based on consensus criteria18–20 (clinical signs and symptoms of heart failure with left ventricular ejection fraction [LVEF] ≥ 50%, and either 1) structural heart disease or diastolic dysfunction on echocardiography 21 with elevated N-terminal pro–B-type natriuretic peptide [NTproBNP] ≥ 125 pg/mL; or 2) hemodynamic evidence of elevated left sided filling pressures (left ventricular end diastolic pressure or pulmonary artery wedge pressure [PAWP] ≥ 15 mmHg at baseline; or ≥ 25 mmHg with exercise). Exclusion criteria are as previously described: history of LVEF <40%, severe valvular disease, infiltrative or restrictive cardiomyopathy (including cardiac amyloidosis on clinical histology), congenital heart disease, constrictive pericarditis, hypertrophic cardiomyopathy, or prior heart transplantation. 8 HFpEF patients were further pre-categorized into those with predominantly systolic hypertension and LV hypertrophy (HFpEF-Ht/Hp; n=15), those with more prominent obesity/DM (HFpEF-Met, n=13), and a group that mixed both features (Mixed; n=17) as previously described. 9
Myocardial tissue and plasma procurement and processing
Endomyocardial tissue from the right ventricular septum was obtained by a standard clinical bioptome procedure (Jawz Bioptome, Argon Medical, Frisco, TX) in patients with HFpEF (n=45) as described. 8 Participants with histologically confirmed cardiac amyloidosis (Congo Red) were excluded. Biopsies were rapidly immersed in liquid nitrogen and stored in liquid nitrogen until analyzed. Plasma obtained simultaneously was placed on ice and stored at −80°C until analyzed.
Right ventricular (RV) septum control tissue (n= 20) was obtained from unused hearts of organ donors with brain death determination and explanted hearts of patients with HFrEF undergoing cardiac transplantation (n=30). 8, 22 In both cases, hearts were removed under cardioplegic perfusion arrest, and rapidly placed in ice cold cardioplegia until dissected, aliquoted, and frozen within 2–4 hours of explant. These were provided in a collaboration with the University of Pennsylvania under an institutional review board-approved protocol. Plasma samples in these participants were obtained, processed, and frozen at the time of cardiac explant.
Targeted Metabolomics
The University of Pennsylvania Metabolomics Core performed the assay. Frozen human heart tissue was homogenized in 50% acidified acetonitrile and 100uL of human plasma was extracted with ice cold methanol. Targeted Liquid Chromatography/Mass Spectrometry metabolomics was performed using validated, optimized protocols as reported. 23, 24 The protocols arrest cellular metabolism using cold conditions and solvents to optimize the stability of metabolites. Classes of metabolites (acyl-carnitines, amino acids, organic acids) were separated using a unique High Performance Liquid Chromatography method to optimize resolution and sensitivity. Multiple reaction monitoring of calibration solutions and study subject samples were performed on an Agilent 1290 Infinity UHPLC/6495 triple quadrupole mass spectrometer for quantitation. 23, 24 Raw data were processed using Mass Hunter quantitative analysis software (Agilent). Calibration curves (R2 = 0.99 or greater) were fitted with a linear or a quadratic curve with a 1/X or 1/X2 weighting. Raw data were normalized to the starting tissue weight or plasma volume as appropriate.
Metabolomics Data Analysis
The raw metabolomic data were filtered, removing metabolites with ≥ 50% of values above or below the limit of quantitation in any disease group (control, HFpEF, HFrEF). This impacted 25 of 97 tissue metabolites (7 amino acids and 18 acylcarnitines) and 28 of 97 plasma metabolites (7 amino acids and 21 acylcarnitines). Tissue in which ≥ 45% of metabolites fell above or below the quantitation limit in heart tissue were removed from analysis (7 participants, all HFpEF), leaving 38 patients with HFpEF for the final analysis. Random forest imputation was used to derive values above or below the limit of quantitation using the R package missForest. 25 The imputed data were log-transformed for normalization prior to agnostic clustering. Agnostic clustering used Principal Component Analysis (PCA) with the R package ggbiplot, and top contributors to each Principal Component were identified. Hierarchical clustering was performed based on correlation of metabolites and displayed heatmaps using R package pheatmap. Tissue and plasma metabolites between the three disease groups were compared, using only raw concentration data for disease group comparisons (i.e. prior to imputation and log transformation), with a three-way Kruskal-Wallis test with Dunn test for pairwise comparisons. Benjamini-Hochberg method was used to adjust for multiple comparisons. Non-imputed, non-normalized concentration data were scaled for display in polar plots or boxplots generated using the R package ggplot2. Non-negative matrix factorization (R package NMF) was used to generate subgroups of HFpEF based on the myocardial tissue metabolome, 26 using scaled, imputed data. Two methods were used to determine the optimal number (k) of clusters; 1) Elbow Method using the sum of squared distance to choose the ideal number of groups; and 2) running the NMF algorithm 250 times for each k between 2 and 5 to determine which k yielded the best model performance. 27 Metabolites were identified that best identified each cluster. Clinical characteristics, tissue metabolites, and plasma metabolites were compared between NMF groups 1 and 2 using Wilcoxon test for continuous variables or Fisher’s exact test for categorical variables, and Benjamini-Hochberg method to adjust for multiple comparisons in the metabolite analysis.
Adjustment for differences in demographics and clinical comorbidities
Multivariable linear regression was performed to determine the relationship between each metabolite (myocardial and plasma) vs. disease group, adjusting for age, body mass index (BMI), diabetes, and self-identified race (African-American or non-African American). These clinical characteristics were chosen due to their significant differences between the disease groups. P values were adjusted for multiple comparisons using the Benjamini-Hochberg method.
Gene expression data
Volcano and box plots for gene expression specific to a given metabolic pathway were generated from previously reported transcriptomic data (RNAseq) from a very similar group of control (n=24), HFpEF (n=41), and HFrEF (n=30). 8 Eleven of the current study HFpEF patients were also in this prior group. Unbiased analysis of differential gene expression has been previously published and so is not reported here. 8 The R package DESeq2 28 determined differential gene expression between control and HFpEF as described. 8 Data are shown by adjusted P values (P-adj; Benjamini-Hochberg method) and log-2-transformed fold change. Genes were from published data or publicly available databases (Kyoto Encyclopedia of Genes and Genomes [KEGG] or Gene Ontology: Biological Processes [GO:BP]).
Clinical Data Analyses
Demographic, clinical, laboratory, hemodynamic, and echocardiographic data were compared by either Kruskal-Wallis/Dunn test or Wilcoxon test for continuous variables, and Fisher’s exact test for categorical variables. Clinical characteristics were compared between 1) HFrEF, HFpEF and control groups; 2) HFpEF included in metabolomics analysis vs. those excluded due to too many missing values; 3) HFpEF subgroups based on both pre-determined clinical groups (e.g. primarily hypertensive/LV hypertrophy, obesity-metabolic, or mixed, and using a de-novo machine learning algorithm to define metabolomics-subgroups; and 4) participants with HFpEF with or without corresponding transcriptomics from the prior study 8.
Results
Clinical Characteristics of Subject Groups
Baseline characteristics of control, HFpEF, and HFrEF are provided in Table 1. Compared with the other two groups, HFpEF patients were older (median [25th-75th percentile]; 65 years [56–69 years], p=0.0002), more frequently female (71%, p=0.004 for overall Kruskal-Wallis test), self-identified as African-American (53%, p=0.0006), and diabetic (74%, p<0.0001), with higher body mass index (BMI, 39.8 kg/m2 [30.5–43.4 kg/m2], p<0.0001). The seven HFpEF patients excluded from the study due to undetectable metabolites (likely inadequate tissue) had similar clinical features to those who were included (Table S1). The eleven HFpEF patients that overlapped with the prior transcriptomics analysis8 had no significant differences in clinical characteristics compared with those who did not overlap (Table S2)
Table 1.
Clinical characteristics of study participants.
| Control (N=20) |
HFpEF (N=38) |
HFrEF (N=30) |
Overall p value |
Control-HFpEF P value |
Control-HFrEF P value |
HFpEF-HFrEF P value |
|
|---|---|---|---|---|---|---|---|
| Sex | 0.0043 | 0.0272 | 0.7654 | 0.0031 | |||
| Female, n (%) | 8 (40%) | 27 (71.1%) | 10 (33.3%) | ||||
| Male, n (%) | 12 (60.0%) | 11 (28.9%) | 20 (66.7%) | ||||
| Self-identified Race/Ethnicity | 0.0006 | 0.0003 | 0.0613 | 0.039 | |||
| African-American, n (%) | 1 (5.0%) | 20 (52.6%) | 8 (26.7%) | ||||
| Caucasian, n (%) | 18 (90.0%) | 17 (44.7%) | 21 (70.0%) | ||||
| Hispanic, n (%) | 1 (5.0%) | 0 (0.0%) | 0 (0.0%) | ||||
| Other, n (%) | 0 (0.0%) | 1 (2.6%) | 1 (3.3%) | ||||
| Age, years | 58.5(53. 8, 63.0) | 64.5(56.2, 68.8) | 50(45.2, 61.8) | 0.0002 | 0.0692 | 0.0882 | <0.0001 |
| HF Hospitalization prior 12mos, n (%) | NA | 22 (59.5%) | NA | NA | NA | NA | NA |
| Medications | |||||||
| ACEi/ARB, n (%) | 5 (25.0%) | 22 (57.9%) | 20 (66.7%) | 0.0114 | 0.0263 | 0.0086 | 0.6158 |
| Beta Blocker, n (%) | 5 (25.0%) | 21 (55.3%) | 28 (93.3%) | <0.0001 | 0.0505 | <0.0001 | 0.0008 |
| Loop Diuretic, n (%) | 0 (0.0%) | 32 (84.2%) | 30 (100.0%) | <0.0001 | <0.0001 | <0.0001 | 0.0306 |
| Past Medical History | |||||||
| Hypertension, n (%) | 9 (45.0%) | 35 (92.1%) | 30 (100.0%) | <0.0001 | 0.0001 | <0.0001 | 0.2493 |
| Diabetes, n (%) | 2 (10.0%) | 28 (73.7%) | 9 (30.0%) | <0.0001 | <0.0001 | 0.163 | 0.0005 |
| Coronary artery disease, n (%) | 1 (5.0%) | 2 (5.3%) | 5 (16.7%) | 0.2511 | 1 | 0.3811 | 0.2272 |
| Atrial fibrillation or flutter, n (%) | 2 (10.0%) | 10 (26.3%) | 19 (63.3%) | 0.0002 | 0.1865 | 0.0003 | 0.0031 |
| Clinical Features | |||||||
| BMI, kg/m2 | 25.8(21. 7, 31.3) | 39.8(30.5, 43.4) | 26.1(23.3, 28.9) | <0.0001 | <0.0001 | 0.7976 | <0.0001 |
| SBP, mmHg | NA | 141.5 (127.8, 165.0) | NA | NA | NA | NA | NA |
| DBP, mmHg | NA | 72 (67.2, 81.0) | NA | NA | NA | NA | NA |
| Echocardiography | |||||||
| LVEF, % | 65(60.0, 65.0) | 65(60.0, 70.0) | 17.8(10.8, 20.0) | <0.0001 | 0.5292 | <0.0001 | <0.0001 |
| LVEDD, cm | 4(3.9, 4.5) | 4.3(3.9, 4.7) | 6.8(6.1, 7.3) | <0.0001 | 0.5759 | <0.0001 | <0.0001 |
| LV mass index, g/m2 | 118(97.6, 137.8) | 91.2(67.9, 113.5) | 145.6(131.3, 157.8) | <0.0001 | 0.0103 | 0.0065 | <0.0001 |
| Laboratory data | |||||||
| NTproBNP, pg/mL | NA | 164 (47.2, 643.0) | NA | NA | NA | NA | NA |
| Hemoglobin A1C, % | NA | 6.7 (5.9, 8.0) | NA | NA | NA | NA | NA |
| Creatinine, mg/dL | 0.8(0.7, 1.2) | 1.3(1.0, 1.8) | 1.1(1.0, 1.3) | 0.0062 | 0.0016 | 0.0186 | 0.4199 |
| eGFR, mL/min/1.73m2 | 90(58.5, 106.5) | 54.5(33.0, 75.8) | 69.5(56.0, 81.8) | 0.0035 | 0.0013 | 0.1929 | 0.0326 |
| HFrEF etiology | |||||||
| Familial, (n%) | NA | NA | 6 (20.0%) | NA | NA | NA | NA |
| Ischemic, (n%) | NA | NA | 6 (20.0%) | NA | NA | NA | NA |
| LV non-compaction, (n%) | NA | NA | 2 (6.7%) | NA | NA | NA | NA |
| NICM, (n%) | NA | NA | 15 (50.0%) | NA | NA | NA | NA |
| Sarcoidosis, (n%) | NA | NA | 1 (3.3%) | NA | NA | NA | NA |
| Invasive Hemodynamics | |||||||
| RA, mmHg | NA | 10(7.0, 13.8) | 7(5.2, 10.5) | NA | NA | NA | 0.064 |
| PASP, mmHg | NA | 42(31.2, 51.5) | 47.5(40.2, 55.8) | NA | NA | NA | 0.1239 |
| PADP, mmHg | NA | 21.5(15.0, 25.8) | 18(15.2, 25.8) | NA | NA | NA | 0.6784 |
| PAmean, mmHg | NA | 30(21.0, 34.0) | 29.3(23.1, 34.0) | NA | NA | NA | 0.9704 |
| PAWP, mmHg | NA | 20(13.2, 23.0) | 20(16.2, 23.8) | NA | NA | NA | 0.5686 |
| CO, L/min | NA | 5.6(4.6, 6.5) | 4.1(3.2, 4.8) | NA | NA | NA | <0.0001 |
| CI, L/min/m2 | NA | 2.5(2.3, 2.7) | 2(1.8, 2.3) | NA | NA | NA | 0.0007 |
| PVR, wu | NA | 1.4(1.1, 2.2) | 1.9(1.3, 3.2) | NA | NA | NA | 0.145 |
| Trans-pulmonary gradient, mmHg | NA | 8(6.2, 11.0) | 8.7(5.8, 11.2) | NA | NA | NA | 0.8673 |
| RA/PAWP | NA | 0.5(0.4, 0.6) | 0.4(0.3, 0.6) | NA | NA | NA | 0.0249 |
| PAPI | NA | 2.1(1.4, 2.9) | 3.8(2.8, 6.0) | NA | NA | NA | 0.0011 |
Data presented as n (%) or median (25th-75th percentile). Fisher’s exact test used for categorical variables. Kruskal-Wallis test used for continuous variables with 3 groups, with Dunn’s test for pairwise comparisons. Mann-Whitney test used for continuous variables with 2 groups. HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; ACEi, angiotensin converting enzyme inhibitor; ARB, angiotensin II receptor blocker; BMI, body mass index; SBP, systolic blood pressure; DBP, diastolic blood pressure; LVEF, left ventricular ejection fraction; LVEDD, left ventricular end diastolic diameter; LV, left ventricle; NTproBNP, N-terminal pro-B type natriuretic peptide; eGFR, estimated glomerular filtration rate; NICM, non-ischemic cardiomyopathy; RA, right atrial pressure; PASP, pulmonary artery systolic pressure; PADP, pulmonary artery diastolic pressure; PAmean, mean pulmonary artery pressure; PAWP, pulmonary artery wedge pressure; CO, cardiac output; CI, cardiac index; PVR, pulmonary vascular resistance; wu, Wood units; PAPI, pulmonary artery pulsatility index; N/A, data not available for Control or HFrEF.
Agnostic clustering separates groups based on myocardial tissue but not plasma metabolome
Figure 1A shows PCA based on all myocardial metabolites from the three patient groups, revealing separable clusters (Figure 1A, ellipses are 1 SD of mean). The top contributors to principal component 1 (PC1) separating control from HFrEF were primarily medium and long-chain acylcarnitines (MLAC) and catabolites of FA oxidation (Figure S1A). Top contributors to PC2 (separating HFpEF from control/HFrEF) were mostly amino acids, including the branched-chain amino acids (BCAA) and their catabolites, and MLFA (Figure S1B). By contrast, the plasma metabolome showed more overlap between groups (Figure 1B). Figure 1C displays hierarchical clustering heatmaps of the myocardial metabolites, demonstrating separation of disease groups. Metabolites formed 3 clusters: 1) those uniquely lower in HFpEF: branched-chain amino acid catabolites (C04-OH Butyryl, C04-OH Isobutyryl), ketone body, tricarboxylic acid cycle (TCA) intermediates; 2) those lower in both HFrEF and HFpEF: medium and long chain acylcarnitines, which were overall lower in HFrEF than HFpEF; 3) Higher in HFpEF: amino acids and pyruvate. Myocardial metabolites are compared between the disease groups in Table S3. Applied to the plasma metabolome (Figure 1D), hierarchical clustering yielded less distinction between groups. In contrast to the myocardium, the plasma metabolome (Table S4) showed the following findings: 1) higher acylcarnitines in both HF groups vs. control, with HFrEF higher than HFpEF; 3) higher ketones in HFrEF vs. control, but not HFpEF. In contrast to the heart, TCA intermediates and BCAA catabolites were not lower in HFpEF plasma, and MLFA were higher in HF plasma vs. control. There were some consistent findings between the myocardium and plasma- namely the pattern of higher amino acids in both HF groups vs. control and higher ketone body in HFrEF but not HFpEF.
Figure 1. Myocardial metabolomic signatures in HFpEF, HFrEF and control.
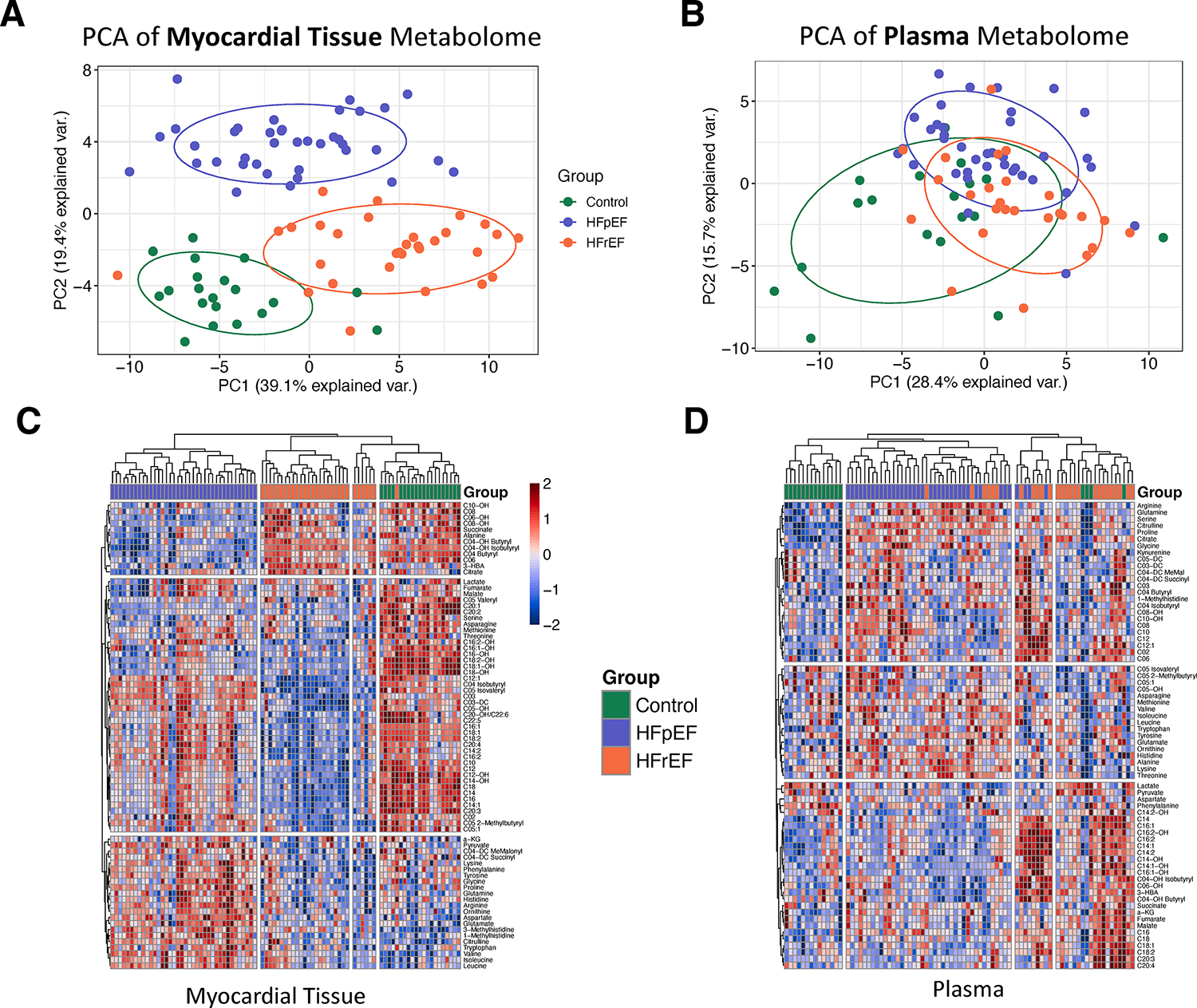
A) Principal component analysis of myocardial metabolomics shows fairly distinct clusters for each subject disease group. B) Principal component analysis of plasma metabolomics shows substantial overlap between disease groups. C) Hierarchical clustering of participants (columns) using the myocardial metabolome demonstrates similar separation of the disease groups. There were 3 main clusters of metabolites (rows): cluster 1 was lower in HFpEF uniquely, cluster 2 lowest in HFrEF and intermediate in HFpEF, cluster 3 was highest in HFpEF. D) Hierarchical clustering of participants using the plasma metabolome yielded 3 clusters with substantial overlap between disease groups. PC: principal component; 3-HBA: 3-hydroxybutyric acid, a-KG: alpha-ketoglutarate, HFpEF: heart failure with preserved ejection fraction; HFrEF: heart failure with reduced ejection fraction.
Adjustment for differences in clinical characteristics does not alter disease group comparisons
Differences in myocardial and plasma metabolite levels between the disease groups persisted even after adjustment for age, BMI, diabetes (presence or absence), and self-identified African American race (Tables S5–S10). Only self-identified African-American race was associated with some of the long-chain acylcarnitines from myocardial tissue in HFpEF vs. control data. None of the plasma metabolites were associated with any comorbidities, and disease group associations generally remained significant.
Genes and metabolites of fatty acid oxidation are reduced in HFpEF compared with control
Figure 2 compares MLAC levels and regulating genes linked to FA uptake and catabolism in the three groups. Somewhat surprisingly given higher BMI in HFpEF, MLAC were similarly lower in both forms of HF compared with control in myocardial tissue (Figure 2A, Table S3), being somewhat lower in HFrEF than HFpEF. In plasma, however, MLAC were generally higher in HFrEF over control or HFpEF (Figure 2B, Table S4). HFpEF myocardium also lower reduced gene expression of proteins involved with FA uptake and oxidation vs. control (list from KEGG Peroxisome proliferator-activated receptor [PPAR] pathway; Figure 2C). Genes regulating FA uptake (CD36, FABP, CPT1A, PLIN2, LPL), lipolysis and FA oxidation (ACSL1, -3, -5), and master regulators of FA metabolism (PPARA, PPARG, PPARGC1A, LPL) were all lower in HFpEF over control. Fewer genes were higher, most prominently the nuclear hormone receptor RXRG involved in transcriptional regulation. HFrEF also exhibited lower FA metabolism genes compared with control (Figure S2A), with several (e.g. PPARA, PPARGC1A, LPL) being significantly lower in HFpEF vs. HFrEF, while FA binding and uptake genes were lower in HFrEF vs. HFpEF (Figure S2B).
Figure 2. Medium and long-chain acylcarnitines (MLAC) and fatty acid metabolism gene expression.
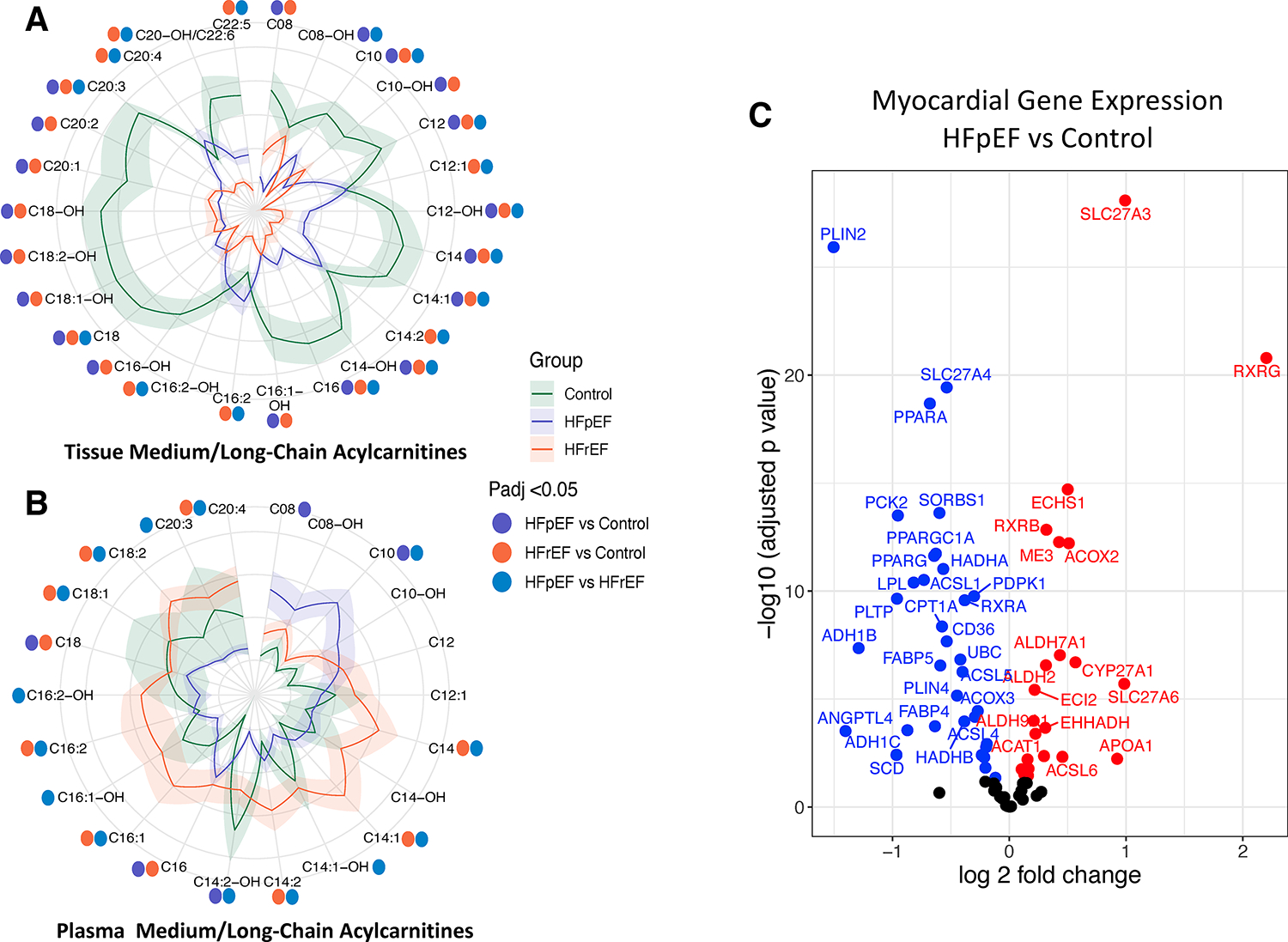
A) MLAC in HFpEF, HFrEF, and control in myocardial tissue are displayed in a polar (flower) plot. The mean (dark line) and ± standard error (shaded region) Z-scores for each metabolite are shown by their distance from the polor plot origin. Statistical differences between each set of group comparisons are denoted by colored circles surrounding the plot, as defined in the figure. B) Same comparison based on plasma MLAC. For both, a Kruskal-Wallis test with post-hoc Dunn’s test for multiple comparisons was used, and adjusted p values (Benjamini-Hochberg) annotated by colored dots corresponding to each comparison. C) Genes related to fatty acid uptake and metabolism are lower in myocardium from HFpEF vs. control. HFpEF: heart failure with preserved ejection fraction; HFrEF: heart failure with reduced ejection fraction.
Glucose metabolism genes and tricarboxylic acid cycle intermediates are reduced in HFpEF
Pyruvate was significantly higher in HFpEF myocardium compared with control or HFrEF (p<0.0001 vs. either, Figure 3A, Table S3). This metabolomic analysis did not include glycolysis or glucose oxidation intermediates. However, gene expression of multiple proteins central to glucose metabolism were generally lower in HFpEF vs. control (Figure 3B), including GLUT1 (SLC2A1) involved in glucose uptake. Mitochondrial pyruvate carrier 1 (MPC1) was 1.24-fold higher in HFpEF over control (not shown, P-adj 6.2e−5) and MPC2 expression was not different. Protein levels were not determined. Compared with control, TCA cycle intermediates succinate and fumarate were significantly less in HFpEF (Figure 3A, Table S3; P-adj = 0.003, P-adj = 0.04, respectively), with malate borderline lower (P-adj = 0.06). For HFrEF, citrate was higher (P-adj = 0.002) whereas fumarate and malate trended lower (P-adj = 0.09 and P-adj = 0.13, respectively) vs. control.
Figure 3. Tricarboxylic acid cycle intermediates and genes related to glucose metabolism.
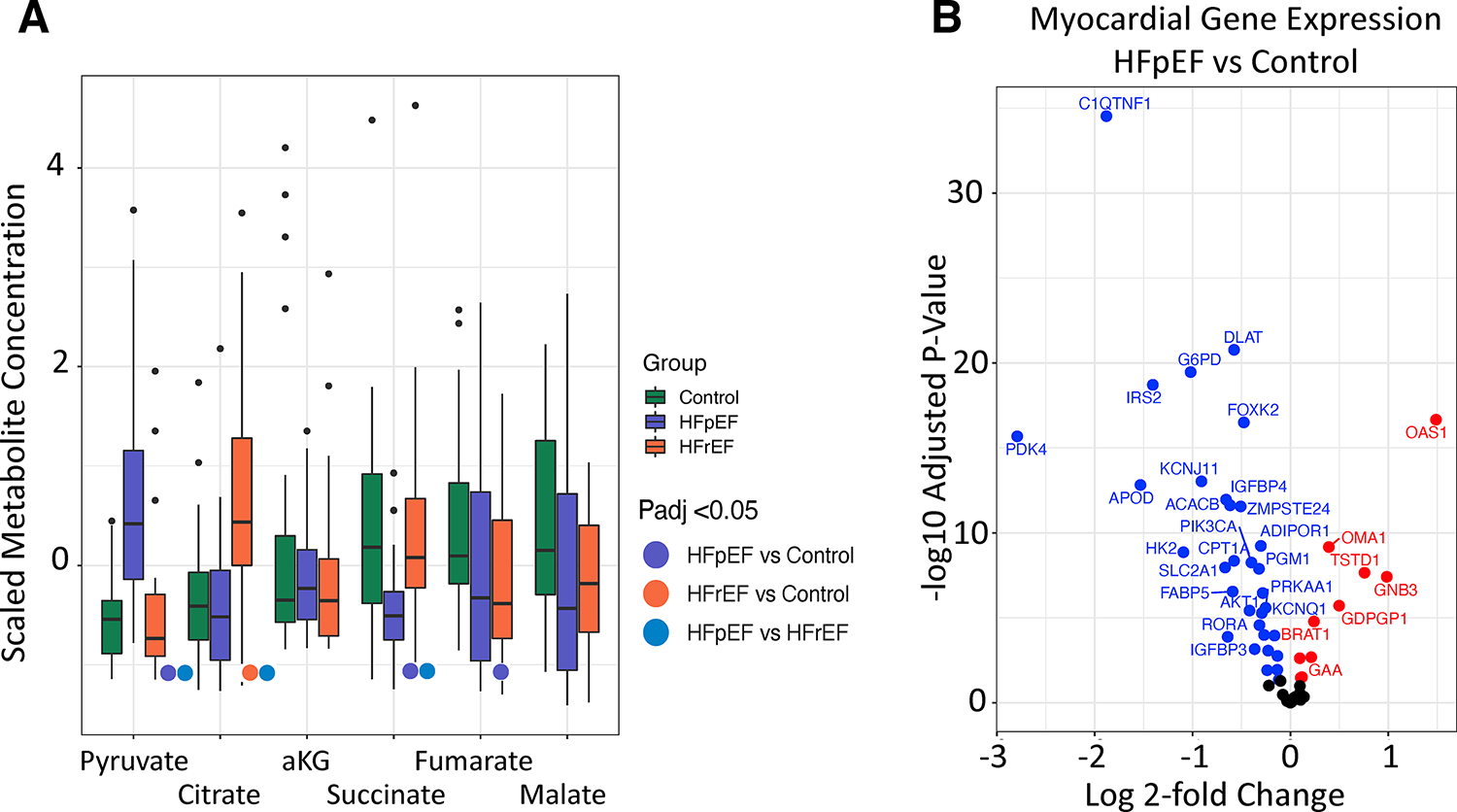
A) Several metabolites (succinate, fumarate, malate) in the tricarboxylic acid cycle are lower in myocardium from HFpEF vs. control. Pyruvate is higher in HFpEF vs. control. Raw data were scaled for visualization purposes. Analyzed using Kruskal-Wallis test with post-hoc Dunn’s test for multiple comparisons, adjusted p-values (Benjamini-Hochberg) provided. B) Myocardial gene expression of genes related to glucose metabolism and its regulation/uptake in HFpEF vs. control. Gene expression adjusted p value determined as described in methods. HFpEF: heart failure with preserved ejection fraction; HFrEF: heart failure with reduced ejection fraction; alpha-KG: alpha-ketoglutarate.
Branched-chain amino acids (BCAA) are higher in HFpEF but metabolites are reduced
Myocardial levels of BCAA - leucine, valine, and isoleucine were higher in HFpEF vs. the other two groups (Figure 4A, Table S3). BCAA were also directionally higher in HFrEF compared with control, but only valine was statistically significant (isoleucine, P-adj = 0.11; leucine, P-adj = 0.28; valine, P-adj = 0.009). However, major BCAA catabolites were reduced in both HF groups (Figure 4A), suggesting impaired catabolism. In plasma, only valine was higher in HFpEF vs. control (P-adj = 0.005) and BCAA catabolites were either higher or not different in both HF groups (Figure 4B, Table S4). Consistent with higher BCAA but lower catabolites, gene expression of key enzymes responsible for BCAA catabolism were also lower in HFpEF compared with control myocardium (BCKDHB, DBT, SLC25A44, Figure 4C). The proximal catabolic enzyme for BCAA is BCAT2, the mitochondrial isoform most abundantly expressed in myocardium, and this was higher in HFpEF vs. control. Figure 4D summarizes these results and pathways, supporting downstream inhibition of BCAA catabolism. Intriguingly, non-BCAA were also generally higher in HFpEF myocardium vs. control (Figure S3A, Table S3), and higher in plasma (Figure S3B, Table S4). This suggests either greater uptake of amino acids into the heart, or more protein turnover in HFpEF myocardium and release into the bloodstream.
Figure 4. Branched-chain amino acid (BCAA) metabolites and amino acids are altered in HFpEF.
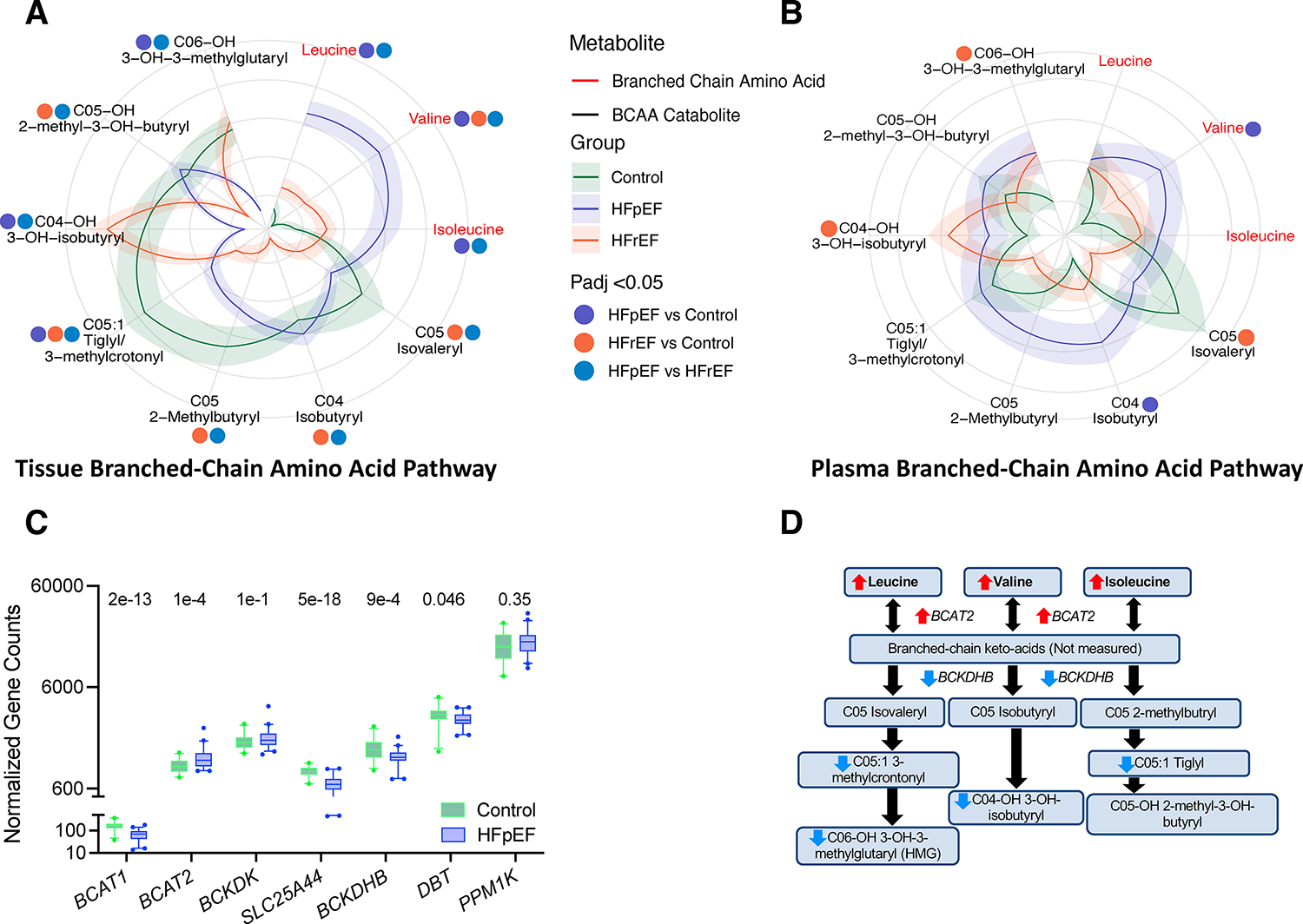
A) Polar – flower plot of myocardial BCAA and their downstream catabolites in all three groups. Data displayed as described for Figure 2A. BCAA were higher in HFpEF, but several corresponding mitochondrial carnitine catabolites were lower. BCKA were not measured. C05:1 includes an unresolved mixture of tiglyl and 3-methylcrontonyl. B) Similar display of these metabolites in plasma shows fewer significant differences and less asymmetry between BCAA and catabolites in HFpEF. P values calculated by Kruskal-Wallis test with post-hoc Dunn’s test for multiple comparisons and adjustment for multiple comparisons (Benjamini-Hochberg), and annotated by colored dots for respective comparisons. C) Box-plot for gene expression of key proteins in the BCAA metabolic pathway. Adjusted P-values from Benjamini-Hochberg multiple comparisons tests are shown for each. D) Summary diagram of differences in metabolites and gene expression of regulating proteins in the BCAA metabolic pathway comparing HFpEF to control. HFpEF: heart failure with preserved ejection fraction; HFrEF: heart failure with reduced ejection fraction. Other abbreviations in text.
Beta-hydroxybutyric acid (ketone body) is increased in HFrEF but not HFpEF myocardium
Ketone body 3-hydroxybutyric acid (or beta-hydroxybutyric acid) was significantly higher in HFrEF myocardium over the other two groups (P-adj <0.0005 for both; Figure S4A) and in plasma (P-adj = 0.01 v control, P-adj = 0.002 v HFpEF, Figure S4B), whereas HFpEF and control myocardial and plasma levels were similar. C4-OH beta-hydroxybutyryl, a downstream metabolite of ketone bodies, was significantly lower in HFpEF vs. control and HFrEF (p<0.0001 for both comparisons), whereas HFrEF and control levels were not significantly different.
Metabolomic-based HFpEF subgroups and clinical-metabolite correlations
The prespecified HFpEF subgroups focused on testing if obesity/DM vs. LV hypertrophy/hypertension had distinctive metabolic features. Despite substantial disparities in clinical characteristics (Table S11), none of the myocardial metabolites were significantly different between these HFpEF groups, and PCA of the metabolome showed substantial overlap between the subgroups (Figure S5, Table S12).
The process was then reversed, generating HFpEF subgroups based on the metabolome using NMF. This yielded 3 groups, and Figure 5A shows their hierarchical clustering based on levels of five metabolites (C03 propionyl, C04 Isobutyryl, C05 2-methylbutyryl, C05 Isovaleryl, and 3-methylhistidine) identified by the NMF algorithm as the least number of metabolites required to distinguish between groups. Figure 5B shows PCA based on the full myocardial metabolome, and groups 1 and 2 were most separated. Key clinical and metabolic features of these two groups are summarized in Figure 5C with further details in Table S13 and Table S14. Plasma metabolites showed no differences between the HFpEF subgroups, while only 2 of the myocardial BCAA catabolites were significantly different between the HFpEF subgroups (after adjustment for multiple comparisons).
Figure 5. HFpEF subgroups identified by non-negative matrix factorization (NMF) finds substantial overlap in metabolome.
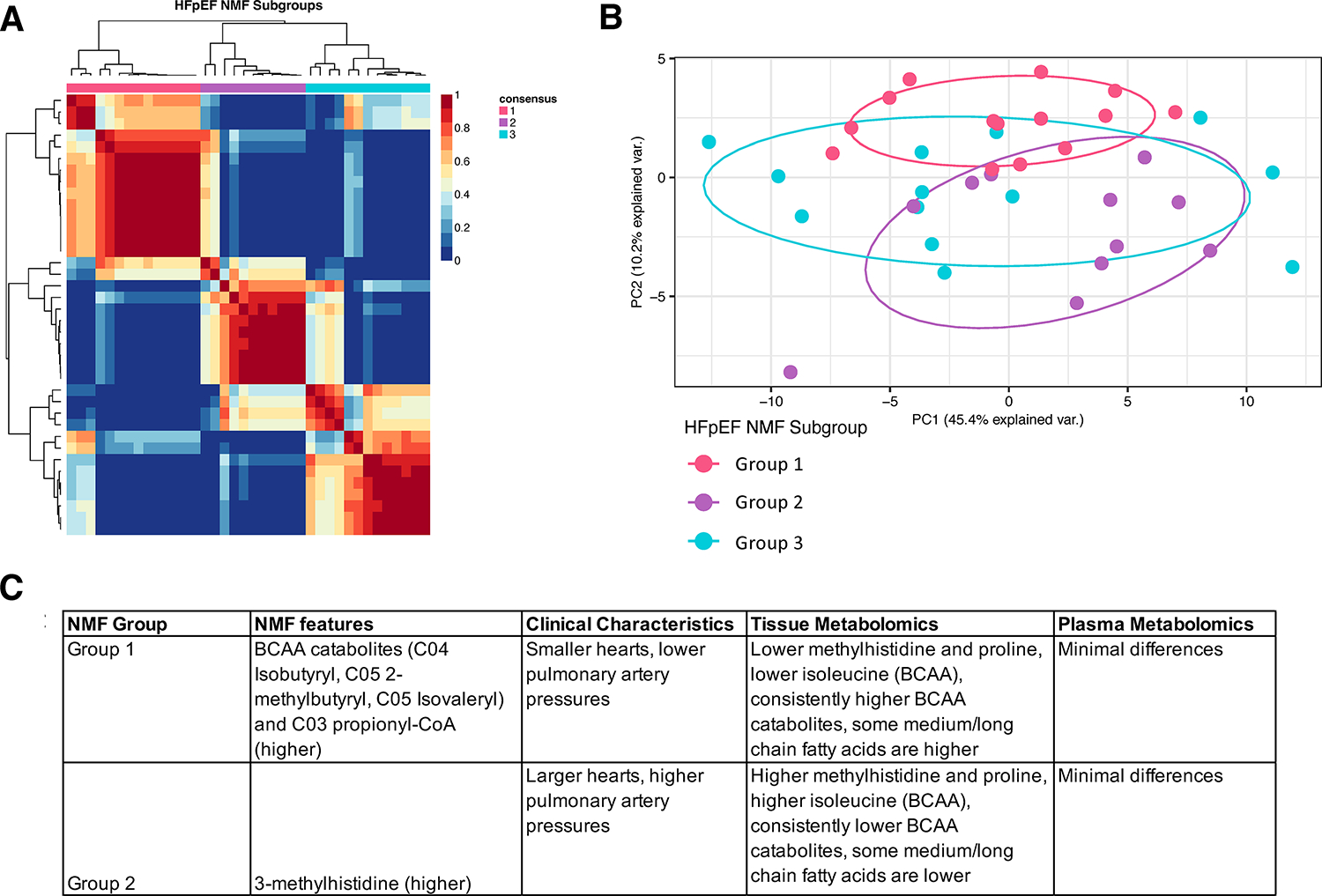
A) Patient-patient correlation plot shows classification into three groups based on 5 metabolites selected by the NMF algorithm (see text and panel C for details). B) Principal component analysis using all myocardial metabolites shows significant overlap between HFpEF NMF subgroups, with Groups 1 and 2 being the most different. C) NMF-metabolite, clinical, and tissue metabolomics differences for NMF-derived Groups 1 and 2.
Lastly, Pearson correlations were determined between the individual metabolites and clinical characteristics of the HFpEF patients (Figure S6). Methylhistidines were positively correlated with measures of pulmonary hypertension and serum natriuretic peptides in both myocardial tissue and plasma. Tissue BCAA metabolites and MLAC negatively correlated whereas plasma BCAA metabolites and MLAC positively correlated with the same two clinical features. Unadjusted P values were used for the clinical characteristics / metabolite correlations.
Discussion
This study reveals several important new findings regarding myocardial tissue and plasma metabolomics in a mostly obese HFpEF cohort, comparing their features with those in HFrEF and non-failing controls. First, myocardial metabolite differences between groups were not generally predicted by corresponding plasma data, and heart vs. plasma results for medium/long-chain acylcarnitines were broadly discrepant. This is important, as to date all reported metabolomics from patients with obesity/DM +/− HFpEF has been from blood. 15, 16, 29, 30 Second, despite their marked obesity (median BMI 40 kg/m2), the HFpEF patients had significantly lower myocardial MLAC levels than control, being similar to the pattern found in HFrEF patients with a median BMI of 26 kg/m2. This difference was not observed in plasma where MLACs tended to be not different or higher in both HF groups vs. control, with HFrEF higher than HFpEF. Third, metabolites uniquely separating HFpEF from control and HFrEF were 1) amino acids, particularly BCAAs (higher in HFpEF) and their catabolites (lower in HFpEF); 2) pyruvate (higher in HFpEF); and 3) 3-hydroxybutyric acid (ketone body, not different in HFpEF vs. control, yet higher in HFrEF vs. control). Gene expression correlates support lower FA and BCAA metabolism in HFpEF. Together these data show that the myocardial metabolome in HFpEF with obesity +/− DM is much closer to HFrEF than might be expected and raise the hypothesis that the HFpEF heart has substantial metabolic inflexibility.
FA utilization in the HFpEF heart
FA metabolism normally accounts for approximately 85% of ATP generation in the normal heart, whereas this declines to 70% in the HFrEF heart. 12 The HFrEF heart compensates by expanding utilization of amino acids, ketones, and lactate. 12 The data presented here are consistent with prior studies in HFrEF myocardium that report lower FAs and acylcarnitines, and higher fatty acids and acylcarnitines in HFrEF plasma vs. control. 22, 31, 32 However, obesity and insulin resistance are thought to shift metabolism to favor FA oxidation, coupled to increased circulating free FA, transcriptional programs to augment fat uptake and catabolism, and concomitant impaired insulin-dependent myocardial glucose uptake14, 33. Human studies34, 35 have used non-invasive measures of FA uptake in small numbers of participants, and data from myocardial metabolomics in obese/DM humans remain lacking. But if anything, the observations that patients with both HFpEF and obesity have lower cardiac fatty acid oxidation than controls without obesity suggests there should be even greater disparities if they were compared with controls with obesity.
In plasma, patients with both obesity/DM have higher long-chain acylcarnitines and those with diabetes, increased short- and medium-chain acylcarnitines, both suggesting incomplete FA oxidation. 29 Yet, despite these co-morbidities being prominent in most of the participants with HFpEF, only two medium-chain acylcarnitines were significantly higher in plasma. Some reports have found plasma MLAC to be higher in HFpEF vs. control, 15, 16 although their patient cohorts were also less obese (BMI ~31) and diabetic (40%). Importantly, our data shows these plasma levels do not reflect MLAC in the myocardium. This disparity could be due to lower myocardial FA uptake, consistent with the lower expression of FA-uptake genes. Plasma MLAC also rise with fasting, exercise, insulin resistance, and obesity, and are secreted/taken-up by other organs including skeletal muscle and liver impacting different tissue and circulating levels. 36
Given the plethora of FA in obese HFpEF, increasing FA oxidation and clearance from abnormal stores in tissues such as heart, 37 liver, 38 and skeletal muscle, 39 may ultimately prove therapeutically beneficial. By contrast, FAO inhibitors that have previously been tried in HFrEF to bias towards glucose oxidation 40 may be less useful in HFpEF given the present data and high probability of co-existing depressed glucose uptake and catabolism. However, this cross-sectional study cannot measure flux and future studies dissecting fatty acid uptake and oxidation in the HFpEF heart in vivo are needed to further clarify the metabolic bottlenecks and thus metabolic therapy to pursue.
Glucose metabolism and TCA cycle intermediates
This study provided limited analysis of glucose metabolism, only measuring the key glycolytic metabolite pyruvate that was higher in HFpEF, but not glycolytic intermediates. However, gene expression for glucose metabolism proteins, including glucose uptake, were often lower in HFpEF vs. control. MPC1 and MPC2 gene expression were not reduced. Protein levels of both are reportedly lower in late-stage human HFrEF, and gene deletion models develop hypertrophic-HF phenotypes41–43. Further work is needed to clarify protein levels in HFpEF.
TCA intermediates were lower in HFpEF myocardium compared with control suggesting anaplerosis was inadequate, however flux could not be measured in this cross-sectional study. Similar intermediates have also been reported to be lower in HFrEF myocardium,22 and while some trended to be lower in HFrEF in this study, they were not statistically significant. TCA intermediates are replenished by anaplerosis where other substrates are metabolized to enter TCA cycle at multiple entry points. Such pathways include breakdown of amino acids, lactate, and pyruvate. Many amino acids and pyruvate were higher in this study, and this may be an indicator for inadequate TCA intermediate replacement. Enzymatic activity of key proteins in the TCA cycle itself is another potential cause and these are under investigation.
Branched-chain and non-branched-chain acid metabolism in HFpEF
Prior studies have reported higher circulating amino acids in HFpEF vs. control16 and several amino acid-derived metabolites correlate with echocardiographic markers of diastolic dysfunction, including methylhistidine. 44 Methylhistidine is not metabolized, but serves as a surrogate marker for myofibrillar protein turnover, 45 skeletal muscle loss, and frailty, 46 and is found in dietary meat. 47 Methylhistidine was markedly higher in both HFpEF plasma and myocardium, and its correlation to pulmonary hypertension and serum NTproBNP, both reflecting higher cardiac load, may reflect myofibrillar turnover associated with higher stress.
Among amino acids, BCAA were quantitatively the highest in HFpEF myocardium, though their catabolites were lower compared with control. BCAA were also borderline higher in HFrEF as previously reported22 but lower than in HFpEF. Increased BCAA levels yet with lower catabolites are consistent with depressed gene expression of several key proteins in the metabolic pathway. These genes are regulated by PPARa (a master regulator of fatty acid metabolism) and their expression declines in models of hypertrophy and early-stage heart failure. 48 In the heart, BCAAs are largely transaminated by mitochondrial branched-chain amino acid aminotransferase BCAT2 into their respective branched-chain keto-acids (BCKA)49. BCAT2 was higher in HFpEF vs. control and as protein levels are primarily transcriptionally regulated 50 it is likely the branched-chain keto acids (BCKA) are also increased though this remains to be confirmed. SLC25A44 transports BCAAs from the cytosol into mitochondria, and this too was lower in HFpEF vs. control, potentially contributing to depressed BCAA oxidation51. The key rate limiting step in BCKA catabolism is at BCKDH, which is inhibited by phosphorylation by BCKD kinase and activated by dephosphorylation by PPM1K 52. BCKDH complex activity remains to be determined in HFpEF myocardium, but it is suspected to be be reduced given less distal catabolites. While these data support reduced BCAA oxidation in HFpEF hearts, direct experimental confirmation is needed. It is worth noting, for example, that cardiac BCAA oxidation appears to increase, rather than decrease, in murine HFrEF induced by myocardial infarction53. This may reflect differences in the HF conditions, as increased BCAAs unique to HFpEF were found in this study. Similar analyses in current obese/hypertensive/diabetic experimental HFpEF models will be of interest.
BCAA are oxidized to fuel the TCA cycle through conversion to acetyl-CoA and propionyl-CoA. They also affect signaling and accumulate in the circulation when tissue oxidation is compromised. For example, leucine activates the mechanistic target of rapamycin (mTOR), a prominent regulator of cellular growth and cardiac hypertrophy and insulin desensitization. 54, 55 Leucine also stimulates insulin secretion in response to protein intake that may further contribute to T2D. 56 Multiple studies in animals and humans find BCAAs contribute to insulin resistance, mTOR being but one mechanism. 57–59 Thus, inhibition of BCAA oxidation and accumulation of BCAAs in HFpEF could both limit a fuel source for the heart, and contribute to cardiac and other organ pathobiology by worsening insulin resistance. Serum metabolomics in patients with diabetes and cardiovascular disease suggest BCAA oxidation is enhanced by SGLT2 inhibitor therapy, 60 potentially contributing to their benefit in HFpEF. 10, 11 Of note, none of the patients in the current study were on SGLT2 inhibitors. Enhancing BCAA oxidation benefits animal models of heart disease 53, 61, 62, suggesting this manipulation might have therapeutic value particularly in the subgroup of HFpEF with higher BCAA and reduced catabolites. Mice with cardiac BCKDK deletion (suppressing BCAA oxidation) with reduced EF from myocardial infarction still displayed benefits in cardiac function from enhanced BCAA oxidation, suggesting indirect effects perhaps related to reduced vascular tone53. Whether this applies as well to human HFpEF or if there are direct cardiac contributions from increased cardiac BCAA oxidation will need to be experimentally tested.
Ketone body oxidation
In vivo studies have found ketone body uptake by the myocardium expands 2- to 3- fold with HFrEF, 12 with its oxidation higher in HF both in mice 63 and in humans. 31 This is considered protective for heart failure providing an important alternative fuel. 64–66 However, 3-hydroxybutyric acid was higher in HFrEF myocardium and plasma, yet in neither for HFpEF. These data suggest this fuel source is less available in HFpEF. Increasing ketones by diet is being explored for HFrEF where it appears to be beneficial, 66 but whether it will be useful for HFpEF remains to be tested. Another approach maybe the SGLT2 inhibitors that increase circulating ketones 67 and now appear beneficial to human HFpEF, 10, 11 though precise mechanisms remain unknown.
Homogeneity of myocardial metabolic profiles despite HFpEF clinical heterogeneity
While the clinical pre-specified HFpEF subgroups had prominent differences in clinical features, there were no differences among their metabolomes. As the cohort overall was very obese, lack of sufficient variance in this key parameter may have masked differences, though there were very few significant correlations between BMI and individual metabolites. The metabolome groupings in HFpEF by NMF highlight BCAA metabolism as a biomarker for two groups, but overall, there were very few differences in metabolites between the HFpEF subgroups. Importantly, groups could not be separated based on the plasma metabolome, suggesting striking homogeneity from a metabolome standpoint across this HFpEF cohort, despite clinical heterogeneity.
Study Limitations
This study has several limitations. Metabolomics analysis was performed on tissue and plasma at one time point, and this snapshot does not characterize flux in the various pathways or causality. Although the HFpEF patients had a high symptom burden, they were not likely as end-stage as those with HFrEF undergoing transplantation. Myocardial samples were studied from the right not left ventricular septum. Control tissue came from organ donors with brain death determination, which while not likely reflecting a truly normal condition, were still harvested under highly controlled settings and processed just as the HFrEF tissues. The patient groups were not matched based on age, sex, race, obesity, and clinical co-morbidities, as many of these co-morbidities are limitations to cardiac transplantation or donor organ suitability. Importantly, metabolite differences persisted even after adjustment for the key differentiating clinical features of each group. Lastly, while metabolomes were correlated to transcriptomic data, this may not match changes in protein expression or activity. Ongoing studies are attempting to fill in more of these knowledge gaps.
In conclusion, obese HFpEF displays impairment in multiple metabolic substrates, including fatty acids, amino acids, ketones, and most likely glucose. The notion that the heart of obese individuals will take up circulating FA to use for oxidation to offset depressed glucose uptake does not seem to apply to obese HFpEF. Fuel alternatives observed with HFrEF that also provide metabolic intermediates by anaplerosis also seem lacking in HFpEF. Together, obese HFpEF would appear to combine metabolic defects from both HF and obesity/DM creating a syndrome with substantial fuel inflexibility. This may well be a factor in why it has been hard to treat, but it points to important features worth future study and ultimately targeting by therapy.
Supplementary Material
Clinical Perspective.
What is new?
While obesity/diabetes is thought to increase myocardial fatty acid uptake/utilization, HFpEF demonstrates evidence of lower fatty acid oxidation similar to that in HFrEF.
Use of alternative fuels, including glucose, ketones, and branched-chain amino acids, and tricarboxylic acid cycle intermediates also appears to be compromised in HFpEF myocardium, suggesting insufficient anaplerosis.
Despite variability in obesity and other co-morbidities in HFpEF, metabolic profiled are generally conserved among subgroups.
What are the clinical implications?
The human HFpEF heart exhibits metabolomics consistent with substantial fuel inflexibility, supporting therapeutic efforts to enhance fat and anaplerotic pathways.
Differential BCAA catabolism is one of the few features of HFpEF myocardium that distinguishes between subgroups within the syndrome, and its enhancement may prove therapeutically useful in the appropriate subgroup.
Acknowledgements
Metabolomics analysis was performed by the Metabolomics Core at the University of Pennsylvania in the Penn Cardiovascular Institute.
Funding Sources
VS.H was supported by NIH 2T32HL007227 and Sarnoff Scholar Award 138828. CP by NIH P30 CA016520 and P30 DK050306; KCB, KBM, and human tissue procurement were supported by NIH R01: HL105993, NIH R01: HL133080 and NIH R01: HL149891; SM by American Heart Associaton CDA 938718; EJY by NIH 2T32HL007227; ZA by R01: HL152446; DPK by NIH R01:HL128349 and NIHR01:HL151345; DAK by American Heart Associaton 16SFRN28620000 and National Heart, Lung, and Blood Institute R35:HL135827; KS by AHA: 16SFRN27870000, National Heart, Lung, and Blood Institute R01:HL61912, and Amgen.
Non-standard Abbreviations and Acronyms
- HFpEF
heart failure with preserved ejection fraction
- HFrEF
heart failure with reduced ejection fraction
- DM
diabetes mellitus
- SGLT2
sodium glucose co-transporter 2
- FA
fatty acids
- MLFA
medium and long-chain fatty acids
- BCAA
branched-chain amino acids
- BCKA
branched-chain keto-acids
- TCA
tricarboxylic acid cycle
- LVEF
left ventricular ejection fraction
- PAWP
pulmonary artery wedge pressure
- HFpEF-Ht/Hp
HFpEF with predominantly hypertension and ventricular hypertrophy
- HFpEF-Met
HFpEF with predominantly metabolic disease and obesity
- NT-proBNP
N-terminal pro–B-type natriuretic peptide
- RV
right ventricular
- PCA
principal component analysis
- NMF
Non-negative matrix factorization
- BMI
Body mass index
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- CD36
cluster of differentiation 36
- FABP
fatty acid binding protein
- CPT1A
Carnitine palmitoyltransferase Ia
- PLIN2
Perilipin 2
- LPL
lipoprotein lipase
- ACSL1, -3, -5
Long-chain-fatty-acid—CoA ligase 1, 3, and 5
- PPARA
Peroxisome proliferator-activated receptor alpha
- PPARG
Peroxisome proliferator-activated receptor gamma
- PPARGC1A
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- RXRG
Retinoid X receptor gamma
- GLUT1
Glucose transporter 1
- SLC2A1
solute carrier family 2, facilitated glucose transporter member 1
- MPC1/MPC2
mitochondrial pyruvate carrier 1 and 2
- BCKDHB
2-Oxoisovalerate dehydrogenase subunit beta, mitochondrial
- DBT
Lipoamide acyltransferase component of branched-chain alpha-keto acid dehydrogenase complex, mitochondrial
- SLC25A44
Solute carrier family 25 member 46
- BCAT2
Branched chain amino acid transaminase 2
- PPM1K
protein phosphatase, Mg2+/Mn2+ dependent 1K
- mTOR
mechanistic target of rapamycin
- BCKDK
branched chain keto acid dehydrogenase kinase
Footnotes
Conflict of Interest Disclosures
VS.H, CP, MSK, KCB, HW, SM, NK, EJY, ZA- none. KBM receives research grant funding from Amgen, Inc. and serves on an advisory board for Bristol-Myers-Squibb. DPK serves on the advisory boards for Pfizer and Amgen, and receives grant funding from Amgen. DAK serves on advisory boards for Amgen, Cytokinetics, Cardurion, Boehringer-Ingelheim, and AstraZeneca. He also receives grant funding from Cytokinetics, Amgen, and Boehringer-Ingelheim. KS serves as an advisory board member and/or consultant to AstraZeneca, Alleviant, Bayer, Boehringer-Ingelheim, Imbria, Novartis, NovoNordisk, RIVUS, and ViCardia. She receives grant support from Amgen.
References
- 1.Savarese G, Becher PM, Lund LH, Seferovic P, Rosano GMC and Coats A. Global burden of heart failure: A comprehensive and updated review of epidemiology. Cardiovasc Res. 2023; 118(17):3272–3287. [DOI] [PubMed] [Google Scholar]
- 2.Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Cheng S, Delling FN, Elkind MSV, Evenson KR, Ferguson JF, Gupta DK, Khan SS, Kissela BM, Knutson KL, Lee CD, Lewis TT, Liu J, Loop MS, Lutsey PL, Ma J, Mackey J, Martin SS, Matchar DB, Mussolino ME, Navaneethan SD, Perak AM, Roth GA, Samad Z, Satou GM, Schroeder EB, Shah SH, Shay CM, Stokes A, VanWagner LB, Wang NY, Tsao CW, American Heart Association Council on E, Prevention Statistics C and Stroke Statistics S. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation. 2021;143:e254–e743. [DOI] [PubMed] [Google Scholar]
- 3.Runte KE, Bell SP, Selby DE, Häußler TN, Ashikaga T, LeWinter MM, Palmer BM and Meyer M. Relaxation and the Role of Calcium in Isolated Contracting Myocardium From Patients With Hypertensive Heart Disease and Heart Failure With Preserved Ejection Fraction. Circ Heart Fail. 2017;10:e004311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Heerebeek L, Hamdani N, Falcao-Pires I, Leite-Moreira AF, Begieneman MP, Bronzwaer JG, van der Velden J, Stienen GJ, Laarman GJ, Somsen A, Verheugt FW, Niessen HW and Paulus WJ. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation. 2012;126:830–9. [DOI] [PubMed] [Google Scholar]
- 5.Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, von Schlippenbach J, Skurk C, Steendijk P, Riad A, Poller W, Schultheiss HP and Tschope C. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail. 2011;4:44–52. [DOI] [PubMed] [Google Scholar]
- 6.Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD, Slater R, Palmer BM, Van Buren P, Meyer M, Redfield MM, Bull DA, Granzier HL and LeWinter MM. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation. 2015;131:1247–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kitzman DW and Shah SJ. The HFpEF Obesity Phenotype: The Elephant in the Room. J Am Coll Cardiol. 2016;68:200–3. [DOI] [PubMed] [Google Scholar]
- 8.Hahn VS, Knutsdottir H, Luo X, Bedi K, Margulies KB, Haldar SM, Stolina M, Yin J, Khakoo AY, Vaishnav J, Bader JS, Kass DA and Sharma K. Myocardial Gene Expression Signatures in Human Heart Failure With Preserved Ejection Fraction. Circulation. 2021;143:120–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aslam MI, Hahn VS, Jani V, Hsu S, Sharma K and Kass DA. Reduced Right Ventricular Sarcomere Contractility in Heart Failure With Preserved Ejection Fraction and Severe Obesity. Circulation. 2021;143:965–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anker SD, Butler J, Filippatos G, Ferreira JP, Bocchi E, Bohm M, Brunner-La Rocca HP, Choi DJ, Chopra V, Chuquiure-Valenzuela E, Giannetti N, Gomez-Mesa JE, Janssens S, Januzzi JL, Gonzalez-Juanatey JR, Merkely B, Nicholls SJ, Perrone SV, Pina IL, Ponikowski P, Senni M, Sim D, Spinar J, Squire I, Taddei S, Tsutsui H, Verma S, Vinereanu D, Zhang J, Carson P, Lam CSP, Marx N, Zeller C, Sattar N, Jamal W, Schnaidt S, Schnee JM, Brueckmann M, Pocock SJ, Zannad F, Packer M and Investigators EM-PT. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N Engl J Med. 2021;385:1451–1461. [DOI] [PubMed] [Google Scholar]
- 11.Nassif ME, Windsor SL, Borlaug BA, Kitzman DW, Shah SJ, Tang F, Khariton Y, Malik AO, Khumri T, Umpierrez G, Lamba S, Sharma K, Khan SS, Chandra L, Gordon RA, Ryan JJ, Chaudhry SP, Joseph SM, Chow CH, Kanwar MK, Pursley M, Siraj ES, Lewis GD, Clemson BS, Fong M and Kosiborod MN. The SGLT2 inhibitor dapagliflozin in heart failure with preserved ejection fraction: a multicenter randomized trial. Nat Med. 2021;27:1954–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murashige D, Jang C, Neinast M, Edwards JJ, Cowan A, Hyman MC, Rabinowitz JD, Frankel DS and Arany Z. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science. 2020;370:364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cardoso AC, Lam NT, Savla JJ, Nakada Y, Pereira AHM, Elnwasany A, Menendez-Montes I, Ensley EL, Petric UB, Sharma G, Sherry AD, Malloy CR, Khemtong C, Kinter MT, Tan WLW, Anene-Nzelu CG, Foo RS, Nguyen NUN, Li S, Ahmed MS, Elhelaly WM, Abdisalaam S, Asaithamby A, Xing C, Kanchwala M, Vale G, Eckert KM, Mitsche MA, McDonald JG, Hill JA, Huang L, Shaul PW, Szweda LI and Sadek HA. Mitochondrial Substrate Utilization Regulates Cardiomyocyte Cell Cycle Progression. Nat Metab. 2020;2:167–178. [PMC free article] [PubMed] [Google Scholar]
- 14.Nakamura M and Sadoshima J. Cardiomyopathy in obesity, insulin resistance and diabetes. J Physiol. 2020;598:2977–2993. [DOI] [PubMed] [Google Scholar]
- 15.Hunter WG, Kelly JP, McGarrah RW 3rd, Khouri MG, Craig D, Haynes C, Ilkayeva O, Stevens RD, Bain JR, Muehlbauer MJ, Newgard CB, Felker GM, Hernandez AF, Velazquez EJ, Kraus WE and Shah SH. Metabolomic Profiling Identifies Novel Circulating Biomarkers of Mitochondrial Dysfunction Differentially Elevated in Heart Failure With Preserved Versus Reduced Ejection Fraction: Evidence for Shared Metabolic Impairments in Clinical Heart Failure. J Am Heart Assoc. 2016;5:e003190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zordoky BN, Sung MM, Ezekowitz J, Mandal R, Han B, Bjorndahl TC, Bouatra S, Anderson T, Oudit GY, Wishart DS, Dyck JR and Alberta H. Metabolomic fingerprint of heart failure with preserved ejection fraction. PLoS One. 2015;10:e0124844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruiz M, Labarthe F, Fortier A, Bouchard B, Thompson Legault J, Bolduc V, Rigal O, Chen J, Ducharme A, Crawford PA, Tardif JC and Des Rosiers C. Circulating acylcarnitine profile in human heart failure: a surrogate of fatty acid metabolic dysregulation in mitochondria and beyond. Am J Physiol Heart Circ Physiol. 2017;313:H768–H781. [DOI] [PubMed] [Google Scholar]
- 18.Pieske B, Tschöpe C, de Boer RA, Fraser AG, Anker SD, Donal E, Edelmann F, Fu M, Guazzi M, Lam CSP, Lancellotti P, Melenovsky V, Morris DA, Nagel E, Pieske-Kraigher E, Ponikowski P, Solomon SD, Vasan RS, Rutten FH, Voors AA, Ruschitzka F, Paulus WJ, Seferovic P and Filippatos G. How to diagnose heart failure with preserved ejection fraction: the HFA-PEFF diagnostic algorithm: a consensus recommendation from the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur J Heart Fail. 2020;22:391–412. [DOI] [PubMed] [Google Scholar]
- 19.Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, Falk V, González-Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GMC, Ruilope LM, Ruschitzka F, Rutten FH and van der Meer P. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37:2129–2200. [DOI] [PubMed] [Google Scholar]
- 20.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ and Wilkoff BL. 2013 ACCF/AHA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128:1810–52. [DOI] [PubMed] [Google Scholar]
- 21.Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, Flachskampf FA, Foster E, Goldstein SA, Kuznetsova T, Lancellotti P, Muraru D, Picard MH, Rietzschel ER, Rudski L, Spencer KT, Tsang W and Voigt JU. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr. 2015;28:1–39 e14. [DOI] [PubMed] [Google Scholar]
- 22.Flam E, Jang C, Murashige D, Yang Y, Morley MP, Jung S, Kanter DS, Pepper H, Bedi KC, Brandimarto J, Prosser BL, Cappola T, Snyder NW, Rabinowitz JD, Margulies KB and Arany Z. Integrated landscape of cardiac metabolism in end-stage human non ischemic dilated cardiomyopathy. Nature Cardiovascular Research. 2022;1:817–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gardell SJ, Hopf M, Khan A, Dispagna M, Hampton Sessions E, Falter R, Kapoor N, Brooks J, Culver J, Petucci C, Ma CT, Cohen SE, Tanaka J, Burgos ES, Hirschi JS, Smith SR, Sergienko E and Pinkerton AB. Boosting NAD(+) with a small molecule that activates NAMPT. Nat Commun. 2019;10:3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lanfear DE, Gibbs JJ, Li J, She R, Petucci C, Culver JA, Tang WHW, Pinto YM, Williams LK, Sabbah HN and Gardell SJ. Targeted Metabolomic Profiling of Plasma and Survival in Heart Failure Patients. JACC Heart Fail. 2017;5:823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stekhoven DJ and Buhlmann P. MissForest--non-parametric missing value imputation for mixed-type data. Bioinformatics. 2012;28:112–8. [DOI] [PubMed] [Google Scholar]
- 26.Gaujoux R and Seoighe C. A flexible R package for nonnegative matrix factorization. BMC Bioinformatics. 2010;11:367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brunet JP, Tamayo P, Golub TR and Mesirov JP. Metagenes and molecular pattern discovery using matrix factorization. Proc Natl Acad Sci U S A. 2004;101:4164–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Love MI, Huber W and Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mihalik SJ, Goodpaster BH, Kelley DE, Chace DH, Vockley J, Toledo FG and DeLany JP. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity (Silver Spring). 2010;18:1695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rutkowsky JM, Knotts TA, Ono-Moore KD, McCoin CS, Huang S, Schneider D, Singh S, Adams SH and Hwang DH. Acylcarnitines activate proinflammatory signaling pathways. Am J Physiol Endocrinol Metab. 2014;306:E1378–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bedi KC Jr, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, Wang LL, Javaheri A, Blair IA, Margulies KB and Rame JE. Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation. 2016;133:706–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldenberg JR, Carley AN, Ji R, Zhang X, Fasano M, Schulze PC and Lewandowski ED. Preservation of Acyl Coenzyme A Attenuates Pathological and Metabolic Cardiac Remodeling Through Selective Lipid Trafficking. Circulation. 2019;139:2765–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schulze PC, Drosatos K and Goldberg IJ. Lipid Use and Misuse by the Heart. Circ Res. 2016;118:1736–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peterson LR, Soto PF, Herrero P, Mohammed BS, Avidan MS, Schechtman KB, Dence C and Gropler RJ. Impact of gender on the myocardial metabolic response to obesity. JACC Cardiovasc Imaging. 2008;1:424–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herrero P, Peterson LR, McGill JB, Matthew S, Lesniak D, Dence C and Gropler RJ. Increased myocardial fatty acid metabolism in patients with type 1 diabetes mellitus. J Am Coll Cardiol. 2006;47:598–604. [DOI] [PubMed] [Google Scholar]
- 36.Xu G, Hansen JS, Zhao XJ, Chen S, Hoene M, Wang XL, Clemmesen JO, Secher NH, Haring HU, Pedersen BK, Lehmann R, Weigert C and Plomgaard P. Liver and Muscle Contribute Differently to the Plasma Acylcarnitine Pool During Fasting and Exercise in Humans. J Clin Endocrinol Metab. 2016;101:5044–5052. [DOI] [PubMed] [Google Scholar]
- 37.Mahmod M, Pal N, Rayner J, Holloway C, Raman B, Dass S, Levelt E, Ariga R, Ferreira V, Banerjee R, Schneider JE, Rodgers C, Francis JM, Karamitsos TD, Frenneaux M, Ashrafian H, Neubauer S and Rider O. The interplay between metabolic alterations, diastolic strain rate and exercise capacity in mild heart failure with preserved ejection fraction: a cardiovascular magnetic resonance study. J Cardiovasc Magn Reson. 2018;20:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Itier R, Guillaume M, Ricci JE, Roubille F, Delarche N, Picard F, Galinier M and Roncalli J. Non-alcoholic fatty liver disease and heart failure with preserved ejection fraction: from pathophysiology to practical issues. ESC Heart Fail. 2021;8:789–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ying W, Sharma K, Yanek LR, Vaidya D, Schär M, Markl M, Subramanya V, Soleimani S, Ouyang P, Michos ED, Shah SJ and Hays AG. Visceral adiposity, muscle composition, and exercise tolerance in heart failure with preserved ejection fraction. ESC Heart Fail. 2021;8:2535–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang L, Lu Y, Jiang H, Zhang L, Sun A, Zou Y and Ge J. Additional use of trimetazidine in patients with chronic heart failure: a meta-analysis. J Am Coll Cardiol. 2012;59:913–22. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, Taufalele PV, Cochran JD, Robillard-Frayne I, Marx JM, Soto J, Rauckhorst AJ, Tayyari F, Pewa AD, Gray LR, Teesch LM, Puchalska P, Funari TR, McGlauflin R, Zimmerman K, Kutschke WJ, Cassier T, Hitchcock S, Lin K, Kato KM, Stueve JL, Haff L, Weiss RM, Cox JE, Rutter J, Taylor EB, Crawford PA, Lewandowski ED, Des Rosiers C and Abel ED. Mitochondrial pyruvate carriers are required for myocardial stress adaptation. Nat Metab. 2020;2:1248–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fernandez-Caggiano M, Kamynina A, Francois AA, Prysyazhna O, Eykyn TR, Krasemann S, Crespo-Leiro MG, Vieites MG, Bianchi K, Morales V, Domenech N and Eaton P. Mitochondrial pyruvate carrier abundance mediates pathological cardiac hypertrophy. Nat Metab. 2020;2:1223–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCommis KS, Kovacs A, Weinheimer CJ, Shew TM, Koves TR, Ilkayeva OR, Kamm DR, Pyles KD, King MT, Veech RL, DeBosch BJ, Muoio DM, Gross RW and Finck BN. Nutritional modulation of heart failure in mitochondrial pyruvate carrier-deficient mice. Nat Metab. 2020;2:1232–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Razavi AC, Bazzano LA, He J, Fernandez C, Whelton SP, Krousel-Wood M, Li S, Nierenberg JL, Shi M, Li C, Mi X, Kinchen J and Kelly TN. Novel Findings From a Metabolomics Study of Left Ventricular Diastolic Function: The Bogalusa Heart Study. J Am Heart Assoc. 2020;9:e015118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sheffield-Moore M, Dillon EL, Randolph KM, Casperson SL, White GR, Jennings K, Rathmacher J, Schuette S, Janghorbani M, Urban RJ, Hoang V, Willis M and Durham WJ. Isotopic decay of urinary or plasma 3-methylhistidine as a potential biomarker of pathologic skeletal muscle loss. J Cachexia Sarcopenia Muscle. 2014;5:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kochlik B, Stuetz W, Peres K, Feart C, Tegner J, Rodriguez-Manas L, Grune T and Weber D. Associations of Plasma 3-Methylhistidine with Frailty Status in French Cohorts of the FRAILOMIC Initiative. J Clin Med. 2019;8:1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kochlik B, Gerbracht C, Grune T and Weber D. The Influence of Dietary Habits and Meat Consumption on Plasma 3-Methylhistidine-A Potential Marker for Muscle Protein Turnover. Mol Nutr Food Res. 2018;62:e1701062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, Kapoor K, Koves TR, Stevens R, Ilkayeva OR, Vega RB, Attie AD, Muoio DM and Kelly DP. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ Heart Fail. 2014;7:1022–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suryawan A, Hawes JW, Harris RA, Shimomura Y, Jenkins AE and Hutson SM. A molecular model of human branched-chain amino acid metabolism. Am J Clin Nutr. 1998;68:72–81. [DOI] [PubMed] [Google Scholar]
- 50.Biswas D, Duffley L and Pulinilkunnil T. Role of branched-chain amino acid-catabolizing enzymes in intertissue signaling, metabolic remodeling, and energy homeostasis. FASEB J. 2019;33:8711–8731. [DOI] [PubMed] [Google Scholar]
- 51.Walejko JM, Christopher BA, Crown SB, Zhang GF, Pickar-Oliver A, Yoneshiro T, Foster MW, Page S, van Vliet S, Ilkayeva O, Muehlbauer MJ, Carson MW, Brozinick JT, Hammond CD, Gimeno RE, Moseley MA, Kajimura S, Gersbach CA, Newgard CB, White PJ and McGarrah RW. Branched-chain alpha-ketoacids are preferentially reaminated and activate protein synthesis in the heart. Nat Commun. 2021;12:1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paxton R and Harris RA. Isolation of rabbit liver branched chain alpha-ketoacid dehydrogenase and regulation by phosphorylation. J Biol Chem. 1982;257:14433–9. [PubMed] [Google Scholar]
- 53.Murashige D, Jung JW, Neinast MD, Levin MG, Chu Q, Lambert JP, Garbincius JF, Kim B, Hoshino A, Marti-Pamies I, McDaid KS, Shewale SV, Flam E, Yang S, Roberts E, Li L, Morley MP, Bedi KC Jr, Hyman MC, Frankel DS, Margulies KB, Assoian RK, Elrod JW, Jang C, Rabinowitz JD and Arany Z. Extra-cardiac BCAA catabolism lowers blood pressure and protects from heart failure. Cell Metab. 2022;34:1749–1764.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, Guertin DA, Sabatini DM and Baur JA. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335:1638–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR and Sabatini DM. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science. 2016;351:43–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sener A and Malaisse WJ. L-leucine and a nonmetabolized analogue activate pancreatic islet glutamate dehydrogenase. Nature. 1980;288:187–9. [DOI] [PubMed] [Google Scholar]
- 57.Harris LLS, Smith GI, Patterson BW, Ramaswamy RS, Okunade AL, Kelly SC, Porter LC, Klein S, Yoshino J and Mittendorfer B. Alterations in 3-Hydroxyisobutyrate and FGF21 Metabolism Are Associated With Protein Ingestion-Induced Insulin Resistance. Diabetes. 2017;66:1871–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lotta LA, Scott RA, Sharp SJ, Burgess S, Luan J, Tillin T, Schmidt AF, Imamura F, Stewart ID, Perry JR, Marney L, Koulman A, Karoly ED, Forouhi NG, Sjogren RJ, Naslund E, Zierath JR, Krook A, Savage DB, Griffin JL, Chaturvedi N, Hingorani AD, Khaw KT, Barroso I, McCarthy MI, O’Rahilly S, Wareham NJ and Langenberg C. Genetic Predisposition to an Impaired Metabolism of the Branched-Chain Amino Acids and Risk of Type 2 Diabetes: A Mendelian Randomisation Analysis. PLoS Med. 2016;13:e1002179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, Lewis GD, Fox CS, Jacques PF, Fernandez C, O’Donnell CJ, Carr SA, Mootha VK, Florez JC, Souza A, Melander O, Clish CB and Gerszten RE. Metabolite profiles and the risk of developing diabetes. Nat Med. 2011;17:448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kappel BA, Lehrke M, Schutt K, Artati A, Adamski J, Lebherz C and Marx N. Effect of Empagliflozin on the Metabolic Signature of Patients With Type 2 Diabetes Mellitus and Cardiovascular Disease. Circulation. 2017;136:969–972. [DOI] [PubMed] [Google Scholar]
- 61.Li T, Zhang Z, Kolwicz SC Jr, Abell L, Roe ND, Kim M, Zhou B, Cao Y, Ritterhoff J, Gu H, Raftery D, Sun H and Tian R. Defective Branched-Chain Amino Acid Catabolism Disrupts Glucose Metabolism and Sensitizes the Heart to Ischemia-Reperfusion Injury. Cell Metab. 2017;25:374–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, Jeyaraj D, Youn JY, Ren S, Liu Y, Rau CD, Shah S, Ilkayeva O, Gui WJ, William NS, Wynn RM, Newgard CB, Cai H, Xiao X, Chuang DT, Schulze PC, Lynch C, Jain MK and Wang Y. Catabolic Defect of Branched-Chain Amino Acids Promotes Heart Failure. Circulation. 2016;133:2038–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, Koves T, Gardell SJ, Kruger M, Hoppel CL, Lewandowski ED, Crawford PA, Muoio DM and Kelly DP. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation. 2016;133:698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Horton JL, Davidson MT, Kurishima C, Vega RB, Powers JC, Matsuura TR, Petucci C, Lewandowski ED, Crawford PA, Muoio DM, Recchia FA and Kelly DP. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. JCI Insight. 2019;4:e124079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nielsen R, Moller N, Gormsen LC, Tolbod LP, Hansson NH, Sorensen J, Harms HJ, Frokiaer J, Eiskjaer H, Jespersen NR, Mellemkjaer S, Lassen TR, Pryds K, Botker HE and Wiggers H. Cardiovascular Effects of Treatment With the Ketone Body 3-Hydroxybutyrate in Chronic Heart Failure Patients. Circulation. 2019;139:2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yurista SR, Matsuura TR, Sillje HHW, Nijholt KT, McDaid KS, Shewale SV, Leone TC, Newman JC, Verdin E, van Veldhuisen DJ, de Boer RA, Kelly DP and Westenbrink BD. Ketone Ester Treatment Improves Cardiac Function and Reduces Pathologic Remodeling in Preclinical Models of Heart Failure. Circ Heart Fail. 2021;14:e007684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ferrannini E, Baldi S, Frascerra S, Astiarraga B, Heise T, Bizzotto R, Mari A, Pieber TR and Muscelli E. Shift to Fatty Substrate Utilization in Response to Sodium-Glucose Cotransporter 2 Inhibition in Subjects Without Diabetes and Patients With Type 2 Diabetes. Diabetes. 2016;65:1190–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The majority of the data presented in this paper are provided in the Figures and Tables. The methods used for the metabolomic analysis are available in detail from the University of Pennsylvania Metabolomics Core. The raw metabolomics data coupled to de-identified metadata and the R script used to perform the analyses will be provided upon reasonable request.