

Abstract
Hydroxyapatite (HAP) constitutes the primary mineral component of bones, and its crystal structure, along with the surface interaction with proteins, significantly influences the outstanding mechanical properties of bone. This study focuses on natural hydroxyapatite, constructing a surface model with calcium vacancy defects. Employing a representative model of aspartic acid residues, we delve into the adsorption mechanism on the crystal surface and scrutinize the adsorption forms of amino acid residues on HAP and calcium-deficient hydroxyapatite (CDHA) surfaces. The research also explores the impact of different environments on adsorption energy. Furthermore, a simplified sandwich structure of crystal-polypeptide-crystal is presented, analyzing the distribution of amino acid residue adsorption sites on the crystal surface of the polypeptide fragment. This investigation aims to elucidate how the stick–slip mechanism of polypeptide molecules on the crystal surface influences the mechanical properties of the system. By uncovering the interface mechanical behavior between HAP and osteopontin peptides, this article offers valuable theoretical insights for the construction and biomimetic design of biocomposites.
Subject terms: Molecular biology, Materials science
Introduction
Bone, a paradigmatic nanocomposite material, is composed of 25% proteinaceous organic matter and primarily features Hydroxyapatite (HAP) crystals in its inorganic components1. Remarkably, the crystal structure undergoes dynamic alterations within living organisms. The mineralization process of HAP in natural bone tissue unfolds in a low-temperature and body fluid environment, resulting in the formation of CDHA due to calcium vacancy defects2,3. During the formation of CDHA, protons in the solution environment bind with hydroxyl and phosphate groups to form H2O and HPO42, leading to increased calcium precipitation and formation of calcium vacancies in the crystal; ultimately, Ca II vacancies are replaced by adjacent H2O and HPO42− to maintain charge neutrality, resulting in the formation of calcium-deficient hydroxyapatite in bone tissues4. In the solution environment of CDHA, proton interactions lead to the formation of water and hydrogen phosphate ions, causing a shift in the Ca/P ratio as the pH decreases5,6. The correction of calcium vacancies by protons results in the formation of crystals with a chemical formula of Ca10−x(HPO4)x(PO4)6−x(OH)2−x (0 < x < 1)7.
Natural apatite in bone tissue, particularly CDHA formed through organism metabolism, significantly contributes to bone reconstruction and growth, imparting strength and supporting physiological activities8,9. Despite this, the mechanical properties, including bone toughness, are intricately linked to the organic component's proteins10. Through an examination of bone tissue microstructure, it is evident that osteopontin (OPN) acts as a binder at the crystal-protein interface, promoting strong collagen adhesion to the crystal surface and enhancing bone toughness11,12. OPN presence reduces structural deformation and fractures in collagen and mineral crystals in the staggered arrangement of bones, also mitigating the loss of interface adsorption energy during bone damage13. Highly phosphorylated OPN, rich in aspartic and glutamic acid, demonstrates diverse regulatory functions concerning HAP crystals14. The analysis of OPN's amino acid residues reveals its robust binding ability to calcium ions, particularly on the crystal's (100) surface15–17. Studies modifying OPN's conformation through phosphorylation enhance its binding affinity to the HAP surface18–21. Amino acid residues containing carboxyl groups, like aspartic acid and glutamic acid, play a pivotal role in the interaction between proteins and crystals, as confirmed by molecular dynamics simulations of OPN adsorption22,23.
In the intricate process of bone formation, HAP crystals embark on their initiation and development journey within the periodic interstitial channels nestled between adjacent collagen molecules24,25. Over time, a prevailing perspective suggests that these crystals adopt a staggered arrangement, contributing to a structural framework endowed with exceptional toughness26. This resilience is primarily attributed to the synergistic effects of proteins releasing fracture energy and the adsorption energy at the two-phase interface27,28. At the nanoscale, the interface interaction between HAP crystals and collagen showcases remarkable resistance to high shear loads, underscoring the imperative need to explore interface mechanical behavior for a comprehensive understanding of bone tissue material properties29.
Within bone tissue, the intricate cross-linking between organic matter and crystals is not solely dictated by adsorption sites but is intricately influenced by factors like the surface morphology of crystals and the dynamic sliding of other protein molecules on the crystal surface30. The biomolecular regulation during HAP nucleation and growth induces calcium vacancy defects, prompting alterations in both internal and surface spatial structures, introducing irregularities and undulations in the crystal lattice31. The atomic morphology on the HAP surface plays a pivotal role in influencing protein adsorption capacity, with research indicating that the concave and convex structure of the crystal surface enhances protein interaction32. However, the characterization of the surface structure based on calcium ion morphology remains an underexplored realm. Moreover, at the nanoscale, shear load governs slip at the interface between protein molecules and crystals, dissipating a substantial amount of energy and efficaciously averting material failure33. The resistance to slip at this interface is multifactorial, encompassing considerations of the viscoelastic properties of proteins, interface adsorption energy, adsorption form, among others.
In this study, we delve into the nanoscale examination of mineralized collagen fibers in bones. During crystal nucleation and growth, HAP displays a specific orientation influenced by the body fluid environment, revealing internal flaws. Limited research exists on the adsorption site mechanism during crystal-protein interaction, particularly in understanding the unclear interface toughening mechanism. Employing molecular dynamics, we constructed comprehensive surface models of HAP (100) and CDHA (100) surfaces. Molecular dynamics simulations investigated the interaction mechanism of aspartic acid residues on the crystal surface, aiming to unveil the protein adsorption site interaction and explore the impact of different environments on adsorption energy. Additionally, crystal models with diverse surface morphologies were examined to understand their influence on the interface behavior of ossification protein polypeptide chains under shear load through molecular dynamics restricted shear simulation. This effort aims to elucidate biological responses under shear load and the anti-deformation mechanism of composite materials, providing theoretical guidance for biocomposite construction and biomimetic design.
Materials and methods
Modeling description
HAP is represented as Ca10(PO4)6(OH)2 with P63/m symmetry, having lattice parameters a = b = 0.9432 nm, c = 0.6881 nm, and angles α = β = 90°, γ = 120°. The crystal contains two types of Ca atoms: Ca I (4 atoms) at z = 0 and z = 1/2, and Ca II (6 atoms) forming a channel structure along the c-axis, creating a triangular arrangement around hydroxyl groups34–36. The CDHA is delineated by the formula Ca10−x(HPO4)x(PO4)6−x(H2O)x (0 < x ≤ 1). Given that the mineralization process of biological bone tissue takes place in the relatively lower-temperature environment of body fluids and is regulated by acidic amino acids37, it is more probable for HAP containing Ca II vacancies to form in bone tissue. Therefore, this work focuses on HAP crystals with Ca II vacancy defects. In the model, one Ca II atom is removed, and the charge is compensated by two protons. The removed Ca II atom is then combined with adjacent PO43− groups and hydroxyl groups, respectively, forming HPO42− groups. In this work, two forms of defect structures were constructed, as illustrated in Fig. 1. These structures are identified as the single defect structure CDHA (CDHA-1) and the double defect structure CDHA (CDHA-2).
Figure 1.

Ca II vacancy model: (a) CDHA-1, and (b) CDHA-2.
The investigation into adsorption sites focused on aspartic acid residues as the subject of study38. Aspartic acid, with a chemical formula of HOOCCH2CH(NH2)COOH, is known for its easy combination with calcium ions to form adsorption sites. The potential energy of aspartic acid is described using COMPASS II force field parameters39. The geometric optimization of the aspartate structure involves ensuring that the initial aspartate residue attains the most stable structural state with the smallest initial potential energy. The optimized spatial structure is illustrated in Fig. 2.
Figure 2.
Aspartic acid residue model.
OPN in humans comprises 298 amino acid residues, with approximately 25% of them being acidic amino acids. Drawing on existing research on OPN polypeptides, the amino acid sequence selected for this experiment is DDSHQSDESHHSDESDEL. The atomic structures of various amino acids are based on the research by Addison et al40. This polypeptide consists of 8 residues, each being either aspartic acid (D) or glutamic acid (E), and its structure is depicted in Fig. 3.
Figure 3.
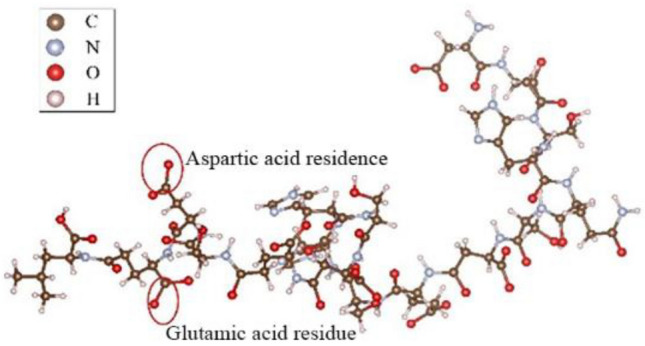
OPN peptide model.
Description of the method
In this study, we utilized the Forcite module within Materials Studio, a comprehensive computational platform for materials science developed by BIOVIA. The Forcite module is particularly adept at performing a range of computational tasks, including rapid energy calculations for single molecules and periodic systems, as well as geometric optimizations to identify stable conformations at minimum energy states. Additionally, Forcite supports molecular dynamics simulations, which are essential for understanding the dynamic behavior of molecules and their interactions within a given environment. For the purposes of this research, the Forcite module was employed for geometric optimization and molecular dynamics simulations to ensure that the modeled structures were in their energetically favorable states and to accurately capture the adsorption and shear behaviors of the materials under investigation.
Adsorption simulation parameters
This study employs molecular dynamics simulation, placing the (100) face of two crystal types amid amino acid residues. The simulation starts from the optimized structural position, introducing 100 water molecules to hydrate the system and immobilizing atoms on the crystal surface. Two simulation environments are utilized: a neutral water molecule setting for comparison and an environment with added HPO42− and H2PO4− in a 1:1 ratio. The Forcite module in Materials Studio is used for simulation, employing the NVT ensemble with Nosé-Hoover for temperature control to mimic the biological environment at 310 K. The Ewald method calculates electrostatic interactions, and the simulation has a time step of 1 fs, a system simulation time of 5 ps, and energy deviation controlled at 50,000 kcal/mol.
Shear simulation settings
The restricted shear calculation in this study utilized the Forcite module in Materials Studio. Initially, the Amorphous Cell module constructed the polypeptide's amorphous structure, added to a simulation box (38 Å × 20 Å × 20 Å). The Build layer feature created a sandwich structure of crystals and polypeptides. The Forcite module's Confined Shear feature simulated with a shear speed of 1 Å/ps, a temperature of 310 K, a time step of 1 fs, and a total simulation time of 10 ps.
Results and discussion
Adsorption mechanism of aspartic acid residue on hydroxyapatite
The molecular dynamics simulations were conducted to achieve the adsorption of aspartic acid residues on the (100) surface of both HAP and CDHA. However, there are differences in the adsorption behaviors between the two, and the atomic snapshots of the adsorption process are illustrated in Fig. 4.
Figure 4.

Atomic snapshots. (a) HAP (H2O). (b) HAP (HPO4). (c) CDHA (H2O). (d) CDHA (HPO4).
In aqueous conditions, molecular dynamics simulations on the HAP surface reveal interactions between the carboxyl group of aspartic acid and calcium ions, forming close contacts with bond lengths ranging from approximately 1.6 to 2.5 Å. Additionally, the amino end of aspartic acid exhibits short-distance contacts with oxygen atoms of adjacent PO43− groups, forming bond lengths between 1.0 and 1.5 Å. By 3 ps, adsorption reaches a stable state, with both the amino and carboxyl groups extending outward. The carboxyl and amino groups elongate by 0.003 Å and 0.009 Å, respectively. In contrast, structural changes on the CDHA crystal surface are more significant, with carboxyl and amino groups extending by 0.009 Å and 0.011 Å, respectively, emphasizing the greater impact of vacancy defects on aspartic acid. In a hydrogen phosphate solution environment, structural changes in aspartic acid are more pronounced than in the aqueous solution.
Observing changes in aspartic acid and HAP during adsorption reveals that it mainly occurs through interactions of –COO (carboxyl) and NH2 (amino) groups. Calcium ions interact with –COO, and phosphate groups with NH2. The adsorption involves oxygen atoms on aspartic acid's carboxyl groups, carrying a negative charge, facilitating binding to Ca cations on the crystal surface. Additionally, the amino group tends to interact with the oxygen atom on the phosphate group. Oxygen atoms in carboxyl groups, having a larger charge, enhance electrostatic interactions. Adsorption between oxygen on the carboxyl group in aspartic acid and Ca ions on the HAP surface crucially strengthens the HAP-protein interface. In a phosphate buffer solution, the structural changes of aspartic acid are more pronounced compared to in water due to the acidic nature of the phosphate buffer. The acidic H2PO4− ions in the solution protonate the functional groups of aspartic acid, forming NH3+ and –COOH2+ groups, leading to an overall positive charge on the molecule. This enhances internal electrostatic interactions and hydrogen bonding, making the molecule more compact and stable.
The examination of the adsorption process highlights the significant influence of the calcium triangular channel on the crystal, shaping the adsorption structure. Figure 5 displays the stable state and atomic snapshots, offering a visual understanding of –COO and calcium ion channel interactions.
Figure 5.

Interactions between –COO and Calcium Ion Channels: (a-1,a-2) depict the structure of the adsorption site on HAP, while (b-1,b-2) show the structure of the adsorption site on CDHA.
In stable-state adsorption, carboxyl groups tightly bind to the HAP surface, with carboxyl group oxygen interacting with three calcium ions, maintaining a central position in a triangular structure. In vacancies, carboxyl groups shift closer to remaining calcium ions, reducing distances by 0.397 Å and 0.486 Å, positioning them near the crystal surface. Amino acid chain deformation reveals the amino group entering the space around HAP phosphate ions, forming hydrogen bonds with PO4 oxygen on the crystal surface. Amino end adsorption causes slight deformation in the corresponding carbon chain. During HAP mineralization, calcium vacancies induce structural changes, resulting in sparse HPO4 distribution on the surface, leading to weaker hydrogen bonding with the amino end. After adding hydrogen phosphate, HPO4 primarily interacts with aspartic acid's amino group. Hydrogen phosphate ionization imparts acidity, simulating HAP mineralization. Phosphate ions tend to adsorb on the crystal surface, mimicking growth along the (100) crystal plane, forming stronger hydrogen bonds than in a conventional solution environment.
Interface behavior of polypeptide at the hydroxyapatite interface
Figures 6, 7 and 8 present atomic snapshots during shear simulations. Observing the first three snapshots (a), (b), (c), it is evident that the crystal's shear displacement is small, ranging from 0.15 to 2 Å. Consequently, there is limited structural change in the central polypeptide. Interface alterations primarily manifest in the configuration of peptide adsorption sites on the crystal. Amino acid residues on the peptide chain are concentrated at both ends, resulting in a "thicker at both ends, narrower in the middle" appearance. The following three snapshots (d), (e), (f) correspond to structural changes within 2–10 ps. Although there are minor energy fluctuations during this period, the overall trend is toward stability. The structure of residue aggregation areas at both ends of the peptide chain remains highly stable, undergoing minimal change during shear, indicating that the adsorption configuration of end residues has reached stability. Energy fluctuations are primarily caused by deformation in the spatial structure of the residue-free section in the middle of the peptide chain, with the transferred shear energy being compensated, ensuring the overall structural integrity of the simulation system.
Figure 6.

Atomic snapshots of deformation processes for HAP-peptide shear simulations.
Figure 7.

Atomic snapshots of deformation processes for CDHA1-peptide shear simulations.
Figure 8.

Atomic snapshots of deformation processes for CDHA2-peptide shear simulations.
This study further elucidates the interaction energy and deformation processes at different shear rates through atomic snapshots, as depicted in Fig. 9. The figure illustrates the interaction energy between the peptide chain and the upper and lower surfaces of HAP and two types of CDHA at shear rates of 0.5 Å/ps, 0.8 Å/ps, and 1.0 Å/ps, along with the structural features of the peptide and crystal when the configuration stabilizes. Among the three crystals, the interaction energy between HAP and the peptide is higher than the other two, but its stability is slightly compromised, showing greater fluctuations and requiring a longer loading process to reach a stable state. The interaction energy of HAP exhibits a staggered decreasing trend, while both CDHA types rapidly decrease to stable values before gradually stabilizing, a pattern similarly observed in (b) and (c). Furthermore, energy analysis reveals that, in the two CDHA crystal types, when the adsorption configuration reaches a stable state, the interaction energy between the single-vacancy crystal and the peptide is lower than that of the double-vacancy structure.
Figure 9.

Shear Rate-dependent interaction energy and deformation processes.
Figure 10 intricately details the interface adsorption characteristics in simulation systems of HAP, CDHA-1, and CDHA-2 under a shear rate of 1.0 Å/ps, specifically focusing on strain values of 0.01 and 0.05. In the context of (a-1), the initial loading phase for the HAP crystal reveals a noteworthy reconstruction of adsorbed residues on the interface. Initially, a solitary amino acid residue (aspartic acid residue) on the lower interface significantly influences interface interactions, while the upper interface exhibits dual configurations. This dynamic is observed during the swift decrease in interaction energy between HAP and peptides as depicted in Fig. 9c. As the loading progresses, the configurations of adsorbed residues undergo a comprehensive reconstruction, as illustrated in (a-2). Nearly all residues participate in the adsorption process on both upper and lower interfaces, contributing to the stabilization of interaction energy. Analogously, in (b-1) and (c-1), configurations of structures with vacancy defects are initially fewer than those of HAP, elucidating their lower interaction energy. Subsequent to continued loading, the configurations on the interface achieve stability after reaching a strain value of 0.05, portrayed in (b-2) and (c-2). Notably, double-vacancy structures exhibit a proclivity for the aggregation of aspartic acid and glutamic acid residues on the peptide chain, resulting in more stable adsorption configurations in comparison to single-vacancy structures. Consequently, despite harboring a greater number of calcium ion vacancies, CDHA-2 manifests higher interaction energy and forms more resilient adsorption configurations compared to CDHA-1, a nuanced observation derived from Fig. 9.
Figure 10.

Shear-induced structural changes at the polypeptide-hydroxyapatite interface from 2-10 ps: (a-1) Initial Loading Phase and Reconstruction of Adsorbed Residues on HAP Interface. (a-2) Comprehensive Reconstruction of Adsorbed Residues on HAP Interface. (b-1) Initial Configurations of CDHA-1 with Vacancy Defects. (b-2) Stability Achieved in Adsorption Configurations of CDHA-1. (c-1) Initial Configurations of CDHA-2 with Vacancy Defects; (c-2) More Resilient Adsorption Configurations in CDHA-2.
Conclusion
In conclusion, this study delves into nanoscale investigations of bone tissue, revealing insights into its mechanical properties and dynamic evolution. Employing comprehensive models and molecular dynamics simulations, the research analyzes stability changes during loading, emphasizing the pivotal -COO and Ca interaction and the influence of amino groups on aspartic acid's structural deformation. Calcium vacancies impact carboxyl group adsorption, and hydrogen phosphate stabilizes the process. The shear simulation highlights energy fluctuations and equilibrium attainment during adsorption, showcasing the efficiency of the double-vacancy structure in enhancing polypeptide adhesion and interface mechanical properties. Overall, the study provides theoretical guidance for biocomposite construction, bionic design, and microscale experiments.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 32271432), the National Key Research and Development Program (No. 2022YFC3601900), the Key Project of Natural Science Foundation of Tianjin (No. 22JCZDJC00340), and the Tianjin Health and Health Technology Project (No. TJWJ2022QN053).
Author contributions
Yadi Sun, Yan Wang and Chunhui Ji: Writing—review & editing, Writing—original draft, Resources, Investigation, Conceptualization. Bingnan He: Writing-original draft, Formal analysis, Data curation, Formal analysis. Jianxiong Ma: Supervision, Project administration, Methodology, Conceptualization.
Data availability
The data generated and/or analyzed during the current study cannot be publicly disclosed due to their intended use in subsequent experiments, but they are available upon reasonable request to the corresponding author.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Yadi Sun and Yan Wang.
Contributor Information
Chunhui Ji, Email: jichunhui@tju.edu.cn.
Jianxiong Ma, Email: yjslwtg@126.com.
References
- 1.Sabater González M. Skeletal cartilage and bone formation, composition, and function in small mammals, birds, and reptiles. Vet. Clin. North Am. 2019;22:123–134. doi: 10.1016/j.cvex.2019.01.001. [DOI] [PubMed] [Google Scholar]
- 2.Chauhan N, Singh Y. L-histidine controls the hydroxyapatite mineralization with plate-like morphology: Effect of concentration and media. Mater. Sci. Eng. C. 2021;120:111669. doi: 10.1016/j.msec.2020.111669. [DOI] [PubMed] [Google Scholar]
- 3.Liou S-C, Chen S-Y, Liu D-M. Synthesis and characterization of needlelike apatitic nanocomposite with controlled aspect ratios. Biomaterials. 2003;24:3981–3988. doi: 10.1016/S0142-9612(03)00303-X. [DOI] [PubMed] [Google Scholar]
- 4.Matsunaga K. Theoretical investigation of the defect formation mechanism relevant to nonstoichiometry in hydroxyapatite. Phys. Rev. B. 2008;77:104106. doi: 10.1103/physrevb.77.104106. [DOI] [Google Scholar]
- 5.Bhat SS, Waghmare UV, Ramamurty U. First-principles study of structure, vibrational, and elastic properties of stoichiometric and calcium-deficient hydroxyapatite. Cryst. Growth Des. 2014;14:3131–3141. doi: 10.1021/cg5004269. [DOI] [Google Scholar]
- 6.Zhang H, Darvell BW. Morphology and structural characteristics of hydroxyapatite whiskers: effect of the initial Ca concentration, Ca/P ratio and pH. Acta Biomater. 2011;7:2960–2968. doi: 10.1016/j.actbio.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 7.Bustos-flores E, Elizondo-álvarez MA, Uribe-salas A. Thermodynamic and experimental studies on removal of calcium and sulfate ions from recycling water of complex sulfide flotation operations. Trans. Nonferrous Metal. Soc. 2021;31:3116–3127. doi: 10.1016/s1003-6326(21)65720-5. [DOI] [Google Scholar]
- 8.Veiga A, Castro F, Rocha F, Oliveira AL. An update on hydroxyapatite/collagen composites: What is there left to say about these bioinspired materials? J. Biomed. Mater. Res. B. 2022;110:1192–1205. doi: 10.1002/jbm.b.34976. [DOI] [PubMed] [Google Scholar]
- 9.Cardoso GBC, et al. In vivo approach of calcium deficient hydroxyapatite filler as bone induction factor. Mater. Sci. Eng. C. 2019;99:999–1006. doi: 10.1016/j.msec.2019.02.060. [DOI] [PubMed] [Google Scholar]
- 10.Gautieri A, Vesentini S, Redaelli A, Buehler MJ. Viscoelastic properties of model segments of collagen molecules. Matrix Biol. 2012;31:141–149. doi: 10.1016/j.matbio.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 11.Nikel O, Laurencin D, McCallum SA, Gundberg CM, Vashishth D. NMR investigation of the role of osteocalcin and osteopontin at the organic-inorganic interface in bone. Langmuir. 2013;29:13873–13882. doi: 10.1021/la403203w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poundarik AA, et al. Dilatational band formation in bone. Proc. Natl. Acad. Sci. U S A. 2012;109:19178–19183. doi: 10.1073/pnas.1201513109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buehler MJ. Molecular nanomechanics of nascent bone: Fibrillar toughening by mineralization. Nanotechnology. 2007;18:295102. doi: 10.1088/0957-4484/18/29/295102. [DOI] [Google Scholar]
- 14.Shapses SA, et al. Osteopontin facilitates bone resorption, decreasing bone mineral crystallinity and content during calcium deficiency. Calcif. Tissue Int. 2003;73:86–92. doi: 10.1007/s00223-002-1090-x. [DOI] [PubMed] [Google Scholar]
- 15.Oldberg A, Franzén A, Heinegård D. Cloning and sequence analysis of rat bone sialoprotein (osteopontin) cDNA reveals an Arg-Gly-Asp cell-binding sequence. Proc. Natl. Acad. Sci. USA. 1986;83:8819–8823. doi: 10.1073/pnas.83.23.8819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giachelli CM, Steitz S. Osteopontin: A versatile regulator of inflammation and biomineralization. Matrix Biol. 2000;19:615–622. doi: 10.1016/S0945-053X(00)00108-6. [DOI] [PubMed] [Google Scholar]
- 17.Ryuichi F, Yoshinori K. Preferential adsorption of dentin and bone acidic proteins on the (100) face of hydroxyapatite crystals. Biochim. Biophys. Acta. 1991;1075:56–60. doi: 10.1016/0304-4165(91)90074-Q. [DOI] [PubMed] [Google Scholar]
- 18.Hoang QQ, Sicheri F, Howard AJ, Yang DSC. Bone recognition mechanism of porcine osteocalcin from crystal structure. Nature. 2003;425:977–980. doi: 10.1038/nature02079. [DOI] [PubMed] [Google Scholar]
- 19.Grohe B, et al. Control of calcium oxalate crystal growth by face-specific adsorption of an osteopontin phosphopeptide. J. Am. Chem. Soc. 2007;129:14946–14951. doi: 10.1021/ja0745613. [DOI] [PubMed] [Google Scholar]
- 20.Hunter GK, O’Young J, Grohe B, Karttunen M, Goldberg HA. The flexible polyelectrolyte hypothesis of protein−biomineral interaction. Langmuir. 2010;26:18639–18646. doi: 10.1021/la100401r. [DOI] [PubMed] [Google Scholar]
- 21.Holzinger J, Kotisch H, Richter K, Konrat R. Binding mode characterization of osteopontin on hydroxyapatite by solution NMR spectroscopy. ChemBioChem. 2021;22:2300–2305. doi: 10.1002/cbic.202100139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Addison WN, et al. Phosphorylation-dependent mineral-type specificity for apatite-binding peptide sequences. Biomaterials. 2010;31(36):9422–9430. doi: 10.1016/j.biomaterials.2010.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hug S, Hunter GK, Goldberg H, Karttunen M. Ab initio simulations of peptide-mineral interactions. Phys. Procedia. 2010;4:51–60. doi: 10.1016/j.phpro.2010.08.008. [DOI] [Google Scholar]
- 24.Lees S, Prostak KS, Ingle VK, Kjoller K. The loci of mineral in turkey leg tendon as seen by atomic force microscope and electron microscopy. Calcif. Tissue Int. 1994;55:180–189. doi: 10.1007/BF00425873. [DOI] [PubMed] [Google Scholar]
- 25.Hassenkam T, et al. High-resolution AFM imaging of intact and fractured trabecular bone. Bone. 2004;35:4–10. doi: 10.1016/j.bone.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 26.Gao H, Ji B, Jäger IL, Arzt E, Fratzl P. Materials become insensitive to flaws at nanoscale: Lessons from nature. Proc. Natl. Acad. Sci. U S A. 2003;100:5597–5600. doi: 10.1073/pnas.0631609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang RZ, Suo Z, Evans AG, Yao N, Aksay IA. Deformation mechanisms in nacre. J. Mater. Res. 2001;16:2485–2493. doi: 10.1557/JMR.2001.0340. [DOI] [Google Scholar]
- 28.Smith BL, et al. Molecular mechanistic origin of the toughness of natural adhesives, fibres and composites. Nature. 1999;399:761–763. doi: 10.1038/21607. [DOI] [Google Scholar]
- 29.Ji B, Gao H. Mechanical properties of nanostructure of biological materials. J. Mech. Phys. Solids. 2004;52:1963–1990. doi: 10.1016/j.jmps.2004.03.006. [DOI] [Google Scholar]
- 30.George A, Veis A. Phosphorylated proteins and control over apatite nucleation, crystal growth, and inhibition. Chem. Rev. 2008;108:4670–4693. doi: 10.1021/cr0782729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ji C, He B, Yun S, Bai X, Lin B. The fracture mechanical behavior simulation of calcium-deficient hydroxyapatite crystals by molecular dynamics and first-principles calculation. J. Mech. Behav. Biomed. Mater. 2023;137:105526. doi: 10.1016/j.jmbbm.2022.105526. [DOI] [PubMed] [Google Scholar]
- 32.Wang Q, et al. Effects of atomic-level nano-structured hydroxyapatite on adsorption of bone morphogenetic protein-7 and its derived peptide by computer simulation. Sci. Rep. 2017;7:15152. doi: 10.1038/s41598-017-15219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Launey ME, Buehler MJ, Ritchie RO. On the mechanistic origins of toughness in bone. Annu. Rev. Mater. Res. 2010;40:25–53. doi: 10.1146/annurev-matsci-070909-104427. [DOI] [Google Scholar]
- 34.Kandori K, Toshima S, Wakamura M, Fukusumi M, Morisada Y. Effects of modification of calcium hydroxyapatites by trivalent metal ions on the protein adsorption behavior. J. Phys. Chem. B. 2010;114:2399–2404. doi: 10.1021/jp911783r. [DOI] [PubMed] [Google Scholar]
- 35.Kandori K, Hamazaki H, Matsuzawa M, Togashi S. Selective adsorption of acidic protein of bovine serum albumin onto sheet-like calcium hydroxyapatite particles produced by microreactor. Adv. Powder Technol. 2014;25:354–359. doi: 10.1016/j.apt.2013.05.016. [DOI] [Google Scholar]
- 36.Franzén A, Hultenby K, Reinholt F, Önnerfjord P, Heinegård D. Altered osteoclast development and function in osteopontin deficient mice. J. Orthop. Res. 2008;26:721–728. doi: 10.1002/jor.20544. [DOI] [PubMed] [Google Scholar]
- 37.Ghiasi B, et al. Hydroxyapatite as a biomaterial: A gift that keeps on giving. Drug Dev. Ind. Pharm. 2020;46:1035–1062. doi: 10.1080/03639045.2020.1776321. [DOI] [PubMed] [Google Scholar]
- 38.Zeng J, et al. Simulation insight into the synergic role of citrate and polyaspartic peptide in biomineralization. Langmuir. 2021 doi: 10.1021/acs.langmuir.0c03626. [DOI] [PubMed] [Google Scholar]
- 39.MacKerell AD, Feig M, Brooks CL. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 40.Addison W, Masica D, Gray J, McKee M. Phosphorylation-dependent inhibition of mineralization by osteopontin ASARM peptides is regulated by PHEX cleavage. J. Bone Miner. Res. 2009;25:695–705. doi: 10.1359/jbmr.090832. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data generated and/or analyzed during the current study cannot be publicly disclosed due to their intended use in subsequent experiments, but they are available upon reasonable request to the corresponding author.