

Abstract
Alzheimer’s disease (AD) is the most common cause of dementia, accounting for an estimated 60–80% of cases, and is the sixth-leading cause of death in the United States. While considerable advancements have been made in the clinical care of AD, it remains a complicated disorder that can be difficult to identify definitively in its earliest stages. Recently, mass spectrometry (MS)-based metabolomics has shown significant potential for elucidation of disease mechanisms and identification of therapeutic targets as well diagnostic and prognostic markers that may be useful in resolving some of the difficulties affecting clinical AD studies, such as effective stratification. In this study, complementary gas chromatography- and liquid chromatography-MS platforms were used to detect and monitor 2080 metabolites and features in 48 postmortem tissue samples harvested from the superior frontal gyrus of male and female subjects. Samples were taken from four groups: 12 normal control (NC) patients, 12 cognitively normal subjects characterized as high pathology controls (HPC), 12 subjects with nonspecific mild cognitive impairment (MCI), and 12 subjects with AD. Multivariate statistics informed the construction and cross-validation (p < 0.01) of partial least squares-discriminant analysis (PLS-DA) models defined by a nine-metabolite panel of disease markers (lauric acid, stearic acid, myristic acid, palmitic acid, palmitoleic acid, and four unidentified mass spectral features). Receiver operating characteristic analysis showed high predictive accuracy of the resulting PLS-DA models for discrimination of NC (97%), HPC (92%), MCI (~96%), and AD (~96%) groups. Pathway analysis revealed significant disturbances in lysine degradation, fatty acid metabolism, and the degradation of branched-chain amino acids. Network analysis showed significant enrichment of 11 enzymes, predominantly within the mitochondria. The results expand basic knowledge of the metabolome related to AD and reveal pathways that can be targeted therapeutically. This study also provides a promising basis for the development of larger multisite projects to validate these candidate markers in readily available biospecimens such as blood to enable the effective screening, rapid diagnosis, accurate surveillance, and therapeutic monitoring of AD. All raw mass spectrometry data have been deposited to MassIVE (data set identifier MSV000087165).
Keywords: Alzheimer’s disease, biomarkers, mass spectrometry, metabolomics, pathogenesis
Graphical Abstract
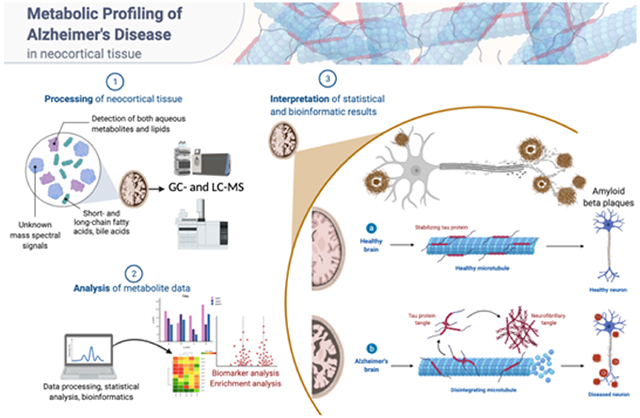
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disorder marked primarily by cognitive decline and dementia,1,2 in addition to the accumulation of extracellular amyloid β (Aβ) plaques and intracellular neurofibrillary tau tangles.3,4 AD is the most common cause of adult dementia, accounting for 60–80% of cases worldwide5 and is the sixth-leading cause of death in the United States.6 Currently, AD affects more than 5.8 million Americans,5,7 with prevalence expected to triple by 2050.8 In the United States, total payments in 2020 for healthcare, long-term care, and hospice services are estimated to be $305 billion.6 Consequently, AD represents a significant threat to human health and exerts a substantial financial and societal impact.
Considerable advancements have been made in the ability to accurately diagnose AD, largely owing to the development of positron emission tomography (PET) scans for detection of plaques and tangles, as well as cerebral spinal fluid (CSF) and plasma tests for AD-associated biomarkers.9 However, treatment of patients and appropriate evaluation of the outcomes of clinical studies remain complicated by the many unknowns involved in AD. Individuals with mild cognitive impairment (MCI) may be in the early stages of AD or may be affected by an unrelated disease process, confounding studies of early interventional treatments. In addition, some individuals with intermediate to high levels of AD-associated pathology (plaques and tangles) do not display cognitive deficits,10 indicating at least a partial disconnect between those specific pathologies and cognitive function. These resilient individuals, known as high pathology controls (HPC), can sustain AD-consistent pathology such as high amyloid loads, synaptic and neuronal demise, demyelination, and atrophy while simultaneously remaining cognitively intact.10,11 Although critical to our understanding of pathology- and cognition-specific metabolic alterations underlying AD,10-12 this group remains understudied. It is also common for AD to be found in conjunction with other pathologies such as synucleinopathy, TDP-43, or microinfarcts.13 Therefore, the ability to more effectively stratify and subgroup individuals would likely produce clearer and more actionable results. Furthermore, provisional diagnosis relies on a combination of mental status testing, neuropsychological tests, interviews with friends and family, laboratory tests, and various brain imaging techniques such as magnetic resonance imaging (MRI), computerized tomography (CT), and PET.14-16 These conventional diagnostic methods show low specificity against other dementias (70%) and only moderate sensitivity (80%).17 Additionally, these criteria are unable to capture early brain pathology that may predate symptoms by as much as 30 years;18 without timely diagnosis, patients are less likely to access appropriate treatment options that may slow disease progression.19,20 Consequently, there is a critical need for highly sensitive and specific markers of AD that may enable early disease detection as well as identification of potential drug targets, improved prognosis, and monitoring of therapeutic response.
A growing body of evidence suggests that perturbations in various metabolic pathways play a significant role in AD.21-25 Most notably, the mitochondrial cascade hypothesis states that widespread mitochondrial metabolic dysfunction is a strong characteristic of AD and plays a role in the accumulation of Aβ plaques.9,24,26 Furthermore, studies have also shown long- and short-chain fatty acids to play an important role in AD pathology, exerting both protective and pathogenic effects.5,21,24,26-28 Alterations in glycerophospholipid29 and phosphatidylcholine metabolism2,5,26,27,29 have also been strongly linked to AD in previous studies utilizing metabolomics-based approaches. More recently, an emerging role of the gut microbiome in AD has been identified,30 and metabolic profiling studies have demonstrated the potential for bile acids in discriminating subclinical forms of AD.31,32 Indeed, with advanced deterioration of cortical vasculature as seen in late-stage AD, it is possible for bile acids to be found in the central nervous system.33-35 Metabolomics, the scientific study of metabolic composition and pathways present in biological systems,26,36-41 has facilitated the accurate characterization of various metabolomes for advances in disease classification, drug therapy, and biomarker discovery. More specifically, mass spectrometry (MS) is an analytical approach in metabolomics that allows for the accurate detection and quantification of metabolites in biological samples.12,42,43 Various forms of untargeted and targeted MS-based metabolomic assays have been implemented in an attempt to profile biochemical processes in AD pathology. Methods mainly include direct infusion-MS,44 ultrahigh resolution-MS enabled by liquid chromatography (LC)-Orbitrap,45 and gas chromatography (GC)-time-of-flight-MS,24 which have been used in many previous studies for therapeutic drug targeting, disease characterization, and potential diagnostic biomarkers.
The current study employs hyphenated MS-based assays that combine both targeted and untargeted metabolomics approaches to detect aqueous metabolites, lipids, fatty acids, and bile acids, in addition to profiling unidentified features. A total of 2080 metabolites/features were detected in 48 samples of superior frontal gyrus tissue taken from four groups of patients: normal control (NC), HPC, MCI, and AD. Multivariate significance testing and model estimation constructed cross-validated partial least squares-discriminant analysis (PLS-DA) models confined to a highly predictive nine-metabolite panel of potential biomarkers capable of distinguishing each clinical group with high sensitivity and specificity. In conjunction with pathway and enrichment analyses, the current study corroborates findings of previous literature and adds to basic knowledge of the metabolome related to neurodegenerative decline as well as the behavioral symptoms of AD-induced dementia; this study offers a large-scale analysis of molecular alterations associated with AD pathogenesis and progression, potentially supporting future drug development and prevention efforts. Importantly, this study provides clinically relevant candidate biomarkers capable of accurate postmortem classification which may, eventually, prove useful to in vivo diagnosis and disease monitoring.
METHODS
Reagents
Acetonitrile (ACN), methanol (MeOH), ammonium acetate (NH4OAc), acetic acid (AcOH), and isopropanol (IPA), all LC–MS grade, were purchased from Fisher Scientific (Pittsburgh, PA). Ammonium hydroxide (NH4OH), methyl tert-butyl ether (MTBE), O-methylhydroxylamine hydrochloride (MeOX), and N-methyl-N-(tert-butyldimethylsilyl) trifluoroacetamide (MTBSTFA) were bought from Sigma-Aldrich (Saint Louis, MO). High-performance LC grade chloroform (CHCl3) was obtained from VWR (Radnor, PA). Deionized water was provided in-house by a water purification system from EMD Millipore (Billerica, MA). Phosphate-buffered saline (PBS) was bought from GE Healthcare Life Sciences (Logan, UT). Standard compounds corresponding to measured aqueous metabolites/features were purchased from Sigma-Aldrich and Fisher Scientific. Lipid standards were purchased from Fisher Scientific, Sigma-Aldrich, and Avanti Polar Lipids (Alabaster, AL).
Clinical Samples
Frozen tissue from the superior frontal gyrus of male and female subjects was obtained from the Arizona Study of Aging and Neurodegenerative Disorders/Brain and Body Donation Program at the Banner Sun Health Research Institute (BSHRI) in Sun City, Arizona.12 Samples were collected under a previously approved institutional review board (IRB) protocol with broad consent for the usage of biospecimens (WIRB Protocol #20120821). All research protocols were conducted in accordance with the principles expressed in the Declaration of Helsinki. Subjects were divided into four groups by clinical status (n = 12 for all groups) based on assessment of postmortem brain pathology and cognitive status before death. These groups were: normal control (NC) subjects with criteria not met for AD neuropathology, cognitively normal subjects with intermediate AD pathology characterized as high pathology controls (HPC), subjects with nonspecific mild cognitive impairment (MCI) and intermediate AD neuropathology, and subjects with dementia and high neuropathology with criteria met for AD. For the definition of groups by AD pathology, the National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease were used.10 Relevant clinical characteristics were provided by BSHRI for each subject such as age, sex, APOE genotype, postmortem interval (PMI), Mini-Mental State Examination (MMSE), and Braak score. Additionally, measures of brain pathology including cerebral amyloid angiopathy (CAA), amyloid plaques, and neurofibrillary tangles were taken from either a frontal area of the brain or a compilation of sampling from various brain regions.
Targeted LC–MS/MS Aqueous Profiling
For tissue lysates, 400 μg pieces of frozen superior frontal gyrus were hand homogenized in 400 μL of ice-cold sterile PBS containing a protease/phosphatase inhibitor cocktail (Halt, Thermo Scientific). Three samples for which less tissue was available were homogenized in equal ratios (weight to volume) of the PBS/inhibitor solution. Homogenized samples were sonicated on ice in a biosafety cabinet at a 40% amplitude for a total time of 1 min, with alternating on/off sequences of 15 s. Samples were then centrifuged for 30 min at 14 000 rpm at 4 °C. The supernatant and pellets were stored separately at −80 °C until analysis.
Prior to LC–MS/MS targeted measurement, frozen tissue supernatant samples were first thawed overnight under 4 °C. Afterward, 50 μL of each sample were placed in a 2 mL Eppendorf vial. The initial step for protein precipitation and metabolite extraction was performed by adding 500 μL of MeOH and 50 μL of internal standard solution (containing 1810.5 μM 13C3-lactate and 142 μM 13C5-glutamic acid). The mixture was then vortexed for 10 s and stored at −20 °C for 30 min, followed by centrifugation at 14 000 rpm for 10 min at 4 °C. The supernatants (450 μL) were collected into new Eppendorf vials and dried using a CentriVap Concentrator. The dried samples were reconstituted in 150 μL of 40% PBS/60% ACN and centrifuged again at 14 000 rpm at 4 °C for 10 min. Afterward, 100 μL of supernatant was collected from each sample into an LC autosampler vial for subsequent analysis. A pooled sample, which was a mixture of all experimental samples, was used as the quality control (QC) sample and injected once every 10 experimental samples.
The targeted LC–MS/MS method used here was modeled after that developed and used in a growing number of studies.46-51 Briefly, all LC–MS/MS experiments were performed on an Agilent 1290 UPLC-6490 QQQ-MS system. Each supernatant sample was injected twice, 10 μL for analysis using negative ionization mode and 4 μL for analysis using positive ionization mode. Both chromatographic separations were performed in hydrophilic interaction chromatography mode on a Waters XBridge BEH Amide column (150 × 2.1 mm2, 2.5 μm particle size, Waters Corporation, Milford, MA). The flow rate was 0.3 mL/min, autosampler temperature was kept at 4 °C, and the column compartment was set to 40 °C. The mobile phase was composed of Solvents A (10 mM NH4OAc, 10 mM NH4OH in 95% H2O/5% ACN) and B (10 mM NH4OAc, 10 mM NH4OH in 95% ACN/5% H2O). After an initial 1 min isocratic elution of 90% B, the percentage of solvent B decreased to 40% at t = 11 min. The composition of solvent B was maintained at 40% for 4 min (t = 15 min), after which the percentage of B gradually went back to 90%, to prepare for the next injection. The mass spectrometer was equipped with an electrospray ionization (ESI) source. Targeted data acquisition was performed in multiple-reaction-monitoring (MRM) mode. For targeted data acquisition, we monitored 118 and 160 MRM transitions in negative and positive mode, respectively (278 transitions in total). The whole LC–MS system was controlled by Agilent MassHunter Workstation software. The extracted MRM peaks were integrated using Agilent MassHunter Quantitative Data Analysis software.
Targeted LC–MS/MS Lipidomics
Tissue pellet samples were thawed under 4 °C. Then, 200 μL 10× diluted PBS and 80 μL of MeOH containing 50 μM PC (17:0, 17:0) and PG (17:0, 17:0) internal standards were added to 20 mg of each thawed sample. A half spoonful of stainless-steel micro beads was added to each sample, which was subsequently homogenized for 20 s. Afterward, 400 μL of MTBE was added to each sample (MTBE/MeOH/H2O = 10:2:5, v/v/v) and vortexed for 30 s followed by sonication in ice bath for 20 min. Finally, samples were centrifuged at 14 000 rpm to separate phases. The upper MTBE layer (300 μL) was extracted, transferred to new 1.5 mL Eppendorf tubes, dried in a Vacufuge Plus Evaporator (Hamburg, Germany), and then reconstituted with 100 μL 1:1 CHCl3/MeOH. Each sample (80 μL) was then transferred to a LC–MS vial for LC–MS/MS targeted lipidomics analysis, while the remaining 20 μL was pooled to create a QC sample.
For lipidomic profiling, all mass spectrometry experiments were done on an Agilent 1290 LC-6490 QQQ-MS (Santa Clara, CA), and 4 μL was injected for positive ionization, whereas 6 μL was used in negative ion mode injections. Both modes used reverse-phase chromatography with a Waters XSelect HSS T3 column (150 × 2.1 mm2, 2.5 μm particle size; Waters Corporation, Milford, MA). The flow rate through the column was maintained at 0.3 mL/min. The mobile phase Solvent A was composed of 10 mM NH4OAc in 60% H2O/40% ACN. Solvent B consisted of 10 mM NH4OAc in 90% IPA/10% ACN. An isocratic elution was used with 50% solvent B for 3 min before its percentage was gradually increased to 100% over the next 12 min. Following 10 min of continued 100% solvent B, at t = 25 min, the percent of B was decreased gradually back to 50% to prepare for the next sample injection. The set of lipids covered in our LC–MS/MS lipidomics assay were the same as those in our previous study,52 and 357 lipids were selected from various lipid classes including fatty acids, glycolipids, glycerophospholipids, sphingolipids, etc.52 Lipid standards were used to test the MRM and retention time (RT) for each individual lipid.
Untargeted GC–MS Aqueous Profiling
The aqueous bottom layer (180 μL) from the MTBE extraction described above was collected into a new Eppendorf tube for derivatization prior to untargeted metabolic profiling with GC–MS. The collected bottom layer was dried under vacuum at 37 °C for 4 h using a CentriVap Concentrator (Labconco, Fort Scott, KS). The residues were first derivatized with 40 μL of 20 mg/mL MeOX solution in pyridine under 60 °C for 90 min. Next, 60 μL of MTBSTFA containing d27-mysristic acid were added, and the mixture was incubated at 60 °C for 30 min. The samples were then vortexed for 30 s, followed by centrifugation at 14 000 rpm for 10 min. Finally, 70 μL of supernatant were collected from each sample into new glass vials for GC–MS analysis.
GC–MS conditions used here were mainly adopted from previous studies.42,43 Briefly, GC–MS experiments were performed on an Agilent 7820A GC-5977B MSD system (Santa Clara, CA) by injecting 1 μL of prepared samples. Helium was used as the carrier gas with a constant flow rate of 1.2 mL/min. The separation of metabolites was achieved using an Agilent HP-5ms capillary column (30 m × 250 μm × 0.25 μm). The column temperature was maintained at 60 °C for 1 min, increased at a rate of 10 °C/min to 325 °C, and then held at this temperature for 10 min. Mass spectral signals were recorded at an m/z range of 50–600. Data extraction was performed using Agilent MassHunter Profinder software. A batch recursive feature extraction algorithm for small molecules was used, and peaks were filtered so that only peaks with absolute height ≥1000 counts were included. An RT tolerance of 0.10 min was established, and extraction was limited to the largest 1000 compound groups. Results were filtered if the overall identification score was less than 75.
Long-Chain Fatty Acids (LCFAs)
Weighed 20 mg samples were added to separate Eppendorf tubes and prepared using the same protocol as that outlined for LC–MS/MS lipidomics. Derivatization was performed using the same protocol as that outlined for GC–MS untargeted profiling. For analysis of LCFAs, 60 μL of supernatant was transferred to a glass vial for GC–MS analysis, while 20 μL was pooled from each sample for QC analysis. The GC–MS method was the same as that for GC–MS untargeted profiling.
Short-Chain Fatty Acids (SCFAs)
Frozen tissue pellet samples were first thawed overnight under 4 °C. Afterward, 20 mg of each sample was homogenized with 5 μL of hexanoic acid-3,3,3 (internal standard), 15 μL of sodium hydroxide (NaOH [0.5 M]), and 500 μL of methanol (MeOH). Following storage at −20 °C for 20 min and centrifugation at 14 000 rpm for 10 min, 450 μL of supernatant were collected and sample pH was adjusted to 10 by adding 30 μL of NaOH/H2O (1:4, v/v). Samples were then dried, and they were measured using the same protocol as that outlined for GC–MS untargeted profiling.53
Bile Acids
Sample preparation techniques used here are well established and described in the previous literature.54-58 Briefly, 50 mg of each tissue sample were homogenized with methanol (500 μL) and then vortexed for 10 s. Samples were stored at −20 °C for 20 min, followed by sonication in an ice bath for 10 min and then centrifugation at 14 000 rpm for 15 min at 4 °C. Supernatants (450 μL) were vacuum-dried and then reconstituted in 100 μL of MeOH/H2O (1:1, v/v). Each prepared sample (2 μL) was injected into the LC–MS system (Agilent 1290 UPLC-6490 QQQ-MS) for analysis using negative ionization mode. The mobile phase was composed of 5 mM NH4OAc in H2O with 0.1% AcOH (A) and ACN with 0.1% AcOH (B). After a 1 min of isocratic elution of 75% solvent A, the content percentage decreased to 5% A at t = 15 min. The composition of solvent A was then maintained at 5% for 10 min, followed by an increase to 75% at t = 25 min. The MS parameters were the same as those reported for targeted LC–MS/MS aqueous profiling, except that 55 bile acids were included in the detection panel.57 Samples were spiked with mixtures of standard compounds to validate bile acid identities.
Data Analysis
Following peak integration, metabolites were filtered for reliability and only those with QC coefficient of variation (CV) <20% and relative abundance of 1000 in >80% of samples were retained for analysis. Data were then normalized by tissue weight and lysate volume as appropriate. The data were log10-transformed and Pareto scaled prior to model construction. Univariate testing was performed using SPSS 22.0 (SPSS Inc., Chicago, IL). Multivariate statistical analyses were performed using open-source R software. Pathway and integrating enzyme enrichment analysis were performed and visualized using MetaboAnalyst v4.0.38
RESULTS
Clinical Characteristics
A total of 2080 metabolites and mass spectral features were reliably detected after signal and QC filtering. Of these, 728 were identified with retention times and/or fragment mass spectra using chemical standards, while 1352 were unidentified m/z values. All raw mass spectrometry data have been deposited to MassIVE (data set identifier MSV000087165). A total of 48 subjects were included in this study: NC (n = 12), HPC (n = 12), MCI (n = 12), and AD (n = 12). Figure 1 shows a graphical schema of the analytical workflow. Table 1 shows the clinical information of subjects, while Table 2 shows the neuropathological characteristics of study subjects. Subjects between all experimental groups were age- and sex-matched such that no statistically significant difference was observed between groups (p > 0.05). Principal component analysis (PCA) conducted with all reliably detected metabolites (i.e., filtered metabolites) between all groups and QC samples was performed, and 95% confidence intervals were evaluated for potential outliers. QC samples were highly clustered, suggesting good system performance. However, the initial PCA revealed one outlier (HPC subject) which, upon confirming extensive nonignorable missingness, was removed from subsequent analyses (Figure S1).
Figure 1.

Overview of the analytical workflow of the current study. Created with BioRender.com.
Table 1.
Clinical Information of Study Subjects
| nondemented normal controls (n = 12) |
high pathology controls (n = 12) |
mild cognitive impairment (n = 12) |
Alzheimer’s disease (n = 12) |
|
|---|---|---|---|---|
| age in years, mean (SD) | 79.7 (12.2) | 90.3 (5.1) | 88.1 (8.6) | 81.3 (8.3) |
| sex of subjects (male/female) | 7/5 | 6/6 | 8/4 | 7/5 |
| postmortem interval mean, hours (SD) | 2.93 (1.0) | 2.84 (0.9) | 3.06 (0.8) | 3.2 (0.5) |
| mini-mental state examination score mean (SD) | 28.8 (0.8)a | 27.6 (1.4) | 26.0 (3.4)b | 10.3 (7.6) |
| APOE alleles: number of subjects with each genotype 2/3, 2/4, 3/3, 3/4 | 0, 0, 8, 4 | 2, 0, 7, 3 | 0, 1, 9, 2 | 0, 0, 6, 6 |
Scores not available for two subjects.
Score not available for one subject.
Table 2.
Neuropathological Characteristics of Study Subjects
| frontal plaque and tangle scores (number of subjects scoring none, sparse, moderate, or frequent) |
nondemented normal controls (n = 12) |
high pathology controls (n = 12) |
mild cognitive impairment (n = 12) |
Alzheimer’s disease (n = 12) |
|---|---|---|---|---|
| plaque score (based on CERAD) | 10, 2, 0, 0 | 0, 0, 0, 12 | 1, 1, 1, 9 | 0, 0, 0, 12 |
| tangle score | 11, 0, 0, 0a | 9, 3, 0, 0 | 5, 6, 1, 0 | 0, 2, 1, 9 |
| Braak staging (number of subjects stage 0 to stage VI) | 0, 5, 3, 4, 0, 0 | 0, 0, 1, 2, 9, 0 | 0, 0, 1, 1, 9, 1, 0 | 0, 0, 0, 0, 6, 6 |
Score not available for one subject.
Case (MCI, AD) vs Control (NC, HPC)
To assess broad differences in metabolic profiles, groups were collapsed among case (MCI and AD) and control (NC and HPC). Initial t-testing between case and control revealed two highly significant and predictive metabolites with p < 0.001 and univariate area under curve (AUC) > 0.90: lauric acid and myristic acid. Box plots of these metabolites are given in Figure S2. In addition, a partial least squares-discriminant analysis (PLS-DA) model was constructed using levels of lauric and myristic acid, and receiver operating characteristic (ROC) analysis was conducted using model-implied values to assess performance. As shown in Figure 2, the resulting PLS-DA score plot showed appreciable separation between collapsed case and control groups, and ROC analysis by 100-fold leave-one-out cross-validation (LOOCV) showed an overall accuracy of 95%, more than either metabolite individually.
Figure 2.

PLS-DA and ROC analysis of case (MCI and AD) and control (NC and HPC) constructed using levels of lauric acid and myristic acid: (A) Score plot of the PLS-DA model (R2X = 0.593, R2Y = 0.814, R2Q = 0.701; 10-fold cross-validated Q2 = −0.183) and (B) ROC analysis by 100-fold leave-one-out cross-validation (LOOCV) of model-implied values showing AUC = 0.95.
NC/HPC vs Other Groups
To analyze differences among groups individually, we first compared metabolic profiles of NC vs HPC/MCI/AD, and HPC vs NC/MCI/AD. Multivariate analysis of variance (MANOVA) testing and associated post-hoc comparisons were performed to identify significant metabolites, while PLS-DA was used to operationalize those metabolites for classification. Well-established risk factors (age, sex, and APOE status)59,60 were included in the MANOVA as covariates and all significance values adjusted accordingly. As outlined in Table 3, four metabolites (lauric acid, myristic acid, stearic acid, palmitic acid) showed significant main effects, as evidenced by LSD-controlled p < 0.05, and were mostly predictive of NC and HPC groups, irrespective of biological risk. Testing for group by age, group by sex, and group by allele interactions revealed no significant effects (p-values = 0.077–0.991). Further analysis of a full-factorial GLM showed no significant main effect of age, sex, or APOE allele nor any significant interactions among them (all p > 0.05). Normalized box plots of these metabolites between all groups are shown in Figure 3. A PLS-DA model was constructed using levels of these four significant metabolites, and internal validation was performed using a 100-iteration permutation test. More than 98% of total variance between the four study groups was explained by the first two components, and permutation testing revealed the model to be statistically sound (observed p < 0.001). To assess the predictive performance of this unified biomarker panel, the resulting PLS-DA model was subjected to ROC analysis with 100-fold LOOCV. ROC curves for each comparison are provided in Figure S3. Evaluation of model accuracy showed high classification performance for discrimination of NC samples (96.6%, Figure S3A) and HPC samples (91.7%, Figure S3B). We further used a random forest (RF) classifier for group predictions (Figure S4) but found that the RF models exhibited poor generalization (OOB error = 0.511) and, therefore, suboptimal predictive performance of case and control (AUC = 0.917), as compared to PLS classification (AUC = 0.95). One potential explanation for this may be the small sample size (48 subjects).
Table 3.
Significant Between-Group Metabolites As Determined by MANOVA Testing, Focusing on NC and HPC Groups
| metabolite | F-value | p-value | LSD p | significant post-hoc comparisons |
|---|---|---|---|---|
| lauric acid | 23.360 | 3.98 × 10−9 | 8.29 × 10−6 | HPC-AD; NC-AD; HPC-MCI; NC-HPC; NC-MCI |
| myristic acid | 16.727 | 3.92 × 10−7 | 2.49 × 10−4 | HPC-AD; NC-AD; HPC-MCI; HPC-NC; NC-MCI |
| stearic acid | 9.911 | 4.34 × 10−5 | 0.036 | HPC-AD; NC-AD; HPC-MCI; HPC-NC; NC-MCI |
| palmitic acid | 9.133 | 8.56 × 10−5 | 0.036 | HPC-AD; HPC-MCI; HPC-NC; NC-MCI |
Figure 3.

Relative abundances of four metabolites found to be significant between groups (LSD p < 0.05) by MANOVA testing. Data were log10-transformed and Pareto scaled prior to plotting.
MCI vs Other Groups
To increase model performance for discrimination of the MCI subgroup (see Figure S3C), other groups were compared sequentially for identification of significant/predictive metabolites and model construction. Comparison of MCI and NC groups revealed lauric acid to be both highly significant (p < 0.001) and predictive (AUC = 0.993) (Figure S5). Comparison of MCI and HPC groups revealed four metabolites (myristic acid, palmitic acid, stearic acid, palmitoleic acid) to have AUC > 0.90 and FDR q < 0.05 (Figure 4), while the comparison of MCI and AD groups revealed four unidentified features (from untargeted GC–MS) with AUC > 0.80 and q < 0.05 (Figure 4). Candidate metabolites for the classification of MCI and HPC samples were used to construct an independent PLS-DA model, while candidate markers for MCI discrimination from AD samples were ported to construct a separate PLS-DA model. ROC analysis showed a predictive accuracy of 96.6% for the identification of MCI samples from high pathology controls (Figure 5A). Meanwhile, ROC analysis of the PLS-DA model constructed using levels of four unidentified features with an average AUC ~ 0.811 showed an appreciable improvement in accuracy relative to each univariate AUC; classification accuracy of the PLS-DA model exhibited an AUC = 0.917 for discrimination of MCI and AD groups (Figure 5B).
Figure 4.

Top row: Relative abundances of myristic acid, palmitic acid, stearic acid, and palmitoleic acid with high predictive accuracy (AUC > 0.90) and significance (FDR q < 0.05) in univariate ROC analysis and t-testing between HPC and MCI groups. Bottom row: Relative abundances of four unidentified features from untargeted GC–MS analysis with good predictive accuracy (AUC > 0.80) and significance (FDR q < 0.05) in univariate ROC analysis and t-testing between MCI and AD groups.
Figure 5.

ROC analysis of classification performance by 100-fold LOOCV: (A) PLS-DA model constructed using levels of myristic acid, palmitic acid, stearic acid, and palmitoleic acid (observed p = 0.005) for classification of MCI and HPC samples (AUC = 0.966) and (B) PLS-DA model constructed using levels of four significant unidentified features (observed p = 0.022) for classification of MCI and AD samples (AUC = 0.917).
AD vs Other Groups
Relevant groups were also compared to AD samples for enhanced identification of disease. Univariate ROC analysis and independent samples t-testing of NC and AD samples showed lauric acid to be highly significant (p < 0.001) and predictive (AUC > 0.99) (see Figure S6). For classification of AD samples from high pathology controls, t-testing revealed the unified biomarker panel in Figure 3 as being significantly altered between groups (p < 0.01) and to have high predictive potential (AUC > 0.90). Direct comparisons between AD and HPC groups for these metabolites are visualized as box plots in Figure S7. For discrimination of these groups, an additional PLS-DA model was constructed using the aforementioned candidate markers, and ROC analysis revealed high predictive accuracy (94.8%) for discrimination of AD and HPC samples (Figure 6).
Figure 6.

(A) PLS-DA model constructed using levels of myristic acid, lauric acid, palmitic acid, and stearic acid for classification of AD and HPC groups (observed p = 0.007) and (B) ROC analysis of the PLS-DA model by 100-fold LOOCV showing AUC = 0.948.
Correlation Analysis of Candidate Markers and Clinical/Neuropathological Characteristics.
To assess relevant associations between the set of candidate markers and measures of brain pathology and disease progression, a correlation analysis was performed, and measures of association strength and significance were evaluated. A visualization of association strength between correlation variables is given in Figure 7. Full details regarding the magnitude of association (r) and significance of association (p) can be found in Table S1. In total, four associations had r > 0.5 or < −0.5 and p < 0.05. Lauric acid showed strong, significant associations with frontal plaque (r = −0.598, p < 0.001), total plaque (r = −0.579, p < 0.001), total tangle (r = −0.507, p < 0.001), and Braak score (r = −0.539, p < 0.001). A PLS-DA model was articulated using this set of four neuropathological characteristics and significant between-group metabolites (lauric acid, myristic acid, stearic acid, and palmitic acid). With the inclusion of these clinical markers, a clear separation of AD, MCI, and HPC groups from normal controls was observed (see Figure S8). Furthermore, correlation analysis between age, sex, APOE allele, and all 2080 reliably detected metabolites/features was also performed; no association was observed to be both strongly correlated (r > |0.5|) and statistically significant (p < 0.05).
Figure 7.

Correlation coefficients among the panel of candidate markers and clinical characteristics.
Pathway and Enzyme Enrichment Analyses of Metabolic Data
Subjects were grouped as case (MCI and AD) and control (NC and HPC) for analysis of significantly impacted pathways in response to Alzheimer’s progression. Pathway and enzyme analysis was conducted using KEGG database searches and metabolite intensities (Figure 8). Pathways were mapped to the human metabolome and only identified metabolites confirmed with authentic standards (i.e., based on retention time and MS2 fragmentation for LC–MS/MS and retention index for GC–MS) were included in the analysis. Three pathways were observed to have large impact coefficients (>0.5): (1) linoleic acid metabolism, (2) alanine, aspartate, and glutamate metabolism, and (3) arginine and proline metabolism. Importantly, three pathways were found to be significantly affected (p < 0.05) as a result of increased AD pathogenesis. Namely, those were lysine degradation, fatty acid metabolism, and valine, leucine, and isoleucine degradation.
Figure 8.

Metabolome view of pathway analysis conducted using levels of all reliably detected metabolites showing significantly altered pathways (p < 0.05) and those with high impact (>0.50).
Subjects were dichotomously grouped as case/control, and enrichment analysis was conducted using a library containing 912 metabolic sets that are predicted to be changed in the case of dysfunctional enzymes using a genome-scale network model of human metabolism (Figure S9). Eleven enzymes were found to be significantly enriched (p < 0.05). Notably, seven of those were mitochondrial enzymes indicated against the background of mitochondrial pathways. Full results of the enzyme enrichment analysis are displayed in Table S2.
DISCUSSION
For the last 2 decades, significant innovations in MS-based metabolic profiling and analysis of disease-related alterations have been made and, in doing so, these efforts have borne highly sensitive and valuable diagnostic information.61-63 In the current study, we explored a combination of targeted and untargeted metabolic profiling in addition to advanced multivariate statistical analysis for the discovery of sensitive and specific metabolite biomarkers for rapid AD classification postmortem. To capture the diversity of metabolites involved in AD pathobiology, we have used this method to detect 2080 metabolites of the superior frontal gyrus from many biologically relevant metabolic pathways. Our multistep biomarker selection, model construction, and cross-validation have demonstrated the robust diagnostic power of this metabolic profiling method in this study of 48 NC, HPC, MCI, and AD subjects. Additionally, we have applied complementary LC/GC–MS approaches for enhanced monitoring of the metabolome related to AD and, cumulatively, our results show clinically relevant disturbances in energy metabolism and substrate utilization.
The metabolite profiling approach presented in this study determined five fatty acids capable of discriminating AD patients from NC and HPC samples with an average AUC of 97%. Recent metabolomics studies have also shown perturbations in fatty acid metabolism across differing Alzheimer pathologies. It was found that the dysregulation of sphingolipids and glycerophospholipids, long-chain fatty acids, and unsaturated fatty acids have been associated with AD.7,21,24 Similarly, significant disturbances in fatty acid metabolism were also observed in the current study (p = 0.008). More specific to our results, a recent serum profiling approach demonstrated the high predictive accuracy of palmitoleic acid, myristic acid, linoleic acid, and palmitic acid in differentiating central cognitive impairment in AD.64 Notably, these metabolites were also flagged as candidate markers in our profiling of neocortical tissue. Medium-chain fatty acids like lauric acid, which is found in high levels in coconut oil, have been proposed as possible nutritional therapies for the treatment of cognitive decline,65-67 and a significant difference in lauric acid was also observed in the AD and NC groups in this study. The markedly reduced levels of these fatty acids observed in our AD subjects could be linked to the impaired glucose metabolism that is well documented in AD patients.68-70 Declines in the levels of the identified fatty acids in conjunction with decreased glucose metabolism might suggest that β-oxidation of fatty acids, which is generally low in the brain, is being upregulated to support the energy needs of the brain in AD patients. Supplementation of the fatty acids that can be rapidly metabolized might help support the energy needs of the brain, potentially ameliorating symptoms. This might account for the data cited in the above-referenced reports, which suggest that the addition of lauric acid to the diet (via coconut oil) may improve some symptoms in AD patients. Lauric acid is known to cross the blood–brain barrier,71 and dietary lauric acid might therefore be accessible as an energy source for the brain.72 The results of the current study warrant further investigation of the therapeutic potential of lauric acid for the treatment and prevention of AD.
Previous studies have shown evidence for brain glucose dysregulation in AD as characterized by higher brain tissue glucose concentration, reduced glycolytic flux, and lower GLUT3 expression as a function of increasing AD pathogenesis.22,68,73 Interestingly, the literature has shown the involvement of the Warburg effect in nontumor disease processes74 and, in the context of AD, loss of brain aerobic glycolysis as a function of normal human aging is associated with increased tau deposition in preclinical AD.75,76 In addition, previous results have shown impaired hypothalamic insulin signaling to be associated with elevated BCAA levels in a mouse model of AD,77 while defects in BCAA metabolism have in-turn been shown to drive primary AD neuropathology.77 A prospective cohort study of over 22 000 participants found significant associations between circulating BCAAs and risk of incident dementia and AD.27 It has been shown that defects in BCAA metabolism, and subsequent accumulation, can lead to the phosphorylation of tau proteins and the incidence of AD.78 Other studies have found post-translational modifications to the stabilizing tau proteins, which were induced by lysine residues. It has been proposed that these modifications may play an integral role in the pathobiology of tau protein.79 Our pathway analysis also revealed similar results with a significant degradation of lysine (p = 0.007) and BCAAs (p = 0.025), potentially signifying the underlying pathophysiology of AD. Given the recent failure of numerous billion-dollar clinical trials targeting traditionally hypothesized AD mechanisms such as reduced acetylcholine, Aβ plaques/neurofibrillary tangles, and tau protein,80 our enzyme and pathway enrichment results further corroborate previous evidence of widespread mitochondrial dysfunction concomitant with Aβ pathology and AD progression,80-82 providing compelling evidence for mitochondrial bioenergetics as a novel therapeutic target for preventing/slowing the onset/progression of AD.
Overall, our findings led to an integrated hypothesis describing the pathophysiology of AD in Figure 9 and are conceptualized with respect to the widespread mitochondrial dysfunction observed in our results. In Figure 9, the darker red areas (to the right) are more increased with AD pathology, and greater enrichment is observed in those pathways. As can be seen, with increased AD pathogenicity (darker red areas), significant metabolic reprogramming is observed. Specifically, a decrease in aerobic glycolysis (lighter red areas) is followed by a shift toward degradation of BCAA for energy production, mostly associated with HPC and MCI subgroups (darker red areas). With even greater disease progression, further metabolic reprogramming is observed; fatty acids are progressively utilized for the generation of ATP via increased β-oxidation activity and generation of FADH2 and NADH for oxidative phosphorylation in the electron transport chain (darker red areas). Preference for fatty acid substrates was most pronounced in the MCI and AD subgroups.
Figure 9.

Conceptual schema articulating observed changes in substrate utilization and energy production as a function of increasing AD pathogenesis. The darker red areas (to the right) are more increased with AD pathology, and greater enrichment is observed in those pathways. Results show reduced aerobic glycolysis and increased degradation of BCAA associated with HPC and MCI groups as compared to NC. Meanwhile, a preference for fatty acid substrates is seen in MCI and AD groups, with further reductions in aerobic glycolysis as compared to NC. ETC, electron transport chain; IMM, inner mitochondrial membrane; OMM, outer mitochondrial membrane; and OXPHOS, oxidative phosphorylation.
Additionally, we evaluated levels of four unidentified features with p < 0.05 and FC > 2, which informed the construction of independent PLS-DA models for enhanced classification of AD from MCI samples. The combination of these four features had a diagnostic sensitivity and specificity of 84.1 and 86.3%, respectively (AUC = 0.917). Although accurate tests for AD pathology with high severity are currently available (i.e., PET amyloid and tau, CSF amyloid and tau, plasma tau), diagnostic tests useful for intermediate (MCI) and low (HPC) pathology levels are still lacking. In this study, diagnosis of HPC and MCI subgroups was achieved with more than 90% overall AUC. In addition to enabling mass screening, a realization of these findings in plasma or CSF may inform clinical trial selection via improved study stratification.
Strengths and Limitations
A major strength of the study lies in the well-characterized BSHRI cohort12 with measures of cognitive status and neuropathological examination at death. Furthermore, the inclusion of traditionally understudied HPC and MCI groups allowed for the metabolic characterization of asymptomatic individuals with AD-consistent pathology and non-AD individuals with cognitive decline, respectively. Cumulatively, our panel of candidate markers shows potential for the classification of individuals with early brain pathology and other dementias. Although this approach is not suited for in vivo diagnosis of AD given the impracticality of brain biopsy for living individuals, the putative metabolite markers/metabolic pathways and associated models reported herein serve as a strong proof-of-principle for their use as therapeutic targets. Furthermore, if validated in readily available biospecimens with minimally invasive sample collection (i.e., from blood draw), this novel panel of candidate markers may enable AD diagnosis in living patients and subsequently enhanced treatment options. Additionally, we applied six distinct metabolomics assays encompassing complementary GC and LC techniques to ensure maximal coverage of the brain metabolome and were able to monitor more than 2000 metabolites and features. Given the known benefit of complementary MS platforms for elucidation of AD pathology,20,83,84 our large-scale multiplatform metabolomics approach utilizing both targeted and untargeted profiling enables comprehensive pathway and enzyme analysis, a key strength of this study to previous literature.
The main limitation of this study is the relatively small sample size. Moreover, our samples were taken cross-sectionally and therefore cannot infer longitudinal changes in metabolite information over time. Also, samples were only taken from a single-brain region; inferences to other AD-associated brain structures are unknown. Nevertheless, conventional power was achieved for all biomarker analyses (β < 0.2), and models were internally validated (p < 0.01). It should also be noted that, while the current study infers many proposed alterations in enzymes and pathways using metabolite-level data, the results cannot determine whether the purported markers and associated pathways are drivers of AD pathology or are themselves effects of other latent pathologies; future studies are warranted to investigate this relationship mechanistically. Additionally, of the 48 subjects considered in this study, there were none with APOE 4/4 genotype. There was only one subject with APOE 2/4 genotype and 15 subjects with APOE 3/4 genotype. As APOE 4/4 individuals have the highest risk for AD, it would be useful for future studies to consider subjects with this genotype and monitor metabolites in this group. Our results merit further investigation in a larger sample with serial cognitive assessments taken during life as well as tissue samples collected from distinct brain regions both resistant and vulnerable to AD pathology to monitor possible differential changes between tissue types.
CONCLUSIONS
This study is part of a growing body of literature in which MS-based metabolomics methods have been utilized for disease biomarker discovery and accurate classification.38,49-51 We performed comparisons of brain tissue metabolites from AD patients, MCI samples, as well as high pathology and normal controls using both targeted LC–MS/MS metabolic profiling and an untargeted GC–MS approach.29 Our results demonstrate significant alterations in a variety of metabolites, mainly fatty acids, which are characteristic of different groups. Furthermore, we evaluated the performance of four unidentified metabolic features and, through multivariate model construction, achieved an overall classification performance of >90% for comparison of AD and MCI patients, which has the potential to fulfill critical clinical needs.85 Application of bioinformatic methods expanded basic knowledge of the metabolome related to AD and showed decreased glycolytic function with increased degradation of BCAAs and β-oxidation of fatty acids associated with increased AD pathogenicity. Results of our fold change analysis, significance testing, and pathway analysis indicate metabolites and pathways previously shown to be crucial in immune response inhibition86 and increased AD severity.87 Likewise, the metabolites and associated metabolic pathways and enzymes identified in this study may inform the development of new therapeutic treatments for AD. In addition, this study provides a strong basis for larger multisite projects to validate our findings across different population groups and further advances the development of improved clinical care for AD patients.
Supplementary Material
Funding
Support from the College of Health Solutions (Jumpstart) at Arizona State University to H.G. is gratefully acknowledged. This work was also supported by funding to G.J. from the Arizona Alzheimer’s Consortium (funded by the Arizona Department of Health Services, Grant no. CTR040636) and matching funds from Midwestern University. Support from the NIH to H.G. and G.J. is kindly appreciated (1R21AG072561-01). Human tissues used in this study were provided at no cost by the Banner Sun Health Research Institute (BSHRI) Brain and Body Donation Program (BBDP). Support for recovery of tissues by the BBDP and provision of subject data collected by the Arizona Study of Aging and Neurodegenerative Disorders (AZSAND) is provided by the following grants: National Institute of Neurological Disorders and Stroke, U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders; National Institute on Aging, P30 AG19610 Arizona Alzheimer’s Disease Core Center; Arizona Department of Health Services, Arizona Alzheimer’s Consortium; Arizona Biomedical Research Commission, Arizona Parkinson’s Disease Consortium; and Michael J. Fox Foundation for Parkinson’s Research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
ABBREVIATIONS
- AcOH
acetic acid
- ACN
acetonitrile
- NH4OAc
ammonium acetate
- NH4OH
ammonium hydroxide
- AD
Alzheimer’s disease
- Aβ
amyloid β
- AUC
area under curve
- BSHRI
Banner Sun Health Research Institute
- BCAA
branched-chain amino acid
- CAA
cerebral amyloid angiopathy
- CSF
cerebrospinal fluid
- CHCl3
chloroform
- CT
computerized tomography
- ETC
electron transport chain
- ESI
electrospray ionization
- GC
gas chromatography
- HPC
high pathology control
- IMM
inner mitochondrial membrane
- IRB
institutional review board
- IPA
isopropanol
- LOOCV
leave-one-out cross-validation
- LC
liquid chromatography
- LCFA
long-chain fatty acid
- MRI
magnetic resonance imaging
- MS
mass spectrometry
- MeOH
methanol
- MTBE
methyl tert-butyl ether
- MCI
mild cognitive impairment
- MMSE
Mini-Mental State Examination
- MRM
multiple reaction monitoring
- MANOVA
multivariate analysis of variance
- MTBSTFA
N-methyl-N-(tert-butyldimethylsilyl) trifluoroacetamide
- NC
normal control
- MeOX
O-methylhydroxylamine hydrochloride
- OMM
outer mitochondrial membrane
- OXPHOS
oxidative phosphorylation
- PLS-DA
partial least squares-discriminant analysis
- PBS
phosphate-buffered saline
- PET
positron emission tomography
- PMI
postmortem interval
- PCA
principal component analysis
- QC
quality control
- ROC
receiver operating characteristic
- RT
retention time
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.1c00290.
Table S1: matrix correlation analysis; Table S2: full list of significantly enriched enzymes; Figure S1: PCA performed using all metabolites between NC, HPC, MCI, AD, and QC; Figure S2: significant metabolites between case and control; Figure S3: ROC analysis of the PLS-DA model for classification of NC, HPC, MCI, and AD; Figure S4: RF analysis of study groups and ROC analysis of case and control; Figure S5: univariate ROC analysis and t-testing between NC and MCI groups; Figure S6: univariate ROC analysis and t-testing between NC and AD groups; Figure S7: univariate ROC analysis and t-testing between HPC and AD groups; Figure S8: PLS-DA model of significant metabolites and correlated clinical markers; and Figure S9: enzyme enrichment analysis between case and control (PDF)
The authors declare no competing financial interest.
All research protocols were conducted in accordance with the principles expressed in the Declaration of Helsinki. Samples were collected under a previously approved IRB protocol with broad consent for usage of biospecimens (WIRB Protocol #20120821).
All raw mass spectrometry data, in addition to deidentified subject demographic and clinical information, have been deposited to MassIVE and are publicly available (data set identifier MSV000087165).
Contributor Information
Paniz Jasbi, Arizona Metabolomics Laboratory, College of Health Solutions, Arizona State University, Phoenix, Arizona 85004, United States.
Xiaojian Shi, Arizona Metabolomics Laboratory, College of Health Solutions, Arizona State University, Phoenix, Arizona 85004, United States; Systems Biology Institute, Cellular and Molecular Physiology, Yale School of Medicine, West Haven, Connecticut 06516, United States.
Ping Chu, Department of Biochemistry and Molecular Genetics, Midwestern University, Glendale, Arizona 85308, United States.
Natalie Elliott, Department of Biochemistry and Molecular Genetics, Midwestern University, Glendale, Arizona 85308, United States.
Haley Hudson, Department of Biochemistry and Molecular Genetics, Midwestern University, Glendale, Arizona 85308, United States.
Douglas Jones, Department of Pharmacology, Midwestern University, Glendale, Arizona 85308, United States.
Geidy Serrano, Banner Sun Health Research Institute, Sun City, Arizona 85351, United States.
Brandon Chow, Arizona Metabolomics Laboratory, College of Health Solutions, Arizona State University, Phoenix, Arizona 85004, United States.
Thomas G. Beach, Banner Sun Health Research Institute, Sun City, Arizona 85351, United States
Li Liu, College of Health Solutions, Biodesign Institute, Arizona State University, Tempe, Arizona 85281, United States; Department of Neurology, Mayo Clinic, Scottsdale, Arizona 85259, United States.
Garilyn Jentarra, Department of Biochemistry and Molecular Genetics, Midwestern University, Glendale, Arizona 85308, United States; Precision Medicine Program, Midwestern University, Glendale, Arizona 85308, United States.
Haiwei Gu, Arizona Metabolomics Laboratory, College of Health Solutions, Arizona State University, Phoenix, Arizona 85004, United States.
REFERENCES
- (1).Qiu C; De Ronchi D; Fratiglioni L The Epidemiology of the Dementias: An Update. Curr. Opin. Psychiatry 2007, 20, 380–385. [DOI] [PubMed] [Google Scholar]
- (2).Costa AC; Joaquim HPG; Forlenza OV; Gattaz WF; Talib LL Three Plasma Metabolites in Elderly Patients Differentiate Mild Cognitive Impairment and Alzheimer’s Disease: A Pilot Study. Eur. Arch. Psychiatry Clin. Neurosci 2020, 270, 483–488. [DOI] [PubMed] [Google Scholar]
- (3).Hölscher C. Development of Beta-Amyloid-Induced Neurodegeneration in Alzheimer’s Disease and Novel Neuroprotective Strategies. Rev. Neurosci 2005, 16, 181–212. [DOI] [PubMed] [Google Scholar]
- (4).Belbin O; Brown K; Shi H; Medway C; Abraham R; Passmore P; Mann D; Smith AD; Holmes C; McGuinness B; et al. A Multi-Center Study of ACE and the Risk of Late-Onset Alzheimer’s Disease. J. Alzheimer’s Dis 2011, 24, 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Yi L; Liu W; Wang Z; Ren D; Peng W Characterizing Alzheimer’s Disease through Metabolomics and Investigating Anti-Alzheimer’s Disease Effects of Natural Products. Ann. N. Y. Acad. Sci 2017, 1398, 130–141. [DOI] [PubMed] [Google Scholar]
- (6).Alzheimer’s Association. Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2020, 16, 391–460. [Google Scholar]
- (7).Snowden SG; Ebshiana AA; Hye A; An Y; Pletnikova O; O’Brien R; Troncoso J; Legido-Quigley C; Thambisetty M Association between Fatty Acid Metabolism in the Brain and Alzheimer Disease Neuropathology and Cognitive Performance: A Nontargeted Metabolomic Study. PLoS Med. 2017, 14, No. e1002266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hendrix JA; Bateman RJ; Brashear HR; Duggan C; Carrillo MC; Bain LJ; DeMattos R; Katz RG; Ostrowitzki S; Siemers E; et al. Challenges, Solutions, and Recommendations for Alzheimer’s Disease Combination Therapy. Alzheimer’s Dement. 2016, 12, 623–630. [DOI] [PubMed] [Google Scholar]
- (9).Hane FT; Robinson M; Lee BY; Bai O; Leonenko Z; Albert MS Recent Progress in Alzheimer’s Disease Research, Part 3: Diagnosis and Treatment. J. Alzheimer’s Dis 2017, 57, 645–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hyman BT; Phelps CH; Beach TG; Bigio EH; Cairns NJ; Carrillo MC; Dickson DW; Duyckaerts C; Frosch MP; Masliah E; et al. National Institute on Aging-Alzheimer’s Association Guidelines for the Neuropathologic Assessment of Alzheimer’s Disease. Alzheimer’s Dement. 2012, 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Maarouf CL; Daugs ID; Kokjohn TA; Walker DG; Hunter JM; Kruchowsky JC; Woltjer R; Kaye J; Castaño EM; Sabbagh MN; et al. Alzheimer’s Disease and Non-Demented High Pathology Control Nonagenarians: Comparing and Contrasting the Biochemistry of Cognitively Successful Aging. PLoS One 2011, 6, No. e27291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Beach TG; Adler CH; Sue LI; Serrano G; Shill HA; Walker DG; Lue L; Roher AE; Dugger BN; Maarouf C; et al. Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program. Neuropathology 2015, 35, 354–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Nelson PT; Abner EL; Schmitt FA; Kryscio RJ; Jicha GA; Smith CD; Davis DG; Poduska JW; Patel E; Mendiondo MS; et al. Modeling the Association between 43 Different Clinical and Pathological Variables and the Severity of Cognitive Impairment in a Large Autopsy Cohort of Elderly Persons. Brain Pathol. 2010, 20, 66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kang S; Jeong H; Baek JH; Lee SJ; Han SH; Cho HJ; Kim H; Hong HS; Kim YH; Yi EC; et al. PiB-PET Imaging-Based Serum Proteome Profiles Predict Mild Cognitive Impairment and Alzheimer’s Disease. J. Alzheimer’s Dis 2016, 53, 1563–1576. [DOI] [PubMed] [Google Scholar]
- (15).Seyfried NT; Dammer EB; Swarup V; Nandakumar D; Duong DM; Yin L; Deng Q; Nguyen T; Hales CM; Wingo T; et al. A Multi-Network Approach Identifies Protein-Specific Co-Expression in Asymptomatic and Symptomatic Alzheimer’s Disease. Cell Syst. 2017, 4, 60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).McKhann G; Drachman D; Folstein M; Katzman R; Price D; Stadlan EM Clinical Diagnosis of Alzheimer’s Disease: Report of the NINCDS-ADRDA Work Group under the Auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [DOI] [PubMed] [Google Scholar]
- (17).Iverson DJ; Gronseth GS; Reger MA; Classen S; Dubinsky RM; Rizzo M Practice Parameter Update: Evaluation and Management of Driving Risk in Dementia: Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2010, 74, 1316–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sancesario GM; Bernardini S Alzheimer’s Disease in the Omics Era. Clin. Biochem 2018, 59, 9–16. [DOI] [PubMed] [Google Scholar]
- (19).Roberts R; Knopman DS Classification and Epidemiology of MCI. Clin. Geriatr. Med 2013, 29, 753–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).González-Domínguez R; Sayago A; Fernández-Recamales Á Metabolomics in Alzheimer’s Disease: The Need of Complementary Analytical Platforms for the Identification of Biomarkers to Unravel the Underlying Pathology. J. Chromatogr. B 2017, 1071, 75–92. [DOI] [PubMed] [Google Scholar]
- (21).Varma VR; Oommen AM; Varma S; Casanova R; An Y; Andrews RM; O’Brien R; Pletnikova O; Troncoso JC; Toledo J; et al. Brain and Blood Metabolite Signatures of Pathology and Progression in Alzheimer Disease: A Targeted Metabolomics Study. PLoS Med. 2018, 15, No. e1002482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).An Y; Varma VR; Varma S; Casanova R; Dammer E; Pletnikova O; Chia CW; Egan JM; Ferrucci L; Troncoso J; et al. Evidence for Brain Glucose Dysregulation in Alzheimer’s Disease. Alzheimer’s Dement. 2018, 14, 318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zetterberg H; Burnham SC Blood-Based Molecular Biomarkers for Alzheimer’s Disease. Mol. Brain 2019, 12, No. 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wang G; Zhou Y; Huang FJ; Tang HD; Xu XH; Liu JJ; Wang Y; Deng YL; Ren RJ; Xu W; et al. Plasma Metabolite Profiles of Alzheimer’s Disease and Mild Cognitive Impairment. J. Proteome Res 2014, 13, 2649–2658. [DOI] [PubMed] [Google Scholar]
- (25).Han X; Rozen S; Boyle SH; Hellegers C; Cheng H; Burke JR; Welsh-Bohmer KA; Doraiswamy PM; Kaddurah-Daouk R Metabolomics in Early Alzheimer’s Disease: Identification of Altered Plasma Sphingolipidome Using Shotgun Lipidomics. PLoS One 2011, 6, No. e21643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wilkins JM; Trushina E Application of Metabolomics in Alzheimer’s Disease. Front. Neurol 2018, 8, No. 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Tynkkynen J; Chouraki V; van der Lee SJ; Hernesniemi J; Yang Q; Li S; Beiser A; Larson MG; Sääksjärvi K; Shipley MJ; et al. Association of Branched-Chain Amino Acids and Other Circulating Metabolites with Risk of Incident Dementia and Alzheimer’s Disease: A Prospective Study in Eight Cohorts. Alzheimer’s Dement. 2018, 14, 723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Peña-Bautista C; Roca M; Hervás D; Cuevas A; López-Cuevas R; Vento M; Baquero M; García-Blanco A; Cháfer-Pericás C Plasma Metabolomics in Early Alzheimer’s Disease Patients Diagnosed with Amyloid Biomarker. J. Proteomics 2019, 200, 144–152. [DOI] [PubMed] [Google Scholar]
- (29).Klavins K; Koal T; Dallmann G; Marksteiner J; Kemmler G; Humpel C The Ratio of Phosphatidylcholines to Lysophosphatidylcholines in Plasma Differentiates Healthy Controls from Patients with Alzheimer’s Disease and Mild Cognitive Impairment. Alzheimer’s Dement. 2015, 1, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).MahmoudianDehkordi S; Arnold M; Nho K; Ahmad S; Jia W; Xie G; Louie G; Kueider-Paisley A; Moseley MA; Thompson JW; et al. Altered Bile Acid Profile Associates with Cognitive Impairment in Alzheimer’s Disease—An Emerging Role for Gut Microbiome. Alzheimer’s Dement. 2019, 15, 76–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Pan X; Elliott CT; McGuinness B; Passmore P; Kehoe PG; Hölscher C; McClean PL; Graham SF; Green BD Metabolomic Profiling of Bile Acids in Clinical and Experimental Samples of Alzheimer’s Disease. Metabolites 2017, 7, No. 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Baloni P; Funk CC; Yan J; Yurkovich JT; Kueider-Paisley A; Nho K; Heinken A; Jia W; Mahmoudiandehkordi S; Louie G; et al. Metabolic Network Analysis Reveals Altered Bile Acid Synthesis and Metabolism in Alzheimer’s Disease. Cell Rep. Med. 2020, 1, No. 100138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Nho K; Kueider-Paisley A; MahmoudianDehkordi S; Arnold M; Risacher SL; Louie G; Blach C; Baillie R; Han X; Kastenmüller G; et al. Altered Bile Acid Profile in Mild Cognitive Impairment and Alzheimer’s Disease: Relationship to Neuroimaging and CSF Biomarkers. Alzheimer’s Dement. 2019, 15, 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Graham SF; Rey NL; Ugur Z; Yilmaz A; Sherman E; Maddens M; Bahado-Singh RO; Becker K; Schulz E; Meyerdirk LK; et al. Metabolomic Profiling of Bile Acids in an Experimental Model of Prodromal Parkinson’s Disease. Metabolites 2018, 8, No. 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Griffiths WJ; Abdel-Khalik J; Yutuc E; Roman G; Warner M; Gustafsson JÅ; Wang Y Concentrations of Bile Acid Precursors in Cerebrospinal Fluid of Alzheimer’s Disease Patients. Free Radical Biol. Med 2019, 134, 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ismail IT; Showalter MR; Fiehn O Inborn Errors of Metabolism in the Era of Untargeted Metabolomics and Lipidomics. Metabolites 2019, 9, No. 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Chen Z; Li Z; Li H; Jiang Y Metabolomics: A Promising Diagnostic and Therapeutic Implement for Breast Cancer. Onco Targets Ther. 2019, 12, 6797–6811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Chong J; Soufan O; Li C; Caraus I; Li S; Bourque G; Wishart DS; Xia J MetaboAnalyst 4.0: Towards More Transparent and Integrative Metabolomics Analysis. Nucleic Acids Res. 2018, 46, W486–W494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Havelund JF; Heegaard NHH; Færgeman NJK; Gramsbergen JB Biomarker Research in Parkinson’s Disease Using Metabolite Profiling. Metabolites 2017, 7, No. 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Jové M; Portero-Otín M; Naudí A; Ferrer I; Pamplona R Metabolomics of Human Brain Aging and Age-Related Neurodegenerative Diseases. J. Neuropathol. Exp. Neurol 2014, 73, 640–657. [DOI] [PubMed] [Google Scholar]
- (41).Patti GJ; Yanes O; Siuzdak G Metabolomics: The Apogee of the Omics Trilogy. Nat. Rev. Mol. Cell Biol 2012, 13, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Gu H; Gowda GAN; Neto FC; Opp MR; Raftery D RAMSY: Ratio Analysis of Mass Spectrometry to Improve Compound Identification. Anal. Chem 2013, 85, 10771–10779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kind T; Wohlgemuth G; Lee DY; Lu Y; Palazoglu M; Shahbaz S; Fiehn O FiehnLib: Mass Spectral and Retention Index Libraries for Metabolomics Based on Quadrupole and Time-of-Flight Gas Chromatography/Mass Spectrometry. Anal. Chem 2009, 81, 10038–10048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).González-Domínguez R; García-Barrera T; Gómez-Ariza JL Using Direct Infusion Mass Spectrometry for Serum Metabolomics in Alzheimer’s Disease. Anal. Bioanal. Chem 2014, 406, 7137–7148. [DOI] [PubMed] [Google Scholar]
- (45).Lin S; Liu H; Kanawati B; Liu L; Dong J; Li M; Huang J; Schmitt-Kopplin P; Cai Z Hippocampal Metabolomics Using Ultrahigh-Resolution Mass Spectrometry Reveals Neuroinflammation from Alzheimer’s Disease in CRND8 Mice. Anal. Bioanal. Chem 2013, 405, 5105–5117. [DOI] [PubMed] [Google Scholar]
- (46).Zhu J; Djukovic D; Deng L; Gu H; Himmati F; Chiorean EG; Raftery D Colorectal Cancer Detection Using Targeted Serum Metabolic Profiling. J. Proteome Res 2014, 13, 4120–4130. [DOI] [PubMed] [Google Scholar]
- (47).Carroll PA; Diolaiti D; McFerrin L; Gu H; Djukovic D; Du J; Cheng PF; Anderson S; Ulrich M; Hurley JB; et al. Deregulated Myc Requires MondoA/Mlx for Metabolic Reprogramming and Tumorigenesis. Cancer Cell 2015, 27, 271–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Gu H; Zhang P; Zhu J; Raftery D Globally Optimized Targeted Mass Spectrometry: Reliable Metabolomics Analysis with Broad Coverage. Anal. Chem 2015, 87, 12355–12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Gu H; Carroll PA; Du J; Zhu J; Neto FC; Eisenman RN; Raftery D Quantitative Method to Investigate the Balance between Metabolism and Proteome Biomass: Starting from Glycine. Angew. Chem., Int. Ed 2016, 55, 15646–15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Li R; Grimm SA; Mav D; Gu H; Djukovic D; Shah R; Merrick BA; Raftery D; Wade PA Transcriptome and DNA Methylome Analysis in a Mouse Model of Diet-Induced Obesity Predicts Increased Risk of Colorectal Cancer. Cell Rep. 2018, 22, 624–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Buas MF; Gu H; Djukovic D; Zhu J; Onstad L; Reid BJ; Raftery D; Vaughan TL Candidate Serum Metabolite Biomarkers for Differentiating Gastroesophageal Reflux Disease, Barrett’s Esophagus, and High-Grade Dysplasia/Esophageal Adenocarcinoma. Metabolomics 2017, 13, No. 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Eghlimi R; Shi X; Hrovat J; Xi B; Gu H Triple Negative Breast Cancer Detection Using LC-MS/MS Lipidomic Profiling. J. Proteome Res 2020, 19, 2367–2378. [DOI] [PubMed] [Google Scholar]
- (53).Gu H; Jasbi P; Patterson J; Yan J Enhanced Detection of Short-Chain Fatty Acids Using Gas Chromatography Mass Spectrometry. Curr. Protoc 2021, 1, No. e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Li CY; Dempsey JL; Wang D; Lee SW; Weigel KM; Fei Q; Bhatt DK; Prasad B; Raftery D; Gu H; et al. PBDEs Altered Gut Microbiome and Bile Acid Homeostasis in Male C57BL/6 Mice. Drug Metab. Dispos 2018, 46, 1226–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Dempsey JL; Wang D; Siginir G; Fei Q; Raftery D; Gu H; Yue Cui J Pharmacological Activation of PXR and CAR Downregulates Distinct Bile Acid-Metabolizing Intestinal Bacteria and Alters Bile Acid Homeostasis. Toxicol. Sci 2019, 168, 40–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Ginos BNR; Navarro SL; Schwarz Y; Gu H; Wang D; Randolph TW; Shojaie A; Hullar MAJ; Lampe PD; Kratz M; et al. Circulating Bile Acids in Healthy Adults Respond Differently to a Dietary Pattern Characterized by Whole Grains, Legumes and Fruits and Vegetables Compared to a Diet High in Refined Grains and Added Sugars: A Randomized, Controlled, Crossover Feeding Stud. Metabolism 2018, 83, 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Gutierrez D; Weinstock A; Antharam VC; Gu H; Jasbi P; Shi X; Dirks B; Krajmalnik-Brown R; Maldonado J; Guinan J; et al. Antibiotic-Induced Gut Metabolome and Microbiome Alterations Increase the Susceptibility to Candida Albicans Colonization in the Gastrointestinal Tract. FEMS Microbiol. Ecol 2019, 96, No. fiz187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Scoville DK; Li CY; Wang D; Dempsey JL; Raftery D; Mani S; Gu H; Cui JY Polybrominated Diphenyl Ethers and Gut Microbiome Modulate Metabolic Syndrome-Related Aqueous Metabolites in Mice. Drug Metab. Dispos 2019, 47, 928–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Riedel BC; Thompson PM; Brinton RD Age, APOE and Sex: Triad of Risk of Alzheimer’s Disease. J. Steroid Biochem. Mol. Biol 2016, 160, 134–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Liu D; Zhou XH ROC Analysis in Biomarker Combination with Covariate Adjustment. Acad. Radiol 2013, 20, 874–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Nagana Gowda GA; Raftery D Biomarker Discovery and Translation in Metabolomics. Curr. Metabolomics 2013, 1, 227–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Andrisic L; Dudzik D; Barbas C; Milkovic L; Grune T; Zarkovic N Short Overview on Metabolomics Approach to Study Pathophysiology of Oxidative Stress in Cancer. Redox Biol. 2018, 14, 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Lawal O; Ahmed WM; Nijsen TME; Goodacre R; Fowler SJ Exhaled Breath Analysis: A Review of ‘Breath-Taking’ Methods for off-Line Analysis. Metabolomics 2017, 13, No. 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Wang J; Wei R; Xie G; Arnold M; Kueider-Paisley A; Louie G; Mahmoudian Dehkordi S; Blach C; Baillie R; Han X; et al. Peripheral Serum Metabolomic Profiles Inform Central Cognitive Impairment. Sci. Rep 2020, 10, No. 14059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Nafar F; Clarke JP; Mearow KM Coconut Oil Protects Cortical Neurons from Amyloid Beta Toxicity by Enhancing Signaling of Cell Survival Pathways. Neurochem. Int 2017, 105, 64–79. [DOI] [PubMed] [Google Scholar]
- (66).Chatterjee P; Fernando M; Fernando B; Dias CB; Shah T; Silva R; Williams S; Pedrini S; Hillebrandt H; Goozee K; et al. Potential of Coconut Oil and Medium Chain Triglycerides in the Prevention and Treatment of Alzheimer’s Disease. Mech. Ageing Dev 2020, 186, No. 111209. [DOI] [PubMed] [Google Scholar]
- (67).de la Rubia Ortí JE; García-Pardo MP; Drehmer E; Sancho Cantus D; Julián Rochina M; Aguilar MA; Hu Yang I Improvement of Main Cognitive Functions in Patients with Alzheimer’s Disease after Treatment with Coconut Oil Enriched Mediterranean Diet: A Pilot Study. J. Alzheimer’s Dis 2018, 65, 577–587. [DOI] [PubMed] [Google Scholar]
- (68).Chen Z; Zhong C Decoding Alzheimer’s Disease from Perturbed Cerebral Glucose Metabolism: Implications for Diagnostic and Therapeutic Strategies. Prog. Neurobiol 2013, 108, 21–43. [DOI] [PubMed] [Google Scholar]
- (69).Szablewski L Glucose Transporters in Brain: In Health and in Alzheimer’s Disease. J. Alzheimer’s Dis 2016, 55, 1307–1320. [DOI] [PubMed] [Google Scholar]
- (70).Burns CM; Chen K; Kaszniak AW; Lee W; Alexander GE; Bandy D; Fleisher AS; Caselli RJ; Reiman EM Higher Serum Glucose Levels Are Associated with Cerebral Hypometabolism in Alzheimer Regions. Neurology 2013, 80, 1557–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Spector R. Fatty Acid Transport Through the Blood-Brain Barrier. J. Neurochem 1988, 50, 639–643. [DOI] [PubMed] [Google Scholar]
- (72).Fernando WMADB; Martins IJ; Goozee KG; Brennan CS; Jayasena V; Martins RN The Role of Dietary Coconut for the Prevention and Treatment of Alzheimer’s Disease: Potential Mechanisms of Action. Br. J. Nutr 2015, 114, 1–14. [DOI] [PubMed] [Google Scholar]
- (73).Butterfield DA; Halliwell B Oxidative Stress, Dysfunctional Glucose Metabolism and Alzheimer Disease. Nat. Rev. Neurosci 2019, 20, 148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Chen Z; Liu M; Li L; Chen L Involvement of the Warburg Effect in Non-Tumor Diseases Processes. J. Cell. Physiol 2018, 233, 2839–2849. [DOI] [PubMed] [Google Scholar]
- (75).Vlassenko AG; Gordon BA; Goyal MS; Su Y; Blazey TM; Durbin TJ; Couture LE; Christensen JJ; Jafri H; Morris JC; et al. Aerobic Glycolysis and Tau Deposition in Preclinical Alzheimer’s Disease. Neurobiol. Aging 2018, 67, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Goyal MS; Vlassenko AG; Blazey TM; Su Y; Couture LE; Durbin TJ; Bateman RJ; Benzinger TLS; Morris JC; Raichle ME Loss of Brain Aerobic Glycolysis in Normal Human Aging. Cell Metab. 2017, 26, 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Ruiz HH; Chi T; Shin AC; Lindtner C; Hsieh W; Ehrlich M; Gandy S; Buettner C Increased Susceptibility to Metabolic Dysregulation in a Mouse Model of Alzheimer’s Disease Is Associated with Impaired Hypothalamic Insulin Signaling and Elevated BCAA Levels. Alzheimer’s Dement. 2016, 12, 851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Li H; Ye D; Xie W; Hua F; Yang Y; Wu J; Gu A; Ren Y; Mao K Defect of Branched-Chain Amino Acid Metabolism Promotes the Development of Alzheimer’s Disease by Targeting the MTOR Signaling. Biosci. Rep 2018, 38, No. BSR20180127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Kontaxi C; Piccardo P; Gill AC Lysine-Directed Post-Translational Modifications of Tau Protein in Alzheimer’s Disease and Related Tauopathies. Front. Mol. Biosci 2017, 4, No. 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Albensi BC Dysfunction of Mitochondria: Implications for Alzheimer’s Disease. Int. Rev. Neurobiol 2019, 145, 13–27. [DOI] [PubMed] [Google Scholar]
- (81).Wilkins HM; Swerdlow RH Relationships Between Mitochondria and Neuroinflammation: Implications for Alzheimer’s Disease. Curr. Top. Med. Chem 2015, 16, 849–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Swerdlow RH Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis 2018, 62, 1403–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Fiehn O Metabolomics by Gas Chromatography-Mass Spectrometry: Combined Targeted and Untargeted Profiling. Curr. Protoc. Mol. Biol 2016, 114, 30.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).González-Domínguez R; González-Domínguez Á; Sayago A; Fernández-Recamales Á Mass Spectrometry-Based Metabolomic Multiplatform for Alzheimer’s Disease Research. In Methods in Molecular Biology; Humana Press Inc., 2018; Vol. 1750, pp 125–137. [DOI] [PubMed] [Google Scholar]
- (85).Sun C; Gao M; Wang F; Yun Y; Sun Q; Guo R; Yan C; Sun X; Li Y Serum Metabolomic Profiling in Patients with Alzheimer Disease and Amnestic Mild Cognitive Impairment by GC/MS. Biomed. Chromatogr 2020, 34, No. e4875. [DOI] [PubMed] [Google Scholar]
- (86).Godyń J; Jończyk J; Panek D; Malawska B Therapeutic Strategies for Alzheimer’s Disease in Clinical Trials. Pharmacol. Rep 2016, 68, 127–138. [DOI] [PubMed] [Google Scholar]
- (87).Blennow K; de Leon MJ; Zetterberg H Alzheimer’s Disease. Lancet 2006, 368, 387–403. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.