

Abstract
In addition to oxidative phosphorylation (OXPHOS), mitochondria perform other functions such as heme biosynthesis and oxygen sensing and mediate calcium homeostasis, cell growth, and cell death. They participate in cell communication and regulation of inflammation and are important considerations in aging, drug toxicity, and pathogenesis. The cell’s capacity to maintain its mitochondria involves intramitochondrial processes, such as heme and protein turnover, and those involving entire organelles, such as fusion, fission, selective mitochondrial macroautophagy (mitophagy), and mitochondrial biogenesis. The integration of these processes exemplifies mitochondrial quality control (QC), which is also important in cellular disorders ranging from primary mitochondrial genetic diseases to those that involve mitochondria secondarily, such as neurodegenerative, cardiovascular, inflammatory, and metabolic syndromes. Consequently, mitochondrial biology represents a potentially useful, but relatively unexploited area of therapeutic innovation. In patients with genetic OXPHOS disorders, the largest group of inborn errors of metabolism, effective therapies, apart from symptomatic and nutritional measures, are largely lacking. Moreover, the genetic and biochemical heterogeneity of these states is remarkably similar to those of certain acquired diseases characterized by metabolic and oxidative stress and displaying wide variability. This biologic variability reflects cell-specific and repair processes that complicate rational pharmacological approaches to both primary and secondary mitochondrial disorders. However, emerging concepts of mitochondrial turnover and dynamics along with new mitochondrial disease models are providing opportunities to develop and evaluate mitochondrial QC-based therapies. The goals of such therapies extend beyond amelioration of energy insufficiency and tissue loss and entail cell repair, cell replacement, and the prevention of fibrosis. This review summarizes current concepts of mitochondria as disease elements and outlines novel strategies to address mitochondrial dysfunction through the stimulation of mitochondrial biogenesis and quality control.
I. Introduction and Background
Eukaryotic mitochondria were once considered static bean-shaped organelles that generate ATP, but are now recognized as an expansive network of organelles that undergoes dimensional and structural changes to meet the specific energy needs of the cell (Fig. 1). Their involvement in heme metabolism, calcium homeostasis, inflammation, cell proliferation, and apoptosis gives them wide-ranging roles in cell survival (Smith et al., 2012). The cell’s mitochondrial mass is closely regulated by complex intracellular and extracellular signaling pathways that respond to energy demand and is adjusted through the inducible process of mitochondrial biogenesis (Scarpulla, 2008; Piantadosi and Suliman, 2012a,b; Scarpulla et al., 2012).
Fig. 1.

Normal mitochondrial structure in the heart. (Left) Representative micrographs of section of the mouse left ventricle showing immunofluorescence staining for TCA cycle enzyme citrate synthase. The mitochondria appear as tiny green, bead-like structures (Alexa 488, green fluorescence). Nuclei are stained blue (DAPI; 4′,6-diamidino-2-phenylindole). Scale bar = 10 µm. (Middle) Isolated rat cardiomyocytes showing the intracellular mitochondrial network by confocal fluorescence microscopy stained with MitoTracker red. The cytoskeleton protein F-actin is green and the cell nucleus is blue (DAPI; 4′,6-diamidino-2-phenylindole). 600×. Scale bar = 0.4 µm. (Right) Electron micrograph of normal mouse heart mitochondria. Cardiomyocyte exhibits abundant electron-dense mitochondria (m) surrounded by smooth endoplasmic reticulum (SER). N = cell nucleus. Scale bar = 0.2 μm.
Mitochondrial biogenesis is defined as the set of molecular instructions by which cells replace or increase their mitochondria through the proliferation of pre-existing organelles (Scarpulla, 2011; Piantadosi and Suliman, 2012a). It involves close cooperation between nuclear and mitochondrial genomes that was originally characterized as part of the process of organelle expansion during mitosis, where the doubling of mitochondrial volume imparts each daughter cell with a roughly equivalent complement of mitochondria. This process is fundamental to growth and development, is regulated by specific hormonal or paracrine signals, and in adult tissues is induced in response to increased energy requirements, for instance, in cardiac and skeletal muscle during exercise training (Holloszy, 2008; Joseph et al., 2012). It is also induced by calorie restriction as well as loss of mitochondrial functional reserve due to damage to the organelles by a range of pathologic events (Piantadosi and Suliman, 2012b).
To maintain its mitochondria and conserve aerobic energy reserve, the cell must integrate three processes near simultaneously: the identification of irreparably damaged mitochondria; their targeted elimination through selective mitochondrial autophagy (mitophagy); and their efficient replacement through mitochondrial biogenesis. If this cycle is compromised, the cell becomes susceptible not just to loss of energy regulation, but to calcium dysregulation, disruption of heme biosynthesis, oxidative damage from excessive generation of reactive oxygen species (ROS) by dysfunctional mitochondria and intrinsic apoptosis (Murphy and Smith, 2007). Thus, like other high-fidelity subcellular processes, for example, the proofreading of nascent protein folding in the endoplasmic reticulum (Kopito, 1997), mitochondria are engaged in an assiduous quality control (QC) system.
Under most circumstances, mitochondria are the main endogenous producers of ROS in the cell but also constitute a major antioxidant defense. Both facets encompass central prosurvival functions involving antiapoptotic and anti-inflammatory pathways that limit tissue loss and hold fibrotic mechanisms in check. The antioxidant role is related both to inducible mitochondrial ROS-scavenging systems and to the fact that cytochrome c oxidase fully reduces molecular O2 to water. The antiapoptotic effect derives from both the calcium storage function and the expression of antiapoptotic mitochondrial proteins, whereas counter-inflammatory mechanisms are related mainly to regulation of inflammasome assembly. Each of these aspects is also coupled to the genetic programming for mitochondrial QC.
Despite extensive work on the molecular regulation of mitochondrial turnover in health and disease, rational mitochondrial-based strategies are only now being formulated for conditions that involve mitochondrial dysfunction and have proven refractory to conventional therapeutics. The conditions that damage mitochondria entail a range of injuries including ischemia-reperfusion, systemic inflammatory states, cardiovascular disease, degenerative diseases of the musculoskeletal and central nervous systems, aging, and toxic injuries to solid organs such as the liver and kidney.
Many of the signaling pathways that maintain energy homeostasis also support the resolution of mitochondrial damage. In addition to ATP production, mitochondria coordinate numerous metabolic and anaplerotic reactions through the Krebs cycle and fatty acid metabolism (Chan, 2007). The capacity to regenerate mitochondria, however, is challenging because of the mitochondrial genome, as each organelle harbors multiple copies of the circular ∼16.6-kb double-stranded DNA molecule (mitochondrial DNA, mtDNA) that encode for 13 electron transport chain proteins, 2 rRNA of the mitochondrial ribosome, and 22 tRNA required for the translation of protein in the matrix (Bonawitz et al., 2006). The remaining ∼80 OXPHOS subunits and 1,000+ other mitochondrial proteins are encoded by nuclear genes, the mRNA translated by cytosolic ribosomes, and the protein products imported via specialized mitochondrial targeting and translocation mechanisms (Becker et al., 2012). This bigenomic origination means that mutations in either mitochondrial or nuclear genes produce mitochondrial dysfunction and contribute to heritable metabolic disorders, cancer, neurodegenerative disease, and to age-related disorders (Wallace, 2005; Shutt and Shadel, 2010).
The genetic organization of mammalian mtDNA is highly conserved (Shadel and Clayton, 1997). Genes are present on both DNA strands, designated by relative weight per nucleotide as the heavy (H)- and light (L)-strands. The H-strand encodes two rRNAs, 12 mRNAs, and 14 tRNAs, whereas the L-strand encodes 1 mRNA and 8 tRNAs (Fig. 2). The noncoding control region or D-loop encompasses cis-acting regulatory elements required for mtDNA transcription and replication (Shadel and Clayton, 1997). Polycistronic RNA generated by mtDNA transcription is processed into individual rRNAs, tRNAs, and mRNAs. Mitochondrial plasticity, however, is largely under nuclear control, and many nuclear-encoded mitochondrial proteins (NEMPs) are expressed in a tissue- or cell-specific manner that allows proteome modifications for specialized function (Balaban, 2012).
Fig. 2.

Schematic of the major steps of mtDNA replication and transcription. The double-stranded mtDNA molecule is replicated in a strand a synchronous manner and L-strand replication starts at its origin of replication (OL) after the nascent H-strand has completed about two-thirds of the circle proceeding in the opposite direction. H-strand replication is initiated using a short L-strand mRNA transcript synthesized by mitochondrial RNA polymerase (POLRMT). POLRMT accesses its DNA template by binding of mitochondrial transcription factor A, B1, and B2 (Tfam, TFB1M, and TFB2M) at the mtDNA duplex. Precursor RNA primer is cleaved by processing endonuclease (RNase) at the H-strand origin of replication (OH). Bases are added to free 3′-termini by mitochondrial DNA polymerase gamma (Polγ). Twinkle is required for preparation of single-stranded templates for Polγ activity. H-strand transcription is initiated from two promoter sites, HSP1 and HSP2. HSP1 transcripts are terminated at the termination sequence (TERM) within the tRNA-Leu (UUR) gene where MTERF1 binds. HSP2 generate near full-length polycistronic transcripts that are processed into individual RNAs. L-strand transcription is initiated from a single promoter site LSP, which generates near full-length polycistronic messages. The replication origins of both the H- and L-strands are indicated (OH and OL, respectively). CSB, conserved sequence block; HSP, heavy-strand promoter; LS, light-strand promoter; MTERF1, mitochondrial termination factor 1; MtSSB, mitochondrial single-stranded binding protein.
Mitochondria, of course, regulate intrinsic programmed cell death, aspects of innate immunity (Hailey et al., 2010), and cellular responses to starvation, such as autophagosomal membrane genesis (West et al., 2011). Specific domains called mitochondria-associated membranes (Vance, 1990) form through interaction and communication with cytoskeleton and endoplasmic reticulum (ER), generating small vesicular carriers called mitochondrial-derived vesicles (MDVs) (Neuspiel et al., 2008) that transport cargo to peroxisomes. Recently, a new aspect is the transport of mitochondrial proteins and lipids to other intracellular organelles, e.g., fusion with multivesicular bodies or the late endosome (Soubannier et al., 2012a; Sugiura et al., 2014). Conceptually, therefore, mitochondrial QC is not only tied to energy production but to ancillary functions for the cell’s homeostatic response to external challenges.
For disruptions in mitochondrial morphology, or movement not to impact cell survival, mitochondrial QC induction opposes limitations on mitochondrial function, distribution, and mass. This implies fine control between the production of functional organelles (biogenesis) and the selective degradation and elimination of dysfunctional ones by mitophagy. Although QC involves a far-reaching set of molecular instructions to protect cell function and viability, the transcriptional regulation of mitochondrial biogenesis is emphasized here because many disease states have been linked to persistent mitochondrial dysfunction and altered redox homeostasis. Interruption of mitochondrial assembly, turnover, or function without cell death contributes to a surprisingly wide range of pathologies, raising the need for highly targeted therapies (Murphy and Smith, 2007; Wallace et al., 2010). The opportunities and challenges of mitochondrial biogenesis as a therapeutic target are outlined here and illustrated with examples from current mitochondrial-based pharmacology.
II. Mitochondrial Biogenesis and Mitochondrial Turnover
Healthy mitochondrial populations are dynamic and exhibit variable turnover rates in an actively regulated transcriptional process that links mitochondrial biogenesis to the degradation of senescent and damaged mitochondria by mitophagy. Because mitochondria cannot be synthesized de novo but must be constantly renewed, the rate of homeostatic mammalian QC in vivo is fairly low (Miwa et al., 2008), on the order of days, whereas it is more rapid in cell culture (Hernandez et al., 2013). In the rat heart, for instance, the mitochondrial population has an estimated half-life of roughly 2 weeks (Rabinowitz and Zak, 1975). Thus, a typical unstressed cardiomyocyte (∼1000 mitochondria) would replace ∼1.5 mitochondria each hour. Moreover, turnover may be regulated by the circadian clock, because a number of OXPHOS enzymes show strong diurnal variation. This may be related in part to dietary intake and to the fasting period during sleep; hence nocturnal turnover may lead to replacement of a few percent of the mitochondrial population daily. Turnover rates also vary with the metabolic status of specific tissues, and can be greatly accelerated to allow cells to rapidly adjust to new energy demands from genetic, metabolic, dietary, or damage events, thereby dampening the impact of sudden changes in local microenvironments (Medeiros, 2008). Hence, the mitochondrial turnover rate is important in the rate of mitochondrial damage resolution.
The importance of intermediate repair and disposal of damaged macromolecules for mitochondrial homeostasis cannot be overstated, particularly for mtDNA and matrix proteins. Mammalian mtDNA repair is mediated by enzymes similar in activity to those in the nucleus, and all are encoded by nuclear genes and must therefore be imported into the organelles (Kazak et al., 2012). The turnover rates of respiratory chain subunit proteins are variable and have recently been shown to correlate with location within the mitochondria, evolutionary origin, site of protein encoding, and degree of ubiquitination (Karunadharma et al., 2015). Mitochondria contain imported proteases that are responsible for the maintenance of protein quality (mitochondrial unfolded protein response) including the Lon protease and other AAA+ superfamily proteases (ATPases associated with diverse cellular activities) (Grisolia et al., 1979), which are required for the energy-dependent remodeling or translocation of macromolecules. These proteases degrade superfluous or damaged subunits into small peptides and act as chaperones to extrude and target them for degradation by proteasomes (Wikstrom et al., 2009). The exact peptide extrusion mechanism is not clear, but it is insufficient to handle large numbers of damaged inner membrane and matrix proteins or to compensate for high damage rates. As mentioned, mitochondria generate MDVs and compartmentalize them differently, including sequestration into lysosomes and peroxisomes. MDV cargo transported to lysosomes is destined for degradation, and MDVs are enriched with oxidized proteins (Soubannier et al., 2012a,b). However, the purpose of targeting MDVs to the peroxisome is unclear (Sugiura et al., 2014), because the peroxisome specializes in detoxification of hydrogen peroxide and the breakdown of purines and certain types of fatty acids. However important these repair or disposal mechanisms are, none is sufficient to eliminate whole mitochondria (Sugiura et al., 2014). Those situations require mitophagy in conjunction with the activation of mitochondrial biogenesis. Although our understanding of this area is evolving rapidly, mitophagy is not a singular event (Lemasters, 2014), and it is not clear how the cell determines when to halt repair and eliminate the organelle.
For many years, our understanding of the regulation of mitochondrial biogenesis was limited to events related to cell division and to increased work. The unique challenge for the cell is the rapid synchronization of mtDNA replication and mitochondrial-encoded gene expression in conjunction with the upregulation of many genes for NEMPs and associated subsets of nonmitochondrial genes. Subsequently, it became known that the program is activated by changes in substrate availability (e.g., starvation) (Moreno-Loshuertos et al., 2006, 2011) and by certain paracrine effectors and hormones (Scarpulla, 2011) necessary for the cell to maintain its functional mitochondrial mass (Onyango et al., 2010). The process is also essential to recovery from oxidative stress, inflammation, and mitochondrial drug toxicity (Suliman et al., 2007a). In each case, signal specificity is maintained through a network of nuclear DNA-binding transcription factors and coregulators upregulated by diverse physiologic conditions and by tissue or cell-specific events that respond to changes in mitochondrial function and/or mass. The response to cell damage is especially challenging because the precise communication between nuclear and mitochondrial genomes may become difficult to maintain, but is critical to support function and avoid apoptosis or necrosis as well as to adjust mitochondrial phenotype to changes in microenvironment or disease pathogenesis.
The data on mitochondrial dysfunction in acquired diseases has at times seemed contradictory because each situation is different and quite complex. For instance, during acute experimental ischemia, inflammation, and/or oxidative stress, various studies have reported impaired (Crouser et al., 2004; Gellerich et al., 1999), unchanged (Mela-Riker et al., 1992; Taylor et al., 1995), or improved (Taylor et al., 1998; Lu et al., 2003) mitochondrial function. Such discrepancies have many causes, such as type of cell injury, duration and severity of injury, the cell type and function, and the experimental situation. There are also uncontrolled variables in the repair responses that promote or impede the resolution of mitochondrial dysfunction (Reynolds et al., 2009). These complexities have made it harder to devise rational mitochondrial-based therapies and to confirm the mode of action of the intervention and its impact on cell damage and resolution.
Nonetheless, there is a clear set of mitochondria-related events that occur after acute sterile or infectious damage to previously healthy cells and tissues. Cells that exhibit mitochondrial damage, but are able to survive, show the induction of mitochondrial biogenesis, activation of autophagy, and eventual repopulation with functional mitochondria (Chang et al., 2015). This sequence of events is now well appreciated morphologically for major disturbances in oxygen delivery to tissues, for instance after ischemia-reperfusion or severe hypoxia-reoxygenation in the heart and the brain (Gutsaeva et al., 2008; Ahuja et al., 2010). Mitochondrial QC is activated by mechanisms involving both energy limitation and redox pathways, which, as discussed later, remain under active investigation.
The early response to oxidative or electrophilic stress involve activation of the transcription factor nuclear factor (erythoid-derived 2)-like2 (NFE2L2 or Nrf2), which binds to antioxidant response (ARE) elements in the promoter regions of target genes (Wakabayashi et al., 2004), coordinates antioxidant and antiapoptotic gene upregulation (Alam et al., 2004), and induces mitochondrial biogenesis (Piantadosi et al., 2008; Athale et al., 2012; MacGarvey et al., 2012). In a chemical stress model of renal ischemia-reperfusion, increased mitochondrial biogenesis has been shown to accelerate the recovery of OXPHOS through p38 and epidermal growth factor receptor activation of PGC-1α (Rasbach and Schnellmann, 2007). Those investigators also found that stimulation of the 5-hydroxytryptamine receptor activated mitochondrial biogenesis and suggested that 5-hydroxytryptamine agonists as a treatment of mitochondrial and cell injury (Rasbach et al., 2010). Because damage by ongoing inflammatory processes is often periodic, mitochondria are frequently damaged not just by the original insult but by subsequent oxidative and nitrosative stress driven by cytokine production. In severe sepsis, for instance, ROS and nitric oxide (NO) overproduction leads to mtDNA damage, but NO also activates mitochondrial biogenesis through complex crosstalk between a mitochondrion and the nucleus that yields a net increase in the synthesis of new organelles (Piantadosi et al., 2007; Medeiros, 2008; Ahuja et al., 2010; Seo et al., 2010).
The transcriptional activation of nuclear and mitochondrial gene expression leads to a nascent protein supply in appropriate quantities at appropriate times for an expanding mitochondrial subpopulation. Because the bulk of the proteins are NEMPs, and mtDNA-encoded mRNAs are involved only in the synthesis of a dozen OXPHOS proteins, nuclear transcription factors play a predominant role in mitochondrial biogenesis. The mitochondrial genome, however, must be transcribed and replicated during mitochondrial biogenesis so that the new population remains at full transcriptional capacity.
A relatively small set of DNA-binding transcriptional regulators are essential for mitochondrial biogenesis including nuclear respiratory factors 1 and 2 (NRF-1 and NRF-2), cAMP response element-binding protein (CREB1), estrogen response receptor alpha (ERRα), and the nuclear coactivators peroxisome proliferator-activated receptor γ coactivator 1-α, 1-β (PGC-1α, β), and PPRC (Peroxisome proliferator-activated receptor gamma coactivator-related protein 1). PGC-1α interacts, for example, with NRF-1 and 2, ERRα, and CREB1 to regulate the transcription of genes involved in mitochondrial biogenesis and energy metabolism (Wu et al., 2006). These coactivators provide a direct link between physiologic events and the regulation of mitochondrial biogenesis and can be induced pathologically as well as pharmacologically.
Several other nuclear transcription factors are linked to the expression of respiratory chain subunits such as YY1 and myocyte enhancer factor-2, and YY1 is an important component of nutrient sensing by the mammalian target of rapamycin (mTOR) pathway. Inhibition of mTOR decreases ERRα and NRF-1/2 expression and prevents the interaction between YY1 and PGC-1α. The oncoprotein Myc also regulates nuclear-encoded mitochondrial programming largely by an interaction with NRF-1 (Morrish et al., 2003) and by increasing PGC-1β expression(Morrish and Hockenbery, 2014).
Although mitochondria are often damaged by ROS, now recognized as both agents of damage and architects of senescence, some ROS, particularly H2O2, act as redox messengers for NEMP expression. Indeed, mitochondrial H2O2 production is an important trigger for mitochondrial proliferation, particularly during acute inflammation where severe mitochondrial damage in organs like the liver, heart, and kidneys must be cleared (Suliman and Piantadosi, 2014). Hence, mitochondria-targeted antioxidant compounds may limit damage but also interfere with the redox sensing of damage that is not prevented by the drug.
The prompt restoration of a highly functional population of mitochondria is fundamental to the recovery of injured cells and tissues, particularly those with delicate bioenergetic equilibria such as cardiomyocytes, hepatocytes, renal tubular cells, and neurons. Moreover, proliferating cells depend on mitochondrial biogenesis to provide daughter cells with an adequate supply of mitochondria for the required aerobic capacity. These concepts have brought mitochondrial biogenesis to the forefront as a therapeutic target for diseases characterized by gradual cell loss that to date have had no effective therapies. The presence of cyclical cell damage leads to the need for additional energy while exposing the proliferating mitochondria to recurrent bouts of damage.
Only a handful of small molecules are known today to induce mitochondrial biogenesis without first causing explicit mitochondrial damage. Therefore, the pharmacological manipulation of mitochondrial biogenesis using noninjurious agents is a priority for injuries and diseases already characterized by mitochondrial dysfunction (Funk et al., 2010). Mitochondrial pharmacology is thus an exciting area with many promising new directions, and in covering a number of approaches to induce mitochondrial biogenesis, including natural products, we have highlighted novel concepts and targets for therapy of mitochondrial genetic disorders, chronic degenerative diseases, and life span extension.
III. Mitochondrial Disorders and Diseases
Mitochondrial diseases are conveniently divided into primary and secondary disorders. Primary disorders are characterized by heritable mitochondrial gene defects such as base substitution or insertion-deletion mutations in mtDNA coding regions or genes for NEMPs, whereas the secondary disorders are due to nongenetic events including mitochondrial toxins, ischemia, infections, or sterile inflammation. The most common inherited disorders, Friedreich's ataxia; Leber’s hereditary optic neuropathy; mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS); myoclonic epilepsy with ragged-red fibers; and Leigh Syndrome show variable clinical profiles and susceptibility to stressors. In addition to defects in energy conservation, they share, to various degrees, oxidative stress as a pathogenic feature due to endogenous ROS production or to a decrease in mitochondrial anti-oxidant protection or both.
The frequency of primary mitochondrial disorders ranges up to 1 in 5,000 (Schaefer et al., 2004), and the organelle a logical pharmacological target in these individuals. Many primary disorders manifest in early childhood and therapeutic options are limited. Conceptually, however, phenotypic commonalities in mtDNA loss-of-function mutations help to inform the pathogenic impact of secondary or acquired disorders. Acquired mitochondrial loss-of-function has been linked with varying levels of evidence to Parkinson’s disease (PD), Alzheimer’s disease (AD), amyotrophic lateral sclerosis (Lin and Beal, 2006; Trushina and McMurray, 2007), schizophrenia, and autism (Manji et al., 2012; Rossignol and Frye, 2012). Secondary mitochondrial defects are best exemplified by severe, persistent inflammatory states but are also represented in metabolic disorders (Nasrallah and Horvath, 2014) and some cancers (Wallace, 2012). For example, mutations in the mitochondrial intermembrane form of superoxide dismutase SOD1, which prevents superoxide damage, is linked to the progression of amyotrophic lateral sclerosis; (Vehvilainen et al., 2014) NADH dehydrogenase 4 to Leber’s hereditary optic neuropathy (Kornmann, 2013); PARKIN to the familial form of PD (Schmidt et al., 2010); and TCA cycle enzymes to oncogenesis (Schaefer et al., 2004).
The induction of mitochondrial biogenesis has also been promoted as a strategy for the treatment of OXPHOS disorders in mouse models (Smith et al., 2012). Hypothetically, boosting mitochondrial quality and mass should increase remaining OXPHOS activity unless the defective protein acts as a dominant-negative. Otherwise, the approach should enhance the cellular ATP synthetic capacity and presumably improve cellular energy deficits and enhance repair mechanisms. Although optimal and specific agents are not yet available, there is promising proof-of-principle evidence. For instance, the mTOR signaling pathway integrates many processes that are involved in the promotion of cell growth (Laplante and Sabatini, 2009). Specific inhibition of the mTOR pathway with rapamycin enhances survival and attenuates disease progression in a mouse model of Leigh syndrome. These mice, which are deficient in mitochondrial Complex I subunit Ndufs4 [NADH dehydrogenase (ubiquinone) Fe-S protein 4] display reduced neurologic symptoms, neuroinflammation, and brain lesions after rapamycin treatment. Although an exact rescue mechanism was not identified, the metabolic shift toward amino acid catabolism was postulated to have alleviated a buildup of glycolytic intermediates (Johnson et al., 2013).
A. Metabolic Disorders
Mitochondrial dysfunction in primary metabolic organs such as the liver has a large impact on systemic metabolic homeostasis (Grattagliano et al., 2012). Studies on patients with obesity, diabetes, nonalcoholic steatohepatitis (NASH) or nonalcoholic fatty liver disease consistently display functional and structural abnormalities in hepatocyte mitochondria, such as OXPHOS impairment or mega-mitochondria (Begriche et al., 2006). Increased and decreased oxidation is reported as features of hepatocyte steatosis and insulin resistance (Fabbrini et al., 2010; Pessayre, 2007; Zhang et al., 2007). A decrease in oxidative activity in the hepatocyte induces diacylglycerol accumulation and steatosis with concurrent PKC activation and inhibition of insulin signaling (Zhang et al., 2007). Insulin-resistant patients show an increase in hepatic oxidation that is correlated with an increase in ROS production (Pessayre and Fromenty, 2005; Fabbrini et al., 2010).
Persistently high hepatic carbon oxidation rates in insulin resistance could be an adaptive mechanism to limit free fatty acid toxicity or to provide large quantities of reduced NADH. This effect may be decoupled from actual energy requirements and the ROS production reflective of respiratory chain damage or dysfunction (Vial et al., 2010). On the other hand, mitochondrial ROS production during OXPHOS facilitates insulin signaling through insulin receptor oxidation and phosphatase inhibition, for instance PTP1B and phosphatase and tensin homolog (PTEN) (Cheng et al., 2010). As such, ROS production would be expected to promote the inflammation by some of the cellular mechanisms discussed earlier.
Insulin resistance is characteristic of type 2 diabetes (DM2) and the inability of insulin to inhibit glucose output from the liver and promote glucose uptake by muscle (Saltiel and Kahn, 2001; Hribal et al., 2002). In mouse models, hepatic PGC-1α expression is elevated with the onset of DM2, where it contributes to constitutive gluconeogenesis and fatty acid oxidation (FAO) through coactivation of nuclear receptors HNF-4 and peroxisome proliferator-activated receptors (PPAR)-α, respectively (Herzig et al., 2001; Rhee et al., 2003). Insulin suppresses gluconeogenesis stimulated by Forkhead box O (FOXO) 1/PGC-1α, whereas a mutant, insulin-insensitive FOXO1 allele completely reverses this suppression in hepatocytes and in transgenic mice. Recent findings support a role for PGC-1α in linking nutrient deprivation to mitochondrial oxidant production through coactivation of ERRα to enhance sirtuin-1 (SIRT1) expression (Kong et al., 2010; Giralt et al., 2011).
SIRT3 is s nicotinamide adenine dinucleotide (NAD)-dependent protein deacetylase localized to mitochondria that allows tissue-specific optimization of ATP levels and metabolic enzyme activities (Verdin et al., 2010). SIRT3 opposes oxidative stress by precipitating a series of reactions beginning with activation of isocitrate dehydrogenase 2 and culminating in detoxification of peroxides by glutathione peroxidase (Someya et al., 2010). It deacetylates SOD2, leading to increased activity and enhanced oxidant scavenging in the matrix (Qiu et al., 2010; Tao et al., 2010). Another key factor is the high expression of PPARα in the liver, heart, and small intestine (Kersten, 2014). Acting as a nutrient sensor, PPARα promotes the adaptation of fatty acid catabolism, lipogenesis, and ketone body synthetic rates in response to feeding and starvation to stimulate hepatic fatty acid oxidation and fasting ketogenesis (Hashimoto et al., 2000; Sugden et al., 2002; Patsouris et al., 2004). An increase in PPARα leads to target gene activation in fatty acid oxidation (e.g., Cpt1a, Cpt2, Acadvl, Hadha) and ketogenesis (Hmgcs2, Hmgcl, Acat1). Furthermore, PPARα negatively regulates proinflammatory and nuclear factor (NF)-κB and AP-1 signaling in systemic inflammation, atherosclerosis, and NASH (Ip et al., 2003; Gervois et al., 2004).
Mitochondrial dysfunction and body energy balance are integral to metabolic disorders such as obesity and metabolic syndrome (Joseph et al., 2012; Szendroedi et al., 2012). In these disorders, there is renewed interest in the role of mitochondria-rich brown fat, which efficiently dissipates energy through nonshivering thermogenesis as a result of high levels of the uncoupling protein 1 (Wu et al., 2013). Attention has also been focused on the role of beige fat, which can be generated from white adipose tissue by thermogenic pathways in which mitochondrial biogenesis has a key role (Schulz et al., 2013). Although strategies to target mitochondria in the treatment of nonalcoholic fatty liver disease and NASH are still in the early stages, the opposite induction of mitochondrial biogenesis is a logical target for treatment of obesity and metabolic syndrome (Szendroedi et al., 2012; Wu et al., 2013). Pharmacological stimulation of mitochondrial biogenesis seems to induce certain positive effects of calorie restriction and exercise on such generic metabolic disorders.
B. Mitochondrial Quality Control and Inflammation
1. Importance of Innate Immunity.
The regulation of mitochondrial QC provides an important duel anti-inflammatory function: the control of inflammasome assembly and the expression of counter-inflammatory genes. This is made possible through connections between nuclear QC network programming and the innate immune system in tissues where host defense must be coordinated with repair mechanisms necessary to control damage from circulatory failure, pro-oxidants, xenobiotics, and pathogens. The connections between mitochondria, inflammation, and metabolism are of great interest, because dysfunctionality is associated with chronic inflammatory diseases, cancer, and fibrosis. The presence of infection is normally sensed by pattern recognition receptors, which, upon activation, trigger an acute inflammatory response and initiate defense and repair programs (Dostert et al., 2008; Schroder and Tschopp, 2010). The offender is sequestered and eliminated while tissue repair is initiated, which occurs over the course of a few days. When the source cannot be effectively removed, progressive cell damage and cell death leads to tissue loss and failure to heal. Such cycles of tissue destruction and healing lead to chronic inflammation and fibrosis and are characteristic of many degenerative diseases, including DM2, arthritis, inflammatory bowel disease, and neurodegenerative conditions. Under such circumstances, the mitochondrial mechanisms discussed here are actively involved (Tschopp, 2011).
The activation of mitochondrial biogenesis by the host immune response was first demonstrated more than a decade ago when lipopolysaccharide was found to stimulate hepatic and cardiac mitochondrial biogenesis in rats (Suliman et al., 2003b, 2004). Subsequently, the resolution of extensive cytokine-induced mtDNA damage after heat-inactivated Escherichia coli exposure was found to be due to compensatory mitochondrial biogenesis initiated in part by activation of toll-like receptor 4 (TLR4) and downstream signaling via CREB1 and NF-κB-dependent induction of NRF-1 (Suliman et al., 2005, 2007b). In murine peritonitis, depressed oxidative metabolism is eventually restored by mitochondrial biogenesis, thus providing a way to affect sepsis outcome (Haden et al., 2007). Indeed, sepsis-induced mitochondrial dysfunction is linked to multiple organ dysfunction syndrome, whereas the rate of resolution and restoration of organ function may depend on the reestablishment of mitochondrial number and function (Singer, 2007). The failure to maintain and/or restore mitochondrial mass not only impairs tissue repair but may foster immune suppression through prolonged upregulation of counter-inflammatory mediator genes such as interleukin (IL)-10 (Suliman and Piantadosi, 2014). Clinical data also support that activation of mitochondrial biogenesis in skeletal muscle influences survival in critical illness (Carre et al., 2010). Accordingly, innovative strategies to promote and protect mitochondrial biogenesis may lead to new ways to prevent organ failure in severe systemic inflammatory states (Piantadosi et al., 2007).
2. Role of the Inflammasome.
The full expression of host inflammation after immune cell exposure to pathogens or to diverse pathogen- and danger-associated molecular patterns or environmental triggers requires assembly of multiprotein inflammasome complexes. Inflammasomes generally contain a Nuclear oligomerization domain (NOD)-like receptor (NLR) molecule, NLRP1 (NOD-, leucine rich repeat-, and pyrin domain-containing 1), NLRP3, or others (Latz et al., 2013). A typical sensor complex consists of three protein components, for example NLRP3 (NALP3), ASC (PYCARD), and caspase-1. The NLRP3 complex activates caspase-1 (and caspase-11 in mice), leading to processing and release of IL-1β and IL-18.
Inflammasomes are also mitochondrial stress monitors and regulators of intrinsic apoptosis (Latz et al., 2013). Mitochondrial stress is sensed by the NLRP3 inflammasome through ROS production (Zhou et al., 2011) and/or the release of oxidized mitochondrial DNA (Shimada et al., 2012). Two lines of evidence favor mitochondria as the main source of ROS in NLRP3 inflammasome activation and hence as a signal-integrating organelle for innate immunity (Schroder and Tschopp, 2010; Nakahira et al., 2011; Zhou et al., 2011). In macrophages, inflammasome activation is impaired by mtDNA depletion or by inactivation of the outer membrane voltage-dependent anion channel (Fabbrini et al., 2010). Also, respiratory chain inhibition activates the NLRP3 inflammasome (Nakahira et al., 2011).
Disturbances in energy homeostasis lead to mitochondrial relocation via the microtubule system, which facilitates NLRP3 interaction with the ASC adaptor protein (Misawa et al., 2013). Optimal NLRP3 activation requires a Retinoic acid-inducible gene-I-like receptor adaptor molecule called MAVS (mitochondrial antiviral signaling protein) localized to the outer mitochondrial membrane (Subramanian et al., 2013). Another mechanism that increases IL-1β output in activated macrophages is through increases in the TCA cycle intermediate succinate, which stabilizes hypoxia-inducible factor-1α and allows sustained IL-1β transcription (Tannahill et al., 2013). The NLRP3 inflammasome is inhibited by the removal of damaged or dysfunctional mitochondria by mitophagy. The cell eliminates high ROS-generating mitochondria to avoid autodestructive inflammation (Nakahira et al., 2011). Inhibition of mitophagy leads to retention of ROS-generating mitochondria and inflammasome activation (Nakahira et al., 2011; Zhou et al., 2011). Mitochondria removed by mitophagy are replaced through mitochondrial biogenesis, thus avoiding persistent inflammasome activation that would lead to energy failure and cell death.
In overwhelming infections, immune dysregulation may lead to tissue damage in which surviving cells undergo cycles of mitochondrial damage and exhibit accelerated mitochondrial turnover, particularly for highly active tissues. Mitochondrial damage is driven by toll-like (TLR) receptor-dependent production of early-phase inflammatory mediators, such as tumor necrosis factor alpha and nitric oxide (NO) (Schulze-Osthoff et al., 1992). Activated macrophages and Kupffer cells generate certain interleukins that also cause mitochondrial damage (Schulze-Osthoff et al., 1992). Damaged mitochondria generate higher levels of ROS and may release calcium and intrinsic apoptosis proteins (Taylor et al., 1995; Kantrow et al., 1997). Excessive oxidant production can compromise mitochondrial function and structure through the direct chemical oxidation of proteins, lipids, and DNA. For example, the NO-superoxide reaction generates the powerful oxidant peroxynitrite (ONOO−) (Kurose et al., 1993), and in the presence of transition metals such as iron, H2O2 and superoxide generate the hydroxyl radical (⋅OH) by the Fenton reaction (Halliwell, 1989).
Transcription factors that initiate the host defense, including NF-κB, are also activated by oxidative stress (Koay et al., 2001). Hence, with the transcriptional activity of PGC-1α, NRF-1 upregulates many antioxidant defenses (Piantadosi and Suliman, 2006; St-Pierre et al., 2006). PGC-1α also promotes the induction for ROS-detoxifying enzymes such as mitochondrial superoxide dismutase (SOD2) and glutathione peroxidase-1. And in neuronal cells, CREB1 is an important activator of the Pargc1a (PGC-1α) gene after oxidant exposure (St-Pierre et al., 2006). In macrophages, PGC-1β overexpression inhibits canonical NF-κB-dependent cytokine production, which mitigates the acute inflammatory response (Vats et al., 2006). In murine Staphylococcus aureus peritonitis, early TLR-mediated events include the rapid upregulation of hepatic PGC-1α and PGC-1β in wild-type mice, whereas both genes are deregulated in TLR2−/− mice and increased in TLR4−/− mice. Meanwhile, PRC is upregulated in all three strains (Sweeney et al., 2010).
An inability to regulate inflammasome activity creates overlapping cycles of inflammatory tissue damage that may partly conceal organized mitochondrial QC processes. In mice, sublethal damage by lipopolysaccharide transiently depletes hepatic mtDNA content and impairs mitochondrial transcription (Suliman et al., 2003a). Compensation involves Akt/PKB phosphorylation of NRF-1, increasing its nuclear translocation and subsequent expression of Tfam and other mitochondrial transcriptome proteins, which after importation, allow mtDNA copy number to be restored (Suliman et al., 2003b). Initial mtDNA content depletion is related to TLR4 and NF-κB activation, and genetic ablation of TLR4 reduces, but does not eliminate this damage; however, recovery of mtDNA copy number is delayed in TLR4 null mice (Suliman et al., 2005).
This mtDNA damage leads to upregulation of repair enzymes such as the base excision glycosylase, OGG1, in part through NRF-1 and NRF-2 binding to promoter elements (Bartz et al., 2011). Notably, mtDNA copy number can be protected by increasing SOD2 in mitochondria, by inhibiting NOS activity, or by scavenging ONOO− (Choumar et al., 2011). In true infection, or after exposure to surrogate damage-associated molecular patterns, there is morphologic evidence of mitophagy and mitochondrial biogenesis, which in survivors restore mitochondrial mass within days (Suliman et al., 2003a; Haden et al., 2007; Watanabe et al., 2009).
If these processes cannot prevent energy compromise, impending energy failure, specifically ATP depletion, activates a key pathway regulated by the serine/threonine kinase AMPK (Bergeron et al., 2001). AMPK stimulates glucose and lipid catabolism and blocks energy-utilizing pathways such as protein and fatty acid biosynthesis. AMPK promotes mitochondrial biogenesis (Bergeron et al., 2001; Zong et al., 2002; Reznick and Shulman, 2006), NO production (Morrow et al., 2003), and autophagy (Kim et al., 2011) and opposes inflammation by interfering with NF-κB-dependent cytokine expression (Bai et al., 2010). The mechanism of NF-κB antagonism is not clear, but may indirectly involve PGC-1α, Forkhead box O (FoxO)-type transcription factors, and/or SIRT1 (Canto et al., 2009; Barroso et al., 2011; Salminen et al., 2011). Conversely, loss of AMPK activity is associated with increased inflammation.
However, the transcriptional activity of relevant nuclear receptors (NRs), particularly estrogen-related receptor ERRα, is blocked by repressors such as RIP140 and others (L'Horset et al., 1996); RIP140 thus suppresses metabolic gene expression and mitochondrial biogenesis (Powelka et al., 2006). In macrophages, RIP140 is actually a cytokine gene coactivator, and RIP140 deficiency inhibits the inflammatory response (Zschiedrich et al., 2008). RIP140 interacts with the RelA subunit of NF-κB and the histone acetylase CREB-binding protein and cooperates with CREB-binding protein coactivator complex on RelA-regulated promoters. RIP140 modulation of inflammatory gene expression is thus a good example of cell-specific integration of control pathways for metabolism and inflammation (Zschiedrich et al., 2008). Accordingly, newer findings on the trained monocyte response, which improves resistance to infection, indicates a requirement for a switch from respiration to glycolysis involving genes of the mTOR-HIF-1 α pathway (Borden, 2014).
C. Mitochondrial Dysfunction Caused by Therapeutic Agents
Well over 300 mitochondria-targeted small molecules have been annotated in DrugBank (Wishart et al., 2008) with a number of recent additions, suggesting that this is an area of growing therapeutic interest. However, drugs are also a frequent cause of mitochondrial dysfunction (Wallace, 2008), and a surprising number of them are taken up by mitochondria and inhibit one or more mitochondrial functions directly or lead to increased ROS generation and the damage produces a clinical phenotype. Thus, mitochondrial dysfunction is often implicated in drug-induced toxicity (Dykens and Will, 2007), yet there is no requirement to screen drugs for mitochondrial toxicity before Food and Drug Administration approval. This issue has been recognized for some time in patients with genetic mitochondrial diseases, particularly for agents that increase mitochondrial ROS production. Hence, molecules that supress mitochondrial ROS production have been an area of strong research interest for a number of years, particularly for CNS applications.
An example is the triaminopyridine flupirtine, a nonopioid analgesic with mitochondria-dependent antioxidant and free radical scavenging activity with efficacy toward ischemic neuronal damage, apoptosis, and age-associated brain disorders (Schluter et al., 2000). Instructively, the commercially available statins (e.g., atorvastatin and simvastatin), which lower cholesterol levels by inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase, have been implicated in neuroprotection against PD, AD, traumatic brain injury, and secondary progressive multiple sclerosis (Kumar et al., 2012; Malkki, 2014). In some instances, neuroprotection has been associated with the lipophilicity of the drug (Haag et al., 2009; Huang et al., 2015), but direct mitochondrial protection is not established either experimentally or clinically, and the well-known adverse effects of statins on mitochondria, such as inhibition of Coenzyme Q synthesis, is an important caveat with these agents (Golomb and Evans, 2008). The 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors cause a myopathy that has been attributed in part to a reduction in CoQ10 levels (Potgieter et al., 2013).
Other notable examples of mitochondrial drug toxicity include cardiac and skeletal myopathy associated clinically with doxorubicin, cisplatin, and nucleoside reverse-transcriptase inhibitors such as zidovudine (Galluzzi et al., 2014). Doxorubicin is notable because it prevents expression of compensatory mitochondrial biogenesis (Suliman et al., 2007a; Finsterer and Ohnsorge, 2013). Certain antibiotics (e.g., tetracycline and aminoglycosides) inhibit mtDNA translation and protein synthesis. Anticonvulsants, such as valproate and barbiturates, inhibit respiration, and the common over-the-counter analgesic acetaminophen (paracetamol) can inhibit mitochondrial oxygen consumption as discussed in Than et al. (2014).
Mitochondrial toxicity has led to withdrawal of a number of previously approved drugs, such as troglitazone, mainly due to hepatotoxicity. A majority of drugs with U.S. Food and Drug Administration black box warnings have been shown to be associated with mitochondrial toxicity (Dykens and Will, 2007). This has led the pharmaceutical industry to begin to screen for mitochondrial toxicity earlier to identify compounds that may too toxic to bring into clinical testing.
IV. Induction of Mitochondrial Biogenesis by the Metabolic Gases
The discovery that the metabolic gases induce mitochondrial biogenesis by specific cell signaling pathways has raised the possibility of capitalizing pharmacologically on them to hasten the resolution of tissue injury during and after episodes of extensive mitochondrial damage. Moreover, many cells activate the production of these gases during periods of severe cell stresses that are associated with mitochondrial damage.
A. Nitric Oxide
The potent paracrine vasodilator gas, nitric oxide (NO), has numerous effects on mitochondrial biology. NO is produced catalytically from l-arginine in an NADPH-dependent reaction by the three isoforms of nitric oxide synthase (NOS), and NO generated in vascular endothelium (by NOS3) transcriptionally activates mitochondrial biogenesis (Nisoli et al., 2004a). Correspondingly, the inducible NOS (NOS2) augments mitochondrial biogenesis during periods of inflammation (Reynolds et al., 2009; Suliman et al., 2010). Physiologic NO activates mitochondrial biogenesis via soluble guanylate cyclase (GC) and cyclic GMP-dependent signals (Nisoli et al., 2003; Gao, 2010); cGMP production upregulates PGC-1α and NRF-1 to initiate the program (Nisoli et al., 2003). The NO-dependent signaling mechanisms are not fully defined, but increased PGC-1α, CREB1, and NRF-1 expression are required for mitochondrial proliferation (Fig. 3) (Nisoli et al., 2004b; Lira et al., 2010).
Fig. 3.

Mitochondrial biogenesis by the endogenous gases. Nitric oxide (NO) and carbon monoxide (CO) are generated enzymatically by the NOS (1-3) and HO (1-2) isoforms, respectively. H2S is generated enzymatically from l-cysteine. The mitochondrion is a common organelle target of all three gases. Pharmaceutical development has leveraged these systems to design exogenous molecules to simulate the endogenous gases. Illustrated are ROS pathways for CO (and H2S), activation of cGMP by NO (or CO), and phosphorylation of peroxisome proliferator-activated receptor gamma coactivator (PGC-1α) via PKA. ROS, mainly H2O2, can activate Akt, leading to NRF-1 phosphorylation and its nuclear translocation (CO, H2S and their releasing molecules). NRF-1, NRF-2, and other nuclear transcription factors (NTFs) are coactivated by PGC-1α, leading to transcription of nuclear-encoded mitochondrial proteins (NEMPs), such as mitochondrial transcription factor A (Tfam), needed to activate mtDNA transcription and replication. NEMPs are imported into mitochondria through multiple outer (Tom)- and inner (Tim)-membrane transport machinery, and the proteins are folded and assembled. For the outer membrane, fission through the dynamin-related protein 1 (DRP1) and for the inner membrane OPA1 allow mitochondrial division, whereas mitofusins (Mfn) regulate fusion. Dynamic control of fusion/fission allows for mitochondrial network organization. Damaged mitochondria are tagged by Park and Pink for ubiquitination (Ub) and elimination by mitophagy; CORM, CO-releasing molecule; HO-1, heme oxygenase-1; NOS, NO synthase; OXPHOS: oxidative phosphorylation; PKA protein kinase A; ROS, reactive oxygen species.
NO-dependent vasodilation improves oxygen and carbon substrate availability for cell respiration and metabolism, but despite this vasodilation, excessive NO production during certain other pathologic conditions directly inhibits respiration through blockade of mitochondrial Complexes I (NADH dehydrogenase) and IV (cytochrome c oxidase, COX), markedly reducing OXPHOS (Antunes et al., 2004; Brown and Borutaite, 2007). NO reacts with superoxide and generates peroxynitrite (ONOO−), which not only diminishes vasodilation but leads to macromolecular damage to mitochondria (Pacher et al., 2007; Poderoso, 2009). This damage may enhance mitochondrial ROS production and stimulate prosurvival responses by AMP kinase (AMPK) and/or Akt/PKB (Piantadosi and Suliman, 2012a).
NO is also involved in myogenic differentiation in which GC-dependent inhibition of the Drp1 mitochondrial fission protein plays an important part (De Palma et al., 2010). Since first reported that mitochondrial content is low in skeletal muscle in NOS3−/− mice (Nisoli et al., 2004b), muscle has been an active site of investigation (Nisoli and Carruba, 2006). Therapeutic NO supplementation has become an objective of treatment of heritable muscle wasting conditions such as Duchenne’s muscular dystrophy (DMD) and mitochondrial myopathies. NO levels can also be influenced by NO-donating drugs or by stimulating its enzymatic synthesis with the amino acid precursors. Treatment of rat primary skeletal muscle cells with the NO donor S-nitroso-N-acetylpenicillamine causes a significant increase in mitochondrial mass (Tengan et al., 2007), an effect also observed in other cells and tissues. For instance, in primary cultures of brown adipocytes, treatment with S-nitroso-N-acetylpenicillamine increases mitochondrial size and mtDNA content (Nisoli et al., 2003). This stimulatory effect is abolished by supplementation of the media with oxyhemoglobin, a potent NO scavenger, implying dependence on NO generation.
Activation of mitochondrial biogenesis by NO donors leads to rapid proliferation of functional mitochondria (Nisoli et al., 2004b; Lira et al., 2010). Several drugs with NO-releasing properties currently used in clinical practice, mainly for cardiovascular diseases, may specifically impact on NOS3 (Dudzinski et al., 2006). Other NO-based therapeutic approaches to improve skeletal muscle performance involve diet, exercise, and pharmacological agents designed to improve mitochondrial content and OXPHOS efficiency (Levine et al., 2012). Aerobic training, a potent stimulus for muscle mitochondrial biogenesis, improves muscle strength and oxidative capacity in patients with mitochondrial myopathy (Murphy et al., 2008) in part through NO-dependent mechanisms. An active area of research is to determine the safety, effectiveness, and the most appropriate conditioning regimens for these patients.
The treatment of mitochondrial disorders exhibiting NO deficiency by using arginine or citrulline supplementation (El-Hattab et al., 2012a) was first tried in MELAS (Koga et al., 2005; El-Hattab et al., 2012b), because data from these patients had indicated low levels of serum arginine (Koga et al., 2005) and citrulline (Naini et al., 2005) accompanied by endothelial dysfunction (Koga et al., 2006). The rationale was to improve the low NO production rates observed in some mitochondrial diseases and not necessarily to directly increase mitochondrial biogenesis. Some encouraging results were reported using arginine and citrulline to treat relative NO deficiency in MELAS patients (Koga et al., 2005; El-Hattab et al., 2012a; Tengan et al., 2012), although mitochondrial proliferation in an effort to compensate for the disease has been proposed as an underlying cause of impaired NO synthesis by these patients in the first place (Tengan et al., 2012).
On the basis of these findings and the NO deficiency observed in muscle fibers of patients with mitochondrial myopathies, it seems reasonable that NO precursor supplementation could have broader applicability in mitochondrial diseases that exhibit muscle weakness (El-Hattab et al., 2012a). The precursor strategy has been tested in the Mdx mouse model of DMD and delayed disease progression was observed (De Luca, 2012; De Palma and Clementi, 2012). Clinically, the success of NO therapy depends on whether the patient can increase the proliferation rate of mitochondria with functional OXPHOS systems, a limitation in hereditary mtDNA disorders with low heteroplasmy.
Other drugs such as HCT 1025, a nonsteroidal anti-inflammatory derivative of flurbiprofen that releases NO, seems to slow disease progression in mice with limb girdle and Duchenne muscular dystrophies (Brunelli et al., 2007). HCT 1026 combined with arterially delivered donor stem cells offered improved efficacy. This also demonstrated better functional gain than l-arginine combined with deflazacort in mdx dystrophic mice (Archer et al., 2006). Another study reported that the NO-releasing drug molsidomine has therapeutic effects in dystrophic mice (Buono et al., 2012). The drug increased muscle fiber regeneration and enhanced muscle function by increasing satellite cell proliferation. These studies raise new prospects for the treatment of both primary and secondary mitochondrial dysfunction, because several NO-based drugs already tested for safety in humans are available for study in clinical trials.
NO is also cardioprotective during ischemia-reperfusion (I/R) injury. By using a mitochondrial-selective S-nitrosating agent, reversible mitochondrial S-nitrosation (S-nitrosylation) of Complex I subunits constitutes a major mechanism of NO protection during I/R and is a potential therapeutic target for the prevention of injury after myocardial and possibly cerebral infarction (Chouchani et al., 2013). In the heart and brain, progression of I/R injury is associated with mitochondrial dysfunction manifested by ATP depletion, calcium-induced opening of the mitochondrial permeability transition pore, and exacerbation of mitochondrial ROS release (Cadenas et al., 2010). Several studies have investigated acute nitrite treatment, a modest NO donor under hypoxic/acidic conditions, and one has shown that nitrite therapy initiated 24 hours after I/R improves tissue and vascular regeneration, and functional recovery (Kapil et al., 2014).
B. The Heme Oxygenase-1/Carbon Monoxide System
Endogenous carbon monoxide (CO) produced by the heme oxygenases (Hmox, HO-1 and -2) acts as a messenger gas that exerts control over mitochondrial QC through its impact on the regulation of mitochondrial biogenesis and presumably mitophagy. Prior to this understanding, there was controversy over the incontrovertible toxicity of the gas versus observations supporting low dose CO as a cell protectant operating through general anti-inflammatory and antiapoptotic properties (Ryter et al., 2006; Suliman and Piantadosi, 2014). Indeed, many protective effects of HO-1 induction by ischemia, hypoxia, oxidative stress, metals, and inflammation are not simply due to heme clearance and in many cases can be recapitulated by CO administration (Maines, 2005; Vieira et al., 2008; Biermann et al., 2010; Lee et al., 2014).
The targets of CO binding in tissue are largely restricted to reduced transition metals, such as enzymatic copper and iron centers in hemoglobin, myoglobin, and cytochrome c oxidase, thereby influencing enzymatic function and redox state (Piantadosi, 2008; Motterlini and Otterbein, 2010). Its affinity for heme proteins is responsible for its potent asphyxia effect. Like NO, CO activates guanylate cyclase but binds with a far lower affinity, whereas its effects on mitochondria are mediated through direct binding to reduced cytochrome c oxidase or indirectly to nearby heme proteins and enzymes of cytochrome P450 systems. An important effect is an increase in mitochondrial ROS production that leads to further HO-1 induction (Piantadosi, 2008). Under such circumstances, CO administration or the use of CO-releasing molecules (CORMs) generates a positive feedback loop that contributes to upregulation of the HO-1/CO pathway and its induction of mitochondrial biogenesis (Fig. 3).
Abundant preclinical evidence has demonstrated beneficial effects of CO administered as authentic gas or as CORMs in experimental models of cardiovascular disease, stroke, sepsis, organ transplant rejection, and various acute injuries in the lungs, heart, kidneys, and liver (Ryter et al., 2006; Bauer and Pannen, 2009; Motterlini and Otterbein, 2010). Early studies found CO modulates local inflammatory responses by promoting anti-inflammatory cytokine production and opposing apoptosis, and a number of these effects appear to be linked to the transcriptional network of mitochondrial biogenesis (Piantadosi et al., 2011). Moreover, the induction of mitochondrial biogenesis leads directly to counter-inflammatory cytokine induction such as interleukin-10 (IL-10), IL-1 receptor antagonist (Piantadosi et al., 2011), and suppressor of cytokine synthesis 3 (Athale et al., 2012).
In resting humans breathing air, CO has a half-life of 4–5 hours and is eliminated unaltered almost exclusively through the lungs (Coburn, 2012) with limited oxidation to CO2 by mitochondria. Moreover, the critical safe upper limit of carboxyhemoglobin [HbCO] in normal individuals is not known, but it is certainly lower in patients with cardiovascular and cerebrovascular disease (Hampson et al., 2012).
CO gas is relatively nonreactive chemically, straightforward to produce, and inexpensive. Like NO, which is in clinical use, it is readily administered by inhalation (Motterlini and Otterbein, 2010). By giving inhaled CO as discrete pulses, tissue CO levels will rise transiently and follow PO2-dependent elimination kinetics. The ease of administration has led to clinical trials focused on the anti-inflammatory effects of CO and set an upper limit of exposure at 8% HbCO, the equivalent of smoking about two packs of cigarettes per day. The first randomized, placebo-controlled Phase I trial to evaluate the clinical safety and pharmacokinetics of inhaled CO in healthy humans has been completed (Motterlini and Otterbein, 2010). Low concentrations of the gas were found to be safe in dose-escalation studies in healthy humans and deemed acceptable for Phase II testing in emphysema and fibrotic lung disease. Inhaled CO is also undergoing evaluation for safety and potential efficacy in patients with advanced pulmonary arterial hypertension. In humans, it has also been shown that low-dose inhaled CO improves muscle mitochondrial density but also regulates myoglobin content, localization of the insulin-regulated glucose transporter (GLUT4), and muscle capillarity (Pecorella et al., 2015).
The application of CO releasing molecules (CORMs) is a useful alternative to inhaled CO because they can be administered via systemic routes and in a tissue-specific manner, thus avoiding generalized pulmonary delivery systems and allowing judicious targeting of organs such as the brain. CORMs also show many of the protective features of authentic CO gas, e.g., suppressing the inflammatory response in glial cells (Bani-Hani et al., 2006), and may therefore offer neuroprotection. The apparent inconsistency of neuroprotection in the literature has caused confusion but may in part reflect the choice of CORM and route of administration.
CORMs all possess a backbone carrier moiety embodied most commonly as organometallic carbonyl complexes that must be stringently characterized from a metabolic and toxicological standpoint. Some agents such as the boranocarbonates avoid metal toxicity but generally release CO rapidly. Several CORMs have been synthesized for therapeutic agents for delivering controlled amounts of CO to tissues and organs (Motterlini et al., 2002). CORM-1 is soluble in water and decomposes rapidly, but under physiologic conditions CO is released with slower kinetics (half-life of 21 min) (Motterlini et al., 2003). CORM-2 is soluble in DMSO and olive oil and releases CO by photo-dissociation (Motterlini et al., 2002). CORM-3 is a water-soluble ruthenium-based agent with a half-life of 1 min (Clark et al., 2003). CORM-3 is effective when given before neuronal injury but not if given shortly afterward (Yabluchanskiy et al., 2012). A molybdenum-based, water-soluble CORM (ALF186) that releases CO in a dose- and oxygen-dependent manner is protective in vivo in models of acute inflammation (Sheikh et al., 2011) and in the regulation of vasomotor tone (Szabó, 2007; Marazioti et al., 2011). ALF186 inhibits I/R-induced neuronal cell death via soluble GC activation and may be useful for acute ischemic insults to the retina and the brain. More recent work has shown utility of water-soluble synthetic carbonyl complexes of rhenium, which release CO and ReO4; the latter is excreted by the kidneys (Zobi et al., 2013). All of these agents relax blood vessels and generally lower blood pressure via GC and potassium channel activation, and some are postulated to improve vascular function. In addition, CO released from CORMs inhibits NF-κΒ-mediated inflammatory gene expression and upregulates adaptive gene expression for oxidative stress. Thus, these compounds are being proposed as novel agents for the therapy of an array of vascular, inflammatory, and oxidative stress-related disorders.
C. Hydrogen Sulfide
A substantial body of evidence implicates H2S as a signaling molecule involved in multiple physiologic and cellular processes that protect organ function against damage (Szabó, 2007; Wagner et al., 2009), including I/R injury (Zhang et al., 2006; Tamizhselvi et al., 2008). It is made endogenously by the conversion of l-cysteine through two pyridoxal-50 phosphate-dependent enzymes, cystathionine b-synthase and cystathionine g-lyase. It has anti-inflammatory properties, such as inhibiting NF-κB-dependent NOS2 expression, but may also inhibit HO-1 expression; nonetheless, its precise role as an endogenous suppressor of inflammation is still under investigation. Marked proinflammatory effects reported for H2S (Collin et al., 2005; Li et al., 2005) stand in contrast with the anti-inflammatory effects observed in different studies (Elrod et al., 2007; Wagner et al., 2011).
H2S is a well-known inhibitor of OXPHOS via cytochrome c oxidase (COX) blockade, a toxic property of many low molecular weight sulfides (Cooper and Brown, 2008). On the other hand, low-dose H2S has been reported to preserve mitochondrial function in the heart muscle of rodents after I/R injury (Elrod et al., 2007; Calvert et al., 2010). Like CO, low level H2S administration or exposure of cells to H2S-releasing compounds increases the phosphorylation of protein serine/threonine kinase B (Akt) and enhances the nuclear localization of NRF-1 and -2 to increase mitochondrial biogenesis, attenuate apoptosis, and increase endogenous antioxidant levels (Calvert et al., 2010). The impact of the gas on mitophagy and overall mitochondrial QC in injury and disease states however is not yet clear.
V. Induction of Mitochondrial Biogenesis by Hormones, Drugs, and Natural Products
A. Estrogens, Erythropoietin, and Thyroid Hormone
Certain hormones modulate metabolism and mitochondrial function through the actions of the nuclear receptor (NR) superfamily of proteins. Upon ligand binding, these receptors enter the nucleus and bind hormone-responsive elements in the promoter regions of target genes, including TFAM, TFB1M, and TFB2M (Psarra and Sekeris, 2011). In various tissues, steroid (type I) and nonsteroid (type II) NRs influence mitochondrial biogenesis and OXPHOS components (Weber et al., 2002). Some NRs, such as the glucocorticoid receptor (GR), estrogen receptor, and thyroid receptor (TR) are also found in mitochondria where certain direct transcriptional effects can occur.
1. Estrogens.
The loss of the main circulating estrogen 17β-estradiol (E2) due to either natural or surgical menopause leads to a prompt reduction in body metabolic rate. Other manifestations may include muscle weakness, fatigue, reduced exercise capacity, and weight gain. Some of these signs and symptoms are clearly associated with changes in aerobic energy metabolism.
Molecular studies have shown a regulatory role for E2 in mitochondrial function (Fig. 4) involving ATP production, generation of membrane potential, mitochondrial biogenesis, and calcium regulation (Wang et al., 2001; Duckles et al., 2006). The mechanisms of these effects, especially in humans, are not well understood. Some evidence suggests that ERs localize to mitochondria and elicit their effects directly. ERα is essential for most of the E2-mediated increase in electron transport chain (ETC) and antioxidant proteins (O'Lone et al., 2007; Chen et al., 2009). On the other hand, ERβ can downregulate mRNA expression of nuclear-encoded subunits of the ETC complexes in vasculature (O'Lone et al., 2007). E2 may also influence mitochondrial function by altering mitochondrial ROS production (Borras et al., 2010) by induction of antioxidant responses (Sugishita et al., 2003). E2 also activates NRF1/2, Tfam, and PGC1α (Mattingly et al., 2008). Further research on E2 is warranted to understand these mitochondrial effects in greater detail.
Fig. 4.

Mechanisms of estrogen action on mitochondria. Binding and dimerization of estrogen receptors (ERs) by estrogen E2 trigger nuclear and mitochondrial genomic effects. These are mediated by nuclear translocation of E2-ER complex and either 1) direct binding with estrogen response elements (ERE) along with co-activators to form a transcription complex or 2) binding to transcriptional coactivators to induce gene transcription indirectly. ERs may also localize to mitochondria to induce potentially genomic and nongenomic actions, the mechanisms of which are not clearly understood.
E2 also stimulates ERα relocation to mitochondria where it interacts with hydroxysteroid (17-β) dehydrogenase 10 (HSD10). HSD10 is involved in steroid metabolism and functions as a core subunit of the mitochondrial RNaseP complex responsible for cleavage of polycistronic mitochondrial transcripts. This interaction results in mitochondrial transcript processing and mature RNA that is available for translation. Conversely, HSD10 inactivates E2 to a weaker form, estrone, the significance of which requires further investigation (Sanchez et al., 2015).
Impaired E2 signaling and subsequent mitochondrial dysfunction may also be involved in insulin resistance. Mitochondrial dysfunction is associated with reduced or partial FAO that can activate stress kinases that inhibit insulin signaling (Koves et al., 2008; Zhang et al., 2010). The expression of the adipokine, adiponectin, and its receptor, AR1, is induced by estrogen in conjunction with mitochondrial biogenesis (Capllonch-Amer et al., 2014), and adiponectin may improve insulin sensitivity.
Estrogen replacement therapy to protect postmenopausal women from diabetes and other metabolic disorders and osteoporosis is offset by the increased cancer risk by E2 and ER agonists. Bazedoxifene with conjugated E2 is a combination of selective tissue E2 complexes and receptor modulator that provides tissue-specific benefits of E2 in menopausal osteoporosis in women (Archer, 2010) and improves cardiovascular disease risk and metabolic syndrome with greater effects in endometrium and breast in animal models (Barrera et al., 2014; Kim et al., 2014). Although selective tissue E2 complexes are still in trials, newer, more innovative, efficient, and tissue-specific E2 receptor agonists are being investigated. An E2 conjugated preparation with glucagon-like peptide-1 is more active than either hormone alone in the reversal of obesity, hyperglycemia, and dyslipidemia in mice and prevents reproductive endocrine toxicity and oncogenicity (Finan et al., 2012). It is not yet clear which patients benefit and which agents would minimize the risks of E2-related therapy.
2. Erythropoietin.
EPO regulates erythrocyte mass in response to hypoxia and anemia, but EPO activity is detectable in some non-erythroid cells that express functional EPO receptor (EpoR). Protective effects of EPO have been demonstrated in various nonerythroid tissues and experimental models of I/R injury and attributed to nonhematopoietic metabolic effects, inhibition of apoptosis, or to stimulation of angiogenesis (Chatterjee, 2005). EPO stimulates cardiac mitochondrial proliferation through mitochondrial biogenesis mediated by NO and NRF-1 and PGC-1α (Carraway et al., 2010). Clinically, EPO reverses cardiac remodeling, improves cardiac function, and enhances exercise tolerance and quality of life of a subset of heart patients by protective effects beyond the correction of anemia (Bergmann et al., 2011). These findings highlight the possibility that EPO-mediated protection may depend on modulatory effects on bioenergetics. EPO can stimulate proliferation of cultured myoblasts through binding to EpoR to expand the progenitor population during differentiation, and it may have a role in muscle repair (Jia et al., 2009; Ogilvie et al., 2000). EPO/EpoR signaling also improves glucose tolerance and protects against dietary obesity in association with skeletal muscle adaptation (Hojman et al., 2009; Teng et al., 2011).
The association of EPO treatment in mice with increased mitochondrial biogenesis and PGC-1α activation in myocytes raises the possibility that EPO may contribute to skeletal muscle plasticity and the balance between slow and fast-twitch fibers. Furthermore, recombinant protein increases OXPHOS in human skeletal muscle (Plenge et al., 2012). The enhancement and recovery of cellular functions through stimulation of hemoglobin production and mitochondrial activity in nonhematopoietic cells by EPO-like drugs might be a testable therapeutic strategy for certain ischemic or mitochondrial diseases in carefully selected patient populations, although the procoagulant effects of EPO administration have raised a legitimate caution.
3. Thyroid Hormone.
The ability of thyroid hormone (T3 and T4) and thyroid hormone receptors (TRs) to regulate oxygen uptake and energy utilization is related to the thyroid system’s multiple effects on mitochondrial function. TH targets several regulatory pathways for gene expression that may contribute to mitochondrial biogenesis (Weitzel and Iwen, 2011). TH binds to nuclear-localized TR belonging to a ligand-dependent transcription factor family that modulates nuclear gene expression via binding to a thyroid response or TRE motif. TH also directly affects mitochondria by binding a mitochondrial-localized TR. However, intermediate factors are also expressed (perhaps via TREs) that enter the nucleus and regulate other TH-target genes. PGC-1α and PGC-1β and NRF-1, NRF-2 have been postulated to serve as these intermediate factors. There is evidence of rapid PGC-1α induction by TH both at the mRNA and protein levels [mediated by a TRE in the gene promoter (Wulf et al., 2008)], but its impact on mitochondrial proliferation seems modest and the therapeutic opportunities limited.
B. Pharmacological Agents
The pharmacological activation of mitochondrial biogenesis is potentially achievable by a broad range of compounds including select natural products and pharmaceutical agents (Fig. 5) (Andreux et al., 2013). Some of these agents may operate through physiologic pathways, whereas others may cause mild mitochondrial damage that then leads to the induction of physiologic QC mechanisms. These distinctions are made below when possible.
Fig. 5.
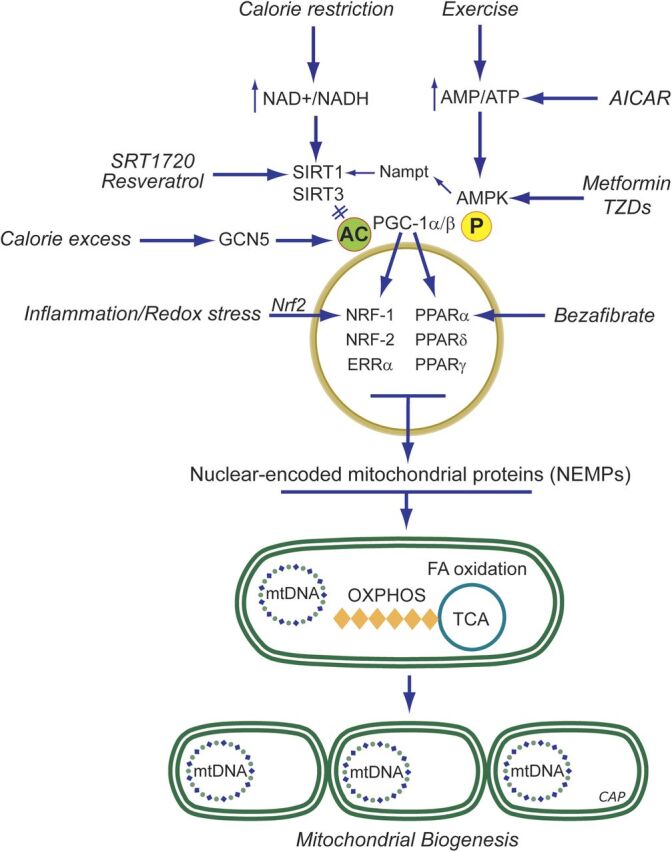
Pharmacological induction of mitochondrial biogenesis. Schematic representation of pathways for exercise, calorie restriction, stress, or small molecules. During caloric excess, PGC-1α, a key coactivator of mitochondrial biogenesis is in an inactive acetylated (ac) state. In times of caloric restriction, or after exercise, energy becomes scarce, resulting in increases in the AMP/ATP and NAD+/NADH ratios. The former has a direct impact on the activation of AMPK as binding of AMP (or ADP) facilitates phosphorylation (p) of AMPK by upstream kinases. Activated AMPK phosphorylates PGC-1α directly, which may either activate this coactivator or prime PGC-1α for activation via deacetylation by SIRT1/SIRT3. SIRT1 requires NAD+ as the nucleotide is a cosubstrate. The deacetylated PGC-1α is able to coactivate nuclear mitochondrial gene transcription via transcription factors including NRF1/2, estrogen-related receptor (ERR) α,β,γ (ERRs), and PPARα,β,γ. This activates mtDNA transcription, translation, and replication (represented by mtDNA); production of OXPHOS subunits, tricarboxylic acid (TCA) cycle enzymes, enzymes of fatty acid oxidation; and leads to increased mitochondrial mass. Resveratrol is a SIRT1 activator by a mechanism that is not well-unresolved but ultimately leads to PGC-1α pathway activation via coactivator deacetylation. AICAR is phosphorylated to an AMP analog, ZMP, that activates AMPK by promoting phosphorylation by upstream kinases. AMPK causes PGC-1α phosphorylation and SIRT1 activation via increases in the NAD+/NADH ratio. Bezafibrate is a pan-PPAR agonist and induces PGC-1α expression via the PPAR-responsive element in its promoter. See section on Pharmacological Agents for details.
1. Peroxisome Proliferator-Activated Receptor Activation by Bezafibrate.
The PPAR (α, β/δ, γ) agonist bezafibrate is commonly used to treat dyslipidemias (Tenenbaum et al., 2005). Although fibrates have long been linked to fatty acid oxidation (FAO) and to peroxisome proliferation, later studies reveal that PPARs via PGC-1α also regulate other aspects of mitochondrial function (Hondares et al., 2011). Bezafibrate stimulates heart and skeletal muscle PGC-1α expression in mice in a PPARδ-dependent manner (Hondares et al., 2007). Moreover, bezafibrate treatment or PGC-1α overexpression upregulates respiratory capacity and OXPHOS function in fibroblasts and myoblasts of OXPHOS-deficient patients (Srivastava et al., 2009). Dietary bezafibrate at a concentration of 0.5% (w/w) induced mitochondrial biogenesis in skeletal muscle of Cox10 knockout mice. Bezafibrate stimulates Ppar and Pgc-1 gene expression and the induction of mtDNA and mitochondrial proteins in muscle. Importantly, bezafibrate treatment in Cox10 knockout mice delays the onset of myopathy and prolongs survival. In the Surf1−/− model of mild COX deficiency, bezafibrate does not replicate the effect of PGC-1α overexpression (Viscomi et al., 2011). Instead, the drug induces both PPARα and δ and causes FAO upregulation in skeletal muscle. It also caused weight loss and severe hepatomegaly, indicating drug toxicity that could be due to PPARα activation (Hays et al., 2005).
Bezafibrate showed benefit in Deletor mice, a model of adult-onset mitochondrial myopathy from accumulation of mtDNA deletions as a result of transgenic expression of mutated Twinkle helicase (Wallace et al., 2010; Yatsuga and Suomalainen, 2012). The drug caused significant induction of hepatic liver FAO, hepatomegaly, and decreases in adipose tissue in this model (Yatsuga and Suomalainen, 2012). In another study, the bezafibrate effect in Deletor mice was limited to delayed accumulation of COX-negative muscle fibers and a reduced mtDNA deletion-load with no change in phenotype and no induction of mitochondrial biogenesis. The treatment strategy differed in that the drug was started at the onset of symptoms at 1 year of age instead of at 5 weeks (Viscomi et al., 2011).
In a mouse model of AD (P301S transgenic mice) bezafibrate decreased disease progression by diminishing locomotor and stress-related abnormalities (Dumont et al., 2012; Noe et al., 2013). A modest increase in mitochondrial proliferation was noted, but mild increases in mtDNA content and Sirt1 and Tfam gene expression was more apparent in brown fat with increased expression of NRF1 and PGC-1α. Small differences between mouse and human PPARα orthologs also seem to influence hepatocyte proliferation by PPARα activators such as fenofibrate, which does not induce hepatomegaly in PPARα-humanized mice as in wild-type mice (Yang et al., 2008). Bezafibrate is relatively safe in humans, although rhabdomyolysis and kidney failure have been reported (Wu et al., 2009). A promising finding is that long-term bezafibrate treatment of patients with the mitochondrial FAO disorder CPT2 deficiency leads to significant clinical improvement (Bonnefont et al., 2009, 2010).
2. β2-Adrenergic Receptor Agonists.
β2-AR receptor agonists are airway smooth muscle relaxants and bronchodilators approved for the treatment of asthma and other airways diseases. These agonists are reported to modulate oxidative metabolism, energy expenditure, lipolysis, glucose transport, and glucose oxidation (Pearen et al., 2008). Mice treated with formoterol, a β2-AR agonist, showed a fivefold induction of PGC-1α mRNA in skeletal muscle. The β2-AR has been successfully targeted for drug discovery using a ligand-based approach, resulting in the creation of multiple receptor-specific drugs (Wishart et al., 2006; Kolb et al., 2009). However, the therapeutic potential of established and novel β2-AR agonists for the induction of mitochondrial biogenesis and as counter-inflammatory agents has not been well explored.
3. Metformin.
This insulin-sensitizing biguanide is a front-line oral hypoglycemic treatment of DM2 (Halimi, 2006). Metformin lowers blood glucose mainly via hepatic glucose output and insulin-stimulated glucose uptake in skeletal muscle and adipocytes (Larsen et al., 2011). The molecular mechanisms involved are not completely clear. Although inhibition of mitochondrial Complex I has been reported in cells, isolated mitochondria, and muscle homogenate (Brunmair et al., 2004); metformin-induced activation of AMPK has been shown in rodent muscle (Zhou et al., 2001) and in human skeletal muscle from patients with DM2 (Musi et al., 2002). Numerous studies have documented a role for AMPK in regulating mitochondrial function and biogenesis, and it too leads to anti-inflammatory effects (O'Neill et al., 2011). On the other hand, effects of metformin on mitochondrial function by decreasing ROS production have been postulated (Hou et al., 2010). Long-term metformin treatment may increase PGC-1α expression in muscle together with increased activity and protein expression of certain mitochondrial marker enzymes (Jager et al., 2007).
4. 5-Aminoimidazole-4-Carboxamide Ribonucleotide.
5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) has been found to increased mitochondrial biogenesis and cell ATP content while decreasing ROS production. The effect is mediated via AMPK and direct regulation of energy expenditure by modulating SIRT1 deacetylase activity, resulting in deacetylation of downstream targets including PGC-1α and Forkhead box O1 and 3 transcription factors (Canto et al., 2009). The drug does have significant toxicity, and the use of AICAR as a cardiac therapeutic is limited by bradycardia and hypoglycemia (Young et al., 2005). AICAR requires phosphorylation inside the cell, and the phosphorylated derivative known as ZMP acts as an AMP mimetic (Merrill et al., 1997). Unlike other presumed AMPK agonists, AICAR promotes AMPK phosphorylation and activation while not disturbing the energy status of the cell, i.e., the AMP/ATP ratio remains the same.
AICAR is orally effective to increase endurance performance in sedentary mice by increasing expression of oxidative metabolism genes (Narkar et al., 2008). This is thought to happen via direct phosphorylation/activation of PGC-1α by activated AMPK and subsequent induction of mitochondrial biogenesis (Jager et al., 2007). Unfortunately, AICAR penetrates the blood-brain barrier poorly, which limits its use in the central nervous system (Marangos et al., 1990). Nevertheless peripheral stimulation of AMPK and mitochondrial biogenesis may have some beneficial effects on brain function as demonstrated in healthy mice (Yu et al., 2015).
In inherited OXPHOS disorders, partial activation of AMPK may naturally occur in certain tissues. Hence, additional stimulation of this pathway with AICAR may or may not be therapeutically useful. Accordingly, in a drug screen using skin fibroblasts from a cohort of Complex I-deficient patients, AICAR has shown promise in ameliorating mitochondrial defects (Golubitzky et al., 2011).
In mice, AICAR’s value for OXPHOS disorders has been shown by the treatment of muscle-specific COX-deficient mouse models of mitochondrial myopathy (Viscomi et al., 2011). Injected for 1 month from 8 weeks of age (0.25 mg⋅day−1⋅g−1 s.c.), AICAR resulted in AMPK phosphorylation and induction of FAO and OXPHOS gene expression in muscle in Surf1−/−, Sco2KO/KI, and ACTA-Cox15−/− mice (Yang et al., 2010; Viscomi et al., 2011). AICAR also increased other OXPHOS enzyme activities, indicating an increase in mitochondrial biogenesis and an ability to improve muscle pathology in the Mdx model of DMD (Ljubicic et al., 2011; Jahnke et al., 2012). This coincided with mitochondrial biogenesis as shown by increases in COX activity in fast-twitch muscles and a switch to more oxidative fiber types. In addition, AICAR induced utrophin and PGC-1α expression plus ETC components cytochrome c and COX IV. However, in the diaphragm, AICAR treatment did not produce changes in fiber type or induction of mitochondrial biogenesis (Pauly et al., 2012). Contrasting data suggested that AICAR caused increased mitophagy in the diaphragm of Mdx mice (Pauly et al., 2012). AICAR had a positive effect on diaphragm structure and contractile capacity, suggesting enhancement of mitophagy may be an alternative treatment of some muscle diseases such as DMD and mitochondrial myopathies (Pauly et al., 2012).
C. Polyphenols and Other Natural Products
Polyphenols are ubiquitously present in fruits and vegetables and have traditionally been viewed as antioxidant molecules. This idea developed mainly from their widely established ability to scavenge carbon and lipid radicals and ROS. The prototype plant-derived polyphenol, resveratrol (3,5,4′-trihydroxystilbene), is enriched in red wine and functions as a caloric restriction mimetic via upregulation of sirtuin-1 (SIRT1) and AMPK (Baur et al., 2006; Lagouge et al., 2006). It also enhances mitochondrial biogenesis through upregulation of PGC-1α, NRF-1 and -2, and Tfam (Biala et al., 2010). Polyphenols act on many mitochondrial processes and some appear to be modulators of mitochondrial biogenesis, mitochondrial membrane potential, mitochondrial electron transport and ATP synthesis, and mitochondrial ROS formation, as well as intrinsic cell death (Fig. 5).
1. Resveratrol.
This polyphenol has received attention for a range of potentially beneficial effects including improvements in mitochondrial function, protection against obesity and obesity-related diseases such as type-2 diabetes, suppression of inflammation and cancer cell growth, and protection against heart disease. Resveratrol alleviates negative effects of a high-fat diet in mice such as weight gain and improves glucose tolerance because of activation of AMPK (Um et al., 2010). Many studies have pointed to the ability of resveratrol to upregulate PGC-1α, SIRT1, and mitochondrial biogenesis (Puigserver et al., 1998; Rodgers et al., 2005). Resveratrol treatment has been shown to increase AMPK activation, increase deacetylation of PGC-1α, and promote its activation (Canto et al., 2009; Um et al., 2010). Currently, this increase in PGC-1α activity is still under investigation.
Resveratrol is modified by glucuronidation and sulfation (Meng et al., 2004), which reduce its bioavailability. It is thus unlikely to reach serum concentrations above 1 μM from dietary consumption or 10 μM from direct oral supplements (Baur and Sinclair, 2006). Two human studies have reported no effect of resveratrol on major metabolic parameters. A double-blind placebo-controlled trial of 12 weeks of resveratrol supplementation (75 mg/day) to nonobese postmenopausal women found no effect of resveratrol on glucose homeostasis, insulin resistance, blood pressure, or inflammation markers (Yoshino et al., 2012). Another trial involving skeletal muscle and fat biopsies from human subjects found no increases in Sirt1, AMPK, NAMPT, or PGC-1α expression. Similarly, euglycemic hyperinsulinemic clamp showed no significant effects of resveratrol treatment (Yoshino et al., 2012). This trial also found that resveratrol did not affect levels of phosphorylated AMPK or the expression levels of PGC-1α, GLUT4, tumor necrosis fact α, or NF-κB (Poulsen et al., 2013).
Resveratrol has anti-inflammatory effects, and two groups have shown the prevention of cardiac dysfunction in hypertensive rats without reductions in blood pressure (Thandapilly et al., 2010; Rimbaud et al., 2011), supporting a direct influence on the heart. Resveratrol seemed to preserve mitochondrial mass, biogenesis, and function in salt-sensitive hypertensive rats (Rimbaud et al., 2011) and in rats transgenic for human renin and angiotensin (Biala et al., 2010). A small human clinical trial reported a minor improvement in diastolic function in 40 patients with acute coronary syndrome receiving 10 mg resveratrol daily during a 3-month trial. In addition, improvement in endothelial function and lower low-density lipoprotein levels were noted in the treatment group, but mitochondrial biogenesis and function were not assessed (Magyar et al., 2012). The cardiac benefits may thus be limited to special situations, such as refractory hypertensive or postinfarction patients, but as mentioned, any therapeutic effects are limited by short half-life and low bioavailability (Baur and Sinclair, 2006). Overall, these clinical studies tell an ambiguous story, and to better differentiate cardioprotective from pleiotropic effects, small molecule analogs of resveratrol are being pursued that directly and potently activate Sirt1 (Wood et al., 2004).
2. Quercetin.
This well-known polyphenol does induce mitochondrial biogenesis. Studies by Davis et al. (2009) reported on the ability of this flavonol (7-day treatment of mice with 12.5 or 25 mg/kg) to induce the expression and activation of SIRT1 and PGC-1α and to increase mtDNA and cytochrome c content in skeletal muscle and brain. As a possible functional effect, the authors showed a concomitant increase in maximal physical endurance capacity of treated animals (Davis et al., 2009).
The influence of quercetin on both mitochondrial biogenesis and exercise endurance has been studied in untrained human subjects (Nieman et al., 2010) where the flavonoid was administered orally (1000 mg/day for 2 weeks). Quercetin slightly increased the number of muscle mtDNA and mRNA copies (P = not significant) and induced a small (yet significant) increase in physical performance in untrained adult men. This agrees with MacRae and Mefferd (2006) where the administration of an antioxidant-rich beverage containing 300 mg of quercetin to trained cyclists significantly increased their physical performance after 6 weeks (no effect was seen in controls given a quercetin-free antioxidant-rich beverage). However, no mitochondrial biogenesis markers were reported.
3. Hydroxytyrosol.
Another polyphenol linked to mitochondrial biogenesis is hydroxytyrosol, a phenolic alcohol present in olives (and in extra virgin olive oils). In vitro, hydroxytyrosol (1–10 μM) activates PGC-1α through SIRT1 deacetylation and induces mitochondrial biogenesis in 3T3-L1 murine adipocytes and in ARPE-19 human retinal pigment epithelial cells (Zhu et al., 2010). The administration of hydroxytyrosol to rats (25 mg/kg daily during 8 weeks) by the same investigators stimulated PGC-1α activity and the expression of mitochondrial biogenesis markers in skeletal muscle of both treadmill-exercised and sedentary animals, and increased their endurance (Feng et al., 2011). The authors noted that in contrast to the mitochondrial biogenesis-inducing effect of moderate exercise, extreme exercise was associated with lower PGC-1α levels. This effect was prevented by hydroxytyrosol administration to the animals (Feng et al., 2011).
Other dietary polyphenols have been implicated in the induction of mitochondrial biogenesis. For instance, isoflavones (genistein, daidzein, and formononetin) in rabbit renal proximal tubular cells (Rasbach and Schnellmann, 2008), flavones (baicalein, its 7-O-glucuronide baicalin, and wogonin) in L6 skeletal muscle cells (Im et al., 2012), and flavan-3-ol epigallocatechin-3-gallate in skin fibroblasts from Down’s syndrome patients (Valenti et al., 2013) all activate the SIRT1/PGC-1α pathway. In contrast to the SIRT1-inducing effect of genistein (Rasbach and Schnellmann, 2008), this isoflavone also reduces SIRT1 mRNA and protein expression and induces SIRT1 nuclear exclusion into the cytosol of the human prostate cancer cell lines LNCaP and PC-3 (Kikuno et al., 2008). However, PGC-1α levels or acetylation were not measured in this study. The SIRT1-mediated actions of genistein could relate to differences in basal levels or activity of SIRT-1 or to mechanisms that modulate its expression in normal compared with cancer cells.
Greater consistency of SIRT-1-inducing effects of polyphenols is seen by comparing in vivo studies. In humans (Taub et al., 2012), the administration of an epicatechin-rich cocoa (total epicatechin was 100 mg/day during 3 months) to patients with type 2 diabetes and heart failure stimulates mitochondrial biogenesis in skeletal muscle biopsies, as expressed by an increment in SIRT1-dependent activation of PGC-1α and as an elevation in mitofilin, porin, and mitochondrial complexes I and V protein levels. The long-term effect of curcumin, a dietary polyphenol derived from turmeric has been investigated on various markers of mitochondrial biogenesis. For instance, curcumin (5-month dietary supplementation at 0.05%) upregulates PGC-1α protein expression in the brain of senescence-accelerated mouse-prone 8, a fast-aging mice strain, improving MMP and ATP levels and restoring mitochondrial fusion (Eckert et al., 2013). Dietary curcumin (3-month dietary supplementation at 0.2%) increases the expression of TFAM and PGC-1α in association with a greater expression of mitochondrial respiratory complexes, especially Complex IV, and an increase in ATP levels in the brains of apolipoprotein E3-targeted gene replacement mice. Comparatively, although ineffective on mitochondrial biogenesis, curcumin did increase ATP levels in high Alzheimer-risk apolipoprotein E4 mice (Chin et al., 2014). Hence, polyphenols belonging to many subclasses (stilbenes, flavonols, phenolic alcohols, isoflavones, flavones, and flava-3-ols), are all potential dietary regulators of mitochondrial QC, but most of the regulatory pathways are not clear.
4. Triterpenoids.
TPs are Nrf2 transcription factor-activating compounds. Normally Nrf2 is activated by cellular oxidative stress or by electrophiles, but for therapeutic purposes readily activated by synthetic TP analogs of olenolic acid. These inhibit oxidative stress and inflammation and are protective in some cancer models (Liby et al., 2007). For decades, many plant-derived chemicals, or phytochemicals, have been known to have chemopreventive activities, and many are Nrf2 inducers (Lau et al., 2008). The most potent also induce HO-1 and include sulforaphanes from cruciferous vegetables (Kensler et al., 2000), curcumin (Balogun et al., 2003), caffeic acid phenethyl ester (Balogun et al., 2003), and wasabi (Morimitsu et al., 2002). A novel Nrf2-activating, synthetic TP, 2-Cyano-3,12-Dioxooleana-1,9-Dien-28-Oic acid (CDDO) methylamide, was found to augment the expression of genes involved in mitochondria biogenesis, glutathione synthesis, and the antioxidant defenses (Yang et al., 2009). These authors also found neuroprotective effects of CDDO methylamide in 3-nitropropionic acid rat and MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) mouse models (Yang et al., 2009). Other intriguing neuroprotective agents that induce Hmox1 expression by this mechanism include the electrophilic neurite outgrowth-promoting prostaglandin compounds (Satoh et al., 2000).
The cell’s redox state is protected by the induction of the antioxidant response, which responds physiologically to a variety of stimuli and entails the upregulation of genes containing the antioxidant response element (ARE) (Hayes et al., 2010; Magesh et al., 2012). The best known inducer of the antioxidant response is a sulforaphane, an isothiocyanate discovered in Brassica oleracea var. italica (Zhang et al., 1992). It binds irreversibly to the Nrf2 docking protein, Keap1, that blocks its activity (Kensler et al., 2013). Inactivation of Keap1 permits Nrf2 to enter the nucleus and bind to promoter AREs (Cheng et al., 2011; Dinkova-Kostova and Abramov, 2015), responsible for antioxidant gene expression as well as the expression of the p62 mitophagy protein (Dinkova-Kostova and Abramov, 2015). Another protein, the p62-mediated mitophagy inducer is reported to induce the antioxidant response specifically by inhibiting Keap1 (East et al., 2014). This protein induces mitophagy via p62 upregulation with a beneficial effect on mitochondrial homeostasis and no major toxicity. Moreover, the antioxidant response also induces neuroprotection in the mitochondrial-damage driven MPTP mouse model (Jazwa et al., 2011; Lavich et al., 2015). The pharmacological exploitation of mitochondrial QC through the ARE is an attractive prospect, but a core pharmacological approach is moving slowly because long-term activation of some of these transcriptional elements is tumorigenic.
A notable mitophagy inducer is the ATP analog kinetin triphosphate, which increases mitophagy by means of PINK1 (Hertz et al., 2013), and increases survival of dopaminergic neurons in culture. Another mitochondrial targeting approach is drugs that inhibit the mitochondrial PTP such as cyclosporine (Tedesco and Haragsim, 2012). This and related calcineurin inhibitors are potent immunosuppressive agents but have side effect profiles that include hepatic, renal, and heart failure and the potential to impact mitochondrial QC, which is unexplored.
5. Thiazolidinedione Drugs.
Thiazolidinediones activate the PPAR (peroxisome proliferator-activated receptors) group of nuclear receptors with high affinity for PPARγ, also a cognate target of PGC-1α. The endogenous ligands (mainly free fatty acids and eicosanoids) allow the receptor to bind to DNA in complex with the retinoid X receptor to increase the transcription of some genes while decreasing the transcription of others. The main effect of this differential gene expression is an increase in free fatty acid storage in adipocytes, thereby decreasing the circulating fatty acid pool. The cells then become more dependent on carbohydrate oxidation, i.e., glucose, for free energy. Thiazolidinediones are approved only for the treatment of DM2, and although their modulation of PGC-1α is well known, thiazolidinedione's effects on mitochondrial biogenesis are controversial. In the heart, for instance, mitochondrial biogenesis has been implicated in thiazolidinedione-mediated postinfarction cardiac remodeling, and the drug reduces infarct size in several species (Liu et al., 2004; Yue et al., 2005), although these findings have also been disputed (Lygate et al., 2003; Xu et al., 2005). Drug toxicity has been an issue in clinical studies.
D. Other Compounds that Protect Mitochondria through Mitochondrial Biogenesis
1. Mitochondrial Nutrients.
Mitochondrial nutrients protect the organelle from oxidative damage and maintain or improve aerobic metabolism. Some are being employed to correct relative or absolute deficiencies of critical metabolites or to increase the levels of critical substrates and enzymes resulting in enhanced cellular metabolic efficiency. Such nutrients can also perform other functions including limiting mitochondrial ROS production and oxidative stress and reducing acute inflammation. Key micronutrients include iron-chelating agents; enzymatic and nonenzymatic free-radical scavengers, such as α-tocopherol (Liu and Ames, 2005); antioxidant enhancers, such as coenzyme Q, lipoic acid, glutathione, and α-tocopherol; and enhancers of metabolism and mechanisms that repair or degrade mitochondria. Some may increase mitochondrial biogenesis, such as lipoic acid and acetyl-l-carnitine, creatine, pyruvate, and choline (Shen et al., 2008a,b), or protect mitochondrial enzymes and/or stimulate matrix enzyme activity by elevating substrate and cofactor levels, such as B vitamins (Liu and Ames, 2005). It is important to reiterate that potent ROS scavengers may not only decrease mitochondrial damage but interfere with redox signals involved in mitochondrial QC.
2. Mitochondrial-Targeted Reactive Oxygen Species Scavengers.
The persistence of damaged mitochondria as a source of ROS production in aerobic cells and has led to many attempts to target them with small molecule antioxidant drugs. The best known endogenous molecule with specific mitochondrial oxidant/antioxidant properties is probably the neurohormone melatonin (Luchetti et al., 2010), but the prototypical synthetic mitochondria-targeted antioxidant is MitoQ, a hydroquinone ROS-scavenging moiety linked to triphenylphosphonium (TPP), a lipophilic compound that crosses membranes and accumulates in the matrix as a function of membrane potential. MitoQ thus mimics the role of endogenous coenzyme Q10 (CoQ10), and also augments the antioxidant capacity of CoQ in a mitochondrial membrane potential-dependent manner.
ROS scavenging is achieved by oxidation of MitoQ to its quinone, which is then recycled to the active hydroquinone by mitochondrial Complex II (Murphy and Smith, 2007). MitoQ is orally bioavailable with no detectable toxicity when administered to mice at ∼20 mg/kg. Tracer studies find the compound to be taken up rapidly by the heart, liver, brain, kidney, and muscle, with highest accumulation in heart and liver (Smith et al., 2003). Long-term administration of MitoQ had no effect on plasma glucose, insulin, free fatty acid, or cholesterol levels but is associated with reduced triglyceride levels. After receiving MitoQ, heart and liver tissues of mice showed no significant treatment-related differences in gene expression profiles by microarray analysis (Rodriguez-Cuenca et al., 2010). MitoQ thus appears to be a relatively safe, orally available small molecule that does not significantly alter basal physiology.
In humans, MitoQ has been studied in Parkinson's disease (PROTECT) and in patients with chronic hepatitis C infection. The PROTECT trial was negative, but it provided good data on oral drug safety for as long as one year (Snow et al., 2010). Major side effects of the MitoQ regimen were not seen in either trial. In the chronic hepatitis C trial, patients receiving 40 and 80 mg MitoQ showed significant improvement in liver function (Gane et al., 2010). Despite the therapeutic potential of MitoQ and other TPP-conjugated antioxidants, they have significant limitations. The uptake of these compounds is governed by the membrane potential, which may be close to collapse in the cells of failing organs, such as the heart. Moreover, the accumulation of cationic TPP in the matrix may depolarize mitochondria, leading to adverse effects. Thus, this strategy requires further investigation.
Mitochondrial targeting with peptide mimetics or lipophilic cationic agents (MitoQ, SkQ1) or manganese superoxide (SOD2) mimetics (Pucheu et al., 1996; van Empel et al., 2006; Kawakami et al., 2009; Iranzo, 2011) may also offer therapy for mitochondrial antioxidant protection. However, strategies to direct these compounds to the organ or cell type of interest at the apposite time are lacking, and their impact on the redox-regulation of mitochondrial biogenesis is unknown.
The deployment of mitochondrial protective agents is ensnared by the bewildering array of molecular targets from which to choose and the difficulty of delivering a candidate to the desired mitochondrial compartment and target (Szeto and Schiller, 2011). There are few methods for noninvasively assessing mitochondrial function, although newer spectroscopic approaches are being developed for monitoring electron transport and OXPHOS in experimental animals and humans (Marcinek and Conley, 2014). Attempts to improve the delivery of CoQ10 into the mitochondrial matrix have been made by conjugating it to the triphenylphosphonium ion (TPP+) (Smith et al., 2012). MitoTEMPO has also been used experimentally to minimize sepsis-induced acute kidney injury by increasing mitochondrial superoxide scavenging and preserving capillary blood flow (Sims et al., 2014).
Targeted peptides are a promising therapeutic approach for treating other causes of mitochondrial dysfunction. Rather thoroughly investigated is SS-31 (Szeto and Birk, 2014), which concentrates in the inner mitochondrial membrane by binding selectively to cardiolipin, causing a conformational change that optimizes mitochondrial electron transport and ATP synthesis while inhibiting cardiolipin peroxidation (Birk et al., 2014). The latter property enables SS-31 to inhibit MPT pore opening in myocardial ischemia and reduce infarct size (Kloner et al., 2012). The therapeutic potential of SS-31 also has been demonstrated in aging animals and in diseases associated with mitochondrial dysfunction (Szeto, 2014).
VI. Targeting Mitochondrial Dynamics and Mitophagy
As mitophagy is often either activated or blocked by pathologic processes, pharmacological approaches to these pathways are receiving considerable interest (Rubinsztein et al., 2012) and highlight the translational potential of this area of research. During an energy crisis, for instance, induction of mitochondrial biogenesis by AMPK may also directly activate autophagy/mitophagy, e.g., via phosphorylation of the ULK1 mitophagy regulator (Egan et al., 2011), which targets the degradation/recycling of dysfunctional and damaged mitochondria.
Certain compounds have been identified as strong modulators of autophagy/mitophagy including sirolimus (rapamycin), an mTORC1 inhibitor that blocks autophagy. The relationship between ROS production and elimination must be delicately balanced to appropriately stimulate mitophagy (Frank et al., 2012). Overwhelming cellular ROS production is not only directly injurious but also disrupts redox signal localization, while ROS over-scavenging may interrupt redox communication. This apparent paradox may explain the unexpectedly weak effects of antioxidant therapies (Benfeito et al., 2013), especially for chronic degenerative diseases (Singh et al., 2011). Still, highly localizable mitochondrial antioxidant compounds hold promise for mitochondrial protection without interference with redox signaling. For instance, targeted antioxidants may limit oxidative damage by protecting lipid-rich mitochondrial membranes while allowing redox signals to stimulate the replacement of badly damaged mitochondria.
There also appear to be redox-controlled genetic mechanisms involved in mitochondrial biogenesis that are involved in the expression of mitophagy proteins (Chang et al., 2015). Using argument by analogy, certain antioxidants may influence mitophagy in accordance with the redox signaling pathways outlined above. This could also be true of mitochondrial dynamics, and although one study has reported that CoQ10 supplementation in DM2 decreased renal mitochondrial fragmentation and mitochondrial size (Persson et al., 2012), it is not clear if the compound prevented alterations in morphology and function by strictly antioxidant effects or also by modulation of mitophagy.
The expanding recognition that mitochondrial dynamics influences function has led to other therapeutic perspectives(Andreux et al., 2013). Mitochondrial mass and morphology are defined by an exquisite balance among fission/fusion, biogenesis, and mitophagy (Fig. 6). Thus, modifications in one or more each of these processes may be required for the treatment of different mitochondrial disorders (Andreux et al., 2013). There are genetic disorders of mitochondrial dynamics including defects of mitochondrial fusion caused by mutations in MFN2 or OPA1 in Charcot-Marie-Tooth type 2A and autosomal dominant optic atrophy, respectively (Zuchner et al., 2004), and mitochondrial fission caused by mutations in DRP1 and MFF (Shamseldin et al., 2012). The discovery of specific inhibitors of mitochondrial fusion (M-hydrazone) and fission (MDIVI-1 and P110) hint at potential therapeutic applications for these disorders (Qi et al., 2013), but rigorous preclinical studies are needed to investigate efficacy and possible adverse effects. Such novel agents may also help regulate mitophagy in mitochondrial disorders not primarily caused by fission/fusion defects (Andreux et al., 2013).
Fig. 6.

Regulation of mitochondrial quality control (QC) via mitochondrial biogenesis and mitophagy. Mitophagy, in conjunction with mitochondrial biogenesis, regulates changes in mitochondrial number required to meet metabolic demand. Activated AMPK triggers ULK1-dependent mitophagy and simultaneously triggers the biogenesis of new mitochondria via effects on PGC-1α-dependent transcription. Conversely, mTOR represses ULK1-dependent mitophagy when nutrients are plentiful. In addition, AMPK can be activated by ROS-mediated opening of calcium release-activated calcium (CRAC) channels, leading to cytosolic calcium flux that activate the AMPK upstream kinase CaMKKβ. These processes controlled by AMPK and mTOR determine the net effect of replacing defective mitochondria with new functional mitochondria. AMPK: AMP-activated protein kinase; mTOR: mammalian target of rapamycin; PGC-1α: PPARγ co-activator 1-alpha; ULK1: the mammalian Atg1 homologs, uncoordinated family member (unc)-51: like kinase 1; ERK2: the extracellular signal-regulated protein kinase 2.
One devastating autosomal dominant disease that displays increased mitochondrial fragmentation, Huntington's disease (HD), is a relentless and fatal neurodegenerative condition characterized by progressive chorea and cognitive impairments. The mutant Huntingtin (mHtt) interacts with Dynamin-related protein 1 (Drp1), causing excessive mitochondrial fission, leading to abnormal mitochondrial dynamics and neuronal damage in HD-affected neurons. In neurons that express mHtt, an imbalance between fission and fusion leads to aberrant mitochondrial structure and function and ultimately neuronal damage. By using mice that express full-length human Htt gene with 97 CAA and CAG mixed repeats (BACHD mice), significant increases in mRNA levels of Drp1, Fis1, and Cyclophilin D and decreased levels of Mfn1 and Mfn2 in 2-month-old BACHD compared with age-matched wild-type mice support abnormal mitochondrial dynamics as an early event in HD progression(Shirendeb et al., 2012). It has also been determined that S-nitrosylation of Drp1 contributes to HD pathogenesis (Haun et al., 2013), because mHtt protein triggers a significant increase in nitric oxide (NO) production in primary neurons. Consistent with this result, increased levels of S-nitrosylation in Drp1 were seen in the striatum of an HD transgenic mouse as well as in the neurons from patients with HD.
Progress is being made in developing therapeutic molecules that can prevent excessive mitochondrial fission and still maintain fission-fusion balance, mitochondrial function, and neuronal activity in diseases where mitochondrial oxidative stress and bioenergetic dysfunction is involved. Several groups have screened chemical libraries and uncovered three novel molecules: Mdivi-1 (Cassidy-Stone et al., 2008), Dynasore (Macia et al., 2006), and P110 (Qi et al., 2013). Among these, Mdivi-1 has been extensively investigated in experimental rodent models of epilepsy (Qiu et al., 2013; Xie et al., 2013), ischemia (Park et al., 2011; Zhang et al., 2013), oxygen-glucose deprivation (Wappler et al., 2013), rhabdomyolysis (Tang et al., 2013), and conditions involving endosome aggregation and vesicle fusion during exocytosis (Chlystun et al., 2013). In all of these states, Mdivi-1 exhibited beneficial effects on cells and tissues by restricting mitochondrial fission and maintaining normal fission-fusion balance and cell function. However, this is not the only role of Mdivi-1, which also independently suppresses the activity of acetylcholine-activated potassium channels in murine cardiomyocytes in a concentration-dependent manner (So et al., 2012),
The Dynasore molecule interferes with the GTPase activities of Dynamin 1, Dynamin 2, and Drp1 but does not interfere with fusion. The protective role of Dynasore in ischemia and/or I/R injury in mice has been studied by Gao et al. (2013) who observed that mice pretreated with Dynasore exhibited less ischemia and/or reperfusion-induced cardiac impairment, suggesting that Dynasore protects cardiomyocytes from excessive mitochondrial fission and oxidative stress.
VII. Conclusions
The enhancement of mitochondrial QC through the pharmacological induction of mitochondrial biogenesis, mitophagy, and/or the dynamic control of fission/fusion balance is emerging as a strategy for the prevention or treatment of primary and secondary mitochondrial disorders. Efforts to develop these strategies have highlighted the complexity of mitochondrial targeting, the limitations of the tools available today, and the many uncertainties of mitochondrial QC regulation. Nonetheless, the biologic elimination of unsalvageable mitochondria prevents excessive intracellular oxidative stress and the pathologic accumulation of oxidized macromolecules that may propagate damage and interfere with redox signaling mechanisms. It should also be kept in mind that induction of mitochondrial biogenesis in some cases may actually lead to increased numbers of dysfunctional mitochondria. For example, patients with heritable OXPHOS disorders have residual activity because the complete loss of OXPHOS is incompatible with life; hence, an increased mass of even low efficiency mitochondria may increase ATP synthesis rates above the threshold of functional sustainability. Moreover, no agent can rescue every cell or tissue, but a measurable impact on a subset of cells and conditions depending on the type of mitochondrial defect and the pathologic phenotype could be beneficial to tissue repair.
Apart from OXPHOS defects, mitochondrial dysfunction commonly increases endogenous ROS generation, and although ROS are necessary in cellular communication, excessive oxidation of constitutive macromolecules compromises cell viability. OXPHOS disorders also do not always show damaging mitochondrial ROS production, and mitochondrial-targeted antioxidants may thus be of little value and may interfere with redox communication in some situations. Many times, however, mitochondrial ROS production is substantially increased and mitochondrial antioxidants may be more useful than the induction of biogenesis, which could increase the number of ROS generating sites in the cell. On the other hand, the re-establishment of a healthy population of working mitochondria through mitochondrial biogenesis may actually ameliorate ROS production by both close regulation of mitochondrial ROS release and by the concurrent antioxidant response, for example the SIRT3-dependent PGC-1α-mediated antioxidant response (Bell and Guarente, 2011). Finally, the natural molecular correspondence between mitochondrial biogenesis and mitophagy, for instance mediated by AMPK during an impending energy crisis, may provide opportunities for targeted protection against persistent mitochondrial dysfunction through overall QC mechanisms. Scientific efforts have prepared this area for innovative molecular pharmacological approaches and clinical protocols that address promising mitochondrial QC targets, but further elucidation of novel pathways that protect the cell through mitochondrial QC may offer unique opportunities for disease therapy where ongoing mitochondrial damage is less obvious as an important factor.
Abbreviations
- AD
Alzheimer’s disease
- AICAR
5-aminoimidazole-4-carboxamide ribonucleotide
- AMPK
AMP kinase
- ARE
antioxidant response element
- CoQ10
coenzyme Q10
- CORM
CO-releasing molecule
- COX
cytochrome c oxidase
- CREB1
cAMP response element-binding protein
- DM2
type 2 diabetes
- DMD
Duchenne’s muscular dystrophy
- Drp1
Dynamin-related protein 1
- E2
17β-estradiol
- EPO
erythropoietin
- EpoR
erythropoietin receptor
- ER
endoplasmic reticulum
- ERRα
estrogen response receptor alpha
- ETT
electron transport chain
- FAO
fatty acid oxidation
- FoxO
Forkhead box O
- GC
guanylate cyclase
- H
heavy
- HD
Huntington's disease
- HO
heme oxygenase
- HSD10
hydroxysteroid (17-β) dehydrogenase 10
- IL
interleukin
- IR
ischemia-reperfusion
- L
light
- MDV
mitochondrial-derived vesicle
- MELAS
mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes
- mHtt
mutant Huntingtin
- mtDNA
mitochondrial DNA
- mTOR
mammalian target of rapamycin
- NASH
nonalcoholic steatohepatitis
- NEMP
nuclear-encoded mitochondrial protein
- NF-κB
nuclear factor κB
- NLR
Nuclear oligomerization domain (NOD)-like receptor
- NO
nitric oxide
- NOS
nitric oxide synthase
- NR
nuclear receptor
- NRF
nuclear respiratory factor
- OXPHOS
oxidative phosphorylation
- PD
Parkinson’s disease
- PGC
peroxisome proliferator-activated receptor γ coactivator
- PPAR
peroxisome proliferator-activated receptors
- QC
quality control
- ROS
reactive oxygen species
- SIRT1
sirtuin-1
- SOD
superoxide dismutase
- TLR
Toll-like receptor
- TPP
triphenylphosphonium
- TR
thyroid receptor
Authorship Contributions: Drafted and edited the manuscript: Suliman and Piantadosi.
Footnotes
References
- Ahuja P, Zhao P, Angelis E, Ruan H, Korge P, Olson A, Wang Y, Jin ES, Jeffrey FM, Portman M, et al. (2010) Myc controls transcriptional regulation of cardiac metabolism and mitochondrial biogenesis in response to pathological stress in mice. J Clin Invest 120:1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam J, Igarashi K, Immenschuh S, Shibahara S, Tyrrell RM. (2004) Regulation of heme oxygenase-1 gene transcription: recent advances and highlights from the International Conference (Uppsala, 2003) on Heme Oxygenase. Antioxid Redox Signal 6:924–933. [DOI] [PubMed] [Google Scholar]
- Andreux PA, Houtkooper RH, Auwerx J. (2013) Pharmacological approaches to restore mitochondrial function. Nat Rev Drug Discov 12:465–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes F, Boveris A, Cadenas E. (2004) On the mechanism and biology of cytochrome oxidase inhibition by nitric oxide. Proc Natl Acad Sci USA 101:16774–16779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer DF. (2010) Tissue-selective estrogen complexes: a promising option for the comprehensive management of menopausal symptoms. Drugs Aging 27:533–544. [DOI] [PubMed] [Google Scholar]
- Archer JD, Vargas CC, Anderson JE. (2006) Persistent and improved functional gain in mdx dystrophic mice after treatment with L-arginine and deflazacort. FASEB J 20:738–740. [DOI] [PubMed] [Google Scholar]
- Athale J, Ulrich A, MacGarvey NC, Bartz RR, Welty-Wolf KE, Suliman HB, Piantadosi CA. (2012) Nrf2 promotes alveolar mitochondrial biogenesis and resolution of lung injury in Staphylococcus aureus pneumonia in mice. Free Radic Biol Med 53:1584–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai A, Ma AG, Yong M, Weiss CR, Ma Y, Guan Q, Bernstein CN, Peng Z. (2010) AMPK agonist downregulates innate and adaptive immune responses in TNBS-induced murine acute and relapsing colitis. Biochem Pharmacol 80:1708–1717. [DOI] [PubMed] [Google Scholar]
- Balaban RS. (2012) Perspectives on: SGP symposium on mitochondrial physiology and medicine: metabolic homeostasis of the heart. J Gen Physiol 139:407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balogun E, Hoque M, Gong P, Killeen E, Green CJ, Foresti R, Alam J, Motterlini R. (2003) Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem J 371:887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bani-Hani MG, Greenstein D, Mann BE, Green CJ, Motterlini R. (2006) Modulation of thrombin-induced neuroinflammation in BV-2 microglia by carbon monoxide-releasing molecule 3. J Pharmacol Exp Ther 318:1315–1322. [DOI] [PubMed] [Google Scholar]
- Barrera J, Chambliss KL, Ahmed M, Tanigaki K, Thompson B, McDonald JG, Mineo C, Shaul PW. (2014) Bazedoxifene and conjugated estrogen prevent diet-induced obesity, hepatic steatosis, and type 2 diabetes in mice without impacting the reproductive tract. Am J Physiol Endocrinol Metab 307:E345–E354. [DOI] [PubMed] [Google Scholar]
- Barroso E, Eyre E, Palomer X, Vázquez-Carrera M. (2011) The peroxisome proliferator-activated receptor β/δ (PPARβ/δ) agonist GW501516 prevents TNF-α-induced NF-κB activation in human HaCaT cells by reducing p65 acetylation through AMPK and SIRT1. Biochem Pharmacol 81:534–543. [DOI] [PubMed] [Google Scholar]
- Bartz RR, Suliman HB, Fu P, Welty-Wolf K, Carraway MS, MacGarvey NC, Withers CM, Sweeney TE, Piantadosi CA. (2011) Staphylococcus aureus sepsis and mitochondrial accrual of the 8-oxoguanine DNA glycosylase DNA repair enzyme in mice. Am J Respir Crit Care Med 183:226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer I, Pannen BH. (2009) Bench-to-bedside review: Carbon monoxide--from mitochondrial poisoning to therapeutic use. Crit Care 13:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, et al. (2006) Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444:337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur JA, Sinclair DA. (2006) Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov 5:493–506. [DOI] [PubMed] [Google Scholar]
- Becker T, Böttinger L, Pfanner N. (2012) Mitochondrial protein import: from transport pathways to an integrated network. Trends Biochem Sci 37:85–91. [DOI] [PubMed] [Google Scholar]
- Begriche K, Igoudjil A, Pessayre D, Fromenty B. (2006) Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion 6:1–28. [DOI] [PubMed] [Google Scholar]
- Bell EL, Guarente L. (2011) The SirT3 divining rod points to oxidative stress. Mol Cell 42:561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfeito S, Oliveira C, Soares P, Fernandes C, Silva T, Teixeira J, Borges F. (2013) Antioxidant therapy: still in search of the ‘magic bullet’. Mitochondrion 13:427–435. [DOI] [PubMed] [Google Scholar]
- Bergeron R, Ren JM, Cadman KS, Moore IK, Perret P, Pypaert M, Young LH, Semenkovich CF, Shulman GI. (2001) Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab 281:E1340–E1346. [DOI] [PubMed] [Google Scholar]
- Bergmann MW, Haufe S, von Knobelsdorff-Brenkenhoff F, Mehling H, Wassmuth R, Münch I, Busjahn A, Schulz-Menger J, Jordan J, Luft FC, et al. (2011) A pilot study of chronic, low-dose epoetin-beta following percutaneous coronary intervention suggests safety, feasibility, and efficacy in patients with symptomatic ischaemic heart failure. Eur J Heart Fail 13:560–568. [DOI] [PubMed] [Google Scholar]
- Biala A, Tauriainen E, Siltanen A, Shi J, Merasto S, Louhelainen M, Martonen E, Finckenberg P, Muller DN, Mervaala E. (2010) Resveratrol induces mitochondrial biogenesis and ameliorates Ang II-induced cardiac remodeling in transgenic rats harboring human renin and angiotensinogen genes. Blood Press 19:196–205. [DOI] [PubMed] [Google Scholar]
- Biermann J, Lagrèze WA, Dimitriu C, Stoykow C, Goebel U. (2010) Preconditioning with inhalative carbon monoxide protects rat retinal ganglion cells from ischemia/reperfusion injury. Invest Ophthalmol Vis Sci 51:3784–3791. [DOI] [PubMed] [Google Scholar]
- Birk AV, Chao WM, Bracken C, Warren JD, Szeto HH. (2014) Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol 171:2017–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonawitz ND, Clayton DA, Shadel GS. (2006) Initiation and beyond: multiple functions of the human mitochondrial transcription machinery. Mol Cell 24:813–825. [DOI] [PubMed] [Google Scholar]
- Bonnefont JP, Bastin J, Behin A, Djouadi F. (2009) Bezafibrate for an inborn mitochondrial beta-oxidation defect. N Engl J Med 360:838–840. [DOI] [PubMed] [Google Scholar]
- Bonnefont JP, Bastin J, Laforêt P, Aubey F, Mogenet A, Romano S, Ricquier D, Gobin-Limballe S, Vassault A, Behin A, et al. (2010) Long-term follow-up of bezafibrate treatment in patients with the myopathic form of carnitine palmitoyltransferase 2 deficiency. Clin Pharmacol Ther 88:101–108. [DOI] [PubMed] [Google Scholar]
- Bordon Y. (2014) Macrophages: innate memory training. Nat Rev Immunol 14:713–713. [DOI] [PubMed] [Google Scholar]
- Borrás C, Gambini J, López-Grueso R, Pallardó FV, Viña J. (2010) Direct antioxidant and protective effect of estradiol on isolated mitochondria. Biochim Biophys Acta 1802:205–211. [DOI] [PubMed] [Google Scholar]
- Brown GC, Borutaite V. (2007) Nitric oxide and mitochondrial respiration in the heart. Cardiovasc Res 75:283–290. [DOI] [PubMed] [Google Scholar]
- Brunelli S, Sciorati C, D’Antona G, Innocenzi A, Covarello D, Galvez BG, Perrotta C, Monopoli A, Sanvito F, Bottinelli R, et al. (2007) Nitric oxide release combined with nonsteroidal antiinflammatory activity prevents muscular dystrophy pathology and enhances stem cell therapy. Proc Natl Acad Sci USA 104:264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunmair B, Staniek K, Gras F, Scharf N, Althaym A, Clara R, Roden M, Gnaiger E, Nohl H, Waldhäusl W, et al. (2004) Thiazolidinediones, like metformin, inhibit respiratory complex I: a common mechanism contributing to their antidiabetic actions? Diabetes 53:1052–1059. [DOI] [PubMed] [Google Scholar]
- Buono R, Vantaggiato C, Pisa V, Azzoni E, Bassi MT, Brunelli S, Sciorati C, Clementi E. (2012) Nitric oxide sustains long-term skeletal muscle regeneration by regulating fate of satellite cells via signaling pathways requiring Vangl2 and cyclic GMP. Stem Cells 30:197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadenas S, Aragonés J, Landázuri MO. (2010) Mitochondrial reprogramming through cardiac oxygen sensors in ischaemic heart disease. Cardiovasc Res 88:219–228. [DOI] [PubMed] [Google Scholar]
- Calvert JW, Elston M, Nicholson CK, Gundewar S, Jha S, Elrod JW, Ramachandran A, Lefer DJ. (2010) Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation 122:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458:1056–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capllonch-Amer G, Sbert-Roig M, Galmés-Pascual BM, Proenza AM, Lladó I, Gianotti M, García-Palmer FJ. (2014) Estradiol stimulates mitochondrial biogenesis and adiponectin expression in skeletal muscle. J Endocrinol 221:391–403. [DOI] [PubMed] [Google Scholar]
- Carraway MS, Suliman HB, Jones WS, Chen CW, Babiker A, Piantadosi CA. (2010) Erythropoietin activates mitochondrial biogenesis and couples red cell mass to mitochondrial mass in the heart. Circ Res 106:1722–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carré JE, Orban JC, Re L, Felsmann K, Iffert W, Bauer M, Suliman HB, Piantadosi CA, Mayhew TM, Breen P, et al. (2010) Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am J Respir Crit Care Med 182:745–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, et al. (2008) Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 14:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. (2007) Mitochondrial dynamics in disease. N Engl J Med 356:1707–1709. [DOI] [PubMed] [Google Scholar]
- Chang AL, Ulrich A, Suliman HB, Piantadosi CA. (2015) Redox regulation of mitophagy in the lung during murine Staphylococcus aureus sepsis. Free Radic Biol Med 78:179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee PK. (2005) Pleiotropic renal actions of erythropoietin. Lancet 365:1890–1892. [DOI] [PubMed] [Google Scholar]
- Chen JQ, Cammarata PR, Baines CP, Yager JD. (2009) Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim Biophys Acta 1793:1540–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Siow RC, Mann GE. (2011) Impaired redox signaling and antioxidant gene expression in endothelial cells in diabetes: a role for mitochondria and the nuclear factor-E2-related factor 2-Kelch-like ECH-associated protein 1 defense pathway. Antioxid Redox Signal 14:469–487. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Zhou M, Tung CH, Ji M, Zhang F. (2010) Studies on two types of PTP1B inhibitors for the treatment of type 2 diabetes: Hologram QSAR for OBA and BBB analogues. Bioorg Med Chem Lett 20:3329–3337. [DOI] [PubMed] [Google Scholar]
- Chin D, Hagl S, Hoehn A, Huebbe P, Pallauf K, Grune T, Frank J, Eckert GP, Rimbach G. (2014) Adenosine triphosphate concentrations are higher in the brain of APOE3- compared to APOE4-targeted replacement mice and can be modulated by curcumin. Genes Nutr 9:397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlystun M, Campanella M, Law AL, Duchen MR, Fatimathas L, Levine TP, Gerke V, Moss SE. (2013) Regulation of mitochondrial morphogenesis by annexin A6. PLoS One 8:e53774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, James AM, Cochemé HM, Reinhold J, Lilley KS, et al. (2013) Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med 19:753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choumar A, Tarhuni A, Lettéron P, Reyl-Desmars F, Dauhoo N, Damasse J, Vadrot N, Nahon P, Moreau R, Pessayre D, et al. (2011) Lipopolysaccharide-induced mitochondrial DNA depletion. Antioxid Redox Signal 15:2837–2854. [DOI] [PubMed] [Google Scholar]
- Clark JE, Naughton P, Shurey S, Green CJ, Johnson TR, Mann BE, Foresti R, Motterlini R. (2003) Cardioprotective actions by a water-soluble carbon monoxide-releasing molecule. Circ Res 93:e2–e8. [DOI] [PubMed] [Google Scholar]
- Coburn RF. (2012) The measurement of endogenous carbon monoxide production. J Appl Physiol (1985) 112:1949–1955. [DOI] [PubMed] [Google Scholar]
- Collin M, Anuar FB, Murch O, Bhatia M, Moore PK, Thiemermann C. (2005) Inhibition of endogenous hydrogen sulfide formation reduces the organ injury caused by endotoxemia. Br J Pharmacol 146:498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper CE, Brown GC. (2008) The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J Bioenerg Biomembr 40:533–539. [DOI] [PubMed] [Google Scholar]
- Crouser ED, Julian MW, Huff JE, Joshi MS, Bauer JA, Gadd ME, Wewers MD, Pfeiffer DR. (2004) Abnormal permeability of inner and outer mitochondrial membranes contributes independently to mitochondrial dysfunction in the liver during acute endotoxemia. Crit Care Med 32:478–488. [DOI] [PubMed] [Google Scholar]
- Davis JM, Murphy EA, Carmichael MD, Davis B. (2009) Quercetin increases brain and muscle mitochondrial biogenesis and exercise tolerance. Am J Physiol Regul Integr Comp Physiol 296:R1071–R1077. [DOI] [PubMed] [Google Scholar]
- De Luca A.(2012) Pre-clinical drug tests in the mdx mouse as a model of dystrophinopathies: an overview. Acta Myol 31:40–47. [PMC free article] [PubMed] [Google Scholar]
- De Palma C, Clementi E. (2012) Nitric oxide in myogenesis and therapeutic muscle repair. Mol Neurobiol 46:682–692. [DOI] [PubMed] [Google Scholar]
- De Palma C, Falcone S, Pisoni S, Cipolat S, Panzeri C, Pambianco S, Pisconti A, Allevi R, Bassi MT, Cossu G, et al. (2010) Nitric oxide inhibition of Drp1-mediated mitochondrial fission is critical for myogenic differentiation. Cell Death Differ 17:1684–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Abramov AY. (2015) The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med; doi: 10.1016/j.freeradbiomed.2015.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. (2008) Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320:674–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckles SP, Krause DN, Stirone C, Procaccio V. (2006) Estrogen and mitochondria: a new paradigm for vascular protection? Mol Interv 6:26–35. [DOI] [PubMed] [Google Scholar]
- Dudzinski DM, Igarashi J, Greif D, Michel T. (2006) The regulation and pharmacology of endothelial nitric oxide synthase. Annu Rev Pharmacol Toxicol 46:235–276. [DOI] [PubMed] [Google Scholar]
- Dumont M, Stack C, Elipenahli C, Jainuddin S, Gerges M, Starkova N, Calingasan NY, Yang L, Tampellini D, Starkov AA, et al. (2012) Bezafibrate administration improves behavioral deficits and tau pathology in P301S mice. Hum Mol Genet 21:5091–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykens JA, Will Y. (2007) The significance of mitochondrial toxicity testing in drug development. Drug Discov Today 12:777–785. [DOI] [PubMed] [Google Scholar]
- East DA, Fagiani F, Crosby J, Georgakopoulos ND, Bertrand H, Schaap M, Fowkes A, Wells G, Campanella M. (2014) PMI: a ΔΨm independent pharmacological regulator of mitophagy. Chem Biol 21:1585–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert GP, Schiborr C, Hagl S, Abdel-Kader R, Müller WE, Rimbach G, Frank J. (2013) Curcumin prevents mitochondrial dysfunction in the brain of the senescence-accelerated mouse-prone 8. Neurochem Int 62:595–602. [DOI] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. (2011) Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331:456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hattab AW, Emrick LT, Craigen WJ, Scaglia F. (2012a) Citrulline and arginine utility in treating nitric oxide deficiency in mitochondrial disorders. Mol Genet Metab 107:247–252. [DOI] [PubMed] [Google Scholar]
- El-Hattab AW, Hsu JW, Emrick LT, Wong LJ, Craigen WJ, Jahoor F, Scaglia F. (2012b) Restoration of impaired nitric oxide production in MELAS syndrome with citrulline and arginine supplementation. Mol Genet Metab 105:607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, et al. (2007) Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci USA 104:15560–15565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbrini E, Sullivan S, Klein S. (2010) Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology 51:679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Bai L, Yan J, Li Y, Shen W, Wang Y, Wertz K, Weber P, Zhang Y, Chen Y, et al. (2011) Mitochondrial dynamic remodeling in strenuous exercise-induced muscle and mitochondrial dysfunction: regulatory effects of hydroxytyrosol. Free Radic Biol Med 50:1437–1446. [DOI] [PubMed] [Google Scholar]
- Finan B, Yang B, Ottaway N, Stemmer K, Müller TD, Yi CX, Habegger K, Schriever SC, García-Cáceres C, Kabra DG, et al. (2012) Targeted estrogen delivery reverses the metabolic syndrome. Nat Med 18:1847–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsterer J, Ohnsorge P. (2013) Influence of mitochondrion-toxic agents on the cardiovascular system. Regul Toxicol Pharmacol 67:434–445. [DOI] [PubMed] [Google Scholar]
- Frank M, Duvezin-Caubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A, Ruonala MO, Priault M, Salin B, Reichert AS. (2012) Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta 1823:2297–2310. [DOI] [PubMed] [Google Scholar]
- Funk JA, Odejinmi S, Schnellmann RG. (2010) SRT1720 induces mitochondrial biogenesis and rescues mitochondrial function after oxidant injury in renal proximal tubule cells. J Pharmacol Exp Ther 333:593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Michels J, Brenner C, Szabadkai G, Harel-Bellan A, Castedo M, Kroemer G. (2014) Systems biology of cisplatin resistance: past, present and future. Cell Death Dis 5:e1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM, Frampton CM, Taylor KM, Smith RA, Murphy MP. (2010) The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int 30:1019–1026. [DOI] [PubMed] [Google Scholar]
- Gao D, Zhang L, Dhillon R, Hong TT, Shaw RM, Zhu J. (2013) Dynasore protects mitochondria and improves cardiac lusitropy in Langendorff perfused mouse heart. PLoS One 8:e60967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y. (2010) The multiple actions of NO. Pflugers Arch 459:829–839. [DOI] [PubMed] [Google Scholar]
- Gellerich FN, Trumbeckaite S, Hertel K, Zierz S, Müller-Werdan U, Werdan K, Redl H, Schlag G. (1999) Impaired energy metabolism in hearts of septic baboons: diminished activities of Complex I and Complex II of the mitochondrial respiratory chain. Shock 11:336–341. [PubMed] [Google Scholar]
- Gervois P, Kleemann R, Pilon A, Percevault F, Koenig W, Staels B, Kooistra T. (2004) Global suppression of IL-6-induced acute phase response gene expression after chronic in vivo treatment with the peroxisome proliferator-activated receptor-alpha activator fenofibrate. J Biol Chem 279:16154–16160. [DOI] [PubMed] [Google Scholar]
- Giralt A, Hondares E, Villena JA, Ribas F, Díaz-Delfín J, Giralt M, Iglesias R, Villarroya F. (2011) Peroxisome proliferator-activated receptor-gamma coactivator-1alpha controls transcription of the Sirt3 gene, an essential component of the thermogenic brown adipocyte phenotype. J Biol Chem 286:16958–16966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golomb BA, Evans MA. (2008) Statin adverse effects: a review of the literature and evidence for a mitochondrial mechanism. Am J Cardiovasc Drugs 8:373–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubitzky A, Dan P, Weissman S, Link G, Wikstrom JD, Saada A. (2011) Screening for active small molecules in mitochondrial complex I deficient patient’s fibroblasts, reveals AICAR as the most beneficial compound. PLoS One 6:e26883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grattagliano I, de Bari O, Bernardo TC, Oliveira PJ, Wang DQ, Portincasa P. (2012) Role of mitochondria in nonalcoholic fatty liver disease--from origin to propagation. Clin Biochem 45:610–618. [DOI] [PubMed] [Google Scholar]
- Grisolía S, Knecht E, Hernández-Yago J, Wallace R. (1979) Turnover and degradation of mitochondria and their proteins. Ciba Found Symp 75:167–188. [DOI] [PubMed] [Google Scholar]
- Gutsaeva DR, Carraway MS, Suliman HB, Demchenko IT, Shitara H, Yonekawa H, Piantadosi CA. (2008) Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J Neurosci 28:2015–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag MD, Hofman A, Koudstaal PJ, Stricker BH, Breteler MM. (2009) Statins are associated with a reduced risk of Alzheimer disease regardless of lipophilicity. The Rotterdam Study. J Neurol Neurosurg Psychiatry 80:13–17. [DOI] [PubMed] [Google Scholar]
- Haden DW, Suliman HB, Carraway MS, Welty-Wolf KE, Ali AS, Shitara H, Yonekawa H, Piantadosi CA. (2007) Mitochondrial biogenesis restores oxidative metabolism during Staphylococcus aureus sepsis. Am J Respir Crit Care Med 176:768–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. (2010) Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141:656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halimi S. (2006) Metformin: 50 years old, fit as a fiddle, and indispensable for its pivotal role in type 2 diabetes management. Diabetes Metab 32:555–556. [DOI] [PubMed] [Google Scholar]
- Halliwell B. (1989) Free radicals, reactive oxygen species and human disease: a critical evaluation with special reference to atherosclerosis. Br J Exp Pathol 70:737–757. [PMC free article] [PubMed] [Google Scholar]
- Hampson NB, Piantadosi CA, Thom SR, Weaver LK. (2012) Practice recommendations in the diagnosis, management, and prevention of carbon monoxide poisoning. Am J Respir Crit Care Med 186:1095–1101. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Cook WS, Qi C, Yeldandi AV, Reddy JK, Rao MS. (2000) Defect in peroxisome proliferator-activated receptor alpha-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J Biol Chem 275:28918–28928. [DOI] [PubMed] [Google Scholar]
- Haun F, Nakamura T, Shiu AD, Cho DH, Tsunemi T, Holland EA, La Spada AR, Lipton SA. (2013) S-nitrosylation of dynamin-related protein 1 mediates mutant huntingtin-induced mitochondrial fragmentation and neuronal injury in Huntington’s disease. Antioxid Redox Signal 19:1173–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT. (2010) Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid Redox Signal 13:1713–1748. [DOI] [PubMed] [Google Scholar]
- Hays T, Rusyn I, Burns AM, Kennett MJ, Ward JM, Gonzalez FJ, Peters JM. (2005) Role of peroxisome proliferator-activated receptor-alpha (PPARalpha) in bezafibrate-induced hepatocarcinogenesis and cholestasis. Carcinogenesis 26:219–227. [DOI] [PubMed] [Google Scholar]
- Hernandez G, Thornton C, Stotland A, Lui D, Sin J, Ramil J, Magee N, Andres A, Quarato G, Carreira RS, et al. (2013) MitoTimer: a novel tool for monitoring mitochondrial turnover. Autophagy 9:1852–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz NT, Berthet A, Sos ML, Thorn KS, Burlingame AL, Nakamura K, Shokat KM. (2013) A neo-substrate that amplifies catalytic activity of parkinson’s-disease-related kinase PINK1. Cell 154:737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, et al. (2001) CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413:179–183. [DOI] [PubMed] [Google Scholar]
- Hojman P, Brolin C, Gissel H, Brandt C, Zerahn B, Pedersen BK, Gehl J. (2009) Erythropoietin over-expression protects against diet-induced obesity in mice through increased fat oxidation in muscles. PLoS One 4:e5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloszy JO. (2008) Regulation by exercise of skeletal muscle content of mitochondria and GLUT4. J Physiol Pharmacol 59 (Suppl 7):5–18. [PubMed] [Google Scholar]
- Hondares E, Pineda-Torra I, Iglesias R, Staels B, Villarroya F, Giralt M. (2007) PPARdelta, but not PPARalpha, activates PGC-1alpha gene transcription in muscle. Biochem Biophys Res Commun 354:1021–1027. [DOI] [PubMed] [Google Scholar]
- Hondares E, Rosell M, Díaz-Delfín J, Olmos Y, Monsalve M, Iglesias R, Villarroya F, Giralt M. (2011) Peroxisome proliferator-activated receptor α (PPARα) induces PPARγ coactivator 1α (PGC-1α) gene expression and contributes to thermogenic activation of brown fat: involvement of PRDM16. J Biol Chem 286:43112–43122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X, Song J, Li XN, Zhang L, Wang X, Chen L, Shen YH. (2010) Metformin reduces intracellular reactive oxygen species levels by upregulating expression of the antioxidant thioredoxin via the AMPK-FOXO3 pathway. Biochem Biophys Res Commun 396:199–205. [DOI] [PubMed] [Google Scholar]
- Hribal ML, Oriente F, Accili D. (2002) Mouse models of insulin resistance. Am J Physiol Endocrinol Metab 282:E977–E981. [DOI] [PubMed] [Google Scholar]
- Huang X, Alonso A, Guo X, Umbach DM, Lichtenstein ML, Ballantyne CM, Mailman RB, Mosley TH, Chen H. (2015) Statins, plasma cholesterol, and risk of Parkinson's disease: a prospective study. Mov Disord 30:552–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im AR, Kim YH, Uddin MR, Lee HW, Chae SW, Kim YH, Jung WS, Kang BJ, Mun CS, Lee MY. (2012) Scutellaria baicalensis extracts and flavonoids protect rat l6 cells from antimycin a-induced mitochondrial dysfunction. Evid Based Complement Alternat Med 2012:517965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip E, Farrell GC, Robertson G, Hall P, Kirsch R, Leclercq I. (2003) Central role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology 38:123–132. [DOI] [PubMed] [Google Scholar]
- Iranzo O. (2011) Manganese complexes displaying superoxide dismutase activity: a balance between different factors. Bioorg Chem 39:73–87. [DOI] [PubMed] [Google Scholar]
- Jäger S, Handschin C, St-Pierre J, Spiegelman BM. (2007) AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA 104:12017–12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnke VE, Van Der Meulen JH, Johnston HK, Ghimbovschi S, Partridge T, Hoffman EP, Nagaraju K. (2012) Metabolic remodeling agents show beneficial effects in the dystrophin-deficient mdx mouse model. Skelet Muscle 2:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazwa A, Rojo AI, Innamorato NG, Hesse M, Fernández-Ruiz J, Cuadrado A. (2011) Pharmacological targeting of the transcription factor Nrf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism. Antioxid Redox Signal 14:2347–2360. [DOI] [PubMed] [Google Scholar]
- Jia Y, Warin R, Yu X, Epstein R, Noguchi CT. (2009) Erythropoietin signaling promotes transplanted progenitor cell survival. FASEB J 23:3089–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, et al. (2013) mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 342:1524–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph AM, Joanisse DR, Baillot RG, Hood DA. (2012) Mitochondrial dysregulation in the pathogenesis of diabetes: potential for mitochondrial biogenesis-mediated interventions. Exp Diabetes Res 2012:642038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantrow SP, Taylor DE, Carraway MS, Piantadosi CA. (1997) Oxidative metabolism in rat hepatocytes and mitochondria during sepsis. Arch Biochem Biophys 345:278–288. [DOI] [PubMed] [Google Scholar]
- Kapil V, Weitzberg E, Lundberg JO, Ahluwalia A. (2014) Clinical evidence demonstrating the utility of inorganic nitrate in cardiovascular health. Nitric Oxide 38:45–57. [DOI] [PubMed] [Google Scholar]
- Karunadharma PP, Basisty N, Chiao YA, Dai DF, Drake R, Levy N, Koh WJ, Emond MJ, Kruse S, Marcinek D, et al. (2015) Respiratory chain protein turnover rates in mice are highly heterogeneous but strikingly conserved across tissues, ages, and treatments. FASEB J 29:3582–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami S, Matsuda A, Sunagawa T, Noda Y, Kaneko T, Tahara S, Hiraumi Y, Adachi S, Matsui H, Ando K, et al. (2009) Antioxidant, EUK-8, prevents murine dilated cardiomyopathy. Circ J 73:2125–2134. [DOI] [PubMed] [Google Scholar]
- Kazak L, Reyes A, Holt IJ. (2012) Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat Rev Mol Cell Biol 13:659–671. [DOI] [PubMed] [Google Scholar]
- Kensler TW, Curphey TJ, Maxiutenko Y, Roebuck BD. (2000) Chemoprotection by organosulfur inducers of phase 2 enzymes: dithiolethiones and dithiins. Drug Metabol Drug Interact 17:3–22. [DOI] [PubMed] [Google Scholar]
- Kensler TW, Egner PA, Agyeman AS, Visvanathan K, Groopman JD, Chen JG, Chen TY, Fahey JW, Talalay P. (2013) Keap1-nrf2 signaling: a target for cancer prevention by sulforaphane. Top Curr Chem 329:163–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten S. (2014) Integrated physiology and systems biology of PPARα. Mol Metab 3:354–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuno N, Shiina H, Urakami S, Kawamoto K, Hirata H, Tanaka Y, Majid S, Igawa M, Dahiya R. (2008) Genistein mediated histone acetylation and demethylation activates tumor suppressor genes in prostate cancer cells. Int J Cancer 123:552–560. [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13:132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Meyers MS, Khuder SS, Abdallah SL, Muturi HT, Russo L, Tate CR, Hevener AL, Najjar SM, Leloup C, et al. (2014) Tissue-selective estrogen complexes with bazedoxifene prevent metabolic dysfunction in female mice. Mol Metab 3:177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloner RA, Hale SL, Dai W, Gorman RC, Shuto T, Koomalsingh KJ, Gorman JH, 3rd, Sloan RC, Frasier CR, Watson CA, et al. (2012) Reduction of ischemia/reperfusion injury with bendavia, a mitochondria-targeting cytoprotective Peptide. J Am Heart Assoc 1:e001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koay MA, Christman JW, Segal BH, Venkatakrishnan A, Blackwell TR, Holland SM, Blackwell TS. (2001) Impaired pulmonary NF-kappaB activation in response to lipopolysaccharide in NADPH oxidase-deficient mice. Infect Immun 69:5991–5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga Y, Akita Y, Junko N, Yatsuga S, Povalko N, Fukiyama R, Ishii M, Matsuishi T. (2006) Endothelial dysfunction in MELAS improved by l-arginine supplementation. Neurology 66:1766–1769. [DOI] [PubMed] [Google Scholar]
- Koga Y, Akita Y, Nishioka J, Yatsuga S, Povalko N, Tanabe Y, Fujimoto S, Matsuishi T. (2005) L-arginine improves the symptoms of strokelike episodes in MELAS. Neurology 64:710–712. [DOI] [PubMed] [Google Scholar]
- Kolb P, Rosenbaum DM, Irwin JJ, Fung JJ, Kobilka BK, Shoichet BK. (2009) Structure-based discovery of beta2-adrenergic receptor ligands. Proc Natl Acad Sci USA 106:6843–6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, Wang R, Xue Y, Liu X, Zhang H, Chen Y, Fang F, Chang Y. (2010) Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS One 5:e11707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopito RR. (1997) ER quality control: the cytoplasmic connection. Cell 88:427–430. [DOI] [PubMed] [Google Scholar]
- Kornmann B. (2013) The molecular hug between the ER and the mitochondria. Curr Opin Cell Biol 25:443–448. [DOI] [PubMed] [Google Scholar]
- Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, et al. (2008) Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab 7:45–56. [DOI] [PubMed] [Google Scholar]
- Kumar A, Sharma N, Gupta A, Kalonia H, Mishra J. (2012) Neuroprotective potential of atorvastatin and simvastatin (HMG-CoA reductase inhibitors) against 6-hydroxydopamine (6-OHDA) induced Parkinson-like symptoms. Brain Res 1471:13–22. [DOI] [PubMed] [Google Scholar]
- Kurose I, Miura S, Fukumura D, Yonei Y, Saito H, Tada S, Suematsu M, Tsuchiya M. (1993) Nitric oxide mediates Kupffer cell-induced reduction of mitochondrial energization in hepatoma cells: a comparison with oxidative burst. Cancer Res 53:2676–2682. [PubMed] [Google Scholar]
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, et al. (2006) Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127:1109–1122. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. (2009) mTOR signaling at a glance. J Cell Sci 122:3589–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen S, Stride N, Hey-Mogensen M, Hansen CN, Andersen JL, Madsbad S, Worm D, Helge JW, Dela F. (2011) Increased mitochondrial substrate sensitivity in skeletal muscle of patients with type 2 diabetes. Diabetologia 54:1427–1436. [DOI] [PubMed] [Google Scholar]
- Latz E, Xiao TS, Stutz A. (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD. (2008) Dual roles of Nrf2 in cancer. Pharmacol Res 58:262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavich IC, de Freitas BS, Kist LW, Falavigna L, Dargél VA, Köbe LM, Aguzzoli C, Piffero B, Florian PZ, Bogo MR, et al. (2015) Sulforaphane rescues memory dysfunction and synaptic and mitochondrial alterations induced by brain iron accumulation. Neuroscience 301:542–552. [DOI] [PubMed] [Google Scholar]
- Lee S, Lee SJ, Coronata AA, Fredenburgh LE, Chung SW, Perrella MA, Nakahira K, Ryter SW, Choi AM. (2014) Carbon monoxide confers protection in sepsis by enhancing beclin 1-dependent autophagy and phagocytosis. Antioxid Redox Signal 20:432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters JJ. (2014) Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol 2:749–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AB, Punihaole D, Levine TB. (2012) Characterization of the role of nitric oxide and its clinical applications. Cardiology 122:55–68. [DOI] [PubMed] [Google Scholar]
- L’Horset F, Dauvois S, Heery DM, Cavaillès V, Parker MG. (1996) RIP-140 interacts with multiple nuclear receptors by means of two distinct sites. Mol Cell Biol 16:6029–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Bhatia M, Zhu YZ, Zhu YC, Ramnath RD, Wang ZJ, Anuar FB, Whiteman M, Salto-Tellez M, Moore PK. (2005) Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J 19:1196–1198. [DOI] [PubMed] [Google Scholar]
- Liby KT, Yore MM, Sporn MB. (2007) Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat Rev Cancer 7:357–369. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795. [DOI] [PubMed] [Google Scholar]
- Lira VA, Brown DL, Lira AK, Kavazis AN, Soltow QA, Zeanah EH, Criswell DS. (2010) Nitric oxide and AMPK cooperatively regulate PGC-1 in skeletal muscle cells. J Physiol 588:3551–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HR, Tao L, Gao E, Lopez BL, Christopher TA, Willette RN, Ohlstein EH, Yue TL, Ma XL. (2004) Anti-apoptotic effects of rosiglitazone in hypercholesterolemic rabbits subjected to myocardial ischemia and reperfusion. Cardiovasc Res 62:135–144. [DOI] [PubMed] [Google Scholar]
- Liu J, Ames BN. (2005) Reducing mitochondrial decay with mitochondrial nutrients to delay and treat cognitive dysfunction, Alzheimer’s disease, and Parkinson’s disease. Nutr Neurosci 8:67–89. [DOI] [PubMed] [Google Scholar]
- Ljubicic V, Miura P, Burt M, Boudreault L, Khogali S, Lunde JA, Renaud JM, Jasmin BJ. (2011) Chronic AMPK activation evokes the slow, oxidative myogenic program and triggers beneficial adaptations in mdx mouse skeletal muscle. Hum Mol Genet 20:3478–3493. [DOI] [PubMed] [Google Scholar]
- Lu SM, Song SM, Liu JC, Yang HM, Li P, Wang ZG. (2003) Changes of proton transportation across the inner mitochondrial membrane and H(+)-ATPase in endotoxic shock rats. Chin J Traumatol 6:292–296. [PubMed] [Google Scholar]
- Luchetti F, Canonico B, Betti M, Arcangeletti M, Pilolli F, Piroddi M, Canesi L, Papa S, Galli F. (2010) Melatonin signaling and cell protection function. FASEB J 24:3603–3624. [DOI] [PubMed] [Google Scholar]
- Lygate CA, Hulbert K, Monfared M, Cole MA, Clarke K, Neubauer S. (2003) The PPARgamma-activator rosiglitazone does not alter remodeling but increases mortality in rats post-myocardial infarction. Cardiovasc Res 58:632–637. [DOI] [PubMed] [Google Scholar]
- MacGarvey NC, Suliman HB, Bartz RR, Fu P, Withers CM, Welty-Wolf KE, Piantadosi CA. (2012) Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2-related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis. Am J Respir Crit Care Med 185:851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. (2006) Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell 10:839–850. [DOI] [PubMed] [Google Scholar]
- MacRae HS, Mefferd KM. (2006) Dietary antioxidant supplementation combined with quercetin improves cycling time trial performance. Int J Sport Nutr Exerc Metab 16:405–419. [DOI] [PubMed] [Google Scholar]
- Magesh S, Chen Y, Hu L. (2012) Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med Res Rev 32:687–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magyar K, Halmosi R, Palfi A, Feher G, Czopf L, Fulop A, Battyany I, Sumegi B, Toth K, Szabados E. (2012) Cardioprotection by resveratrol: A human clinical trial in patients with stable coronary artery disease. Clin Hemorheol Microcirc 50:179–187. [DOI] [PubMed] [Google Scholar]
- Maines MD. (2005) The heme oxygenase system: update 2005. Antioxid Redox Signal 7:1761–1766. [DOI] [PubMed] [Google Scholar]
- Malkki H. (2014) Multiple sclerosis: could simvastatin slow down secondary progressive MS? Nat Rev Neurol 10:241. [DOI] [PubMed] [Google Scholar]
- Manji H, Kato T, Di Prospero NA, Ness S, Beal MF, Krams M, Chen G. (2012) Impaired mitochondrial function in psychiatric disorders. Nat Rev Neurosci 13:293–307. [DOI] [PubMed] [Google Scholar]
- Marangos PJ, Loftus T, Wiesner J, Lowe T, Rossi E, Browne CE, Gruber HE. (1990) Adenosinergic modulation of homocysteine-induced seizures in mice. Epilepsia 31:239–246. [DOI] [PubMed] [Google Scholar]
- Marazioti A, Bucci M, Coletta C, Vellecco V, Baskaran P, Szabó C, Cirino G, Marques AR, Guerreiro B, Gonçalves AM, et al. (2011) Inhibition of nitric oxide-stimulated vasorelaxation by carbon monoxide-releasing molecules. Arterioscler Thromb Vasc Biol 31:2570–2576. [DOI] [PubMed] [Google Scholar]
- Marcinek DJ, Conley KE. (2014) In vivo metabolic spectroscopy identifies deficits in mitochondrial quality and capacity in aging skeletal muscle. Clin Pharmacol Ther 96:669–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattingly KA, Ivanova MM, Riggs KA, Wickramasinghe NS, Barch MJ, Klinge CM. (2008) Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis. Mol Endocrinol 22:609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros DM. (2008) Assessing mitochondria biogenesis. Methods 46:288–294. [DOI] [PubMed] [Google Scholar]
- Mela-Riker L, Bartos D, Vlessis AA, Widener L, Muller P, Trunkey DD. (1992) Chronic hyperdynamic sepsis in the rat. II. Characterization of liver and muscle energy metabolism. Circ Shock 36:83–92. [PubMed] [Google Scholar]
- Meng X, Maliakal P, Lu H, Lee MJ, Yang CS. (2004) Urinary and plasma levels of resveratrol and quercetin in humans, mice, and rats after ingestion of pure compounds and grape juice. J Agric Food Chem 52:935–942. [DOI] [PubMed] [Google Scholar]
- Merrill GF, Kurth EJ, Hardie DG, Winder WW. (1997) AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol 273:E1107–E1112. [DOI] [PubMed] [Google Scholar]
- Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S. (2013) Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol 14:454–460. [DOI] [PubMed] [Google Scholar]
- Miwa S, Lawless C, von Zglinicki T. (2008) Mitochondrial turnover in liver is fast in vivo and is accelerated by dietary restriction: application of a simple dynamic model. Aging Cell 7:920–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Loshuertos R, Acín-Pérez R, Fernández-Silva P, Movilla N, Pérez-Martos A, Rodriguez de Cordoba S, Gallardo ME, Enríquez JA. (2006) Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat Genet 38:1261–1268. [DOI] [PubMed] [Google Scholar]
- Moreno-Loshuertos R, Ferrín G, Acín-Pérez R, Gallardo ME, Viscomi C, Pérez-Martos A, Zeviani M, Fernández-Silva P, Enríquez JA. (2011) Evolution meets disease: penetrance and functional epistasis of mitochondrial tRNA mutations. PLoS Genet 7:e1001379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimitsu Y, Nakagawa Y, Hayashi K, Fujii H, Kumagai T, Nakamura Y, Osawa T, Horio F, Itoh K, Iida K, et al. (2002) A sulforaphane analogue that potently activates the Nrf2-dependent detoxification pathway. J Biol Chem 277:3456–3463. [DOI] [PubMed] [Google Scholar]
- Morrish F, Giedt C, Hockenbery D. (2003) c-MYC apoptotic function is mediated by NRF-1 target genes. Genes Dev 17:240–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrish F, Hockenbery D. (2014) MYC and mitochondrial biogenesis. Cold Spring Harb Perspect Med 4: doi: 10.1101/cshperspect.a014225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow VA, Foufelle F, Connell JM, Petrie JR, Gould GW, Salt IP. (2003) Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. J Biol Chem 278:31629–31639. [DOI] [PubMed] [Google Scholar]
- Motterlini R, Clark JE, Foresti R, Sarathchandra P, Mann BE, Green CJ. (2002) Carbon monoxide-releasing molecules: characterization of biochemical and vascular activities. Circ Res 90:E17–E24. [DOI] [PubMed] [Google Scholar]
- Motterlini R, Mann BE, Johnson TR, Clark JE, Foresti R, Green CJ. (2003) Bioactivity and pharmacological actions of carbon monoxide-releasing molecules. Curr Pharm Des 9:2525–2539. [DOI] [PubMed] [Google Scholar]
- Motterlini R, Otterbein LE. (2010) The therapeutic potential of carbon monoxide. Nat Rev Drug Discov 9:728–743. [DOI] [PubMed] [Google Scholar]
- Murphy JL, Blakely EL, Schaefer AM, He L, Wyrick P, Haller RG, Taylor RW, Turnbull DM, Taivassalo T. (2008) Resistance training in patients with single, large-scale deletions of mitochondrial DNA. Brain 131:2832–2840. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Smith RA. (2007) Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol 47:629–656. [DOI] [PubMed] [Google Scholar]
- Musi N, Hirshman MF, Nygren J, Svanfeldt M, Bavenholm P, Rooyackers O, Zhou G, Williamson JM, Ljunqvist O, Efendic S, et al. (2002) Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes 51:2074–2081. [DOI] [PubMed] [Google Scholar]
- Naini A, Kaufmann P, Shanske S, Engelstad K, De Vivo DC, Schon EA. (2005) Hypocitrullinemia in patients with MELAS: an insight into the “MELAS paradox”. J Neurol Sci 229-230:187–193. [DOI] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12:222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, et al. (2008) AMPK and PPARdelta agonists are exercise mimetics. Cell 134:405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrallah CM, Horvath TL. (2014) Mitochondrial dynamics in the central regulation of metabolism. Nat Rev Endocrinol 10:650–658. [DOI] [PubMed] [Google Scholar]
- Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, Andrade-Navarro MA, McBride HM. (2008) Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol 18:102–108. [DOI] [PubMed] [Google Scholar]
- Nieman DC, Williams AS, Shanely RA, Jin F, McAnulty SR, Triplett NT, Austin MD, Henson DA. (2010) Quercetin’s influence on exercise performance and muscle mitochondrial biogenesis. Med Sci Sports Exerc 42:338–345. [DOI] [PubMed] [Google Scholar]
- Nisoli E, Carruba MO. (2006) Nitric oxide and mitochondrial biogenesis. J Cell Sci 119:2855–2862. [DOI] [PubMed] [Google Scholar]
- Nisoli E, Clementi E, Moncada S, Carruba MO. (2004a) Mitochondrial biogenesis as a cellular signaling framework. Biochem Pharmacol 67:1–15. [DOI] [PubMed] [Google Scholar]
- Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, et al. (2003) Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 299:896–899. [DOI] [PubMed] [Google Scholar]
- Nisoli E, Falcone S, Tonello C, Cozzi V, Palomba L, Fiorani M, Pisconti A, Brunelli S, Cardile A, Francolini M, et al. (2004b) Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc Natl Acad Sci USA 101:16507–16512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noe N, Dillon L, Lellek V, Diaz F, Hida A, Moraes CT, Wenz T. (2013) Bezafibrate improves mitochondrial function in the CNS of a mouse model of mitochondrial encephalopathy. Mitochondrion 13:417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Lone R, Knorr K, Jaffe IZ, Schaffer ME, Martini PG, Karas RH, Bienkowska J, Mendelsohn ME, Hansen U. (2007) Estrogen receptors alpha and beta mediate distinct pathways of vascular gene expression, including genes involved in mitochondrial electron transport and generation of reactive oxygen species. Mol Endocrinol 21:1281–1296. [DOI] [PubMed] [Google Scholar]
- O’Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jørgensen SB, Schertzer JD, Shyroka O, Kiens B, van Denderen BJ, Tarnopolsky MA, et al. (2011) AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci USA 108:16092–16097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogilvie M, Yu X, Nicolas-Metral V, Pulido SM, Liu C, Ruegg UT, Noguchi CT. (2000) Erythropoietin stimulates proliferation and interferes with differentiation of myoblasts. J Biol Chem 275:39754–39761. [DOI] [PubMed] [Google Scholar]
- Onyango IG, Lu J, Rodova M, Lezi E, Crafter AB, Swerdlow RH. (2010) Regulation of neuron mitochondrial biogenesis and relevance to brain health. Biochim Biophys Acta 1802:228–234. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. (2007) Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87:315–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SW, Kim KY, Lindsey JD, Dai Y, Heo H, Nguyen DH, Ellisman MH, Weinreb RN, Ju WK. (2011) A selective inhibitor of drp1, mdivi-1, increases retinal ganglion cell survival in acute ischemic mouse retina. Invest Ophthalmol Vis Sci 52:2837–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsouris D, Mandard S, Voshol PJ, Escher P, Tan NS, Havekes LM, Koenig W, März W, Tafuri S, Wahli W, et al. (2004) PPARalpha governs glycerol metabolism. J Clin Invest 114:94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauly M, Daussin F, Burelle Y, Li T, Godin R, Fauconnier J, Koechlin-Ramonatxo C, Hugon G, Lacampagne A, Coisy-Quivy M, et al. (2012) AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am J Pathol 181:583–592. [DOI] [PubMed] [Google Scholar]
- Pearen MA, Myers SA, Raichur S, Ryall JG, Lynch GS, Muscat GE. (2008) The orphan nuclear receptor, NOR-1, a target of beta-adrenergic signaling, regulates gene expression that controls oxidative metabolism in skeletal muscle. Endocrinology 149:2853–2865. [DOI] [PubMed] [Google Scholar]
- Pecorella SR, Potter JV, Cherry AD, Peacher DF, Welty-Wolf KE, Moon RE, Piantadosi CA, Suliman HB. (2015) HO-1/CO regulation of mitochondrial-capillary density relationships in human skeletal muscle. Am J Physiol Lung Cell Mol Physiol 309:L857–L871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson MF, Franzén S, Catrina SB, Dallner G, Hansell P, Brismar K, Palm F. (2012) Coenzyme Q10 prevents GDP-sensitive mitochondrial uncoupling, glomerular hyperfiltration and proteinuria in kidneys from db/db mice as a model of type 2 diabetes. Diabetologia 55:1535–1543. [DOI] [PubMed] [Google Scholar]
- Pessayre D. (2007) Role of mitochondria in non-alcoholic fatty liver disease. J Gastroenterol Hepatol 22 (Suppl 1):S20–S27. [DOI] [PubMed] [Google Scholar]
- Pessayre D, Fromenty B. (2005) NASH: a mitochondrial disease. J Hepatol 42:928–940. [DOI] [PubMed] [Google Scholar]
- Piantadosi CA. (2008) Carbon monoxide, reactive oxygen signaling, and oxidative stress. Free Radic Biol Med 45:562–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piantadosi CA, Carraway MS, Babiker A, Suliman HB. (2008) Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res 103:1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piantadosi CA, Carraway MS, Haden DW, Suliman HB. (2007) Protecting the permeability pore and mitochondrial biogenesis. Novartis Found Symp 280:266–276, discussion 276–280. [DOI] [PubMed] [Google Scholar]
- Piantadosi CA, Suliman HB. (2006) Mitochondrial transcription factor A induction by redox activation of nuclear respiratory factor 1. J Biol Chem 281:324–333. [DOI] [PubMed] [Google Scholar]
- Piantadosi CA, Suliman HB. (2012a) Redox regulation of mitochondrial biogenesis. Free Radic Biol Med 53:2043–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piantadosi CA, Suliman HB. (2012b) Transcriptional control of mitochondrial biogenesis and its interface with inflammatory processes. Biochim Biophys Acta 1820:532–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piantadosi CA, Withers CM, Bartz RR, MacGarvey NC, Fu P, Sweeney TE, Welty-Wolf KE, Suliman HB. (2011) Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. J Biol Chem 286:16374–16385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenge U, Belhage B, Guadalupe-Grau A, Andersen PR, Lundby C, Dela F, Stride N, Pott FC, Helge JW, Boushel R. (2012) Erythropoietin treatment enhances muscle mitochondrial capacity in humans. Front Physiol 3:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poderoso JJ. (2009) The formation of peroxynitrite in the applied physiology of mitochondrial nitric oxide. Arch Biochem Biophys 484:214–220. [DOI] [PubMed] [Google Scholar]
- Potgieter M, Pretorius E, Pepper MS. (2013) Primary and secondary coenzyme Q10 deficiency: the role of therapeutic supplementation. Nutr Rev 71:180–188. [DOI] [PubMed] [Google Scholar]
- Poulsen MM, Vestergaard PF, Clasen BF, Radko Y, Christensen LP, Stødkilde-Jørgensen H, Møller N, Jessen N, Pedersen SB, Jørgensen JO. (2013) High-dose resveratrol supplementation in obese men: an investigator-initiated, randomized, placebo-controlled clinical trial of substrate metabolism, insulin sensitivity, and body composition. Diabetes 62:1186–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powelka AM, Seth A, Virbasius JV, Kiskinis E, Nicoloro SM, Guilherme A, Tang X, Straubhaar J, Cherniack AD, Parker MG, et al. (2006) Suppression of oxidative metabolism and mitochondrial biogenesis by the transcriptional corepressor RIP140 in mouse adipocytes. J Clin Invest 116:125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psarra AM, Sekeris CE. (2011) Glucocorticoids induce mitochondrial gene transcription in HepG2 cells: role of the mitochondrial glucocorticoid receptor. Biochim Biophys Acta 1813:1814–1821. [DOI] [PubMed] [Google Scholar]
- Pucheu S, Boucher F, Sulpice T, Tresallet N, Bonhomme Y, Malfroy B, de Leiris J. (1996) EUK-8 a synthetic catalytic scavenger of reactive oxygen species protects isolated iron-overloaded rat heart from functional and structural damage induced by ischemia/reperfusion. Cardiovasc Drugs Ther 10:331–339. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. (1998) A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92:829–839. [DOI] [PubMed] [Google Scholar]
- Qi X, Qvit N, Su YC, Mochly-Rosen D. (2013) A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci 126:789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. (2010) Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab 12:662–667. [DOI] [PubMed] [Google Scholar]
- Qiu X, Cao L, Yang X, Zhao X, Liu X, Han Y, Xue Y, Jiang H, Chi Z. (2013) Role of mitochondrial fission in neuronal injury in pilocarpine-induced epileptic rats. Neuroscience 245:157–165. [DOI] [PubMed] [Google Scholar]
- Rabinowitz M, Zak R. (1975) Mitochondria and cardiac hypertrophy. Circ Res 36:367–376. [DOI] [PubMed] [Google Scholar]
- Rasbach KA, Funk JA, Jayavelu T, Green PT, Schnellmann RG. (2010) 5-hydroxytryptamine receptor stimulation of mitochondrial biogenesis. J Pharmacol Exp Ther 332:632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasbach KA, Schnellmann RG. (2007) Signaling of mitochondrial biogenesis following oxidant injury. J Biol Chem 282:2355–2362. [DOI] [PubMed] [Google Scholar]
- Rasbach KA, Schnellmann RG. (2008) Isoflavones promote mitochondrial biogenesis. J Pharmacol Exp Ther 325:536–543. [DOI] [PubMed] [Google Scholar]
- Reynolds CM, Suliman HB, Hollingsworth JW, Welty-Wolf KE, Carraway MS, Piantadosi CA. (2009) Nitric oxide synthase-2 induction optimizes cardiac mitochondrial biogenesis after endotoxemia. Free Radic Biol Med 46:564–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznick RM, Shulman GI. (2006) The role of AMP-activated protein kinase in mitochondrial biogenesis. J Physiol 574:33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee J, Inoue Y, Yoon JC, Puigserver P, Fan M, Gonzalez FJ, Spiegelman BM. (2003) Regulation of hepatic fasting response by PPARgamma coactivator-1alpha (PGC-1): requirement for hepatocyte nuclear factor 4alpha in gluconeogenesis. Proc Natl Acad Sci USA 100:4012–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimbaud S, Ruiz M, Piquereau J, Mateo P, Fortin D, Veksler V, Garnier A, Ventura-Clapier R. (2011) Resveratrol improves survival, hemodynamics and energetics in a rat model of hypertension leading to heart failure. PLoS One 6:e26391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. (2005) Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434:113–118. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Cuenca S, Cochemé HM, Logan A, Abakumova I, Prime TA, Rose C, Vidal-Puig A, Smith AC, Rubinsztein DC, Fearnley IM, et al. (2010) Consequences of long-term oral administration of the mitochondria-targeted antioxidant MitoQ to wild-type mice. Free Radic Biol Med 48:161–172. [DOI] [PubMed] [Google Scholar]
- Rossignol DA, Frye RE. (2012) Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry 17:290–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Codogno P, Levine B. (2012) Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 11:709–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter SW, Alam J, Choi AM. (2006) Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 86:583–650. [DOI] [PubMed] [Google Scholar]
- Salminen A, Hyttinen JM, Kaarniranta K. (2011) AMP-activated protein kinase inhibits NF-κB signaling and inflammation: impact on healthspan and lifespan. J Mol Med (Berl) 89:667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltiel AR, Kahn CR. (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414:799–806. [DOI] [PubMed] [Google Scholar]
- Sanchez MI, Shearwood AM, Chia T, Davies SM, Rackham O, Filipovska A. (2015) Estrogen-mediated regulation of mitochondrial gene expression. Mol Endocrinol 29:14–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Furuta K, Tomokiyo K, Nakatsuka D, Tanikawa M, Nakanishi M, Miura M, Tanaka S, Koike T, Hatanaka H, et al. (2000) Facilitatory roles of novel compounds designed from cyclopentenone prostaglandins on neurite outgrowth-promoting activities of nerve growth factor. J Neurochem 75:1092–1102. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. (2008) Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 88:611–638. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. (2011) Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta 1813:1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC, Vega RB, Kelly DP. (2012) Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab 23:459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer AM, Taylor RW, Turnbull DM, Chinnery PF. (2004) The epidemiology of mitochondrial disorders--past, present and future. Biochim Biophys Acta 1659:115–120. [DOI] [PubMed] [Google Scholar]
- Schlüter T, Struy H, Schönfeld P. (2000) Protection of mitochondrial integrity from oxidative stress by the triaminopyridine derivative flupirtine. FEBS Lett 481:42–46. [DOI] [PubMed] [Google Scholar]
- Schmidt O, Pfanner N, Meisinger C. (2010) Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol 11:655–667. [DOI] [PubMed] [Google Scholar]
- Schroder K, Tschopp J. (2010) The inflammasomes. Cell 140:821–832. [DOI] [PubMed] [Google Scholar]
- Schulz TJ, Huang P, Huang TL, Xue R, McDougall LE, Townsend KL, Cypess AM, Mishina Y, Gussoni E, Tseng YH. (2013) Brown-fat paucity due to impaired BMP signalling induces compensatory browning of white fat. Nature 495:379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W. (1992) Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J Biol Chem 267:5317–5323. [PubMed] [Google Scholar]
- Seo AY, Joseph AM, Dutta D, Hwang JC, Aris JP, Leeuwenburgh C. (2010) New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J Cell Sci 123:2533–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadel GS, Clayton DA. (1997) Mitochondrial DNA maintenance in vertebrates. Annu Rev Biochem 66:409–435. [DOI] [PubMed] [Google Scholar]
- Shamseldin HE, Alshammari M, Al-Sheddi T, Salih MA, Alkhalidi H, Kentab A, Repetto GM, Hashem M, Alkuraya FS. (2012) Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J Med Genet 49:234–241. [DOI] [PubMed] [Google Scholar]
- Sheikh SZ, Hegazi RA, Kobayashi T, Onyiah JC, Russo SM, Matsuoka K, Sepulveda AR, Li F, Otterbein LE, Plevy SE. (2011) An anti-inflammatory role for carbon monoxide and heme oxygenase-1 in chronic Th2-mediated murine colitis. J Immunol 186:5506–5513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Hao J, Tian C, Ren J, Yang L, Li X, Luo C, Cotma CW, Liu J. (2008a) A combination of nutriments improves mitochondrial biogenesis and function in skeletal muscle of type 2 diabetic Goto-Kakizaki rats. PLoS One 3:e2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Liu K, Tian C, Yang L, Li X, Ren J, Packer L, Cotman CW, Liu J. (2008b) R-alpha-lipoic acid and acetyl-L-carnitine complementarily promote mitochondrial biogenesis in murine 3T3-L1 adipocytes. Diabetologia 51:165–174. [DOI] [PubMed] [Google Scholar]
- Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, et al. (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36:401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirendeb UP, Calkins MJ, Manczak M, Anekonda V, Dufour B, McBride JL, Mao P, Reddy PH. (2012) Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum Mol Genet 21:406–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutt TE, Shadel GS. (2010) A compendium of human mitochondrial gene expression machinery with links to disease. Environ Mol Mutagen 51:360–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims CR, MacMillan-Crow LA, Mayeux PR. (2014) Targeting mitochondrial oxidants may facilitate recovery of renal function during infant sepsis. Clin Pharmacol Ther 96:662–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer M. (2007) Mitochondrial function in sepsis: acute phase versus multiple organ failure. Crit Care Med 35(9, Suppl)S441–S448. [DOI] [PubMed] [Google Scholar]
- Singh DK, Winocour P, Farrington K. (2011) Oxidative stress in early diabetic nephropathy: fueling the fire. Nat Rev Endocrinol 7:176–184. [DOI] [PubMed] [Google Scholar]
- Smith RA, Hartley RC, Cochemé HM, Murphy MP. (2012) Mitochondrial pharmacology. Trends Pharmacol Sci 33:341–352. [DOI] [PubMed] [Google Scholar]
- Smith RA, Porteous CM, Gane AM, Murphy MP. (2003) Delivery of bioactive molecules to mitochondria in vivo. Proc Natl Acad Sci USA 100:5407–5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O’Sullivan JD, Fung V, Smith RA, Murphy MP, Taylor KM, Protect Study Group (2010) A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov Disord 25:1670–1674. [DOI] [PubMed] [Google Scholar]
- So EC, Hsing CH, Liang CH, Wu SN. (2012) The actions of mdivi-1, an inhibitor of mitochondrial fission, on rapidly activating delayed-rectifier K⁺ current and membrane potential in HL-1 murine atrial cardiomyocytes. Eur J Pharmacol 683:1–9. [DOI] [PubMed] [Google Scholar]
- Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. (2010) Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143:802–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, McBride HM. (2012a) A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol 22:135–141. [DOI] [PubMed] [Google Scholar]
- Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA, McBride HM. (2012b) Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS One 7:e52830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Diaz F, Iommarini L, Aure K, Lombes A, Moraes CT. (2009) PGC-1alpha/beta induced expression partially compensates for respiratory chain defects in cells from patients with mitochondrial disorders. Hum Mol Genet 18:1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, Handschin C, Zheng K, Lin J, Yang W, et al. (2006) Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127:397–408. [DOI] [PubMed] [Google Scholar]
- Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. (2013) The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153:348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden MC, Bulmer K, Gibbons GF, Knight BL, Holness MJ. (2002) Peroxisome-proliferator-activated receptor-alpha (PPARalpha) deficiency leads to dysregulation of hepatic lipid and carbohydrate metabolism by fatty acids and insulin. Biochem J 364:361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugishita K, Li F, Su Z, Barry WH. (2003) Anti-oxidant effects of estrogen reduce [Ca2+]i during metabolic inhibition. J Mol Cell Cardiol 35:331–336. [DOI] [PubMed] [Google Scholar]
- Sugiura A, McLelland GL, Fon EA, McBride HM. (2014) A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J 33:2142–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suliman HB, Babiker A, Withers CM, Sweeney TE, Carraway MS, Tatro LG, Bartz RR, Welty-Wolf KE, Piantadosi CA. (2010) Nitric oxide synthase-2 regulates mitochondrial Hsp60 chaperone function during bacterial peritonitis in mice. Free Radic Biol Med 48:736–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suliman HB, Carraway MS, Ali AS, Reynolds CM, Welty-Wolf KE, Piantadosi CA. (2007a) The CO/HO system reverses inhibition of mitochondrial biogenesis and prevents murine doxorubicin cardiomyopathy. J Clin Invest 117:3730–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suliman HB, Carraway MS, Piantadosi CA. (2003a) Postlipopolysaccharide oxidative damage of mitochondrial DNA. Am J Respir Crit Care Med 167:570–579. [DOI] [PubMed] [Google Scholar]
- Suliman HB, Carraway MS, Tatro LG, Piantadosi CA. (2007b) A new activating role for CO in cardiac mitochondrial biogenesis. J Cell Sci 120:299–308. [DOI] [PubMed] [Google Scholar]
- Suliman HB, Carraway MS, Welty-Wolf KE, Whorton AR, Piantadosi CA. (2003b) Lipopolysaccharide stimulates mitochondrial biogenesis via activation of nuclear respiratory factor-1. J Biol Chem 278:41510–41518. [DOI] [PubMed] [Google Scholar]
- Suliman HB, Piantadosi CA. (2014) Mitochondrial biogenesis: regulation by endogenous gases during inflammation and organ stress. Curr Pharm Des 20:5653–5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suliman HB, Welty-Wolf KE, Carraway M, Tatro L, Piantadosi CA. (2004) Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis. Cardiovasc Res 64:279–288. [DOI] [PubMed] [Google Scholar]
- Suliman HB, Welty-Wolf KE, Carraway MS, Schwartz DA, Hollingsworth JW, Piantadosi CA. (2005) Toll-like receptor 4 mediates mitochondrial DNA damage and biogenic responses after heat-inactivated E. coli. FASEB J 19:1531–1533. [DOI] [PubMed] [Google Scholar]
- Sweeney TE, Suliman HB, Hollingsworth JW, Piantadosi CA. (2010) Differential regulation of the PGC family of genes in a mouse model of Staphylococcus aureus sepsis. PLoS One 5:e11606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó C. (2007) Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov 6:917–935. [DOI] [PubMed] [Google Scholar]
- Szendroedi J, Phielix E, Roden M. (2012) The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol 8:92–103. [DOI] [PubMed] [Google Scholar]
- Szeto HH. (2014) First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol 171:2029–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto HH, Birk AV. (2014) Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin Pharmacol Ther 96:672–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto HH, Schiller PW. (2011) Novel therapies targeting inner mitochondrial membrane--from discovery to clinical development. Pharm Res 28:2669–2679. [DOI] [PubMed] [Google Scholar]
- Tamizhselvi R, Moore PK, Bhatia M. (2008) Inhibition of hydrogen sulfide synthesis attenuates chemokine production and protects mice against acute pancreatitis and associated lung injury. Pancreas 36:e24–e31. [DOI] [PubMed] [Google Scholar]
- Tang WX, Wu WH, Qiu HY, Bo H, Huang SM. (2013) Amelioration of rhabdomyolysis-induced renal mitochondrial injury and apoptosis through suppression of Drp-1 translocation. J Nephrol 26:1073–1082. [DOI] [PubMed] [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. (2013) Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496:238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, et al. (2010) Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell 40:893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taub PR, Ramirez-Sanchez I, Ciaraldi TP, Perkins G, Murphy AN, Naviaux R, Hogan M, Maisel AS, Henry RR, Ceballos G, et al. (2012) Alterations in skeletal muscle indicators of mitochondrial structure and biogenesis in patients with type 2 diabetes and heart failure: effects of epicatechin rich cocoa. Clin Transl Sci 5:43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DE, Ghio AJ, Piantadosi CA. (1995) Reactive oxygen species produced by liver mitochondria of rats in sepsis. Arch Biochem Biophys 316:70–76. [DOI] [PubMed] [Google Scholar]
- Taylor DE, Kantrow SP, Piantadosi CA. (1998) Mitochondrial respiration after sepsis and prolonged hypoxia. Am J Physiol 275:L139–L144. [DOI] [PubMed] [Google Scholar]
- Tedesco D, Haragsim L. (2012) Cyclosporine: a review. J Transplant 2012:230386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenenbaum A, Motro M, Fisman EZ. (2005) Dual and pan-peroxisome proliferator-activated receptors (PPAR) co-agonism: the bezafibrate lessons. Cardiovasc Diabetol 4:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng R, Gavrilova O, Suzuki N, Chanturiya T, Schimel D, Hugendubler L, Mammen S, Yver DR, Cushman SW, Mueller E, et al. (2011) Disrupted erythropoietin signalling promotes obesity and alters hypothalamus proopiomelanocortin production. Nat Commun 2:520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tengan CH, Kiyomoto BH, Godinho RO, Gamba J, Neves AC, Schmidt B, Oliveira AS, Gabbai AA. (2007) The role of nitric oxide in muscle fibers with oxidative phosphorylation defects. Biochem Biophys Res Commun 359:771–777. [DOI] [PubMed] [Google Scholar]
- Tengan CH, Rodrigues GS, Godinho RO. (2012) Nitric oxide in skeletal muscle: role on mitochondrial biogenesis and function. Int J Mol Sci 13:17160–17184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Than TA, Win S, Kaplowitz N. (2014) In vitro assays of mitochondrial function/dysfunction. Clin Pharmacol Ther 96:665–668. [DOI] [PubMed] [Google Scholar]
- Thandapilly SJ, Wojciechowski P, Behbahani J, Louis XL, Yu L, Juric D, Kopilas MA, Anderson HD, Netticadan T. (2010) Resveratrol prevents the development of pathological cardiac hypertrophy and contractile dysfunction in the SHR without lowering blood pressure. Am J Hypertens 23:192–196. [DOI] [PubMed] [Google Scholar]
- Trushina E, McMurray CT. (2007) Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 145:1233–1248. [DOI] [PubMed] [Google Scholar]
- Tschopp J. (2011) Mitochondria: Sovereign of inflammation? Eur J Immunol 41:1196–1202. [DOI] [PubMed] [Google Scholar]
- Um JH, Park SJ, Kang H, Yang S, Foretz M, McBurney MW, Kim MK, Viollet B, Chung JH. (2010) AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes 59:554–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti D, De Rasmo D, Signorile A, Rossi L, de Bari L, Scala I, Granese B, Papa S, Vacca RA. (2013) Epigallocatechin-3-gallate prevents oxidative phosphorylation deficit and promotes mitochondrial biogenesis in human cells from subjects with Down’s syndrome. Biochim Biophys Acta 1832:542–552. [DOI] [PubMed] [Google Scholar]
- Vance JE. (1990) Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem 265:7248–7256. [PubMed] [Google Scholar]
- van Empel VP, Bertrand AT, van Oort RJ, van der Nagel R, Engelen M, van Rijen HV, Doevendans PA, Crijns HJ, Ackerman SL, Sluiter W, et al. (2006) EUK-8, a superoxide dismutase and catalase mimetic, reduces cardiac oxidative stress and ameliorates pressure overload-induced heart failure in the harlequin mouse mutant. J Am Coll Cardiol 48:824–832. [DOI] [PubMed] [Google Scholar]
- Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, Chawla A. (2006) Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab 4:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vehviläinen P, Koistinaho J, Gundars G. (2014) Mechanisms of mutant SOD1 induced mitochondrial toxicity in amyotrophic lateral sclerosis. Front Cell Neurosci 8:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E, Hirschey MD, Finley LW, Haigis MC. (2010) Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci 35:669–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vial G, Dubouchaud H, Leverve XM. (2010) Liver mitochondria and insulin resistance. Acta Biochim Pol 57:389–392. [PubMed] [Google Scholar]
- Vieira HL, Queiroga CS, Alves PM. (2008) Pre-conditioning induced by carbon monoxide provides neuronal protection against apoptosis. J Neurochem 107:375–384. [DOI] [PubMed] [Google Scholar]
- Viscomi C, Bottani E, Civiletto G, Cerutti R, Moggio M, Fagiolari G, Schon EA, Lamperti C, Zeviani M. (2011) In vivo correction of COX deficiency by activation of the AMPK/PGC-1α axis. Cell Metab 14:80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner F, Asfar P, Calzia E, Radermacher P, Szabó C. (2009) Bench-to-bedside review: Hydrogen sulfide--the third gaseous transmitter: applications for critical care. Crit Care 13:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner F, Wagner K, Weber S, Stahl B, Knöferl MW, Huber-Lang M, Seitz DH, Asfar P, Calzia E, Senftleben U, et al. (2011) Inflammatory effects of hypothermia and inhaled H2S during resuscitated, hyperdynamic murine septic shock. Shock 35:396–402. [DOI] [PubMed] [Google Scholar]
- Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW, Talalay P. (2004) Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci USA 101:2040–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39:359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. (2012) Mitochondria and cancer. Nat Rev Cancer 12:685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Fan W, Procaccio V. (2010) Mitochondrial energetics and therapeutics. Annu Rev Pathol 5:297–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace KB. (2008) Mitochondrial off targets of drug therapy. Trends Pharmacol Sci 29:361–366. [DOI] [PubMed] [Google Scholar]
- Wang J, Green PS, Simpkins JW. (2001) Estradiol protects against ATP depletion, mitochondrial membrane potential decline and the generation of reactive oxygen species induced by 3-nitroproprionic acid in SK-N-SH human neuroblastoma cells. J Neurochem 77:804–811. [DOI] [PubMed] [Google Scholar]
- Wappler EA, Institoris A, Dutta S, Katakam PV, Busija DW. (2013) Mitochondrial dynamics associated with oxygen-glucose deprivation in rat primary neuronal cultures. PLoS One 8:e63206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe E, Muenzer JT, Hawkins WG, Davis CG, Dixon DJ, McDunn JE, Brackett DJ, Lerner MR, Swanson PE, Hotchkiss RS. (2009) Sepsis induces extensive autophagic vacuolization in hepatocytes: a clinical and laboratory-based study. Lab Invest 89:549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber K, Brück P, Mikes Z, Küpper JH, Klingenspor M, Wiesner RJ. (2002) Glucocorticoid hormone stimulates mitochondrial biogenesis specifically in skeletal muscle. Endocrinology 143:177–184. [DOI] [PubMed] [Google Scholar]
- Weitzel JM, Iwen KA. (2011) Coordination of mitochondrial biogenesis by thyroid hormone. Mol Cell Endocrinol 342:1–7. [DOI] [PubMed] [Google Scholar]
- West AP, Shadel GS, Ghosh S. (2011) Mitochondria in innate immune responses. Nat Rev Immunol 11:389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikstrom JD, Twig G, Shirihai OS. (2009) What can mitochondrial heterogeneity tell us about mitochondrial dynamics and autophagy? Int J Biochem Cell Biol 41:1914–1927. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, Gautam B, Hassanali M. (2008) DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res 36:D901–D906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J. (2006) DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 34:D668–D672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D. (2004) Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 430:686–689. [DOI] [PubMed] [Google Scholar]
- Wu J, Cohen P, Spiegelman BM. (2013) Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev 27:234–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Song Y, Li H, Chen J. (2009) Rhabdomyolysis associated with fibrate therapy: review of 76 published cases and a new case report. Eur J Clin Pharmacol 65:1169–1174. [DOI] [PubMed] [Google Scholar]
- Wu Z, Huang X, Feng Y, Handschin C, Feng Y, Gullicksen PS, Bare O, Labow M, Spiegelman B, Stevenson SC. (2006) Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1alpha transcription and mitochondrial biogenesis in muscle cells. Proc Natl Acad Sci USA 103:14379–14384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulf A, Harneit A, Kröger M, Kebenko M, Wetzel MG, Weitzel JM. (2008) T3-mediated expression of PGC-1alpha via a far upstream located thyroid hormone response element. Mol Cell Endocrinol 287:90–95. [DOI] [PubMed] [Google Scholar]
- Xie N, Wang C, Lian Y, Zhang H, Wu C, Zhang Q. (2013) A selective inhibitor of Drp1, mdivi-1, protects against cell death of hippocampal neurons in pilocarpine-induced seizures in rats. Neurosci Lett 545:64–68. [DOI] [PubMed] [Google Scholar]
- Xu Y, Gen M, Lu L, Fox J, Weiss SO, Brown RD, Perlov D, Ahmad H, Zhu P, Greyson C, et al. (2005) PPAR-gamma activation fails to provide myocardial protection in ischemia and reperfusion in pigs. Am J Physiol Heart Circ Physiol 288:H1314–H1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabluchanskiy A, Sawle P, Homer-Vanniasinkam S, Green CJ, Foresti R, Motterlini R. (2012) CORM-3, a carbon monoxide-releasing molecule, alters the inflammatory response and reduces brain damage in a rat model of hemorrhagic stroke. Crit Care Med 40:544–552. [DOI] [PubMed] [Google Scholar]
- Yang H, Brosel S, Acin-Perez R, Slavkovich V, Nishino I, Khan R, Goldberg IJ, Graziano J, Manfredi G, Schon EA. (2010) Analysis of mouse models of cytochrome c oxidase deficiency owing to mutations in Sco2. Hum Mol Genet 19:170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Calingasan NY, Thomas B, Chaturvedi RK, Kiaei M, Wille EJ, Liby KT, Williams C, Royce D, Risingsong R, et al. (2009) Neuroprotective effects of the triterpenoid, CDDO methyl amide, a potent inducer of Nrf2-mediated transcription. PLoS One 4:e5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Nagano T, Shah Y, Cheung C, Ito S, Gonzalez FJ. (2008) The PPAR alpha-humanized mouse: a model to investigate species differences in liver toxicity mediated by PPAR alpha. Toxicol Sci 101:132–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsuga S, Suomalainen A. (2012) Effect of bezafibrate treatment on late-onset mitochondrial myopathy in mice. Hum Mol Genet 21:526–535. [DOI] [PubMed] [Google Scholar]
- Yoshino J, Conte C, Fontana L, Mittendorfer B, Imai S, Schechtman KB, Gu C, Kunz I, Rossi Fanelli F, Patterson BW, et al. (2012) Resveratrol supplementation does not improve metabolic function in nonobese women with normal glucose tolerance. Cell Metab 16:658–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young LH, Li J, Baron SJ, Russell RR. (2005) AMP-activated protein kinase: a key stress signaling pathway in the heart. Trends Cardiovasc Med 15:110–118. [DOI] [PubMed] [Google Scholar]
- Yu HY, Cai YB, Liu Z. (2015) Activation of AMPK improves lipopolysaccharide-induced dysfunction of the blood-brain barrier in mice. Brain Inj 29:777–784. [DOI] [PubMed] [Google Scholar]
- Yue TL, Bao W, Gu JL, Cui J, Tao L, Ma XL, Ohlstein EH, Jucker BM. (2005) Rosiglitazone treatment in Zucker diabetic Fatty rats is associated with ameliorated cardiac insulin resistance and protection from ischemia/reperfusion-induced myocardial injury. Diabetes 54:554–562. [DOI] [PubMed] [Google Scholar]
- Zhang D, Liu ZX, Choi CS, Tian L, Kibbey R, Dong J, Cline GW, Wood PA, Shulman GI. (2007) Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc Natl Acad Sci USA 104:17075–17080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhi L, Moore PK, Bhatia M. (2006) Role of hydrogen sulfide in cecal ligation and puncture-induced sepsis in the mouse. Am J Physiol Lung Cell Mol Physiol 290:L1193–L1201. [DOI] [PubMed] [Google Scholar]
- Zhang L, Keung W, Samokhvalov V, Wang W, Lopaschuk GD. (2010) Role of fatty acid uptake and fatty acid beta-oxidation in mediating insulin resistance in heart and skeletal muscle. Biochim Biophys Acta 1801:1–22. [DOI] [PubMed] [Google Scholar]
- Zhang N, Wang S, Li Y, Che L, Zhao Q. (2013) A selective inhibitor of Drp1, mdivi-1, acts against cerebral ischemia/reperfusion injury via an anti-apoptotic pathway in rats. Neurosci Lett 535:104–109. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Talalay P, Cho CG, Posner GH. (1992) A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci USA 89:2399–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108:1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J. (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469:221–225. [DOI] [PubMed] [Google Scholar]
- Zhu L, Liu Z, Feng Z, Hao J, Shen W, Li X, Sun L, Sharman E, Wang Y, Wertz K, et al. (2010) Hydroxytyrosol protects against oxidative damage by simultaneous activation of mitochondrial biogenesis and phase II detoxifying enzyme systems in retinal pigment epithelial cells. J Nutr Biochem 21:1089–1098. [DOI] [PubMed] [Google Scholar]
- Zobi F, Quaroni L, Santoro G, Zlateva T, Blacque O, Sarafimov B, Schaub MC, Bogdanova AY. (2013) Live-fibroblast IR imaging of a cytoprotective PhotoCORM Activated with Visible Light. J Med Chem 56:6719–6731. [DOI] [PubMed] [Google Scholar]
- Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI. (2002) AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci USA 99:15983–15987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zschiedrich I, Hardeland U, Krones-Herzig A, Berriel Diaz M, Vegiopoulos A, Müggenburg J, Sombroek D, Hofmann TG, Zawatzky R, Yu X, et al. (2008) Coactivator function of RIP140 for NFkappaB/RelA-dependent cytokine gene expression. Blood 112:264–276. [DOI] [PubMed] [Google Scholar]
- Züchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, et al. (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet 36:449–451. [DOI] [PubMed] [Google Scholar]