

Abstract
Sepsis and septic shock are global healthcare problems associated with mortality rates of up to 40% despite optimal standard-of-care therapy and constitute the primary cause of death in intensive care units worldwide. Circulating biomarkers of septic shock severity may represent a clinically relevant approach to individualize those patients at risk for worse outcomes early in the course of the disease, which may facilitate early and more precise interventions to improve the clinical course. However, currently used septic shock biomarkers, including lactate, may be non-specific and have variable impact on prognosis and/or disease management. Activation of the renin-angiotensin-aldosterone system (RAAS) is likely an early event in septic shock, and studies suggest that an elevated level of renin, the early and committed step in the RAAS cascade, is a better predictor of worse outcomes in septic shock, including mortality, than the current standard-of-care measure of lactate. Despite a robust increase in renin, other elements of the RAAS, including endogenous levels of Ang II, may fail to sufficiently increase to maintain blood pressure, tissue perfusion, and protective immune responses in septic shock patients. We review the current clinical literature regarding the dysfunction of the RAAS in septic shock and potential therapeutic approaches to improve clinical outcomes.
1. Introduction
Sepsis and septic shock are global healthcare problems associated with 20% of all global deaths every year despite optimal therapy and are a leading cause of death in intensive care units (ICU) worldwide. The identification of circulating biomarkers of septic shock severity may constitute a clinically relevant approach to individualize those patients at risk for worse outcomes early in the course of the disease, which may facilitate early and more precise interventions to improve the clinical course. However, currently used septic shock biomarkers, including lactate, may be non-specific and have variable impact on prognosis and/or disease management. The renin-angiotensin-aldosterone system (RAAS) is an endocrine system essential for the regulation of blood pressure and fluid homeostasis. The RAAS influences various target organs and cell systems, including the vasculature, kidney, heart, brain, adrenals, and immune cells [1–4]. The activation of the RAAS is likely an early event in septic shock, and recent data reveal that elevated levels of renin, the early and committed step in the activation of the RAAS cascade, are a better predictor of worse outcomes in septic shock than lactate, as well as a more relevant biomarker for mortality [5–9]. However, despite a robust increase in renin, the response of other elements of the RAAS, particularly the ACE-Ang II-AT1 receptor (AT1R) axis, may be insufficient to maintain blood pressure, tissue perfusion or to stimulate the innate immune system in septic shock patients, which overall may contribute to disease severity and increased mortality. In the present review, we critically examine the clinical literature regarding the possible dysfunction of the RAAS in septic shock as an underlying mechanism that may exacerbate the course of the disease syndrome. We further propose a hypothesis for future investigations that will allow for further evolution of therapeutic targets in this area.
2. Renin-Angiotensin-Aldosterone System (RAAS)
Classical RAAS pathway.
The RAAS is an endocrine system that evolved to maintain blood pressure and tissue perfusion primarily through the generation in the circulation of the octapeptide Angiotensin II (Ang II) by the dipeptidyl carboxypeptidase ACE. Ang II then activates the G-protein coupled receptor AT1R that constitutes the functional arm of the classical RAAS axis (Figure 1) [1–4]. Key downstream effectors of the ACE-Ang II-AT1R axis include the mineralocorticoid aldosterone, the potent vasoconstrictor peptide endothelin and a myriad of AT1R-dependent signaling pathways that promote vasoconstriction, sympathetic activation, an increase in oxidative stress and stimulation of immune cells (Figure 1) [1–4]. Angiotensinogen is the sole precursor to Ang II, and circulating levels of the protein are in the high nanomolar range (~200 nM) [3,10]. The stimulated renal release of the aspartyl protease renin generates Ang I in blood, which is rapidly converted to Ang II by highly abundant levels of ACE, particularly in the pulmonary endothelium (Figure 1). Although the plasma levels of angiotensinogen are approximately 10,000-fold higher than that of Ang II (~20 picomolar, pM), the concentration of angiotensinogen approaches the KD for renin (600 nM) such that alterations in renin or the angiotensinogen substrate directly influence the generation of Ang I and subsequent conversion to Ang II in humans [2,3,10]. Indeed, circulating levels of angiotensinogen positively correlate with blood pressure and a greater prevalence of hypertension; novel anti-hypertensive therapies that chronically reduce angiotensinogen expression are currently in clinical trials [10]. The sole enzymatic function of renin is the hydrolysis of angiotensinogen directly to Ang I; however, circulating renin and its inactive precursor prorenin may bind to the (pro)renin receptor (PRR) to elicit inflammatory and pro-fibrotic actions mediated by MAP kinase activation [11]. Binding of prorenin to the PRR also induces a conformational change in the precursor that fully activates the enzyme without the requisite hydrolysis of the N-terminal handle peptide that resides in the active site of prorenin [11]. A controversial aspect of the PRR is that it functions not only as a receptor for (pro)renin, but as an accessory protein (ATPase accessory protein 2, ATP6ap2) for the vacuolar ATPase H+ pump complex (V-ATPase), which is essential for lysosomal acidification and the maintenance of cellular pH. Moreover, the PRR and V-ATPase were shown to be essential for β-catenin/Wnt signaling, a key cellular pathway involved in the regulation of the RAAS [12,13]. There is also evidence for a secreted form of the PRR, termed the soluble PRR (sPRR), which retains the capacity to bind and activate prorenin, as well as exhibits additional cellular actions that may reflect the interaction of (pro)renin-sPRR with other cellular partners [14].
Figure 1. Proposed activation of the Renin-Angiotensin-Aldosterone System pathway.
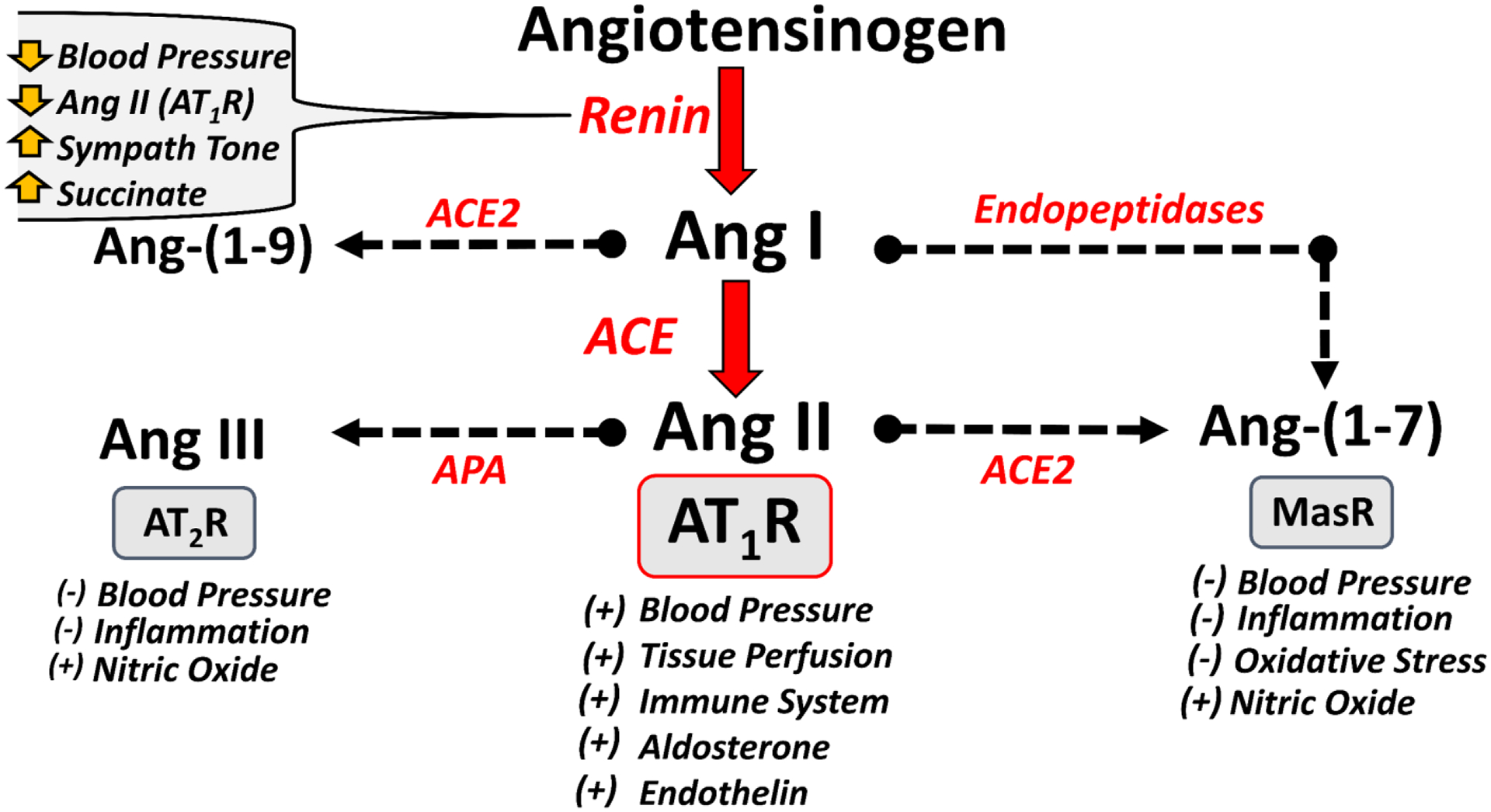
Activation of the renin-angiotensin-aldosterone system (RAAS) occurs by an increase in circulating renin. Angiotensinogen is cleaved by renin to generate angiotensin I (Ang I), which is rapidly hydrolyzed by ACE to Ang II. Ang II binds to the AT1 receptor (AT1R) to increase blood pressure and tissue perfusion, stimulate aldosterone and endothelin, as well as activate the immune system. Ang II is metabolized to Ang-(1–7) by ACE2 that stimulates the MasR to reduce blood pressure by releasing nitric oxide and reducing oxidative stress and inflammation. Ang I undergoes processing by endopeptidases (neprilysin, prolyl endopeptidase, thimet oligopeptidase) to Ang-(1–7). Ang II also undergoes N-terminal metabolism by aminopeptidase A (APA) to Ang III that can activate the AT2R to reduce blood pressure through nitric oxide and reducing inflammation. Various stimuli including reduced blood pressure, lower Ang II-AT1R but high sympathetic tone and increased metabolic levels of succinate stimulate the renal release of renin to increase Ang II. Although not shown, Ang II is also metabolized by dipeptidyl peptidase 3 (DPP3) to Ang-(3–8) and then Ang-(5–8) while Ang I is processed to Ang-(1–9) by ACE2 and Ang-(1–7) is metabolized to Ang-(1–5) by ACE. Symbols (+) and (−) connotate increase and decrease, respectively. Adapted from Chappell [84].
Counter-regulatory RAAS pathway:
Distinct from the classical RAAS, numerous studies over the past 30 years have revealed an alternative or non-classical axis of the RAAS that may oppose or antagonize the actions of the classical axis [15–18]. A key component of the alternative axis is the heptapeptide Ang-(1–7) that differs from Ang II by deletion of a single amino acid (Phe at the 8th position) and which binds to a different class of receptors, termed the Mas or Mas-related receptors (Figure 1) [15]. A homolog of ACE, termed ACE2, exhibits high but not exclusive specificity to hydrolyze Ang II to Ang-(1–7) [3,18]. Therefore, ACE2 may directly influence the balance of endogenous Ang II and Ang-(1–7) levels in the circulation and various tissues by a single catalytic step (Figure 1). In addition, other enzymes termed endopeptidases (neprilysin, prolyl endopeptidase and thimet oligopeptidase) cleave the Pro7-Phe8 bond of Ang I directly to Ang-(1–7); this derivation of Ang-(1–7) may predominate under conditions of ACE inhibition that divert the processing of Ang I to Ang-(1–7), as well as attenuate the metabolism of Ang-(1–7) to Ang-(1–5) [3,15,16]. Additional metabolism occurs by the N-terminal processing of Ang II through aminopeptidases (aspartyl aminopeptidase, APA), generating Ang-(2–8) or Ang III, which may stimulate the AT2R, resulting in antagonism of the actions of the Ang II-AT1R axis (Figure 1) [17]. Moreover, the C-terminal processing of Ang I to Ang-(1–9) by ACE2 potentially generates another AT2R agonist, although Ang-(1–9) may also serve as a substrate for Ang-(1–7) [17,18] (Figure 1).
In addition to the endocrine or circulating RAAS, there is evidence that local or tissue-based systems generate/secrete Ang II or Ang-(1–7) and express associated Ang receptors that may convey autocrine or paracrine effects [3,16]. Key evidence in support of various tissue RAAS were the demonstration of mRNA levels for angiotensinogen and renin in tissues distinct from the liver and kidney, which were historically considered the primary if not the sole tissue sources of angiotensinogen and renin, respectively. Recent studies, however, suggest that circulating liver-derived angiotensinogen is taken up in multiple tissues and may constitute the primary source for both angiotensinogen and Ang II in these tissues [19,20]. Matsusaka et al [19] initially demonstrated that knockdown of liver angiotensinogen markedly reduced the renal content of both angiotensinogen and Ang II, while deletion of renal angiotensinogen reduced urinary levels of the protein that likely reflect the tubular synthesis/secretion of the precursor protein. Kukida and colleagues [20] confirmed these findings for liver-derived angiotensinogen as the primary source of renal angiotensinogen in a non-human primate model. Moreover, we find that renal proximal tubules take up angiotensinogen that distributes to the nucleus and mitochondria, which may provide a source of Ang II or Ang-(1–7) ligands for their intracellular receptors [21]. Finally, the role of kidney-derived renin versus the contribution of local renin systems to the generation of Ang II remains an area of debate. Similar to the tissue uptake of circulating angiotensinogen, there is evidence for the internalization of renin through a non-PRR pathway that may contribute to the intracellular generation of Ang II and Ang-(1–7) [2,3].
3. RAAS and Immune Responses
In addition to maintaining blood pressure and tissue perfusion, the RAAS contributes in diverse ways to immune responses and inflammation. The widespread administration of renin-angiotensin blockers for the treatment of hypertension has revealed effects of the RAAS on the production of reactive oxygen species (ROS), tissue remodeling, fibrosis, and benefits in solid organ cancers [1–4].
The physiological response to severe infection includes bone marrow myelopoiesis, leading to increases in circulating neutrophils and monocytes that are then available to phagocytose and kill pathogens in infected tissue. Multiple components of the RAAS are widely expressed on bone marrow progenitor cells [22,23]. Under physiological conditions, Ang II promotes myeloid proliferation, differentiation, and maturation by signaling through the AT1R [24]. In addition to promoting myelopoiesis, the RAAS contributes to the optimal function of immune cells. Administration of ACE inhibitors or AT1R antagonists reduce infiltration of immune cells in experimental animal models of renal injury. In a rat model of interstitial nephritis, infiltration of macrophages in response to renal injury depends on Ang II-dependent induction of the chemoattractant CCL2 via the NF-κB pathway [24]. In mice, the production of antibacterial ROS and neutrophil extracellular traps and the release of interleukin-1β (IL-1β) in response to infection depend on neutrophil ACE [25,26]. Ang II is required for myeloid cell lineage ROS, chemotaxis, recruitment of monocytes and neutrophils to vascular endothelium in response to injury, and phagocytosis and for tissue inflammation, granulation, and healing. Many of these Ang II effects occur via signaling through the AT1R [27–30]. The physiological actions of Ang II mediated through AT1R are generally opposed by Ang II activation of AT2R or MasR, the latter of which is thought to impact AT1R signaling by complexing with the AT1R to form Mas-AT1R heterodimers [15–18, 31]. Inhibition of renin by the specific agent aliskerin in septic rats was associated with increased ROS buffering (increased superoxide dismutase and glutathione) and decreased levels of the proinflammatory cytokines TNF-α, IL-1β, and IL-6 in lung tissue, indicating that the direct effects of Ang II on ROS, chemotaxis, and inflammation [32]. However, the cytokine responses to Ang II stimulation of AT1R on certain non-myeloid cells differs from that on myeloid cells. For example, disruption of AT1R receptors on T lymphocytes exacerbates TNF-α production and renal injury during hypertension or after cisplatin treatment, whereas disruption of AT1R receptors on kidney epithelium ameliorates these phenotypes [33,34].
4. RAAS and Septic Shock
Multiple stimuli in septic shock likely enhance circulating levels of active renin, including a reduction in arterial blood pressure and renal perfusion, a marked increase in sympathetic stimulation of β−1 receptors on renal juxtaglomerular (JG) cells and elevated levels of the metabolite succinate (Figure 1) [3]. The buffering or negative feedback to elevated renin is the expected increase in circulating Ang II that activates AT1Rs on JG cells to reduce cAMP levels and subsequently attenuates renin secretion/synthesis (Figure 1). However, the failure to stimulate a significant Ang II response may attenuate activation of the short feedback loop and amplify the overall renal renin response in septic shock.
Our recent study characterizing the response of the circulating RAAS in septic shock patients within 24 hours of admission revealed an exceptionally wide range of renin values [5]. As shown in Figure 2A, circulating renin content in septic shock subjects at baseline varied over 1,000-fold from 0.065 pM to 70 pM, which reflects the remarkable capability of the kidney to synthesize renin and activate the RAAS [5]. Importantly, renin values above the median value of this patient cohort of 5.1 pM (Quartiles 3–4) were strongly associated with increasing mortality within 30 days of admission and exhibited a linear response of renin to mortality (Figure 2B) [5]. Moroever, patients with higher renin had fewer ventilator- and vasopressor-free days (VVFD) over 30 days in this cohort (median [interquartile range] VVFD for high renin: 23 [1–29] vs. lower renin: 29 [23–30]; p=0.025) [5]. The high renin values associated with increase mortality may reflect a desperate attempt to maintain blood pressure and adequate renal perfusion in this subset of patients. However, despite the marked rise in active renin, there appeared to be a blunted Ang II response that was not proportionally commensurate with the increase in renin in most of the high renin subjects (Figure 3A) [5]. Moreover, the ratio of plasma Ang II to Ang-(1–7) declined in the high renin groups, particularly in the comparison of the peptide ratios between Q1 and Q4 quartiles (Figure 3B). The lower ratio of Ang II to Ang-(1–7) could reflect alterations in the overall processing of Ang II and Ang-(1–7). Indeed, the shift in the balance of the vasoconstrictor Ang II to the vasodilator peptide Ang-(1–7) may not be beneficial in maintaining adequate blood pressure and tissue perfusion in septic shock since Ang-(1–7) induces vasorelaxation through nitric oxide (NO) release [15].
Figure 2. Active renin content and renin quartiles in septic shock patients.

Left panel: Variable levels of active renin in the circulation of septic shock patients ranged over a 1,000-fold (N=97 subjects). Dotted line is detection limit of the renin assay at 0.065 pM. Right panel: Expression of median renin values by quartiles Q1 (N=25), Q2 (N=25), Q3 (N=23) and Q4 (24) that range over 60-fold. Hashed line is the median renin value of 5.1 pM for entire cohort. Data are adapted from Busse et al [5].
Figure 3. Angiotensin II versus Renin values and Angiotensin II to Angiotensin-(1–7) ratios in septic shock.

Left panel: Circulating Angiotensin II (Ang II) did not associate with renin values in septic shock patients (N=94). Shaded area is the expected range of Ang II from 5 to 50 pM in healthy control subjects. Dotted line is the detection limit of the Ang II assay (2.0 pM) and hashed line is median renin value of 5.1 pM. Right panel: Plasma Ang II to Ang-(1–7) (Ang 7) ratios (median values, 10th-90th range) show a significant decline between renin quartiles Q1 and Q4 in septic shock subjects. Peptide ratios for renin quartiles Q1 (N=23), Q2 (N=22), Q3 (N=21) and Q4 (N=24) were analyzed by one-way ANOVA with Kruskal-Wallis post-test, **P=0.026. Data adapted from Busse et al [5].
Although both Ang II ang Ang-(1–7) levels were assessed in the VICTAS cohort, we did not determine serum levels of aldosterone, a key target of the Ang II-AT1R pathway to regulate sodium and fluid balance [1–4, 35]. Studies have suggested a decoupling between elevated renin and inappropriately low levels of aldosterone termed hyperreninemic hypoaldosteronism in a subset of critically ill patients that is typically defined as a plasma aldosterone to plasma renin activity ratio (PA/PRA) less than 2 [35–38]. A blunted aldosterone response may reflect the reduced generation of Ang II or the downregulation of adrenal AT1Rs, as well as tissue injury of the adrenal gland that may impair aldosterone synthesis. However, there are concerns with immunological approaches to accurately quantify circulating levels of aldosterone, as well as diurnal fluctuations of the steroid that may change up to 2-fold requiring a similar time period for collection of patent samples [35,39].
Our findings that septic shock patients with high circulating renin are at greater risk of poor clinical outcomes including death and fewer VVFD support previous studies in pediatric septic shock [45], severe sepsis [6,36], adult septic shock [7], catecholamine-resistant vasodilatory shock [8], and a heterogeneous group of critically ill patients [9, 41,44]. Similar to the renin values reported in our study [5], Bellomo et al [8] determined a median renin value of 4.7 pM (173 pg/mL) in their patient cohort with vasodilatory shock and renin values above the median were also strongly predictive of increased risk. Jeyaraju et al [7] reported a baseline median renin value of 4.3 pM (160 pg/mL) as a predictor for in-hospital mortality of septic shock patients and that renin levels determined over 72 hours were a stronger predictor of mortality than serum lactate. Gleason et al [9] also demonstrated that assessment of renin was a better predictor of ICU mortality than lactate. These studies all utilized immunoassays (ELISAs) from either DG International (Springfield, NJ USA) [5,7,8] or DiaSorin (Vercelli, Italy) [9] to quantify renin protein levels rather than the traditional measurement of PRA. Longstanding concerns with PRA are that the assay is dependent on endogenous intact angiotensinogen as the renin substrate, which may vary in critically ill patients, the additional steps to generate and quantify Ang I typically by immunoassays and the time required for this multi-stage assay [3]. Moreover, other renin ELISAs may recognize prorenin, which is catalytically inactive, circulates at levels up to 10-fold higher than renin, and differs from renin regarding their temporal regulation; these features could preclude the establishment of a strong association between PRA or PA/PRA and disease severity.
We note that the wide distribution of renin values in the VICTAS study was not due to potential alterations in the angiotensinogen substrate as renin protein levels were quantified rather than renin activity. The direct renin assay allows for a wider detection range of renin than other assays, which may facilitate the identification of distinct patient subgroups based on the renin response, although factor(s) contributing to these differing responses are not well-defined. It should be emphasized that improper sample handling in the clinic or ICU may be an issue as renin can be cyro-activated by the thawing of samples on ice or prolonged storage at 4°C that result in a conformational change in the handle region of renin that normally blocks the active of the enzyme [2,3]. Use of anti-hypertensive agents including ACE inhibitors (ACEIs) and AT1R antagonists (ARBs) also increase renin levels, although ACEIs would lower Ang II while ARBS have either no effect or may increase Ang II levels [3]. The abrupt discontinuation and clearance of ACEIs, however, may transiently increase Ang II due to the prior accumulation of circulating Ang [3]. Moreover, the onset of septic shock and hospital admittance varies among these patients. Caveats to our characterization of the RAAS components in the VICTAS study are that patient data on the use and type of anti-hypertensive therapies, as well as time of disease onset and discontinuation of anti-hypertensive medications are not known. The Bellomo study [8] also demonstrated the correlation of a reduced ratio of Ang II to Ang I to disease severity, which may confirm the higher renin levels observed, as well as potentially a blunted Ang II response that would contribute to the high renin response. Finally, Zhang et al [44] documented that lower plasma Ang II levels and reduced ACE activity were associated with increased mortality in patients with severe sepsis.
5. Septic Shock and Acute Kidney Injury
A major complication of septic shock is the increased risk of acute kidney injury (AKI), a pathology that reflects a reduction in glomerular filtration rate (GFR) and renal perfusion as well as other deleterious factors [40–41]. AKI is associated with a markedly higher rate of mortality in critically ill patients, particularly those on renal replacement therapy [40]. Reduced perfusion pressure may activate renal mechanisms to stimulate renin release and increased renin levels may be predictive of AKI [9, 42–46]. Flannery and colleagues [46] reported that patients with a renin value greater than 1.1 pM were at greater risk of adverse renal events (mortality, renal replacement therapy and reduced GFR) as compared to those with a lower renin content (< 0.2 pM). These investigators also noted that the high renin response was not correlated to blood pressure that again suggest other factors influence renin expression such as adrenergic tone, succinate levels and tissue perfusion [46]. An attenuated Ang II response to elevated renin may contribute to the development of AKI in septic shock, which may also reflect a reduction in either renal AT1R expression or receptor-coupled signaling. Flannery et al [47] find that patients with premorbid use of ACEIs or ARBs had more severe AKI than those not on RAS blockers, but there were no overall differences in renal function or mortality at the time of hospital discharge. Chou and colleagues [48] report that the use of RAS blockers was associated with a higher risk of AKI in septic patients. In a metanalysis of 15 clinical studies (>95,000 patients), Hasegawa et al [49] also find that premorbid RAS inhibition was associated with a higher risk of AKI but lower short-term mortality while other studies suggest that premorbid RAS inhibition may reduce the risk or severity of AKI in septic shock [50] Conversely, therapeutic treatment with Ang II may be beneficial in AKI by increasing glomerular pressure to promote glomerular filtration and renal perfusion thus improving renal outcomes. In a post-hoc analysis of the ATHOS3 trial, patients who received Ang II and had acute kidney injury requiring renal replacement therapy experienced a 28-day survival and mean arterial pressure response that was better, and rate of renal replacement therapy liberation that was greater compared to the group that did not receive the study drug [52].
Preclinical studies have revealed that treatment with exogenous Ang-(1–7) improves renal outcomes in septic shock. Garica et al [53] demonstrate that Ang-(1–7) administration increased creatinine clearance and reduced renal inflammation but this treatment was not associated with a significant reduction in blood pressure or change in urine volume in a sheep model of sepsis. Zhu et al [54] also reported that Ang-(1–7) attenuated AKI in a LPS mouse model that associated with a reduction in renal NF-kB activation. In both studies, treatment with Ang-(1–7) began prior to the initiation of sepsis, which clearly differs from the expected time course for treatment of patients in the later periods of sepsis. Moroever, RAS blockade with ACE inhibitors or AT1R antagonists may have variable effects on endogenous levels (circulation and tissue) of Ang-(1–7) which could contribute to their overall actions regarding the renal response to septic shock [3].
6. Mechanisms for RAAS Dysfunction in Septic Shock
The underlying mechanism for RAAS dysfunction in septic shock patients may result from multiple factors that influence the synthesis and metabolism of Ang II, as well as the Ang II-AT1R response (Figure 4). It is well-documented that in septic shock, pulmonary and circulating ACE activities are reduced, which could contribute to an attenuated conversion of Ang I to Ang II [44–46, 55, 56]. Orfanose and colleagues [55] demonstrated reduced ACE activity across the lung in critically ill patients suggesting that lower expression of endothelial ACE due to vascular injury. Pode-Shakked et al [45] recently reported that most patients (69%) in their pediatric septic shock cohort had no detectable circulating ACE activity despite elevated protein levels of the peptidase, which suggests the presence of an endogenous peptidase inhibitor. The reduced expression of ACE due to vascular endothelial injury or inhibition of ACE activity from endogenous substances may also lead to higher levels of Ang-(1–7) by diverting Ang I to endopeptidase processing, as well as decreasing the metabolism of Ang-(1–7) by ACE (Figure 1) [3]. Increased levels of dipeptidyl aminopeptidase 3 (DAP3 or DPP3), an intracellular peptidase released into the circulation, is also associated with septic shock and may contribute to Ang II metabolism [57]. DPP3 hydrolyzes Ang II to Ang-(3–8) also known as Ang IV and then rapidly generates Ang-(5–8) given the enzyme’s preference for shorter peptides [57,58]. Moreover, treatment with the DPP3 antibody Procizumab that blocked catalytic activity of the peptidase, enhanced cardiac function in sepsis-induced heart failure [59]. Although ACE activity is reduced in septic shock, a potential increase in ACE2 would also contribute to a reduced ratio of Ang II to Ang-(1–7), and potentially higher levels of Ang-(1–9) (Figure 1).
Figure 4: Potential mechanisms that may contribute to an attenuated Angiotensin II response in septic shock patients.

In septic shock with high renin, lower expression of ACE combined with potentially higher levels of metabolizing enzymes (ACE2, DPP3) may reduce circulating levels of Ang II. The reduced expression of angiotensinogen (Aogen) with increased consumption of intact angiotensinogen by high renin levels, as well as the potential oligomerization of the precursor protein may also contribute to an attenuated Ang II response. Septic shock may reduce expression of AT1 receptors (AT1R) and/or associated AT1R signaling pathways to reduce Ang II tone. The potential expression of Ang II autoantibodies (AutoAbs) that bind and sequester circulating Ang II may further attenuate the Ang II response in septic shock. The overall loss of Ang II tone in septic shock would reduce the feedback inhibition of renin by Ang II leading to the increased release of renin. Symbols (+) and (−) connotate increase and decrease, respectively.
In addition to the reduced generation of Ang II coupled to increased metabolism of the peptide, it is possible that a lower capacity to maintain circulating levels of angiotensinogen may contribute to the attenuated Ang II response. The liver is particularly susceptible to septic shock in lieu of its role in bacterial clearance and regulation of innate immune responses, as well as receiving 25% of total cardiac output [60,61]. Reduced tissue perfusion and potentially other factors in septic shock may impair the capacity of hepatocytes to meet the demand for sufficient levels of angiotensinogen in the circulation and tissues. Moreover, the reduced synthesis and/or release of angiotensinogen in septic shock may be compounded by the high levels of renin that essentially deplete the Ang I-intact forms of angiotensinogen. In this regard, Corvol and colleagues [62] demonstrated that high circulating renin was associated with reduced levels of intact angiotensinogen and lower serum levels of retinol binding hormone 4 (RBH4) and prealbumin in patients with severe heart failure and liver dysfunction. Additionally, there is evidence that circulating angiotensinogen may homodimerize or heterodimerize with other proteins to form high molecular weight oligomers under conditions of increased oxidative stress [63,64]. Oligomeric forms of angiotensinogen are poor substrates for renin likely due to steric hinderance of the renin-binding site on angiotensinogen; the potential formation of these complexes could contribute to a reduced Ang II response in septic shock [64]. The oligomerization of circulating angiotensinogen may also interfere with ELISA-based detection of the protein that are likely dependent on the angiotensinogen epitopes recognized by the assay antibodies. Although the presence of autoantibodies to Ang II has not been determined in septic shock, Briquez et al [65] reported circulating Ang II antibodies capable of sequestering Ang II in COVID-19 that may contribute to lower blood pressure in these patients. The potential generation of autoantibodies in septic shock that bind Ang II may prevent AT1R activation and downstream signaling actions of Ang II, including the feedback inhibition of renin.
Finally, downregulation of the renal AT1R, as well as attenuated AT1R signaling is evident in a murine model of sepsis [66,67]. We found that treatment of septic mice with Ang II rescued myeloid immune dysfunction, modulating the inflammatory response, enhancing bacterial clearance, and rescuing sepsis-associated decreases in monocyte phagocytosis and ROS production [68]. These effects of Ang II on myeloid function were mediated through AT1R on myeloid lineage cells, suggesting that Ang II enhanced immune responses in a specific manner [68].
The reduced AT1R responsiveness in target organs may further exacerbate conditions of inappropriately low levels of circulating Ang II in septic shock (Figure 4). In contrast to the inhibition of renal renin, Ang II functions in a positive feedback manner to stimulate angiotensinogen synthesis and release [69–71]. Thus, an attenuated ACE-Ang II-AT1R axis may further impact circulating levels of angiotensinogen in septic shock patients. Finally, hepatocytes also express a functional ACE2-Ang-(1–7)-MasR axis, as well as the PRR, which may portend for the direct regulation of liver-derived angiotensinogen by Ang-(1–7) and/or (pro)renin [72–74]. Additional studies are required to address the specific effects of Ang-(1–7) and PRR activation on the synthesis and release of angiotensinogen by hepatocytes.
7. Therapeutic approaches
Following the progress in understanding the behavior of RAAS pathways in human septic shock, the most important and obvious next step is appropriate therapeutic interventions and targeted choice of drugs that would correct the dysfunctional RAAS and possibly lead to improved clinical outcomes. Khanna and colleagues [75] demonstrated a blood pressure benefit with the use of exogenous Ang II in high output shock, and those receiving Ang II had a significant reduction in serum renin in the first three hours where the intervention or placebo were rapidly up titrated to achieve a blood pressure response. The post hoc analysis of the Khanna study by Tumlin et al [52] revealed that a higher Ang I/Ang II ratio, indicative of high renin in these patients, was associated with greater norepinephrine requirements to maintain blood pressure and is an independent predictor of mortality. Furthermore, Bellomo and colleagues [8] find that patients with a serum renin greater than population median for the Angiotensin II in High Output Shock (ATHOS3) study population and randomized to Ang II treatment had a significant survival benefit compared to the use of placebo in the high renin group. Moreover, in the subset of septic shock patients that responded clinically and with a blood pressure response to exogenous Ang II, there was a significant reduction in renin levels [8]. This reset of the increased renin would portend a better prognosis as evident in our post-hoc analysis of the Vitamin C, Thiamine, Steroids in Sepsis (VICTAS) clinical trial data where a decreasing trend in renin was associated with a survival benefit compared with an increase in renin over time [5]. An essential corollary would be a modification of the decision tree for early vasopressor choices at the bedside in septic shock and the possible use of exogenous Ang II as a primary vasopressor in the setting of high renin septic shock [75–77]. This would, however, necessitate the ubiquitous availability of a well-validated, accurate, and rapid point of care test for active circulating renin or a renin-peptide ratio that would facilitate evidence-based vasopressor choices.
There remain, however, several unanswered questions regarding the use of Ang II as a vasopressor treatment. Firstly, what is the role of a low endogenous Ang II and high renin state compared with high endogenous Ang-(1–7) and other metabolites of the non-classical pathway and how does the use of synthetic Ang II impact the non-classical pathway? Pre-clinical studies in septic shock suggest that modulation of the alternative RAAS, particularly treatment with Ang-(1–7) or ACE2 has beneficial effects including increased survival [78–82]. However, rodent septic shock models may not parallel the responses in patients, and a large clinical trial for Ang-(1–7) treatment of COVID-19 patients showed no overall benefit (reduced mortality, ventilator free days) as compared to the placebo group [83,84]. One issue with Ang-(1–7) administration is the very short half-life of the peptide as it is rapidly metabolized by ACE and achieving adequate therapeutic levels of the peptide is challenging [3,15].
Secondly, does the correction of blood pressure in of itself restore the dysfunctional RAAS or does synthetic Ang II play a more primary and specific role effect? Furthermore, is the high renin signal of mortality and survival in case of Ang II administration to these patients confounded by unaccounted confounders? An obvious next step would be trials with a high renin septic shock population (predictive enrichment) where the effect of Ang II would be compared with standard of care vasopressors and followed with timed biomarkers. Thirdly, if the metabolism of Ang II is indeed elevated in septic shock patients (by ACE2, DPP3, APA or other peptidases), should an Ang II treatment regimen constitute more stable Ang II analogs that exhibit greater affinity and selectivity to the AT1R and prevent activation of MasR- or AT2R-dependent pathways? An important area of potential concern is the effect of Ang II on organ remodeling and induction of fibrotic changes. However, most data comes from a variety of animal models where Ang II was administered in prolonged infusions (2– 4 weeks) and typically at doses that far exceed endogenous peptide levels that stimulate a marked increase in blood pressure [85]. Moreover, there is debate on the tissue response to Ang II regarding whether the peptide is given directly into the circulation or applied subcutaneously, and we do not have adequate long term follow-up data in human clinical exposure to this agent [85]. Clearly, the duration of exposure may be an important protective factor since most clinical use of Ang II is in brief 24 to 48-hour infusions for the correction of hypotension. Finally, does the accurate assessment of other RAAS markers or their ratios (i.e., Ang II/Ang-(1–7), renin/aldosterone, prorenin/renin, ACE/ACE2) provide a more precise prediction of outcomes in septic shock compared with renin alone (or Ang I/Ang II ratio) and compared with the traditional standard of care bedside measurement of lactate currently done world-wide? Specifically, knowing the state of relative adrenal exhaustion in septic shock, the dependence of aldosterone secretion on an intact RAAS pathway, and the benefit of glucocorticoids and mineralocorticoids in clinical care of septic shock, the renin/aldosterone test may have a role in decision making for the use of steroids in vasopressor dependent septic shock states. Assessment of relevant RAAS biomarkers for septic shock is also contingent on a rapid and accurate bedside point of care assays for renin and other components that could guide choice and dose range titration of exogenous vasopressors including Ang II.
8. Conclusion
The activation of the RAAS in septic shock is the expected response of this powerful endocrine system to maintain blood pressure and adequate tissue perfusion; however, the ACE-Ang II-AT1R axis of the RAAS may be functionally attenuated through various mechanisms in a subset of patients that contributes to disease severity and mortality. Indeed, there may also be an imbalance in the RAAS favoring the ACE2-Ang-(1–7)-MasR pathway that further contributes to the vasodilatory response in septic shock. Rationale for the therapeutic use of exogenous Ang II in septic shock is compelling, particularly during the acute phase given the numerous pathways stimulated by Ang II to promote hemodynamic stability, as well as augment cardiac, renal, and immune function. Further investigation is clearly needed to identify biomarkers that most accurately reflect the health and function of the RAAS in septic shock and those that might optimally guide the use of therapeutic Ang II to restore RAAS function.
Highlights.
Review summarizes clinical data on the possible dysfunction of the renin-angiotensin-aldosterone system (RAAS) in septic shock.
Review discusses the potential influence of reduced ACE and angiotensinogen but increased ACE2 and DPP3 to attenuate Ang II response.
Review discusses potential therapeutic approaches involving both Ang II and Ang-(1–7) in septic shock patients.
Reviews discusses the role of reduced or attenuated aldosterone response in high renin septic shock or hyperreninemic hypoaldosteronism.
ACKNOWLEDGEMENTS
Support for this study was provided by National Institutes of Health (NIH) grants R01HL14877 (MCC), K01AG073581 (CLS) and R01AI153142 (MBG, MRF), American Heart Association (AHA) grant TPA34170522 (MCC), WFUSM Cardiovascular Science Center (CVSC) pilot awards (AKK and MCC), funding from Innoviva (DEL, MBG) and funding from La Jolla Pharmaceuticals for the Angiotensin II in High Output Shock trial (AKK). We acknowledge additional funding to the Hypertension Center investigators from the Groskert Heart Fund, the Wake Forest Venture Fund and the Farley-Hudson Foundation (Jacksonville, NC, USA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors declare that there are no competing financial interests in the work described.
Literature Cited
- 1.Forrester SJ, Booz GW, Sigmund CD, Coffman TM, Kawai T, Rizzo V, Scalia R, Eguchi S. Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol Rev 98 (2018) 1627–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Te RL, van Esch JH, Roks AJ, van den Meiracke AH, Danser AH. Hypertension: renin-angiotensin-aldosterone system alterations. Circ Res 116 (2015) 960–975. [DOI] [PubMed] [Google Scholar]
- 3.Chappell MC. Biochemical evaluation of the renin-angiotensin system – the good, bad and absolute? Am. J. Physiol. Heart. Circ. Physiol 310 (2016) H137–H152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bitker L, Burrell LM. Classic and nonclassic Renin-Angiotensin Systems in the critically ill. Critical Care Clinics 35 (2019) 213–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Busse LW, Schaich CL, Chappell MC McCurdy MT, Staples EM, Teri Lohuis CD, Hinson JK, Severansky JE, Rothman RE, Wright DW, Marin GS, Khanna AK. Association of active renin content with mortality in critically ill patients: A post-hoc analysis of the VICTAS trial. Crit Care Med 50 (2023) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lesnik P, Lysenko L, Korpacka MK, Niesobska EW, Pasierb MM, Janc J. Renin as a marker of tissue perfusion, septic shock and mortality in septic patients. In J Mol Sci 23 (2022) 9133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeyaraju M, McCurdy MT, Levine AR, Devarajan P, Mazzeffi MA, Mullins KE, Reif M, Yim DN, Parrino C, Lankford AS. Renin kinetics are superior to lactate kinetics for predicting in-hospital mortality in hypotensive critically ill patients. Crit Care Med 50 (2022) 50–60. [DOI] [PubMed] [Google Scholar]
- 8.Bellomo R, Forni LG, Busse LW, McCurdy MT, Ham KR, Boldt DW, Hästbacka J, Khanna AK, Albertson TE, Tumlin J, Storey K, Handisides D, Tidmarsh GF, Chawla LS, Ostermann M. Renin and survival in patients given angiotensin ii for catecholamine-resistant vasodilatory shock. A clinical trial. Am J Respir Crit Care Med 202 (2020) 1253–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gleeson PJ, Crippa IA, Mongkolpun W, Cavicchi FZ, Van Meerhaeghe T, Brimioulle S, Taccone FS, Vincent JL, Creteur J. Renin as a marker of tissue-perfusion and prognosis in critically ill patients. Crit Care Med 2019, 47(2):152–158. [DOI] [PubMed] [Google Scholar]
- 10.Kahlon T, Carlisle S, Moscatero DO, Williams N. Trainor P, DeFilippis AP. Angiotensinogen: More than its downstream products: evidence from population studies and novel therapeutics. J Am Coll Cardiol 10 (2022) 699–713. [DOI] [PubMed] [Google Scholar]
- 11.Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. . J. Clinical Invest 109 (2002) 1417–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou L, Li Y, Hao S, Zhou D, Tan RJ, Nie J, Hou FF, Kahn M, Liu Y. Multiple genes of the renin-angiotensin system are novel targets of Wnt/β-catenin signaling. J Am Soc Nephrol. 2015, 26:107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Z, Zhou L, Wang Y, Miao J, Hong X, Hou FF, Liu Y. (Pro)renin receptor Is an amplifier of Wnt/β-Catenin signaling in kidney injury and fibrosis. J Am Soc Nephrol 28 (2017) 2393–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang T. Soluble (Pro)Renin Receptor in hypertension. Nephron 147 (2023) 234–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santos RAS, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M, Campagnole-Santos MJ. The ACE2/Angiotensin-(1–7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1–7). Physiol Rev 98 (2018):505–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chappell MC, Marshall AC, Alzayadneh EM, Shaltout HA, Diz DI. Update on the Angiotensin converting enzyme 2-Angiotensin (1–7)-MAS receptor axis: fetal programing, sex differences, and intracellular pathways. Front Endocrinol (Lausanne) 4 (2014) 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steckelings UM, Widdop RE, Sturrock ED, Lubbe L, Hussain T, Kaschina E, Unger T, Hallberg A, Carey RM, Sumners C. The Angiotensin AT(2) Receptor: from a binding site to a novel therapeutic target. Pharmacol Rev 74 (2022) 1051–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong JC, Turner AJ, Raizada MK, Grant MB, Oudit GY. Angiotensin-Converting Enzyme 2: SARS-CoV-2 receptor and regulator of the Renin-Angiotensin System: celebrating the 20th anniversary of the discovery of ACE2. Circ Res 126 (2020) 1456–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsusaka T, Niimura F, Shimizu A, Pastan I, Saito A, Kobori H, Nishiyama A, Ichikawa I. Liver angiotensinogen is the primary source of renal Angiotensin II. J Am Soc Nephrol 23 (2012) 1181–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kukida M, Cai L, Ye D, Sawada H, Katsumata Y, Franklin MK, Hecker PI, Campbell KS, Danser AH, Mullick AE, Daugherty A, Temel RE, Lu HS. Renal angiotensinogen is predominantly liver-derived in nonhuman primates. Arterioscler Thromb Vasc Biol 41 (2021) 2851–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson BA, Cruz-Diaz N, Su Y, Rose JC, Gwathmey TM, Chappell MC. Angiotensinogen import in isolated proximal tubules: evidence for mitochondrial trafficking and uptake. Am J Physiol Renal Physiol 312 (2017) F879–F866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strawn WB, Richmond RS, Ann Tallant E, Gallagher PE, Ferrario CM. Renin-angiotensin system expression in rat bone marrow hematopoietic and stromal cells. Br J Haematol 126 (2004) 120–126. [DOI] [PubMed] [Google Scholar]
- 23.Lin C, Datta VD, Okwan-Duodu X, Chen S, Fuchs R, Alsabeh R, Billet S, Bernstein KE, Shen XZ. Angiotensin-converting enzyme is required for normal myelopoiesis. FASEB J 25 (2011) 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruiz-Ortega M, Bustos C, Hernandez-Presa MA, Lorenzo O, Plaza JJ. Egido J. Angiotensin II participates in mononuclear cell recruitment in experimental immune complex nephritis through nuclear factor-kappa B activation and monocyte chemoattractant protein-1 synthesis. J Immunol 161 (1998) 430–439. [PubMed] [Google Scholar]
- 25.Khan Z, Shen XZ, Bernstein EA, Giani JF, Eriguchi M, Zhao TV, Villaloboz RAG, Fuchs S, Liu GY, Bernsteiin KE. Angiotensin-converting enzyme enhances the oxidative response and bactericidal activity of neutrophils. Blood 130 (2017) 328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Okwan-Duodu D, Datta V, Shen XZ, Goodridge HS, Bernstein EA, Fuchs S, Liu GY, Bernstein KE. Angiotensin-converting enzyme overexpression in mouse myelomonocytic cells augments resistance to Listeria and methicillin-resistant Staphylococcus aureus. J Biol Chem 285 (2010) 39051–3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hagiwara S, Iwasaka H, Hidaka S, Hasegawa A, Koga H, Noguchi T Antagonist of the type-1 ANG II receptor prevents against LPS-induced septic shock in rats. Intensive Care Med 35 (2009)1471–1478. [DOI] [PubMed] [Google Scholar]
- 28.Miyoshi M, Nagata K, Imoto T, Goto O, Ishida A, Watanabe T. Ang II is involved in the LPS-induced production of proinflammatory cytokines in dehydrated rats. Am J Physiol Regul Integr Comp Physiol 284 (2003) R1092–R1097. [DOI] [PubMed] [Google Scholar]
- 29.Mateo T, Abu Nabah YN, Abu Taha M, Mata M, Nicolas MC, Proudfoot AEI, Stahl RAK, Issekuttz AC, Cortijo J, Morcillo EJ Jose PJ, Sanz MJ. Angiotensin II-induced mononuclear leukocyte interactions with arteriolar and venular endothelium are mediated by the release of different CC chemokines. J Immunol 176 (2006) 5577–5586. [DOI] [PubMed] [Google Scholar]
- 30.Nabah YN, Mateo T, Estellés R, Mata M, Zagorski J, Sarau H, Cortijo J, Morcillo EJ, Jose PJ, Sanz MJ. Angiotensin II induces neutrophil accumulation in vivo through generation and release of CXC chemokines. Circulation 110 (2004) 3581–3586. [DOI] [PubMed] [Google Scholar]
- 31.Kostenis E, Milligan G, Christopoulos A, Sanchez-Ferrer CF, Heringer-Walther S, Sexton PM, Gembardt F, Kellett E, Martini L, Vanderheyden P, Schultheiss HP, Walther T. G-protein-coupled receptor Mas is a physiological antagonist of the Angiotensin II Type 1 receptor. Circulation 111 (2005) 1806–1813. [DOI] [PubMed] [Google Scholar]
- 32.Akpinar E, Halici Z, Cadirci E, Bayir Y, Karakus E, Calik M, Topcu A, Polat B. What is the role of renin inhibition during rat septic conditions: preventive effect of aliskerin on sepsis-induced lung injury. Naunyn Schmiedebergs Arch Pharmacol 387 (2014) 969–978. [DOI] [PubMed] [Google Scholar]
- 33.Zhang J, Rudemiller NP, Patel MB, Wei Q, Karlovich NS, Jeffs AD, Wu M, Sparks MA, Privratsky JR, Herrera M, Gurley SB, Nedospasov SA, Crowley SD. Competing Actions of Type 1 Angiotensin II Receptors Expressed on T Lymphocytes and Kidney Epithelium during Cisplatin-Induced AKI. J Am Soc Nephrol 27 (2016) 2257–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang JD, Patel MB, Song YS, Griffiths R, Burchette J, Ruiz P, Sparks MA, Yan M, Howell DN, Gomez JA, Spurney RF, Coffman TM, Crowley SD. A novel role for type 1 angiotensin receptors on T lymphocytes to limit target organ damage in hypertension. Circ Res. 110 (2012) 1604–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nethathe GD, Cohen J, Lipman J, Anderson R, Feldman C. Mineralocorticoid dysfunction during critical illness: A Review of the Evidence. Anesthesiology 133.(2020) 439–457. [DOI] [PubMed] [Google Scholar]
- 36.Chung KS, Song JH, Jung WJ, Kim YS, Kim EK, Chang J, Park MS. Implication of plasma renin activity and plasma aldosterone concentration in critically ill patients with septic shock. Korean J Crit Care Med 32 (2017)142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davenport MW, Zipser RD. Association of hypotension with hyperreninemic hypoaldosteronism in the critically ill patient. 143:Arch Intern Med. (1983); 735–757. [PubMed] [Google Scholar]
- 38.du Cheyron D, Lesage A, Daubin C, Ramakers M, Charbonneau P. Hyperreninemic hypoaldosteronism: a possible etiological factor of septic shock-induced acute renal failure. Intensive Care Med 29 (2003) 1703–1709. [DOI] [PubMed] [Google Scholar]
- 39.Thosar SS. Rueda JF, Berman AM, Lasarev MR, Herzig MX, Clemons NA, Roberts SA, Bowles NP, Emens JS, Ellison DH, Shea SA. Separate and interacting effects of the endogenous circadian system and behaviors on plasma aldosterone in humans. Am J Physiol Regul Integr Comp Physiol 316 (2019) R157–R164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu J,Xie H, Ye Z, Li F, Wang L. Rates, predictors, and mortality of sepsis-associated acute kidney injury: a systematic review and meta-analysis. BMC Nephrol 21 (2020) 318–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Poston JT & Koyner JL Sepsis associated acute kidney injury. BMJ 364 (2019) k4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Du Cheyron D, Fradin S, Ramakers M, Terzi N, Guillotin D, Bouchet B, Daubin C, Charbonneau P. Angiotensin converting enzyme insertion/deletion genetic polymorphism: its impact on renal function in critically ill patients. Crit Care Med 36 (2008):3178–3183 [DOI] [PubMed] [Google Scholar]
- 42.Stanski NL, Shakked NP, Zhang B, Cvijanovich NZ, Fitzgerald JC, Jain PN, Schwarz AJ, Nowak J, Weiss SL, Allen GL, Thomas NJ, Haileselassie B, Goldstein SL. .Serum renin and prorenin concentrations predict severe persistent acute kidney injury and mortality in pediatric septic shock. Pediatr Nephrol. 38 (2023) 3099–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nguyen M, Denimal D, Dargent A, Guinot PG, Duvillard L, Quenot JP, Bouhemad B. Plasma renin concentration is associated with hemodynamic deficiency and adverse renal outcome in septic shock. 52Shock. (2019) :e22–30. [DOI] [PubMed] [Google Scholar]
- 44.Zhang W, Chen X, Huang L, Lu N, Zhou L, Wu G, Chen Y. Severe sepsis: low expression of the renin-angiotensin system is associated with poor prognosis. Exp Ther Med 7 (2014) 1342–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pode-Shakked N, Ceschia G, Rose JE, Goldstein SL, Stanski NL. Genomics of Pediatric Septic Shock Investigators. Increasing angiotensin-converting enzyme concentrations and absent angiotensin-converting enzyme activity are associated with adverse kidney outcomes in pediatric septic shock. Crit Care Lond Engl 12 (2023) 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Flannery AH, Ortiz-Soriano V, Li X, Gianella FG, Toto RD, Moe OW, Devarajan P, Goldstein SL, Neyra JA. Serum renin and major adverse kidney events in critically ill patients: a multicenter prospective study. Crit Care 25 (2021) 294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flannery AH, Kiser AS, Behal ML, Li X, Neyra JA. RAS inhibition and sepsis-associated acute kidney injury. J Crit Care 69 (2022) 153986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chou RH, Yang SF, Wu CH, Tsai YL, Lu YW, Guo JY, Huang PH, Lin SJ. Association between premorbid renin-angiotensin-aldosterone system blockade and the risk of acute kidney injury in critically ill patients. Acta Cardiol Sin. 39 (2023) 709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hasegawa D, Lee YI, Prasitlumkum N, Chopra L, Nishida K, Smith RL, Sato R. Premorbid angiotensin converting enzyme inhibitors or angiotensin II receptor blockers in patients with sepsis. The American Journal of Emergency Medicine. 62 (2022) 69–77. [DOI] [PubMed] [Google Scholar]
- 50.Tibi S, Zeynalvand G, Mohsin H. Role of the Renin Angiotensin Aldosterone System in the pathogenesis of sepsis-Induced acute kidney injury: A systematic review J Clin Med 12 (2023) 4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu X, Xue J, Liu Z, Dai W, Xiang J, Xu H, Zhou Q, Zhou Q, Wei X, Chen W. The effects of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers in critically ill patients with acute kidney injury: An observational study using the MIMIC database. .Front Pharmacol. 13 (2022) 918385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tumlin JA, Murugan R, Deane AM, Ostermann M, Busse LW, Ham KR, Kashani K, Szerlip HM, Prowle JR, Bihorac A, Finkel KW, Zarbock A, Forni LG, Lynch SJ, Jensen J, Kroll S, Chawla LS, Tidmarsh GF, Bellomo R. Angiotensin II for the Treatment of High-Output Shock 3 (ATHOS-3) Investigators (2018). Outcomes in patients with vasodilatory shock and renal replacement therapy treated with intravenous angiotensin II. Crit Care Med 46 (2018) 949–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia B, Su F, Manicone F, Dewachter L, Favory R, Khaldi A, Moiroux-Sahrou A, Moreau A, Herpain A, Vincent JL, Creteur J, Taccone FS, Annoni F. Angiotensin-(1–7) in an experimental septic shock model. Crit Care 27 (2023) 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu Y, Xu D, Deng F, Yan Y, Li J, Zhang C, Chu J. Angiotensin-(1–7) attenuates sepsis-induced acute kidney injury by regulating the NF-κB pathway. Front Pharmacol 12 (2021) 601909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Orfanos SE, Armaganidis A, Glynos C, Psevdi E, Kaltsas P, Sarafidou P, Catravas JD, Dafni UG, Langleben D, Roussos C. Pulmonary capillary endothelium-bound angiotensin-converting enzyme activity in acute lung injury. Circulation 102 (2000) 2011–2018. [DOI] [PubMed] [Google Scholar]
- 56.Chawla LS, Chen S, Bellomo R, Tidmarsh GF. Angiotensin converting enzyme defects in shock: implications for future therapy. Critical Care London Engl 22 (2018), 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deniau B, Picod A, Van Lier D, Ayar PV, Santos K, Hartmann O, Gayat E, Mebazaa A, Blet A, Azibani F. High plasma dipeptidyl peptidase 3 levels are associated with mortality and organ failure in shock: results from the international, prospective and observational FROG-ICU cohort. Br J Anaesth 128 (2022) e54. [DOI] [PubMed] [Google Scholar]
- 58.Jha S, Taschle U, Domenig O, Poglitsch M, Bourgeois B, Pollheimer M, Pusch LM, Malovan G, Frank S, Madl T, Gruber K, Zimmermann R, Macheroux P. Dipeptidyl peptidase 3 modulates the renin–angiotensin system in mice. J Biol. Chem 295 (2020) 13711–13723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deniau B, Blet A, Santos K, Ayar PV, Genest M, Kästorf M, Sadoune M, de Sousa Jorge A, Samuel JL, Vodovar N, Bergmann A, Mebazaa A, Azibani F. Inhibition of circulating dipeptidyl-peptidase 3 restores cardiac function in a sepsis-induced model in rats: A proof of concept study. PLOS One 15 (2020) e0238039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Srrnad P, Tacke F Koch A, Trautwein C. Liver - guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol 14 (2017) 55–66. [DOI] [PubMed] [Google Scholar]
- 61.Kim TS, Choi DH. Liver dysfunction in sepsis. Korean J Gastroenterol 25 (2020) 182–187. [DOI] [PubMed] [Google Scholar]
- 62.Arnal JF, Cudek P, Plouin PF, Guenne TT, Michel JB, Corvol P. Low angiotensinogen levels are related to the severity and liver dysfunction of congestive heart failure: implications for renin measurements. Am J Medi 90 (1991) 17–22. [DOI] [PubMed] [Google Scholar]
- 63.Stanley P, Serpell LG, Stein PE. Polymerization of human angiotensinogen: insights into its structural mechanism and functional significance. Biochem J 400 (2006) 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kløverpris S, Skov LL, Glerup S, Pihl K, Christiansen M, Oxvig C. Formation of high-molecular-weight angiotensinogen during pregnancy is a result of competing redox reactions with the proform of eosinophil major basic protein. Biochem J 449 (2013) 209–217. [DOI] [PubMed] [Google Scholar]
- 65.Briquez PS, Rouhani SJ, Yu J, Pyzer AR, Trujillo J, Dugan HL, Stamper CT, Changrob S, Sperling AI, Wilson PC, Gajewski TF, Hubbell JA, Swartz MA. Severe COVID-19 induces autoantibodies against angiotensin II that correlate with blood pressure dysregulation and disease severity. Sci Adv 8 (2022) eabn3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bucher M, Ittner KP, Hobbhahn J, Taeger K, Kurtz A. Downregulation of angiotensin II type 1 receptors during sepsis. Hypertension 38 (2001) 177–182. [DOI] [PubMed] [Google Scholar]
- 67.Leisman DE, Fernandes TD, Bijol V, Abraham MN, Lehman JR, taylor MD, Capone C, Yaipan O, Bellomo R, Deutschman CS. Impaired angiotensin II type 1 receptor signaling contributes to sepsis-induced acute kidney injury. Kidney Int 99 (2021) 148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leisman DE, Privratsky JR, Lehman JR, Abraham MN, Yaipan OY, Brewer MR, Nedeljkovic-Kurepa A, Capone CC, Fernandes TD, Griffith R, Stein WJ, Goldberg MB, Crowley SD, Bellomo R, Deutschman CS, Taylor MD. Angiotensin II enhances bacterial clearance via myeloid signaling in a murine sepsis model. Proc Natl Acad Sci USA. 119 (2022) e2211370119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li J, Brasier AR. Angiotensinogen gene activation by angiotensin II is mediated by the rel A (nuclear factor-kappaB p65) transcription factor: one mechanism for the renin angiotensin system positive feedback loop in hepatocytes. Mol Endo 10 (1996) 252–264. [DOI] [PubMed] [Google Scholar]
- 70.Hilgenfeldt U, Schwind S. Angiotensin II is the mediator of the increase in hepatic angiotensinogen synthesis after bilateral nephrectomy. Am J Physiol 265 (1993) E414–E418. [DOI] [PubMed] [Google Scholar]
- 71.Klett C, Nobiling R, Gierschik P, Hackenthal E. Angiotensin II stimulates the synthesis of angiotensinogen in hepatocytes by inhibiting adenylyl cyclase activity and stabilizing angiotensinogen mRNA. J Biol Chem 268 (1993) 25095–25103. [PubMed] [Google Scholar]
- 72.Cai SM, Yang RQ, Li Y, Ning ZW, Zhang LL, Zhou GS, Luo W, Li DH, Chen Y, Pan MX, Li X. Angiotensin-(1–7) improves liver fibrosis by regulating the NLRP3 inflammasome via redox balance modulation. Antioxid Redox Signal 24 (2016) 795–812. [DOI] [PubMed] [Google Scholar]
- 73.Hsieh YC, Lee KC, Lei HJ, Lan KH, Huo TI, Lin YT, Chan CC, Schnabl B, Huang YH, Hou MC, Lin HC. (Pro)renin receptor knockdown attenuates liver fibrosis through inactivation of ERK/TGF-β1/SMAD3 pathway. Cell Mol Gastroenterol Hepatol 12 (2021) 813–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rajapaksha IG, Gunarathne LS, Angus PW, Herath CB. Update on new aspects of the Renin-Angiotensin System in hepatic fibrosis and portal hypertension: implications for novel therapeutic options. J Clin Med 10 (2021) 702–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Khanna A, English SW, Wang XS, Ham K, Tumlin J, Szerlip H, Busse LW, Altaweel L, Albertson TE, Mackey C, McCurdy MT, Boldt DW, Chock S, Young PJ, Krell K, Wunderink RG, Ostermann M, Murugan R, Gong MN, Panwar R, Hästbacka J, Favory R, Venkatesh B, Thompson BT, Bellomo R, Jensen J, Kroll S, S Chawla L, Tidmarsh GF, Deane AM. ATHOS-3 Investigators (2017) Angiotensin II for the treatment of vasodilatory shock. N Engl J Med 377 (2017) 419–430. [DOI] [PubMed] [Google Scholar]
- 76.Wieruszewski PM, Khanna AK. Vasopressor choice and timing in vasodilatory shock. Crit Care 22 (2022) 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.See EJ, Clapham C, Liu J, Khasin M, Liskaser G, Chan JW, Serpa Neto A, Costa Pinto R, Bellomo R. A pilot study of angiotensin II as primary vasopressor in critically ill adults with vasodilatory hypotension: the ARAMIS study. Shock 59 (2023) 691–696. [DOI] [PubMed] [Google Scholar]
- 78.Garcia B, Zarbock A, Bellomo R, Legrand M. The alternative renin angiotensin system in critically ill patients: pathophysiology and therapeutic implications. Crit Care 27 (2023) 453–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Garcia B, Su F, Manicone F, Dewachter L, Favory R, Khaldi A, Moiroux-Sahrou A, Moreau A, Herpain A, Vincent JL, Creteur J, Taccone FS, Annoni F. Angiotensin-(1–7) in an experimental septic shock model. Crit Care 27 (2023) 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhu Y, Xu D, Deng F, Yan Y, Li J, Zhang C, Chu J. Angiotensin-(1–7) attenuates sepsis-induced acute kidney injury by regulating the NF-κB pathway. Front Pharmacol 12 (2021) 601909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li JX, Xiao X, Teng F, Li HH. Myeloid ACE2 protects against septic hypotension and vascular dysfunction through Ang-(1–7)-Mas-mediated macrophage polarization. Redox Biol 18 (2023) 103004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu C, Chen Y, Zhou P, Hu Z. Recombinant human angiotensin-converting enzyme 2 plays a protective role in mice with sepsis-induced cardiac dysfunction through multiple signaling pathways dependent on converting angiotensin II to angiotensin-(1–7). Ann Transl Med 11 (2023) 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Self WH, Shotwell MS, Gibbs KW, DeWit M, Files DC, Harkins M, Hudock K, Merkc L, Moskowitz A, Apocada KD, et al. Effect of renin-angiotensin system modulation with synthetic angiotensin (1–7) [TXA-127] and an AT1R biased ligand [TRV-027] among adults with COVID-19-induced acute lung injury: randomized clinical trials. JAMA. 29 (2023) 1170–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chappell MC. Renin-angiotensin system and sex differences in COVID-19: A critical assessment. Circ Res 132 (2023) 1320–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Campbell DJ. Do intravenous and subcutaneous angiotensin II increase blood pressure by different mechanisms? Clin Exp Pharmacol Physiol. 40 (2013):560–570. [DOI] [PubMed] [Google Scholar]