

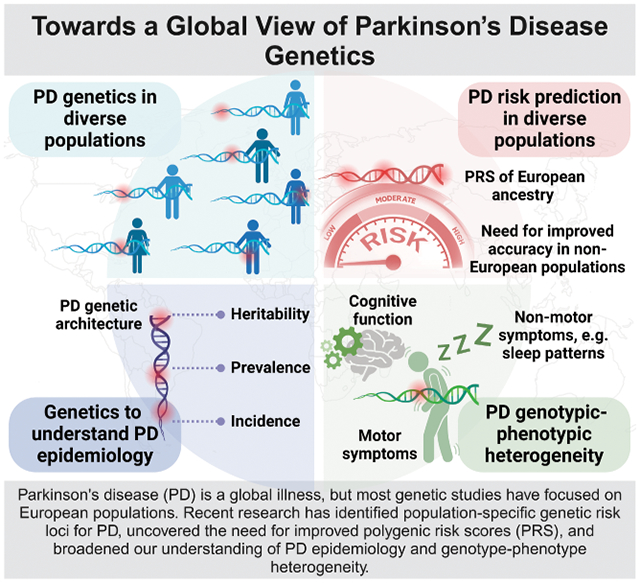
Abstract
Parkinson's disease (PD) is a global health challenge, yet historically studies of PD have taken place predominantly in European populations. Recent genetics research conducted in non-European populations has revealed novel population-specific genetic loci linked to PD risk, highlighting the importance of studying PD globally. These insights have broadened our understanding of PD etiology, which is crucial for developing disease-modifying interventions. This review comprehensively explores the global genetic landscape of PD, emphasizing the scientific rationale for studying underrepresented populations. It underscores challenges like genotype-phenotype heterogeneity and inclusion difficulties for non-European participants, emphasizing the ongoing need for diverse and inclusive research in PD.
Keywords: Parkinson’s disease, non-European populations, diversity, genetics
Graphical Abstract
Introduction
Parkinson's disease (PD) is a neurodegenerative condition characterized by Lewy body inclusions in the brain and the death of dopaminergic neurons in the midbrain, especially in the substantia nigra.1 PD is a widespread global condition affecting various ethnic groups and representing a growing social and economic burden worldwide.2 However, PD in non-European populations remains understudied. Various factors have hindered the inclusion of understudied populations in PD genetics research. These include financial and logistic limitations, a shortage of trained scientific workforce, and potential cultural or religious barriers.3 A solid genetic understanding plays a crucial role in developing models with the capacity to accurately predict the risk, onset, and progression of PD and can further elucidate disease mechanisms in individual patients. Without a comprehensive genetic foundation, our capacity to develop and implement treatments is constrained.
In recent years, various national and international efforts have emerged, such as The Global Parkinson’s Genetics Program (GP2) supported by the Aligning Science Across Parkinson’s (ASAP) initiative, which aims to expand our current understanding of PD genetics in a global context.4 These efforts have helped to increase the number of well-powered research studies on PD genetics in populations from diverse ancestral backgrounds. The identification of both novel disease-causing mutations involved in monogenic forms of disease and genetic risk factors underlying complex inheritance across diverse ancestries offers potential insights into unknown disease etiology in a global context. Inclusive genetics research and a global understanding of disease manifestation lend themselves well to early diagnosis and the prevention of misdiagnosis, drug development, and the overall future of precision medicine. Inclusive research is important for reducing health disparities across diverse populations.5
In this review, we summarize current knowledge of genetic studies of PD in a global context. We describe the ongoing necessity to broaden our research across diverse ancestry populations. We discuss how this approach is providing insights into the diverse genetic foundations of the disease worldwide and how current research is assisting in refining the mapping of identified loci. We anticipate genetic findings in combination with multi-omics data will significantly enhance our capacity to improve disease prediction, inform prognosis, and contribute to defining specific disease subtypes–representing crucial advancements for precision medicine in PD.
Diversification in genetics research has led to novel insights into Parkinson’s disease etiology
Our present knowledge of PD is primarily derived from research conducted in populations of European ancestry. By studying underrepresented populations, we can address the notable gap of knowledge in the genetics, clinical features, and pathophysiology of the disease in non-European populations. Findings obtained from studying diverse populations have proven to be key for the development of novel effective treatments and for the identification of population-specific features pertaining to PD etiology. Recent genetic studies conducted in non-European populations,2,6-9 in addition to having confirmed some previously reported findings, have indeed led to the identification of novel PD-relevant genetic loci linked to disease risk (Supplementary Table 1, Fig 1).
Figure 1-. Schematic of genetic insights gained from genome-wide association studies across diverse populations.

The figure illustrates identified risk loci across five distinct ancestry populations: European, East Asian, South Asian, Latin American, and African/African admixed. Additionally, it highlights the novel identified loci through multi-ancestry analysis. Novel loci are shown in bold. “X pattern” denotes countries that participated in more than one population-specific GWAS. Orange color denotes East Asian ancestry origin including individuals from Singapore, Malaysia, Hong Kong, Taiwan, mainland China and Korea. Blue denotes African and African admixed ancestries and consists of individuals from the International Parkinson’s Disease Genomics Consortium-Africa, and participants from 23andMe. Green denotes Latin American ancestry consisting of LARGE PD participants from Uruguay, Peru, Chile, Brazil, Colombia, and Hispanic/Latino subjects from 23andMe. Purple denotes individuals recruited in India of South Asian ancestry.
New studies have revealed both similarities and differences across genetic risk factors for PD between Asian and European populations. In 2020, the first East Asian meta-genome-wide association study (GWAS), with 6,724 PD cases and 24,851 controls, identified eleven genome-wide significant loci. Nine of these had previously been observed in European populations (PARK16, ITPKB, MCCC1, SNCA, FAM47E, SCARB2, DLG2, LRRK2, RIT2, and FYN) while two were novel and population-specific (SV2C and WBSCR17). A replication study that included 1,926,361 individuals of European ancestry and 3,509 Japanese individuals confirmed a robust association between SV2C (rs246814) and PD. This study also suggested potential genetic heterogeneity between the two populations with regards to the WBSCR17 locus (rs9638616).6
Some studies have also compared genetic factors pertaining to PD risk among individuals of European descent and Latino populations. In 2021, a GWAS on 442,253 PD cases and controls from South America identified one locus (SNCA) as one of the major risk factors of PD in the Latino population, in concordance with the European population.2 Additionally, admixture analysis revealed the presence of two potential novel PD risk loci, one on chromosome 14 containing the STXBP6 gene and another on chromosome 6 spanning the RPS6KA2 gene, that warrant further study.2
Recently, researchers performed the first GWAS of PD in the African and African-admixed populations. The study included 197,918 individuals, of which 1,488 were cases and 196,430 were controls. The study identified a novel risk factor at the GBA1 locus (rs3115534-G) that was found to be very common among PD patients of African ancestry. Strikingly, this intronic association has not been observed in other populations, and it is independent of the well-known GBA1 variants identified in European and Ashkenazi populations, p.E365K and p.N409S.7
The largest genetic investigation of PD in a South Asian population, comprising a total of 674 cases of early-onset PD and 1376 controls, identified a genome-wide significant signal in the SNCA region. This signal strongly colocalized with the SNCA region signal from European PD GWAS. Additionally, gene burden studies identified BSN, which has been previously associated with neurodegenerative diseases.9
Expanding on these efforts, the first large-scale multi-ancestry GWAS meta-analysis of PD included 49,049 cases, 18,785 proxy cases, and 2,458,063 controls from European, East Asian, Latin American, and African populations. This GWAS revealed 78 independent genome-wide significant loci, of which 12 were novel (MTF2, RP11-360P21.2, ADD1, SYBU, IRS2, USP8:RP11-562A8.5, PIGL, FASN, MYLK2, AJ006998.2, Y_RNA, PPP6R2). Remarkably, cross-ancestry fine-mapping improved the identification of causal variants. In this study, for the first time, fine mapping isolated six potential causal variants within previously identified loci. 8
Multi-ancestry meta-analyses have helped define and refine PD-associated genetic risk factors, shedding new light on the intricate genetic landscape of the disease and yielding insights that could benefit individuals with PD from all ancestral groups. Further efforts will facilitate fine-mapping to identify the causal variants underlying these associations. Multi-ancestry studies can provide a deeper understanding of the underlying biology of disease and yield insights into relevant biological pathways that can be leveraged to develop personalized therapeutic interventions.
Risk prediction models are inherently biased toward European populations
Given the complex nature of PD, it’s important to develop polygenic risk scores (PRSs) as a means to summarize the cumulative effect of one’s genetic background on an individual’s disease risk.10,11 The importance of early detection for the treatment of PD also cannot be overstated. Predictive analytics, using PRS and multimodal machine-learning studies12 such as deep learning, can help identify complex patterns and interactions that may not be apparent based solely on individual data modalities, which in turn is likely to enhance the precision and applicability of risk assessment models for PD.
PRSs are mathematical predictions that leverage information from multiple genetic variants to estimate disease risk. However, when applied across ancestries, the predictive accuracy can be compromised. The greater the ancestral distance, the poorer the performance of these risk scores.13 Most studies have been conducted exclusively on populations of European descent, with few evaluating population-level applicability to non-Europeans. Studies that have examined global transferability have found that the predictive utility of PRSs decreases significantly when applied across ancestral groups.14 For instance, PRSs calculated in European samples have been shown to be about one-third as informative when applied to samples of African ancestry.15
PRSs computed via current standard methods with summary statistics from a single-ancestry discovery cohort face numerous challenges, including the fact that the genetic architecture of disease across populations is different and unpredictable. Simulations performed by Martin et al., recapitulate these results and show that across replicates (i.e., traits, and thus not necessarily within a single trait), cross-population prediction accuracy is diminished with increasing divergence from the discovery cohort. These simulations provide further insight into directional inconsistencies in inferred PRSs with the same demographic model across replicate simulations, indicating that different traits are likely to suffer from genetic biases that cannot be adjusted, e.g., using principal components alone.15 It is critical to explore how PRSs differ across various ethnic backgrounds.
Recently, Saffie et al. evaluated the performance of polygenic risk scoring in PD in a multi-ancestry context16 by comparing the performance of PRSs across seven ancestry populations, including European, African admixed, African, Ashkenazi Jewish, Latino/Admixed American, Central Asian, and East Asian ancestry populations. By examining 90 risk variants identified in latest and largest European GWAS meta-analysis in PD across these ancestries, the authors conducted a proof of concept study and demonstrated major differences in the directionalities of these predictors across different ancestries with limited applicability.11 These findings highlight the difference that exists in the genetic architecture of PD across populations.
Current PD PRS models based on genetic risk unraveled from European GWAS restrict our ability to generalize their applicability to other ancestral groups. Moreover, when using base and target data from the same ancestral population, Pan et al. explored the performance of PRS in the Chinese population. The authors observed that individuals with the highest burden of disease risk had a 3.9-fold higher risk of developing PD than those clustering in the lowest quartile.17 Additionally, a recent PRS study conducted in the Latino population concluded that the European PD PRS had potential for disease risk prediction in Latinos, but variability caused by admixture patterns and bias in a European-ancestry PD PRS data limited its utility.18 Furthermore, simulations show that the magnitude of bias rises with increasing divergence from European ancestry, and this is attributed to population differences in linkage disequilibrium and allele frequencies of European-discovered variants, likely as a result of genetic drift.19
As the field moves forward, there are some critical next steps that should be prioritized. First of all, genetic analyses in admixed populations substantially benefit from appropriate proxies for the ancestral populations, applying to local and global ancestry estimation, admixture mapping, and genotype imputation. 20 It is essential that future research efforts prioritize larger sample sizes, ancestry-specific replication datasets, and the use of well-powered ancestry-specific summary statistics. Secondly, studying biomarker-defined PD cohorts with definite diagnosis as opposed to those relying on clinical diagnostic criteria only, will improve PRS accuracy, since at least 5% of individuals with PD do not exhibit neuronal alpha-synuclein.21 Lastly, collaboration across different research groups and data sharing initiatives can help ensure that data harmonization is consistent and that ancestries from various geographic locations and ethnicities are adequately represented. In recent years, several initiatives such as the Parkinson's Progression Markers Initiative (PPMI), the Parkinson's Disease Biomarkers Program (PDBP), PREDICT PD (https://predictpd.com/en), and the Accelerating Medicines Partnership for Parkinson's disease (AMP-PD)22 have emerged as valuable resources to aggregate and harmonize efforts in PD.1
To conclude, by accounting for the genetic makeups specific to each ancestral background, improved risk scores may enhance the accuracy of prediction models, providing opportunities for personalized medicine tailored to diverse populations.
Leveraging the power of genetics to understand epidemiology in Parkinson’s disease
The Global Burden of Disease Study estimated a PD prevalence of around 94 per 100,000 globally, representing a 22% age-standardized increase between 1990 and 2016.23 Though this estimate represents the global prevalence of PD, it is well known that regional differences in PD incidence and prevalence exist.24,25 The exact source of this heterogeneity is not perfectly understood but can likely be attributed to a variety of factors including the breadth and depth of epidemiological data, diagnostic criteria and health record management, access to specialized care, cultural beliefs, distinct environmental exposures, and unique genetic architecture of disease across populations, among others.26-28
PD heritability–defined as the proportion of disease attributable to genetic factors–is currently estimated at ~16–36% in European populations 11,14 and around 38% in Latino populations.2 A large portion of PD etiology has yet to be explained. Heritability estimates in other non-European populations are largely unknown, leaving uncertainty about the extent to which genetics influence disease etiology relative to other factors, such as environmental exposures, in these populations. A sophisticated understanding of the global genetic architecture of PD will help clarify the heritability of PD between different ancestral populations and will help elucidate the contribution and heterogeneity of environmental risk factors for PD. It is clear that gene-environment interactions play an important role in PD risk and its progression 29-31 and thereby likely contribute to the variation in both local and global incidence and prevalence rates.
The leading risk factor for PD is age. By 2030, roughly 80% of older adults are predicted to reside in low- and middle-income countries.32 Thus, understanding the global epidemiology of PD is becoming increasingly important. A comprehensive pan-ancestral map of PD genetics combined with global allele frequencies could enable the prediction of PD incidence and prevalence. Such a model may be particularly relevant in regions where it has been difficult to accurately assess the epidemiology of PD24 due to insufficient infrastructure, deferment of care, dearth of specialty care, and lack of public awareness.33-35
Recent genetic studies have begun to yield important insights into overlapping genetic etiologies between PD and a number of other conditions.7,36-38 Although the genetics of these conditions are known to vary between different ancestral populations,39-41 little is known about potential shared pleiotropic intricacies. This further underscores the importance of understanding the global architecture of PD, as pleiotropic loci may facilitate the discovery of the etiology and epidemiology of related diseases with shared genetic risk factors.
Parkinson’s disease heterogeneity on the phenotypic and genotypic level across different populations
The genotypic variability in PD, encompassing common and rare variants, is increasingly expanding. Monogenic forms of PD only account for approximately ~5% to 15% of PD cases.42 Next-generation sequencing techniques widely integrated in research have identified a multitude of genetic variants potentially associated with PD across more than 80 genes, albeit most of them lack replication in subsequent studies.43 Despite these advancements, unraveling the genetic mechanisms contributing to the heterogeneity of PD phenotypes remains a daunting challenge, especially as we extend our exploration to other ancestral backgrounds. While some of the most frequent pathogenic variants have been linked to specific phenotypic patterns, clinical presentations associated with the same mutation can vary widely across diverse populations.44 These differences underscore the complex interactions of polygenic and environmental factors that modify penetrance and influence disease severity.29,45 Moreover, the inability to replicate many initially identified genetic variants in subsequent studies highlights the critical need for conducting research in diverse populations, as confirming pathogenicity across ancestries strengthens the evidence that an association was not made in error. As the number of identified loci associated with PD continues to grow 11,46, only a few specific genes have been unequivocally linked to monogenic forms of disease: SNCA, LRRK2, VPS35 for autosomal-dominant PD, and PARKIN, PINK1, PARK7 for autosomal-recessive forms.44 Furthermore, mutations in GBA1 have emerged as PD's most significant genetic risk factor.47
LRRK2, one of the most implicated genes in autosomal-dominant PD, has been extensively studied in diverse populations.48-50 Over 26 pathogenic missense variants in LRRK2, thought to enhance the kinase activity of its protein, have been associated with PD, positioning it as a significant target for therapeutic intervention.51,52 The LRRK2 p.G2019S mutation is the most prevalent, found in about 4% of familial PD cases and 1% of sporadic cases worldwide.50 This variant is particularly frequent in North African Arab and Ashkenazi Jewish populations, representing 36-67% 50,53-55 and 26-30% 50,56,57 of familial cases, respectively. These differences are closely related to the significant age-dependent variation in the penetrance of LRRK2 mutations across different ethnicities, establishing this gene as the most common monogenic cause of late-onset PD.58 In this instance, up to 43% of Ashkenazi Jewish carriers are thought to develop parkinsonism by the age of 80, whereas penetrance in Tunisian Arab-Berbers can reach 86% by 70.59-61 Additionally, other less frequent LRRK2 pathogenic variants have also shown significant population-specific prevalence, such as p.R1441G in the Basque region in Spain 62 and p.G2385R in East Asian populations.63 In Latin America, frequency varies across different countries, directly correlating with the amount of European ancestry64, with prevalence of p.G2019S ranging from 3.75 to 5.5% in Uruguay and Argentina (in a study with a higher Ashkenazi Jewish ancestry) to less than 0.5% in Peru.64-66 The clinical presentation of LRRK2 mutations generally parallels that of idiopathic late-onset PD 67 but with a slower progression of motor symptoms and higher survival rates.68-70 Converging evidence, particularly from research on well-studied European and North American cohorts, suggests that LRRK2 carriers may exhibit better cognitive function 71,72 and fewer non-motor symptoms, such as hyposmia and rapid eye movement sleep behavior disorder (RBD).73,74
The SNCA gene, which encodes alpha-synuclein, was the first gene to be linked to PD upon the identification of a missense mutation (p.A53T) in a large kindred.75 Subsequently, additional pathogenic variants including duplications and triplications have been documented as an important cause of autosomal-dominant PD, predominantly in populations of European ancestry.76-79 SNCA duplications and triplications have also been identified in Asian populations 80,81 and SNCA p.A53V has been found exclusively in this ancestry.82 In contrast, pathogenic variants in the SNCA gene have not been frequently reported as a cause of PD in Latin Americans.83 Even so, the prevalence of SNCA mutations in the general population is very rare, and the penetrance is variable.84 While most of the reported carriers develop the disease before the age of 50, some individuals with duplications may persist as asymptomatic even into older age.85 Despite such heterogeneity, the majority of mutation carriers demonstrate a severe parkinsonian phenotype initially responsive to levodopa, but progressively developing a variable range of atypical features, including postural instability, antecollis, pyramidal signs, and non-motor symptoms such as autonomic dysfunction, sleep disorders, psychiatric disturbances, and dementia.51,86 Conversely, a few carriers have been reported to develop milder phenotypes resembling idiopathic PD, mostly linked to duplications or specific point mutations, whereas the more severe clinical presentations have been associated with gene triplications.80,85,87
VPS35 was the initial gene identified through whole exome sequencing as a causative factor in late-onset PD.88,89 Further research revealed the VPS35 p.D620N mutation in additional PD families and sporadic cases among individuals of European ancestry, as well as in Japanese patients with East Asian ancestry.90,91 Although other potential disease-causing variants have been reported, only the p.D620N variant has been definitively classified as pathogenic.51 A small number of studies including VPS35 have been conducted in underrepresented populations and mainly focus on specific mutations such as p.D620N and p.R524W 83, making it more challenging to confirm the pathogenicity of less-studied variants in these groups. Penetrance studies of the VPS35 p.D620N mutation show that 25% of carriers manifest PD by 45 years of age or younger, the median age of onset is 49 years, and 75% of individuals develop symptoms by the age of 59 or older.92 The phenotype typically associated with this variant commonly presents classic PD symptoms and an excellent response to levodopa. However, it is characterized by fewer cognitive and neuropsychiatric features and hyposmia in approximately 50% of patients.93
Similarly, autosomal-recessive PD, primarily associated with mutations in the PRKN (Parkin), PINK1, and PARK7 genes, presents distinct clinical characteristics. These mutations are predominantly found in early-onset PD (EOPD) individuals across various ethnicities. The frequency of homozygous or compound heterozygous PRKN mutations varies, showing rates ranging from less than 2% to approximately 13% in EOPD cohorts of European 94-98 and Latin American descent.42,99-101 This variability is attributed to differences in case detection, ancestry, and the proportion of familial cases and consanguinity. In Asian and North African populations, PRKN gene mutations contribute to EOPD, accounting for approximately 3-7% of cases.82,102-105 PINK1 mutations, though rare, are observed more frequently in Asians and African Berbers compared to Europeans and Latin Americans.96,98 The MDSGene review of 958 PRKN mutation carriers indicated that around 80% had a disease onset before 40 years of age and 16% by 20 years.106
The clinical presentation of PRKN, PINK1, and PARK7 mutations typically include classic parkinsonian motor symptoms that manifest at an earlier age, generally before or around 30 years, and are associated with a slower disease progression.106,107 Common features, regardless of genetic background, include dyskinesias and dystonia, particularly prevalent in early-onset PD cases.106,108 Compared to idiopathic PD, non-motor symptoms are less frequent, especially with PRKN mutations, and neuropathological examinations often reveal fewer or almost absent Lewy bodies.109 A systematic review showed variable levels of cognitive impairment associated with these mutations. Cognitive impairment was present in less than 10% of PRKN cases, while PARK7 and PINK1 cases displayed rates on the order of 29% and 58%, respectively.110 However, the availability of comprehensive clinical information in reports of these mutations remains largely incomplete. As indicated by Kasten et al. in the MDSGene Systematic Review, there is a significant degree of missing phenotypic data across publications, encompassing both non-motor symptoms and cardinal motor features.106 Despite significant advances in genetic research5,42, this gap is even more pronounced in studies concerning underrepresented populations, highlighting an urgent need for expanded research efforts to uncover phenotypic variability of these mutations in diverse ethnic backgrounds.
On the other hand, heterozygous mutations in the GBA1 gene, associated with Gaucher's disease (GD) in homozygous individuals, have emerged as a significant risk factor for PD and Lewy body dementia.111,112 These pathogenic variants impair glucocerebrosidase function, reducing enzymatic activity and potentially heightening alpha-synuclein aggregation.113 The frequency of GBA1 mutations in PD varies considerably across ethnicities, with up to 20% prevalence in Ashkenazi Jewish patients111, contrasting with 4-5% found in Asian and Latin American studies.42,114,115 The spectrum of GBA1 variants across populations is also highly variable, with specific mutations conferring differing levels of risk.116 For example, the p.N409S mutation, prevalent in Europeans and Ashkenazi Jews, is associated with a milder PD phenotype, whereas the p.L483P mutation is linked to a more severe PD phenotype.47 Both mutations are associated with GD when present in a homozygous state. In contrast, the p.E365K variant, also frequently found in Europeans, is not linked to GD but has been associated with worse clinical PD symptoms.47 Meanwhile, the GBA1 rs3115534-G variant, identified as a novel PD risk factor in a recent GWAS study in African and African-admixed populations7, has not been observed in GWAS studies of European, Latin American, or Asian ancestry.2,6,11 Interestingly, this non-coding variant was recently associated with RBD in individuals of Nigerian origin, suggesting that it could have downstream effects on par with traditional GBA1 coding variants.117 While the rs3115534-G does not appear to be linked with GD,7 larger cohort studies are needed to uncover the phenotypic relationship of this locus in this population and its impact on glucocerebrosidase activity and PD pathogenesis. Furthermore, studies have identified specific variants unique to certain populations, such as GBA1 p.K237E, reported in some Latin American cohorts, which have not been found in other groups and whose phenotype is not yet widely described.118,119 In the broader context, individuals carrying the most frequently described GBA1 mutations often exhibit a phenotype characterized by more rapid disease progression, impaired visual cognitive tasks and olfaction, increased psychosis, and a higher likelihood of developing dementia compared to non-carriers.120,121 The estimated disease penetrance in heterozygous carriers is low, ranging from 8% to 30% in studies performed on individuals of European and Ashkenazi Jewish ancestry.122-124 However, considering the more than 300 variants identified, clinical investigations have primarily focused on the most common mutations observed in European ancestry populations, highlighting the need to explore trait associations, penetrance, and pathogenesis of all variants in a broader range of populations.
Discussion
This review provides a comprehensive overview of the current knowledge of PD genetics research worldwide, delineating both strengths and limitations. By exploring the scientific rationale for conducting genetics research in underrepresented populations, we underscore the significance of identifying novel, population-specific risk and highlight the potential to enhance prediction models. We draw attention to the complexity of genotype-phenotype heterogeneity across diverse ancestries and the challenges associated with including participants of non-European backgrounds (Fig 2). By following this course, we anticipate that genetic discoveries in PD will markedly boost our ability to predict disease, provide prognosis insights, and potentially prevent healthcare disparities among individuals with PD.
Figure 2: Schematic diagram of challenges in Parkinson's disease genetic research.

Worldwide collaborative initiatives have been crucial for the inclusion of diverse populations in PD genetic studies and for reducing disparities in research representation. Identifying new risk variants, age at onset, and progression markers across various ethnic groups will also advance the understanding of PD pathogenesis and likely contribute to the development of targeted therapies. One limitation is that we are only beginning to explore GWAS versus disease risk in a global context, and other clinical manifestations, such as age at onset and progression, have only been explored in European populations. Improving generalizability across diverse ancestries and cohorts will only be possible to the extent that larger sample sizes and underrepresented groups are included.
The development of PRSs that can accurately and reliably predict disease risk and age of onset across diverse populations is important to the advancement of precision medicine that is both effective and equitable. In turn, addressing gaps in genotype-phenotype correlations in monogenic PD is imperative125, especially for less studied variants. Key opportunities include refining criteria for designating causative genes, seeking replication across diverse ancestries, addressing clinical data gaps by standardizing clinical data collection, and expanding the phenotypic spectrum of diverse variants across different populations in larger cohorts.
In general, genetic tools in PD have not yet reached the level of clinical utility, hindered in part by the limited inclusion of diverse populations in the existing body of research. As our methods improve and approach clinical utility, it is important we ensure the tools we develop work well for all populations–not just European populations–to prevent the exacerbation of existing health disparities or creation of new ones. As part of this effort, it is crucial that genetic research studies move toward a standardized model in which returning results to participants is a baseline expectation. Return of results is particularly relevant in genetic studies involving underrepresented populations to promote transparency and provide value to participants. Notably, return of genetic results to study participants is a priority clearly articulated by various communities, and as researchers and clinicians, is a research outcome we should enthusiastically strive to achieve. As PD clinical care moves toward precision medicine, enhancing access to genetic testing and training diverse genetic counselors and clinicians who are representative of the population and understand the utility and limitations of genetic tools will be crucial to the equitable implementation of precision medicine.
In the future, our aim should be to not only better understand how genetics impact PD at a global level, but also to translate that understanding into action. Multimodal data integration will facilitate the translation of genetic maps to mechanisms and will improve our ability to develop more accurate models of disease prediction, prognosis, and treatment.3 While significant progress has been made in researching diverse populations in PD, there is still substantial work to be done. In addition to ongoing genetics research in underrepresented populations and increasing diversity among researchers, a critical next step is to ensure the inclusion of diverse populations in clinical trials.
Supplementary Material
Supplementary Table 1- Parkinson’s disease known risk loci across diverse populations.
Acknowledgments
This work was supported in part by the Intramural Research Program of the NIH, the National Institute on Aging (NIA), National Institutes of Health, Department of Health and Human Services; project number ZO1 AG000535 and ZIA AG000949. This work was carried out with the support and guidance of the GP2 Trainee Network which is part of the Global Parkinson's Genetics Program and funded by the Aligning Science Across Parkinson's (ASAP) initiative.
Footnotes
Potential Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- 1.Bandres-Ciga S, Diez-Fairen M, Kim JJ, Singleton AB. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis 2020;137:104782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loesch DP, Horimoto ARVR, Heilbron K, et al. Characterizing the Genetic Architecture of Parkinson’s Disease in Latinos. Ann. Neurol 2021;90(3):353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elsayed I, Martinez-Carrasco A, Cornejo-Olivas M, Bandres-Ciga S. Mapping the Diverse and Inclusive Future of Parkinson’s Disease Genetics and Its Widespread Impact [Internet]. Genes 2021;12(11)Available from: 10.3390/genes12111681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Global Parkinson’s Genetics Program. GP2: The Global Parkinson’s Genetics Program. Mov. Disord 2021;36(4):842–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schumacher-Schuh AF, Bieger A, Okunoye O, et al. Underrepresented Populations in Parkinson’s Genetics Research: Current Landscape and Future Directions. Mov. Disord 2022;37(8):1593–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foo JN, Chew EGY, Chung SJ, et al. Identification of Risk Loci for Parkinson Disease in Asians and Comparison of Risk Between Asians and Europeans: A Genome-Wide Association Study. JAMA Neurol. 2020;77(6):746–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rizig M, Bandres-Ciga S, Makarious MB, et al. Identification of genetic risk loci and causal insights associated with Parkinson’s disease in African and African admixed populations: a genome-wide association study. Lancet Neurol. 2023;22(11):1015–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim JJ, Vitale D, Véliz Otani D, et al. Multi-ancestry genome-wide meta-analysis in Parkinson’s disease [Internet]. bioRxiv 2022;Available from: https://www.medrxiv.org/content/10.1101/2022.08.04.22278432v1 [Google Scholar]
- 9.Andrews SV, Kukkle PL, Menon R, et al. The Genetic Drivers of Juvenile, Young, and Early-Onset Parkinson’s Disease in India [Internet]. Mov. Disord 2023;Available from: 10.1002/mds.29676 [DOI] [PubMed] [Google Scholar]
- 10.Koch S, Laabs B-H, Kasten M, et al. Validity and Prognostic Value of a Polygenic Risk Score for Parkinson’s Disease [Internet]. Genes 2021;12(12)Available from: 10.3390/genes12121859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18(12):1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makarious MB, Leonard HL, Vitale D, et al. Multi-modality machine learning predicting Parkinson’s disease. NPJ Parkinsons Dis 2022;8(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding Y, Hou K, Xu Z, et al. Polygenic scoring accuracy varies across the genetic ancestry continuum. Nature 2023;618(7966):774–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020;19(2):170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin AR, Gignoux CR, Walters RK, et al. Human Demographic History Impacts Genetic Risk Prediction across Diverse Populations. Am. J. Hum. Genet 2017;100(4):635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saffie Awad P, Elsayed I, Sanyaolu A, et al. Evaluating the performance of polygenic risk profiling across diverse ancestry populations in Parkinson’s disease [Internet]. bioRxiv 2023;Available from: https://www.medrxiv.org/content/10.1101/2023.11.28.23299090v1 [Google Scholar]
- 17.Pan H, Liu Z, Ma J, et al. Genome-wide association study using whole-genome sequencing identifies risk loci for Parkinson’s disease in Chinese population. NPJ Parkinsons Dis 2023;9(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loesch DP, Horimoto ARVR, Sarihan EI, et al. Polygenic risk prediction and SNCA haplotype analysis in a Latino Parkinson’s disease cohort. Parkinsonism Relat. Disord 2022;102:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cavazos TB, Witte JS. Inclusion of variants discovered from diverse populations improves polygenic risk score transferability [Internet]. HGG Adv 2021;2(1)Available from: 10.1016/j.xhgg.2020.100017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thornton TA, Bermejo JL. Local and global ancestry inference and applications to genetic association analysis for admixed populations. Genet. Epidemiol 2014;38 Suppl 1(0 1):S5–S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siderowf A, Concha-Marambio L, Lafontant D-E, et al. Assessment of heterogeneity among participants in the Parkinson’s Progression Markers Initiative cohort using α-synuclein seed amplification: a cross-sectional study. Lancet Neurol. 2023;22(5):407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Accelerating Medicines Partnership (AMP) [Internet]. National Institutes of Health (NIH) [date unknown];[cited 2023 Nov 19 ] Available from: https://www.nih.gov/research-training/accelerating-medicines-partnership-amp [Google Scholar]
- 23.GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(5):459–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schiess N, Cataldi R, Okun MS, et al. Six Action Steps to Address Global Disparities in Parkinson Disease: A World Health Organization Priority. JAMA Neurol. 2022;79(9):929–936. [DOI] [PubMed] [Google Scholar]
- 25.Ou Z, Pan J, Tang S, et al. Global Trends in the Incidence, Prevalence, and Years Lived With Disability of Parkinson’s Disease in 204 Countries/Territories From 1990 to 2019. Front Public Health 2021;9:776847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walker R, Fothergill-Misbah N, Kariuki S, et al. Transforming Parkinson’s Care in Africa (TraPCAf): protocol for a multimethodology National Institute for Health and Care Research Global Health Research Group project. BMC Neurol. 2023;23(1):373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okubadejo NU, Bower JH, Rocca WA, Maraganore DM. Parkinson’s disease in Africa: A systematic review of epidemiologic and genetic studies. Mov. Disord 2006;21(12):2150–2156. [DOI] [PubMed] [Google Scholar]
- 28.Mele B, Goodarzi Z, Hanson HM, Holroyd-Leduc J. Barriers and facilitators to diagnosing and managing apathy in Parkinson’s disease: a qualitative study. BMC Neurol. 2019;19(1):101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Periñán MT, Brolin K, Bandres-Ciga S, et al. Effect Modification between Genes and Environment and Parkinson’s Disease Risk. Ann. Neurol 2022;92(5):715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynoso A, Torricelli R, Jacobs BM, et al. Gene-environment interactions for Parkinson disease [Internet]. bioRxiv 2023;Available from: https://www.medrxiv.org/content/10.1101/2023.06.15.23291423v1 [DOI] [PubMed] [Google Scholar]
- 31.Ong Y-L, Deng X, Li H-H, et al. Caffeine intake interacts with Asian gene variants in Parkinson’s disease: a study in 4488 subjects. Lancet Reg Health West Pac 2023;40:100877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goodman-Palmer D, Ferriolli E, Gordon AL, et al. Health and wellbeing of older people in LMICs: a call for research-informed decision making. Lancet Glob Health 2023;11(2):e191–e192. [DOI] [PubMed] [Google Scholar]
- 33.Dekker MCJ, Coulibaly T, Bardien S, et al. Parkinson’s Disease Research on the African Continent: Obstacles and Opportunities. Front. Neurol 2020;11:512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okubadejo NU, Ojo OO, Oshinaike OO. Clinical profile of parkinsonism and Parkinson’s disease in Lagos, Southwestern Nigeria. BMC Neurol. 2010;10:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dotchin CL, Msuya O, Walker RW. The challenge of Parkinson’s disease management in Africa. Age Ageing 2007;36(2):122–127. [DOI] [PubMed] [Google Scholar]
- 36.Kung P-J, Elsayed I, Reyes-Pérez P, Bandres-Ciga S. Immunogenetic Determinants of Parkinson’s Disease Etiology. J. Parkinsons. Dis 2022;12(s1):S13–S27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nalls MA, Saad M, Noyce AJ, et al. Genetic comorbidities in Parkinson’s disease. Hum. Mol. Genet 2014;23(3):831–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim K, Kim S, Myung W, et al. Shared Genetic Background between Parkinson’s Disease and Schizophrenia: A Two-Sample Mendelian Randomization Study [Internet]. Brain Sci 2021;11(8)Available from: 10.3390/brainsci11081042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bigdeli TB, Genovese G, Georgakopoulos P, et al. Contributions of common genetic variants to risk of schizophrenia among individuals of African and Latino ancestry. Mol. Psychiatry 2020;25(10):2455–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramos PS, Shedlock AM, Langefeld CD. Genetics of autoimmune diseases: insights from population genetics. J. Hum. Genet 2015;60(11):657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mahajan A, Spracklen CN, Zhang W, et al. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat. Genet 2022;54(5):560–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saffie Awad P, Teixeira-Dos-Santos D, Santos-Lobato BL, et al. Frequency of Hereditary and GBA1-Related Parkinsonism in Latin America: A Systematic Review and Meta-Analysis [Internet]. Mov. Disord 2023;Available from: 10.1002/mds.29614 [DOI] [PubMed] [Google Scholar]
- 43.Lange LM, Gonzalez-Latapi P, Rajalingam R, et al. Nomenclature of Genetic Movement Disorders: Recommendations of the International Parkinson and Movement Disorder Society Task Force - An Update. Mov. Disord 2022;37(5):905–935. [DOI] [PubMed] [Google Scholar]
- 44.Jia F, Fellner A, Kumar KR. Monogenic Parkinson’s Disease: Genotype, Phenotype, Pathophysiology, and Genetic Testing [Internet]. Genes 2022;13(3)Available from: 10.3390/genes13030471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iwaki H, Blauwendraat C, Makarious MB, et al. Penetrance of Parkinson’s Disease in LRRK2 p.G2019S Carriers Is Modified by a Polygenic Risk Score. Mov. Disord 2020;35(5):774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang D, Nalls MA, Hallgrímsdóttir IB, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet 2017;49(10):1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith L, Schapira AHV. GBA Variants and Parkinson Disease: Mechanisms and Treatments [Internet]. Cells 2022;11(8)Available from: 10.3390/cells11081261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paisán-Ruíz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004;44(4):595–600. [DOI] [PubMed] [Google Scholar]
- 49.Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004;44(4):601–607. [DOI] [PubMed] [Google Scholar]
- 50.Healy DG, Falchi M, O’Sullivan SS, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;7(7):583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trinh J, Zeldenrust FMJ, Huang J, et al. Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov. Disord 2018;33(12):1857–1870. [DOI] [PubMed] [Google Scholar]
- 52.Leschziner AE, Reck-Peterson SL. Structural Biology of LRRK2 and its Interaction with Microtubules. Mov. Disord 2021;36(11):2494–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lesage S, Dürr A, Tazir M, et al. LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N. Engl. J. Med 2006;354(4):422–423. [DOI] [PubMed] [Google Scholar]
- 54.Ishihara L, Gibson RA, Warren L, et al. Screening for Lrrk2 G2019S and clinical comparison of Tunisian and North American Caucasian Parkinson’s disease families. Mov. Disord 2007;22(1):55–61. [DOI] [PubMed] [Google Scholar]
- 55.Bouhouche A, Tibar H, Ben El Haj R, et al. LRRK2 G2019S Mutation: Prevalence and Clinical Features in Moroccans with Parkinson’s Disease. Parkinsons Dis. 2017;2017:2412486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ozelius LJ, Senthil G, Saunders-Pullman R, et al. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med 2006;354(4):424–425. [DOI] [PubMed] [Google Scholar]
- 57.Orr-Urtreger A, Shifrin C, Rozovski U, et al. The LRRK2 G2019S mutation in Ashkenazi Jews with Parkinson disease: is there a gender effect? Neurology 2007;69(16):1595–1602. [DOI] [PubMed] [Google Scholar]
- 58.Haugarvoll K, Wszolek ZK. Clinical features of LRRK2 parkinsonism. Parkinsonism Relat. Disord 2009;15 Suppl 3:S205–8. [DOI] [PubMed] [Google Scholar]
- 59.Hentati F, Trinh J, Thompson C, et al. LRRK2 parkinsonism in Tunisia and Norway: a comparative analysis of disease penetrance. Neurology 2014;83(6):568–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marder K, Wang Y, Alcalay RN, et al. Age-specific penetrance of LRRK2 G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology 2015;85(1):89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee AJ, Wang Y, Alcalay RN, et al. Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry. Mov. Disord 2017;32(10):1432–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ruiz-Martínez J, Gorostidi A, Ibañez B, et al. Penetrance in Parkinson’s disease related to the LRRK2 R1441G mutation in the Basque country (Spain). Mov. Disord 2010;25(14):2340–2345. [DOI] [PubMed] [Google Scholar]
- 63.Simpson C, Vinikoor-Imler L, Nassan FL, et al. Prevalence of ten LRRK2 variants in Parkinson’s disease: A comprehensive review. Parkinsonism Relat. Disord 2022;98:103–113. [DOI] [PubMed] [Google Scholar]
- 64.Cornejo-Olivas M, Torres L, Velit-Salazar MR, et al. Variable frequency of LRRK2 variants in the Latin American research consortium on the genetics of Parkinson’s disease (LARGE-PD), a case of ancestry. NPJ Parkinsons Dis 2017;3:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mata IF, Cosentino C, Marca V, et al. LRRK2 mutations in patients with Parkinson’s disease from Peru and Uruguay. Parkinsonism Relat. Disord 2009;15(5):370–373. [DOI] [PubMed] [Google Scholar]
- 66.Gatto EM, Parisi V, Converso DP, et al. The LRRK2 G2019S mutation in a series of Argentinean patients with Parkinson’s disease: clinical and demographic characteristics. Neurosci. Lett 2013;537:1–5. [DOI] [PubMed] [Google Scholar]
- 67.Aasly JO, Toft M, Fernandez-Mata I, et al. Clinical features of LRRK2-associated Parkinson’s disease in central Norway. Ann. Neurol 2005;57(5):762–765. [DOI] [PubMed] [Google Scholar]
- 68.Alcalay RN, Mirelman A, Saunders-Pullman R, et al. Parkinson disease phenotype in Ashkenazi Jews with and without LRRK2 G2019S mutations. Mov. Disord 2013;28(14):1966–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saunders-Pullman R, Mirelman A, Alcalay RN, et al. Progression in the LRRK2-Asssociated Parkinson Disease Population. JAMA Neurol. 2018;75(3):312–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thaler A, Kozlovski T, Gurevich T, et al. Survival rates among Parkinson’s disease patients who carry mutations in the LRRK2 and GBA genes. Mov. Disord 2018;33(10):1656–1660. [DOI] [PubMed] [Google Scholar]
- 71.Alcalay RN, Mejia-Santana H, Mirelman A, et al. Neuropsychological performance in LRRK2 G2019S carriers with Parkinson’s disease. Parkinsonism Relat. Disord 2015;21(2):106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Somme JH, Molano Salazar A, Gonzalez A, et al. Cognitive and behavioral symptoms in Parkinson’s disease patients with the G2019S and R1441G mutations of the LRRK2 gene. Parkinsonism Relat. Disord 2015;21(5):494–499. [DOI] [PubMed] [Google Scholar]
- 73.Ruiz-Martínez J, Gorostidi A, Goyenechea E, et al. Olfactory deficits and cardiac 123I-MIBG in Parkinson’s disease related to the LRRK2 R1441G and G2019S mutations. Mov. Disord 2011;26(11):2026–2031. [DOI] [PubMed] [Google Scholar]
- 74.Pont-Sunyer C, Iranzo A, Gaig C, et al. Sleep Disorders in Parkinsonian and Nonparkinsonian LRRK2 Mutation Carriers. PLoS One 2015;10(7):e0132368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997;276(5321):2045–2047. [DOI] [PubMed] [Google Scholar]
- 76.Krüger R, Kuhn W, Müller T, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet 1998;18(2):106–108. [DOI] [PubMed] [Google Scholar]
- 77.Singleton AB, Farrer M, Johnson J, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003;302(5646):841. [DOI] [PubMed] [Google Scholar]
- 78.Chartier-Harlin M-C, Kachergus J, Roumier C, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004;364(9440):1167–1169. [DOI] [PubMed] [Google Scholar]
- 79.Ibáñez P, Bonnet A-M, Débarges B, et al. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 2004;364(9440):1169–1171. [DOI] [PubMed] [Google Scholar]
- 80.Nishioka K, Hayashi S, Farrer MJ, et al. Clinical heterogeneity of alpha-synuclein gene duplication in Parkinson’s disease. Ann. Neurol 2006;59(2):298–309. [DOI] [PubMed] [Google Scholar]
- 81.Sekine T, Kagaya H, Funayama M, et al. Clinical course of the first Asian family with Parkinsonism related to SNCA triplication. Mov. Disord 2010;25(16):2871–2875. [DOI] [PubMed] [Google Scholar]
- 82.Chen Y-P, Yu S-H, Zhang G-H, et al. The mutation spectrum of Parkinson-disease-related genes in early-onset Parkinson’s disease in ethnic Chinese. Eur. J. Neurol 2022;29(11):3218–3228. [DOI] [PubMed] [Google Scholar]
- 83.Koros C, Bougea A, Simitsi AM, et al. The Landscape of Monogenic Parkinson’s Disease in Populations of Non-European Ancestry: A Narrative Review [Internet]. Genes 2023;14(11)Available from: 10.3390/genes14112097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Blauwendraat C, Makarious MB, Leonard HL, et al. A population scale analysis of rare SNCA variation in the UK Biobank. Neurobiol. Dis 2021;148:105182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Book A, Guella I, Candido T, et al. A Meta-Analysis of α-Synuclein Multiplication in Familial Parkinsonism. Front. Neurol 2018;9:1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Konno T, Ross OA, Puschmann A, et al. Autosomal dominant Parkinson’s disease caused by SNCA duplications. Parkinsonism Relat. Disord 2016;22 Suppl 1(Suppl 1):S1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ibáñez P, Lesage S, Janin S, et al. Alpha-synuclein gene rearrangements in dominantly inherited parkinsonism: frequency, phenotype, and mechanisms. Arch. Neurol 2009;66(1):102–108. [DOI] [PubMed] [Google Scholar]
- 88.Vilariño-Güell C, Wider C, Ross OA, et al. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet 2011;89(1):162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zimprich A, Benet-Pagès A, Struhal W, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet 2011;89(1):168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Deng H, Gao K, Jankovic J. The VPS35 gene and Parkinson’s disease. Mov. Disord 2013;28(5):569–575. [DOI] [PubMed] [Google Scholar]
- 91.Ando M, Funayama M, Li Y, et al. VPS35 mutation in Japanese patients with typical Parkinson’s disease. Mov. Disord 2012;27(11):1413–1417. [DOI] [PubMed] [Google Scholar]
- 92.Trinh J, Guella I, Farrer MJ. Disease penetrance of late-onset parkinsonism: a meta-analysis. JAMA Neurol. 2014;71(12):1535–1539. [DOI] [PubMed] [Google Scholar]
- 93.Guadagnolo D, Piane M, Torrisi MR, et al. Genotype-Phenotype Correlations in Monogenic Parkinson Disease: A Review on Clinical and Molecular Findings. Front. Neurol 2021;12:648588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brooks J, Ding J, Simon-Sanchez J, et al. Parkin and PINK1 mutations in early-onset Parkinson’s disease: comprehensive screening in publicly available cases and control. J. Med. Genet 2009;46(6):375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Alcalay RN, Caccappolo E, Mejia-Santana H, et al. Frequency of known mutations in early-onset Parkinson disease: implication for genetic counseling: the consortium on risk for early onset Parkinson disease study. Arch. Neurol 2010;67(9):1116–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kilarski LL, Pearson JP, Newsway V, et al. Systematic review and UK-based study of PARK2 (parkin), PINK1, PARK7 (DJ-1) and LRRK2 in early-onset Parkinson’s disease. Mov. Disord 2012;27(12):1522–1529. [DOI] [PubMed] [Google Scholar]
- 97.Milanowski ŁM, Lindemann JA, Hoffman-Zacharska D, et al. Frequency of mutations in PRKN, PINK1, and DJ1 in Patients With Early-Onset Parkinson Disease from neighboring countries in Central Europe. Parkinsonism Relat. Disord 2021;86:48–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lesage S, Lunati A, Houot M, et al. Characterization of Recessive Parkinson Disease in a Large Multicenter Study. Ann. Neurol 2020;88(4):843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Aguiar P de C, Lessa PS, Godeiro C Jr, et al. Genetic and environmental findings in early-onset Parkinson’s disease Brazilian patients. Mov. Disord 2008;23(9):1228–1233. [DOI] [PubMed] [Google Scholar]
- 100.Camargos ST, Dornas LO, Momeni P, et al. Familial Parkinsonism and early onset Parkinson’s disease in a Brazilian movement disorders clinic: phenotypic characterization and frequency of SNCA, PRKN, PINK1, and LRRK2 mutations. Mov. Disord 2009;24(5):662–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marder KS, Tang MX, Mejia-Santana H, et al. Predictors of parkin mutations in early-onset Parkinson disease: the consortium on risk for early-onset Parkinson disease study. Arch. Neurol 2010;67(6):731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Choi JM, Woo MS, Ma H-I, et al. Analysis of PARK genes in a Korean cohort of early-onset Parkinson disease. Neurogenetics 2008;9(4):263–269. [DOI] [PubMed] [Google Scholar]
- 103.Li N, Wang L, Zhang J, et al. Whole-exome sequencing in early-onset Parkinson’s disease among ethnic Chinese. Neurobiol. Aging 2020;90:150.e5–150.e11. [DOI] [PubMed] [Google Scholar]
- 104.Ton ND, Thuan ND, Thuong MTH, et al. Rare and novel variants of PRKN and PINK1 genes in Vietnamese patients with early-onset Parkinson’s disease. Mol Genet Genomic Med 2020;8(10):e1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tay YW, Tan AH, Lim JL, et al. Genetic study of early-onset Parkinson’s disease in the Malaysian population. Parkinsonism Relat. Disord 2023;111:105399. [DOI] [PubMed] [Google Scholar]
- 106.Kasten M, Hartmann C, Hampf J, et al. Genotype-Phenotype Relations for the Parkinson’s Disease Genes Parkin, PINK1, DJ1: MDSGene Systematic Review. Mov. Disord 2018;33(5):730–741. [DOI] [PubMed] [Google Scholar]
- 107.Khan NL, Brooks DJ, Pavese N, et al. Progression of nigrostriatal dysfunction in a parkin kindred: an [18F]dopa PET and clinical study. Brain 2002;125(Pt 10):2248–2256. [DOI] [PubMed] [Google Scholar]
- 108.Puschmann A. Monogenic Parkinson’s disease and parkinsonism: clinical phenotypes and frequencies of known mutations. Parkinsonism Relat. Disord 2013;19(4):407–415. [DOI] [PubMed] [Google Scholar]
- 109.Doherty KM, Silveira-Moriyama L, Parkkinen L, et al. Parkin disease: a clinicopathologic entity? JAMA Neurol. 2013;70(5):571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Piredda R, Desmarais P, Masellis M, Gasca-Salas C. Cognitive and psychiatric symptoms in genetically determined Parkinson’s disease: a systematic review. Eur. J. Neurol 2020;27(2):229–234. [DOI] [PubMed] [Google Scholar]
- 111.Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med 2009;361(17):1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nalls MA, Duran R, Lopez G, et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. 2013;70(6):727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Alcalay RN, Levy OA, Waters CC, et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain 2015;138(Pt 9):2648–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Huang C-L, Wu-Chou Y-H, Lai S-C, et al. Contribution of glucocerebrosidase mutation in a large cohort of sporadic Parkinson’s disease in Taiwan. Eur. J. Neurol 2011;18(10):1227–1232. [DOI] [PubMed] [Google Scholar]
- 115.Lim JL, Lohmann K, Tan AH, et al. Glucocerebrosidase (GBA) gene variants in a multi-ethnic Asian cohort with Parkinson’s disease: mutational spectrum and clinical features. J. Neural Transm. 2022;129(1):37–48. [DOI] [PubMed] [Google Scholar]
- 116.Huh YE, Usnich T, Scherzer CR, et al. GBA1 Variants and Parkinson’s Disease: Paving the Way for Targeted Therapy. J Mov Disord 2023;16(3):261–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ojo OO, Bandres-Ciga S, Makarious MB, et al. The non-codingGBA1rs3115534 variant is associated with REM sleep behavior disorder in Nigerians [Internet]. bioRxiv 2023;Available from: https://www.medrxiv.org/content/10.1101/2023.11.07.23298092v2 [Google Scholar]
- 118.Velez-Pardo C, Lorenzo-Betancor O, Jimenez-Del-Rio M, et al. The distribution and risk effect of GBA variants in a large cohort of PD patients from Colombia and Peru. Parkinsonism Relat. Disord 2019;63:204–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tipton PW, Soto-Beasley AI, Walton RL, et al. Prevalence of GBA p.K198E mutation in Colombian and Hispanic populations. Parkinsonism Relat. Disord 2020;73:16–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Creese B, Bell E, Johar I, et al. Glucocerebrosidase mutations and neuropsychiatric phenotypes in Parkinson’s disease and Lewy body dementias: Review and meta-analyses. Am. J. Med. Genet. B Neuropsychiatr. Genet 2018;177(2):232–241. [DOI] [PubMed] [Google Scholar]
- 121.Toffoli M, Chohan H, Mullin S, et al. Phenotypic effect of GBA1 variants in individuals with and without Parkinson’s disease: The RAPSODI study. Neurobiol. Dis 2023;188:106343. [DOI] [PubMed] [Google Scholar]
- 122.Anheim M, Elbaz A, Lesage S, et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 2012;78(6):417–420. [DOI] [PubMed] [Google Scholar]
- 123.Alcalay RN, Dinur T, Quinn T, et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol. 2014;71(6):752–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Balestrino R, Tunesi S, Tesei S, et al. Penetrance of Glucocerebrosidase (GBA) Mutations in Parkinson’s Disease: A Kin Cohort Study. Mov. Disord 2020;35(11):2111–2114. [DOI] [PubMed] [Google Scholar]
- 125.Klein C, Hattori N, Marras C. MDSGene: Closing Data Gaps in Genotype-Phenotype Correlations of Monogenic Parkinson’s Disease. J. Parkinsons. Dis 2018;8(s1):S25–S30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1- Parkinson’s disease known risk loci across diverse populations.