

Abstract
Morbidity and mortality rates in patients with autosomal recessive, congenital generalized lipodystrophy type 4 (CGL4), an ultra-rare disorder, remain unclear. We report on 30 females and 16 males from 11 countries with biallelic null variants in CAVIN1 gene (mean age, 12 years; range, 2 months - 41 years). Hypertriglyceridemia was seen in 79% (34/43), hepatic steatosis in 82% (27/33) but diabetes mellitus in only 21% (8/44). Myopathy with elevated serum creatine kinase levels (346–3325 IU/L) affected all of them (38/38). 39% had scoliosis (10/26) and 57% had atlantoaxial instability (8/14). Cardiac arrhythmias were detected in 57% (20/35) and 46% had ventricular tachycardia (16/35). Congenital pyloric stenosis was diagnosed in 39% (18/46), 9 had esophageal dysmotility and 19 had intestinal dysmotility. Four patients suffered from intestinal perforations. Seven patients died at mean age of 17 years (range: 2 months - 39 years). The cause of death in four patients was cardiac arrhythmia and sudden death, while others died of prematurity, gastrointestinal perforation, and infected foot ulcers leading to sepsis. Our study highlights high prevalence of myopathy, metabolic abnormalities, cardiac and gastrointestinal problems in patients with CGL4. CGL4 patients are at high risk of early death mainly caused by cardiac arrhythmias.
Keywords: Congenital generalized lipodystrophy, CAVIN1, gastrointestinal disease, metabolic abnormalities, myopathy, ventricular tachycardia
1. Introduction
Congenital generalized lipodystrophies (CGL) are a heterogeneous group of autosomal recessive disorders characterized by near-total loss of adipose tissue at birth or soon thereafter and predisposition to metabolic complications of insulin resistance such as diabetes mellitus, hypertriglyceridemia, hepatic steatosis and others (Akinci et al., 2000; Brown et al., 2016; Garg, 2011). In the past two decades, four genetic loci have been identified for various subtypes of CGL: 1-acylglycerol-3-phosphate-O-acyltransferase 2 (AGPAT2) for CGL1 (Agarwal et al., 2002; Garg et al., 1999); Berardinelli-Seip congenital lipodystrophy 2 (BSCL2) for CGL2 (Magre et al., 2001); caveolin 1 (CAV1) for CGL3 (Kim et al., 2008); and caveolae associated protein 1 (CAVIN1; formerly known as polymerase I and transcript release factor, PTRF) for CGL4 (Hayashi et al., 2009). While most of the CGL patients have either CGL1 or CGL2, only 5 patients have been reported with CGL3 and ~30 patients with CGL4 (Hayashi et al., 2009).
The anecdotal case reports of patients with CGL4 suggest some unique clinical features such as congenital myopathy, cardiomyopathy, congenital pyloric stenosis, and skeletal abnormalities compared to other CGL subtypes. Congenital myopathy in CGL4 patients is characterized by muscle weakness, elevated serum creatine kinase (CK) levels and percussion induced muscle mounding (Akinci et al., 2017). Cardiac manifestations of CGL4 can be seen early in life and include atrial and ventricular arrhythmias, prolonged QT interval, and sudden death, mostly due to catecholaminergic polymorphic ventricular tachycardia (CPVT) induced after excessive physical activity (Akinci et al., 2016). A few patients have reportedly died of cardiac disease, mainly due to development of ventricular arrhythmias but not congestive heart failure (Akinci et al., 2016; Patni et al., 2019; Rajab et al., 2010). While congenital pyloric stenosis requiring early surgery has been reported in some patients, other gastrointestinal complications are not well recognized (Akinci et al., 2016; Munoz et al., 2019; Nilay et al., 2020; Shastry et al., 2010; Sorkina et al., 2020).
Further, whether patients with CGL4 develop as severe metabolic complications as seen in CGL1 and CGL2 is not clear (Akinci et al., 2000). Metabolic abnormalities, such as insulin-resistant diabetes, severe hypertriglyceridemia, and hepatic steatosis, can cause serious morbidity and early mortality in CGL (Akinci et al., 2019). Previous case reports indicate absence of or only mild metabolic abnormalities at the time of diagnosis of CGL4 (Akinci et al., 2016; Rajab et al., 2010; Shastry et al., 2010) and lack of overt diabetes (Hussain et al., 2019).
Due to paucity of case reports, the prevalence of various morbidities and causes of mortality in CGL4 remain unclear so far. Therefore, we report metabolic and other complications and causes of mortality among a large cohort of 46 patients from 27 independent families with CGL4 from 11 different countries.
2. Methods
CGL4 was diagnosed clinically by treating physicians. In addition to physical examination, whole-body magnetic resonance imaging (MRI) or dual-energy x-ray absorptiometry (DXA) was used to study fat distribution in selected cases. The diagnosis was genetically confirmed in all cases. Genetic tests were completed at contributing centers. The study protocol was reviewed and approved by the Dokuz Eylul University Ethics Committee, Izmir, Turkey, and by the Institutional Review Board of UT Southwestern Medical Center, Dallas, Texas, United States. The protocol was further reviewed at individual centers as necessary. Clinical and laboratory data on some patients have been published previously and we report follow up information on these patients (Acar et al., 2022; Adiyaman et al., 2022; Akinci et al., 2016; Akinci et al., 2017; Babalola et al., 2022; Mancioppi et al., 2023; Nilay et al., 2020; Patni et al., 2019; Shastry et al., 2010; Sorkina et al., 2020) as well as the new patients.
Data for the study were obtained through a retrospective review of medical charts at different centers. To follow a common algorithm for data collection, dedicated template tables were used. These template tables included information regarding sex, ethnicity, consanguinity, age at diagnosis of CGL4, age at the final visit, weight and height, duration of follow-up, genetic testing, puberty development, gonadal functions, neuromuscular disease (e.g., myopathy, percussion-induced rapid contractions (PIRCs), atlantoaxial instability, scoliosis), cardiac disease (e.g., arrhythmia and other cardiac findings, sudden death history in the family, pacemaker implantation, sympathectomy), gastrointestinal disease (e.g., pyloric stenosis, gastrointestinal dysmotility, esophageal disease, intestinal perforations), umbilical prominence, lytic bone lesions, laboratory assessments including serum leptin, creatine kinase, glucose, total cholesterol, triglyceride, low density lipoprotein (LDL) cholesterol, high density lipoprotein (HDL) cholesterol, alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood hemoglobin A1c, acanthosis nigricans, age at detection of components of metabolic disease (e.g., diabetes, low HDL cholesterol, high triglycerides, hepatic steatosis), organ complications, age at detection of organ abnormalities, medications (including daily insulin dose), and mortality (including causes of death).
Diabetes mellitus was defined according to the criteria defined by the American Diabetes Association (American Diabetes Association Professional Practice, 2022). Hypertriglyceridemia and low level of HDL cholesterol were defined according to the National Cholesterol Education Program Adult Treatment Panel III guidelines (Expert Panel on Detection & Treatment of High Blood Cholesterol in, 2001). Hypertriglyceridemia was defined as serum triglyceride level 150 mg/dL or higher. Low HDL cholesterol was defined as less than 40 mg/dL for men and less than 50 mg/dL for women. Age-specific thresholds were used for children and adolescents (Daniels et al., 2008).
Statistical analysis was performed using Statistical Package of Social Science (SPSS Inc), version 24.0, for Windows. Data are expressed as mean ± SD and range as necessary. Kaplan-Meier curves were plotted using GraphPad Prism 9 (GraphPad Software, Boston, MA) to analyze the time to a diagnosis of metabolic abnormality or organ complication. Kaplan-Meier curves were also generated to describe survival.
3. Results
A total of 46 patients (mean age: 12 ± 9 years; range: 2 months - 41 years; 30 females and 16 males) from 27 independent families were included. Patients were evaluated by treating physicians at 18 different international referral centers from 11 countries (Turkey, Oman, United States (US), Canada, Germany, Italy, Russia, Austria, Egypt, and India). There were a total of 23 patients from 9 Omani families; two families with one affected member each; four families with two affected members each, two families with three affected members each, and one family with seven affected members. There were two Mexican families, one Syrian, one Indian, and a Romanian family, each with two affected patients. Seven Turkish patients were distributed across seven families. Moreover, there were one Syrian, one Indian, one Egyptian, one Tatar, one Pakistani, and one Thai family, each with one affected member. Representative images showing generalized fat loss are presented in Figure 1A. A whole-body magnetic resonance (MR) image from a 9-year-old female with CGL4 shows severe generalized loss of fat but sparing of bone marrow fat (Figure 1B). Ethnic origins of patients are reported in Table 1. The mean duration of follow-up was 68 ± 55 months (range 1 to 214 months). No longitudinal follow-up data were available for three patients. Follow-up was short in four patients (less than a year); others had at least 12 months of observation. Seven patients died during the follow-up period.
Figure 1: Phenotypic features and metabolic and organ complications in CGL4.
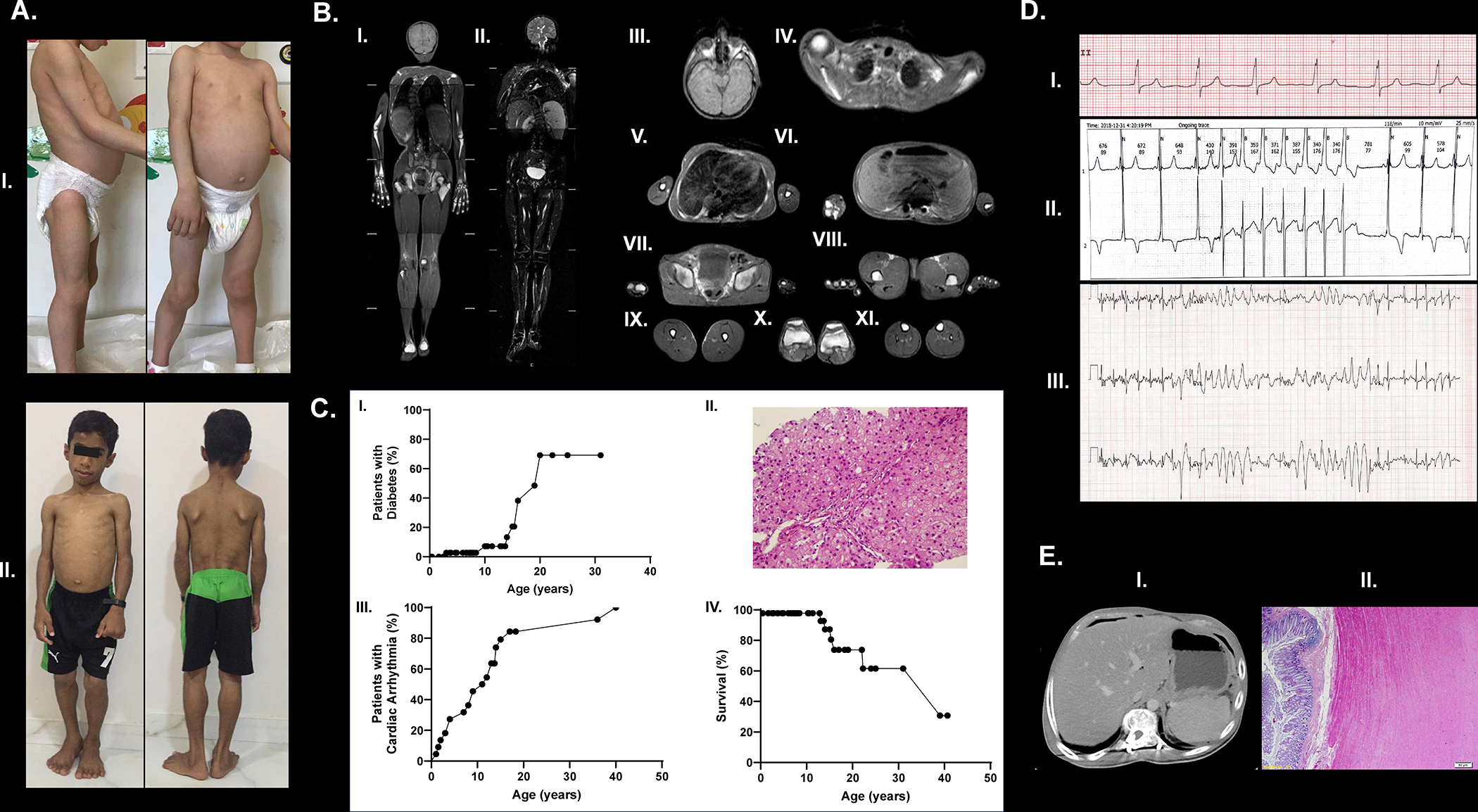
A. Patient pictures showing generalized fat loss; patient 7 (panel I) and patient 8 (panel II).
B. Whole-body MRI (T1-weighted) demonstrating near total fat loss in a patient with CGL4 (patient 1; MRI performed at age of 16 years). Bone marrow fat is preserved along with orbital fat. Scalp fat is lost. Palms and soles show no significant fat signal. Reduced amounts of supraclavicular fat can be visualized. (I), whole body, coronal T1-weighted imaging; (II), whole body, coronal T2-weighted fat suppressed imaging; (III), orbital and scalp, axial T1-weighted imaging; (IV), shoulders, axial T1-weighted imaging; (V), breasts, axial T1-weighted imaging; (VI), visceral and retroperitoneal, axial T1-weighted imaging; (VII-VIII), external genital region and palms, axial T1-weighted imaging; (IX-XI), limbs, axial T1-weighted imaging.
C. Kaplan Meier curves showing time to diabetes (panel I). A liver biopsy specimen (panel II) showing early changes of hepatic steatosis at age 1 in a patient with CGL4 (patient 3). Mild swelling of parenchymal cells. and macro- and microvesicular lipid depositions are shown (H&EX400). Kaplan Meier curves showing time to cardiac disease (panel III) and survival (panel IV)
D. ECG showing Wolff-Parkinson-White syndrome (panel I) and an episode of ventricular tachycardia (panel II) in patient 8, and non-sustained polymorphic ventricular tachycardia (panel III) during treadmill exercise test in patient 2.
E. Axial computerized tomography (CT) image showing air-fluid level due to intestinal perforation (panel I). Histopathology shows muscularis propria hypertrophy along with loss of orientation of circular and longitudinal layers of muscularis propria (panel II). Patient 1 had complaints of abdominal distention and pain that usually happened after meals (a feeling of fullness and early satiety and episodes of constipation and diarrhea when she first presented). Her symptoms were first treated with prokinetic agents, but no significant relief was observed. A small bowel follow-through study using barium showed slow transit time. At age 21, she presented with severe abdominal pain and tenderness. A CT showed colonic perforation and she underwent a colonic resection with colostomy. She did not have recurrent perforations but still lives with a colostomy bag.
Table 1:
Characteristics of subjects with CGL4
| Family ID | Patient ID | Current age | Age at initial diagnosis | Sex | CAVIN1 variant Nucleotide (amino acid) | Homozygosity vs. compound heterozygosity | Ethnicity | Known Consanguinity |
|---|---|---|---|---|---|---|---|---|
| 1 | P1 | 22 y | 11 | F | c.481_482insGTGA (p.K161Sfs*51) | Homozygote | Turkish | Yes |
| 2 | P2 | 19 y | 13 | F | c.259C>T (p.Q87*) | Homozygote | Turkish | Yes |
| 3 | P3 | 8 y and 5 m | 2 y 3 m | M | c.259C>T (p.Q87*) | Homozygote | Turkish | Yes |
| 4 | P4 | Died at age 16 y | 9 | F | c.259C>T (p.Q87*) | Homozygote | Turkish | Yes |
| 5 | P5 | 10 y and 6 m | 9 | F | c.214C>T (p.Q72*) | Homozygote | Syrian | Yes |
| 6 | P6 | 15 y | 7 | F | c.471G>C (p.G157Hfs*52) | Homozygote | Egyptian | Yes |
| 7 | P7 | 2 y and 10 m | 15 m | F | c.698delA (p.K233Rfs*42) | Homozygote | Turkish | Yes |
| 8 | P8 | 8 y | 18 m | M | c.160del (p.V54Cfs*2) | Homozygote | Omani | Yes |
| P9 | 4 y and 8 m | 2 y | F | c.160del (p.V54Cfs*2) | Homozygote | Omani | Yes | |
| 9 | P10 | 7 y and 10 m | 2 y | M | c.160del (p.V54Cfs*2) | Homozygote | Omani | Yes |
| P11 | 2 y and 10 m | 3 m | F | c.160del (p.V54Cfs*2) | Homozygote | Omani | Yes | |
| 10 | P12 | 6 y | 20 m | F | c.160del (p.V54Cfs*2) | Homozygote | Omani | Yes |
| P13 | 5 m | 5 m | F | c.160del (p.V54Cfs*2) | Homozygote | Omani | Yes | |
| 11 | P14 | Died at age 39 y | 36 y | F | c.631G>T (p.E211*) | Homozygote | Tatar | Yes |
| 12 | P15 | 14 y | 14 y | F | c.648delC (p.R217Afs*58) | Homozygote | Pakistani | Yes |
| 13 | P16 | 17 y | 17 y | F | c.192delC (p.V65*) | Homozygote | Thai | Yes |
| 14 | P17 | 6 y and 7 m | 4 y | M | c.T21A (p.Y7*) | Homozygote | Romanian | No* |
| P18 | 3 y and 8 m | 18 m | F | c.T21A (p.Y7*) | Homozygote | Romanian | No* | |
| 15 | P19 | 2 y and 6 m | 2 y | F | c.1061_1091 del (p.E354Gfs*55) | Homozygote | Turkish | Yes |
| 16 | P20 | Died at 22 y 3 m | 11 y | M | c.135delG (p.K45Dfs*6) | Homozygote | Mexican | No* |
| P21 | Died at 13 y 1 m | 1 y | F | c.135delG (p.K45Dfs*6) | Homozygote | Mexican | No* | |
| 17 | P22 | 25 y | 12 y | F | c.518-521delAAGA/471+1 G>T (p.K173Sfs*101/D158Vfs*50) | Compound heterozygote | Mexican | No* |
| P23 | Died at 15 y 4 m | 7 y | M | c.518-521delAAGA/471+1 G>T (p.K173Sfs*101/D158Vfs*50) | Compound heterozygote | Mexican | No* | |
| 18 | P24 | 19 y | 13 y | F | c.613G>T (p.E205*) | Homozygote | Indian | Yes |
| 19 | P25 | 24 y | 13 y | F | c.259C>T (p.Q87*) | Homozygote | Turkish | Yes |
| 20 | P26 | 10 y 4 m | 9 y 6 m | M | c.1061_1109del (p.E354Gfs*55) | Homozygote | Syrian | Yes |
| P27 | 7 y 7 m | 7 y 5 m | F | c.1061_1109del (p.E354Gfs*55) | Homozygote | Syrian | Yes | |
| 21 | P28 | 12 y 10 m | 2 y | F | c160delG (p.V54Cfs*2) | Homozygote | Omani | Yes |
| P29 | 7 y 4 m | 2 y | M | c160delG (p.V54Cfs*2) | Homozygote | Omani | Yes | |
| P30 | 40 y 8 m | 25 y | F | c160delG (p.V54Cfs*2) | Homozygote | Omani | NA | |
| P31 | 8 y 5 m | 5 y | M | c160delG (p.V54Cfs*2) | Homozygote | Omani | Yes | |
| P32 | 2 y 8 m | 1 d | F | c160delG (p.V54Cfs*2) | Homozygote | Omani | Yes | |
| P33 | 31 y | 28 y | F | c160delG (p.V54Cfs*2) | Homozygote | Omani | NA | |
| P34 | 1 y 8 m | 2 m | F | c160delG (p.V54Cfs*2) | Homozygote | Omani | Yes | |
| 22 | P35 | 14 y | 11 m | M | c160delG (p.V54Cfs*2) | Homozygote | Omani | Yes |
| P36 | 11 y 4 m | 3 m | M | c160delG (p.V54Cfs*2) | Homozygote | Omani | NA | |
| P37 | Died at 2 m | 1 m | F | c160delG (p.V54Cfs*2) | Homozygote | Omani | Yes | |
| 23 | P38 | 7 y 2 m | 15 m | M | c160delG (p.V54Cfs*2) | Homozygote | Omani | Yes |
| P39 | 4 y 11 m | 3 m | F | c160delG (p.V54Cfs*2) | Homozygote | Omani | Yes | |
| 24 | P40 | 7 y 2 m | 11 m | F | c160delG (p.V54Cfs*2) | Homozygote | Omani | Yes |
| 25 | P41 | 3 y 10 m | 9 m | M | c160delG (p.V54Cfs*2) | Homozygote | Omani | Yes |
| P42 | 2 y 8 m | 6 m | M | c160delG (p.V54Cfs*2) | Homozygote | Omani | Yes | |
| P43 | Died at 14 y | 1 y | F | c160delG (p.V54Cfs*2) | Homozygote | Omani | Yes | |
| 26 | P44 | 6 y 9 m | 5 y | M | c160delG (p.V54Cfs*2) | Homozygote | Omani | No* |
| 27 | P45 | 18 y 4 m | 6 m | F | c.613G>T (p.E205*) | Homozygote | Indian | Yes |
| P46 | 13 y 8 m | 9 y | M | c.613G>T (p.E205*) | Homozygote | Indian | Yes |
F: female, M: male, d: days, m: months, y: years
apparently.
Mean age at the time of diagnosis was 7 ± 8 years (range: birth to 36 years). Serum leptin levels were low (Table 2). Four unrelated Turkish subjects shared the CAVIN1 p.Q87* homozygous variant. All Omani subjects had the same p.V54Cfs*2 homozygous variant. Other variants are listed in Table 1. A schematic of the location of various variants in the CAVIN1 gene and corresponding CAVIN1 protein sequence are shown in Figure 2. Parental consanguinity was reported in 36 patients (Table 1). Information on gonadal function was available in 27 patients (20 females and 7 males), excluding 17 patients from one of the centers from Oman and 2 patients from one center in India. Primary amenorrhea was an ongoing problem in 5 females. Late puberty was reported in additional 5 patients (3 females and 2 males). Two females had normal pubertal development and 14 patients were prepubertal (age < 13 years in males and 10 years and 6 months in females). An additional girl had started puberty but had not yet attained menarche. Previous autopsy study had revealed lack of spermatogenesis in testis of a 15-year-old Mexican male with CGL4 (Patni et al., 2019). Unfortunately, none of the other males with CGL4 in our report are post-pubertal (except 2 males with delayed puberty) to further substantiate whether males with CGL4 are infertile.
Table 2:
Demographic and metabolic variables of patients with CGL4 at the most recent visit
| n | Mean ± SD | Range | |
|---|---|---|---|
| Current age | 46 | 12 ± 9 years | 2 months - 41 years |
| Gender (female/male) | 46 | 30/16 | NA |
| Current BMI (kg/m2) | 40 | 16.46 ± 2.59 | 12.23 – 21.76 |
| Age at diagnosis | 44 | 7 ± 8 years | At birth - 41 years |
| Fasting glucose (mg/dL) | 35 | 114 ± 69 | 75 – 443 |
| HbA1c (%) | 36 | 5.7 ± 1.7 | 4.3 – 14.2 |
| Total cholesterol (mg/dL) | 41 | 174 ± 49 | 86 – 286 |
| Triglyceride (mg/dL) | 43 | 361 ± 322 | 71 – 1431 |
| LDL cholesterol (mg/dL) | 35 | 80 ± 32 | 18 – 147 |
| HDL cholesterol (mg/dL) | 42 | 27 ± 8 | 12 – 52 |
| ALT (IU/L) | 41 | 63 ± 32 | 7 – 144 |
| AST (IU/L) | 27 | 64 ± 34 | 21 – 176 |
| Creatine kinase (IU/L) | 38 | 1434 ±749 | 346 – 3325 |
| Leptin (ng/mL) | 17 | 0.56 ± 0.95 | 0.07 – 4.20 |
ALT: alanine aminotransferase, AST: aspartate aminotransferase, BMI: body mass index, HDL: high density lipoprotein, LDL: low density lipoprotein, NA: not applicable.
Figure 2: Pathogenic variants reported in the CAVIN1 gene.

A. Schematic drawing of the CAVIN1 gene and the pathogenic variants. Black rectangles are the coding regions of the exons and white rectangles indicate the non-coding regions. Solid line between the exons indicates the intronic region. CAVIN1 constitutes only two exons shown with numbers. The 5’ and 3’ ends are shown. Schematic is not drawn to scale. The numbering is as per the coding DNA.
B. Schematic of the CAVIN1 protein and the pathogenic variants. CAVIN1 constitutes 390 amino acids. Modified from Nassar. International Review of Cell and Molecular Biology, Volume 320, 2015. The disordered regions are shown in light blue and helical regions in gray. The PEST (proline–glutamic acid–serine–threonine) domains are shown in black, and the Leucine zipper domains in green. The nuclear localization signals are shown in dark blue.
Variants above the schematics are previously reported and those below are novel. Variants in orange color are reported in the paper and those in black color are not reported in the paper.
Myopathy with elevated serum CK levels (1434 ± 749 IU/L, range: 346–3325 IU/L) were present in all subjects (n = 38) at the time of diagnosis. Scoliosis was detected in 10 of 26 subjects (39%). Atlantoaxial instability was detected in 8 of 14 subjects (57%) in whom a radiological assessment was available. A representative image is shown in Figure 1F. Although not systematically investigated, we noted additional neuromuscular abnormalities (such as shortening of the Achilles tendon, pes cavus, leg pain, and gait abnormalities) in at least 4 patients.
Metabolic profiles of patients at the final data collection visit are shown in Table 2. Individual laboratory values are presented in Supplemental Table 1. Out of 43 patients with an available lipid profile, elevated serum triglycerides were detected in 34 (79%) patients and low HDL cholesterol in 40 (93%) patients. In 27 patients with available information regarding the age at onset of lipid abnormalities (excluding 17 patients from one center from Oman and 2 patients from one center in India with no available data), the mean age at diagnosis of hypertriglyceridemia was 9 ± 6 years (range: 16 months - 26 years). Kaplan-Meier estimate of the median time-to-diagnosis of hypertriglyceridemia was 9 years. Of 44 patients with serum glucose and/or blood HbA1c levels available, 9 (21%) had diabetes. The age at onset of diabetes was 15 ± 5 years, ranging from 3 to 20 years. Kaplan-Meier estimate of the median time-to-diagnosis of diabetes was 20 years (Figure 1C, panel I). Acanthosis nigricans was reported in 9 patients. Hepatic steatosis was detected in 27 of 33 patients (82%) in whom imaging was available. The mean age at the diagnosis of hepatic steatosis was 8 ± 6 years (range: from 1 to 24 years). Kaplan-Meier estimate of the median time-to-diagnosis of hepatic steatosis was 7 years. A representative image of a liver biopsy showing micro- and macrovesicular steatosis in a subject with CGL4 at age 1 is presented in Figure 1C, panel II.
Cardiac arrhythmias were detected in 20 (57%) subjects with a cardiac evaluation (n = 35). No cardiac work-up was done in 11 patients at the time of this report, and no exercise test was available in 15 subjects. Cardiac arrhythmias were detected in all 20 subjects in whom a full detailed cardiac evaluation was completed (Table 3). Sixteen patients had ventricular tachycardia. Ventricular tachycardia with syncope was reported in two patients. Other arrhythmias/conduction abnormalities were atrial fibrillation (n = 3), supraventricular and atrial tachycardias (n = 5), Wolff-Parkinson-White syndrome (n =1), premature ventricular and atrial contractions (n = 6), ventricular couplets (n = 1), and sinus tachycardia (n =2). Representative electrocardiography images showing various arrhythmias are presented in Figure 1D. Kaplan-Meier estimates of the median time-to-diagnosis of cardiac disease was 11.5 years (Figure 1C, panel III). Four patients received implantable cardioverter defibrillator (ICD) and one of these patients also underwent a cervical sympathectomy for CPVT. Sixteen patients were treated with beta-adrenergic blockers. ICD was able to prevent sudden death by detecting and treating ventricular arrhythmias with a shock in three patients, but one patient died due to sudden cardiac arrest despite ICD and cervical sympathectomy.
Table 3:
Cardiac manifestations in subjects with CGL4.
| Family ID | ID | Age of onset | Type of arrhythmia and other cardiac findings/ comments | Sudden death history in the siblings/family | Pacemaker implantation/sympathectomy |
|---|---|---|---|---|---|
| 1 | P1 | At age 15† | Non-sustained episodes of VT and polymorphic VES beats; catecholaminergic polymorphic VT; episodes of rapid atrial fibrillation | No | ICD at age 16 |
| 2 | P2 | At age 13 | Non-sustained VT and supraventricular tachycardia episodes consistent with atrial tachycardia; VT with syncope | No | No |
| 3 | P3 | At age 4 | Polymorphic VT (age 6); HCM (age 4) | No | No |
| 4 | P4 | At age 13 | Polymorphic VT (age 15); HCM (age 13) | No | No |
| 5 | P5 | At age 9 | VT | No | No |
| 6 | P6 | No cardiac work up | No cardiac work up | Yes | No |
| 7 | P7 | No | Screened by Holter ECG | No | No |
| 8 | P8 | At 18 months | Wolff-Parkinson-White syndrome (18 months); non-sustained VT and supraventricular tachycardia episodes (age 5); preexited atrial tachycardia | No | No |
| P9 | No | Screened by ECG | No | No | |
| 9 | P10 | At age 2 | Short runs of VT (age 2); VES and PAC (age 7) | No | No |
| P11 | No | Screened by ECG | No | No | |
| 10 | P12 | No | Screened by ECG | No | No |
| P13 | No | Screened by ECG | No | No | |
| 11 | P14 | At age 36 | Extra systoles; sinus tachycardia | Yes | No |
| 12 | P15 | At age 14 | Non-sustained VT | Unknown | ICD at age 14 |
| 13 | P16 | At age 17 | Polymorphic premature VES. ventricular couplets; non-sustained VT | No | No |
| 14 | P17 | No | Screened by ECG | No | No |
| P18 | At infancy | Non-sustained VT and supraventricular tachycardia episodes consistent with atrial tachycardia | No | No | |
| 15 | P19 | No | Screened by ECG | No | No |
| 16 | P20 | At 12 years | Biphasic catecholaminergic polymorphic VT; supraventricular tachycardia | Self | ICD at age 16; cervical sympathectomy at age 17 |
| P21 | At 4 years | Nonsustained ventricular ectopy | Self. brother at age 22 | No | |
| 17 | P22 | At 14 years | Catecholaminergic polymorphic VT | Brother at age 15 | No |
| P23 | At 8 years | Catecholaminergic polymorphic VT | Self | No | |
| 18 | P24 | No | No cardiac work up | No | No |
| 19 | P25 | At age 11 | VT with syncope; intermittent atrial fibrillation (age 13) | Sister at age 14 | ICD at age 13 |
| 20 | P26 | At age 9 | Non-sustained supraventricular tachycardia with occasional ventricular extrasystoles | No | No |
| P27 | No cardiac work up | No cardiac work up | No | No | |
| 21 | P28 | No | No exercise test | Yes | No |
| P29 | At 7 y | Sinus dysrhythmia | No | No | |
| P30 | At 40 y | Atrial fibrillation | No | No | |
| P31 | No cardiac work up | No cardiac work up | No | No | |
| P32 | No cardiac work up | No cardiac work up | No | No | |
| P33 | No cardiac work up | No cardiac work up | No | No | |
| P34 | No cardiac work up | No cardiac work up | No | No | |
| 22 | P35 | No | No exercise test | No | No |
| P36 | No | No exercise test | No | No | |
| P37 | No cardiac work up | No cardiac work up | No | No | |
| 23 | P38 | No cardiac work up | No cardiac work up | NA | No |
| P39 | No | No exercise test | No | No | |
| 24 | P40 | No | Sudden death at age of 14 | Self | No |
| 25 | P41 | At 3 y | PAC | No | No |
| P42 | No cardiac work up | No cardiac work up | No | No | |
| P43 | No cardiac work up | No cardiac work up | No | No | |
| 26 | P44 | No | No exercise test | No | No |
| 27 | P45 | No | Normal ECG and ECHO | NA | No |
| P46 | No | Normal ECG and ECHO | NA | No |
ECG: electrocardiogram, HCM: hypertrophic cardiomyopathy, PAC: premature atrial contractions, VES: ventricular extra systoles, VT: ventricular tachycardia
patient described palpitations first at age 12 but formally diagnosed with arrhythmia later at the age of 15.
Gastrointestinal abnormalities were observed in 29 patients (63%; Table 4). Of note, motility studies were not available in most patients. Eighteen patients (39%) were diagnosed with pyloric stenosis. Age at diagnosis was not reported in all patients; however, it ranged from first month of life to age of 20 years. Six patients underwent surgery for pyloric stenosis. Esophageal dysmotility was observed in 9 patients (20%) and intestinal dysmotility in 19 patients (45%). Four patients (9%) suffered from intestinal perforations (Figure 1E) (Acar et al., 2022). An additional patient developed colitis and extraluminal abscess and required a colostomy at age 21 years. Also, another male patient died of a cardiac event but was found to have a friable colon with pericolonic edema/abscess at autopsy (Table-4). Furthermore, one female patient developed severe iron deficiency, which was not responsive to oral iron. A pathologic oral iron absorption test indicated a gastrointestinal iron resorption issue in this patient. Iron refractory iron deficiency anemia was genetically excluded.
Table 4:
Gastrointestinal manifestations in subjects with CGL4
| Family ID | ID | Pyloric stenosis | Esophageal dysmotility or dysphagia | Intestinal dysmotility | Intestinal perforation |
|---|---|---|---|---|---|
| 1 | P1 | Yes (diagnosed at age 20) | No | Yes (age 11) | Yes (age 21) |
| 2 | P2 | No | No | Yes (age 13) | No |
| 3 | P3 | Yes (at 27 months; operated) | No | Yes | No |
| 4 | P4 | No | No | Yes (age 9) | Yes (age 13) |
| 5 | P5 | Yes (operated) | No | Yes | No |
| 6 | P6 | No | No | Yes (age 9) | No |
| 7 | P7 | No | No | Yes (age 2 years 8 months) | No |
| 8 | P8 | No | No | No | No |
| P9 | No | No | No | No | |
| 9 | P10 | Yes (operated) | No | Yes | No |
| P11 | Yes (12 weeks of age; operated) | No | No | No | |
| 10 | P12 | No | No | Yes | No |
| P13 | Yes (6 weeks of age) | No | No | No | |
| 11 | P14 | Yes | Yes | Yes (age 20) | Yes (age 20) |
| 12 | P15 | Yes | No | No | No |
| 13 | P16 | No | No | No | No |
| 14 | P17 | Yes (first month of life; operated) | No | Yes | No |
| P18 | No | No | No | No | |
| 15 | P19 | Yes (at 24 months) | No | Yes | No |
| 16 | P20 | Yes | No | Yes | No (autopsy showed friable colon with pericolonic cavity) |
| P21 | Yes | No | No | No | |
| 17 | P22 | No | Yes (in neonatal period) | Yes | No (colitis and extraluminal abscess requiring colostomy at age 21) |
| P23 | No | Yes (in infancy) | No | No | |
| 18 | P24 | No | Yes | Yes | No |
| 19 | P25 | Yes | Yes | Yes | No |
| 20 | P26 | No | Yes | Yes | No |
| P27 | Yes (operated in infancy) | Yes | Yes | No | |
| 21 | P28 | No | No | No | No |
| P29 | Yes | No | No | No | |
| P30 | No | Yes | Yes (age 39) | Yes (age 29) | |
| P31 | No | Yes | No | No | |
| P32 | No | No | No | No | |
| P33 | No | No | NA | No | |
| P34 | Yes | No | No | No | |
| 22 | P35 | No | No | No | No |
| P36 | No | No | No | No | |
| P37 | No | No | NA | No | |
| 23 | P38 | No | No | No | No |
| P39 | No | No | NA | No | |
| 24 | P40 | Yes | No | No | No |
| 25 | P41 | No | No | No | No |
| P42 | No | No | No | No | |
| P43 | No | No | NA | No | |
| 26 | P44 | Yes | No | No | No |
| 27 | P45 | No | No | No | No |
| P46 | No | No | No | No |
Signs and symptoms of intestinal dysmotility include early satiety, nausea, vomiting, bloating, diarrhea, and constipation. Signs and symptoms of esophageal dysmotility include heartburn, regurgitation, chest pain, dysphagia, hiccups, malnutrition, recurrent episodes of pneumonia or asthma.
The majority of patients in our cohort were young (12 patients younger than 5 years, 26 individuals younger than 12 years, and 35 patients younger than 18 years old). Only two patients were older than 35 years. One patient, whose initial presentation was previously reported by Sorkina et al.(Sorkina et al., 2020) developed diabetes at the age 19, had multiple surgeries for dolichosigmoid, perforation of diverticula and peritonitis between the ages of 17 and 20, and then developed foot ulcers on the right lower extremity at 35 years of age. Her condition deteriorated significantly after being affected by a severe COVID-19 infection that required a month-long stay in the intensive care unit. Subsequently, she developed deep leg ulcers and eventually passed away at age of 39 years after developing sepsis. The other patient was a 40-year-old female from Oman who developed diabetes at the age of 20 years and was diagnosed with CGL4 afterwards at the age of 25 years. Her glycemic control was satisfactory with intensive insulin treatment. She had low HDL cholesterol levels and effectively managed hypertriglyceridemia through dietary measures. At the age of 29, she experienced an intestinal perforation. Later, she was formally diagnosed with intestinal dysmotility. Subsequently, at the age of 40, she developed atrial fibrillation.
Seven patients died during follow-up. Mean age at death was 17 ± 11 years, ranging from 2 months to 39 years. Kaplan-Meier estimate of median time-to-death was 39 years (Figure 1C, panel IV). Cause of death was cardiac arrhythmia in three patients (age at death 22, 13, and 15 years in patients 20, 21, and 23, respectively. Another patient died suddenly at age of 14 years (patient 40). Cardiac disease was unknown in this patient when she died suddenly at school. An infant died at age of 2 months (patient 34) due to complications of prematurity complicated with severe infections and pulmonary hypertension. Patient 4 received abdominal surgery due to acute appendicitis when she was 9 years old. No formal testing was done to screen for gastrointestinal dysmotility at that time or thereafter. She developed recurrent gastrointestinal perforations starting at age 13 years, which required multiple operations. These events became complicated by intraabdominal infections and the development of enterocutaneous fistulas. Her general status did not improve, and she was started on total parenteral nutrition. At age 16, she developed sepsis and died due to multi-organ dysfunction (Nilay et al., 2020). Patient 14 died at age 39 due to nonhealing diabetic foot ulcers complicated with decubitus ulcers and sepsis as described above (Sorkina et al., 2020).
The list of medications used to treat complication of CGL4 is presented in Supplemental Table 2. A total of 11 patients (25%) with CGL4 were treated with metreleptin replacement; however, for this paper, their data were collected just prior to metreleptin initiation. Seven patients with CGL4 were on insulin (four in combination with metformin). The average daily insulin dose was 86 ± 47 units (2.09 ± 0.85 units/kg/day). For high triglycerides, 4 patients were treated with fish oil preparations, 2 with medium-chain triglyceride (MCT) oil, 1 with fibrates, and 1 patient a statin fibrate combination. It is worth mentioning that while myotoxicity has been reported with fibrates (Davidson et al., 2007), the patient who underwent long-term, full-dose fibrate treatment did not experience any clinical deterioration in myopathy, nor did we observe any significant elevation in muscle enzyme levels. However, lipid-lowering drugs were not able to normalize triglyceride levels in these patients. MCT oil helped control triglyceride levels in two young patients (6 years and 7 months, and 3 years and 8 months).
All patients had either homozygous or compound heterozygous null variants. We did not recognize any genotype-phenotype relationships, for example, 7 patients who died prematurely did not have a specific variant. Also, no clear genotype-phenotype association could be documented regarding development of metabolic abnormalities or cardiac and gastrointestinal manifestations.
4. Discussion
This case series highlights severe morbidity and risk of early mortality in patients with CGL4. This international case study is the largest reporting clinical outcomes in CGL4. Also, many centers were able to submit longitudinal data that helped us describe the natural history of CGL4 better than the previous case reports. CGL4 is a complex, ultra-rare disease with several organ system dysfunction contributing to morbidity and mortality. Our results reveal cardiomyopathy, gastrointestinal disorders, myopathy, and metabolic abnormalities as the major causes of morbidity, and cardiac arrhythmias and gastrointestinal complications as the major causes of mortality in CGL4.
Cardiac disease is a major risk factor for mortality in CGL4, which is characterized by life-threatening arrhythmias (Patni et al., 2022). Exercise-induced CPVT is the typical cardiac arrhythmia observed in patients with CGL4 (Rajab et al., 2010). Ventricular arrhythmias were identified in the majority of patients in our cohort. Three patients died due to cardiomyopathy and another patient developed sudden death likely due to a ventricular arrhythmia. Life expectancy is reduced in CGL4 and seems to be shorter than the general GL population. Cardiac arrhythmias were reported in only 5% of patients with GL in the previous International Chart Review Study that enrolled 81 patients with GL at five treatment centers from three counties (Akinci et al., 2019). Kaplan-Meier estimates of mean age to death was 51 years in the overall GL cohort.
CAVIN1 encodes a cytoplasmic protein essential for caveolae formation that regulate substrate trafficking through the cytoplasm and acts as a mechanoprotective element and signal transduction platform (Hill et al., 2008). Cavin1−/− mice show reduction of caveolae in cardiomyocytes and develop cardiomyocyte hypertrophy accompanied by progressive interstitial/perivascular fibrosis (Taniguchi et al., 2016). Autopsy findings from a young male with CGL4 showed near occlusion of the sinoatrial nodal artery, with adipose tissue and strands of myocardium surrounding it. The atrioventricular node showed mild fibro-fatty infiltration near the coronary sinus area (Patni et al., 2019). Ventricular arrhythmias are usually triggered by exercise in CGL4, thus a normal baseline ECG and Holter testing do not rule out predisposition to CPVT. Recordings during exercise can document ventricular arrhythmia. Because these arrhythmias can be life-threatening, all precautions should be taken during the testing procedure. Small children can only be diagnosed when they are old enough to do an exercise test but may still suffer from sudden cardiac death before the cardiac disease is diagnosed. In addition to ventricular arrhythmias, we observed atrial fibrillation, atrial tachycardia, Wolff-Parkinson-White syndrome, supraventricular tachycardia, premature ventricular and atrial contractions, ventricular couplets, and sinus tachycardia in our cohort, highlighting the complex nature of cardiac disease in CGL4. The management of cardiac disease in CGL4 requires assistance from a cardiologist with expertise in treating such arrhythmias. Beta-adrenergic blockers are usually the first choice and can be combined with other antiarrhythmics such as flecainide. These patients should avoid heavy exercise that can easily trigger a life-threatening arrhythmia. ICD implementation can be performed to prevent sudden cardiac death (Akinci et al., 2016; Rajab et al., 2010). Cervical sympathectomy can be helpful to treat CPVT (Kochav et al., 2022). However, due to limited clinical experience with the medications as well as ICD and cervical sympathectomy, the efficacy of these procedures in preventing CPVT in patients with CGL4 remains unclear. Echocardiography was available only in a subset of patients in our series. Although we did not observe any decrease in ejection fraction, hypertrophic cardiomyopathy was found in a 4-year-old boy (current age: 8 years and 5 months old) and 13 years old girl (later died at age 16).
Our study highlights severe morbidity and mortality due to gastrointestinal disease in CGL4. Gastrointestinal abnormalities are characterized by dysmotility that can affect several segments of the gastrointestinal tract. Pyloric stenosis is an early complication that can be detected during the first years of life. Esophageal and intestinal dysmotility are quite common. The etiology of gastrointestinal disease largely remains unknown in CGL4, although myopathy seems to play a central role in the pathogenesis. Previous studies reported smooth muscle hypertrophy and thick muscularis mucosa in patients with CGL4 who suffered from pyloric stenosis (Nilay et al., 2020; Rajab et al., 2002; Rajab et al., 2010; Shastry et al., 2010). Autopsy findings from a patient with CGL4 revealed a thick muscularis mucosa in the esophagus and at the gastro-duodenal junction (Patni et al., 2019). Similarly, histopathology specimens from a patient with colonic perforation in our cohort showed muscularis propria hypertrophy along with a loss of orientation of circular and longitudinal layers of muscularis propria (Acar et al., 2022). Symptoms of intestinal dysmotility may not be obvious in all patients. However, deep symptom questioning may reveal the abnormalities that can be further confirmed by diagnostic tests. Development of intestinal perforations in four patients, leading to death in one case, and a colostomy procedure in another patient in our cohort demonstrates a high risk for serious intestinal disease in CGL4. Change in bowel habits, blood in stool, and unexplained fever should prompt consideration of bowel perforation and early diagnosis can prevent catastrophic complications.
Myopathy is a distinctive feature of CGL4 and patients may exhibit generalized or distal weakness (Hayashi et al., 2009). An elevated serum CK level in a subject with generalized fat loss should always prompt genetic testing for CGL4. While not systematically examined in the entire cohort, our observations among Turkish cases indicated consistently stable CK elevations during a relatively short follow-up period. Percussion induced muscle mounding is a simple maneuver that can reveal myopathy in CGL4. Our study demonstrated that neuromuscular disease can be debilitating in CGL4. Although our study lacked an extensive dataset for tracking disease progression, a subset of patients underwent surgery for neuromuscular issues, such as scoliosis, during the follow-up period. Also, shortening of the Achilles tendon, pes cavus, and gait abnormalities can cause mobility problems and may affect patients’ daily life. Furthermore, the identification of atlantoaxial instability is important in CGL4; this abnormality was frequently found when investigated. Although most cases are asymptomatic, patients with atlantoaxial instability may suffer from neck pain, neck movement restriction, weakness, and numbness. Although rare, atlantoaxial instability may lead to serious spinal cord compression resulting in neurological deficits (Yang et al., 2014). These risks can be especially important for patients undergoing surgical procedures that would require intubation.
Like other subtypes of CGL, patients with CGL4 manifest with near total lack of adipose tissue and low serum leptin levels leading to a high risk of developing severe metabolic abnormalities (Yildirim Simsir et al., 2023). These metabolic abnormalities potentially lead to organ complications and may affect survival rates (Brown et al., 2016). Although some heterogeneity exists, our data suggest that metabolic abnormalities usually develop in childhood and adolescence in CGL4. The timing of the onset of lipid abnormalities and hepatic steatosis seems to be similar to other major CGL subtypes. A previous international chart review study reported hypertriglyceridemia in 82% and hepatic steatosis in 68% of patients with GL (Akinci et al., 2019). Similarly, hypertriglyceridemia was reported in 90% of patients included in the Turkish GL registry and the Kaplan-Meier estimate of the median time to diagnosis of hypertriglyceridemia was 14 years (Yildirim Simsir et al., 2023). The benefit to risk ratio of treating patients with CGL4 with fibrates is not clear as fibrates have a potential side effect of inducing myopathy (Davidson et al., 2007). However, it is not known that fibrates given for hyperlipidemia worsen myopathy in CGL4 patients. One patient in our cohort was treated with fibrates for a long time due to severe hypertriglyceridemia, and no progression in clinical findings of myopathy or a further increase in CK values was observed. Nevertheless, this potential side effect of fibrates should be considered in the treatment of CGL4 patients, and the benefit-risk ratio should be carefully evaluated in the use of these agents. An initial option in CGL4 patients to improve hypertriglyceridemia may be a low-fat diet and long chain n-3 polyunsaturated fatty acids from fish oils. Statin alone or in combination with fibrates should be in general avoided for potential high risk of myopathy and rhabdomyolysis (Enger et al., 2010).
Diabetes, on the other hand, was relatively rarely reported in previous case reports of CGL4. In our cohort, one-fifth of the patients had diabetes. The age at onset of diabetes was 15 years, ranging from 3 to 20 years, and Kaplan-Meier estimate of the median time-to-diagnosis of diabetes was 20 years which seems to be slightly later than other major CGL subtypes. Diabetes was reported in 58% of the patients with generalized lipodystrophy included in the International Chart Review Study which reported that Kaplan-Meier estimates of mean time to diagnosis of diabetes and/or insulin resistance was 13 years in GL (Akinci et al., 2019). Because most patients in our cohort are young, it is difficult to make assumptions regarding the natural history of metabolic disease in CGL4. However, several case examples in our cohort highlight the metabolic burden of CGL4 and the need to treat these complications. For example, we observed rapid deterioration of metabolic health after puberty in two adolescent girls. Diabetes complicated with extremity ulcers leading to sepsis caused the death of a patient in her thirties. Diabetes was diagnosed at age 3 years in an Asian Indian female of consanguineous parentage. She had an episode of pancreatitis at age 6 years, likely due to hypertriglyceridemia. She has also been diagnosed with proteinuria causing hypoalbuminemia and compensated cirrhosis. Her metabolic disease is still not controlled despite treatment with basal bolus insulin regimen (2.5 units/kg/day) along with metformin and bromocriptine and a statin fibrate combination for hypertriglyceridemia. It is not clear why patients with CGL4 develop diabetes relatively later than other major CGL subtypes. Whether pyloric stenosis, esophageal and intestinal dysmotility result in reduced energy intake and thereby a delay in development of DM in CGL4 remains unknown. Nevertheless, we suggest that all patients with CGL4 should be monitored for metabolic disease, and treatment strategies should follow international guideline recommendations for patients with generalized lipodystrophy.
Interestingly, all patients had biallelic null variants of the CAVIN1 gene in our cohort. This raised the possibility that there may be milder versions of CGL4 due to missense variants that may exhibit mild lipodystrophy, myopathy, and cardiomyopathy. Our literature search did not reveal any missense variants; however, we identified a previously reported case showing milder fat loss due to a homozygous single base pair deletion (c.947delA) that led to a frameshift and introduction of the first stop codon 27 residues downstream of the correct stop codon, producing a protein which was predicted to be larger than the wild type protein (Ardissone et al., 2013). Although current evidence is lacking to demonstrate milder versions of CGL4, clinicians should be aware of the potential existence of milder CGL4 cases due to less harmful variants. Interestingly, the GnomAd database reveals no common missense variant with minor allele frequency exceeding 0.01 in CAVIN1, the most prevalent missense variant being p.Val119Asp (rs146547678) with minor allele frequency of 0.002088.
Our study has several limitations. First, this is a retrospective chart review study that collected data from medical charts at different international centers. It is likely that different centers have different monitoring algorithms that may affect our results. However, in an ultra-rare disease setting, achieving relatively large numbers are only possible through international collaboration. Second, our discussion compares our cohort to data from International Chart Review Study that includes data from three counties (Turkey, US, and Brazil). Unfortunately, GL registries are not widely available. It was not possible to generate a comparison group that may reflect all ethnicities.
5. Conclusion
We identified major causes of serious morbidity and mortality in a relatively large group of patients with CGL4 from multiple international centers. Our results highlight the importance of regular monitoring for cardiac, gastrointestinal, neuromuscular, and metabolic complications in CGL4 and the urgency to treat the consequences of these abnormalities as they can be presently serious or may easily progress to life-threatening clinical presentations.
Supplementary Material
Acknowledgments
The authors express their gratitude to Gonca Topuz, Suleyman C. Adiyaman, Canan Altay, Irina Ulyanova, Liubov Bolotskaya, Anica Bulic, Dominic Ng, Nilay Gunes, Tahir Atik, Samim Ozen, and Huseyin Onay for their valuable contributions to the clinical care of the patients.
Funding statement
This work was supported by grants from the National Institutes of Health, R01-DK105448 and Southwestern Medical Foundation, and by the intramural research program of the National Institute of Diabetes and Digestive and Kidney Diseases. This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors. No sponsor is involved in study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
Footnotes
Conflict of interest
BA run projects for and/or served as a consultant, board member, steering committee member, and/or speaker to Alnylam, Amryt, Regeneron, ThirdRock Ventures, Astra Zeneca, Novonordisk, Boehringer Ingelheim, Sanofi, Bilim Ilac, ARIS, and Servier. AG consults for Amryt Pharma plc, Regeneron, Kyttaro Limited, Third Rock Ventures, and has received grant support from Amryt Pharma plc, Regeneron, Quintiles, Akcea Pharmaceuticals, and Intercept Pharmaceuticals. AG is a co-holder of patents for use of leptin for treating human lipoatrophy and method of determining predisposition to said treatment but receives no financial benefit. RJB reports research support from Amryt Pharmaceuticals and Regeneron Pharmaceuticals. EAO reports the following conflicts: Grant support: Aegerion Pharmaceuticals (now Amryt Pharmaceuticals), Ionis Pharmaceuticals, Akcea Therapeutics, Gemphire Therapeutics, GI Dynamics (current), AstraZeneca (past two years). Consultant or Advisor: AstraZeneca, Thera Therapeutics, and BMS (past), Aegerion Pharmaceuticals (now Amryt Pharmaceuticals), Regeneron Pharmaceuticals (current). Drug support: Aegerion Pharmaceuticals (now Amryt Pharmaceuticals), Akcea Therapeutics, Rhythm Pharmaceuticals (all current). Other support: Aegerion Pharmaceuticals (now Amryt Pharmaceuticals), Regeneron Pharmaceuticals (current). TS consulted for Amryt and received grant support for an investigator-initiated trial from Aegerion Pharmaceuticals (now Amryt Pharmaceuticals). MB received an Honorarium as a speaker for Amryt Pharmaceuticals. CK received fees for Lecture and Advisory Board Membership from Amryt Pharmaceuticals. MW has served as consultant for Alnylam, Amryt, LG Chem, Regeneron, ThirdRock Ventures and is PI in clinical studies sponsored by Amryt. NP served as board member and consultant for Amryt Pharmaceutical and Ionis. ES attended advisory board/steering committee meetings organized by Amryt Pharmaceuticals and Regeneron Pharmaceuticals, Inc. Other authors report no conflicts of interest.
Data Availability Statement
Some or all datasets generated during and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.
References
- Acar N, Acar T, Suataman B, Ekinci N, & Tatar F (2022). Coexistence of Colon Perforation and Congenital Lipodystrophy. J Coll Physicians Surg Pak, 32(9), 1222–1224. 10.29271/jcpsp.2022.09.1222 [DOI] [PubMed] [Google Scholar]
- Adiyaman SC, J VS, De Laffolie J, Hahn A, Siebert R, Wabitsch M, & Kamrath C (2022). Congenital generalized lipodystrophy type 4 due to a novel PTRF/CAVIN1 pathogenic variant in a child: effects of metreleptin substitution. J Pediatr Endocrinol Metab, 35(7), 946–952. 10.1515/jpem-2022-0022 [DOI] [PubMed] [Google Scholar]
- Agarwal AK, Arioglu E, De Almeida S, Akkoc N, Taylor SI, Bowcock AM, . . . Garg A (2002). AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat Genet, 31(1), 21–23. 10.1038/ng880ng880 [pii] [DOI] [PubMed] [Google Scholar]
- Akinci B, Oral EA, Neidert A, Rus D, Cheng WY, Thompson-Leduc P, . . . Brown RJ (2019). Comorbidities and Survival in Patients With Lipodystrophy: An International Chart Review Study. J Clin Endocrinol Metab, 104(11), 5120–5135. 10.1210/jc.2018-02730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinci B, Sahinoz M, & Oral E (2000). Lipodystrophy Syndromes: Presentation and Treatment. In Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, Dungan K, Hershman JM, Hofland J, Kalra S, Kaltsas G, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, Morley JE, New M, Purnell J, Sahay R, Singer F, Sperling MA, Stratakis CA, Trence DL, & Wilson DP (Eds.), Endotext. https://www.ncbi.nlm.nih.gov/pubmed/29989768 [PubMed]
- Akinci G, Topaloglu H, Akinci B, Onay H, Karadeniz C, Ergul Y, . . . Garg A (2016). Spectrum of clinical manifestations in two young Turkish patients with congenital generalized lipodystrophy type 4. Eur J Med Genet, 59(6–7), 320–324. 10.1016/j.ejmg.2016.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinci G, Topaloglu H, Demir T, Danyeli AE, Talim B, Keskin FE, . . . Akinci B (2017). Clinical spectra of neuromuscular manifestations in patients with lipodystrophy: A multicenter study. Neuromuscul Disord, 27(10), 923–930. 10.1016/j.nmd.2017.05.015 [DOI] [PubMed] [Google Scholar]
- American Diabetes Association Professional Practice, C. (2022). 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2022. Diabetes Care, 45(Suppl 1), S17–S38. 10.2337/dc22-S002 [DOI] [PubMed] [Google Scholar]
- Ardissone A, Bragato C, Caffi L, Blasevich F, Maestrini S, Bianchi ML, . . . Mora M (2013). Novel PTRF mutation in a child with mild myopathy and very mild congenital lipodystrophy. BMC Med Genet, 14, 89. 10.1186/1471-2350-14-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babalola F, Ng D, Bulic A, & Curtis J (2022). Successful treatment of severe hypertriglyceridemia with icosapent ethyl in a case of congenital generalized lipodystrophy type 4. J Pediatr Endocrinol Metab, 35(7), 968–972. 10.1515/jpem-2021-0718 [DOI] [PubMed] [Google Scholar]
- Brown RJ, Araujo-Vilar D, Cheung PT, Dunger D, Garg A, Jack M, . . . Yorifuji T (2016). The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J Clin Endocrinol Metab, 101(12), 4500–4511. 10.1210/jc.2016-2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels SR, Greer FR, & Committee on N (2008). Lipid screening and cardiovascular health in childhood. Pediatrics, 122(1), 198–208. 10.1542/peds.2008-1349 [DOI] [PubMed] [Google Scholar]
- Davidson MH, Armani A, McKenney JM, & Jacobson TA (2007). Safety considerations with fibrate therapy. Am J Cardiol, 99(6A), 3C–18C. 10.1016/j.amjcard.2006.11.016 [DOI] [PubMed] [Google Scholar]
- Enger C, Gately R, Ming EE, Niemcryk SJ, Williams L, & McAfee AT (2010). Pharmacoepidemiology safety study of fibrate and statin concomitant therapy. Am J Cardiol, 106(11), 1594–1601. 10.1016/j.amjcard.2010.07.041 [DOI] [PubMed] [Google Scholar]
- Expert Panel on Detection, E., & Treatment of High Blood Cholesterol in, A. (2001). Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA, 285(19), 2486–2497. 10.1001/jama.285.19.2486 [DOI] [PubMed] [Google Scholar]
- Garg A (2011). Clinical review#: Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab, 96(11), 3313–3325. 10.1210/jc.2011-1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A, Wilson R, Barnes R, Arioglu E, Zaidi Z, Gurakan F, . . . Bowcock AM (1999). A gene for congenital generalized lipodystrophy maps to human chromosome 9q34. J Clin Endocrinol Metab, 84(9), 3390–3394. [DOI] [PubMed] [Google Scholar]
- Hayashi YK, Matsuda C, Ogawa M, Goto K, Tominaga K, Mitsuhashi S, . . . Nishino I (2009). Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest, 119(9), 2623–2633. 10.1172/JCI38660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MM, Bastiani M, Luetterforst R, Kirkham M, Kirkham A, Nixon SJ, . . . Parton RG (2008). PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell, 132(1), 113–124. 10.1016/j.cell.2007.11.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain I, Patni N, & Garg A (2019). Lipodystrophies, dyslipidaemias and atherosclerotic cardiovascular disease. Pathology, 51(2), 202–212. 10.1016/j.pathol.2018.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CA, Delepine M, Boutet E, El Mourabit H, Le Lay S, Meier M, . . . Magre J (2008). Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab, 93(4), 1129–1134. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18211975 [DOI] [PubMed] [Google Scholar]
- Kochav SM, Garan H, Gorenstein LA, Wan EY, & Yarmohammadi H (2022). Cardiac Sympathetic Denervation for the Management of Ventricular Arrhythmias. J Interv Card Electrophysiol, 65(3), 813–826. 10.1007/s10840-022-01211-2 [DOI] [PubMed] [Google Scholar]
- Magre J, Delepine M, Khallouf E, Gedde-Dahl T Jr., Van Maldergem L, Sobel E, . . . Capeau J (2001). Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet, 28(4), 365–370. [DOI] [PubMed] [Google Scholar]
- Mancioppi V, Daffara T, Romanisio M, Ceccarini G, Pelosini C, Santini F, . . . Prodam F (2023). A new mutation in the CAVIN1/PTRF gene in two siblings with congenital generalized lipodystrophy type 4: case reports and review of the literature. Front Endocrinol (Lausanne), 14, 1212729. 10.3389/fendo.2023.1212729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz A, Radulescu A, Baerg J, Mendez Y, & Khan FA (2019). Bowel perforation in a pediatric patient with congenital generalized lipodystrophy type 4. Journal of Pediatric Surgery Case Reports, 48. 10.1016/j.epsc.2019.101257 [DOI] [Google Scholar]
- Nilay G, Kutlu T, Tekant GT, Eroglu AG, Ustundag NC, Ozturk B, . . . Tuysuz B (2020). Congenital generalized lipodystrophy: The evaluation of clinical follow-up findings in a series of five patients with type 1 and two patients with type 4. Eur J Med Genet, 63(4), 103819. 10.1016/j.ejmg.2019.103819 [DOI] [PubMed] [Google Scholar]
- Patni N, Hegele RA, & Garg A (2022). Caveolar dysfunction and lipodystrophies. Eur J Endocrinol, 186(3), C1–C4. 10.1530/EJE-21-1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patni N, Vuitch F, & Garg A (2019). Postmortem Findings in a Young Man With Congenital Generalized Lipodystrophy, Type 4 Due to CAVIN1 Mutations. J Clin Endocrinol Metab, 104(3), 957–960. 10.1210/jc.2018-01331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajab A, Heathcote K, Joshi S, Jeffery S, & Patton M (2002). Heterogeneity for congenital generalized lipodystrophy in seventeen patients from Oman. Am J Med Genet, 110(3), 219–225. 10.1002/ajmg.10437 [DOI] [PubMed] [Google Scholar]
- Rajab A, Straub V, McCann LJ, Seelow D, Varon R, Barresi R, . . . Schuelke M (2010). Fatal cardiac arrhythmia and long-QT syndrome in a new form of congenital generalized lipodystrophy with muscle rippling (CGL4) due to PTRF-CAVIN mutations. PLoS Genet, 6(3), e1000874. 10.1371/journal.pgen.1000874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shastry S, Delgado MR, Dirik E, Turkmen M, Agarwal AK, & Garg A (2010). Congenital generalized lipodystrophy, type 4 (CGL4) associated with myopathy due to novel PTRF mutations. Am J Med Genet A, 152A(9), 2245–2253. 10.1002/ajmg.a.33578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkina E, Makarova P, Bolotskaya L, Ulyanova I, Chernova T, & Tiulpakov A (2020). Unusual clinical features associated with congenital generalized lipodystrophy type 4 in a patient with a novel E211X CAVIN1 gene variant. Clin Diabetes Endocrinol, 6, 7. 10.1186/s40842-020-00095-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T, Maruyama N, Ogata T, Kasahara T, Nakanishi N, Miyagawa K, . . . Ueyama T (2016). PTRF/Cavin-1 Deficiency Causes Cardiac Dysfunction Accompanied by Cardiomyocyte Hypertrophy and Cardiac Fibrosis. PLoS One, 11(9), e0162513. 10.1371/journal.pone.0162513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SY, Boniello AJ, Poorman CE, Chang AL, Wang S, & Passias PG (2014). A review of the diagnosis and treatment of atlantoaxial dislocations. Global Spine J, 4(3), 197–210. 10.1055/s-0034-1376371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildirim Simsir I, Tuysuz B, Ozbek MN, Tanrikulu S, Celik Guler M, Karhan AN, . . . Akinci B (2023). Clinical features of generalized lipodystrophy in Turkey: A cohort analysis. Diabetes Obes Metab. 10.1111/dom.15061 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Some or all datasets generated during and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.