

Abstract
Hydrogen-bonded organic frameworks (HOFs) represent an interesting type of polymeric porous materials that can be self-assembled through H-bonding between organic linkers. To realize permanent porosity in HOFs, stable and robust open frameworks can be constructed by judicious selection of rigid molecular building blocks and hydrogen-bonded units with strong H-bonding interactions, in which the framework stability might be further enhanced through framework interpenetration and other types of weak intermolecular interactions such as interactions. Owing to the reversible and flexible nature of H-bonding connections, HOFs show high crystallinity, solution processability, easy healing and purification. These unique advantages enable HOFs to be used as a highly versatile platform for exploring multifunctional porous materials. Here, the bright potential of HOF materials as multifunctional materials is highlighted in some of the most important applications for gas storage and separation, molecular recognition, electric and optical materials, chemical sensing, catalysis, and biomedicine.
1. Introduction
Porous materials or porous media have long been the preeminent platforms for global scientists and engineers to explore novel multifunctional materials,1 which are widely applied for filtration, separation, purification, extraction, cooling, drying and catalysis, involving materials science, engineering, mechanics, geosciences and biology. Many common substances, such as charcoal, zeolites and ceramics, can be regarded as porous media, featuring permanent and interconnected voids for gas/liquid permeability. Zeolites are microporous silicates derived from tetrahedral orthosilicate connected by strong Si–O and Al–O bonds, including over 200 unique zeolite frameworks with a Brunauer–Emmett–Teller (BET) surface area of 100–1000 m2 g−1.2–4 Zeolite materials have been well explored as commercial adsorbents and catalysts for many important industrial applications in heterogeneous catalysis, gas separation and ion exchange, giving an annual worldwide market of millions of tonnes. For instance, synthetic mesoporous zeolites, such as MCM-41, have been widely used as catalysts for fluid catalytic cracking and hydrocracking in the petrochemical industry. It should be noted that zeolites have already made a great impact on the current society.
Other important types of porous crystalline materials including metal–organic frameworks (MOFs),5 porous coordination polymers (PCPs),6,7 or covalent organic frameworks (COFs)8–10 have been emerging over the past two decades. MOFs and COFs are crystalline porous materials that can be straightforwardly self-assembled from various molecular building blocks through strong coordinative or covalent bonds, featuring exceptional porosity, high modularity and diverse functionality.11,12 The initial efforts to construct coordination polymers with potential open pore structures can be dated back to 1989.13 However, it took almost one decade to realize the establishment of permanent porosity for the very first MOFs by gas sorption measurements.14,15 By virtue of secondary building units (SBUs) featuring high rigidity and directionality combined with organic struts, thousands and thousands of MOFs with exceptional porosity far superior to zeolites have been developed, showing a BET surface area of 1000–10 000 m2 g−1.5 The implementation of the building block approach and isoreticular principle, especially the discovery of open metal sites, to construct functional MOFs further spurred more extensive studies in this field,16 making it become one of the most rapidly expanding fields among the communities of chemistry, materials science and chemical engineering.17,18 MOFs have been envisioned as versatile porous materials for gas storage and separation,19–23 chemical sensing,24 heterogeneous catalysis25 and biomedicine.26,27 In fact, the commercialization of MOFs has been launched by business entities like BASF, NuMat Technologies and MOF Technologies.
Another type of extended porous frameworks (referred to as hydrogen-bonded organic frameworks,28–32 HOFs), from the self-assembly of discrete organic molecules via intermolecular hydrogen-bonding interactions, have also been proposed as potential porous materials more than two decades ago. The inherent features of H-bonding connections (weak, flexible, poorly directional, and reversible) mean that HOFs have some intriguing differences from zeolites, MOFs and COFs, such as solution processability and characterization, easy purification and healing by simple recrystallization. Although the initial studies to construct HOFs with guest inclusions have been presented since the early 1990s,33–50 it was not until 2010 that examples of HOFs with permanent porosity established by gas sorption isotherms have been reported.28,51 A similar but shorter development process can be found when looking back at the early stage of exploring MOFs with permanent porosity in the 1990s.13–15 Based on the pioneering work on the construction of extended organic networks by Wuest and others,33–50 we demonstrated the permanent porosity of HOFs for their potential applications in gas separation. It is well understood that the activation of HOFs is more challenging than that of other porous frameworks including MOFs if taking the labile nature of H-bonds into account, as they are indeed much weaker than the covalent and coordinative ones. The removal of included guest molecules typically results in denser isomers, so most organic extended frameworks are not sufficiently robust to retain porosity upon guest removal. Only HOFs featuring rigid and directional building units can be demonstrated to show permanent porosity. Therefore, the establishment of permanent porosity for HOFs represented a turning-point in this field. By virtue of rigid backbones, the record BET surface area for HOFs reached over 2700 m2 g−1 by Oppel in 2012,52 and exceeds 3400 m2 g−1 as the current benchmark.53 The achievement of high porosity of HOFs imparted momentum to the development of multifunctional HOFs and spurred interest in the exploitation of new HOFs with recorded porosity. To date, HOFs have shown great potential as a versatile platform to explore novel porous materials for gas storage and separation, molecular recognition, conductive and optical applications, heterogeneous catalysis, and biomedicine.
Since the early development of hydrogen-bonded organic frameworks, the scope for this type of porous materials has broadened rapidly, in terms of structures and applications. It is now time to reassess HOFs, not as a new area but as a more mature field. In this Review, we focus on selected HOF materials as examples to provide broader concepts for readers who are new to this field. We discuss the unique features that set HOFs apart from other porous frameworks. We have highlighted the progress in some of the most important applications of HOF materials.
2. Chemistry of HOFs
HOFs are assembled from organic molecules based on certain H-bonding interactions. Compared to covalent linkages (, , ) in COFs, coordination bonds (, ) in MOFs, and Si–O and Al–O bonds in zeolites, hydrogen bonds in HOFs are relatively weak, which enables reversible reactions to occur during the crystallization of HOFs, giving highly crystalline structures. But the other side of the coin is that challenges lie in the construction of stable, rigid and permanently porous HOFs. Several approaches have been successfully applied for the concerns involving polymorph formation during crystallization, functional sites, and framework stability after guest removal and establishment of porosity.
2.1. Conceptual basis
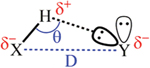
The prototypical H-bond refers to interaction between two water molecules (), which can be further extended to other systems including a hydrogen atom between two electronegative atoms (, and being mainly , , ). According to the IUPAC definition, the hydrogen bond is an attractive interaction between a hydrogen atom from a molecule or a molecular fragment X–H in which X is more electronegative than H and an atom or a group of atoms in the same or a different molecule, in which there is evidence of bond formation. Hydrogen bonded organic frameworks are defined as frameworks connected through H-bonding interactions between the organic units including both pure organic and metal-containing organic moieties (Table 1 and Fig. 1), which can be further enforced by other weak interactions such as the interactions, van der Waals interactions, dipole–dipole interactions, halogen bonds, the cation⋯ interactions and so on.55
Table 1.
Criteria for H-bonds adapted from Steiner and Jeffrey55
|
Strong | Moderate | Weak |
|---|---|---|---|
| 2.2–2.5 | 2.5–3.2 | >3.2 | |
| 1.2–1.5 | 1.5–2.2 | >2.2 | |
| 170–180 | >130 | >90 | |
| X-H vs. H⋯Y | X-H-H⋯Y | X-H > H⋯Y | X-H » H⋯Y |
| Bond energy/kJ mol−1 | 63–167 | 17–63 | <17 |
Fig. 1.

Various types of hydrogen bonds. This sketch is not exactly quantitative but the coloring attempts to give a visual scale of bonding energies. Data from Steiner’s paper.55
The H-bond energies in most systems are usually within 10–40 kJ mol−1, which extend to 1–170 kJ mol−1 when considering other extreme cases, but still lower than those of coordination bonds (90–350 kJ mol−1) and covalent bonds (300–600 kJ mol−1).8 Hence, the rigidity and directionality of H-bonds are lower than those of coordination bonds and covalent bonds, which makes the rational design of extended frameworks more challenging. According to several criteria including the bonding energies of H-bonds, these interactions were proposed to be catalogued into weak, moderate and strong H-bonds. In terms of constructing stable and rigid HOFs, only relatively strong H-bond interactions are feasible, as labile H-bonds may lead to polymorph problem, flexibility or framework collapse after desolvation. Weak H-bonds feature long bonding distances and poor directionality, giving different polymorphs with negligible thermal energy difference. Given that close-packed structures are thermodynamically favoured, once the kinetically formed open structures cannot be stabilized by H-bond interactions, any external stimulus like vacuum would lead to their solid-state transformation to dense structures. Therefore, applying weak H-bonds for HOF construction can be problematic, and make it more complicated.
On the other hand, highly directional and strong H-bonds, mainly intermolecular interactions, often dominate and widely exist in many supramolecular systems. Based on the detailed analyses of crystal structures from the Cambridge Structural Database (CSD), interactions from nearly 140 000 structures show a mean bonding distance of 2.77 Å, while interactions from nearly 120 000 structures show a mean bonding distance of 2.88 Å (Fig. 2). The stronger H-bond with a shorter bonding distance is accompanied by a better directionality (i.e. bond angle, distribution centred at 1711 vs. 1681). From the perspective of rational design, compared to weak H-bonds, highly directional and strong H-bond interactions help to simplify the chemistry of extended networks from the self-assembly of organic molecules.
Fig. 2.

Distributions of (a) and bond-length and (b) corresponding directionality, based on analyses of crystal structures from the Cambridge Structural Database with updates by August 2018. Structures obtained from the database were applied with a bonding cutoff of 3.04 (for ) and 3.07 Å (for ), i.e. the sum of van der Waals radii. The bond angle cutoff is 4120°.
Although discrete organic compounds especially cage like molecules can also exhibit intrinsic, shape-persistent voids, the absence of distinct intermolecular H-bonds makes these porous molecules zero-dimensional, suffering from polymorphism upon aggregation. This class of compounds has long been developed as porous solids, e.g. tris(-phenylenedioxy)cyclophosphazene (TPP),56–59 Dianin’s compound,60–62 calixarenes,63–68 cucurbiturils,69,70 Noria,71 a series of imine cages,72–74 and so on.75–79 Actually, porous organic cages are unique for their intrinsic porosity that exists even in the discrete state,80 which makes them another type of porous organic solids.81
Combining the above together, we here give a working definition for HOFs as porous crystals of extended structures composed of organic molecular building units, with moderate to strong H-bond linkage that specifically involves the H-atom. In most cases, HOFs mainly feature nitrogen or oxygen moieties as donors or acceptors for highly directional H-bonding.
2.2. General design principle
The basic concerns for constructing HOFs by rational design should focus on porosity, framework rigidity and material stability. In this regard, certain approaches demonstrated in the construction of other crystalline materials might be applicable to HOF synthesis. But, the intrinsic nature of organic molecules with corresponding H-bonding makes the self-assembly of HOFs more complicated. Accordingly, the design principles for HOF synthesis need extra insights. Moreover, additional concerns for constructing HOFs with functional sites should also be taken into account.
Given the diversified organic groups of H-bonding potential, many molecular moieties can be applied for H-bonding interactions (Fig. 3), including carboxylic acid,82–87 pyrazole,88 2,4-diaminotriazine,89,90 amide,91 benzimidazolone, imide, imidazole, amidinium,92 boronic acid,42 resorcinol,38,93 pyridine, 2,6-diaminopurine,94 and so on.95,96 It should be noted that, owing to the flexible nature of the H-bond, these organic groups can give enormous polymorphs by rational assembly in different geometry. But only energetically favourable frameworks can be crystallized from molecular building blocks. Cooper and Day et al. presented a good example to identify highly porous HOFs with high performance for target applications, by using energy–structure–function maps of vast computational crystal structures.53 Their work demonstrated that the experience and principles proved successful in MOFs and COFs cannot simply be applied to the design of HOFs.
Fig. 3.

Various O/N containing organic groups for potential H-bonding units, including carboxylic acid, pyrazole, 2,4-diaminotriazine, amide, benzimidazolone, imide, imidazole, amidinium, and so on.
In principle, organic groups with an equal number of H-bonding donors and acceptors are more suitable for HOF generation because these H-bonding donors/acceptors can distinctly form certain inherent H-bonding units, which can be dimers, trimers, and even chain structures (Fig. 4). Apparently, the H-bond building units are more rigid and directional than single H-bonding donor/acceptor pairs, which facilitate the construction of HOFs. Therefore, combining H-bond building units with suitable organic backbones (Fig. 5), expanded frameworks with various topologies and pore structures can be generated. It should be noted that the concept of H-bonding units is critical for the construction of HOFs with higher porosity, as their geometry can be transmitted into the net while the length of organic backbones usually dominates the pore size. Actually, certain isoreticular series of HOFs can be generated based on isomorphic organic molecules of different length via the same connection style. For instance, H-HexNet HOFs from a series of -symmetric hexacarboxylic acids,97–102 fluorinated trispyrazole HOFs,88,103 and tribenzimidazolone HOFs have been successfully synthesized.52,53 Besides single-component HOFs, there are also a few examples of the mixed-ligand approach to construct porous HOFs,104–106 as exemplified by SOF-7,107 guanidinium-sulfonate salts,108 and ammonium-sulfonate salts (Fig. 5),109 which is more challenging. But the implementation of such an approach can enable the community of this field to construct diverse porous materials.
Fig. 4.

The geometry of typical H-bonding units assembled from common organic groups through multiple intermolecular H-bonds, serving as the building blocks for HOF construction.
Fig. 5.

Schematic representation of various organic ligands for the construction of HOFs. Roughly categorized based on the types of components.
In fact, with the template effect of guest molecules, the theoretical porosity of HOFs can reach extremely high values, even for those porous solids featuring very weak intermolecular interactions. But, in most cases, the open framework cannot be well retained after guest removal owing to the fragile connections. To generate HOFs with high stability and framework rigidity, several approaches as listed below can be employed. (a) Get stronger intermolecular interactions involved during the molecular assembly to HOFs. To achieve this, multiple H-bonding interactions between organic ligands, or charge-assisted H-bonds between cations and anions especially highly charged ionic ones are favourable for constructing stable HOFs. Therefore, multi-branched organic molecules would be very promising candidates. For charged H-bonds, the acidity and basicity of involved ligands are proposed to be directly related to the H-bonding strength.109 The combination of strong acids and strong bases can result in high permanent porosity. (b) Employ rigid organic ligands with a stereoscopic backbone for HOF construction. Although the linkage of H-bonded units in most of the porous organic solids featuring weak H-bonds might be sensitive to desolvation, some of these supermolecular frameworks still can exhibit permanent porosity, which originated from insufficient intermolecular stacking owing to the stereosteric effect. For example, Aida and Yamagishi et al. reported a porous molecular crystal from the assembly of polypyridine molecules through extremely weak interactions, yet showing thermally stable porosity with a BET surface area of 219 m2 g−1.110 (c) Introduce suitable interpenetrations into the HOFs. It is well known that interpenetration would reduce the pore size but can stabilize the open framework. This is because interpenetrated frameworks are of a lower energy and thermodynamically favorable. Interpenetration can be readily realized for suitable H-bonded frameworks like polycarboxylic acid by controlling their crystallization conditions involving changes in solvents and/or temperatures. (d) Get extra intermolecular interactions like stacking and van der Waals forces involved for stable HOF construction. It should be noted that intermolecular interactions like stacking are important driving forces for other stable and rigid organic porous solids, especially for two dimensional COFs, which usually contain -conjugate systems. Actually, the existence of aromatic moieties enhances the chemical resistance for solvents, acids and bases owing to their inert reactivity. Thus, large planar aromatic molecules should be employed for constructing stable HOFs. (e) Avoid forming any terminal H-bonding donor and acceptor, as they may interact with polar solvent molecules, which raises the activation challenge and complexity. Therefore, if possible, large polar and high boiling point solvents should also be avoided during the synthesis of HOFs.
In short, based on the combination of directional H-bond building units with rigid organic backbones, highly porous HOFs featuring strong intermolecular interactions can be realized to show permanent porosity, in which the architectural stability plays an important role.
2.3. Stability of HOFs
The structure stability is another important prerequisite for HOFs to exhibit permanent porosity. In terms of thermal and chemical stability, HOFs are comparable to MOFs and COFs. HOFs also exhibit additional unique features. For example, the healing of collapsed HOFs can be readily realized by simple recrystallization,111,112 as their bonding manners are relatively reversible.
Water stability is a very important aspect that needs to be considered for applying porous materials in practical processes.11,113 In this context, many HOFs show remarkable water stability owing to the insolubility of organic ligands in water and the hydrophobic nature of pure organic moieties, especially that of aromatic groups. In contrast, many MOFs are sensitive to humidity or water, as water molecules are good alterative ligands toward metal ions.114
It should be noted that HOFs often exhibit poor stability in highly polar organic solvents like dimethyl sulfoxide. This is because solvents of high polarity are usually good H-bonding donors/acceptors, which can form strong H-bonds with the organic ligands, resulting in the leaching and etching of organic molecules from HOFs. But the good reversibility of H-bonding enables recrystallization to original HOFs after the removal of binding solvent molecules. Hence, HOFs show good solution processability, and can be easily regenerated. Also, besides common organic solvents, some types of HOFs like those of carboxylic acids can be stable even in acids.115
Compared to zeolites, MOFs and COFs, the aforementioned bonding interactions in HOFs are somewhat flexible and weak,8 thus showing relatively low architectural stability. Nevertheless, increasing the coplanarity and/or geometric rigidity of H-bonding units can give relatively rigid HOFs with permanent porosity.
2.4. Permanent porosity in HOFs
Permanent porosity is a direct target when constructing HOFs, which is essential for other further applications. Since the construction of crystalline organic supermolecular solids with an open framework in the 1990s,33–50 the establishment of microporosity with surface area and pore volume determination has been a long-standing challenge. As mentioned, most open framework in HOFs would not give effective permanent porosity in the absence of guest molecules, owing to the fragile nature of H-bonds. Therefore, the guest removal of this type of porous materials needs to be performed under mild conditions. On the other hand, suitable strategies listed in Section 2.2 can be employed to construct relatively robust HOFs. The establishment of permanent porosity was achieved in the early 2010s.28,51 To date, although lots of organic extended frameworks have been reported, only a few dozen HOFs can exhibit microporosity with surface area (Tables 2 and 3).116–120 Except for some robust HOFs, guest removal under vacuum with direct heating is too harsh for most labile HOFs, which would lead to corresponding collapse. To facilitate activation under mild conditions, solvent-exchange of solvent molecules of high boiling-point with volatile ones seems to be a feasible approach.52,53 However, many HOFs are subject to certain solubility in common exchange solvents, which are not applicable for solvent exchange. Therefore, the development of HOFs is significantly lagged behind that of other porous materials including MOFs and COFs. Hence, more suitable activation strategies are required for labile HOFs to show porosity, which need continuous intensive endeavors.
Table 2.
Summary of porosity and functionality in single-component and permanently porous HOFs (until October 2018)
| Types | HOFs | Formula | Void/% | Pore size/Å | SBET/m2 g−1 | Functionality | Ref. |
|---|---|---|---|---|---|---|---|
| DAT derivatives | HOF-1a | 42 | ~8.2 | 359 | / (56/3.9)e | 28 | |
| HOF-2a | 54.3 | ~4.8 | 238 | Separation of chiral secondary alcohols | 138 | ||
| HOF-3a | 75 | b7 | 165 | / (47/21)e | 133 | ||
| HOF-4a | 42.5 | 3.8 × 8.1 | 312 | / (11/3.6)e | 117 | ||
| HOF-5a | 41.1 | ~3.9 × 5.5, × 3.9 × 6.8 | 1101 | / (90/—)e, /, and / (102/—)e | 130 | ||
| HOF-6a | 63.4 | ~6.4, ~7.5 | 130 | Proton conduction, / | 145 | ||
| HOF-7a | — | 3.2 × 4.7, 4.2 × 6.7 | 124 | / (11/—)e | 131 | ||
| HOF-9a | — | 3.2 × 4.7, 4.2 × 6.7 | 124 | Py/BTX, / (40/6)e | 132 | ||
| HOF-lOa | 55 | 3.2 × 4.7, 4.2 × 6.7 | 187 | Fluorescence sensing | 154 | ||
| Carboxylic acid | IISERP-HOF1 | ~34 | 9.1 × 9.4a | 412 | / (65/—)e | 121 | |
| HOF-11 | 33.2 | 6.2 × 6.8 | 687 | /, / (30/2)e, and / (45/8)e | 123 | ||
| Tcpb/H3BTB | 38 | 18.5c | 1095 | and adsorption | 134 | ||
| HOF-BTB | 12/16.6c | 955 | / (64/8.7)e, /, and / | 136 | |||
| HOF-TCBP | 56 | 17.8 × 26.3 | 2066 | / and / | 112 | ||
| PFC-1/ | — | 18 × 23 | 2122 | Chemo-photodynamic therapy | 115 | ||
| CoTCPp | ~19 | 4.0 × 6.5 | 98 | Alkylbenzene oxidation | 160 | ||
| TCF-1 | 17.3 | 2.9 × 5.5 to 4.2 × 6.4 | — | , , and adsorption | 119 | ||
| Tp-1 | 54 | ~11, 15.8a | 788 | Fluorescence and hydrocarbon | 97 | ||
| T12–1 | 41 | ~11, 4.6 × 15.8a | 557 | adsorption | |||
| T18–1 | 58 | ~11, 7.1 × 15.8a | — | ||||
| Ex-1 | 59 | ~11, 11.4 × 15.8a | — | ||||
| TpMe-1 | 45.2 | 6.4 | 561 | Fluorescence | 151 | ||
| TpF-1 | 50.4 | 14.5 | 219 | ||||
| CPHAT-1a | 31 | 6.4 | 649b | Fluorescence | 98 | ||
| CBPHAT-la | 45 | 14.5 | 1288 | Fluorescence | 99 | ||
| PETHOF-1 | 80 | ~11c | 1150 | / (32.6/10.6)e | 85 | ||
| PETHOF-2 | 49 | ~11c | 1140 | / (43.4/4.2)e | |||
| Azole derivatives | Trispyrazole 1 | 56 | 16.5 | 1159 | Fluorocarbon and hydrocarbon adsorption | 88 | |
| Trispyrazole 16 | — | 18 | 903 | — | 103 | ||
| Tristetrazole 23 | — | 11.9 | 222 | — | |||
| Trispyrazole 25 | 58.1 | 26.4 | 1821 | — | |||
| Benzotrisimidazole | 21.7 | 2.9 × 4.6 | 131 | /, / | 137 | ||
| FDM-15 | — | b11.5 | 749 | p-Xylene/, capture | 139 | ||
| Amide derivatives | TTBI | 60 | 14.5, 3.8 × 5.8 | 2796 | and adsorption | 52 | |
| — | — | 1665 | storage, / | 53 | |||
| — | 19.9 | 3425 | |||||
| — | — | 365 | |||||
| NPSU-3 | 16.1 | 10.8 | 29 | Organic dyes adsorption | 144 | ||
| Pyridine derivatives | HOF-8 | 24 | 4.5 × 6.8 | — | /, / and adsorption | 91 | |
| SOF-1a | 34 | ~7.8 | 474 | and adsorption | 51 | ||
| Benzoin | Cyclotetrabenzoin | 10 | — | 42 | and adsorption | 118 | |
| Porous molecular | CB[6] | () | 14.4 | 6 | 210 | adsorption | 70 |
| solids | TTP | 25 | 4.6 | 240d 59 | and adsorption | 58 | |
| CC3 | — | 5.8, 7.2 | 624 | , and adsorption | 72 | ||
| TBC[4]DHQ | — | 2.2, 11.2 | 230 | adsorption | 67 |
Distance of atom centers including vdW radii.
Estimated by PLATON.
Pore size distribution from 77 K .
Langmuir surface area.
The adsorption amount (cm3 g−1) at room temperature and 1 bar is presented in parentheses.
Table 3.
Summary of porosity and functionality in mixed-ligands and permanently porous HOFs
| Types | HOFs | Formula | Void/% | Pore size/Å | SBET/m2 g−1 | Functionality | Ref. |
|---|---|---|---|---|---|---|---|
| Pyridine-carboxylic derivatives | SOF-7a | ()() | 48 | 13.5 × 14.0 | 900 | / | 107 |
| SOF-9a | ()() | 34.2 | 12.0 × 12.3 | 181 | /, / | 120 | |
| SOF-10a | ()() | 29.9 | 12.1 × 14.2 | 221 | |||
| Ammonium sulfonate derivatives | HOF-GS-10 | ( | 16 | 3.9 × 5.7 | — | Proton conduction | 146 |
| d-POS-l | 31.5 | 5.9 × 7.0, 9.1 × 10.4 | 398 | Fluorescence sensing | 116 | ||
| CPOS-1 | — | — | 216 | Proton conduction | 109 | ||
| CPOS-2 | — | — | 129 | ||||
| CPOS-3 | — | — | 12 | ||||
| CPOS-4 | — | — | 29 | ||||
| CPOS-5 | 14.6 | 5.3 × 6.8 | — | /, / | 147 |
Additionally, HOFs can be easily crystallized through solution evaporation/cooling, liquid/vapor diffusion, sublimation,88 and even a solvothermal method121 that is frequently applied for MOFs. Since HOFs can be subject to the polymorph issue, their crystallization is sensitive to solvents, templates, variations in concentrations and temperatures and so on, giving kinetic and thermodynamic products.53 In principle, crystallization at high concentrations or for short reaction time might give kinetic isomers. To obtain a thermodynamic phase, it is better to slow down the rate of crystallization or increase the thermal energy for overcoming barriers of activation energy between various isomers (i.e. at high temperatures). The application of a solvothermal method represents a good step in this field, as it provides a feasible approach to assemble stable HOFs.
Overall, to obtain HOFs with accessible porosity for further applications, organic frameworks based on H-bonds can be successfully constructed based on framework chemistry. The architectural stability is important for HOFs to show permanent porosity, whereas the guest removal of HOFs is important for them to serve as porous materials.
3. HOFs for gas storage and separation
The establishment of permanent porosity enables HOFs to serve as novel porous media for the enrichment of gas molecules. The confined pore spaces in HOFs are suitable to capture or encapsulate various important gas molecules, including , , , , , , , , and so on. In particular, as HOFs are composed of light elements (mainly , , and ) and have high surface areas, their high gravimetric uptake capacities for gases can be expected. On the other hand, highly selective separation of gas molecules based on HOFs can also be realized via precise control of their pore size.
3.1. Gas storage
Portable storage and delivery of gases in a convenient, cheap and safe way is very challenging, especially for energy gases like and . In specific, methane (the main component of natural gas) is highly attractive as a clean fuel considering its natural abundance and low carbon dioxide emission. The potential of hydrogen-bonded frameworks for storage is well demonstrated by a potentially important future fuel source, methane clathrate (or natural gas hydrate),122 which is a solid similar to ice featuring a large amount of trapped by water molecules (Fig. 6). One volume of fully saturated hydrate would dissociate into about 180 volumes (STP) of , corresponding to about 15 wt% (by mass). But reversible and rechargeable storage under mild conditions is more desirable. Given that HOFs show larger surface areas and lower densities, it may be promising to apply these materials for gas storage. HOFs have exhibited high storage capacities for certain important gases such as hydrogen, methane, and carbon dioxide, even at ambient temperature.
Fig. 6.

The cage structure of methane clathrate (Structure I). Structure from Ojamäe.122
The potential of HOFs for gas storage has been proven right after the discovery of their permanent porosities. In 2010, Schröder et al. reported a stable HOF ()_2.5DMF 3MeOH (SOF-1; , 9,10-bis(4-((3,5-dicyano-2,6-dipyridyl)-dihydropyridyl)-phenyl)anthracene) showing high gas uptake capacities for , , and (Fig. 7).51 In this HOF, the bulky dihydropyridyl ligands assemble via multiple strong hydrogen bonds ( 2.87 Å), forming layered networks, which further stack into a 3D structure through weak and interactions. The 1D pyridyl-decorated channel along the crystallographic [010] axis in SOF-1 is ~7.8 Å, with solvent-accessible voids ~34.0% of the total cell volume. The activation of SOF-1 at 403 K gave SOF-1a, and its decomposition temperature is at >673 K. Interestingly, the desolvated SOF-1a showed temperature-dependent porosity, with a significant increase in adsorption for from 77 K to 125 K, implying certain framework flexibility. The BET surface area of SOF-1a was estimated to be 474 m2 g−1 based on isotherm at 125 K (143 cm3 g−1). SOF-1a shows considerable methane uptake at 10 bar and 195 K of 106 cm3 (STP) g−1. SOF-1a also absorbs a large amount of (124 cm3 g−1) at 1 bar and 195 K. At ambient temperature, SOF-1a shows certain uptake capacity for (69 cm3 g−1) at 16 bar and 298 K. The storage performance of SOF-1a is superior to that of many crystalline molecular organic solids owing to its high porosity/surface area. In addition, SOF-1a exhibits certain selective adsorption for different gases.
Fig. 7.

(a) The 3D porous structure of SOF-1 with 1D channels. (b) Comparison of gas adsorption on SOF-1a. (black), (red), (blue), and (magenta) at 195 K (triangles), 270 (or 273) K (circles) and 298 K (stars). Reprinted with permission.51 Copyright 2010 American Chemical Society.
A very efficient approach for boosting the gas storage capacity of HOFs is to increase their porosity and surface area. In 2012, Mastalerz and Oppel synthesized a highly porous HOF triptycene trisbenzimidazolone (, TTBI) with exceptional surface area (Fig. 8).52 In this HOF, the benzimidazolone units are assembled into ribbon-like structures through multiple H-bonding ( 2.74 and 2.88 Å), giving a 3D framework with 1D cylindrical (14.5 Å) and slit-like channels (3.8–5.8 Å). The solvent-accessible void of this HOF is B60% of the total cell volume. The desolvated TTBI is permanently porous, showing an experimental BET surface area of 2796 m2 g−1. Such high porosity enables this HOF to show significant uptake at 1 bar and 77 K of 243 cm3 (STP) g−1 (2.2 wt%), which is even comparable to that of some well-known MOFs with open metal sites (e.g. Mg-MOF-74, 2.2 wt% ) under the same condition. This TTBI HOF also shows a large uptake capacity for (80.7 cm3 g−1, 15.9 wt%) at 1 bar and 273 K.
Fig. 8.

(a) Schematic diagram of the structure of triptycene trisbenzimidazolone (TTBI). (b) Nitrogen sorption isotherm of activated TTBI at 77 K. The inset shows the measured pore-size distribution. Reproduced with permission.52 Copyright 2012 Wiley-VCH.
To discover HOFs with higher porosity, Day et al. reported an impressive way via building energy–structure–function maps to identify highly porous HOFs, which is based on the combination of computational framework structure prediction and property prediction.53 Lattice energy surface analyses for several expanded benzimidazolone and imide molecules were carefully conducted. Multiple highly porous and low-density framework structures that are superior to the above TTBI HOF ( in this work) can be targeted from substantial predicted polymorph structures (Fig. 9). Among these structures, a new large-pore polymorph of the TTBI HOF was predicted to be a superior HOF for methane storage, which was verified by further crystallization of and the corresponding sorption experiments. Benzimidazolone contains hexagonal pore channels with an exceptional diameter of 19.9 Å, showing the lowest density (0.412 g cm−3) of any reported molecular solid. The experimental BET surface area was estimated to be 3425 m2 g−1. The saturation uptake capacity at 115 K for was measured to be 47.4 mol kg−1 (437.4 v(STP)/v). Small-pore and denser polymorphs and were also realized for related hydrocarbon separation. Furthermore, by applying such an approach of energy–structure–function maps, an extended benzimidazolone analogue was obtained, showing a hexagonal pore channel of 28 Å in diameter, which is the largest pore size observed for any HOFs so far.
Fig. 9.

Crystal structures and gas adsorption isotherms for polymorphs of T2. (a) Overlays of predicted (red) and experimental (blue) structures for , and , ordered by increasing predicted density. The transformation conditions for interconverting these polymorphs were as follows: (A) loss of solvent at room temperature, heating at 340 K or mechanical grinding at room temperature; (B) heating at 358–383 K. (b) Nitrogen isotherms (77 K); (c) methane isotherms (115 K); filled circles, adsorption experiments; unfilled circles, desorption experiments; filled triangles, adsorption simulations. Reprinted with permission.53 Copyright 2017 Nature Publishing Group.
These studies indicate the potential to further increase gas storage and working capacities of HOF materials through the design of isoreticular frameworks. Overall, further endeavors for highly porous structures will facilitate eventual implementation of some promising HOFs for energy gas storage in the near future.
3.2. Carbon dioxide capture
The removal of carbon dioxide is heavily involved during many processes owing to significant environmental, energy and health concerns, such as in, for example, natural gas upgrading, flue gas treatment and concentration control in closed air spaces. In particular, the removal of acidic gaseous components including is an industrial scale process in the natural gas industry because the presence of will not only lower the energy level of natural gas but also corrode the pipeline. Natural gas is an important energy source owing to its abundant reserves and high energy intensity, giving a worldwide gas consumption of over 3.5 trillion cubic meters in 2015. molecules in different gas sources, ranging from casing-head gas, shale gas, and biogas to coal-bed methane, are often removed by amine scrubbing, while the corresponding regeneration is corrosive and energy-intensive. In contrast, removal at ambient temperature based on physically adsorptive porous materials is a very promising approach for related processes. As emerging porous materials, HOFs have exhibited high capture capacity and selectivity for the / mixture.
Certain interpenetration is beneficial for the structure rigidity of HOFs. To control the degree of interpenetration, Vaidhyanathan et al. utilized 4,4′,4″-nitrilotribenzoic acid () with a near-propeller shape to construct a 3D permanently porous HOF () (IISERP-HOF1,121 also HOF-11123) with 11-fold interpenetrated networks of 3-connected ths topology. Each carboxylic acid group of forms a dimeric synthon with another one via strong intermolecular H-bonding ( 2.58–2.64 Å). This HOF contains 1D cylindrical channels with an aperture size of 6.2 × 6.8 Å2 (Fig. 10). The solvent-accessible void of this HOF is ~33% of the total cell volume. This HOF can retain its crystallinity until about 553 K. Depending on the activation conditions or synthesis methods, the experimental BET surface area of this HOF ranges from 412 m2 g−1 to 687 m2 g−1. This HOF shows high uptake capacity of 4.8 and 2.9 mmol g−1 at 273 and 303 K, respectively, resulting in high / selectivity of 350 at 303 K. Notably, this HOF was synthesized by a solvothermal method, involving crystallization in acetic acid at 423 K. And, this HOF exhibits remarkable stability towards acid and water. Solvent diffusion of hexane into the tetrahydrofuran solution of this tricarboxylic acid gave the same structure, HOF-11. The single-crystal structure of desolvated HOF-11 can be successfully obtained after mild activation, demonstrating its framework robustness that originated from interpenetration.
Fig. 10.

(a) The crystal structure of IISERP-HOF1 with 1D channels. (b) and isotherms of IISERP-HOF1. (c) The IAST plots of IISERP-HOF1 for the 15 : 85 / mixture. The inset shows the HOA plot calculated using virial methods. Reproduced with permission.121 Copyright 2016, Royal Society of Chemistry.
Another relatively stable HOF () 0.15DMF (HOF-8) is assembled from a tri-pyridine ligand , , -tris-(pyridin-4-yl)benzene-1,3,5-tricarboxamide (TPBTC) that integrates amide and pyridyl groups to serve as H-bonding donors and acceptors.91 Each tri-pyridine ligand contains 6 H-bonding sites to connect with three other ligands via multiple intermolecular H-bonding ( 2.96–3.02 Å), giving a 2D honey-comb layered structure (Fig. 11), which is further stacked through interlayer interactions. This HOF contains 1D channels with a cavity size of 4.5 × 6.8 Å2. The solvent-accessible void of this HOF is estimated to be ~24%. This HOF exhibits excellent stability upon immersion in water and certain organic solvents (benzene and hexane). Variable-temperature PXRD and thermogravimetric analysis demonstrated the good thermal stability of this HOF. The activation of its partially deuterated structure HOF-8d enables this HOF to show permanent porosity and selective adsorption toward over and . The uptake capacity of HOF-8d for at 298 K and 1 atm is 57.3 cm3 (STP) g−1, which is far higher than those negligible ones for and . In addition, this HOF also shows selective adsorption of benzene over -hexane, cyclohexane, toluene, or -xylene.
Fig. 11.

(a) Crystal structure of HOF-8, showing a 2D supermolecular layer. (b) , , and sorption isotherms for HOF-8d at 298 K. Reprinted with permission.91 Copyright 2013 American Chemical Society.
The use of metal complexes as H-bonding units for the construction of HOFs is expected to largely extend the diversity of this type of porous materials.125–129 Based on a discrete dinuclear copper complex, Zaworotko et al. reported a robust HOF [Cu2(ade)4(TiF6)2]·2CH3CN (MPM-1-TIFSIX, ade = adenine) synthesized from solvent diffusion, showing high uptake capacity and selectivity under ambient conditions (Fig. 12).124 In this HOF, each paddle-wheel copper complex interacts with eight adjacent molecules via (2.99 Å) and (2.73–2.94 Å) interactions. This HOF contains hourglass shaped channels with a diameter of about 7.0 Å, featuring a solvent-accessible volume of 49.4%. The experimental BET surface area of this HOF is 840 m2 g−1 calculated from the sorption isotherm at 195 K. At 298 K and 1 atm, this HOF shows high uptake capacity (89.6 cm3 g−1) that is superior to that for (18.5 cm3 g−1) and (8.0 cm3 g−1), resulting in high adsorption selectivities for 10/90 / (74.1) and 50/50 / (20.3), which can be attributed to its high affinity toward at low loading (44.4 kJ mol−1). In addition, MPM-1-TIFSIX shows certain thermal stability and water stability.
Fig. 12.

(a) Views of the paddle-wheel complexes and the assembled networks along the crystallographic [001] axis in MPM-1-TIFSIX. (b) Low-pressure , , and isotherms collected at 298 K and (inset) for MPM-1-TIFSIX. (c) IAST selectivities for 50 : 50 / (green; left ordinate) and 10 : 90 / (blue; right ordinate) binary mixtures predicted at 298 K for MPM-1-TIFSIX. Reprinted with permission.124 Copyright 2013 American Chemical Society.
In terms of constructing new HOFs, the mixed-ligand approach is a very promising strategy based on the known ligands without further design of new ligands. Schröder et al. reported a robust binary HOF (SOF-7) by incorporating the complementary pyridyl and carboxyl groups from independent ligands, which shows high adsorption and selectivity (Fig. 13).107 In SOF-7, ()()·7DMF, each carboxylic acid (5,5′-bis-(azanediyl)-oxalyldiisophthalic acid) connects with four adjacent pyridine ligands (1,4-bis-(4-(3,5-dicyano-2,6-dipyridyl)-pyridyl)-benzene) via multiple (~2.60 Å) interactions, giving a 3D network of four-fold interpenetrating cds topology. After guest removal, the solvent-accessible void space was estimated to be 48% for SOF-7. This HOF contains 1D pore channels with an aperture size of 13.5 × 14.0 Å2, featuring cyano and amide groups on the pore surface for potential guest binding. This HOF shows good thermal stability and chemical stability. The BET surface area of SOF-7a was estimated to be 900 m2 g−1 based on the adsorption isotherm at 195 K. At 1 bar and 298 K, the uptake capacity of SOF-7a is 6.53 wt% (1.49 mmol g−1), further increasing to 24.1 wt% (5.48 mmol g−1) at 20 bar, which is superior to those of 0.35 wt% (0.22 mmol g−1) and 2.74 wt% (1.71 mmol g−1) for . Based on Henry’s law constants, the / selectivity was estimated to be 9.1 at 298 K and 1 bar. The heat of adsorption of is calculated to be 21.6 kJ mol−1. The amide and cyano groups on the pore surface are supposed to account for such selective separation as revealed by modeling studies.
Fig. 13.

(a) The 3D porous structure of SOF-7 with 1D channels viewed from the crystallographic [100] axis. (b) and isotherms for SOF-7a at 273 K and 298 K in the pressure range 0–20 bar. Reprinted with permission.107 Copyright 2014 American Chemical Society.
Compared to other pure organic porous materials, the crystallinity and permanent porosity of HOFs enable certain technologies for crystallographic diffraction to be applicable for the determination of adsorption location. In 2015, we reported a flexible microporous HOF (, HOF-5) that showed reversible structural transformation during solvent removal and exhibited high adsorption capacity (Fig. 14).130 The DAT-based (DAT = 2,4-diamino-1,3,5-triazine) ligand 4,4′,4″,4‴-tetra(2,4-diamino-1,3,5-triazin-6-yl)tetraphenylethene connects with eight adjacent ligands via multiple (2.92–3.38 Å) interactions, giving a 3D network of binodal (4,6)-connected topology. HOF-5 contains 2D intersected channels with aperture sizes of ~3.9 × 5.4 and ~4.0 × 6.8 Å2, in which the solvent-accessible void is 55.3%. Desolvation of the acetone-exchanged sample gave guest-free HOF-5a, accompanied by a contraction of B21% in unit cell volume, as revealed by structural refinement of its PXRD pattern. More and strengthened (2.92–3.01 Å) interactions can be observed in HOF-5a. After guest removal, the solvent-accessible void space reduced to 41.1% in HOF-5a. The sorption isotherm of HOF-5a at 77 K exhibits a stepwise shape with a hysteresis, giving a pore volume of 0.44 cm3 g−1 () that is consistent with that of HOF-5a, and a total pore volume of 0.55 cm3 g−1 (). The BET surface area of HOF-5a was estimated to be 1101 m2 g−1. The uptake capacity of HOF-5a for under ambient conditions is up to 117.1 and 90.0 cm3 g−1 (at 273 and 296 K, respectively), which represents the benchmark of HOFs for capture under mild conditions. The adsorption enthalpy of HOF-5a is 22.8 kJ mol−1 for . In contrast, HOF-5a shows much lower uptake capacity for (32.0 cm3 g−1) and (8.2 cm3 g−1) at 296 K and 1 atm, giving high adsorption selectivities for 10/90 / (22.4) and 50/50 / (5.0). The adsorption location of in this HOF was successfully observed from the powder neutron diffraction data of HOF-5a·1.5, showing high packing density of the array. In addition, this HOF also exhibits unique sorption response to with high uptake capacity.
Fig. 14.

(a) Schematic diagram of structural transformation from HOF-5 to HOF-5a and (b) sorption in the pore structure of HOF-5a. Reprinted with permission.130 Copyright 2015 American Chemical Society.
Another well-known functional moiety, porphyrin, was also employed to construct porous HOFs for capture. For example, the self-assembly of zinc 5,10,15,20-tetrakis(4-(2,4-diaminotriazinyl)-phenyl)porphyrin (ZnTDPP, ) via solvent diffusion gave HOF-7 with a 2D double-layered structure.131 In such a layered structure, each ZnTDPP in HOF-7 forms multiple (2.92–3.06 Å) and (2.88 Å) interactions with five different ZnTDPP molecules using their DAT moieties and the coordinated water molecule. HOF-7 contains 3D intersected pore channels with a pore size of 3.2 4.7 and 4.2 6.7 Å2. The BET surface area of activated HOF-7a is only 124 m2 g−1 calculated from the sorption isotherm at 195 K. HOF-7a exhibits high adsorption selectivity for 15/85 / (40) at 273 K and 1 atm (Fig. 15).
Fig. 15.

(a) The 3D porous structure of HOF-7 viewed from the crystallographic [100] axis. (b) Gas sorption isotherms of HOF-7a (solid symbols: adsorption; open symbols: desorption). Reprinted with permission.131 Copyright 2015 American Chemical Society.
Ultramicroporous HOFs are also explored for separation. A transformed HOF (), HOF-9a,132 showing a binodal (3,9)-connected topology with 1D cylindrical channels (~3.4 × 6.8 Å2, void: ~22%, BET: 286 m2 g−1), can selectively adsorbs over and with an uptake capacity of 40 cm3 g−1 under ambient conditions.
The aforementioned examples have demonstrated that HOFs can serve as promising sorbents like other porous media. Importantly, the control of pore structure in HOF materials can be achieved by means of the design of organic moieties, facilitating the development of this class of materials as novel porous materials for adsorptive applications.
3.3. Hydrocarbon separation
The separation of hydrocarbon mixtures is a very important, challenging and energy-intensive process in the petrochemical industry. Among various hydrocarbons, the light hydrocarbons including methane, ethylene, ethane, acetylene, propylene and propane take the most important part. However, the separation of these gases of low boiling points through cryogenic distillation involves tremendous energy consumption. Adsorptive separation technologies thus have been proposed as alternative technologies for potential energy saving. The high porosity of HOFs enables them to show high gas uptake. But high selectivity is another important aspect, which requires strong binding sites. It should be noted that the flexibility and solution processability set HOFs apart from other polymeric porous frameworks, which provides new possibilities toward further membrane fabrication for advanced gas separation and purification.
Although the establishment of HOFs with permanent porosity was not realized until 2010, their potential in the realm of hydrocarbon separation/purification has been proven almost at the same time as that of MOFs. In 2011, we reported the first microporous HOF for selective / separation under ambient conditions (Fig. 16).28 In this studied HOF, each ligand 4,4′,4″,4‴-tetra(4,6-diamino--triazin-2-yl)tetraphenylmethane connects with eight adjacent ligands via multiple (3.03–3.08 Å) interactions, giving a 3D network of 8-connected bcu topology. HOF-1 contains 1D pore channels with cavity sizes of ~8.2 Å in diameter, in which the solvent-accessible void is 42%. Half of the amino groups in the ligand, which are exposed on the pore surface, are accessible for guest binding. This HOF shows a certain degree of framework flexibility as indicated by the cell contraction after partial solvent removal reported by Wuest. HOF-1 lost all its guest molecules at 443 K, giving HOF-1a, which shows no weight loss until >693 K. The sorption isotherm of HOF-1a at 195 K exhibits a stepwise shape with a hysteresis. The BET surface area of HOF-1a was estimated to be 359 m2 g−1. Owing to the framework flexibility, HOF-1a shows unique gate-opening sorption behavior under ambient conditions for . At 273 K, HOF-1a can take up of 63.2 cm3 g−1 (at 800 mmHg), while only 8.3 cm3 g−1 for , giving a high / uptake ratio of 7.6. The adsorption enthalpies of HOF-1a for and at zero coverage were estimated to be 58.1 and 31.9 kJ mol−1, respectively. The strong binding affinity of HOF-1a for results in a high Henry selectivity of 19.3 at 273 K.
Fig. 16.

(a) The X-ray crystal structure of HOF-1 showing 1D channels along the crystallographic [001] axis. (b) The bcu network topology of HOF-1. (c) sorption isotherm at 196 K and (d) and sorption isotherms at 273 K. Reprinted with permission.28 Copyright 2011 American Chemical Society.
The implementation of establishing permanent porosity initiated great research endeavours in HOFs for the challenging separation of important hydrocarbons. Another example of using HOFs for purification was revealed by HOF-3, showing recorded-high selectivity of HOFs for / separation (Fig. 17).133 In this triangular ligand of symmetry, there are three DAT groups on the 1,3,5-triphenylbenzene scaffold. Each ligand connects with six adjacent ligands via multiple (2.90 and 3.11 Å) interactions, giving a 3D rod-packing network of srs topology. HOF-3 contains a 3D pore channel with sizes of ~7.0 Å in diameter, in which the solvent-accessible void is about 75%. The BET surface area based on the sorption isotherm of activated HOF-1a at 195 K was estimated to be 165 m2 g−1. HOF-3a shows selective adsorption of (47 cm3 g−1) over (21 cm3 g−1) at 296 K and 1 atm, giving high adsorption selectivity for 50/50 /(21). Notably, for the first time, in this work, experimental column breakthrough was used to evaluate the separation performance of porous materials for the / mixture.
Fig. 17.

(a) The X-ray crystal structure of HOF-3 in three-dimensional packing showing the 1D hexagonal channels of about 7.0 Å in diameter along the crystallographic [001] axis. (b) Sorption isotherms of and of HOF-3a at 296 K. (c) Comparison of the heat of adsorption of in HOF-3a and various MOFs. (d) IAST adsorption selectivities of / in an equimolar mixture in HOF-3a and various MOFs at 296 K. (e) Experimental column breakthrough curve for an equimolar / mixture (296 K, 1 bar) in an adsorber bed packed with HOF-3a. Reproduced with permission.133 Copyright 2015 Wiley-VCH.
Stable and robust HOFs are highly favourable for the further development of this type of porous materials. In this context, Wu, Yuan and Hong et al. reported an ultrastable and robust together via strong interactions. These interactions jointly contribute to the high stability of HOF-TCBP. Considering the ligand of distorted tetrahedral configuration as a 4-connected node, HOF-TCBP can be simplified as five-fold interpenetrated dia topology, which contains 1D rhombic channels (17.8 × 26.3 Å2). The solvent-accessible void was estimated as 56%. Based on the sorption isotherm at 77 K, the BET surface area of this HOF was estimated to be 2066 m2 g−1, which is superior to those of most reported HOFs. HOF-TCBP shows excellent water stability as revealed by corresponding PXRD patterns and BET surface area of water-treated samples. Also, this HOF can retain most of its permanent porosity at high temperatures (about 473 K). Notably, the porosity of HOF-TCBP can be easily regenerated by simple recrystallization. This HOF shows selective adsorption of and (158–172 cm3 g−1) over (7.4 cm3 g−1) at 295 K and 1 bar, giving high adsorption selectivity for 50/50 / (147–241), which is promising for the separation and purification of light hydrocarbons.
HOFs can be extended to metal-complex based structures as long as their linkages of networks are through H-bonding. Incorporating metal-complexes with geometric rigidity into HOFs not only facilitates the retention of permanent porosity, but also introduces functional sites. Recently, a microporous hydrogen-bonded metal-complex framework [Cu2(ade)4(O)2](SiF6)2 (HOF-21) was realized for selective separation of / at room temperature (Fig. 19).111 In HOF-21, through H-bonding, each metal cluster [Cu2(ade)4(H2O)2]4+ connects with two adjacent HOF (HOF-TCBP, H4TCBP = 3,3′,5,5′-tetrakis-(4-carboxyphenyl)-1,1′-biphenyl), showing selective separation of several light hydrocarbons over methane under ambient conditions (Fig. 18).112 Each ligand connects with four adjacent ligands through four pairs of intermolecular –COOH⋯HOOC–hydrogen bonds, with strong (2.62 Å) interactions, which further stack clusters and eight SiF62− ions, while each free SiF62− ion forms contacts with four metal clusters. There are three types of H-bonding interactions, including (3.06 Å), (2.75–3.04 Å) and (2.89–2.91 Å) interactions. HOF-21 contains 1D pore channels with aperture sizes of ~3.6 Å in diameter. The BET surface area of HOF-21a was estimated to be 339 m2 g−1 based on the sorption isotherm at 195 K. At 298 K and 1 bar, HOF-21a can take up of 1.98 mmol g−1, while 1.27 mmol g−1 for , giving a / IAST selectivity of 7.1. Such selective sorption of over was attributed to the strong interactions between acetylene molecules and SiF62− binding sites, which is validated by the binding energy of and its adsorption location from neutron powder diffraction studies. Column breakthrough experiments demonstrated that can be successfully removed from by using HOF-21a. This HOF also shows considerable water stability and easy healing.
Fig. 19.

(a) Neutron crystal structure of viewed along the crystallographic [001] axis. (b) Adsorption isotherms of (solid) and (hollow) on HOF-21a (blue) and MPM-1-TIFSIX (red) at 298 K. (c) Experimental column breakthrough curves for the 50 : 50 / binary mixture at 298 K and 1 bar in an adsorber bed packed with HOF-21a (blue) or MPM-1-TIFSIX (red). The hollow dot is for , and the solid dot is for . Reprinted with permission.111 Copyright 2018 American Chemical Society.
Fig. 18.

(a) The X-ray crystal structure of HOF-TCBP showing a 5-fold interpenetrated dia framework. (b) Connolly representation of the 1D channels along the crystallographic [100] axis. (b) Sorption isotherms of and of HOF-3a at 296 K. (c and d) The sorption isotherms of HOF-TCBP for the light hydrocarbons at 295 K. Reproduced with permission.112 Copyright 2017 Wiley-VCH.
In certain framework geometry, the existence of intrinsic pore space might be possible but usually would be minimized by accompanied interpenetrations, which can in turn stabilize the framework. Zentner and Lai et al. reported several HOFs from 1,3,5-tris(4-carboxyphenyl)benzene and its derivatives, showing high and robust porosity after multiple interpenetrations.134,135 Recently, Bae and Kim have demonstrated such HOFs can be applied in the separation of light hydrocarbons.136 To be specific, HOF-BTB based on 4,4′,4″-benzene-1,3,5-triyl-tris(benzoic acid) was studied for selective adsorption of , and over , which contains eight-fold interpenetration of 2D hexagonal sheets. Based on the sorption isotherm at 77 K, the BET surface area of this HOF was estimated to be 955 m2 g−1, with measured pore size of 12–16.6 Å. At 295 K and 1 bar, this HOF takes up of 2.87 mmol g−1, of 2.48 mmol g−1, and of 3.09 mmol g−1, which gives a / (50 : 50) IAST selectivity of 14.
Indeed, HOFs can be applied as porous adsorbents for hydrocarbon separation, showing high uptake capacity and considerable selectivity. In terms of separating gas molecules of small dipole and/or quadrupole moments, strong binding sites often dominate. Hence, to improve separation performance, the introduction of strong binding sites should be taken into account for future design of HOF materials.
3.4. Other volatile adsorbates
Besides the carbon dioxide capture and hydrocarbon separation, there are also few examples of using HOFs for other gas separation, including fluorocarbons over nitrogen and air separation. In fact, the capture of harmful volatile gas/vapour of environmental concern is an important application of porous materials, in which HOFs are still in their early stage. Therefore, there is great potential for HOF materials to capture volatile gaseous molecules.
Miljaníc and co-workers reported a stable HOF for the adsorption of fluorocarbons (FCs) and chlorofluorocarbons (CFCs), which is constructed from a fluorinated trispyrazole ligand (, Fig. 20).88 In this HOF, each organic ligand connects to six adjacent ligands via pyrazole trimer units with multiple (2.78–2.87 Å) interactions, giving 2D honeycomb-like lattices of hnb topology, which further stack together via strong interactions with centroid–centroid distances of 3.42–3.69 Å. This HOF contains 1D hexagonal pore channels (16.5 Å) with a solvent-accessible void ratio of 56%. As demonstrated by PXRD analysis, this HOF exhibits remarkable chemical stability to many organic solvents, water and acid/base aqueous solutions, and high thermal stability upon exposure at 4523 K. The BET surface area of this HOF was determined to be 1159 m2 g−1 based on the sorption isotherm at 77 K. This HOF takes up a negligible amount of vapour owing to its hydrophobicity, but can favorably capture large amounts of FCs and CFCs, including chloroform, dichloromethane, (CFC-113), and (HCFC-225ca). In particular, this HOF adsorbs large amounts of perfluorohexane (74 wt%) at room temperature with good reversibility and fast adsorption dynamics. Later, in their follow-up work,103 several isostructural HOFs extended from this prototypal HOF were successfully obtained by increasing the length of the linker, showing tunable porosity.
Fig. 20.

(a) The X-ray crystal structure of fluorinated trispyrazole showing a hexagonal network, with infinite fluorine-lined channels protruding throughout the structure along the crystallographic [001] axis. (b) Gas sorption isotherms for , and . (c) Uptake of perfluorohexane as a function of time, upon exposure to the flow of -enriched nitrogen. Reprinted with permission.88 Copyright 2014 Nature Publishing Group.
Zhang et al. reported a 3D highly symmetric HOF, , that selectively adsorbs over Ar and (Fig. 21).137 This HOF is constructed based on trifluoromethyl substituted benzotrisimidazole, which connects with three adjacent organic ligands through three pairs of (3.01 Å) interactions in this framework. This HOF can undergo a water-induced reversible crystal-to-crystal transformation. This HOF shows a 3-connected srs topology, containing 3D intersected channels (void ratio of 21.7%) with an aperture size of 4.6 × 2.9 Å2. The BET surface area of this HOF was estimated to be 131 m2 g−1 based on the sorption isotherm at 195 K. At 77 K, this HOF can take up of 50 cm3 g−1, while it shows negligible capacity for Ar and (3.3 and 4.8 cm3 g−1, respectively), giving a Henry’s law selectivity of 119 and 59 for /Ar and /, respectively. In addition, by changing the size of the substituent group, another 2D HOF with a honeycomb-like network can be obtained.
Fig. 21.

(a) The 3D channel (orange/gray surfaces) defined by the hydrogen-bonded network and (b) the /Ar/ sorption isotherms at 77 K for trifluoromethyl benzotrisimidazole. The curves with solid symbols represent adsorption isotherms, while desorption isotherms are represented by open symbols. Reproduced with permission.137 Copyright 2016, Royal Society of Chemistry.
As presented above, HOFs are very promising porous materials for various challenging gas storage and separation. The establishment of permanent porosity enables HOFs to serve as novel porous materials for gas sorption, which is particularly important in advancing the HOF chemistry. Despite the fact that the retention of porosity during the removal of guest molecules is challenging, many HOFs constructed from different H-bonding units can still show remarkable thermal stability, solvent stability and even water stability, affording good modeled structures for further development of permanently porous HOFs. Continuous endeavours to explore porous HOFs are expected, as stimulated by the aforementioned achievements in realizing HOF materials for various gas separations.
4. HOFs for molecular recognition
Molecular recognition plays an important role in biological systems, involving specific intermolecular interactions through noncovalent binding including H-bonding, interactions and van der Waals forces. The implementation of establishing permanent porosity renders HOFs a good platform to understand similar recognition processes. In specific, the following features are particularly important: (i) the immobilization of functional sites in the porous framework can improve specific recognition; (ii) the relatively flexible and adaptive framework that facilitates potential host–guest collaborative interactions; (iii) the high crystallinity that provides the possibility to visualize the host–guest interactions via powerful crystallographic tools. For instance, the introduction of chiral centers affords an asymmetric pore environment for enantioselective separation, while extra H-bonding donors/acceptors offer binding sites for complement analytes.
In 2014, we reported the first example of a chiral HOF based on a 1,1′-bi-2-naphthol (BINOL) derivative, ()-4,4′,6,6′-tetra-(2,4-diamino-1,3,5-triazin-6-yl)-2,2′-diethoxy-1,1′-binaphthalene (, HOF-2, Fig. 22).138 This new organic ligand is exclusively designed by covalently incorporating the DAT H-bonding units onto the ()-BINOL scaffold. In this homochiral HOF, each organic ligand connects to six adjacent ligands via DAT dimers with multiple (2.98–3.34 Å) interactions, giving a 3D uninodal 6-connected network. This HOF contains 3D intersected pore channels (pore size: 4.8 Å) with chiral centers exposed on the pore surface, which facilitates the enantioselective recognition of small molecules. The solvent-accessible void ratio of this HOF was estimated to be 54.3%. The BET surface area of HOF-2a was determined to be 238 m2 g−1 based on the sorption isotherm at 195 K. Utilizing this homochiral HOF, we realized enantioselective separation of racemic secondary alcohols for the first time. Interestingly, this HOF shows higher enantioselectivity for aromatic secondary alcohols than that for aliphatic secondary alcohols. In particular, the enantiomeric excess (e.e.) value for 1-phenylethanol (1-PEA) in this HOF is up to 92%, as determined by HPLC. Single-crystal X-ray diffraction of 1-PEA including samples was applied to gain in-depth insight into the origin of high enantioselectivity. It is because -1-PEA molecules form strong H-bonding ( 2.58 Å) with the diethoxy groups of the BINOL scaffold, while -1-PEA molecules show weaker (3.28–3.57 Å) interactions with the framework. In addition, the permanent porosity of this HOF can also realize gas storage/separation as revealed by its adsorption at 273 K. Overall, this work represents the first example of homochiral porous HOFs for the enantioselective separation of small molecules as another critical potential application.
Fig. 22.
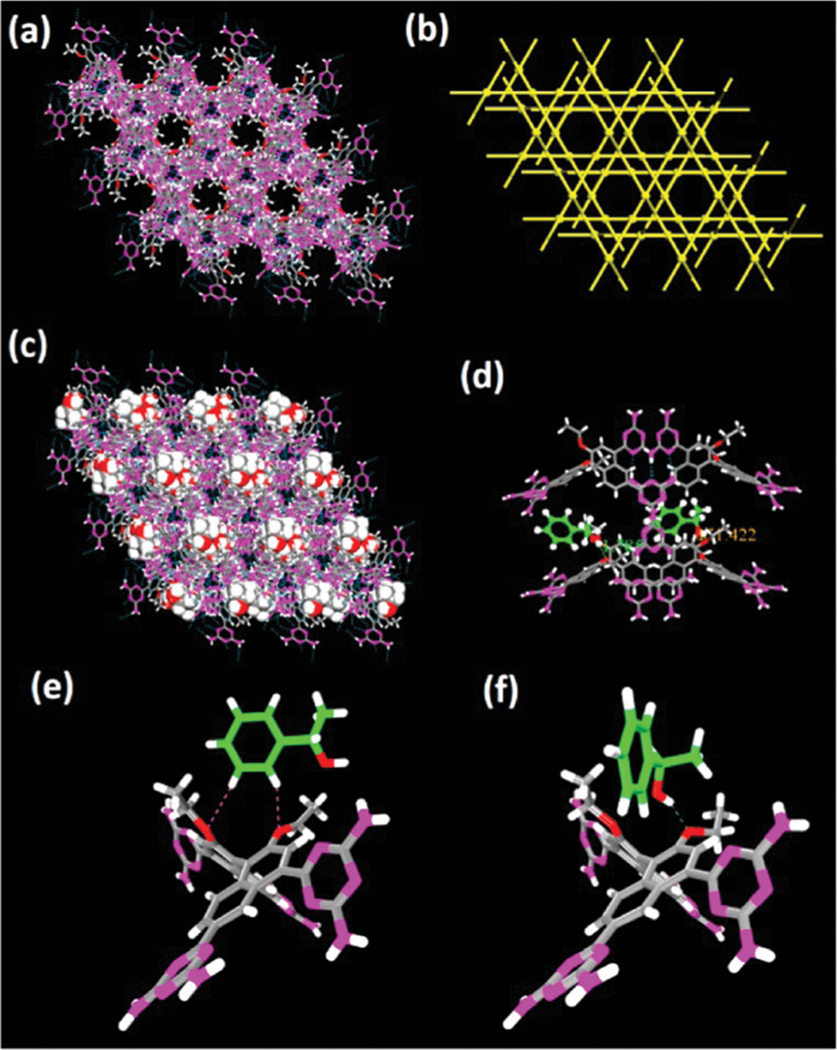
X-ray crystal structure of HOF-2 featuring (a) a three-dimensional hydrogen-bonded organic framework exhibiting 1D hexagonal pores along the crystallographic [001] axis and (b) the uninodal 6-connected network topology. X-ray crystal structure of indicating (c) the enantiopure -1-PEA molecules residing in the channels of the framework along the crystallographic [001] axis and (d) the chiral cavities of the framework for the specific recognition of -1-PEA. Comparison of X-ray crystal structures of (e) HOF-2⊂-1-PEA and (f) . Reprinted with permission.138 Copyright 2014 American Chemical Society.
Introducing functional sites into porous materials like MOFs is well recognized as an efficient approach for the selective recognition of small molecules. But it is challenging to reserve similar sites during the assembly of HOFs. Recently, we reported a DAT-derived HOF (TDTTB)· (HOF-9, TDTTB = 1,3,5-tris(2,4-diamino-1,3,5-triazin-6-yl)-2,4,6-trimethyl-benzene) containing unbonded amine groups (Fig. 23), which exhibits highly selective recognition for pyridine (Py) over BTX aromatic compounds (BTX refers to benzene, toluene, and -, -, -xylene).132 This HOF is constructed from TDTTB dimers, which further connects with six other dimers through multiple (2.94–3.47 Å) interactions, giving a 3D open framework of pcu topology. There are molecules located between TDTTB dimers that can further stabilize the framework through (2.80–2.89 Å) and (2.86–3.07 Å) interactions. HOF-9 contains 1D pore channels (6.9 × 8.8 Å2) along the crystallographic [100] axis, with free amino groups exposed on the pore surface, which facilitates the selective recognition of Py molecules. Simple adsorption studies under ambient conditions demonstrated that HOF-9 can take up 1 Py molecule per formula while no BTX molecules, as shown by the corresponding NMR spectra. Single-crystal structural analysis of HOF-9·2Py revealed that strong (3.20 Å) interactions form between unbonded amino groups and Py molecules. The binding energy of Py in HOF-9 was calculated to be B67 kJ mol−1, which is higher than that of benzene (B41 kJ mol−1). Further selective adsorption of Py from different binary equimolar mixtures confirms the efficient recognition of Py from BTX aromatic molecules.
Fig. 23.

(a) Packing diagram of HOF-9 along the crystallographic [100] axis showing the pore surfaces of 1D channels highlighted as yellow/grey (inner/outer) curved planes; (b) a uninodal 6-connected -Po net. (c) The crystal structure of indicating the hydrogen-bonding interactions between Py and the HOF-9 framework (yellow dashed line), the interaction between the DAT group and the Py molecule (red dashed line) and packed Py molecules residing in the channel of the framework along the crystallographic [100] axis. Reproduced with permission.132 Copyright 2017, Royal Society of Chemistry.
To maximize the accessible surface of aromatic rings, Li et al. employed a rigid and non-coplanar triptycene derivative to construct a HOF, showing selective adsorption for aromatic compounds and highly efficient enrichment for the fullerene molecule (Fig. 24).139 This HOF is based on an imidazole ligand, 2,6,12-trihydrotripty[2,3-:6,7-:12,13-]triimidazole (), which connects with six adjacent ligands through six (2.80 Å, 170.4°) interactions, giving a 3D open framework ()· 3DEF (FDM-15). This HOF contains hexagonal honeycomb-like channels with aperture sizes of ~11.5 Å in diameter. With a very low density of 0.436 g cm−3, this HOF exhibits permanent porosity with a BET surface area of 749 m2 g−1. Given the suitable pore size and desirable pore surface featuring aromatic rings, this HOF was applied to enrich fullerene (, 10.5 Å in diameter) from toluene solution. The adsorbed in this HOF is up to ~12 wt% of the final solid, giving a concentration 420 times higher than that of the original solution. In addition, simple adsorption studies under ambient conditions demonstrated that this HOF can take up 1.3 -xylene molecules per molecule while 0.74 molecule for toluene, as examined by the corresponding NMR spectra. Further selective adsorption of -xylene from different binary equimolar mixtures confirms the selective adsorption of -xylene over toluene, -xylene and ethylbenzene.
Fig. 24.

Illustration of fullerene molecules distributed in the channels of FDM-15. Reproduced with permission.139 Copyright 2017, Royal Society of Chemistry.
The flexibility of HOFs enables them to show adaptive framework transformation during different guest inclusion, which allows the retention of single crystallinity, facilitating direct visualization of corresponding host–guest interactions via single crystal X-ray diffraction. Ward et al. reported interesting guest discrimination from different pore channels based on single crystal analyses of various guest inclusion compounds of a guanidinium-sulfonate HOF (Fig. 25), guanidinium 1,2,4,5-tetra(4-sulfonatophenyl)benzene ().140–142 In these compounds, each TSPB4− connects with 16 adjacent guanidinium cations using all its oxygen atoms through charge assisted (2.6–3.1 Å) interactions, while each guanidinium cation connects with four TSPB4−, giving 3D HOFs with 1D infinite H-bonding cylinders. This HOF contains three types of 1D channels, with cross-section areas of 18, 39 and 52 Å2, respectively. This HOF can retain its single crystallinity upon the exchange of various solvents, owing to its sufficient framework flexibility. Reversible guest exchange between inclusion compounds for dioxane (), tetrahydrofuran (), and toluene () gives single crystals of (, (, ( and (, respectively. For dioxane and tetrahydrofuran molecules, they can take up all the three pore channels. But the toluene molecules show only partial guest exchange toward inclusion compounds of dioxane and tetrahydrofuran, because the smallest pore channel is inaccessible for the larger toluene molecule. All three pore channels exhibit certain expansion and shrinkage after these solvent exchange processes. Notably, the related solvent exchange processes within this HOF are all single crystal to single crystal transformations, which is rarely reported for HOFs.
Fig. 25.

Schematic illustration of reversible single crystal–single crystal transformations based on the G4TSPB framework. Reprinted with permission.141 Copyright 2014 American Chemical Society.
In addition to the variation of the ligand length to get isoreticular structures with tunable pore size, another approach involving replacing the dangling functional groups on the internal pore surface was also proposed, which can not only obtain isostructural HOFs with tunable pore size, but also afford binding sites for guest inclusions. The prototypal HOFs are based on steroidal bis-(-phenyl)ureas (nanoporous steroidal ureas NPSU-2 to 4), derivatives of cholic acid as anion receptors,143 which exhibit 1D chiral channels with pore sizes of 12–14 Å.144 In these prototypes, the carbonyl groups act as H-bonding acceptors while the urea groups act as H-bonding donors. Each hydrated steroidal bis-(-phenyl)urea connects to four adjacent steroids through multiple and interactions, giving helical steroids chains. In 2013, Davis et al. systematically studied a series of steroidal bis-(-phenyl)ureas by altering the terminal groups on the interior pore surface, giving over 20 HOFs with tunable pore size ranging from B0 to 13.1 Å. Among these HOFs, NPSU-3 exhibits certain permanent porosity as demonstrated by adsorption at 77 K, giving a relatively low BET surface area of 29 m2 g−1. Some variants with functional groups exposed on the pore surface afford the potential to bind guest molecules. Organic dyes including aniline, chlorobenzene to squalene can be directly adsorbed by these materials from their liquid state (Fig. 26). For example, the aniline molecules in NPSU-3 form hexamers, binding to ester carbonyl groups of the framework with (3.33 Å) interactions. In addition to adsorption for organic dyes, the HOF crystals exhibit an interesting dichroism feature originated from the alignment of the chromophores.
Fig. 26.

(a) Methyl ,,-tris[(phenylaminocarbonyl)amino]--cholan-24-oate. (b) Interior surfaces of NPSU-3 viewed along the crystallographic [001] axis. (c) X-ray crystal structure of NPSU-3 with adsorbed aniline, viewed along the crystallographic [001] axis. The aniline is shown in the space-filling mode. (d) Optical crystals of NPSU-3 with included dyes upon polarized light irradiation. For each pair of images the plane of polarization is rotated through 90° between top and bottom. Reprinted with permission.144 Copyright 2013 American Chemical Society.
Overall, for molecular recognition, the accessible binding sites in HOFs can only interact with substrates through either H-bonds or weak van der Waals forces, showing good reversibility that facilitates the guest release during the regeneration step. On the other hand, porous materials like MOFs with open metal sites can show high selectivity for recognition of different molecules, owing to their strong binding affinity. Apparently, the lack of strong binding sites in HOFs limits their application in this aspect, which needs to be taken into account for future HOF construction.
5. HOFs for conductive applications
The proton exchange membrane fuel cell (PEMFC) technology for alternative sources of energy is of particular interest, owing to its highly efficient transportation and low-cost maintenance. Porous materials like MOFs have been demonstrated as good proton conductors in related applications, as they are highly porous and easy to functionalize that enable them to provide and accommodate various proton carriers. From a structural viewpoint, the building units and H-bonding connections in HOFs make them ideal conducting materials. To be specific, the H-bonded donor/acceptor groups of building units can serve as proton sources or carriers, while the widespread H-bonds provide diverse proton transportation pathways. The features of flexibility and solution processability of HOFs open up new possibilities for membrane fabrication, giving light-weight proton-conducting solid electrodes.
To facilitate water-mediated proton conduction, it is suggested to construct ionic backbones as a good proton source, while the open channels can accommodate water molecules as proton carriers. Several types of porous organic salts can be utilized, namely guanidinium-sulfonate HOFs and organic ammonium-sulfonate HOFs. Another advantage of HOFs as conductors is that most HOFs are humidity stable, facilitating proton conduction. In contrast, most MOFs are unstable upon humidity exposure.
To realize high proton conduction in HOFs, permanently porous frameworks composed of potential proton carriers are applicable. In 2016, we reported a porphyrin-based porous HOF for the application of proton conduction (Fig. 27), which is assembled from 5,10,15,20-tetrakis(4-(2,4-diaminotriazinyl)-phenyl)porphyrin ().145 In this HOF, ()·6DMF· 5THF (HOF-6), each connects with eight adjacent ligands through two types of binding models between DAT–DAT moieties with (B3.0 Å) interactions, giving a 3D 4,4-connected network of two-fold interpenetrations. This HOF contains 3D intersected pore channels (pore size: B6.4 and 7.5 Å) with neutral porphyrins exposed on the pore surface, which can serve as proton donors/acceptors. The solvent-accessible void ratio of HOF-6 was estimated to be 63.4%. The BET surface area of desolvated HOF-6a was determined to be 130 m2 g−1 based on the sorption isotherm at 195 K, confirming its permanent porosity. The proton conductivity of HOF-6a was examined based on its solid sample. At 97% RH and 300 K, the proton conductivity of HOF-6a is measured to be 3.4 × 10−6 S cm−1, which demonstrates that HOFs are applicable for proton conducting materials. In addition, HOF-6a also shows selective adsorption of over .
Fig. 27.

(a) X-ray crystal structure of HOF-6 indicating a 3D packing supramolecular structure along the crystallographic [101] direction with a channel size of 6.4 Å. (b) Nyquist plots of HOF-6a at 300 K (black), 303 K (red), 308 K (green), and 313 K (blue) at a relative humidity (RH) of 97%. Reprinted with permission.145 Copyright 2016 American Chemical Society.
Porous guanidinium sulfonate salts might also be good candidates considering their ionic backbones featuring a proton source. Although guanidinium sulfonate can form a wide variety of H-bonded structures ranging from clusters, chains to layers, it is still feasible to control their pore structures based on the variation of aromatic moieties. By virtue of guanidinium aryl sulfonate salts, Ghosh et al. reported two bilayered porous HOFs,146 HOF-GS-10 for ()·xG (G = guanidinium, NDS2− = 1,5-naphthalenedisulfonate) and HOF-GS-11 for previously reported ()·xG140 (BPDS = 4,4′-biphenyldisulfonate), that show high proton conduction under humidified conditions (Fig. 28). In HOF-GS-10, each disulfonate connects with six adjacent guanidinium cations from two guanidinium–sulfonate sheets by using all its oxygen atoms through charge assisted (2.90–2.94 Å) interactions, while each guanidinium cation connects with three disulfonates, giving a 2D honeycomb-like double-layered network. HOF-GS-11 contains a similar bilayer structure with shifted ribbon H-bonding sheets rather than quasihexagonal sheets. Both HOFs exhibit certain permanent porosity as demonstrated by their adsorption isotherms at 195 K. Water sorption studies revealed that HOF-GS-10 shows a water uptake of 3.47 mmol g−1, while that for HOF-GS-11 is up to 11.6 mmol g−1. Given the coherent H-bonding network and porosity, both HOFs are applied to solid-state proton conduction under humid conditions. At 95% RH and 303 K, the proton conductivity of HOF-GS-10 and HOF-GS-11 is up to 0.75 × 10−2 and 1.8 × 10−2 S cm−1, which is comparable to that of MOFs. In addition, these compounds also show low activation energy (0.13 eV for HOF-GS-11 vs. 0.16 eV for PCMOF-5),146 further highlighting HOFs as promising lightweight materials for fuel-cell technologies.
Fig. 28.

(a and b) Hydrogen-bonded 2D frameworks of HOF-GS-10 and HOF-GS-11 showing the hydrogen-bonding interaction between the sulfonate groups and the guanidinium cations in both the compounds. (c) (dots), (stars), and (diamonds) adsorption isotherms of HOF-GS-10 at 195 K ( and ) and at 77 K (). (d) Proton conduction values of HOF-GS-10 and HOF-GS-11 at varying humidity and at 303 K. Reproduced with permission.146 Copyright 2016 Wiley-VCH.
The binding strength of organic linkers is also important for the application of HOFs in proton conduction. For the construction of stable porous organic salts, the acidity/basicity of organic ligands has been proposed as an important factor, referring to the favorable combination of strong acids with strong bases. Recently, Ben et al. reported several crystalline porous organic salts (CPOSs)109,147 featuring high proton conductivity by the combinations of different tetra-acids and diamines (Fig. 29), which reveals that such a synthesis strategy shows high correlation with the stability of HOFs. All these four HOFs show a total net charge of zero owing to their composition of acid and base in a 1 : 2 ratio, namely CPOS-1 for , CPOS-2 for , CPOS-3 for and CPOS-4 for . In these HOFs, two types of ligands are connected to each other by 1D H-bonding chains, giving 3D networks that contain one dimensional chains, clusters or isolated molecules of water. The guest water molecules in these HOFs can be removed below 473 K. The surface area of desolvated CPOS-1 to CPOS-4 was determined to be 216, 129, 12 and 29 m2 g−1 based on the sorption isotherm at 273 K by the Dubinin–Astakhov method, which demonstrates that the combination of relatively strong acid and base helps to improve the framework stability for high pore surface area. The H-bonding chains of charge-assisted interactions in the framework and/or guest water molecules can dramatically facilitate proton conductivity. At 98% RH and 333 K, the proton conductivity of CPOS-1 to CPOS-4 is up to 1.0 × 10−2, 2.2 × 10−2, 3.3 × 10−4 and 7.4 × 10−4 S cm−1, which is proportional to their water contents and the number of corresponding proton carriers. The activation energies of these HOFs reveal that the vehicular mechanism accounts for their proton conduction.
Fig. 29.

(a) The 3D porous structure of CPOS-2 showing 1D channels and (b) temperature dependent proton conductivity of CPOS-2 at 98% RH. Reproduced with permission.109 Copyright 2018 Wiley-VCH.
As demonstrated by the above examples, for water-media proton conduction, the proton conductivity of HOFs is comparable to that of the highly conductive MOFs and commercialized Nafion membranes. The first pioneering works provide some inspirations to further explore highly conductive HOFs. HOFs for proton conduction are still rarely explored, and many aspects in such application have never been involved. Given the intrinsic advantages of low density, crystalline nature, good processability, and especially high intensity of proton carriers, HOFs can serve as new lightweight organic materials for fuel-cell technologies in the field of alternative energy.
6. HOFs for optical applications
As pure organic materials, HOFs are assembled from rationally designed H-bonded motifs. To construct HOFs with permanent porosity, their building blocks are usually designed to be highly rigid molecules featuring aromatic moieties of a large -conjugated system, which usually are excellent fluorescent and phosphorescent dyes, highlighting HOFs as very promising luminescent materials. Depending on the degree of -conjugation, the emission wavelengths of these organic molecules can distribute over a wide range, facilitating the rational design of luminescent HOF materials with tunable colors. Furthermore, the highly crystalline nature of HOFs indicates the highly ordered arrangement of organic chromophores, giving significantly different emission behaviours as compared to their solution states, such as aggregation induced emission (AIE). In the crystalline state, the lumophores are subject to constraints from the lattice and certain intermolecular close contacts, resulting in spectral shifts, loss of vibronic structure, broadening in the emission, and increased emission lifetimes. In addition, the widely involved intermolecular interactions including interactions enable electronic interactions (e.g. charge transfer) between the lumophores, giving a change in luminescence. Combining the permanent porosity of HOFs accessible for guest species, luminescence sensing can be readily achieved on the basis of host–guest interactions. Consequently, HOFs show great potential for optical applications, which have been rarely shown and only by a few examples.
Large -conjugated planar moieties have been widely explored for the construction of permanently porous HOFs owing to their high rigidity, which are also good lumophores of highly luminescent materials.148,149 Hisaki and co-workers have developed a series of robust HOFs with remarkable porosity and surface area using various -symmetric polycarboxylic acids,100–102,150,151 such as triphenylene (Tp), hexadehydrotribenzo[12]annulene (T12), dodecadehydro-tribenzo-[18]annulene (T18), and expanded cyclic phenylene ethynylene (Ex) derivatives (Fig. 30).97 Each hexacarboxylic acid connects to six adjacent ligands via six –COOH dimers with multiple interactions, giving a 2D hexagonal network. Each H-bonded layer further stacks with adjacent layers through and interactions, showing an open framework with two types of accessible pores (triangular and nonregular hexagonal shapes) with sizes of ~11 Å in diameter and 2–11.4 × 15.8 Å2, respectively. The solvent-accessible void ratios of these HOFs were estimated to be 54% for Tp-1, 41% for T12–1, 58% for T18–1, and 59% for Ex-1. These HOFs show high thermal stability up to 513–633 K. After desolvation, these HOFs exhibit certain structural transformations. Their BET surface areas were estimated to be 557–788 m2 g−1. These HOFs show different fluorescence spectra with maxima at 416–546 nm, with quantum yields of 5.5–25%. These HOFs also show high uptake capacity for several hydrocarbons.
Fig. 30.

(a) Crystal structures of H-HexNet motifs of Tp-1, T12-1, T18-1, and Ex-1. Normalized solid state fluorescence spectra of (b) Tp-apo (solid line) and Tp-2Ds (dashed line), (c) T12-apo (solid line) and T12-1 (dashed line), (d) T18-apo-II (solid line) and T18-1 (dashed line), and (e) Ex-apo (solid line) and Ex-1 (dashed line). Reprinted with permission.97 Copyright 2016 American Chemical Society.
Another rigid p-conjugated heterocyclic moiety, hexaazatriphenylene (HAT), was also applied to construct rigid and stable HOFs, showing interesting anisotropic single-crystal fluorescence. Using a HAT derived ligand, hexakis(4-carboxyphenyl)-hexaazatriphenylene (CPHAT), with a twisted conformation, a 3D rigid HOF (CPHAT-1) can be assembled.98 This HOF can be simplified as pcu topology with 4-fold interpenetration, showing a solvent-accessible void ratio of 31% with a pore size of 6.7–8.8 Å in diameter. The BET surface area of CPHAT-1a was estimated to be 649 m2 g−1 based on the sorption isotherm at 195 K. This HOF shows remarkable thermal and chemical stability even in hot water and acid aqueous solution. CPHAT-1a shows a fluorescence emission maximum at 460 nm, and the quantum efficiency is 1.6%. CPHAT-1a shows highly anisotropic emission with an anisotropy of 0.65 for perpendicular orientation and −0.32 for parallel orientation, which reveals preferential orientation of the molecular dipole moments in the specific direction.
Recently, Hisaki and Douhal et al. reported an expanded HAT derivative HOF (CBPHAT-1) showing an isoreticular structure with CPHAT-1 (Fig. 31).99 CBPHAT-1 exhibits six-fold interpenetrated frameworks with pcu topology, with a solvent-accessible void ratio of 45% and a pore size of 14.5 Å in diameter. The framework of this HOF can be retained even up to 578 K as revealed by its variable temperature PXRD patterns. This HOF is stable in boiling water and conc. HCl. The BET surface area of CBPHAT-1a was estimated to be 1288 m2 g−1 based on the sorption isotherm at 77 K. CBPHAT-1a shows a green-yellow emission with an emission maximum at 500 nm, and the quantum efficiency is 5.7%. CBPHAT-1a also shows highly anisotropic emission with an anisotropy of 0.45 and −0.30 for perpendicular and parallel orientation, respectively, which is owing to direction-dependent intermolecular interactions originating from oriented molecular stacking. The crystal of CBPHAT-1a shows position-dependent emissions caused by structural defects. These works from the same group well demonstrated that the highly ordered crystalline structure in HOFs can generate unique organic luminescence that is distinct from that of dispersed molecules. The above two examples also represent another isoreticular example for the construction of HOFs.
Fig. 31.

(a) Packing diagram of CBPHAT-1. (b) Gas sorption isotherms of CBPHAT-1a: (77 K), (77 K), (195 K), (77 K). (c) Emission spectra at different points of a CBPHAT-1a crystal. The inset shows an image of the crystal and the points of measurement. (d) Fluorescence decays of a CBPHAT-1a crystal at different spectral regions of the emission spectrum. Reproduced with permission.99 Copyright 2018 Wiley-VCH.
To obtain long-lifetime organic phosphorescence, an efficient approach is to embed phosphors into a rigid crystalline lattice and thus reduce the nonradiative decay of related triplet excitons. Recently, An and Huang et al. reported two phosphorescent HOFs (MA-IPA and MA-TPA) composed of melamine (MA) and aromatic acids, which show high phosphorescence efficiencies and ultralong lifetimes (Fig. 32).152 In both HOFs, the MA molecules are protonated as , which connect with deprotonated dicarboxylate moieties and water molecules through strong , , and interactions, giving H-bonded ribbons and sheets. Extra interlayer H-bondings, van der Waals force, and electrostatic interactions between these secondary structures enable dense packing of these organic phosphors. Notably, all the H-bonding donors and acceptors are fully involved in H-bonding interactions. Therefore, strong photo-luminescence of MA-IPA can be observed upon UV excitation, showing emission peaks at B356 and 466, and 488 nm that can be assigned to fluorescence and phosphorescence, respectively. Typically, under suitable UV light, the excitation of organic molecules in HOFs occurs through the spin-allowed singlet–singlet transition, followed by fluorescence emission as direct radiative decay from the lowest singlet excited state and/or phosphorescence emission after intersystem crossing to triplet excitons. The latter usually features a rapid nonradiative decay rate, resulting in poor efficiency. With multiple constraints from the lattice of HOFs including H-bonding interactions, the nonradiative transition can be suppressed, thus giving highly efficient phosphorescence. The phosphorescence lifetime of MA-IPA is up to 1.91 s, accompanied by a high quantum efficiency of 24.3%, while those of MA-TPA were measured to be 1.09 s and 19.4% (maximum at 509 nm). This type of HOF materials were also applied for barcode identification in darkness. This work shows that HOF materials are superior in the development of long-lifetime organic phosphors.
Fig. 32.

Schematic representation of the supramolecular architecture in MA-IPA showing ultralong organic phosphorescence. Reprinted with permission.152 Copyright 2018 American Chemical Society.
Tetraphenylethylene (TPE) is a promising organic chromophore as it can give aggregation induced emission (AIE) for various applications ranging from chemical sensing to cell imaging. Xie and Chen et al. reported a fluorescent HOF (HOF-1111) that was composed of a DAT-derived ligand with a TPE moiety, for fluorescence sensing of aromatic compounds.153 In this HOF, each ligand connects to four adjacent intralayer ligands via multiple (2.93–3.15 Å) interactions, giving 2D H-bonded layers. Each layer is further linked to an adjacent layer by DMSO and water molecules through and interactions, giving a double-layered structure. This HOF shows a potential solvent-accessible void ratio of ~40% with a pore size of 6.8 × 10.9 Å2. This HOF exhibits fluorescence emission with a maximum at 524 nm. Upon exposure to the vapours of several nitroaromatics, the fluorescence of this HOF was quenched with efficiency up to 73%, showing the potential for detection of nitroaromatic explosives. In contrast, for the vapours of benzene, toluene, -xylene, and trimethylbenzene, enhancement of fluorescence intensity can be observed.
By virtue of a suitable binding site, ion species can also be detected by luminescent HOFs. We have demonstrated that selective fluorescence sensing of metal ions can be achieved by using two fluorescent HOFs (HOF-5 and HOF-10) functionalized with DAT moieties.154 In both HOFs, besides those H-bonding donors/acceptors for the construction of the framework, there are residual sites binding with solvent guest molecules through interactions, which are proposed to be binding sites for metal ions during fluorescence sensing. Each TPE-derived ligand in HOF-10 connects to six adjacent ligands and eight DMSO molecules, while that in HOF-5 connects to eight adjacent ligands, two DMF molecules and two DMSO molecules. Theoretically, the potential solvent-accessible void ratio can be up to 55% in HOF-10 and 56% in HOF-5. The BET surface area of desolvated HOF-10a was determined to be 187 m2 g−1 based on the sorption isotherm at 195 K. Upon excitation at 360 nm, HOF-10 and HOF-5 display blue fluorescence emissions with maxima at 468 and 474 nm, respectively, with quantum yields of 0.52 for HOF-10 and 0.63 for HOF-5. After immersion in the solution of 5.0 mM , both samples exhibit yellow-green fluorescence with maxima at 502 and 504 nm, accompanied by a color change to pale yellow. In contrast, for , , , , , , and , negligible emission shifts can be observed, demonstrating the highly selective sensing for ions. The binding of ions to the N atoms of DAT moieties is regarded to account for such selective fluorescence sensing behavior.
Overall, HOFs show great potential in various optical applications, considering that they integrate the merits of organic chromophores and permanent porosity into the frameworks. The rigid -conjugated moieties can serve not only as scaffolds of pore structure but also as luminescent centers. On the other hand, the highly crystalline nature of HOFs facilitates the reduction of nonradiative decays. The initial endeavors including the applications of HOFs as light emitting materials and for fluorescence sensing have demonstrated these advantages. Strong progress of HOFs in this important application can be expected upon continuous research endeavors.
7. HOFs for other applications
Although HOFs are still in their early stage, some attempts for very promising applications have been carried out, which dramatically highlighted HOFs as emerging porous materials. The advantages that distinguish HOFs from other porous materials are not fully revealed. Given that HOFs are assembled from organic motifs in a reversible binding manner, further applications to make use of such a feature can be expected. Presumably, the major challenge lies in the stability of HOFs.
Considering that HOFs are intrinsically metal-free porous media, their considerable biocompatibility and high porosity make them excellent candidates for drug delivery and biomedical applications. Recently, Liu and Cao et al. reported a stable HOF PFC-1 constructed from 1,3,6,8-tetrakis(-benzoic acid)pyrene () for chemo-photodynamic therapy, which shows good therapeutic efficacy and low cytotoxicity (Fig. 33).115 The organic ligand was targeted owing to its planar core of a large p-conjugated system for extra interactions, which contribute greatly to the framework stability. In this HOF, each ligand connects to six adjacent ligands via four –COOH dimers with multiple strong (2.60 Å) interactions, giving a 2D uninodal 4-connected sql network. Each H-bonded layer further stacks with adjacent layers through strong interactions (3.34 Å), showing an open framework with 1D channels of 18 × 23 Å2. The BET surface area of PFC-1 was estimated to be 2122 m2 g−1 based on the sorption isotherm at 77 K. Based on the strong multiple intermolecular interactions, this HOF exhibits remarkable chemical stability upon immersion in water, organic solvents and acidic solutions, retaining its porosity under harsh conditions. Notably, this HOF can be readily healed from potential thermal damage by simple acid leaching. The metal-free nature, inherent porosity, and chemical stability make PFC-1 a good carrier for the delivery of doxorubicin, showing a loading capacity of 26.5 wt%, which facilitates chemotherapy for cancer. Simultaneously, the suitable arrangement of the pyrene moiety enables this HOF to serve as a good candidate photodynamic therapy (PDT), as it can generate singlet oxygen species monitored by chemical trapping with 9,10-diphenylanthracene. In vitro PDT studies of HeLa cells show that doxorubicin@Nano-PFC-1 exhibits synergetic chemo-photodynamic effects with the merits of low cytotoxicity, good biocompatibility, and high therapeutic efficacy.
Fig. 33.

Schematic diagram of PFC-1 for synergetic chemo-photodynamic therapy by combining the delivery of doxorubicin and generation of singlet oxygen species. Reproduced with permission.115 Copyright 2018 Wiley-VCH.
HOFs have also been explored as volatile drug vessels, involving the storage and delivery of general inhalation anesthetics such as enflurane, isoflurane, and halothane. In 2015, Miljaníc and co-workers reported a fluorinated trispyrazole HOF () as adsorbent for the capture of fluorinated anesthetics, which is aforementioned for the adsorption of fluorocarbons and chlorofluorocarbons.155 Simple adsorption studies revealed that this HOF can take up 2.14–2.67 moles of anesthetic molecules per mole adsorbent, corresponding to 56.7–73.4 wt%. Such adsorption and capture is fast and can be saturated within 3 minutes.
Later, biocompatible and biodegradable porous sorbents157 were applied to related applications in medicine. Recently, Sozzani and Comotti et al. reported several known nanoporous crystalline peptides for biomedical application involving selective adsorption of a family of volatile anesthetics (Fig. 34).156 In particular, crystalline dipeptides l-alanyl-l-isoleucine (AI), l-isoleucyl-l-alanine (IA), l-isoleucyl-l-valine (IV), l-valyl-l-alanine (VA), and l-valyl-l-valine (VV) are investigated, which feature 1D hydrophobic pore channels with aliphatic side groups exposed on the pore surface. In these HOFs, the amphiphilic dipeptide motifs connect with each other via NH +⋯−OOC dimers with multiple charge assisted (2.70–3.00 Å) and weak (3.20–3.41 Å) interactions, giving a honeycomb-shaped H-bonded network that contains double helices of dipeptides. These HOFs contain 1D channels with sizes ranging from 3.5 to 5.3 Å2, as modulated by aliphatic groups. The hydrophobic pore structure in these HOFs is beneficial for adsorption of volatile anesthetics namely halogenated ethers and alkanes as revealed by their high heat of adsorption. Related vapor adsorption isotherms show that these HOFs can take up 170–200 mmol anesthetic molecules per mole adsorbent at 273 K and 80–100 Torr, corresponding to 20 wt%. The heat of adsorption of these HOFs for fluorinated anesthetics is in the range of 35–50 kJ mol−1. Also, 1H, 13C and 19F MAS NMR coupled with modeling studies were applied to detect the arrangement of adsorbed anesthetics. Thus, porous HOFs with good biodegradability and biocompatibility are very promising in biomedical applications.
Fig. 34.

(a) Chemical structures of the halogenated ethers used as guests (above); portion of the crystal structure of l-valyl-l-alanine (VA) along the channel axis (below). (b) Anesthetics adsorption isotherms in different dipeptides. Reproduced with permission.156 Copyright 2018, Royal Society of Chemistry.
The internal pore cavities of HOFs are also suitable as reaction vessels as long as they possess exchangeable guestfilled voids. Since the arrangement of reactant molecules in porous solids is relatively easy to control compared to those in liquid solutions, controlled reactions with configurational selectivity can be readily expected by using porous solids as reaction vessels. In this respect, HOFs show exceptional ability for cocrystallization or recrystallization with various guest molecules, which highlights them as promising reaction media. An early attempt at stereoselective Diels–Alder reactions promoted by a 2D square grid organic network was performed in 1997.158 This H-bonded network is composed of an anthracene-bisresorcinol derivative linked with four adjacent ligands through (2.73 Å) interactions, showing supermolecular sheets with pore cavities. Prior to Diels–Alder reaction, the recrystallization of alkyl acrylates and cyclohexadiene into this network gives single crystals of the reactant adduct, showing a suitable arrangement of both reactants with an ordered interval. Therefore, the acrolein–cyclohexadiene Diels–Alder reaction was performed to show enhanced stereoselectivity.
To improve the crystalline order of polymer, Sozzani et al. used nanoporous crystalline peptides as reaction vessels for controlled solid-state polymerization of acrylic or diene monomers in an in situ -ray-induced manner (Fig. 35).159 Several porous crystals, namely l-alanyl-l-valine (Ala-Val), l-isoleucyl-l-valine (Ile-Val), l-valyl-l-alanine (Val-Ala), and l-valyl-l-isoleucine (Val-Ile), are involved, which contain 1D pore channels with sizes of 5.0, 3.9, 4.7 and 3.7 Å in diameters, respectively. Prior to polymerization, the monomer vapors of acrylonitrile, pentadiene, and isoprene were adsorbed into guest-free crystalline peptides, followed by exposure upon g-ray irradiation for generation of the initiating radicals to promote polymerization. The polymerization at room temperature after one week gives poly(acrylonitrile) (PAN), poly(pentadiene) (PPD) and poly(isoprene) (PI) with a molecular weight of 50–150, 20–68 and 100–156 kDa, respectively. Notably, using porous crystals with a suitable pore size, the resultant polymer can be isotactic with high stereoregularity demonstrated from the corresponding 13C NMR spectra. For isotactic PAN, such topochemical polymerization is achieved under mild conditions of temperature and pressure, which is superior to conventional polymerization methods.
Fig. 35.

(a) Crystal structure of the porous Ala-Val compound showing the empty channels along the crystallographic [001] axis in blue and yellow. (b) Schematic representation of the monomers and dipeptides used for the polymerization process. Reproduced with permission.159 Copyright 2012 Wiley-VCH.
Besides serving as reaction vessels, the introduction of functional sites into HOFs makes them catalysts for organic reactions. Li et al. reported a cobalt(II) porphyrin HOF (CoTCPp, Co(II) 5,10,15,20-tetra(4-(4-acetateethyl)phenoxy)phenyl-porphyrin), ()·2DMF, for oxidation of alkylbenzenes.160 In this HOF, each CoTCPp ligand connects to four adjacent ligands via four –COOH dimers with multiple strong (2.64–2.68 Å) interactions, giving a 2D uninodal 4-connected sql network. This HOF contains 1D pore channels (4.0 6.5 Å2), showing a solvent-accessible void ratio of 18.9% and a BET surface area of 98 m2 g−1 estimated from the sorption isotherm at 77 K. At 353 K in with tertbutylhydroperoxide, the quantitative oxidation of ethylbenzene to acetophenone was achieved with a conversion yield of 83%.
Overall, the intrinsic nature of HOFs renders this type of porous materials attractive for various applications, serving as adsorbents to reaction vessels. There are also attempts to apply HOFs for other applications, e.g. sequestration of radioactive waste like iodine isotopes from the aqueous environment.98 Some interesting dimensions and regions of research have also been explored, involving the post-modification of HOFs to COFs by irreversible organic reactions, which results in new porous organic frameworks of improved stability.161 Nevertheless, the application potential of HOFs is far more than the above examples, which requires more intersections of different disciplines, involving more practical applications.
8. Conclusions
In this review, we have highlighted the current state of the art for HOFs, including important progress in their broad application for gas storage and separation, molecular recognition, conductive and optical applications, heterogeneous catalysis, and biomedicine. HOFs are extended porous crystalline frameworks that are composed of light elements (mainly C, H, O, N) linked by typical H-bonds. The construction of HOFs with open frameworks can be achieved by the combination of rigid molecular backbones and hydrogen-bonded units with strong intermolecular interactions, while the resultant structures can be further stabilized by introducing interpenetration and other types of intermolecular interactions. The reversible and flexible nature of H-bonding connections enables HOFs to show high crystallinity, solution processability, easy healing and purification. HOFs are intrinsically metal-free and low-density porous media, which can even be biocompatible and biodegradable and serve as promising candidates for drug delivery and biomedical applications. These features make HOFs a new platform for exploring light-weight functional materials.
As H-bonds are essentially weak interactions featuring poor rigidity and directionality, compared to zeolites, MOFs and COFs, it is too hard for HOFs to retain their porous framework after the removal of guest molecules. The establishment of microporosity with surface area and pore volume determination has been a long-standing challenge since their early developments, and it took a long time to reveal the permanent porosity of HOFs, which initiated the applications of this class of materials as functional porous media. Since the original inception of HOFs, this field rapidly developed, especially in the last few years. In terms of porosity and surface area, substantial progress has been witnessed, affording the current benchmark HOFs that are even comparable to some MOFs of extraordinarily high porosity. Some HOFs show impressive framework stability upon external stimulus like heat and chemicals, which is even superior to most MOFs. Notably, the healing of HOFs from structural damage can be readily achieved by simple solution processing. Following continuous research endeavours on HOF chemistry, the appearance of this field is expected to change dramatically.
Several challenges remain for HOFs. Precise control over pore size and pore chemistry of HOFs rather than empirical approaches is needed to rationally design new functional HOF materials, in the same manner as MOFs and COFs. Simultaneously, the polymorphism issue during the construction of HOFs needs to be overcome, not only for the phase purity but also for directional synthesis in a predictable way. Also, there are challenges in the construction of outperforming HOFs that integrate the merits of ultra-high surface area and high framework rigidity, where new strategies might be required. It should be noted that strong binding sites especially open metal sites make MOFs unique host materials for some important applications involving various catalysis reactions and highly selective recognition of molecules. In contrast, the lack of functional sites especially strong Lewis base/acid sites has limited the application of HOFs, which needs to be taken into account during future synthesis and design of HOFs. The last but not the least, intensive efforts will be required for the co-crystallization of mixed organic ligands into single HOFs, which can be either of the same type or different types; though this is very challenging it can drastically boost the diversity of HOF structures.
More and more research interests have been directed to this field, owing to the unique features of this class of porous materials. Remarkable evolutions will be brought in the near future. New dimensions and regions of research on HOFs will enrich and supplement the HOF chemistry, which in turn affords diverse functionalities and applications. We see an exciting future for these unique materials.
Acknowledgements
The support from the National Science Foundation (DMR-1606826, B. C.) and the Welch Foundation under Grant AX-1730 (B. C.) is gratefully acknowledged.
Biographies

Rui-Biao Lin obtained his BSc in Chemistry from Sun Yat-Sen University (SYSU) in 2009. After studying the synthesis and crystal engineering of functional coordination polymers under the supervision of professors Xiao-Ming Chen and Jie-Peng Zhang, he obtained his PhD in 2014 (SYSU). He currently works with Prof. Banglin Chen at the University of Texas at San Antonio as a postdoctoral fellow, where he is working on multifunctional porous materials.

Yabing He earned his PhD in organic chemistry from Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, under the direction of Prof. Lianxun Gao in 2010. After that, he worked in the group of Prof. Banglin Chen at the University of Texas at San Antonio as a postdoctoral Research Fellow during 2010–2012. In 2012, he joined the Zhejiang Normal University, where he is a professor of chemistry. His current research focuses on the design and synthesis of porous materials and study of their gas adsorption and separation properties.

Peng Li was born in Hunan, China. He received his BS (2006) and MS (2009) degrees from Fudan University in China, and PhD (2014) from the University of Texas at San Antonio under the supervision of Prof. Banglin Chen. Then he worked with Professors Joseph Hupp and Omar K. Farha at Northwestern University as a postdoctoral fellow during 2014–2018. He will join Fudan University as a professor of chemistry in 2019. His research interest is related to functional porous materials for gas storage, separation, catalysis, and enzyme immobilization.

Hailong Wang was born in Jilin, P. R. China, in 1984. He received his BSc (2007), MSc (2010), and PhD (2013) degrees from Northeast Normal University, Shangdong University, and University of Science and Technology Beijing, respectively, under the supervision of Prof. Jianzhuang Jiang. Then he worked with Prof. Banglin Chen at the University of Texas at San Antonio and Prof. Qiang Xu at National Institute of Advanced Industrial Science and Technology as a postdoctoral fellow during 2013–2018. Now, he is a professor at the University of Science and Technology Beijing. His interest is in the synthesis and application of novel photoactive porous materials, including organic cages, HOFs, COFs, and MOFs.

Wei Zhou received his PhD in 2005 from the University of Pennsylvania, under the supervision of Prof. John E. Fischer. From 2005 to 2007, he was a postdoctoral researcher at the NIST Center for Neutron Research (NCNR), working with Dr Taner Yildirim. After that, he was appointed to his current position as a research scientist at NCNR. His research interests are in the areas of novel porous materials, computational materials design, and neutron diffraction/spectroscopy.

Banglin Chen was born in Zhejiang, China. He received BS (1985) and MS (1988) degrees in Chemistry from Zhejiang University in China, and his PhD from National University of Singapore in 2000. He worked with professors Omar M. Yaghi at University of Michigan, Stephen Lee at Cornell University, and Andrew W. Maverick at Louisiana State University as a postdoctoral fellow during 2000–2003 before joining the University of Texas-Pan American in 2003.He moved to the University of Texas at San Antonio in August 2009, where he is a Dean’s Distinguished Chair Professor of Chemistry, working on multifunctional metal–organic framework and hydrogen bonded organic framework materials.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Davis ME, Nature, 2002, 417, 813. [DOI] [PubMed] [Google Scholar]
- 2.Corma A, Chem. Rev, 1997, 97, 2373–2420. [DOI] [PubMed] [Google Scholar]
- 3.Zhao D, Feng J, Huo Q, Melosh N, Fredrickson GH, Chmelka BF and Stucky GD, Science, 1998, 279, 548. [DOI] [PubMed] [Google Scholar]
- 4.Li J, Corma A. and Yu J, Chem. Soc. Rev, 2015, 44, 7112–7127. [DOI] [PubMed] [Google Scholar]
- 5.Furukawa H, Cordova KE, O’Keeffe M. and Yaghi OM, Science, 2013, 341, 1230444. [DOI] [PubMed] [Google Scholar]
- 6.Kitagawa S, Kitaura R. and Noro S.-i, Angew. Chem., Int. Ed, 2004, 43, 2334–2375. [DOI] [PubMed] [Google Scholar]
- 7.Kitagawa S, Angew. Chem., Int. Ed, 2015, 54, 10686–10687. [DOI] [PubMed] [Google Scholar]
- 8.Jiang J, Zhao Y. and Yaghi OM, J. Am. Chem. Soc, 2016, 138, 3255–3265. [DOI] [PubMed] [Google Scholar]
- 9.Diercks CS and Yaghi OM, Science, 2017, 355, eaal1585. [DOI] [PubMed] [Google Scholar]
- 10.Feng X, Ding X. and Jiang D, Chem. Soc. Rev, 2012, 41, 6010–6022. [DOI] [PubMed] [Google Scholar]
- 11.Yuan S, Feng L, Wang K, Pang J, Bosch M, Lollar C, Sun Y, Qin J, Yang X, Zhang P, Wang Q, Zou L, Zhang Y, Zhang L, Fang Y, Li J. and Zhou H-C, Adv. Mater, 2018, 30, 1704303. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J-P, Zhou H-L, Zhou D-D, Liao P-Q and Chen X-M, Natl. Sci. Rev, 2018, 5, 907–919. [Google Scholar]
- 13.Hoskins BF and Robson R, J. Am. Chem. Soc, 1989, 111, 5962–5964. [Google Scholar]
- 14.Kondo M, Yoshitomi T, Matsuzaka H, Kitagawa S. and Seki K, Angew. Chem., Int. Ed, 1997, 36, 1725–1727. [Google Scholar]
- 15.Li H, Eddaoudi M, Groy TL and Yaghi OM, J. Am. Chem. Soc, 1998, 120, 8571–8572. [Google Scholar]
- 16.Lin R-B, Xiang S, Li B, Cui Y, Zhou W, Qian G. and Chen B, Isr. J. Chem, 2018, 58, 949–961. [Google Scholar]
- 17.Li B, Wen H-M, Cui Y, Zhou W, Qian G. and Chen B, Adv. Mater, 2016, 28, 8819–8860. [DOI] [PubMed] [Google Scholar]
- 18.Hendon CH, Rieth AJ, Korzyński MD and Dincă M, ACS Cent. Sci, 2017, 3, 554–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J-R, Kuppler RJ and Zhou H-C, Chem. Soc. Rev, 2009, 38, 1477–1504. [DOI] [PubMed] [Google Scholar]
- 20.Bao Z, Chang G, Xing H, Krishna R, Ren Q. and Chen B, Energy Environ. Sci, 2016, 9, 3612–3641. [Google Scholar]
- 21.Adil K, Belmabkhout Y, Pillai RS, Cadiau A, Bhatt PM, Assen AH, Maurin G. and Eddaoudi M, Chem. Soc. Rev, 2017, 46, 3402–3430. [DOI] [PubMed] [Google Scholar]
- 22.Lin R-B, Xiang S, Xing H, Zhou W. and Chen B, Coord. Chem. Rev, 2019, 378, 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao X, Wang Y, Li D-S, Bu X. and Feng P, Adv. Mater, 2018, 30, 1705189. [DOI] [PubMed] [Google Scholar]
- 24.Lin R-B, Liu S-Y, Ye J-W, Li X-Y and Zhang J-P, Adv. Sci, 2016, 3, 1500434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiao L, Wang Y, Jiang H-L and Xu Q, Adv. Mater, 2017, 30, 1703663. [DOI] [PubMed] [Google Scholar]
- 26.Simon-Yarza T, Mielcarek A, Couvreur P. and Serre C, Adv. Mater, 2018, 30, 1707365. [DOI] [PubMed] [Google Scholar]
- 27.Lu K, Aung T, Guo N, Weichselbaum R. and Lin W, Adv. Mater, 2018, 30, 1707634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He Y, Xiang S. and Chen B, J. Am. Chem. Soc, 2011, 133, 14570–14573. [DOI] [PubMed] [Google Scholar]
- 29.Lu J¨ and Cao R, Angew. Chem., Int. Ed, 2016, 55, 9474–9480. [DOI] [PubMed] [Google Scholar]
- 30.Hashim MI, Hsu CW, Le HTM and Miljaníc OS, Synlett, 2016, 1907–1918.
- 31.Han Y-F, Yuan Y-X and Wang H-B, Molecules, 2017, 22, 226.28157169 [Google Scholar]
- 32.Luo J, Wang J-W, Zhang J-H, Lai S. and Zhong D-C, CrystEngComm, 2018, 20, 5884–5898. [Google Scholar]
- 33.Simard M, Su D. and Wuest JD, J. Am. Chem. Soc, 1991, 113, 4696–4698. [Google Scholar]
- 34.Venkataraman D, Lee S, Zhang J. and Moore JS, Nature, 1994, 371, 591. [Google Scholar]
- 35.Wang X, Simard M. and Wuest JD, J. Am. Chem. Soc, 1994, 116, 12119–12120. [Google Scholar]
- 36.Endo K, Sawaki T, Koyanagi M, Kobayashi K, Masuda H. and Aoyama Y, J. Am. Chem. Soc, 1995, 117, 8341–8352. [Google Scholar]
- 37.Kolotuchin SV, Fenlon EE, Wilson SR, Loweth CJ and Zimmerman SC, Angew. Chem., Int. Ed, 1996, 34, 2654–2657. [Google Scholar]
- 38.Aoyama Y, Endo K, Anzai T, Yamaguchi Y, Sawaki T, Kobayashi K, Kanehisa N, Hashimoto H, Kai Y. and Masuda H, J. Am. Chem. Soc, 1996, 118, 5562–5571. [Google Scholar]
- 39.Bhyrappa P, Wilson SR and Suslick KS, J. Am. Chem. Soc, 1997, 119, 8492–8502. [Google Scholar]
- 40.Brunet P, Simard M. and Wuest JD, J. Am. Chem. Soc, 1997, 119, 2737–2738. [Google Scholar]
- 41.Kobayashi K, Shirasaka T, Sato A, Horn E. and Furukawa N, Angew. Chem., Int. Ed, 1999, 38, 3483–3486. [DOI] [PubMed] [Google Scholar]
- 42.Fournier J-H, Maris T, Wuest JD, Guo W. and Galoppini E, J. Am. Chem. Soc, 2003, 125, 1002–1006. [DOI] [PubMed] [Google Scholar]
- 43.Brunet P, Demers E, Maris T, Enright GD and Wuest JD, Angew. Chem., Int. Ed, 2003, 42, 5303–5306. [DOI] [PubMed] [Google Scholar]
- 44.Kobayashi K, Sato A, Sakamoto S. and Yamaguchi K, J. Am. Chem. Soc, 2003, 125, 3035–3045. [DOI] [PubMed] [Google Scholar]
- 45.Perron ME, Monchamp F, Duval H, Boils-Boissier D. and Wuest JD, Pure Appl. Chem, 2004, 76, 1345–1351. [Google Scholar]
- 46.Wuest JD, Chem. Commun, 2005, 5830–5837. [DOI] [PubMed]
- 47.Malek N, Maris T, Simard M. and Wuest JD, J. Am. Chem. Soc, 2005, 127, 5910–5916. [DOI] [PubMed] [Google Scholar]
- 48.Saied O, Maris T, Wang X, Simard M. and Wuest JD, J. Am. Chem. Soc, 2005, 127, 10008–10009. [DOI] [PubMed] [Google Scholar]
- 49.Malek N, Maris T, Perron M-È and Wuest JD, Angew. Chem., Int. Ed, 2005, 44, 4021–4025. [DOI] [PubMed] [Google Scholar]
- 50.Görbitz CH, Nilsen M, Szeto K. and Tangen LW, Chem. Commun, 2005, 4288–4290. [DOI] [PubMed]
- 51.Yang W, Greenaway A, Lin X, Matsuda R, Blake AJ, Wilson C, Lewis W, Hubberstey P, Kitagawa S, Champness NR and Schröder M, J. Am. Chem. Soc, 2010, 132, 14457–14469. [DOI] [PubMed] [Google Scholar]
- 52.Mastalerz M. and Oppel IM, Angew. Chem., Int. Ed, 2012, 51, 5252–5255. [DOI] [PubMed] [Google Scholar]
- 53.Pulido A, Chen L, Kaczorowski T, Holden D, Little MA, Chong SY, Slater BJ, McMahon DP, Bonillo B, Stackhouse CJ, Stephenson A, Kane CM, Clowes R, Hasell T, Cooper AI and Day GM, Nature, 2017, 543, 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arunan E, Desiraju GR, Klein RA, Sadlej J, Scheiner S, Alkorta I, Clary DC, Crabtree RH, Dannenberg JJ, Hobza P, Kjaergaard HG, Legon AC, Mennucci B. and Nesbitt DJ, Pure Appl. Chem, 2011, 83, 1637–1641. [Google Scholar]
- 55.Steiner T, Angew. Chem., Int. Ed, 2002, 41, 48–76. [Google Scholar]
- 56.Allcock HR and Siegel LA, J. Am. Chem. Soc, 1964, 86, 5140–5144. [Google Scholar]
- 57.Sozzani P, Comotti A, Simonutti R, Meersmann T, Logan JW and Pines A, Angew. Chem., Int. Ed, 2000, 39, 2695–2699. [DOI] [PubMed] [Google Scholar]
- 58.Sozzani P, Bracco S, Comotti A, Ferretti L. and Simonutti R, Angew. Chem., Int. Ed, 2005, 44, 1816–1820. [DOI] [PubMed] [Google Scholar]
- 59.Couderc G, Hertzsch T, Behrnd NR, Krämer K. and Hulliger J, Microporous Mesoporous Mater., 2006, 88, 170–175. [Google Scholar]
- 60.Barrer RM and Shanson VH, J. Chem. Soc., Chem. Commun, 1976, 333–334.
- 61.Lee F, Gabe E, Tse JS and Ripmeester JA, J. Am. Chem. Soc, 1988, 110, 6014–6019. [DOI] [PubMed] [Google Scholar]
- 62.Lloyd GO, Bredenkamp MW and Barbour LJ, Chem. Commun, 2005, 4053–4055. [DOI] [PubMed]
- 63.Atwood JL, Barbour LJ, Jerga A. and Schottel BL, Science, 2002, 298, 1000. [DOI] [PubMed] [Google Scholar]
- 64.Atwood JL, Barbour LJ and Jerga A, Angew. Chem., Int. Ed, 2004, 43, 2948–2950. [DOI] [PubMed] [Google Scholar]
- 65.Thallapally PK, Dobrzańska L, Gingrich TR, Wirsig TB, Barbour LJ and Atwood JL, Angew. Chem., Int. Ed, 2006, 45, 6506–6509. [DOI] [PubMed] [Google Scholar]
- 66.Dalgarno SJ, Thallapally PK, Barbour LJ and Atwood JL, Chem. Soc. Rev, 2007, 36, 236–245. [DOI] [PubMed] [Google Scholar]
- 67.Thallapally PK, McGrail BP, Atwood JL, Gaeta C, Tedesco C. and Neri P, Chem. Mater, 2007, 19, 3355–3357. [Google Scholar]
- 68.Thallapally PK, Peter McGrail B, Dalgarno SJ, Schaef HT, Tian J. and Atwood JL, Nat. Mater, 2008, 7, 146. [DOI] [PubMed] [Google Scholar]
- 69.Freeman WA, Mock WL and Shih NY, J. Am. Chem. Soc, 1981, 103, 7367–7368. [Google Scholar]
- 70.Lim S, Kim H, Selvapalam N, Kim K-J, Cho SJ, Seo G. and Kim K, Angew. Chem., Int. Ed, 2008, 47, 3352–3355. [DOI] [PubMed] [Google Scholar]
- 71.Kudo H, Hayashi R, Mitani K, Yokozawa T, Kasuga NC and Nishikubo T, Angew. Chem., Int. Ed, 2006, 45, 7948–7952. [DOI] [PubMed] [Google Scholar]
- 72.Tozawa T, Jones JTA, Swamy SI, Jiang S, Adams DJ, Shakespeare S, Clowes R, Bradshaw D, Hasell T, Chong SY, Tang C, Thompson S, Parker J, Trewin A, Bacsa J, Slawin AMZ, Steiner A. and Cooper AI, Nat. Mater, 2009, 8, 973. [DOI] [PubMed] [Google Scholar]
- 73.Giri N, Del Pópolo MG, Melaugh G, Greenaway RL, Rätzke K, Koschine T, Pison L, Gomes MFC, Cooper AI and James SL, Nature, 2015, 527, 216. [DOI] [PubMed] [Google Scholar]
- 74.Hasell T. and Cooper AI, Nat. Rev. Mater, 2016, 1, 16053. [Google Scholar]
- 75.Ogoshi T, Yamagishi T.-a. and Nakamoto Y, Chem. Rev, 2016, 116, 7937–8002. [DOI] [PubMed] [Google Scholar]
- 76.Tian J, Chen L, Zhang D-W, Liu Y. and Li Z-T, Chem. Commun, 2016, 52, 6351–6362. [DOI] [PubMed] [Google Scholar]
- 77.Cai S, Shi H, Zhang Z, Wang X, Ma H, Gan N, Wu Q, Cheng Z, Ling K, Gu M, Ma C, Gu L, An Z. and Huang W, Angew. Chem., Int. Ed, 2018, 57, 4005–4009. [DOI] [PubMed] [Google Scholar]
- 78.Yu T, Ou D, Yang Z, Huang Q, Mao Z, Chen J, Zhang Y, Liu S, Xu J, Bryce MR and Chi Z, Chem. Sci, 2017, 8, 1163–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hisaki I, Senga H, Shigemitsu H, Tohnai N. and Miyata M, Chem. – Eur. J, 2011, 17, 14348–14353. [DOI] [PubMed] [Google Scholar]
- 80.Mastalerz M, Chem. – Eur. J, 2012, 18, 10082–10091. [DOI] [PubMed] [Google Scholar]
- 81.Mastalerz M, Acc. Chem. Res, 2018, 51, 2411–2422. [DOI] [PubMed] [Google Scholar]
- 82.Zhou Y, Liu B, Sun X, Li J, Li G, Huo Q. and Liu Y, Cryst. Growth Des, 2017, 17, 6653–6659. [Google Scholar]
- 83.Sanchez-Sala M, Vallcorba O, Domingo C. and Ayllón JA, Cryst. Growth Des, 2018, 18, 6621–6626. [Google Scholar]
- 84.Wahl H, Haynes DA and le Roex T, Cryst. Growth Des, 2017, 17, 4377–4383. [Google Scholar]
- 85.Li P, Li P, Ryder MR, Liu Z, Stern CL, Farha OK and Stoddart F, Angew. Chem., Int. Ed, 2018, 57, DOI: 10.1002/anie.201811263. [DOI] [Google Scholar]
- 86.Hisaki I, Nakagawa S, Suzuki Y. and Tohnai N, Chem. Lett, 2018, 47, 1143–1146. [Google Scholar]
- 87.Castells-Gil J, Padial NM and Martı´-Gastaldo C, New J. Chem, 2018, 42, 16138–16143. [Google Scholar]
- 88.Chen T-H, Popov I, Kaveevivitchai W, Chuang Y-C, Chen Y-S, Daugulis O, Jacobson AJ and Miljanić OŠ, Nat. Commun, 2014, 5, 5131. [DOI] [PubMed] [Google Scholar]
- 89.Maly KE, Gagnon E, Maris T. and Wuest JD, J. Am. Chem. Soc, 2007, 129, 4306–4322. [DOI] [PubMed] [Google Scholar]
- 90.Maly KE, Buck W. and Dawe LN, CrystEngComm, 2017, 19, 6401–6405. [Google Scholar]
- 91.Luo X-Z, Jia X-J, Deng J-H, Zhong J-L, Liu H-J, Wang K-J and Zhong D-C, J. Am. Chem. Soc, 2013, 135, 11684–11687. [DOI] [PubMed] [Google Scholar]
- 92.Morshedi M, Thomas M, Tarzia A, Doonan CJ and White NG, Chem. Sci, 2017, 8, 3019–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chaumont C, Mobian P, Kyritsakas N. and Henry M, CrystEngComm, 2013, 15, 6845–6862. [Google Scholar]
- 94.Li P, Alduhaish O, Arman HD, Wang H, Alfooty K. and Chen B, Cryst. Growth Des, 2014, 14, 3634–3638. [Google Scholar]
- 95.Liu L, Zhang Y, Wang X-L, Luo G-G, Xiao Z-J, Cheng L. and Dai J-C, Cryst. Growth Des, 2018, 18, 1629–1635. [Google Scholar]
- 96.Makowski SJ, Köstler P. and Schnick W, Chem. – Eur. J, 2012, 18, 3248–3257. [DOI] [PubMed] [Google Scholar]
- 97.Hisaki I, Nakagawa S, Ikenaka N, Imamura Y, Katouda M, Tashiro M, Tsuchida H, Ogoshi T, Sato H, Tohnai N. and Miyata M, J. Am. Chem. Soc, 2016, 138, 6617–6628. [DOI] [PubMed] [Google Scholar]
- 98.Hisaki I, Ikenaka N, Gomez E, Cohen B, Tohnai N. and Douhal A, Chem. – Eur. J, 2017, 23, 11611–11619. [DOI] [PubMed] [Google Scholar]
- 99.Hisaki I, Suzuki Y, Gomez E, Cohen B, Tohnai N. and Douhal A, Angew. Chem., Int. Ed, 2018, 57, 12650–12655. [DOI] [PubMed] [Google Scholar]
- 100.Hisaki I, Nakagawa S, Tohnai N. and Miyata M, Angew. Chem., Int. Ed, 2015, 54, 3008–3012. [DOI] [PubMed] [Google Scholar]
- 101.Hisaki I, Toda H, Sato H, Tohnai N. and Sakurai H, Angew. Chem., Int. Ed, 2017, 56, 15294–15298. [DOI] [PubMed] [Google Scholar]
- 102.Hisaki I, Nakagawa S, Sato H. and Tohnai N, Chem. Commun, 2016, 52, 9781–9784. [DOI] [PubMed] [Google Scholar]
- 103.Hashim MI, Le HTM, Chen T-H, Chen Y-S, Daugulis O, Hsu C-W, Jacobson AJ, Kaveevivitchai W, Liang X, Makarenko T, Miljanić OS, I. Popovs, Tran HV, Wang X, Wu C-H and Wu JI, J. Am. Chem. Soc, 2018, 140, 6014–6026. [DOI] [PubMed] [Google Scholar]
- 104.Luo Y-H, He X-T, Hong D-L, Chen C, Chen F-H, Jiao J, Zhai L-H, Guo L-H and Sun B-W, Adv. Funct. Mater, 2018, 28, 1804822. [Google Scholar]
- 105.Morshedi M, Ward JS, Kruger PE and White NG, Dalton Trans., 2018, 47, 783–790. [DOI] [PubMed] [Google Scholar]
- 106.Manjare Y, Nagarajan V. and Pedireddi VR, Cryst. Growth Des, 2014, 14, 723–729. [Google Scholar]
- 107.Lu J¨, Perez-Krap C, Suyetin M, Alsmail NH, Yan Y, Yang S, Lewis W, Bichoutskaia E, Tang CC, Blake AJ, Cao R. and Schröder M, J. Am. Chem. Soc, 2014, 136, 12828–12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li Y, Handke M, Chen Y-S, Shtukenberg AG, Hu CT and Ward MD, J. Am. Chem. Soc, 2018, 140, 12915–12921. [DOI] [PubMed] [Google Scholar]
- 109.Xing G, Yan T, Das S, Ben T. and Qiu S, Angew. Chem., Int. Ed, 2018, 57, 5345–5349. [DOI] [PubMed] [Google Scholar]
- 110.Yamagishi H, Sato H, Hori A, Sato Y, Matsuda R, Kato K. and Aida T, Science, 2018, 361, 1242. [DOI] [PubMed] [Google Scholar]
- 111.Bao Z, Xie D, Chang G, Wu H, Li L, Zhou W, Wang H, Zhang Z, Xing H, Yang Q, Zaworotko MJ, Ren Q. and Chen B, J. Am. Chem. Soc, 2018, 140, 4596–4603. [DOI] [PubMed] [Google Scholar]
- 112.Hu F, Liu C, Wu M, Pang J, Jiang F, Yuan D. and Hong M, Angew. Chem., Int. Ed, 2017, 56, 2101–2104. [DOI] [PubMed] [Google Scholar]
- 113.Duan J, Jin W. and Kitagawa S, Coord. Chem. Rev, 2017, 332, 48–74. [Google Scholar]
- 114.Low JJ, Benin AI, Jakubczak P, Abrahamian JF, Faheem SA and Willis RR, J. Am. Chem. Soc, 2009, 131, 15834–15842. [DOI] [PubMed] [Google Scholar]
- 115.Yin Q, Zhao P, Sa R-J, Chen G-C, Lu J¨, Liu T-F and Cao R, Angew. Chem., Int. Ed, 2018, 57, 7691–7696. [DOI] [PubMed] [Google Scholar]
- 116.Yamamoto A, Hirukawa T, Hisaki I, Miyata M. and Tohnai N, Tetrahedron Lett., 2013, 54, 1268–1273. [Google Scholar]
- 117.Li P, He Y, Arman HD, Krishna R, Wang H, Weng L. and Chen B, Chem. Commun, 2014, 50, 13081–13084. [DOI] [PubMed] [Google Scholar]
- 118.Ji Q, Le HTM, Wang X, Chen Y-S, Makarenko T, Jacobson AJ and Š O. Miljanić, Chem. – Eur. J, 2015, 21, 17205–17209. [DOI] [PubMed] [Google Scholar]
- 119.Bassanetti I, Bracco S, Comotti A, Negroni M, Bezuidenhout C, Canossa S, Mazzeo PP, Marchio L´ and Sozzani P, J. Mater. Chem. A, 2018, 6, 14231–14239. [Google Scholar]
- 120.Lu J¨, Perez-Krap C, Trousselet F, Yan Y, Alsmail NH, Karadeniz B, Jacques NM, Lewis W, Blake AJ, Coudert F-X, Cao R. and Schröder M, Cryst. Growth Des, 2018, 18, 2555–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nandi S, Chakraborty D. and Vaidhyanathan R, Chem. Commun, 2016, 52, 7249–7252. [DOI] [PubMed] [Google Scholar]
- 122.Lenz A. and Ojamäe L, J. Phys. Chem. A, 2011, 115, 6169–6176. [DOI] [PubMed] [Google Scholar]
- 123.Yang W, Wang J, Wang H, Bao Z, Zhao JC-G and Chen B, Cryst. Growth Des, 2017, 17, 6132–6137. [Google Scholar]
- 124.Nugent PS, Rhodus VL, Pham T, Forrest K, Wojtas L, Space B. and Zaworotko MJ, J. Am. Chem. Soc, 2013, 135, 10950–10953. [DOI] [PubMed] [Google Scholar]
- 125.Dalrymple SA and Shimizu GKH, J. Am. Chem. Soc, 2007, 129, 12114–12116. [DOI] [PubMed] [Google Scholar]
- 126.Dechambenoit P, Ferlay S, Kyritsakas N. and Hosseini MW, J. Am. Chem. Soc, 2008, 130, 17106–17113. [DOI] [PubMed] [Google Scholar]
- 127.Wei W, Li W, Wang X. and He J, Cryst. Growth Des, 2013, 13, 3843–3846. [Google Scholar]
- 128.Wilson BH, Scott HS, Qazvini OT, Telfer SG, Mathonière C, Clérac R. and Kruger PE, Chem. Commun, 2018, 54, 13391–13394. [DOI] [PubMed] [Google Scholar]
- 129.Taylor JM, Dwyer PJ, Reid JW, Gelfand BS, Lim D.-w., Donoshita M, Veinberg SL, Kitagawa H, Vukotic VN and Shimizu GKH, Chem, 2018, 4, 868–878. [Google Scholar]
- 130.Wang H, Li B, Wu H, Hu T-L, Yao Z, Zhou W, Xiang S. and Chen B, J. Am. Chem. Soc, 2015, 137, 9963–9970. [DOI] [PubMed] [Google Scholar]
- 131.Yang W, Li B, Wang H, Alduhaish O, Alfooty K, Zayed MA, Li P, Arman HD and Chen B, Cryst. Growth Des, 2015, 15, 2000–2004. [Google Scholar]
- 132.Wang H, Wu H, Kan J, Chang G, Yao Z, Li B, Zhou W, Xiang S, Zhao JC-G and Chen B, J. Mater. Chem. A, 2017, 5, 8292–8296. [Google Scholar]
- 133.Li P, He Y, Zhao Y, Weng L, Wang H, Krishna R, Wu H, Zhou W, O’Keeffe M, Han Y. and Chen B, Angew. Chem., Int. Ed, 2014, 54, 574–577. [DOI] [PubMed] [Google Scholar]
- 134.Zentner CA, Lai HWH, Greenfield JT, Wiscons RA, Zeller M, Campana CF, Talu O, FitzGerald SA and Rowsell JLC, Chem. Commun, 2015, 51, 11642–11645. [DOI] [PubMed] [Google Scholar]
- 135.Lai HWH, Wiscons RA, Zentner CA, Zeller M. and Rowsell JLC, Cryst. Growth Des, 2016, 16, 821–833. [Google Scholar]
- 136.Yoon T-U, Baek SB, Kim D, Kim E-J, Lee W-G, Singh BK, Lah MS, Bae Y-S and Kim KS, Chem. Commun, 2018, 54, 9360–9363. [DOI] [PubMed] [Google Scholar]
- 137.Zhou D-D, Xu Y-T, Lin R-B, Mo Z-W, Zhang W-X and Zhang J-P, Chem. Commun, 2016, 52, 4991–4994. [DOI] [PubMed] [Google Scholar]
- 138.Li P, He Y, Guang J, Weng L, Zhao JC-G, Xiang S. and Chen B, J. Am. Chem. Soc, 2014, 136, 547–549. [DOI] [PubMed] [Google Scholar]
- 139.Yan W, Yu X, Yan T, Wu D, Ning E, Qi Y, Han Y-F and Li Q, Chem. Commun, 2017, 53, 3677–3680. [DOI] [PubMed] [Google Scholar]
- 140.Swift JA, Reynolds AM and Ward MD, Chem. Mater, 1998, 10, 4159–4168. [Google Scholar]
- 141.Xiao W, Hu C. and Ward MD, J. Am. Chem. Soc, 2014, 136, 14200–14206. [DOI] [PubMed] [Google Scholar]
- 142.Adachi T. and Ward MD, Acc. Chem. Res, 2016, 49, 2669–2679. [DOI] [PubMed] [Google Scholar]
- 143.Sisson AL, del Amo Sanchez V, Magro G, Griffin AME, Shah S, Charmant JPH and Davis AP, Angew. Chem., Int. Ed, 2005, 44, 6878–6881. [DOI] [PubMed] [Google Scholar]
- 144.Natarajan R, Bridgland L, Sirikulkajorn A, Lee J-H, Haddow MF, Magro G, Ali B, Narayanan S, Strickland P, Charmant JPH, Orpen AG, McKeown NB, Bezzu CG and Davis AP, J. Am. Chem. Soc, 2013, 135, 16912–16925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yang W, Yang F, Hu T-L, King SC, Wang H, Wu H, Zhou W, Li J-R, Arman HD and Chen B, Cryst. Growth Des, 2016, 16, 5831–5835. [Google Scholar]
- 146.Karmakar A, Illathvalappil R, Anothumakkool B, Sen A, Samanta P, Desai AV, Kurungot S. and Ghosh SK, Angew. Chem., Int. Ed, 2016, 55, 10667–10671. [DOI] [PubMed] [Google Scholar]
- 147.Xing G, Bassanetti I, Bracco S, Negroni M, Bezuidenhout C, Ben T, Sozzani P. and Comotti A, Chem. Sci, 2019, 10, DOI: 10.1039/C8SC04376K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gomez E, Gutiérrez M, Cohen B, Hisaki I. and Douhal A, J. Mater. Chem. C, 2018, 6, 6929–6939. [Google Scholar]
- 149.Hisaki I, Emilya Affendy NQ and Tohnai N, CrystEngComm, 2017, 19, 4892–4898. [Google Scholar]
- 150.Hisaki I, Ikenaka N, Tohnai N. and Miyata M, Chem. Commun, 2016, 52, 300–303. [DOI] [PubMed] [Google Scholar]
- 151.Hisaki I, Ikenaka N, Tsuzuki S. and Tohnai N, Mater. Chem. Front, 2018, 2, 338–346. [Google Scholar]
- 152.Bian L, Shi H, Wang X, Ling K, Ma H, Li M, Cheng Z, Ma C, Cai S, Wu Q, Gan N, Xu X, An Z. and Huang W, J. Am. Chem. Soc, 2018, 140, 10734–10739. [DOI] [PubMed] [Google Scholar]
- 153.Sun Z, Li Y, Chen L, Jing X. and Xie Z, Cryst. Growth Des, 2015, 15, 542–545. [Google Scholar]
- 154.Wang H, Bao Z, Wu H, Lin R-B, Zhou W, Hu T-L, Li B, Zhao JC-G and Chen B, Chem. Commun, 2017, 53, 11150–11153. [DOI] [PubMed] [Google Scholar]
- 155.Chen T-H, Kaveevivitchai W, Jacobson AJ and Š O. Miljanić, Chem. Commun, 2015, 51, 14096–14098. [DOI] [PubMed] [Google Scholar]
- 156.Bracco S, Asnaghi D, Negroni M, Sozzani P. and Comotti A, Chem. Commun, 2018, 54, 148–151. [DOI] [PubMed] [Google Scholar]
- 157.Teng P, Niu Z, She F, Zhou M, Sang P, Gray GM, Verma G, Wojtas L, van der Vaart A, Ma S. and Cai J, J. Am. Chem. Soc, 2018, 140, 5661–5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Endo K, Koike T, Sawaki T, Hayashida O, Masuda H. and Aoyama Y, J. Am. Chem. Soc, 1997, 119, 4117–4122. [Google Scholar]
- 159.Distefano G, Comotti A, Bracco S, Beretta M. and Sozzani P, Angew. Chem., Int. Ed, 2012, 51, 9258–9262. [DOI] [PubMed] [Google Scholar]
- 160.Zhang Z, Li J, Yao Y. and Sun S, Cryst. Growth Des, 2015, 15, 5028–5033. [Google Scholar]
- 161.Lin Y, Jiang X, Kim ST, Alahakoon SB, Hou X, Zhang Z, Thompson CM, Smaldone RA and Ke C, J. Am. Chem. Soc, 2017, 139, 7172–7175. [DOI] [PubMed] [Google Scholar]