

ABSTRACT
Chaperone-mediated autophagy (CMA) is a major pathway of lysosomal proteolysis critical for cellular homeostasis and metabolism, and whose defects have been associated with several human pathologies. While CMA has been well described in mammals, functional evidence has only recently been documented in fish, opening up new perspectives to tackle this function under a novel angle. Now we propose to explore CMA functions in the rainbow trout (RT, Oncorhynchus mykiss), a fish species recognized as a model organism of glucose intolerance and characterized by the presence of two paralogs of the CMA-limiting factor Lamp2A (lysosomal associated membrane protein 2A). To this end, we validated a fluorescent reporter (KFERQ-PA-mCherry1) previously used to track functional CMA in mammalian cells, in an RT hepatoma-derived cell line (RTH-149). We found that incubation of cells with high-glucose levels (HG, 25 mM) induced translocation of the CMA reporter to lysosomes and/or late endosomes in a KFERQ- and Lamp2A-dependent manner, as well as reduced its half-life compared to the control (5 mM), thus demonstrating increased CMA flux. Furthermore, we observed that activation of CMA upon HG exposure was mediated by generation of mitochondrial reactive oxygen species, and involving the antioxidant transcription factor Nfe2l2/Nrf2 (nfe2 like bZIP transcription factor 2). Finally, we demonstrated that CMA plays an important protective role against HG-induced stress, primarily mediated by one of the two RT Lamp2As. Together, our results provide unequivocal evidence for CMA activity existence in RT and highlight both the role and regulation of CMA during glucose-related metabolic disorders.
Abbreviations: AREs: antioxidant response elements; CHC: α-cyano -4-hydroxycinnamic acid; Chr: chromosome; CMA: chaperone-mediated autophagy; CT: control; DMF: dimethyl fumarate; Emi: endosomal microautophagy; HG: high-glucose; HMOX1: heme oxygenase 1; H2O2: hydrogen peroxide; KFERQ: lysine-phenylalanine-glutamate-arginine-glutamine; LAMP1: lysosomal associated membrane protein 1; LAMP2A: lysosomal associated membrane protein 2A; MCC: Manders’ correlation coefficient; Manders’ correlation coefficient Mo: morpholino oligonucleotide; NAC: N-acetyl cysteine; NFE2L2/NRF2: NFE2 like bZIP transcription factor 2; PA-mCherry: photoactivable mCherry; PCC: Pearson’s correlation coefficient; ROS: reactive oxygen species; RT: rainbow trout; siRNAs: small interfering RNAs; SOD: superoxide dismutase; Tsg101: tumor susceptibility 101; TTFA: 2-thenoyltrifluoroacetone; WGD: whole-genome duplication.
KEYWORDS: CMA, fish, glucose intolerance, Lamp2A, metabolism, NFE2L2/NRF2
Introduction
Chaperone-mediated autophagy (CMA) is a type of autophagy that selectively recognizes cytosolic proteins bearing a pentapeptide sequence sharing biochemical similarities to KFERQ (lysine-phenylalanine-glutamate-arginine-glutamine) [1]. In detail, these KFERQ-like domain containing proteins are first recognized by HSPA8/HSC70 (heat shock protein family A (Hsp70) member 8) and co-chaperones [2,3]. The substrate-chaperone(s) complex then docks at the lysosomal membrane through specific binding to the cytosolic tail of LAMP2A (lysosomal associated membrane protein 2A), the only one of the three spliced isoforms of the LAMP2 gene recognized to be essential and limiting for CMA activity [4,5]. This is followed by LAMP2A multimerization to form a translocation complex required for internalization of substrates, and subsequent lysosomal degradation [6].
While initially described as a basic protein quality control system [7], it is now firmly established that CMA is essential for the selective uptake and degradation of key components involved in transcriptional regulation, immune response, cell cycle progression, and cellular energetics [8], making CMA a critical function for the maintenance of cellular homeostasis and metabolism [9–13]. Accordingly, CMA is now considered as a sentinel pathway to be scrutinized in pathological situations when metabolic dysregulations are involved or suspected [8,14–17]. In this sense, CMA malfunction has been reported in rat models of diabetic-induced renal hypertrophy [18], and a recent study showed that pharmacological CMA activation induces protective effects during early diabetic retinopathy [19], further pointing to the importance of exploring the roles that CMA plays in such disorders.
While CMA is well described in mammals, we recently provided evidence for the existence of a CMA-like process in a fish species, the medaka (Oryzias latipes) [20]. lamp2a knockout in fish induced severe alterations in carbohydrate and lipid metabolisms, similar to the observations in the liver of CMA deficient mice [9]. These results, which imply a conservation of the metabolic role of CMA across phyla and species, opened up new and exciting perspectives for now appreciating this function under a novel angle. Besides, they brought new opportunities on the use of complementary model organisms to expand our knowledge on both the regulation and the physiopathological roles of CMA [21]. In that way, we now propose to explore the CMA function in a fish species considered as an evolutionary model system to study natural cases of impaired glucose homeostasis [22], namely the rainbow trout (RT, Oncorhynchus mykiss), as this species displays persistent hyperglycemia after glucose tolerance tests or the intake of carbohydrate-enriched meals [23–29]. In addition, unlike the most commonly used model species (i.e., rodents and zebrafish) in biomedical research, the RT has two paralogs of the CMA-limiting factor Lamp2A. These two Lamp2A, which most likely originate from the whole-genome duplication event that occurred in the common ancestor of salmonids about 100 million years ago [20], show divergence in some residues within the cytosolic domain (C-terminal), which is considered as a recognition signal for lysosomal targeting and therefore supposedly critical for protein function [1]. Taken together, RT represents an interesting model to explore the roles of CMA during impaired-glucose homeostasis and to gain new insights into the evolution of this selective autophagic pathway.
Our study first addressed the existence of functional CMA in RT. For that purpose, we generated a RT hepatoma-derived cell line (RTH-149) stably expressing a fluorescent reporter (KFERQ-PA-mCherry1) routinely used to track CMA in mammalian cells [30] and more recently validated in medaka fibroblasts [20]. Our results showed that upon either high-glucose (HG, 25 mM) exposure or mild-oxidative stress (H2O2, 25 µM) the CMA reporter accumulates in characteristic puncta that co-occurred together with the lysosomes, in a KFERQ- and Lamp2A-dependent manner. Moreover, the half-life of the reporter was substantially shortened under these same conditions compared to the control medium. Together, this set of experiments univocally established that RT, like medaka, de facto exhibits a functional CMA activity. Moreover, it also emphasizes, for the first time, the strong responsiveness of this autophagic pathway to HG treatment. We next investigated the underlying mechanisms and evidenced the reactive oxygen species (ROS)-Nfe2l2/Nrf2 (nfe2 like bZIP transcription factor 2)-Lamp2A regulatory axis as a critical mechanism involved in the HG-mediated CMA activation. Finally, we highlighted that CMA plays an important protective role against HG-induced stress, essentially mediated by one of the two Lamp2As expressed in RT. Together, our results offer novel insights into both the role and regulation of CMA during glucose-related metabolic disorders.
Results
Of the existence of high-glucose- and mild-oxidative stresses- inducible CMA responses in RT hepatocytes
To characterize the CMA process in RT, we first established RTH-149 cells stably expressing the KFERQ-PA-mCherry1 construct made of the N-terminal 21 amino acids of bovine RNASE1/RNase A containing its KFERQ CMA-targeting motif fused to a photoactivable-mCherry1 (PA-mCherry1) protein (Figure 1A). Originally developed for tracking CMA activity in mammalian cells [30], the usage of this reporter was more recently adapted and validated in fish cells [20]. These RTH-149 cells were then photoactivated for 10 min and incubated for 16 h with either control medium (CT, glucose 5 mM), high-glucose medium (HG, 25 mM), or exposed to a mild-oxidative stress, a condition known to trigger CMA in mammalian cells [5,31], and induced here by hydrogen peroxide (H2O2, 25 µM) (Figure 1B). In CT condition the CMA-reporter showed a diffuse distribution throughout the cytoplasm while only discrete and scattered puncta could be detected (Figure 1C; quantification in Figure 1 1D). In contrast, cells incubated with HG or H2O2 displayed a significantly higher number of KFERQ-PA-mCherry1 puncta (Figure 1C; quantification in Figure 1 1D), with a clear dose-response correlation for the HG treatment (Figure S1A; quantification in Figure S1B).
Figure 1.
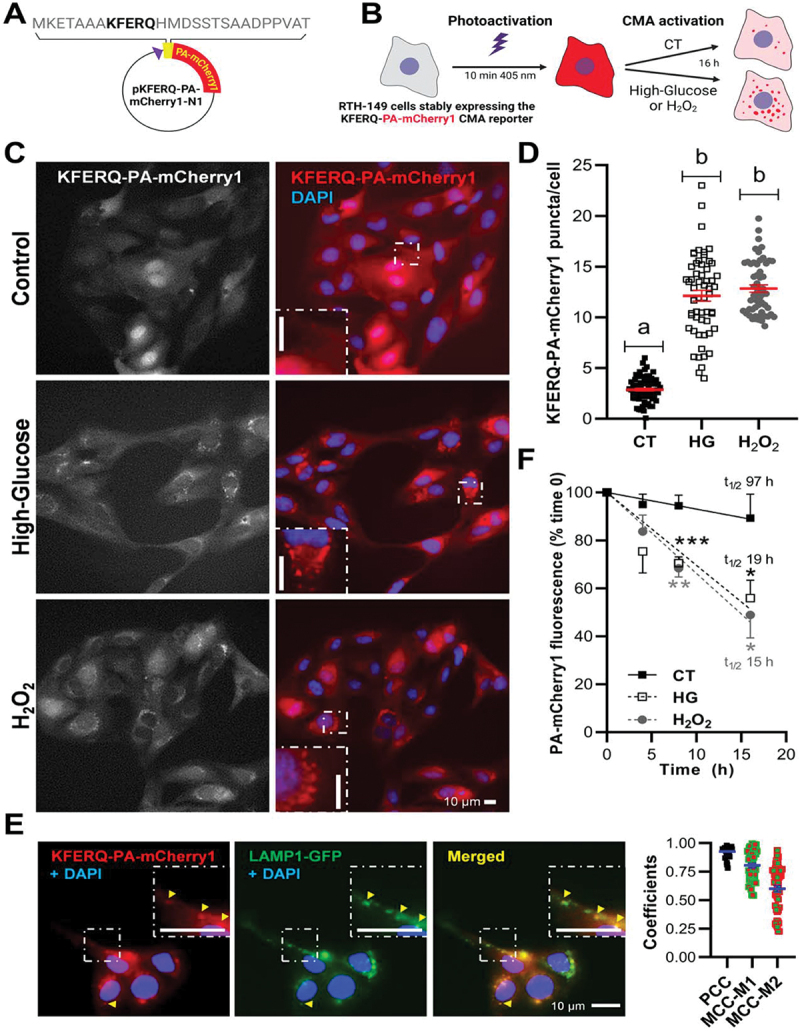
Both elevated glucose (25 mM) and mild-oxidative stress (H2O2 25 µM) induced relocalization of the fluorescence diffuse pattern of the KFERQ-PA-mCherry1 reporter to form puncta colocalizing with lysosomes, and reduced the half-life of the reporter. (A) Schematic representation of the pKFERQ-PA-mCherry1-N1 construction and amino acid sequence including the KFERQ-CMA targeting motif. (B) Simplified scheme of the experimental design. (C) Representative images of RTH-149 cells stably expressing a KFERQ-PA-mCherry1 reporter visualized by fluorescence microscopy after 16 h exposure to control (glucose 5 mM), high-glucose (glucose 25 mM) or mild-oxidative stress (H2O2 25 µM and glucose 5 mM). Images were originally acquired in grey, and nuclei were stained with DAPI (blue). Both experimental conditions induced the translocation of the CMA reporter to form fluorescent puncta, as can be observed in the insets in comparison with the control group, where the reported showed a diffuse pattern. (D) Quantification of KFERQ-PA-mCherry1 reporter number of puncta per cell. All values correspond to individual images (CT 70; HG 57; H2O2 55), with > 15 images/experiment in a total of 3 independent experiments (>900 cells for condition). Different letters denote significant differences between groups compared by one-way ANOVA (p < 0.0001) followed by Bonferroni’s multiple comparisons tests. (E) Representative images of HG-induced KFERQ-PA-mCherry1 puncta (red) in cells transiently transfected with LAMP1-GFP (green) visualized by fluorescence microscopy. Most of the KFERQ-PA-mCherry1 puncta colocalized with the green lysosomal marker LAMP1-GFP, as indicated by the arrowheads and amplified in the insets, and as supported in the plot by the PCC and MCCs results of the global analysis of 50 single cells using the BIOP version of JACoP pluging for Fiji. (F) The lysosomal internalization of the CMA reporter and its decay in fluorescence was quantified to infer CMA flux as the difference between the total fluorescence after photoactivation (0 h, 100%) and the remaining signal after 4, 8, or 16 h incubations in a minimum of 4 independent experiments. The higher reduction in the red fluorescence intensity upon HG or H2O2 exposure compared with cells incubated in the CT medium supports its enhanced degradation under these conditions, which is confirmed by the shortened half-lives (t1/2) of the reporter (19 h for HG and 15 h for H2O2; vs. 97 h for CT). Unpaired Student’s T-test was used to compare each condition to the CT (*p < 0.05; **p < 0.01; ***p < 0.001). All data are presented as Mean ± SEM; scale bars: 10 µm.
To gather more evidence on the CMA specificity of these observations, we then assessed the co-occurrence of the HG-induced KFERQ-PA-mCherry1 puncta with the LAMP1 (lysosomal associated membrane protein 1)-GFP label, which is commonly used to reveal late endosomes and lysosomes (hereafter referred to as endolysosomes) [10,13]. Our results showed a high level of co-occurrence between the CMA reporter’s HG-induced puncta and endolysosomes (Figure 1E) that was further validated by a positive Pearson’s correlation coefficient (PCC, Mean ± SEM: 0.925 ± 0.006), which highlights the correlation between the pixel-intensity of the two channels [32]. Manders’ correlation coefficients were additionally calculated (MCC-M1 and MCC-M2). The former indicates the fraction of PA-mCherry1 signal overlapping the LAMP1-GFP signal, while the latter indicates the opposite. These coefficients range from 0 (no colocalization) to 1 (100% overlapping) [32,33]. MCC-M1 revealed that most of the red signal (i.e., CMA reporter) overlapped with the endolysosomes (Mean ± SEM: 80.44% ± 0.01), whereas only 60% of the endolysosomal green signal colocalized with the red channel (MCC-M2, Mean ± SEM: 60.00% ± 0.03). Hence, these results showed and supported that the observed KFERQ-PA-mCherry1 puncta indeed co-occurred together with endolysosomes. To further test whether the observed CMA puncta require the KFERQ-targeting motif, WT RTH-149 cells were transfected with a PA-mCherry1 plasmid lacking the KFERQ CMA-targeting motif and then treated as described above. In contrast to cells expressing the KFERQ-PA-mCherry1 reporter that displayed clearly observable puncta after either HG or H2O2 exposure, the PA-mCherry1 signal remained stably diffuse in all conditions (Figure S1C; quantification in Figure S1D), demonstrating the dependence of the KFERQ-motif for puncta formation. To determine whether the formation of KFERQ-reporter puncta also require Lamp2A, we next assessed the reporter’s behavior in cells transfected with an antisense morpholino oligonucleotide (Mo) designed to reduce the endogenous level of lamp2a transcripts (Molamp2a). This Mo specifically impaired the splicing of exon a of the two lamp2 genes present in the genome of the RT (located in chromosomes [Chrs] 14 and 31, respectively; Figure S1E). RTH-149 cells transiently transfected with Molamp2a displayed significantly reduced levels of the two lamp2a paralogs mRNAs at 24 h compared to negative standard control (Figure S1F). Transcription of the other splice variants (i.e. lamp2b and lamp2c) remained unaffected. That Molamp2a effects were still observable 48 h post transfection (Table S1-F). Under these conditions of Lamp2A’s knockdown, we no longer observed KFERQ-PA-mCherry1 puncta formation after cells have been incubated with either HG or H2O2 (Figure S1G; quantification in Figure S1H). Finally, in order to quantify the lysosomal internalization of the CMA reporter, we next monitored the changes for total cellular mCherry1 fluorescence intensity, which is expected to decrease as the protein is internalized and degraded within this compartment. To that end, we first measured the total fluorescence after photoactivation (0 h, 100%) to then be able to calculate the fluorescence’s decay after either 4, 8 or 16 h of incubation (Figure 1F). The higher reduction in the fluorescence intensity upon HG or H2O2 exposure compared to cells incubated in the CT medium supports an enhanced degradation of the reporter under these conditions, as revealed by shortened half-lives (t1/2; 19 h for HG and 15 h for H2O2; Vs. 97 h for CT).
Together, these results univocally establish that RT hepatocytes do exhibit, like previously demonstrated in the medaka fish [20], a functional CMA activity, and emphasize, for the first time, the strong responsiveness of the RT to HG treatment.
HG-induced CMA activation rely on generation of mitochondrial ROS
Acute or chronic high-glucose in diabetes models increases the production of ROS and generates cellular oxidative stress [34–40], whose strong stimulatory effect on CMA is well documented in mammalian cells [5,31] and now demonstrated in RTH-149 cells (Figures 1C–F). To test whether oxidative stress is involved in the observed HG-induced activation of CMA, we first assessed the levels of oxidized proteins in RTH-149 cells incubated with CT, HG or H2O2 media using immunoblot analysis. Interestingly, like H2O2, HG exposure also resulted in the significant accumulation of oxidized proteins compared to CT condition (Figure 2A), supporting the generation of a cellular oxidative stress by HG treatment in RTH-149 cells. To determine more directly the specific contribution of the observed oxidative stress upon HG treatment to CMA activation, we then supplemented the HG medium with the antioxidant N-acetyl cysteine (NAC) [39]. In presence of NAC, total recovery of both the HG-compromised oxidative status (Figure 2B) and the HG-induced KFERQ-PA-mCherry1 puncta (Figure 2C; quantification in Figure 2D) was observed, indicating that activation of CMA is mediated through ROS levels upon HG.
Figure 2.
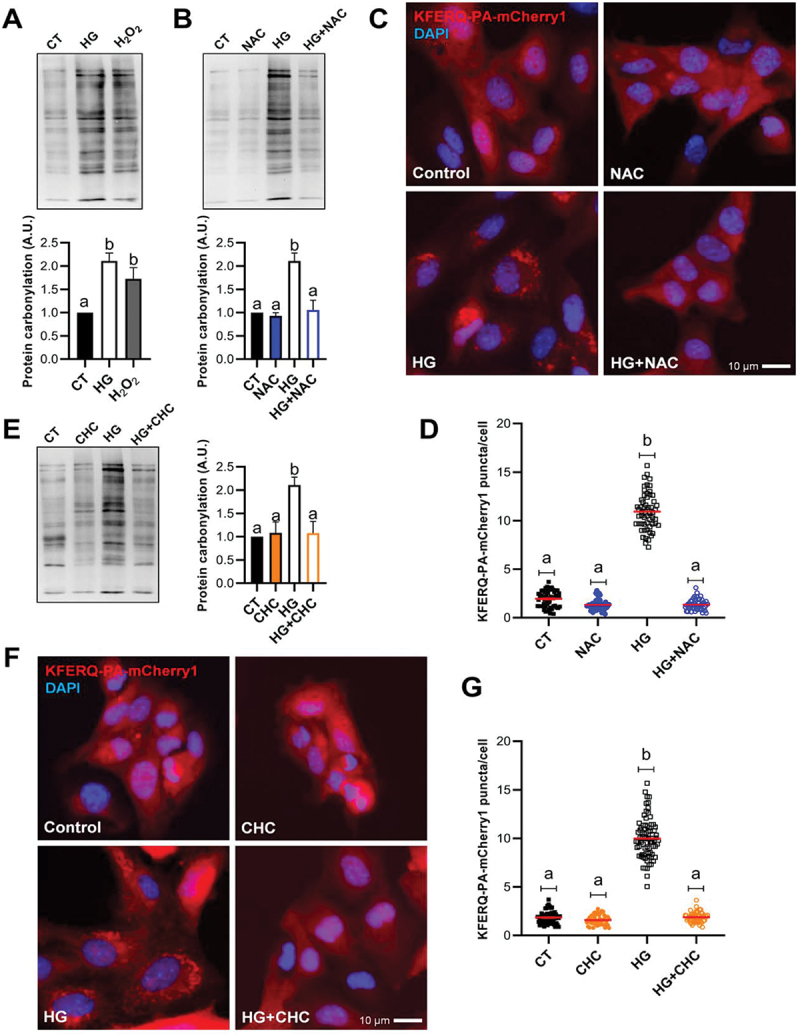
Oxidative stress induced by the use of high-glucose at the mitochondria was associated with the formation of CMA-puncta. (A) Protein carbonylation levels in RTH-149 cells after 8 h exposure to control (CT; glucose 5 mM), high-glucose (HG; glucose 25 mM), or mild-oxidative stress (H2O2; hydrogen peroxide 25 µM and glucose 5 mM) analyzed using OxyBlot western blot. Different letters denote significant differences between groups compared by one-way ANOVA (p < 0.004) followed by Tukey’s multiple comparisons tests in 4 independent experiments. (B) The presence of the antioxidant NAC (10 mM) prevented the generation of carbonylated proteins by HG. One-way ANOVA (p < 0.0005) followed by Bonferroni’s multiple comparisons tests was used to compare between groups in 3–4 independent experiments. (C and D) The presence of NAC prevented the formation of KFERQ-PA-mCherry1 puncta after 16 h exposure to high-glucose. The values in (D) correspond to individual images (CT 56; NAC 45; HG 57; HG+NAC 48), with ≥ 15 images/experiment in a total of 3 independent experiments (>1,000 cells for condition). (E) The inhibition of the pyruvate mitochondrial transporter using CHC (100 µM) reduced the HG-induction of carbonylated proteins. One-way ANOVA (p < 0.003) followed by Bonferroni’s multiple comparisons tests was used to compare between groups in 3–4 independent experiments. (F and G) the presence of CHC prevented the formation of KFERQ-PA-mCherry1 puncta after 16 h exposure to high-glucose. All values in (G) correspond to individual images (CT 62; NAC 48; HG 76; HG+NAC 55), with ≥ 15 images/experiment in a total of 3 independent experiments (>1,000 cells for condition). In D and G, different letters denote significant differences between groups compared by the non-parametric kruskal-wallis test (p < 0.0001) followed by Dunn’s multiple comparisons tests. All data are presented as Mean ± SEM; scale bars: 10 µm.
A critical driver of ROS overproduction during hyperglycemia (or glucose overload) is the increase of glucose metabolism [41,42]. To test whether glucose catabolism is necessary to activate CMA under HG, we first used the stereoisomer L-glucose that cannot be metabolized by cells [40] (Figure S2A). We found no increase in KFERQ-PA-mCherry1 puncta formation after 25 mM L-glucose treatment (Figure S2B; quantification Figure S2C) compared to the CT, as a first hint that HG-induced CMA activation requires glucose catabolism. We next used an inhibitor of the mitochondrial pyruvate transport, α-cyano-4-hydroxycinnamate (CHC) [40,43], to test whether mitochondrial uptake of pyruvate (the end product of glycolysis) is necessary for ROS increase and CMA activation in the HG conditions. Pre-incubation of RTH-149 cells with 100 µM CHC blocked the HG-induced accumulation of oxidized proteins (Figure 2E). Under the same conditions, CHC-treated cells displayed no HG-induced CMA puncta (Figure 2F; quantification Figure 2G). These results indicate that mitochondrial pyruvate uptake is necessary for both HG-induced ROS increase, and CMA activation. Based on these facts, it is tempting to assume that inhibition of the mitochondrial respiratory chain (the site of ROS production) under HG condition would results in impaired activation of CMA. As anticipated, inhibiting the electron transport chain complex II with 10 µM 2-thenoyltrifluoroacetone (TTFA) [42] hampered the increase of KFERQ-PA-mCherry1 puncta formation upon HG exposure (Figure S2D; quantification Figure S2E). Similarly, preloading of RTH-149 cells with the mitochondria-targeted Mito-Tempo antioxidant [44,45] prevented the HG-induction of puncta (Figure S2B; quantification Figure S2C).
Taken together, these results demonstrated that, the observed induction of CMA upon HG treatment relies on the generation of ROS from the mitochondria, and highlight the importance of glucose metabolism during this process.
The HG-dependent activation of CMA is mediated by the Nfe2l2 transcription factor
Exposure to oxidants activates the antioxidant machinery (i.e., detoxifying enzymes and antioxidant proteins) in order to limit and protect cells from the consequences associated with oxidative stress [46]. The transcription factor NFE2L2/NRF2 is considered as a master regulator of this antioxidant response [46,47]. When ROS levels rise, the activity of the main NFE2L2 negative regulator known as KEAP1 (kelch like ECH associated protein 1) is altered, resulting in NFE2L2 stabilization and nuclear translocation [48]. Interestingly, immunofluorescence analysis revealed that the Nfe2l2 signal was restricted to the nuclear area in RTH-149 cells treated with either HG or H2O2 media, but not in CT cells (Figure 3A; quantification Figure 3B), supporting stimulation of Nfe2l2 translocation to the nucleus. There, NFE2L2 binds to cis-acting enhancer sequences, called antioxidant response elements (AREs), and modulate the transcription of the antioxidant machinery [50]. AREs have been identified in more than 250 genes in mammals but also in fish. These include, for example, HMOX1 (heme oxygenase 1), SOD1 (superoxide dismutase 1) and SOD2, or NFE2L2 itself [48,50–52]. Accordingly, we found that HG treatment significantly upregulated the mRNA levels of nfe2l2, hmox1, sod1 and sod2 genes compared to the CT condition (Figure 3C), further indicating that Nfe2l2 pathway is activated in RTH-149 cells upon HG exposure.
Figure 3.
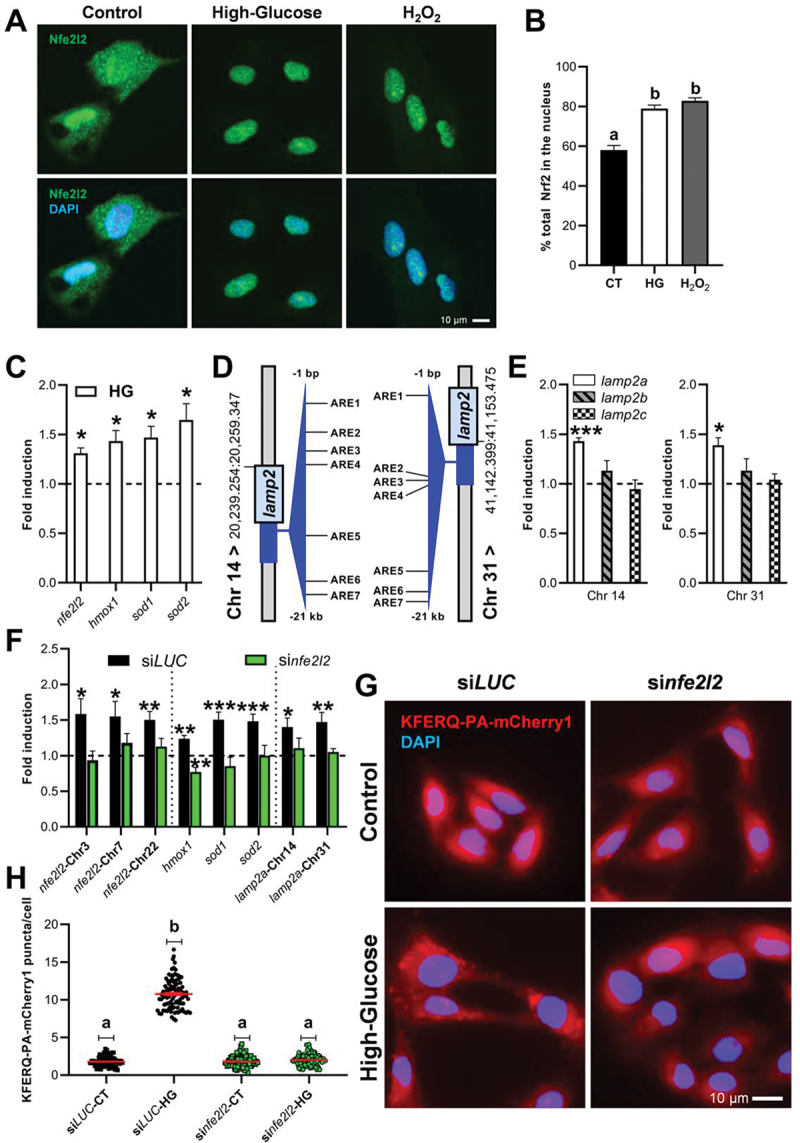
The anti-oxidative stress Nfe2l2 transcription factor mediated both the transcriptional modulation of lamp2a’s and the activation of CMA by high-glucose. (A) Representative images of Nfe2l2 immunofluorescence in RTH-149 cells after incubation for 4 h with either HG or H2O2. (B) Percentage of the total staining that colocalizes with the nucleus in a minimum of 45 individual images, with ≥ 15 images/experiment in a total of 3 independent experiments. Different letters denote significant differences between groups compared by one-way ANOVA (p < 0.003) followed by Bonferroni’s multiple comparisons tests. (C) mRNA levels fold induction of the Nfe2l2 target genes nfe2l2, hmox1, sod1, and sod2 after 16 h incubation with high-glucose compared to the CT supported activation of the Nfe2l2 transcriptional program by HG. Differences with respect to the CT condition were assessed using Unpaired Student’s T-test (*p < 0.05) in three replicates of three independent experiments. (D) Schematic representation of the location of the 7 putative antioxidant response elements (AREs, threshold > 85%) identified within the 21 kilobases preceding the ATG start codon from the DNA sequence of both RT lamp2 paralogs located in Chrs 14 and 31 using the Homo sapiens matrix of NFE2L2/NRF2 in JASPAR [49]. The mRNA levels fold induction of the two RT lamp2a paralogs (E Chr 14 left; Chr 31 right) was upregulated after the incubation with HG compared to the CT, while neither lamp2b nor lamp2c mRnas, independently of the Chr, were altered. Differences with respect to the CT condition were assessed using Unpaired Student’s T-test (*p < 0.05; ***p < 0.001) in three replicates of three independent experiments. (F) siRNA-mediated knock-down of the three RT nfe2l2 paralogs prevented the enhancement of their mRNA levels by HG treatment, as well as hampered the effects of HG on the transcription of Nfe2l2-target genes, including the two RT lamp2a paralogs. Differences with respect to the CT condition were assessed using Unpaired Student’s T-test (*p < 0.05; **p < 0.01; ***p < 0.001) in duplicates of four independent experiments. On the other hand, similarly to the observations at the transcriptional level, the silencing of RT nfe2l2 paralogs prevented the formation of puncta in RTH-149 cells after 16 h exposure to high-glucose (G; quantification in H). All values correspond to individual images (siLUC-CT 86; siLUC-HG 94; sinfe2l2-CT 85; sinfe2l2-HG 87), with > 20 images/experiment in a total of 4 independent experiments (>1,200 cells for condition). Different letters denote significant differences between groups compared by the non-parametric kruskal-wallis test (p < 0.0001) followed by Dunn’s multiple comparisons tests. All data are presented as Mean ± SEM; scale bars: 10 µm.
Recently, it has been reported that NFE2L2 can also modulate CMA activity through the binding of at least two different AREs in the mammalian LAMP2 gene [53]. We therefore searched for putative AREs in the upstream region of the two RT lamp2 paralogs using the JASPAR tool and its CORE collection [49]. We identified seven putative AREs with a relative score higher than 85% (Figure 3D, Figure S3A, and Figure S3B), a threshold commonly used for transcription factor binding-site analysis [54]. Notably, among the potential Nfe2l2 binding sites identified, ARE4 from Lamp2 of Chr 14 and ARE5 from Lamp2 of Chr 31 present a score greater than 91% (Figs. S3A and S3B), suggesting a potential involvement of Nfe2l2 in the transcription of both lamp2a genes in RT as well. In this regard, we found that HG-incubated RTH-149 cells displayed elevated mRNA levels of both lamp2a’s compared to their CT counterparts (Figure 3E), suggesting that Nfe2l2 (shown in Figures 3A–C to be activated by HG) may have a role in the observed HG-dependent activation of CMA. To assess that hypothesis, we first treated RTH-149 with dimethyl fumarate (DMF), a well-known pharmacological activator of NFE2L2 [52,53]. We found an increase of mRNA levels of several Nfe2l2-target genes as well as a comparable induction of both lamp2a’s (Figure S3C). Interestingly, this effect was accompanied by increased numbers of KFERQ-PA-mCherry1 puncta (Figure S3D; quantification Figure S3E), as a first clue that Nfe2l2 indeed regulates CMA in our model as well. To go further and validate the involvement of Nfe2l2 in the HG-dependent activation of CMA observed in RTH-149 cells, small interfering RNAs (siRNA)-induced silencing was used to knockdown the three nfe2l2 paralogs present in the genome of the RT (in Chrs 3, 7, and 22, respectively). We found that the siRNA (hereafter referred to as sinfe2l2) was effective in preventing the inducing effect of HG on the mRNA levels of nfe2l2 paralogs, as well as those of Nfe2l2 target genes (hmox1, sod1 and sod2), and both lamp2a’s in a similar degree (Figure 3F). In contrast, neither lamp2b nor lamp2c mRNAs from Chrs 14 or 31 were affected by sinfe2l2 (Figure S3F). In this Nfe2l2 knockdown condition, HG treatment no longer induced KFERQ-PA-mCherry1 puncta formation (Figure 3G; quantification Figure 3H).
Together, these results identified the ROS-Nfe2l2-Lamp2A regulatory axis as being critically involved in the HG-mediated activation of CMA.
Functional divergence between the two paralogous lamp2a genes present in RT
An important specificity between RT and the different model species commonly employed for biological and biomedical research (i.e., mouse, rat, zebrafish) is the presence of two Lamp2A proteins in the salmonid. Interestingly, these two Lamp2A diverge in the sequences of some motifs previously shown to be critical for protein function (Figure S4A and Figure S4B). To determine whether these two Lamp2A exert the same function (or not), we first designed siRNAs to specifically target the 3’UTR region of each of the lamp2a paralogs (hereafter referred to as si14 and si31, respectively) for discriminating between them (Figure S4C), what was not possible to achieve through morpholino oligos due to sequence homology restrictions. A siRNA targeting GL2 luciferase (siLUC), not expressed in RTH-149 cells, was used as negative control. Then, in RTH-149 cells transfected with these siRNAS, we assessed the KFERQ-PA-mCherry1 reporter behavior upon incubation with either the control or the HG medium. Expression of each lamp2a could be efficiently reduced by its respective siRNA without affecting the mRNA levels of their two splicing variant counterparts (lamp2b and lamp2c from Chr 14 and Chr 31, respectively), which was critical to ensure that any effect observed in these cells was directly related to the Lamp2A isoforms (Figure S4D). Interestingly, reduction in mRNA levels of Chr 14 lamp2a did not prevent the increase of KFERQ-PA-mCherry1 puncta formation upon HG exposure (Figure 4A; quantification Figure 4B). Conversely, reduced expression of either the Chr 31 or both lamp2a isoforms strongly abolished the HG-induced CMA puncta formation, suggesting that protein sequence disparities between the two Lamp2A’s could be associated with functional divergences (Figure S4A and Figure S4B).
Figure 4.
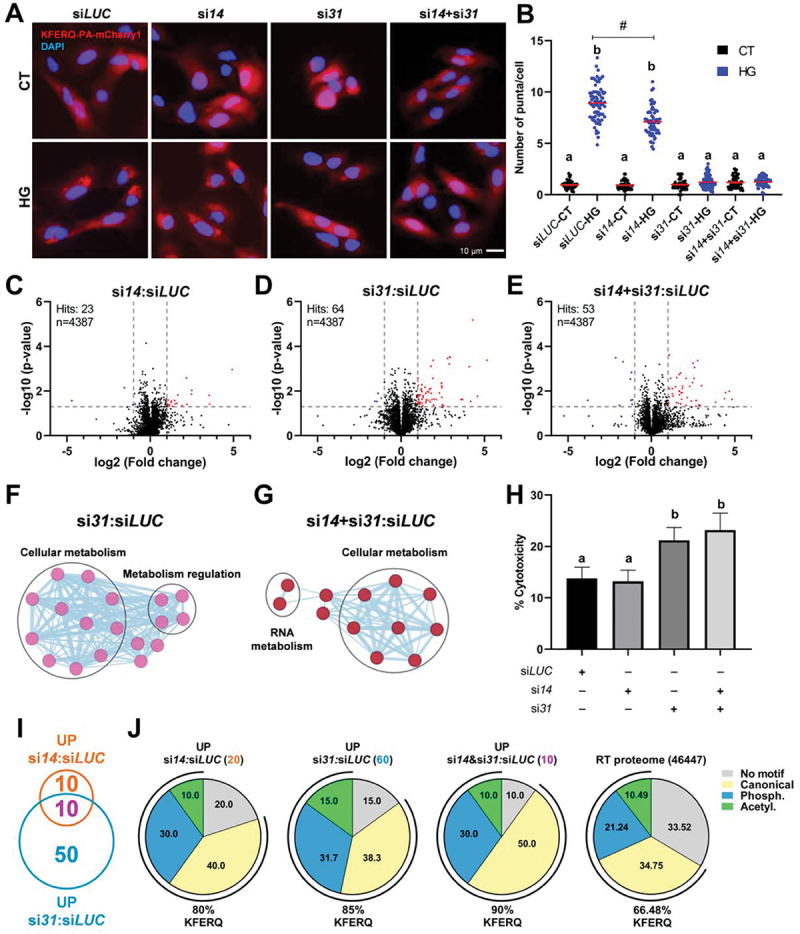
CMA plays a protective role to maintain cellular homeostasis during hyperglycemic stress, mainly mediated by the Lamp2A paralog encoded by Chr 31. (A) Effects of siRNA-mediated knock-down of either (si14 and si31) or both (si14+si31) RT Lamp2A’s on CMA activity. The reduction of the lamp2a isoform from Chr 14 mRnas did not prevent the increase of KFERQ-PA-mCherry1 puncta formation upon 16 h HG exposure (A; quantification in B). In contrast, decreased expression of either the Chr 31 or both lamp2a isoforms strongly abolished the HG-induced CMA puncta formation. All values correspond to individual images (siLUC-CT 55; siLUC-HG 76; si14-CT 62; si14-HG 59; si31-CT 67; si31-HG 75; si14+si31-CT 60; si14+si31-HG 61), with > 18 images/experiment in a total of 3 independent experiments (>600 cells for condition). Different letters denote significant differences between groups compared by the non-parametric kruskal-wallis test (p < 0.0001) followed by Dunn’s multiple comparisons tests; # denotes differences between siLUC-HG and si14-CT groups compared by the Mann Whitney test (p < 0.0001). All data are presented as Mean ± SEM. (C) volcano plot of the quantitative proteomic analysis of RTH-149 cells transfected with si14, (D) si31, or (E) si14+si31, incubated in HG media for 48 h and compared to siLUC. In the top left is presented the number of significant hits and the number of proteins identified. Blue dots indicate differentially downregulated proteins, and red dots correspond to upregulated proteins (p < 0.05 with ≥ 2 fold change (FC)). Four replicated samples for each condition were analyzed. Network representation of the gene ontology biological process enrichment analysis of proteins modified in si31 (F) or si14+si31 (G) transfections compared with siLUC, generated in CytoScape (3.9.1) using EnrichmentMAP (3.3.5), and annotated with AutoAnnotate (1.4.0) apps. Thresholds were set as 0.2 (connectivity cutoff); 0.25 (edge cutoff); p < 0.05; FDR < 0.05. (H) Cell cytotoxicity measured as the activity of lactate dehydrogenase (LDH) released from cells transfected with siLUC, si14, si31, or si14+si31 and incubated, 24 h post-transfection, with HG medium during 48 h. Different letters denote significant differences between groups compared by one-way ANOVA (p < 0.003) followed by Tukey’s multiple comparisons tests in triplicates of three independent experiments. Data are presented as Mean ± SEM. (I) Venn diagram of significantly upregulated proteins in si14 or si31 compared with siLUC, showing the number of overlapping and non-overlapping proteins between these conditions, and (J) percentage of proteins from those three groups and of the total RT proteome containing KFERQ-like motifs (bottom values), as well as of the proteins ranked to each motif type (i.e., canonical, phosphorylation- or acetylation-dependent motifs) according to [55].
Nonetheless, the lack of effect of Chr 14 Lamp2A deficiency in HG-induced KFERQ-PA-mCherry puncta formation does not necessarily imply that this protein is no longer functional. Indeed, the evolution of its protein sequence could have reshaped the nature of the motif recognized and, in turn, of the proteins targeted. As a result, the absence of Chr 14 Lamp2A may have affected other proteins (non-KFERQ) and/or cellular functions than those commonly ascribed to CMA. To gain insight into the specific contribution of each of the two Lamp2A’s to the overall proteostasis of RTH-149 cells under a HG condition, we therefore performed comparative quantitative proteomics in siLUC, si14 and/or si31 transfected cells incubated with the HG medium (Table S2). In the whole cell extracts, out of 4387 proteins identified, only 23 were differentially expressed between si14 and siLUC conditions (Figure 4C). In contrast, up to 64 and 53 significant hits were found after si31 (Figure 4D) or si14+si31 (Figure 4E) transfections, respectively. Gene ontology and network analysis using the differentially expressed proteins did not reveal significant changes of any biological process in the cells transfected with si14. On the other hand, different processes associated with cellular metabolism and its regulation were identified in si31 transfected cells (Figure 4F), as well as linked with cellular and RNA metabolism in the case of si14+si31 condition (Figure 4G). Notably, among these processes several clusters associated with the metabolism of proteins in both conditions stand out (Table S3). To decipher these apparent functional divergences between both Lamp2A’s on overall cellular homeostasis under HG-induced stress, we then measured the activity of lactate dehydrogenase (LDH) released from si14 and/or si31 transfected cells as a proxy of cell cytotoxicity (Figure 4H). While deficiency for Lamp2A from Chr 14 had no effect compared to the control (siLUC) condition, deficiency for either the Chr 31 or both Lamp2A isoforms strongly hampered the viability of RTH-149 cells upon HG exposure (Figure 4H), further supporting a functional divergence between the two Lamp2A’s.
Finally, to take a first glimpse into the ability of each Lamp2A variant to recognize KFERQ-like motifs bearing proteins, we compared the proteins up-regulated under the si14:siLUC (20 hits) or the si31:siLUC (60 hits) conditions (Figure 4I). The presence of KFERQ motifs in protein sequences was analyzed in silico using the KFERQ finder app V0.8 [55] (Table S4). We found a lower percentage of proteins containing at least one KFERQ-like motif in the si14:siLUC group (80%) compared to si31:siLUC (85%; Figure 4J and Figure S4E) condition, which notably included the experimentally validated CMA substrate citrate synthase CS [9] (Table S2 and Table S4). Moreover, when compared with the overall RT proteome (Figure 4J), only the si31:siLUC group exhibited a significant enrichment of proteins containing CMA targeting motifs (chi-squared, p < 0.05). These results further support that Chr 31 Lamp2A display a greater intrinsic ability to recognize KFERQ-bearing proteins compared to Chr 14 Lamp2A. Interestingly, despite this apparent functional deviation between the two Lamp2As, some proteins (10 hits) were jointly affected by both treatments (Figure 4I). We found that 90% of these common proteins displayed KFERQ motifs (Figure 4J and Table S4), a percentage significantly higher than that observed in the full RT proteome (66.48%; chi-squared, p < 0.001), supporting that some of these proteins could indeed be bona fide CMA substrates. Into that direction, among those common upregulated proteins (Figure S4F) we could identify Eif4a1 (eukaryotic translation initiation factor 4a1), which has been recently confirmed as a CMA substrate [56,57]. Noteworthy, we also found Tsg101 (tumor susceptibility 101), which is a component of the ESCRT-1 machinery involved in the selective targeting of KFERQ-like containing-proteins into late endosome or multivesicular bodies that occurs during endosomal microautophagy (eMI) [1,58]. Although the existence of eMI has not been yet reported in fish, most of the core proteins/components of its machinery are nevertheless conserved amongst eukaryotes [1]. Therefore, their ability to carry out this autophagic process cannot be ruled out. Accordingly, the rise of Tsg101 levels in Lamp2A-silenced RT cells might account for the existence of this process in this species, and be indicative of a potential compensatory mechanism for degradation of KFERQ-containing proteins by eMI. Of interest, the KFERQ-like motif present in this protein is also conserved in its human and mouse orthologues (Figure S4F), supporting an evolutionary conservation of this mechanism across vertebrates.
RT fed with a high-carbohydrate diet display a diabetic-like phenotype associated with increased levels of lamp2a transcripts
To evaluate the physiological relevance of the observed HG-dependent activation of CMA, we next analyzed the impact of high dietary carbohydrates intake in RT in vivo. As expected, RTs fed with a high-carbohydrate diet (HCD) for 9 weeks exhibited the typical signs of glucose intolerance characterized by (i) slower growth rates (Figure 5A), (ii) increased liver weight (Figures 5B,C), and (iii) elevated blood glucose levels (Figure 5D), which persisted at least until 24 hours after the last meal. Interestingly, we found that these effects were also accompanied by an increase in the levels of carbonylated proteins (Figure 5E), as well as by higher expression of Nfe2l2 target genes including sod1, sod2 and the two lamp2a paralogs (Figure 5F). Notwithstanding, future studies are needed to explore to what extent the increased lamp2a mRNAs observed with the HCD correlates with an increased CMA activity. Similarly, the protective role of CMA during hyperglycemia reported in vitro in this study deserves further validation in vivo with genetically invalidated Lamp2As RT models. Collectively, these results clearly show the steadily establishment of a diabetic-like phenotype in RTs fed a HCD and the stimulation of the antioxidant protective Nfe2l2 pathway, which likely triggers the CMA activity to preserve cellular homeostasis.
Figure 5.
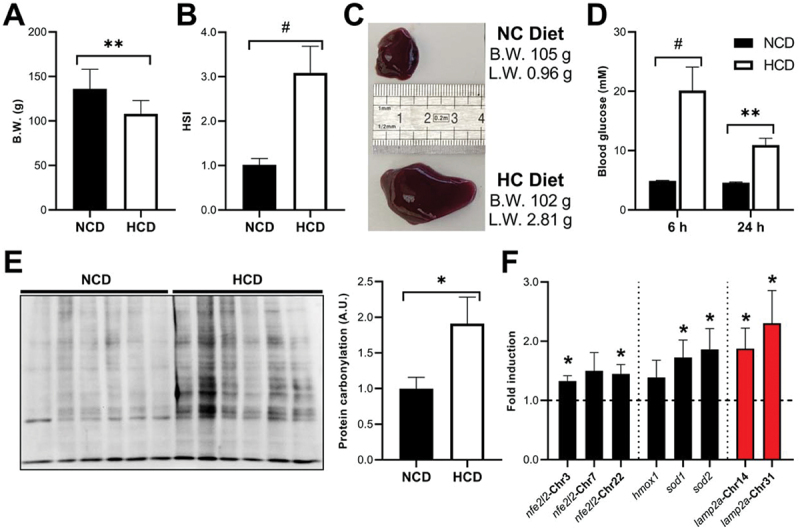
Rts fed a high-carbohydrate diet (HCD) for 9 weeks exhibited signs of glucose intolerance characterized by decreased growth, increased liver weight, and elevated post-prandial blood glucose levels. They also presented signs of oxidative stress and upregulated lamp2a mRnas, compared to fish fed a diet free of carbohydrates (NCD). (A) Final body weight (B.W.) at 9 weeks of the experiment. Unpaired Student’s T-test was used to compare between groups (*p < 0.05; **p < 0.01). (B) The hepatosomatic (HSI) index was calculated as HSI = (liver weight/animal weight) × 100 at 9 weeks of the experiment. Unpaired Student’s T-test was used to compare between groups (#p < 0.0001). (C) Representative images of livers from similar-B.W. fish fed with the NCD (top) or the HCD (bottom) during 9 weeks. (D) Blood glucose levels at both 6 and 24 h post-prandial in NCD and HCD-fed fish after 9 weeks of the experiment. Differences between groups at each time point were assessed using the Mann Whitney test (**p < 0.01; #p < 0.0001). (E) Protein carbonylation levels in the liver of fish fed with the NCD or the HCD for 9 weeks were analyzed using OxyBlot. Unpaired Student’s T-test was used to compare between groups (*p < 0.05). Mean ± SEM. (F) Hepatic mRNA levels of the three RT nfe2l2 paralogs, Nfe2l2-target genes, and the two RT lamp2a paralogs in fish fed with the NCD or the HCD for 9 weeks. Unpaired Student’s T-test was used to compare between groups (*p < 0.05). All data are presented as Mean ± SEM of 9 fish/group.
Discussion
In the present study, we first reported that RT hepatocytes are CMA- or CMA-like- competent. This result strongly support our previous findings in medaka, which revealed for the first time the existence of CMA-like activity in a non-tetrapod species [20]. We now show that the presence of CMA in fish is not restricted to the medaka (which might have acquired or retained this function independently of other species), but is spread to all clades if not all vertebrates. Indeed, the recent analysis of the evolutionary history of the LAMP2 gene in vertebrates revealed that this critical gene for CMA activity appeared after the second round of whole-genome duplication (WGD) at the root of the vertebrate lineage around 500 Mya [20]. More precisely, phylogenetic analyses and synteny conservation data strongly suggested that a single copy of a LAMP gene was already present in the common ancestor of vertebrates, and that the two successive WGDs that occurred at the root of the vertebrate lineages [59] gave rise to both LAMP1/2 and LAMP3/4 genes (from WGD1), and then LAMP1, LAMP2, LAMP3, and LAMP4 (from WGD2), which are common to all vertebrates. Besides, recent analysis of the PhyloFish RNA-seq database [60], which gathers comprehensive gene expression data from 23 different species of ray-finned fish, provided evidence for the expression of the lamp2a splice form in most if not all fish species, supporting that CMA likely appeared much earlier during evolution than initially thought [61]. Overall, these findings, as well as the results presented in the present study confirm the existence of CMA activity in fish, opening new perspectives to tackle CMA under a novel angle, by using complementary models for studying this fundamental function from both evolutionary and comparative perspectives.
The RT is a typical model of glucose-intolerance [22], which is characterized by persistent hyperglycemia [23–28], increased liver weight [26–28] and decreased growth [29], after intake of diet containing more than 30% carbohydrate. An obvious similarity in glucose intolerance to the human pre-diabetic condition has recently drawn numerous scholars to explore trout as an evolutionary model system to study natural cases of impaired glucose homeostasis and the underlying mechanisms [22]. Into that direction, it is attractive to expand our knowledge on both the regulation and the roles of CMA in pre-diabetic-like conditions. Here, we found for the first time that exposure of RTH-149 cells to 25 mM glucose, which is close to maximum postprandial values reported for this species (i.e., 27.7 mM) [26], leads to an increase in ROS production by mitochondria alongside with a strong induction of CMA. Previous findings demonstrated that hyperglycemia-induced generation of ROS at the mitochondrial level is the initial trigger of the vicious cycle of oxidative stress observed in diabetes [35–38,41,42]. Increased CMA activity under these conditions would thus contribute to the removal of oxidized proteins from the cytosol and allow cells to limit oxidative stress-induced subjected to mild oxidative stress [31]. Interestingly, an earlier study in experimental rat models of diabetes reported instead reduced CMA activity in the kidney, damages, as previously reported in rats finally contributing to accumulation of specific proteins in diabetic-induced renal hypertrophy [18]. These contrasted findings regarding the activation status of CMA described in our study can obviously be related to the specificities of the models (kidneys from streptozotocin-treated rats vs. RTH-149 cell line incubated in HG medium). However, it also certainly reflects the existence of several distinct mechanisms of CMA regulation, highlighting the imperative need of developing new approaches and/or models to acquire a better comprehensive picture of the whole processes involved in controlling this cellular function. In any case, CMA emerges as an appealing target for alleviating the pathological manifestations of diabetes, and the recently developed pharmacological activator of CMA, AR7 [62], along with its improved derivatives CA77.1 [12] and QX77 [63], represent promising tools for this objective. In this line, a recent study showed that chemical CMA activation with QX77 is effective in preventing early diabetic retinopathy and thus its consequences in the visual function [19].
At the mechanistic level, our results demonstrate that the oxidative stress induced by HG treatment leads to the activation of CMA through an Nfe2l2-mediated control of the transcription of both RT lamp2a’s. Of interest, such an induction of both lamp2a’s by HG or the chemical or genetic modulation of Nfe2l2 caused a similar fold-change, suggesting that the transcriptional regulation by Nfe2l2 is conserved between the two paralogs. The involvement of NFE2L2 in the regulation of the expression of LAMP2A has also been reported in different human and mouse cell types [53]. This ROS-NFE2L2-LAMP2A regulatory axis thus appears as an evolutionarily conserved cell protection process to cope with oxidative stress through CMA. It mainly relies on the control of lamp2a’s transcription and not on the regulation of its/their degradation and/or relocation at the lysosomal membrane, as described for CMA activation during serum deprivation [64]. Of note, although encoded by the same gene, expressions of the lamp2b and lamp2c splice variants were not significantly impacted upon modulation of Nfe2l2 activity. Previous findings in mammals also reported differential regulation of the three isoforms in response to different situations. For instance, Kiffin and coauthors demonstrated in rats that in vivo administration of the oxidant agent paraquat led to an enhancement in both mRNA and protein levels of LAMP2A, without altering those of LAMP2B and LAMPC2 splice variants [31,65]. Similarly, a more recent study by Pajares et al [53] showed that nfe2l2 KO immortalized mouse hepatocytes displayed reduced Lamp2a mRNA levels compared to WT cells, while Lamp2b and Lamp2c remained stable. On the contrary, when NFE2L2 was overexpressed, Lamp2a was upregulated, and the expression of the other variants remained unchanged. Considering that LAMP2A is the only variant required for CMA [4,64,66], these findings collectively suggest that there may be additional unknown post-transcriptional regulatory mechanisms that would favor splicing of the Lamp2 pre-mRNAs toward the production of LAMP2A, and not of the others alternatives (i.e., B and C), in situations where CMA plays an essential role such as oxidative stress. Further investigations are warranted to unravel the intricacies of these post-transcriptional controls and their significance in coordinating cellular responses to oxidative stress and other conditions requiring robust CMA activity.
WGD events led to the generation in RT of multiple copies of Lamp2A, which display divergences in some motifs critical for protein localization and/or function. For example, the canonical GYXXF motif present in the C-terminal end of Lamp proteins and identified as a recognition signal for lysosomal targeting [64], is fully conserved in the Chr 31 Lamp2A protein sequence. In contrast, Lamp2A from Chr 14 carries the variant form SYXXF (Figure S4A and Figure S4B). As previously discussed in Schnebert et al. [1], while the presence of the YXXF residues seems to be sufficient to ensure the targeting of LAMP2A to the lysosomal membrane, the lack of the glycine (G) could alter the routing of the protein to its destination. Whether or not this variation has an impact on Lamp2A function is therefore worth investigating and would certainly be informative to further improve our knowledge of key residues necessary for the structure function relationship of this protein. Nevertheless, in the present study, we clearly show that while inhibition of Chr 31 lamp2a expression abolished the HG-induced CMA puncta formation, silencing of Chr 14 lamp2a mRNAs caused little if no change on CMA. Likewise, we find that deficiency for the Chr 31 Lamp2A strongly hampered the viability of RTH-149 cells upon HG treatment, while deficiency for Lamp2A from Chr 14 had no effect compared to the control condition. Together, these results supported a functional divergence between the two RT Lamp2A’s, which is further revealed by the strong divergence in the number and nature of proteins affected by the deficiency of either Lamp2A’s, as shown by quantitative proteomic analysis. In this regard, the lack of Chr 31 Lamp2A resulted in the differential expression of nearly three times as many proteins compared to the lack of Chr 14. This larger set of proteins is associated with cellular metabolism and its regulation and is enriched in proteins containing different types of KFERQ-like motifs compared to the one altered by si14, which in addition, does not significantly fit any defined biological process. These results suggest that the evolution of the Chr 14 Lamp2A has reduced its capacity to perform CMA when compared to Chr 31 Lamp2A that seems to present a greater ability to recognize the different types of KFERQ motifs. One hypothesis is that the lack of the G residue in the canonical GYXXF C-terminal motif of Chr 14 Lamp2A, compared to Chr 31 Lamp2A, could be altering the routing to the lysosomal membrane and ending up in the impairment of the CMA activity of this paralog. In this sense, Chr 14 lamp2a could be undergoing a pseudogenization process, although the opposite, that the coding protein is involved in different unknown functions in other cell types or tissues, cannot be ruled out.
Interestingly, some proteins bearing a KFERQ-like motif were found to be jointly up-regulated by inhibiting both Lamp2A isoforms. Among these proteins, Tsg101 caught our attention. This protein is a component of the ESCRT-1 machinery and has been shown to be a critical factor for eMI in mammals [58]. Thus, elevated levels of Tsg101 in CMA-deficient conditions suggest its potential role as a molecular determinant in the interplay between the two types of autophagy. According to this scenario, CMA activity would directly drive eMI via the regulation of Tsg101 levels, which certainly deserves our attention in future studies. This view is supported by previous observations, such as the reciprocal regulation of eMI and CMA during sustained starvation [67,68], and the rerouting of acetylated tau protein to eMI during CMA blockage [69].
In summary, our results provide new insights into the regulation and function of CMA in a natural model organism with impaired glucose tolerance. We discovered that CMA is activated through an Nfe2l2-dependent pathway during hyperglycemic stress and plays a crucial protective role, primarily mediated by the Chr 31 Lamp2A protein. Accordingly, targeting CMA appears as a promising approach to mitigate the pathological symptoms of glucose-metabolic disorders. These findings expand our understanding of CMA’s physiological role and shed light on its evolutionary history in vertebrates.
Materials and methods
Laboratory ware and reagents
All laboratory plastic ware, unless otherwise stated, were purchased from Sarstedt AG & Co. KG. D-(+)-Glucose (G7021), L-(−)-Glucose (G5500), hydrogen peroxide solution (H1009), NAC (A8199), TTFA (T27006) and DMF (242926) were all purchased from Sigma-Aldrich. Instead, CHC (HY-107641) and Mito-TEMPO (HY-112879) were procured by MedChemExpress.
Plasmids, Oligos and bioinformatics analysis
Three different plasmids were used in the present work. The PA-mCherry1-N1 vector was a gift from Michael Davidson (Addgene 54,507; http://n2t.net/addgene:54507; RRID:Addgene_54507; deposited by Michael Davidson), and from this we generated a KFERQ-PA-mCherry1 construction composed by the N-terminal 21 amino acids of bovine RNASE1/RNase A containing its KFERQ CMA-targeting motif, fused to the photoactivable-mCherry1 (PA-mCherry1) protein, as originally reported by Koga et al [30]. The functionality of our construct has recently been validated in fish cells [20]. Moreover, LAMP1-GFP construct was a gift from Benjamin Dehay (Univ. Bordeaux, INSERM, France).
RNA Multiple Sequence Alignments in Clustal Omega [70] was used to assess sequence homology between paralogs and to choose the most appropriate strategy for gene silencing in each case depending on the characteristics of each target sequence and the technical aspects [71]. Hence, a morpholino anti-lamp2a oligonucleotide (5’- CTCCGCTGGATGACAGTAAAGAAAG-3’) was designed in collaboration with GeneTools customer support to target the splice acceptor sites of the exon A (Molamp2a) of the two RT lamp2 paralogs at the same time (located in Chrs 14 and 31 of USDA_OmykA_1.1). This oligo, as well as a standard negative control (5’-CCTCTTACCTCAGTTACAATTTATA-3’; MoSTD-) targeting an intron of human beta-globin associated with beta-thalassemia, were purchased from GeneTools. On the other hand, in order to discriminate between RT lamp2a paralogs, which was impossible to achieve using morpholino oligos due to constrictions of the two lamp2a target sequences, three different siRNAs against the 3’-UTR of each lamp2a were designed using the siDESIGN Center. Moreover, other six different siRNAs against high-homology regions of the RNA sequence between the three rainbow trout nfe2l2 paralogs (located in Chrs 3, 7 and 22 of USDA_OmykA_1.1) were also designed using the same online software. The different siRNAs, including a negative control siRNA against LucGL2 (siLUC: 5’-CGUACGCGGAAUACUUCGA-3’), not expressed in RT cells, were purchased from Horizon Discovery. In a first study, the efficiency of the corresponding siRNA designs in reducing the mRNA levels of either lamp2a paralog in separate, or of the three nfe2l2 paralogs at the same time, was tested by qPCR (data not shown), and the most effectives ones were selected for the experiments presented in this work (for lamp2a’s, si14: 5’-UAAAGAAAUUGCUCGGCUC-3’, and si31: 5’- UGGAGAAGCGGCUGUGUUA-3’; for nfe2l2, sinfe2l2: 5’-UGUCCAAGCACCAGCUCAA-3’).
The JASPAR tool (https://jaspar.genereg.net) was used for the in silico prediction of putative AREs in the promoter region of the two RT lamp2 paralogs. To do that, the 21 kilobase preceding the ATG start codon from the DNA sequence of both paralogs were scanned using the frequency matrix profile for NFE2L2/NRF2 binding sites in Homo sapiens (MA0150.1) from the CORE collection [49].
Cells and general experimental procedures
Rainbow trout RTH-149 hepatoma cell line (ATCC, CRL-1710), recently validated as an useful and reliable model for nutrition and autophagy research in this species [72], was cultured at 18°C without CO2 in supplemented Minimum Essential Medium (Gibco 61,100–053) containing essential amino acids and 10% fetal bovine serum, as previously reported [72]. This complete medium, hereafter referred as control condition (CT), contains 5 mM of glucose.
Nucleofector 2b Device (Lonza, Colmar, France) and the Cell line Nucleofector Kit T (Lonza, VCA-1002) were used for all the cells transfections with either plasmids (1–5 µg), morpholino oligos (5 µM) or siRNAs (1 µM). RTH-149 cells stably transfected with the KFERQ-PA-mCherry1 construction were selected for the resistance to the selective geneticin antibiotic (Gibco 11,811) before cell sorting using a FACS Aria 2-Blue 6-Violet 3-Red 5-YelGr, 2 UV laser configuration (BD Biosciences, Le Pont de Claix Cedex, France) in biosafety cabinet. Prior to experiments, cells were counted using a Cellometer K2 (Nexcelom Bioscience LLC, Lawrence, MA, USA) and plated at a density of 60,000 cells/well onto 4-well culture slides (FALCON 354,114) for microscope imaging experiments, in 24-well plates (90,000 cells/well) for fluorescence decay analysis, in 12-well plates (175,000 cells/well) for cytotoxicity assay, and in 6-cm dishes (400,000 and 500,000 cells/dish) for RNA extraction and protein collection, respectively. In the cases in which the fluorescence of PA-mCherry1 was analyzed, the cells were photoactivated with a 405-nm light source for 10 min prior to starting the treatments.
At the beginning of each experiment cells were washed twice with PBS (ThermoFisher Scientific, BP2944) before incubation with CT (i.e., D-glucose 5 mM) or high-glucose (HG, CT medium supplemented to have 25 mM of D-glucose) media, or instead, exposed to mild-oxidative stress using CT medium supplemented with hydrogen peroxide (H2O2 25 µM). The incubations lasted 4 h for Nfe2l2 immunolocalization studies, 8 h for OxyBlot tests, and 16 h for all the PA-mCherry1 fluorescence analysis with the exception of fluorescent decay experiments for which samples were also taken at 0, 4, and 8 h. The rest of the treatments were prepared as follows, L-glucose (H-LG) and DMF were diluted in CT medium to 25 mM and 30 µM final concentration. DMF dose was chosen according to the literature [53]. NAC (10 mM), CHC (100 µM) and TTFA (10 µM) were diluted in either CT or HG media and doses were selected according to previous studies [39,40,42]. In the case of Mito-TEMPO experiments, cells were first pre-incubated for 60 min with the compound diluted in CT medium at 10 µM as reported before [45], and then exposed to the same concentration of the compound but diluted in HG medium (HG-MitoT) for 16 h. Cell cytotoxicity was measured as the activity of LDH (lactate dehydrogenase) released from cells using the CyQUANT LDH Cytotoxicity Assay Kit (Invitrogen, C20301) following the manufacturer’s recommendations. Briefly, 24 h after cell transfection with siRNAs cells were exposed to HG medium for 48 h before collection of the media and cell lysis. Measurements of absorbance were performed at 490 nm and 680 nm using a microplate reader PowerWaveX Select Spectrophotometer and the KC4 software v3.0 (Bio-Tek Instruments, Winooski, VT, USA).
Cell fluorescence and imaging procedures
Cells were fixed for 20 min using paraformaldehyde 4% fixative (4% PFA; Biotium 22,023) before mounting with an antifade mounting medium including DAPI (Vector Laboratories, Inc., H-2000). All cells images of PA-mCherry1 and LAMP1-GFP fluorescence were acquired with a widefield Microscope Leica DM 5000 (Leica Microsystems, Wetzlar, Germany) equipped with a 40X/1.25 oil objective, 4 filter cubes (DAPI/GFP/TRITC/CY5) and a Greyscale sCMOS camera (Hamamatsu Flash 4.0 V2, Shizuoka, Japan) in the Bordeaux Imaging Center (CNRS-INSERM and Bordeaux University, member of the national infrastructure France BioImaging). Counting of CMA puncta was performed manually by the same researcher using the Cell Counter plugin of Fiji (https://fiji.sc/RRID:SCR_002285 [73]) with images in gray (red channel) and blue (DAPI), in blind and random order and excluding cells on the edges or very superposed, as reported by others [74]. For each image, the number of puncta was normalized by the number of nuclei within the image, and the results showed represent the number of puncta per cell in a minimum of 45 images coming from 3–4 independent experiments. In all cases, more than 600 cells/condition were counted.
The co-occurrence between CMA-puncta and lysosomes was assessed in LAMP1-GFP transiently transfected cells on Fiji using the BIOP version of JACoP pluging [32] (https://c4science.ch/w/bioimaging_and_optics_platform_biop/image-processing/imagej_tools/jacop_b/). First, the PCC between CMA-puncta and LAMP1-GFP was analyzed in 48 single cells by selecting one region of interest (ROI) from 44 different merged images. A PCC ranges from −1 to 1, where −1 indicates a negative correlation and + 1 highlights a complete positive correlation between the pixel-intensity of the two channels [32]. Then, each of the ROIs was manually thresholded for both red (>450) and green (>725) channels considering the background found in negative controls before calculating MCCs (MCC-M1 and MCC-M2). MCC-M1 indicates the fraction of PA-mCherry1 signal overlapping the LAMP1-GFP signal, while MCC-M2 indicates the opposite. These coefficients range from 0 (no colocalization) to 1 (100% overlapping) [32,33].
The immunolocalization of Nfe2l2 was performed following the procedure previously described [75]. Briefly, fixed cells were permeabilized and blocked before overnight incubation with a rabbit anti-NFE2L2 polyclonal antibody (Abcam, ab31163; 1:300) as primary antibody, and then with a goat anti-Rabbit Alexa Fluor 488 (Invitrogen, A-11008; 1:500) as secondary antibody. Finally, preparations were mounted before imaging with a widefield AXIO Imager M2 (Carl Zeiss Microscopy, Oberkochen, Germany) microscope equipped with a 20X objective, 3 filter cubes (DAPI/FITC/TRITC) and a SONY XCD-SX910CR Color Raw camera in the Ecology and Fish Population Biology Facility [76]. The percentage of nuclear Nfe2l2 localization was calculated on Fiji using the Intensity Ratio Nuclei Cytoplasm Tool (RRID:SCR_018573; https://github.com/MontpellierRessourcesImagerie/imagej_macros_and_scripts/wiki/Intensity-Ratio-Nuclei-Cytoplasm-Tool) in a minimum of 43 independent images corresponding to 3 independent experiments.
For PA-mCherry1 fluorescence decay analysis, cell incubations were stopped by rapid washing with ice-cold PBS followed by trypsinization. After washing, cells were fixed with 4% PFA for 15 min, washed and resuspended in a total of 150 µL of PBS from which 120 µL were used to measure fluorescence intensity (excitation at 554–15 nm and emission at 600–20 nm) using a microplate reader CLARIOstar Plus (BMG LABTECH GmbH, Ortenberg, Germany), and the other 30 µL were destined to counting the number of cells using a Cellometer K2. The results were first calculated as the fluorescence intensity of a sample divided by the number of cells of this sample, and then normalized respect the results of the corresponding group at T0 (100%), in a minimum of 3 replicates and 4 independent experiments. The half-life (t1/2) of the reporter was estimated considering the average amount of fluorescence disappeared in 16 h and the amount remaining for each of the conditions.
In vivo feeding trial
RTs (Oncorhynchus mykiss) with an initial weight of 27.35 ± 0.12 g were reared under natural photoperiod and at a constant water temperature of 17.5 ± 0.5°C. Fish were separated into two groups of 3 tanks and fed twice a day to visual satiation with two custom isolipidic and isoenergetic diets (3 tanks/diet), but presenting a significantly different proportion in the gelatinized wheat starch content (Non-Carbohydrate Diet (NCD): 0%; High-Carbohydrate Diet (HCD): 25%), over a 9-week period with routine daily monitoring of the animal welfare. After 9 weeks of the experiment, blood samples were collected from 3 fish/tank (9 fish/diet) at either 6 h or 24 h after the last meal, and subsequently, the 6 h post-prandial fish were euthanized by immersion in a lethal dose of benzocaine before the collection of liver tissue samples. Samples were immediately frozen in liquid nitrogen and stored at − 80°C until further analysis. All experimental procedures were conducted at the INRAe NuMeA facilities (permit number A64.495.1, delivered by French veterinary services) in strict accordance with the legal frameworks of France and the European Union. They respect the directive 2010/63/EU relating to the protection of animals used for scientific purposes as well as the decree No 2013–118, 1 February 2013, of the French legislation governing the ethical treatment of animals.
Protein extraction, western blotting and quantitative proteomics
Proteins were collected using RIPA buffer (ThermoFisher Scientific 89,901) supplemented with protease and phosphatases inhibitor cocktail (ThermoFisher Scientific 78,422) as explained elsewhere [72]. Protein concentration was determined using the Qubit protein assay kit (ThermoFisher Scientific, Q33211). In the case of samples intended to the use of OxyBlot Protein Oxidation Kit (Merck-Millipore, S7150), the lysis buffer was supplemented with a 2% of 2-mercaptoethanol (Thermo Scientific, J66742) to prevent further oxidation of samples. Then, the carbonyl groups from a total of 20 µg of protein/sample were derivatized following the OxyBlot manufacturer’s recommendations before the samples were electrophoresed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Afterwards, proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Merk-Millipore, IPFL00010), blocked with SuperBlock Blocking Buffer in PBS (Thermo Scientific 37,515), and immunoblotted following the manufacturer’s protocol and the antibodies included within the OxyBlot Kit. The different immunoreactive bands were developed using the SuperSignal West Pico Plus Chemiluminescent Substrate (ThermoFisher Scientific 34,578). Signal acquisition was performed using Smart Exposure in an iBright FL1500 Imaging System (ThermoFisher Scientific, Illkirch Cedex, France) to assure signal linearity, and then images were quantified with the iBright Analysis Software (ThermoFisher Scientific, RRID:SCR_017632).
For proteomic analysis, 24 h after cell transfection with siRNAs cells were exposed to HG medium for 48 h before collecting the total protein extracts and its shipping to the ProteoToul Services – Proteomics Facility of Toulouse (https://proteotoul.ipbs.fr/proteotoul-services/) for posterior analysis. Briefly, 50 µg of dried protein extracts from each sample were solubilized with 25 µl of 5% SDS. Proteins were submitted to reduction and alkylation of cysteine residues by addition of TCEP and chloroacetamide to a final concentration respectively of 10 mM and 40 mM. Protein samples were then processed for trypsin digestion on S-trap Micro devices (Protifi) according to manufacturer’s protocol, with the following modifications: precipitation was performed using 216 µl S-Trap buffer, 4 µg trypsin was added per sample for digestion in 25 µl ammonium bicarbonate 50 mM. After that, tryptic peptides were resuspended in 35 µl of 2% acetonitrile and 0.05% trifluoroacetic acid and analyzed by nano-liquid chromatography (LC) coupled to tandem mass spectrometry (MS), using an UltiMate 3000 system (NCS-3500RS Nano/Cap System; ThermoFisher Scientific) coupled to an Orbitrap Exploris 480 mass spectrometer equipped with a FAIMS Pro device (ThermoFisher Scientific). 1 µg of each sample was injected into the analytical C18 column (75 µm inner diameter × 50 cm, Acclaim PepMap 2 µm C18 ThermoFisher Scientific) equilibrated in 97.5% solvent A (5% acetonitrile, 0.2% formic acid) and 2.5% solvent B (80% acetonitrile, 0.2% formic acid). Peptides were eluted using a 2.5% − 40% gradient of solvent B over 62 min at a flow rate of 300 nL/min. The mass spectrometer was operated in data-dependent acquisition mode with the Xcalibur software. MS survey scans were acquired with a resolution of 60,000 and a normalized AGC target of 300%. Two compensation voltages were applied (−45 v/-60 v). For 0.8 s most intense ions were selected for fragmentation by high-energy collision-induced dissociation, and the resulting fragments were analyzed at a resolution of 30,000, using a normalized AGC target of 100%. Dynamic exclusion was used within 45 s to prevent repetitive selection of the same peptide. Thereafter, raw mass spectrometry (MS) files were processed with the Mascot software (version 2.7.0) for database search and Proline55 for label-free quantitative analysis (version 2.1.2). Data were searched against Rainbow trout entries of Uniprot protein database (46,650 sequences; 17861,404 residues). Carbamidomethylation of cysteines was set as a fixed modification, whereas oxidation of methionine was set as variable modifications. Specificity of trypsin/P digestion was set for cleavage after K or R, and two missed trypsin cleavage sites were allowed. The mass tolerance was set to 10 ppm for the precursor and to 20 mmu in tandem MS mode. Minimum peptide length was set to 7 amino acids, and identification results were further validated in Proline by the target decoy approach using a reverse database at both a PSM and protein false-discovery rate of 1%. For label-free relative quantification of the proteins across biological replicates and conditions, cross-assignment of peptide ions peaks was enabled inside group with a match time window of 1 min, after alignment of the runs with a tolerance of ±600 s. Median Ratio Fitting computes a matrix of abundance ratios calculated between any two runs from ion abundances for each protein. For each pair-wise ratio, the median of the ion ratios is then calculated and used to represent the protein ratio between these two runs. A least-squares regression is then performed to approximate the relative abundance of the protein in each run in the dataset. This abundance is finally rescaled to the sum of the ion abundances across runs. A Student T-test (two-tailed t-test, equal variances) was then performed on log2 transformed values to analyze differences in protein abundance in all biologic group comparisons. Significance level was set at p < 0.05, and ratios (Fold change, FC) were considered relevant if higher than ±2, as in previous studies using LAMP2A siRNA [56,57]. The list of proteins identified in the present study is provided in Table S2 along with the FC (log2 transformed) and p-value (log10 transformed) results of the significant differentially expressed (DE) proteins detected between the different comparisons. The lists of DE proteins were then imported to STRING V11.5 (https://string-db.org/) and the networks directly exported to CytoScape (3.9.1) software. Then, Gene Ontology enrichment maps were generated using the EnrichmentMAP (3.3.5) plugin with the thresholds set as 0.2 (connectivity cutoff); 0.25 (edge cutoff); p < 0.05; and FDR < 0.05. The detailed list of significant biological processes detected is provided in Table S3. The maps were annotated using the AutoAnnotate (1.4.0) app. Finally, the presence of KFERQ motifs within the protein sequences was assessed using the KFERQ finder app V0.8 [55], and data is presented in Table S4.
mRNA levels analysis: Quantitative RT-PCR
Total RNA was extracted and purified using an RNeasy Mini Kit (Qiagen 74,106) following the manufacturer’s recommendations. The protocol for sample preparation and the conditions for quantitative RT-PCR have been detailed in previous publications [72,77]. cDNA synthesis was performed in duplicate and the analysis of those samples was also performed in duplicate. The relative quantification of target genes was calculated using the ΔCT method described by Pfaffl [78] and eef1a1 (eukaryotic translation elongation factor 1 alpha 1) as a housekeeping gene, as it was the most stable among the three genes tested (including actb [actin beta] and rna18s1 [RNA, 18S ribosomal 1], data not shown). Primers used are listed in Table S5.
Statistical analysis
All data are reported as means ± SEM, percentages ± SEM or fold induction respect the CT group ± SEM, from a minimum of 3 independent experiments. Normality of data was verified before performing parametric or non-parametric tests. To evaluate the differences between more than two groups when normality was met, one-way ANOVA followed by Tukey’s multiple comparison (same sample size) or Bonferroni’s multiple comparison (different sample size) post-hoc test were used. Otherwise, Kruskal-Wallis followed by Dunn’s multiple comparisons tests were utilized. On the other hand, for comparison between two groups we used a parametric two-tailed unpaired Student’s T-test, or a non-parametric Mann Whitney test. The significant enrichment of proteins containing CMA targeting motifs compared with the overall RT proteome was evaluated by Chi-Square Test. All statistical analyses were performed using GraphPad Prism version 8.0.1 for Windows (GraphPad Software, Inc., www.graphpad.com) and a p-value <0.05 was set as a level of significance. All data generated in this study and presented in the figures are provided along with their statistic report in the Table S1.
Supplementary Material
Acknowledgements
We thanks Atika Zouine and Vincent Pitard for technical assistance at the Flow cytometry facility, Centre National de la Recherche Scientifique (CNRS) unité mixte de service (UMS) 3427, Institut National de la Santé Et de la Recherche Médicale (INSERM) US 005, University of Bordeaux, F-33000 Bordeaux, France. We thanks Christel Poujol for technical assistance at the Bordeaux Imaging Center (BIC), CNRS-INSERM and Bordeaux University, member of the national infrastructure France BioImaging and supported by the French National Research Agency (ANR-10-INBS-04). We thanks Alexandre Stella from the ProteoToul Services - Proteomics Facility of Toulouse for the proteomics analysis. We also want to thanks F. Terrier, and A. Lanuque for the preparation of diets and care of fish, L. Peron for manufacturing the light emitting device, and the IE ECP Ecology and Fish Population Biology Facility for the access to the widefield AXIO Imager M2 microscope. Schematic figures were created with BioRender.com tools.
Funding Statement
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 101030643 and the Aquaexcel3.0 grant agreement No 871108, and by the European Maritime and Fisheries Fund [PFEA470019FA1000005].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary data
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2267415
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its supplementary materials.
References
- [1].Schnebert S, Goguet M, Vélez EJ, et al. Diving into the evolutionary history of HSC70-linked selective autophagy pathways: endosomal microautophagy and chaperone-mediated autophagy. Cells. 2022 Jun 16;11(12):1945. PubMed PMID: 35741074. doi: 10.3390/cells11121945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chiang H-L, Terlecky SR, Plant CP, et al. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science. 1989 Oct 20;246(4928):382–385. PubMed PMID: 2799391. doi: 10.1126/science.2799391. [DOI] [PubMed] [Google Scholar]
- [3].Agarraberes FA, Dice JF.. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci. 2001. Jul;114(Pt 13):2491–2499. PubMed PMID: 11559757. doi: 10.1242/jcs.114.13.2491 [DOI] [PubMed] [Google Scholar]
- [4].Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996 Jul 26;273(5274):501–503. PubMed PMID: 8662539. doi: 10.1126/science.273.5274.501 [DOI] [PubMed] [Google Scholar]
- [5].Kaushik S, Cuervo AM. Autophagy as a cell-repair mechanism: activation of chaperone-mediated autophagy during oxidative stress. Mol Aspects Med. 2006. Oct-Dec;27(5–6):444–454. PubMed PMID: 16978688. doi: 10.1016/j.mam.2006.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bandyopadhyay U, Kaushik S, Varticovski L, et al. The chaperone-mediated autophagy receptor Organizes in Dynamic protein Complexes at the lysosomal membrane. Mol Cell Biol. 2008. Sep;28(18):5747–5763. PubMed PMID: 18644871. doi: 10.1128/MCB.02070-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Arias E, Cuervo AM. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol. 2011. Apr;23(2):184–189. PubMed PMID: 21094035. doi: 10.1016/j.ceb.2010.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018. Jun;19(6):365–381. PubMed PMID: 29626215. doi: 10.1038/s41580-018-0001-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schneider JL, Suh Y, Cuervo AM. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. 2014 Sep 2;20(3):417–432. PubMed PMID: 25043815. doi: 10.1016/j.cmet.2014.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kaushik S, Juste YR, Lindenau K, et al. Chaperone-mediated autophagy regulates adipocyte differentiation. Sci Adv. 2022 Nov 18;8(46):eabq2733. PubMed PMID: 36383673. doi: 10.1126/sciadv.abq2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kaushik S, Cuervo AM. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol. 2015. Jun;17(6):759–770. PubMed PMID: 25961502. doi: 10.1038/ncb3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bourdenx M, Martín-Segura A, Scrivo A, et al. Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell. 2021 May 13;184(10):2696–2714.e25. PubMed PMID: 33891876. doi: 10.1016/j.cell.2021.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gomez-Sintes R, Xin Q, Jimenez-Loygorri JI, et al. Targeting retinoic acid receptor alpha-corepressor interaction activates chaperone-mediated autophagy and protects against retinal degeneration. Nat Commun. 2022 Jul 21;13(1):4220. PubMed PMID: 35864098. doi: 10.1038/s41467-022-31869-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tasset I, Cuervo AM. Role of chaperone-mediated autophagy in metabolism. FEBS J. 2016. Jul;283(13):2403–2413. PubMed PMID: 26854402. doi: 10.1111/febs.13677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jafari M, McCabe M, Cuervo AM. Chaperone-mediated autophagy: mechanisms and physiological relevance. Curr Opin Physiol. 2022. Dec;30:100597. doi: 10.1016/j.cophys.2022.100597 [DOI] [Google Scholar]
- [16].Arias E, Cuervo AM. Pros and Cons of chaperone-mediated autophagy in cancer Biology. Trends Endocrinol Metab. 2020. Jan;31(1):53–66. PubMed PMID: 31699565. doi: 10.1016/j.tem.2019.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Madrigal-Matute J, Cuervo AM, Sluimer JC. Chaperone-mediated autophagy protects against atherosclerosis. Autophagy. 2022. Oct;18(10):2505–2507. PubMed PMID: 35787098. doi: 10.1080/15548627.2022.2096397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sooparb S, Price SR, Shaoguang J, et al. Suppression of chaperone-mediated autophagy in the renal cortex during acute diabetes mellitus. Kidney Int. 2004. Jun;65(6):2135–2144. PubMed PMID: 15149326. doi: 10.1111/j.1523-1755.2004.00639.x [DOI] [PubMed] [Google Scholar]
- [19].Liu C, Sun W, Zhu T, et al. Glia maturation factor-β induces ferroptosis by impairing chaperone-mediated autophagic degradation of ACSL4 in early diabetic retinopathy. Redox Biol. 2022. Jun;52:102292. PubMed PMID: 35325805. doi: 10.1016/j.redox.2022.102292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lescat L, Véron V, Mourot B, et al. Chaperone-mediated autophagy in the light of evolution: insight from fish. Mol Biol Evol. 2020 Oct 1;37(10):2887–2899. PubMed PMID: 32437540. doi: 10.1093/molbev/msaa127. [DOI] [PubMed] [Google Scholar]
- [21].Tesseraud S, Avril P, Bonnet M, et al. Autophagy in farm animals: Current knowledge and future challenges. Autophagy. 2021. Aug;17(8):1809–1827. PubMed PMID: 32686564. doi: 10.1080/15548627.2020.1798064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Krishnan J, Rohner N. Sweet fish: fish models for the study of hyperglycemia and diabetes. J Diabetes. 2019. Mar;11(3):193–203. PubMed PMID: 30264455. doi: 10.1111/1753-0407.12860 [DOI] [PubMed] [Google Scholar]
- [23].Bergot F. Effects of dietary carbohydrates and of their mode of distribution on glycaemia in rainbow trout (salmo gairdneri richardson). Comp Biochem Physiol A Mol Integr Physiol. 1979;64(4):543–547. doi: 10.1016/0300-9629(79)90581-4 [DOI] [Google Scholar]
- [24].Bergot F. Specific problems posed by carbohydrate utilization in the rainbow trout. Ann Nutr Aliment. 1979;33(2):247–257. PubMed PMID: 496176. [PubMed] [Google Scholar]
- [25].Kamalam BS, Medale F, Panserat S. Utilisation of dietary carbohydrates in farmed fishes: new insights on influencing factors, biological limitations and future strategies. Aquaculture. 2017 Jan 20;467:3–27. doi: 10.1016/j.aquaculture.2016.02.007 [DOI] [Google Scholar]
- [26].Palmer TN, Ryman BE. Studies on oral glucose intolerance in fish. J Fish Biol. 1972. Apr;4(2):311–319. doi: 10.1111/j.1095-8649.1972.tb05680.x [DOI] [Google Scholar]
- [27].Wilson RP. Utilization of dietary carbohydrate by fish. Aquaculture. 1994. Jul;124(1–4):67–80. doi: 10.1016/0044-8486(94)90363-8 [DOI] [Google Scholar]
- [28].Kamalam BS, Medale F, Kaushik S, et al. Regulation of metabolism by dietary carbohydrates in two lines of rainbow trout divergently selected for muscle fat content. J Exp Biol. 2012 Aug 1;215(Pt 15):2567–2578. PubMed PMID: 22786633. doi: 10.1242/jeb.070581. [DOI] [PubMed] [Google Scholar]
- [29].Skiba-Cassy S, Panserat S, Larquier M, et al. Apparent low ability of liver and muscle to adapt to variation of dietary carbohydrate: protein ratio in rainbow trout (Oncorhynchus mykiss). Br J Nutr. 2013 Apr 28;109(8):1359–1372. PubMed PMID: 22951215. doi: 10.1017/S0007114512003352. [DOI] [PubMed] [Google Scholar]
- [30].Koga H, Martinez-Vicente M, Maclan F, et al. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat Commun. 2011 Jul 12;2:386. PubMed PMID: 21750540. doi: 10.1038/ncomms1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kiffin R, Christian C, Knecht E, et al. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004. Nov;15(11):4829–4840. PubMed PMID: 15331765. doi: 10.1091/mbc.e04-06-0477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bolte SB, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006. Dec;224(Pt 3):213–232. PubMed PMID: 17210054. doi: 10.1111/j.1365-2818.2006.01706.x [DOI] [PubMed] [Google Scholar]
- [33].Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol - Cell Physiol. 2011. Apr;300(4):C723–C742. PubMed PMID: 21209361. doi: 10.1152/ajpcell.00462.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lucchesi AN, de Freitas NT, Cassettari LL, et al. Diabetes mellitus triggers oxidative stress in the liver of alloxan-treated rats: a mechanism for diabetic chronic liver disease. Acta Cir Bras. 2013. Jul;28(7):502–508. PubMed PMID: 23842931. doi: 10.1590/S0102-86502013000700005 [DOI] [PubMed] [Google Scholar]
- [35].Jiang Z, Lu W, Zeng Q, et al. High glucose-induced excessive reactive oxygen species promote apoptosis through mitochondrial damage in rat cartilage endplate cells. J Orthop Res. 2018. Sep;36(9):2476–2483. PubMed PMID: 29663489. doi: 10.1002/jor.24016 [DOI] [PubMed] [Google Scholar]
- [36].Yu T, Jhun BS, Yoon Y. High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxid Redox Signal. 2011 Feb 1;14(3):425–437. PubMed PMID: 20518702. doi: 10.1089/ars.2010.3284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Russell JW, Golovoy D, Vincent AM, et al. High glucose‐induced oxidative stress and mitochondrial dysfunction in neurons. FASEB J. 2002. Nov;16(13):1738–1748. PubMed PMID: 12409316. doi: 10.1096/fj.01-1027com [DOI] [PubMed] [Google Scholar]
- [38].Gao CL, Zhu C, Zhao YP, et al. Mitochondrial dysfunction is induced by high levels of glucose and free fatty acids in 3T3-L1 adipocytes. Mol Cell Endocrinol. 2010 May 14;320(1–2):25–33. PubMed PMID: 20144685. doi: 10.1016/j.mce.2010.01.039. [DOI] [PubMed] [Google Scholar]
- [39].Alnahdi A, John A, Raza H, et al. N-acetyl cysteine attenuates oxidative stress and glutathione-dependent redox imbalance caused by high glucose/high palmitic acid treatment in pancreatic rin-5F cells. PLoS One. 2019 Dec 20;14(12):e0226696. PubMed PMID: 31860682. doi: 10.1371/journal.pone.0226696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A. 2006 Feb 21;103(8):2653–2658. PubMed PMID: 16477035. doi: 10.1073/pnas.0511154103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001 Dec 13;414(6865):813–820. PubMed PMID: 11742414. doi: 10.1038/414813a [DOI] [PubMed] [Google Scholar]
- [42].Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000 Apr 13;404(6779):787–790. PubMed PMID: 10783895. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- [43].Halestrap AP, Denton RM. Specific inhibition of pyruvate transport in rat liver mitochondria and human erythrocytes by alpha-cyano-4-hydroxycinnamate. Biochem J. 1974. Feb;138(2):313–316. PubMed PMID: 4822737. doi: 10.1042/bj1380313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].McCarthy C, Kenny LC. Therapeutically targeting mitochondrial redox signalling alleviates endothelial dysfunction in preeclampsia. Sci Rep. 2016 Sep 8;6(1):32683. PubMed PMID: 27604418. doi: 10.1038/srep32683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tian Y, Guo H, Miao X, et al. Nestin protects podocyte from injury in lupus nephritis by mitophagy and oxidative stress. Cell Death Dis. 2020 May 5;11(5):319. PubMed PMID: 32371936. doi: 10.1038/s41419-020-2547-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bayo Jimenez MT, Frenis K, Hahad O, et al. Protective actions of nuclear factor erythroid 2-related factor 2 (NRF2) and downstream pathways against environmental stressors. Free Radic Biol Med. 2022. Jul;187:72–91. doi: 10.1016/j.freeradbiomed.2022.05.016 PubMed PMID: 35613665. [DOI] [PubMed] [Google Scholar]
- [47].Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014. Apr;39(4):199–218. PubMed PMID: 24647116. doi: 10.1016/j.tibs.2014.02.002 [DOI] [PubMed] [Google Scholar]
- [48].Zhu L, He S, Huang L, et al. Chaperone-mediated autophagy degrades Keap1 and promotes Nrf2-mediated antioxidative response. Aging Cell. 2022. Jun;21(6):e13616. PubMed PMID: 35535673. doi: 10.1111/acel.13616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022 Jan 7;50(D1):D165–D173. PubMed PMID: 34850907. doi: 10.1093/nar/gkab1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Raghunath A, Sundarraj K, Nagarajan R, et al. Antioxidant response elements: discovery, classes, regulation and potential applications. Redox Biol. 2018. Jul;17:297–314. doi: 10.1016/j.redox.2018.05.002 PubMed PMID: 29775961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Raghunath A, Nagarajan R, Sundarraj K, et al. Genome-wide identification and analysis of Nrf2 binding sites – antioxidant response elements in zebrafish. Toxicol Appl Pharmacol. 2018 Dec 1;360:236–248. PubMed PMID: 30243843. doi: 10.1016/j.taap.2018.09.013 [DOI] [PubMed] [Google Scholar]
- [52].Cuadrado A. Brain-protective mechanisms of transcription factor NRF2: toward a common strategy for neurodegenerative diseases. Annu Rev Pharmacol Toxicol. 2022 Jan 6;62(1):255–277. PubMed PMID: 34637322. doi: 10.1146/annurev-pharmtox-052220-103416 [DOI] [PubMed] [Google Scholar]
- [53].Pajares M, Rojo AI, Arias E, et al. Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy. 2018;14(8):1310–1322. PubMed PMID: 29950142. doi: 10.1080/15548627.2018.1474992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kwon AT, Arenillas DJ, Hunt RW, et al. Opossum-3: advanced analysis of regulatory motif over-representation across genes or chip-seq datasets. G3 (Bethesda). 2012. Sep;2(9):987–1002. PubMed PMID: 22973536. doi: 10.1534/g3.112.003202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kirchner P, Bourdenx M, Madrigal-Matute J, et al. Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol. 2019 May 31;17(5):e3000301. PubMed PMID: 31150375. doi: 10.1371/journal.pbio.3000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hao Y, Kacal M, Ouchida AT, et al. Targetome analysis of chaperone-mediated autophagy in cancer cells. Autophagy. 2019. Sep;15(9):1558–1571. PubMed PMID: 30821613. doi: 10.1080/15548627.2019.1586255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kacal M, Zhang B, Hao Y, et al. Quantitative proteomic analysis of temporal lysosomal proteome and the impact of the KFERQ-like motif and LAMP2A in lysosomal targeting. Autophagy. 2021. Nov;17(11):3865–3874. PubMed PMID: 33446043. doi: 10.1080/15548627.2021.1876343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Sahu R, Kaushik S, Clement CC, et al. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. 2011 Jan 18;20(1):131–139. PubMed PMID: 21238931. doi: 10.1016/j.devcel.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Dehal P, Boore JL, Holland P. Two Rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol. 2005. Oct;3(10):e314. PubMed PMID: 16128622. doi: 10.1371/journal.pbio.0030314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Pasquier J, Cabau C, Nguyen T, et al. Gene evolution and gene expression after whole genome duplication in fish: the PhyloFish database. BMC Genomics. 2016 May 18;17(1):368. PubMed PMID: 27189481. doi: 10.1186/s12864-016-2709-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lescat L, Herpin A, Mourot B, et al. CMA restricted to mammals and birds: myth or reality? Autophagy. 2018;14(7):1267–1270. PubMed PMID: 29929419. doi: 10.1080/15548627.2018.1460021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Anguiano J, Garner TP, Mahalingam M, et al. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat Chem Biol. 2013. Jun;9(6):374–382. PubMed PMID: 23584676. doi: 10.1038/nchembio.1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zhang J, Johnson JL, He J, et al. Cystinosin, the small GTPase Rab11, and the Rab7 effector RILP regulate intracellular trafficking of the chaperone-mediated autophagy receptor LAMP2A. J Biol Chem. 2017 Jun 23;292(25):10328–10346. PubMed PMID: 28465352. doi: 10.1074/jbc.M116.764076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Cuervo AM, Dice JF. Unique properties of lamp2a compared to other lamp2 isoforms. J Cell Sci. 2000. Dec;113(Pt 24):4441–4450. PubMed PMID: 11082038. doi: 10.1242/jcs.113.24.4441 [DOI] [PubMed] [Google Scholar]
- [65].Kiffin R, Kaushik S, Zeng M, et al. Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J Cell Sci. 2007 Mar 1;120(Pt 5):782–791. PubMed PMID: 17284523. doi: 10.1242/jcs.001073. [DOI] [PubMed] [Google Scholar]
- [66].Qiao L, Hu J, Qiu X, et al. LAMP2A, LAMP2B and LAMP2C: similar structures, divergent roles. Autophagy. 2023 Jul 21;1–16:2837–2852. PubMed PMID: 37469132. doi: 10.1080/15548627.2023.2235196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Cuervo AM, Knecht E, Terlecky SR, et al. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am J Physiol. 1995. Nov;269(5 Pt 1):C1200–C1208. PubMed PMID: 7491910. doi: 10.1152/ajpcell.1995.269.5.C1200 [DOI] [PubMed] [Google Scholar]
- [68].Krause GJ, Diaz A, Jafari M, et al. Reduced endosomal microautophagy activity in aging associates with enhanced exocyst-mediated protein secretion. Aging Cell. 2022. Oct;21(10):e13713. PubMed PMID: 36116133. doi: 10.1111/acel.13713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Caballero B, Bourdenx M, Luengo E, et al. Acetylated tau inhibits chaperone-mediated autophagy and promotes tau pathology propagation in mice. Nat Commun. 2021 Apr 14;12(1):2238. PubMed PMID: 33854069. doi: 10.1038/s41467-021-22501-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol Syst Biol. 2011 Oct 11;7(1):539. PubMed PMID: 21988835. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Summerton JM, Compared S-D. Impact of structure and mechanism of action on off-target effects and sequence specificity. Curr Top Med Chem. 2007;7(7):651–660. PubMed PMID: 17430206. doi: 10.2174/156802607780487740. [DOI] [PubMed] [Google Scholar]
- [72].Morin G, Pinel K, Dias K, et al. RTH-149 Cell line, a Useful Tool to Decipher molecular mechanisms related to fish Nutrition. Cells. 2020 Jul 22;9(8):1754. PubMed PMID: 32707879. doi: 10.3390/cells9081754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012 Jun 28;9(7):676–682. PubMed PMID: 22743772. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Endicott SJ, Ziemba ZJ, Beckmann LJ, et al. Inhibition of class I PI3K enhances chaperone-mediated autophagy. J Cell Bio. 2020 Dec 7;219(12):e202001031. PubMed PMID: 33048163. doi: 10.1083/jcb.202001031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Vélez EJ, Lutfi E, Jiménez-Amilburu V, et al. IGF-I and amino acids effects through TOR signaling on proliferation and differentiation of gilthead sea bream cultured myocytes. Gen Comp Endocrinol. 2014 Sep 1;205:296–304. PubMed PMID: 24882593. doi: 10.1016/j.ygcen.2014.05.024 [DOI] [PubMed] [Google Scholar]
- [76].ECP IE. Ecology and fish Population Biology Facility. Saint-Pée-sur-Nivelle, France: INRAE; 2018. [Google Scholar]
- [77].Seiliez I, Belghit I, Gao Y, et al. Looking at the metabolic consequences of the colchicine-based in vivo autophagic flux assay. Autophagy. 2016;12(2):343–356. PubMed PMID: 26902586. doi: 10.1080/15548627.2015.1117732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001 May 1;29(9):e45. PubMed PMID: 11328886. doi: 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its supplementary materials.