

ABSTRACT
Autophagosomes fuse with lysosomes, forming autolysosomes that degrade engulfed cargo. To maintain lysosomal capacity, autophagic lysosome reformation (ALR) must regenerate lysosomes from autolysosomes using a membrane tubule-based process. Maintaining lysosomal capacity is required to maintain cellular health, especially in neurons where lysosomal dysfunction has been repeatedly implicated in neurodegenerative disease. The DNA-J domain HSC70 co-chaperone RME-8/DNAJC13 has been linked to endosomal coat protein regulation and to neurological disease. We report new analysis of the requirements for the RME-8/DNAJC13 protein in neurons, focusing on intact C. elegans mechanosensory neurons, and primary mouse cortical neurons in culture. Loss of RME-8/DNAJC13 in both systems results in accumulation of grossly elongated autolysosomal tubules. Further C. elegans analysis revealed a similar autolysosome tubule accumulation defect in mutants known to be required for ALR in mammals, including mutants lacking bec-1/BECN1/Beclin1 and vps-15/PIK3R4/p150 that regulate the class III phosphatidylinositol 3-kinase (PtdIns3K) VPS-34, and dyn-1/dynamin that severs ALR tubules. Clathrin is also an important ALR regulator implicated in autolysosome tubule formation and release. In C. elegans we found that loss of RME-8 causes severe depletion of clathrin from neuronal autolysosomes, a phenotype shared with bec-1 and vps-15 mutants. We conclude that RME-8/DNAJC13 plays a previously unrecognized role in ALR, likely affecting autolysosome tubule severing. Additionally, in both systems, loss of RME-8/DNAJC13 reduced macroautophagic/autophagic flux, suggesting feedback regulation from ALR to autophagy. Our results connecting RME-8/DNAJC13 to ALR and autophagy provide a potential mechanism by which RME-8/DNAJC13 could influence neuronal health and the progression of neurodegenerative disease.
Abbreviation: ALR, autophagic lysosome reformation; ATG-13/EPG-1, AuTophaGy (yeast Atg homolog)-13; ATG-18, AuTophaGy (yeast Atg homolog)-18; AV, autophagic vacuole; CLIC-1, Clathrin Light Chain-1; EPG-3, Ectopic P Granules-3; EPG-6, Ectopic P Granules-6; LGG-1, LC3, GABARAP and GATE-16 family-1; MAP1LC3/LC3, microtubule-associated protein 1 light chain 3; PD, Parkinson disease; PtdIns3P, phosphatidylinositol-3-phosphate; PtdIns(4,5)P2, phosphatidylinositol-4,5-bisphosphate; RME-8, Receptor Mediated Endocytosis-8; SNX-1, Sorting NeXin-1; VPS-34, related to yeast Vacuolar Protein Sorting factor-34
KEYWORDS: Autophagy, clathrin, endocytosis, lysosomes, neurodegeneration, trafficking
Introduction
Organelle integrity and cellular proteostasis depend heavily upon a variety of degradative mechanisms for macromolecular turnover. Among these, one key pathway is macroautophagy (hereafter referred to as autophagy), a mechanism that can remove whole defective organelles and cytoplasmic protein aggregates as they build up, ameliorating the cellular dysfunction that can occur if such components accumulate. Autophagy begins with the phagophore, a de novo membrane which expands to completely engulf a region of cytoplasm, often including a specific degradative target, forming a double-membraned autophagosome. Once autophagosomes form, they fuse with lysosomes, creating autolysosomes to degrade their contents.
Lysosomes are reformed from autolysosomes using a specific mechanism, termed autophagic lysosome reformation (ALR) [1]. This ALR process is required to maintain an active lysosome pool, allowing degradation of autophagic contents to continue uninterrupted [1]. During ALR, recycling tubules are drawn out from buds on the autolysosome limiting membrane and are then severed to release protolysosomes. The mechanism of ALR tubule formation and release is not well understood, but appears to require phosphoinositide lipids phosphatidylinositol-3-phosphate (PtdIns3P) and phosphatidylinositol-4,5-bisphosphate (PtdIns[4,5]P2), a clathrin coat, the severing enzyme dynamin, and kinesin motor activity [2–5]. ALR failure is typically characterized by the accumulation of enlarged autolysosomes and grossly elongated autolysosomal tubules [2,6]. Interestingly, in some cases ALR defects have been linked to impaired autophagy, suggesting feedback from ALR to autophagy initiation [5].
Autophagy is thought to be particularly important in neurons, with neurons displaying higher levels of constitutive autophagy compared to other cell types [7,8]. This higher level of basal autophagy implies a necessary requirement for higher level basal ALR to maintain lysosomal capacity. Neurodegenerative diseases such as Alzheimer and Parkinson diseases (PD) are strongly linked to impaired degradation in general, especially decreased autophagy that is thought to contribute to a buildup of deleterious protein aggregates, such as tau, amyloid beta, and SNCA/α-synuclein, especially as aging occurs (reviewed in [9,10]). Since most neurons are post-mitotic, they are unable to dilute out protein aggregates via mitosis, an option available for non-terminally differentiated cell types. Neurons also have extremely long, thin axons that proteins and cargoes need to traverse. This architecture makes neurons especially sensitive to neurodegeneration should autophagy and other aggregate clearance mechanisms go awry. This likely includes ALR, as in several cases failure in ALR has been linked to neurodegenerative disease [11,12].
We became interested in the potential role of RME-8 in neuronal health when a Parkinson disease-linked variant of DNAJC13, the only human RME-8 homolog, was identified in a multi-incident Mennonite family [13]. The PD-linked mutation, N855S, alters a residue that is absolutely conserved within organisms that possess a nervous system [14,15]. Patients with this allele displayed late onset PD with Lewy body pathology [13,16]. Other alleles of DNAJC13/RME-8 have also been implicated in PD pathogenesis, and DNAJC13/RME-8 variants have been reported as associated with other neurological disorders, including familial Essential Tremor and Tourette Syndrome [17]. Association of DNAJC13/RME-8 with multiple neurologic diseases and syndromes highlights a crucial and unexplored role for RME-8-family proteins in neuronal health.
RME-8/DNAJC13 proteins are widely conserved DNAJ-domain Hsp70-family co-chaperones that were first discovered in our C. elegans screens for endosomal regulators [15,18–20]. RME-8 contains at least one N-terminal PtdIns3P lipid binding domain prior to the central DNAJ-domain that recruits RME-8 to endosomes [15,21]. On sorting endosomes, RME-8 and its binding partner SNX-1/Snx1 (Sorting Nexin 1) localize to a microdomain functioning in cargo recycling, adjacent to a microdomain functioning in cargo degradation on the same endosome [15,22–24]. The degradative domain is marked by the ESCRT complex component HGRS-1, an HRS homolog, and a flat clathrin lattice [25,26–28]. In the absence of RME-8 and its binding partner SNX-1/Snx1, both HGRS-1 and clathrin overaccumulate on endosomal membranes, causing dysfunction of cargo sorting within endosomes [23,24,29]. Sorting endosomes mature into late endosomes. Like autophagosomes, late endosomes must fuse with lysosomes to degrade their contents.
Here we investigated the requirements for RME-8 in C. elegans mechanosensory neurons in the intact animal, and in primary mouse cortical neurons in culture. Depletion of RME-8/DNAJC13 caused neurons in both systems to accumulate LMP-1/LAMP1- and LGG-1/LC3-positive autolysosomes bearing grossly elongated membrane tubules. In mouse cortical neurons, we also found that depletion of DNAJC13 resulted in gross enlargement of the autolysosomes, and loss of most LC3-negative lysosomes. Further C. elegans analysis revealed a similar autolysosome tubule elongation defect in mutants known to be required for ALR in mammals, including bec-1/BECN1/Beclin1 and vps-15/PIK3R4/p150 that regulate the VPS-34 class III PtdIns3K, and dyn-1/DNM that severs ALR tubules. Furthermore, we found that loss of RME-8 causes severe depletion of clathrin from autolysosomes, a phenotype also shared with snx-1, bec-1 and vps-15 mutants. Taken together, we conclude that RME-8/DNAJC13 plays a conserved but previously unrecognized role in autophagic lysosome reformation, likely affecting ALR tubule severing. Additionally, in both systems, we found that loss of RME-8/DNAJC13 reduced autophagic flux, suggesting feedback regulation from ALR to autophagy. These results connecting RME-8/DNAJC13 to ALR and autophagy provide a potential mechanism by which RME-8/DNAJC13 could influence neuronal health and the progression of neurodegenerative diseases, including Parkinson disease.
Results
Loss of RME-8 function results in accumulation of long lysosomal tubules
Given the association of DNAJC13 with neurological disease [13,17,30–33], we sought to better understand RME-8 function in neurons of C. elegans where RME-8-mediated trafficking mechanisms were first elucidated [15,19,23,24]. Here we focused on the mechanosensory touch neurons, in particular analyzing the two centrally located touch neurons referred to as ALM right and ALM left (Figure 1A). All six touch neurons are embedded in the hypodermis (skin), very close to the cuticle, where they sense gentle mechanical stimuli [34]. The stereotyped location of the ALM neurons close to the exterior of the animal, and their simple architecture with one main process that extends anteriorly to make synapses in the head, make these neurons ideal for quantitative imaging in the living, intact animal. To allow subcellular analysis specifically in these neurons, we expressed a variety of fluorescently tagged proteins from single copy transgenes, driven by the mechanosensory touch neuron-specific mec-7 promoter [35].
Figure 1.
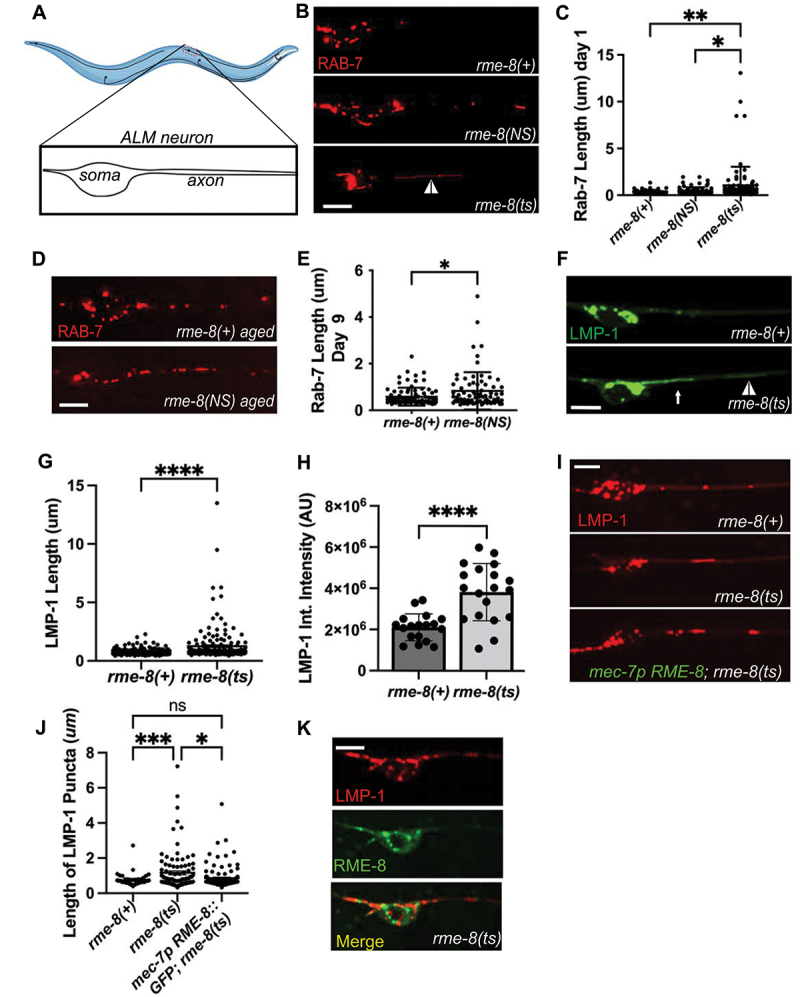
Loss of RME-8 results in accumulation of elongated lysosomal tubules.(A) Representative drawing of ALM neurons in C. elegans. One of two ALM neurons is shown (other is located laterally opposite). Red dashed box represents the approximate area that is shown in example micrographs. (B) fluorescent micrographs showing mec-7p promoter-driven mScarlet::RAB-7 labeled (late endosome/lysosome marker) puncta in soma (left) and proximal axon (right) in ALM neurons in rme-8(+), rme-8(N861S), and rme-8(ts) day 1 adult backgrounds. White arrowhead indicates grossly elongated tubule. (C) length of RAB-7 puncta/tubule in the proximal axon in rme-8(+), rme-8(N861S) and rme-8(ts) is graphed in day 1 adult animals. Each data point represents an individual puncta/tubule. A minimum of 15 animals were analyzed per strain. (D) mec-7 promoter-driven mScarlet::RAB-7 labeled puncta in rme-8(+) and rme-8(N861S) backgrounds in day 9 adult. (E) length of RAB-7 puncta/tubule in the proximal axon in rme-8(+), and rme-8(N861S) is graphed in day 9 adults. Each data point represents an individual puncta/tubule. (F) mec-7p LMP-1::mNeonGreen (NG) labeled (lysosome marker) puncta. A severely elongated LMP-1-positive tubule can be seen emanating into the axon from puncta in the soma (white arrow), and another dimmer tubule can be seen further out in the axon (white arrowhead). (G) quantification of LMP-1::mNG axonal puncta/tubule length seen in conditions shown in (F). Each data point represents an individual puncta/tubule. A minimum of 15 animals were analyzed per strain. (H) graph depicting LMP-1::mNG Integrated intensity (arbitrary Units; AU) for the same experiment as (F) and (G). Each data point represents the integrated intensity of all thresholded puncta in one soma in one animal. (I) micrographs of mec-7p RME-8::GFP rescue of LMP-1::mScarlet length in rme-8(ts) background. (J) graph quantifying elongation of LMP-1::mScarlet in rme-8(+), rme-8(ts), and mec-7p RME-8::GFP; rme-8(ts). (K) Example image showing localization of mec-7p RME-8::GFP and mec-7p LMP-1::mScarlet in rme-8(ts) background. Scale bars: 5 µm. Abbreviations: rme-8(NS)= rme-8(pw22[N861S]); rme-8(ts)= rme-8(b1023ts). (C) and (J): one-way ANOVA followed with Tukey’s multiple comparisons test. (E), (G), (H): Student’s unpaired t-test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
rme-8 is an essential gene, so for most experiments we measured the effects of loss of RME-8 function on neuronal trafficking using the well-characterized temperature sensitive allele, rme-8(b1023ts) [19]. Temperature shifting this strain from the permissive temperature (15°C) to the non-permissive temperature (25°C) is known to destabilize the RME-8 protein, and results in clear phenotypes within 24 h [19]. For most experiments we temperature shifted rme-8(b1023ts) animals at the L4 stage and conducted imaging experiments in young adults 24–30 h later. For some experiments we also analyzed the effects of rme-8(pw22[N861S]), an allele we generated by CRISPR-based genome engineering to mimic the human late onset Parkinson associated allele N855S [13]. rme-8(pw22[N861S]) bears an equivalent point mutation to N855S, changing asparagine 861 to serine (N861S).
Given the known roles of RME-8 on endosomes, and the association of neurodegenerative disease with membrane trafficking pathway dysfunction, we initially analyzed the effects of the rme-8(ts) mutant on late endosomes and lysosomes using a RAB-7 fusion to mScarlet (mSc), expressed in the mechanosensory neurons. Importantly, in young adult (adult day 1) rme-8(ts) mutants we noted the appearance of RAB-7 vesicles bearing highly elongated tubules, a morphology not observed in control animals (Figure 1B, white arrowhead; quantified in 1C). We also assayed for this phenotype in the PD-allele mimic rme-8(NS). Interestingly, we observed a similar accumulation of elongated RAB-7-positive tubules in aged animals in late adulthood (adult day 9), but not in young adults (Figure 1D,E). This mimics the late-onset nature of the homologous PD-linked allele in human patients, suggesting that the N861S allele confers a similar but more subtle defect than the rme-8(ts) loss of function allele. Tubules emanating from endosomes and lysosomes are generally associated with recycling processes, with accumulation of exaggerated tubules often associated with defects in tubule release.
To better define the nature of these vesicles, we extended our analysis using LMP-1/LAMP1 fused to mNeonGreen (mNG), a transmembrane marker of lysosomes (Figure 1F). We noted a very similar phenotype using the lysosome marker in rme-8(ts) mutants, measuring a highly significant increase in LMP-1 tubule length (Figure 1G). We noted that in many cases these long thin tubules appeared connected to a larger vesicle located in the neuronal soma (white arrow, Figure 1F; Movie S1, https://figshare.com/s/f97fd157433ba3a624a5), and in others the vesicle and tubule were located in the soma-proximal region of the neuronal process (white arrowhead, Figure 1F). These persistent tubules were not observed in rme-8(+) backgrounds (Figure 1F, Movie S2, https://figshare.com/s/f97fd157433ba3a624a5). We extended this analysis, measuring the intensity of LMP-1::mNG labeling lysosomes in the soma and proximal axon. Interestingly, LMP-1 total intensity of thresholded puncta (integrated intensity) was increased upon loss of RME-8, indicating a significant abnormal accumulation of LMP-1 labeled lysosomes within neurons upon loss of RME-8 (Figure 1H). These data indicate a key requirement for RME-8 in neuronal lysosome homeostasis.
RME-8 function is touch neuron autonomous
To confirm that the lysosomal tubule phenotype is caused by loss of RME-8, and test if RME-8 is required autonomously within the touch neurons to maintain lysosome homeostasis, we tested for rescue of the neuronal rme-8(ts) phenotype in animals expressing a touch neuron-specific rme-8(+) minigene. Indeed, we observed that expressing wild-type RME-8 in just the six touch neurons rescued touch neuron lysosomal tubule accumulation (Figure 1I–K).
RME-8 affects autolysosomes
Lysosomes receive cargo via membrane fusion, fusing with endosomes, phagosomes, and autophagosomes to form endolysosomes, phagolysosomes, and autolysosomes respectively (reviewed in [36–38]). Neurons in particular display high levels of basal autophagy compared to other cell types [8]. Thus, we sought to determine to what extent the LMP-1 positive structures we observed in the touch neuron somata represent autolysosomes. To achieve this, we created strains expressing the autophagosome marker mNG::LGG-1 along with LMP-1::mSc in touch neurons (Figure 2A). In the somata of ALM neurons, we measured a 75% pixel overlap of mNG:LGG-1 and LMP-1::mSc, with virtually all LMP-1-positive lysosomes containing at least some mNG::LGG-1, indicating that they are autolysosomes (Figure 2B). We only rarely observed mNG::LGG-1 labeled structures lacking LMP-1::mSc (Figure 2A, white arrow), indicating that at steady state most autophagosomes in the soma and soma-proximal region of the axon have already fused with lysosomes.
Figure 2.
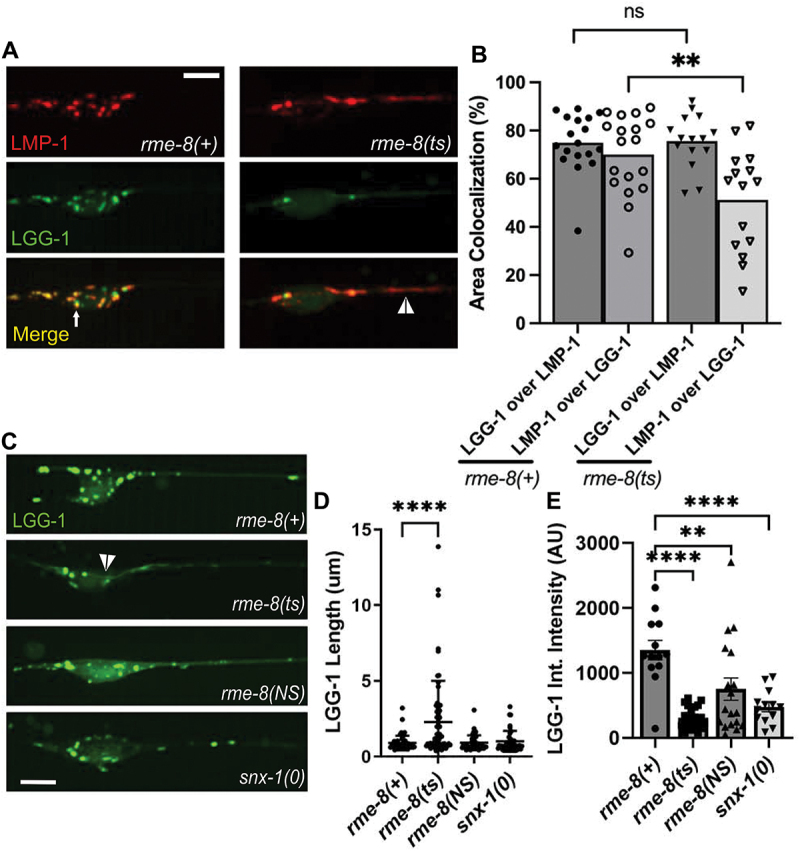
rme-8 mutants accumulate abnormal autolysosomes. (A) micrographs are shown for single channels as well as merged images in double-labeled LMP-1::mScarlet (lysosome marker); mNg::LGG-1 (autophagosome marker) in rme-8(+) and rme-8(ts) backgrounds in ALM somata and proximal axons. White arrow labels mNg::LGG-1+/LMP-1::msc- puncta. White arrowhead labels tubules labeled by LMP-1 and dimly labeled by LGG-1 in the rme-8(ts) background. (B) graphs quantifying the colocalization of LMP-1::mSc and mNg::LGG-1, measured by % area colocalization. Each data point represents average % area colocalization for one animal. *P < 0.01, ns > 0.05 by two-tailed unpaired t-test. (C) micrograph showing mec-7p::mNG::LGG-1 in neurons of rme-8(+), rme-8(ts), rme-8(N861S), and snx-1(0) backgrounds. Scale bar: 5 µm. (D) graph quantifying elongation of mNG::LGG-1 in rme-8(+), rme-8(ts), and rme-8(N861S); snx-1(0) backgrounds. Each data point represents an individual puncta/tubule. (E) graph depicting mNg::LGG-1 integrated intensity (arbitrary Units; AU) for the same experiment as 2C and 2D. Each data point represents the integrated intensity of all thresholded puncta in one soma in one animal. (D), (E): **P < 0.0001, ***P <.01 by one-way ANOVA followed with Tukey’s multiple comparisons test.
We observed the grossly elongated tubules (white arrowhead) in the somata and proximal axons of rme-8(ts) mutants in LMP-1::mSc; mNG::LGG-1 double labeled strains (Figure 2A) and in single labeled mNG::LGG-1 strains (Figure 2C, quantified in 2D). We noted that the overall mNG::LGG-1 signal was weaker in rme-8 mutants, with mNG::LGG-1 fluorescence particularly weak and difficult to detect in the tubules. mNG::LGG-1 fluorescence was much more prominent in vesicular regions of the neuronal autolysosomes (observed in Figure 2A,C). This was reflected in a maintained 76% overlap of LGG-1-positive area that colocalized with LMP-1 in rme-8(ts) mutants, but a reduced percentage of LMP-1-positive area that colocalized with LGG-1 signal in rme-8(ts) mutants (Figure 2B). We also measured a reduced overall integrated (total) intensity for LGG-1::mNG puncta in the neuronal soma in rme-8(ts), rme-8(NS), and snx-1(0) mutants (Figure 2E), although elongated tubules were not observed in rme-8(NS) and snx-1(0) mutants under these conditions (Figure 2D).
We also analyzed colocalization of LGG-1 and LMP-1 in rme-8(+) and rme-8(ts) mutants in ALM distal axons near the synapse-rich nerve ring (Figure S1). We find that in both genetic backgrounds LMP-1 puncta are only very rarely observed in the distal axon, with most LGG-1 puncta lacking LMP-1 signal (Figure S1A, S1C). These results are consistent with neuronal models in which autophagosomes formed in the distal axon must travel back to the soma to fuse with lysosomes and undergo degradation.
Taken together our results indicate that C. elegans touch neurons maintain active autophagy under well-fed conditions, and most lysosomes in the neuronal soma have previously fused with autophagosomes, with loss of RME-8 leading to accumulation of abnormal autolysosome-derived membrane tubules.
Loss of RME-8 phenocopies autophagic lysosome reformation mutants
The strikingly elongated LMP-1-positive tubules emanating from autolysosome puncta were highly reminiscent of autophagic lysosome reformation tubules as reported in mammalian cells [1,2,4,6,39]. ALR tubules will persist and elongate if the mechanism by which the tubules are severed is impaired [2,4,6,39,40]. Our observations that LMP-1/LAMP1 intensity in the autolysosomes increased in rme-8 mutants suggests a failure to recycle LMP-1 out of the autolysosome, further supporting the hypothesis that the elongated tubules accumulating in rme-8 mutant neurons represent an accumulating ALR intermediate (Figure 2).
If the rme-8 mutant phenotype indicates a defect in neuronal ALR, we would expect to find similar phenotypes in C. elegans mutants lacking proteins previously identified as ALR regulators in other systems. In particular, enzymes that regulate lysosomal PtdIns(4,5)P2, PtdIns3P, as well as the coat protein clathrin and the membrane severing GTPase dynamin, have been suggested to function in ALR, in addition to their more well-known roles in other trafficking steps [2,3,6]. For instance, mammalian cells inhibited for class III phosphoinositide 3-kinase VPS34 activity display lysosomal tubule accumulation during ALR [6]. To impair the VPS-34 complex in C. elegans, we used mutants in bec-1/beclin and vps-15/PIK3R4/p150, key regulators of VPS-34 activity [41–43]. We observed severely elongated LMP-1::mNG tubules in bec-1 and vps-15 mutants that strongly resemble those we found in rme-8 mutants (Figure 3A, white arrowheads, quantified in 3B). We also examined the tubule accumulation phenotype in neurons impaired for dynamin [44]. Previous work showed that mammalian dynamin 2 binds to autolysosome tubules along their length, and pharmacologic inhibition of dynamin 2 in hepatocytes produced enlarged autolysosomes bearing extremely long, thin LAMP1-positive tubules [3]. Importantly, we also found that dyn-1(ts) mutants displayed dramatic tubulation of LMP-1-positive autolysosomes in the C. elegans touch neurons (Figure 3C).
Figure 3.
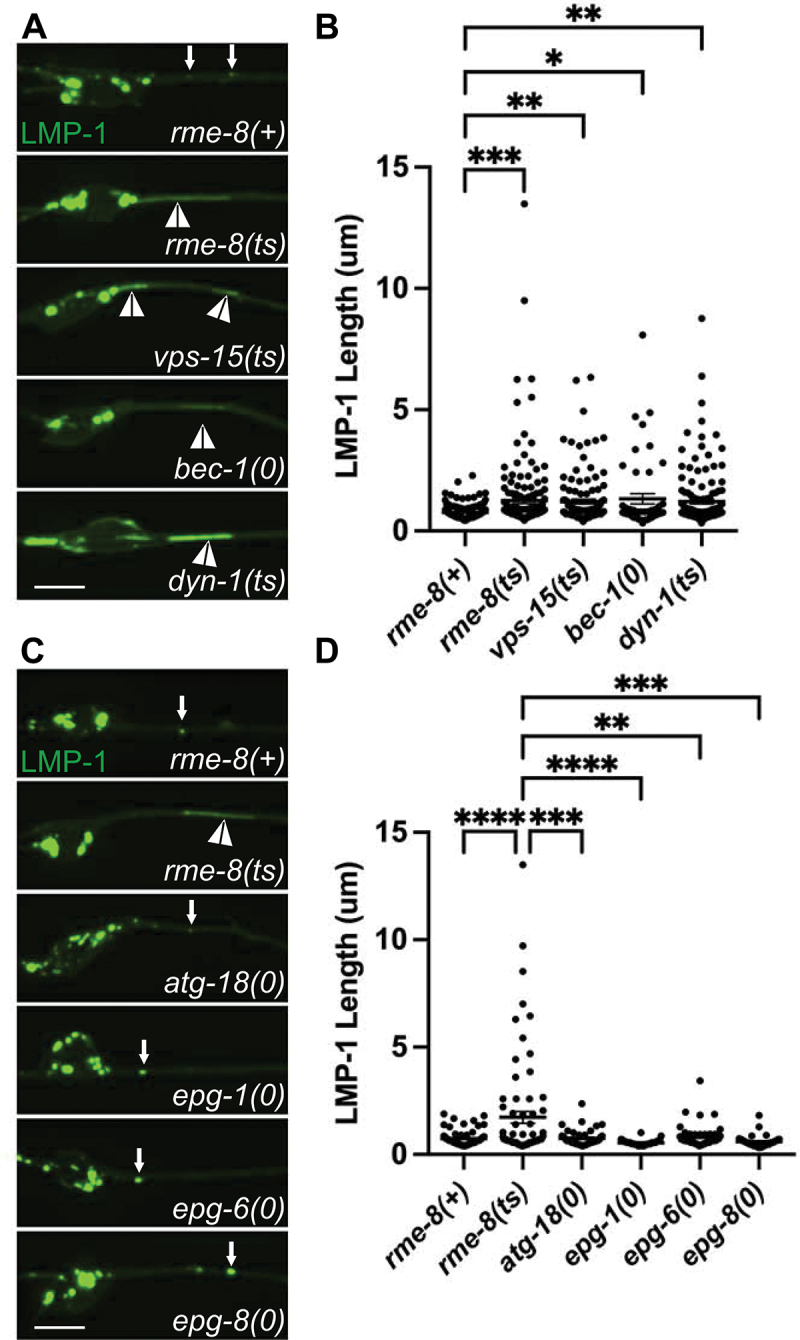
Loss of RME-8 phenocopies autophagic lysosome reformation mutants. (A) micrographs are shown of mec-7p LMP-1::mNG in ALM neurons in rme-8(+), rme-8(ts), vps-15(ts), bec-1(0), and dyn-1(ts) backgrounds. (B) quantification of LMP-1 tubule length (um) in ALM proximal axons in the genetic backgrounds shown in A). Each data point represents an individual puncta/tubule. (C) micrographs are shown of mec-7p LMP-1::mNG in ALM neurons in rme-8(+), rme-8(ts), atg-18(0), epg-1(0), epg-6(0), epg-8(0), backgrounds. Example images and graphs are separate from A) and B) because data was obtained during separate experiments. (D) quantification of LMP-1 tubule length (µm) in ALM proximal axons in the genetic backgrounds shown in (C). Each data point represents an individual puncta/tubule. (B), (D): *P < 0.001, **P < 0.01, ***P < 0.05, ns > 0.05 by one-way ANOVA followed with Tukey’s multiple comparisons test.
Given our results with PtdIns3P generation mutants (vps-15, bec-1), we sought to determine if binding of RME-8 to PtdIns3P was important for ALR. To test this, we assayed the functionality of an RME-8 W20A mutant previously published as PtdIns3P-binding defective [45]. Not only did RME-8 W20A not rescue the rme-8(ts) LMP-1 tubule accumulation phenotype (Figure S2A, S2B), RME-8 W20A exacerbated the phenotype. RME-8 W20A may exacerbate the rme-8(ts) phenotype by producing nonfunctional complexes with RME-8 binding partners such as SNX-1 and HSP-1/Hsc70 [21], or by interfering with any residual activity of RME-8(ts) protein in the cells. Taken together our results indicate phylogenetic conservation of function in autolysosome tubule severing mechanisms from nematode to human and suggest that RME-8 is important for autolysosome tubule release.
Mammalian DNAJC13/RME-8 is also required for neuronal ALR
We also sought to determine if DNAJC13/RME-8 is required for ALR in mammalian neurons. To test this, we analyzed primary cortical neurons derived from the embryonic mouse brain, visualizing autolysosomes with GFP-LAMP1 and mRFP-LC3, knocking down mouse DNAJC13 using shRNAs. Knockdown efficiency of DNAJC13 is shown in Figure S3. Importantly, in neurons transfected with Dnajc13 shRNA we observed strikingly elongated LAMP1-labeled tubules emanating from vesicles positive for mRFP-LC3, identifying them as autolysosomes (Figure 4A). These tubules are strongly labeled for LAMP1 and weakly labeled for LC3 (see insets in Panel A). Under these conditions LAMP1-positive tubule length increased from a mean of <4 µm in control shRNA neurons, to ~18 µm in Dnajc13 shRNA neurons (Figure 4B). Elongated LAMP1-positive tubules in DNAJC13 knockdown conditions can be seen in Movie S3 (https://figshare.com/s/f97fd157433ba3a624a5), and compared to the dynamics of lysosomes in neurons expressing control shRNA (Movie S4, https://figshare.com/s/f97fd157433ba3a624a5). The proportion of long tubules was much greater upon knockdown of DNAJC13, with 39% of tubules having a length of greater than 20 µm upon loss of DNAJC13, while 0% of tubules in control cells were longer than 20 µm (Figure 4C).
Figure 4.
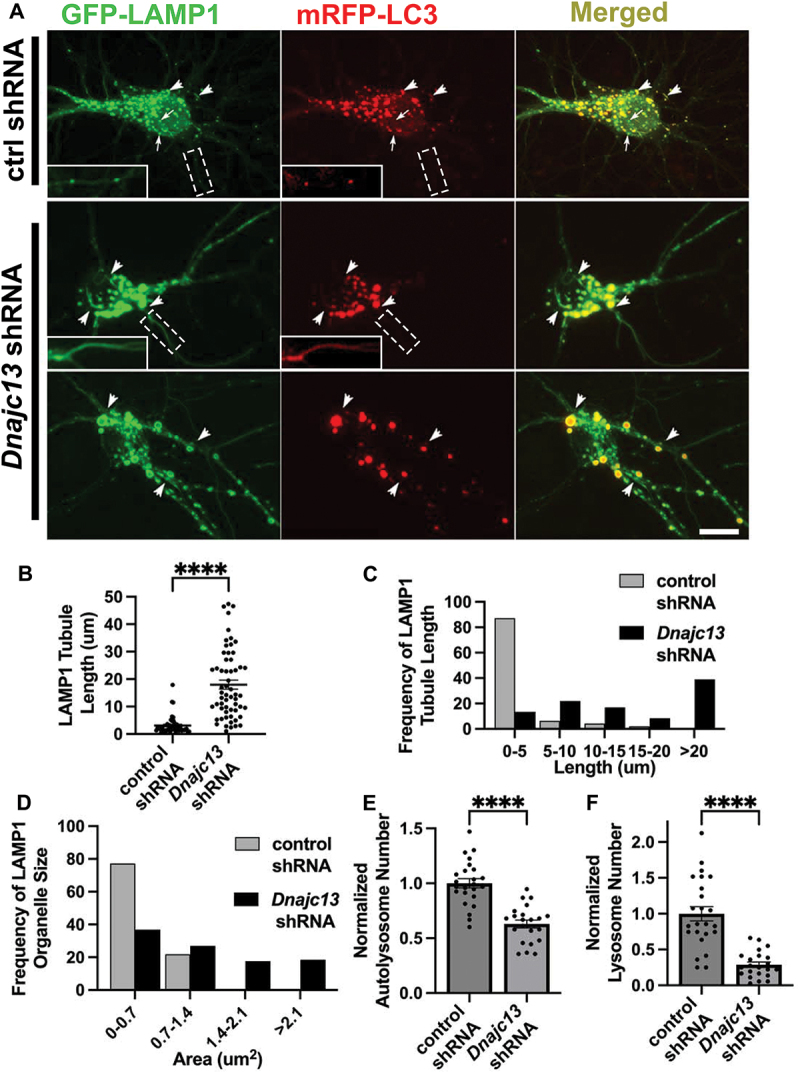
DNAJC13/RME-8 knockdown in mouse cortical neurons causes elongated autolysosomal tubules and enlarged autolysosomes. (A) micrographs of double-labeled GFP-LAMP1, mRFP-LC3 in primary mouse cortical neurons transfected with control (top panel) or Dnajc13 (bottom two panels) shRNA. Images are shown for single channel labeling and merged. Arrows (top panel, control shRNA) indicate lysosomes (GFP-LAMP1 positive; mRFP-LC3 negative), arrowheads indicate autolysosomes (GFP-LAMP1 positive; mRFP-LC3 positive). Insets in white boxes depict magnified portions of axonal processes (boxes with dotted white lines) scaled to better show presence of axonal puncta or tubules in both channels. Scale bars: 10 µm. (B) the average LAMP1 tubules length of each soma is quantified in control and Dnajc13 shRNA conditions. Each puncta represents the average length in one neuron. (C) frequency distribution of average tubule length per soma in control and Dnajc13 shRNA. (D) frequency distribution of LAMP1 positive (lysosomes and autolysosomes) vesicle area per soma in control and Dnajc13 shRNA. (E) autolysosome number in soma, defined as LAMP1 positive, LC3 positive puncta, normalized to control shRNA conditions, in control shRNA conditions and Dnajc13 shRNA conditions. (F) lysosome number in soma, defined as LAMP1 positive, LC3 negative puncta, normalized to control shRNA conditions, in control shRNA conditions and Dnajc13 shRNA conditions. (B), (E), (F) Student’s unpaired t-test. ****P < 0.0001.
We also noted that the autolysosomes in DNAJC13 knockdown neurons were greatly enlarged, in many cases displaying areas > 3 times greater than in controls (Figure 4D), and were fewer in number (Figure 4E). Additional examples of enlarged lysosomes and elongated tubules labeled with GFP-LAMP1 are shown in Figure S4. Our results are similar to previously described ALR defects found upon loss of known ALR regulators, conditions under which enlarged LAMP1-positive autolysosomes form due to an inability to recycle material out of the lysosomes after fusion with autophagosomes [1–4]. We also observed a significantly reduced number of LAMP1-positive lysosomes that lack LC3 in DNAJC13 knockdown neurons (Figure 4F), further indicating a defect in lysosome reformation from autolysosomes. Taken together our results indicate a clear requirement for DNAJC13/RME-8 in completion of ALR in C. elegans and mammalian neurons.
RME-8 localization in neurons
RME-8 in neurons could function in ALR directly via a role on autolysosomes, or it could affect ALR indirectly, for instance through endosomes which also fuse with lysosomes, potentially providing regulatory molecules in addition to degradative cargo. To assay RME-8 subcellular localization in C. elegans we expressed a rescuing touch-neuron-specific RME-8::GFP transgene. We observed a high degree of colocalization of RME-8::GFP and endosome marker mSc::SNX-1 in the neuronal soma, similar to our previous results in other cell types (Figure S5A-B). At steady state we also observed a significant degree of RME-8::GFP overlap with LMP-1::mSc-positive lysosomes in the C. elegans touch neurons (Figure 5A,D). In dyn-1 but not vps-15 mutants, we observed that the percentage of RME-8 signal that overlapped with LMP-1-positive lysosomes increased (RME-8 over LMP-1), consistent with RME-8 residing on LMP-1 lysosomes more frequently or for a longer duration when dynamin-mediated severing is impaired, supporting a potential direct role for RME-8 on autolysosomes (Figure 5A,D). This increased overlap of RME-8 with LMP-1-labeled lysosomes in dyn-1 mutants occurred despite an overall reduction in RME-8-positive area (Figure 5B) and increase in LMP-1-positive area in dyn-1 mutants (Figure 5C). Interestingly, LMP-1-positive lysosome area also increases in vps-15 mutants (Figure 5B), but the percentage of RME-8 signal that overlaps with LMP-1-positive lysosomes in vps-15 mutants decreases (Figure 5A,D). In summary, our results are consistent with a potential direct function of RME-8 on autolysosomes during ALR. If so, RME-8 is likely to be transiently associated with a small region of the autolysosome, as has been proposed for proteins like dynamin that participate in autolysosome tubule fission [3].
Figure 5.
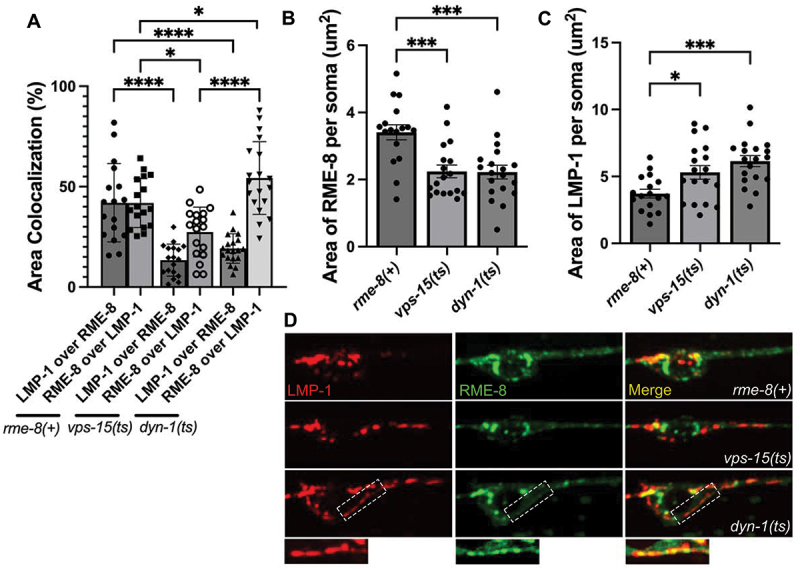
Colocalization of RME-8 with lysosomes. (A) quantification of % area colocalization of double labeled strains LMP-1::mScarlet; RME-8::GFP in rme-8(+), vps-15(ts), and dyn-1(ts) backgrounds. (B) quantification of thresholded area of RME-8 in the somas of rme-8(+), vps-15(ts), and dyn-1(ts) strains. (C) quantification of thresholded area of LMP-1 in the somas of rme-8(+), vps-15(ts), and dyn-1(ts) strains. (D) fluorescent single channel and merged micrographs of double labeled strains quantified in (A). Scale bars: 5 µm. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by one-way ANOVA followed with Tukey’s multiple comparisons test.
Reduced neuronal autophagy in C. elegans rme-8 mutants
Given the apparent reduced intensity of mNG::LGG-1 we observed in rme-8 mutants, we asked if known ALR-relevant proteins are important for maintaining autophagic flux in C. elegans neurons. We first investigated this by comparing mNG::LGG-1 levels in touch neuron somata of rme-8, bec-1, vps-15, and dyn-1 mutants (Figure 6A). Beyond ALR, VPS-15 and BEC-1 also impinge upon PtdIns3P generation at the omegasome, a pre-autophagosomal structure, but dynamin has not been linked to any direct role in autophagosome formation [46,47]. We observed a similar decrease in both the integrated and average intensity of mNG::LGG-1 puncta in rme-8, dyn-1/DNM, bec-1/beclin, and vps-15 mutants (Figure 6B,C). These results are consistent with recent studies linking impaired ALR to decreased autophagy initiation [39].
Figure 6.
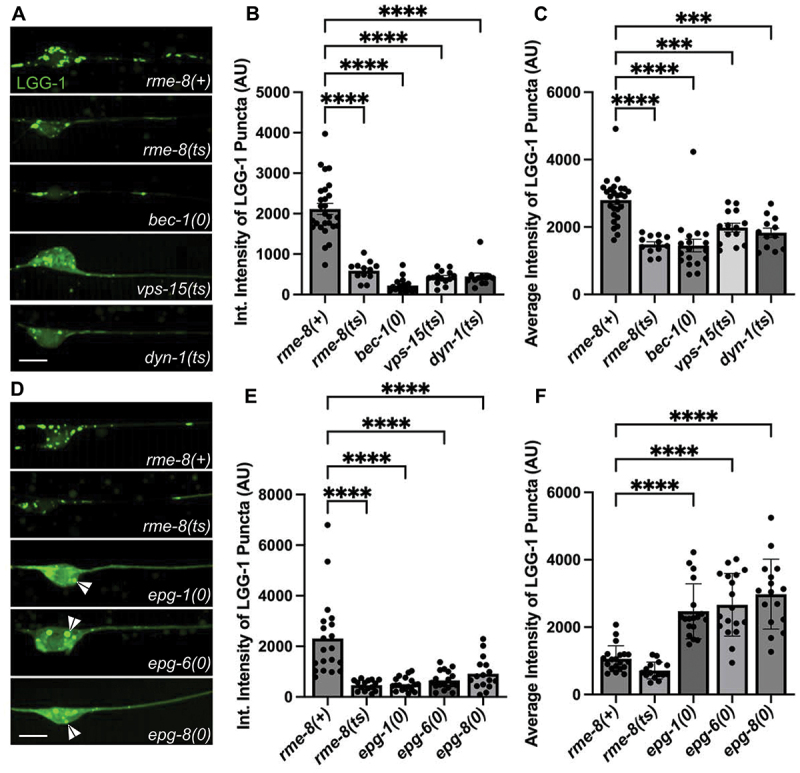
Loss of RME-8 causes decreased autophagosome levels similar to known ALR mutants. (A), (D) micrographs of mec-7p LGG-1::mNG in C. elegans ALM neurons. Area shown includes soma (left) and proximal axon (right). Example images and graphs are separate due to data being obtained during separate experiments. Scale bars: 5 µm. (B), (E) quantification of integrated intensity of mNG::LGG-1 puncta in backgrounds shown in (A) and (D) (AU; arbitrary units). (C), (F) average intensity of LGG-1 positive puncta in axons and somata of backgrounds shown in (A) and (D). *P < 0.001, **P < 0.0001 by one-way ANOVA followed with Tukey’s multiple comparisons test.
We also examined mNG::LGG-1 levels and distribution in epg-1/atg13-like [48], epg-6/atg18-like [49], and epg-8 mutants [50]. These proteins have clearly defined roles within the canonical autophagic initiation pathway, but without known effects on ALR. We found that epg-1, epg-6, and epg-8 mutants displayed a strongly diffusive mNG:LGG-1 signal in the touch neuron soma, with two or three apparent large aggregates near the nucleus (Figure 6D; white arrowheads). While epg-1, epg-6, and epg-8 all have decreased integrated (total) intensity of mNG::LGG-1 puncta (Figure 6E), similar to ALR mutants, the average intensity of the puncta is significantly higher than wild-type puncta (Figure 6F), in stark contrast to the decreased average intensity of ALR mutants (Figure 6C). This aggregation phenotype has been previously observed in cells expressing tagged LGG-1 in which autophagy initiation is strongly blocked, as expected in these mutants, resulting in overexpressed LGG-1 accumulating in an unprocessed form in aggregates [48,51,52]. This is quite different from the reduced levels of mNG::LGG-1 fluorescence we observed in rme-8, dyn-1, bec-1, and vps-15 mutants, where dimmer mNG::LGG-1 is found in more numerous small puncta within the touch neuron soma (Figure 6A). Taken together, we interpret the rme-8 mutant phenotype as reduced but not blocked neuronal autophagy, potentially as an indirect consequence of defective ALR.
Blocking autophagy initiation does not cause accumulation of lysosomal tubules
Another possibility is that defective autophagy initiation is sufficient to produce an RME-8-like lysosome phenotype with accumulation of long lysosomal tubules. However, when we measured LMP-1::mNG tubule length in epg-1(0), epg-6(0), epg-8(0) and atg-18(0) mutants in the touch neurons, we did not find any significant elongation of LMP-1 tubules (Figure 3C–D). These results suggest that a simple block in autophagy initiation is not sufficient to produce this phenotype.
Neuronal autophagic flux requires DNAJC13/RME-8 in mammalian neurons
Given our results in C. elegans, we examined the effects of DNAJC13 knockdown on autophagic flux in primary mouse cortical neurons. Similar to our observations in C. elegans neurons, we found that autophagic vacuole (AV) density per soma, as visualized by GFP-LC3, was decreased in Dnajc13 shRNA neurons compared to controls (Figure 7A, quantified in 7B). We also measured AV density in the presence of trehalose, an autophagy inducer [53,54], as well as trehalose and lysosomal protease inhibitors pepstatin A and E64d [55,56]. Lysosomal protease inhibitors block the autolysosome-mediated degradation of autophagic cargo, including GFP-LC3. This type of analysis is often used to assess levels of autophagic flux, since reduced GFP-LC3 levels can result from reduced autophagy, or increased lysosomal degradation of autophagic cargo. If autophagic flux is reduced, then GFP-LC3 levels should also be reduced, even when lysosomal degradation is blocked. However, if GFP-LC3 levels are reduced due to increased lysosomal activity, blocking such lysosomal activity should block the observed GFP-LC3 reduction. We found GFP-LC3 AV density was still decreased in RME-8 shRNA neurons under basal autophagy conditions, as well as when treated with trehalose, and trehalose and lysosomal protease inhibitors together (Figure 7A,B). There was a consistent ~ 2.5-fold reduction in AV-density under all conditions in which DNAJC13 was depleted. We interpret these results to indicate reduced autophagic flux in DNAJC13-deficient mammalian neurons, potentially at an early stage in autophagy initiation or autophagosome formation.
Figure 7.
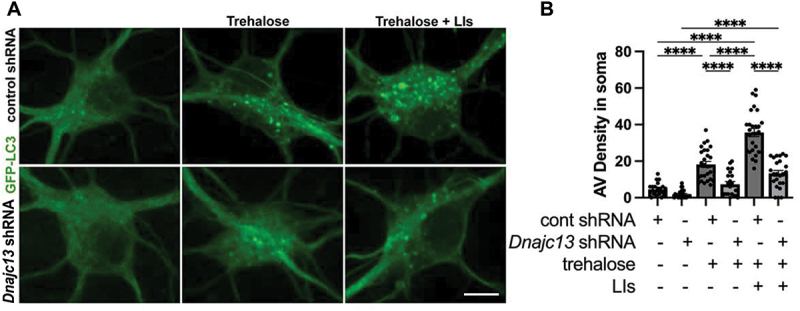
DNAJC13 knockdown in mouse cortical neurons reduces autophagic flux. (A) micrographs are shown of in vitro mouse cortical neurons transfected with GFP-LC3. A 2 × 3 factorial design was used. Conditions shown: basal autophagy, trehalose treatment, and trehalose + lysosome inhibitors (LIs) (pepstatin A and E64d) when cells were treated with either control shRNA or Dnajc13 shRNA. Scale bars: 10 µm. (B) autophagic vacuole (AV) density is quantified in the soma of neurons for the 2 × 3 factorial design shown in (A). ****P < 0.0001 by one-way ANOVA followed with Tukey’s multiple comparisons test.
RME-8, BEC-1/BECN1/beclin, and VPS-15 are required for efficient clathrin recruitment to autolysosomes
One key molecule associated with the biogenesis of autophagic lysosome reformation tubules is clathrin [2,4,39]. Previously, we and other groups had identified an important role for RME-8 in controlling clathrin levels on endosomes in non-neuronal cells [23,45]. Thus, we hypothesized that RME-8 activity might be important for controlling lysosomal clathrin levels relevant to ALR in neurons. To test this idea, we established touch neuron-specific lines expressing clathrin light chain tagged with mNeonGreen (CLIC-1::mNG). We found that CLIC-1::mNG was clearly enriched on LMP-1::mSc labeled lysosomes, supporting a conserved role for clathrin in lysosome homeostasis (Figure 8A,B). Furthermore, we found that bec-1 and vps-15, mutants that accumulate long autolysosomal tubules like rme-8(ts), displayed significantly less recruitment of CLIC-1::mNG to neuronal LMP-1-positive lysosomes than controls. Importantly, we found that rme-8(ts) mutants display an even more severe deficit in CLIC-1::mNG recruitment to LMP-1-positive lysosomes than bec-1 or vps-15 mutants (Figure 8A,B). Notably, the severely elongated LMP-1::mSc tubules in rme-8(ts) mutants completely lack CLIC-1::mNG labeling. We also identified a defect in clathrin recruitment to neuronal lysosomes in snx-1 mutants (Figure 8A,B). These mutants are missing RME-8 partner protein SNX-1 that promotes RME-8 activity [15,24]. BEC-1 and VPS-15 may also be required for efficient RME-8 function, since RME-8 has been reported to have an N-terminal PtdIns3P binding domain [21,45]. Interestingly, dyn-1 mutants do not show a defect in clathrin recruitment onto lysosomes, while dyn-1; rme-8(ts) double mutants show defective clathrin recruitment equivalent to rme-8(ts) single mutants (Figure S6). This supports the hypothesis that the role of DYN-1/dynamin in ALR occurs downstream of RME-8-mediated clathrin recruitment, likely at the severing step. vps-15; rme-8(ts) double mutants display more intermediate levels of impaired clathrin recruitment (Figure S6). Overall, we find that RME-8, SNX-1, BEC-1, and VPS-15, but not DYN-1 are required for efficient recruitment of clathrin to neuronal lysosomes. We also found significant overlap of intracellular clathrin with RME-8, consistent with a direct role for RME-8 in this regulation (Figure 8C,D). Since clathrin has been reported to be a key molecule in ALR tubule biogenesis, reduced levels of clathrin on lysosomes may lead to defective ALR.
Figure 8.
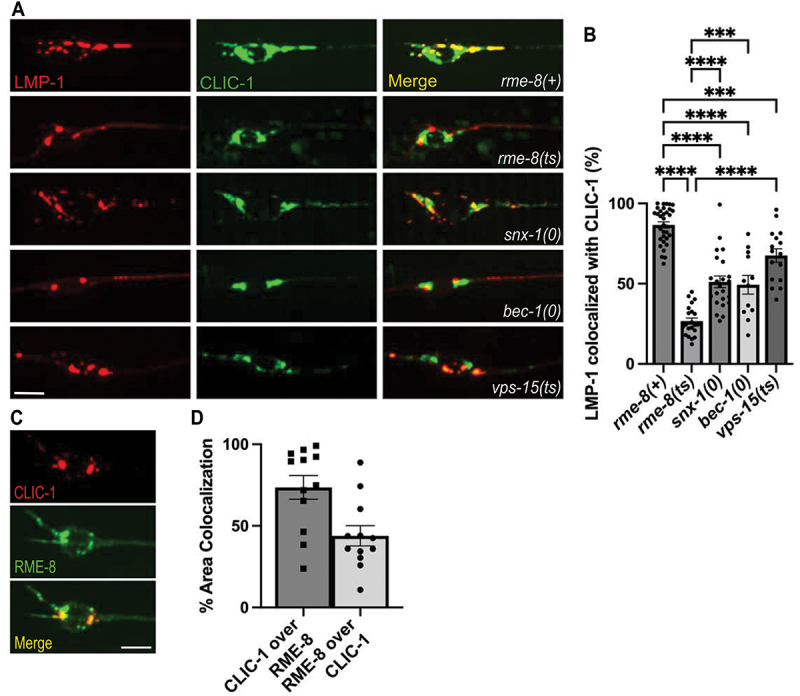
RME-8 is required for efficient clathrin recruitment to lysosomes. (a) single-channel and merge images of double-labeled LMP-1::mSc and CLIC-1::mNG in C. elegans’ ALM neurons. (b) quantification of the percent of LMP-1::mSc thresholded area that has CLIC-1::mNG present on it. One-way ANOVA followed with Tukey’s multiple comparisons test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (c) fluorescent single channel and merged micrographs of double labeled strains CLIC-1::mScarlet; RME-8::GFP wild-type background. (d) quantification of colocalization between strains shown in (C) using % area colocalization. Scale bars: 5 µm.
Discussion
Autophagic engulfment of cytoplasmic content represents a major pathway required for maintaining cellular proteostasis and organelle integrity, as well as survival under nutrient deprivation conditions. Autophagy is likely to be especially important in neurons, which tend to be extremely long-lived and cannot divide, and thus lack an effective mechanism to dilute protein aggregates and defective organelles as rapidly dividing cells can. Consistent with this idea, neurons maintain a relatively high basal rate of autophagy, and autophagic dysfunction has been repeatedly implicated in neurodegenerative disease (reviewed in [7,9,38,57,58]).
Once autophagosomes engulf their cargo they must fuse with lysosomes, producing autolysosomes that degrade their contents. Maintaining a pool of active lysosomes to receive autophagic cargo requires efficient recovery of membrane and other machinery from autolysosomes to reform fresh lysosomes. Defective autophagic lysosome reformation is emerging as an important disease mechanism in its own right, associated with inflammatory and liver disease, muscular dystrophy, metabolic syndrome, Charcot-Marie-Tooth disease, lysosome storage disease, and neurodegenerative disorders such as hereditary spastic paraplegia and Parkinson disease.
Given the genetic connections suggesting linkage of human DNAJC13/RME-8 to Parkinson disease and essential tremor, we sought to analyze the requirement for RME-8 and DNAJC13 in C. elegans and mouse neurons. Our experiments identified an important and conserved role of RME-8/DNAJC13 in ALR in both systems. Loss of RME-8 or DNAJC13 produced enlarged autolysosomes bearing persistently elongated membrane tubules indicative of a defect in ALR tubule fission to release protolysosomes. In the case of DNAJC13-depleted mouse cortical neurons we could document a loss of LC3-negative lysosomes, further indicating a failure in lysosome reformation. We also noted in C. elegans bearing an endogenous rme-8(N861S) allele, equivalent to human PD-associated allele N855S, an age-dependent effect on RAB-7-positive tubule accumulation, suggesting that disease effects could derive from weak dysfunction in ALR.
Extended analysis in the C. elegans system revealed a striking loss of clathrin from neuronal autolysosomes, suggesting that clathrin malfunction may underlie the ALR defects when RME-8 is missing. Interestingly we also observed reduced lysosome-associated clathrin in mutants lacking RME-8 partner SNX-1, as well as mutants lacking PtdIns3K complex proteins VPS-15/P150 and BEC-1/BECN1/Beclin1. While PtdIns3P generated by the PtdIns3K complex, which includes proteins VPS-15 and BEC-1, was previously linked to ALR [6], an effect on clathrin was unexpected, since autolysosomal clathrin is thought to assemble with the AP2 clathrin adapter on PtdIns(4,5)P2 enriched membranes. However, an N-terminal PH-like domain in mammalian DNAJC13/RME-8 binds to PtdIns3P, and C. elegans RME-8 and mammalian DNAJC13/RME-8 both affect clathrin dynamics [21,23,24]. The effects of RME-8 and DNAJC13 are likely due to their function as co-chaperones for members of the HSPA8/Hsc70 family that control clathrin dynamics, so RME-8 could be a key effector of PtdIns3P during ALR [21,23,24]. This possibility is supported by our observation that a PtdIns3P-binding defective form of RME-8 fails to rescue neuronal ALR-associated rme-8 mutant defects, and previous work that indicated that bec-1 mutants affected RME-8 recruitment to membranes in the C. elegans intestine [43]. A complex containing ZFYVE26, SPG11, and AP-5 has been proposed as a PtdIns3P effector during ALR, but C. elegans lacks homologs for any of these proteins, so they cannot explain the conserved requirement from C. elegans to mammals for PtdIns3P during ALR [11,59].
Elegant experiments by Rong et al. suggested dual roles for clathrin in ALR [2]. These authors found that when they depleted PtdIns(4,5)P2 and clathrin from the vesicular body of the autolysosome, tubule budding failed. Alternatively, when PtdIns(4,5)P2 and clathrin were depleted from the tubule, tubule fission failed, blocking protolysosome release. Other work also implicates clathrin regulation in ALR. For instance, abnormally high levels of PtdIns(4,5)P2 on autolysosomes were found to result in increased clathrin on elongated ALR tubules, suggesting that general dysregulation of clathrin on tubules may be sufficient to inhibit tubule release [39].
Neuronal autophagy is compartmentalized, with high levels of autophagosome formation in synaptic regions, followed by long range transport along the axon to the neuronal soma required for fusion with lysosomes. We observed most RME-8 protein in the neurons associated with the soma where lysosomes are abundant. While we did not observe high-level constitutive residence of RME-8 protein on lysosomes, RME-8 colocalization with lysosomes increased in dyn-1/DNM mutants that accumulate ALR-tubule containing intermediates. Under normal circumstances RME-8 could be present quite transiently, like some proteins that act in late stages of vesicle budding. If so, RME-8 could play a direct role in clathrin dynamics regulating ALR. While DNAJ-domain co-chaperone AUXN (auxilin) is best known for uncoating clathrin from clathrin-coated vesicles, in vitro AUXN promotes clathrin assembly, and has also been proposed to provide co-chaperone activity that helps rearrange clathrin coats from flat to curved during budding [60,61]. RME-8 could act similarly on autolysosomes. RME-8 could also play an indirect role via global clathrin metabolism, as our previous work identified a requirement for RME-8 in uncoating clathrin on endosomes, such that loss of RME-8 activity could reduce soluble, cytosolic clathrin levels in the cell needed for assembly on autolysosomes. More analysis will be required to distinguish between these possibilities.
While it is not clear how clathrin contributes to tubule release, clathrin/AP2 assembly likely acts as a precursor to the recruitment of dynamin, the pinchase thought to directly sever ALR tubules, as it does during clathrin-mediated endocytosis [62]. Effects on dynamin may also underlie some of the effects on ALR that we observed in mutants defective in PtdIns3-kinase regulators BEC-1/beclin and VPS-15/P150. Munson et al. found that when a lysosomal pool of PtdIns3P is reduced by inhibition of the Ptdins3K complex, long ALR tubules accumulate [6,63]. PtdIns(3,5)P2 produced from PtdIns3P on late endosomes by the PIKFYVE complex is also important for neuronal lysosome function and may contribute to ALR [64,65]. It is unclear to what extent dynamin requires PtdIns3P and/or PtdIns(3,5)P2 for its function, but in vitro the dynamin PH-domain binds multiple phosphoinositides, including PtdIns3P, PtdIns(3,5)P2, and PtdIns(4,5)P2 [66]. Furthermore, in vitro PtdIns3P and PtdIns(3,5)P2 show high efficiency in stimulating assembly-dependent GTPase activity by dynamin in complex with its partner SNX9, which also displays broad phosphoinositide binding [67]. Phagocytosis studies have expanded upon this idea in vivo, indicating a need for precise and sequential regulation of PtdIns(4,5)P2 and PtdIns3P in phagosome release from the plasma membrane mediated by dynamin and SNX9/LST-4 [68–70]. Similar regulation may be required on ALR tubules for dynamin-mediated protolysosome release.
In addition to effects on ALR, we also noted an apparent defect in autophagy, with strongly reduced levels of LGG-1/LC3 in the neuronal soma of rme-8, bec-1/BECN1/Beclin1, vps-15/P150, and dyn-1/DNM mutants. This was particularly surprising in the case of DYN-1, since dynamin has not been proposed to have any role in autophagosome formation. Our data argue against the ALR tubule accumulation defects we observed for these mutants being an indirect effect of reduced autophagic flux, as we did not observe tubule accumulation in mutants that affect autophagy initiation but that have no known role in ALR. Results vary in the literature as to whether ALR is required to maintain autophagosome formation rates, but our data supports such a requirement. It seems likely that ALR is required to recycle some component(s) or regulator(s) of autophagy from autolysosomes, and that extended failure in ALR leads to reduced rates of autophagy. An interesting candidate for a recycling cargo during ALR is Atg9, the only transmembrane regulator of autophagy initiation. Atg9 was recently shown to be recycled from autolysosomes by the recycler complex, and RME-8 has also been implicated in Atg9 trafficking [71,72]. It will be of great interest to further analyze the mechanism of RME-8 function in ALR, and to gain further insight into mechanisms by which ALR could influence autophagic flux.
Materials and methods
Strains
All C. elegans strains were derived originally from the wild-type Bristol strain N2. Worm cultures, genetic crosses, and other C. elegans husbandry were performed according to standard methods [73]. Since the experiments utilized temperature sensitive strains, all strains were maintained at 15°C. A complete list of strains used in this study can be found in Table S1. Every strain listed was created in the Grant Lab for use in this study.
Construction of C. elegans plasmids
C. elegans expression plasmids utilized the mec-7 promoter (mec-7p) from the mec-7 gene for touch neuron expression [35]. Vector details are available upon request. Cloning was performed using the Gateway in vitro recombination system (Invitrogen, Carlsbad, CA) using Grant lab-modified versions of MiniMos enabled vectors pCFJ1662 (Hygromycin resistant; Addgene 51482) and pCFJ910 (G418 resistant; Addgene 44,481; gifts of Erik Jorgensen, University of Utah): pCFJ1662 mec-7p GTWY mNeonGreen let858 (34F6); pCFJ1662 mec-7p mNeonGreen GTWY let858 (34D4); pCFJ1662 mec-7p GTWY oxGFP let858 (36G3); pCFJ910 mec-7p mScarleti GTWY let858 (33B6); and pCFJ910 mec-7p GTWY mScarleti (35D2). pDONR221 entry vectors containing coding regions for rab-7, lmp-1, lgg-1, clic-1, snx-1, rme-8, were recombined into neuronal destination vectors by Gateway LR clonase II reaction to generate C-/N- terminal fusions. Single-copy integrations were obtained by MiniMOS technology [74]. The RME-8 minigene contains rme-8 genomic DNA including the first 20 exons and introns, with rme-8 cDNA substituted at the 3-prime end, removing the last two introns. This minigene was first used in Shi 2009 [23].
Image acquisition and analysis in C. elegans
Prior to imaging, L4 hermaphrodites maintained at 15°C were picked to fresh OP50 plates and plates were temperature shifted to 25°C (nonpermissive temperature for rme-8[b1023ts]) for 24–30 h prior to imaging. Live animals were picked into 2 µL of 10 mM levamisole on the center of a cover slip. After 2 min, 5% agarose pads were placed on top. Fluorescence images were obtained using a spinning-disk confocal imaging system: Zeiss Axiovert Z1 microscope equipped with X-Light V2 Spinning Disk Confocal Unit (CrestOptics), 7-line LDI Laser Launch (89 North), Prime 95B Scientific CMOS camera (Photometrics) and oil-immersion objective (100X). Fluorescence images were captured using Metamorph 7.10 software. Z series of 25–30 optical sections were acquired using a 0.2-μm step size in order to image through the entire soma. Exposure times ranged from 50 ms to 300 ms based on the fluorescent protein being imaged such that images acquired were at minimum in the 12-bit and did not become saturated in the 16-bit range in any genetic background.
Intensity and length measurements
All data quantification was done using Metamorph 7.10 and data was recorded into Excel. Maximum projections were generated from Z series captured under identical acquisition parameters for each experiment. For each experiment, maximum projections were scaled identically prior to thresholding or length analysis. Puncta within the soma, dendrite, and proximal axon (within ~30 µm of the soma) were manually thresholded and the average, maximum, and integrated intensity measurements recorded using Metamorph’s “Region Measurements” tool. Each worm was treated as one data point.
Maximum projections were also used for length measurements. Lengths were measured and recorded using Metamorph’s “region measurements” and using the line tool to trace the length of every puncta/tubule in the axon. The length of every visible puncta/tubule within each axon was measured and counted as a data point. Analysis of tubule elongation was restricted to axons due to the poor spatial resolution of puncta within the soma. For each experiment and genetic background, 15–25 animals were analyzed.
Colocalization analysis was performed using MetaMorph 7.7 colocalization plugin, whereby intensities in each channel were thresholded and analyzed.
Mouse cortical neuron plasmids and reagents
DNAJC13 shRNA plasmid (sc-77968-SH) and Control shRNA Plasmid-A(sc-108060) were from Santa Cruz Biotechnology. GFP-LC3B, GFP-LAMP1, and mRFP-LC3 constructs were prepared as previously described [54,75–80]. E64d (330005), and pepstatin A (516481) were from Sigma.
Transfection of cultured cortical neurons
Cortices were dissected from E18–19 mouse embryos as described [81–83]. Cortical neurons were dissociated with papain (Worthington, LK003178) and plated at a density of 100,000 cells per cm2 on polyornithine- and fibronectin-coated coverslips. Neurons were grown overnight in a plating medium (5% FBS, insulin, glutamate, G5, and B27) supplemented with 100× L-glutamine in Neurobasal medium (Invitrogen 21103049). Starting at DIV2, cultures were maintained in a conditioned medium with half-feed changes of neuronal feed (B27 in Neurobasal medium) every 3 days. Neurons were co-transfected with Control shRNA (sc-108060) or DnaJC13 (RME-8) shRNA (Santa Cruz Biotechnology, sc-77968-SH) with GFP, GFP-LC3, GFP-LAMP1, or GFP-LAMP1 and mRFP-LC3 at DIV5 using Lipofectamine 2000 (Invitrogen) before live-cell imaging 7–12 days after transfection before quantification analysis. For some experiments, 24 h incubation with lysosomal inhibitors E64d (40 µM) and pepstatin A (40 µM) was applied to suppress lysosomal degradation and/or 24 h incubation with Trehalose (100 mM) to induce autophagy. For immunostaining of RME-8, cultured neurons 7 days after transfection were fixed with 4% formaldehyde (Polyscience, Inc., 1881410) and 4% sucrose (Sigma, S0389) in 1× phosphate-buffered saline (PBS; Quality Biological, 119-069-131) at room temperature (RT) for 20 min, washed three times with PBS for 5 min each, and then incubated in 0.4% saponin (Sigma, S4521), 5% normal goat serum (NGS; Sigma, G9023) and 2% bovine serum albumin (BSA; Sigma, A9647) in PBS for 1 h. Fixed cultures were incubated with primary antibodies RME-8 (Sigma, ABN1657) and MAP2 (BD Biosciences 556,320) in PBS with 2% BSA and 0.4% saponin at 4°C overnight. Cells were washed four times with PBS at RT for 5 min each, incubated with Alexa Fluor 546 anti-rabbit (A-11071) and 647 anti-mouse (A-21235)-conjugated secondary antibodies (Invitrogen) at 1:400 dilution in PBS with 2% BSA and 0.4% saponin for 30 min, re-washed with PBS, and then mounted with Fluor-Gel antifade mounting medium (EMS, H-5000-60) for imaging.
Image acquisition and quantification in cultured neurons
For live-cell imaging, cells were transferred to Tyrode’s solution containing 10 mM HEPES, 10 mM glucose, 1.2 mM CaCl2, 1.2 mM MgCl2, 3 mM KCl, and 145 mM NaCl, pH 7.4. The temperature was maintained at 37°C with an air stream incubator. Confocal images were obtained using an Olympus FV3000 oil immersion 60× oil immersion lens (1.3 numerical aperture) with a sequential-acquisition setting, using 488-nm excitation for GFP-LC3 or GFP-LAMP1 and 543 nm for mRFP-LC3. Images were acquired using the same settings below saturation at a resolution of 1,024 × 1,024 pixels (8-bit). Eight to ten sections were taken from the top-to-bottom of the specimen and brightest point projections were made. Time-lapse sequences of 1,024 × 1,024 pixels (8 bit) were collected at 2 s intervals with 1% intensity of the laser to minimize laser-induced bleaching and cell damage while maximizing pinhole opening. Time-lapse images were captured at a total of 100 frames. All recordings started 6 min after the coverslip was placed in the chamber. Images were imported to Adobe Photoshop and morphometric measurements were performed using NIH ImageJ. The thresholds in all images were set to similar levels. The 100 µm of axon most proximal to the soma were analyzed for tubule length. Measured data were imported into Excel software for analysis. Data were obtained from at least three independent experiments.
Graphing and statistics
All results were graphed using GraphPad Prism 9 software, generating scatter plots depicting individual data points. Bars represent mean with SEM. Significance was measured by Student’s unpaired t-test when comparing 2 strains/conditions or Ordinary one-way ANOVA when comparing > 2 strains, unless otherwise noted. Data were considered statistically different at P < 0.05. P < 0.05 is indicated with single asterisks, P < 0.01 with double asterisks, P < 0.001 with triple asterisks, and P < 0.0001 with quadruple asterisks.
Supplementary Material
Acknowledgements
We would like to thank the Grant lab for critical comments and suggestions. We thank Peter Schweinsberg and Ge Bai for technical assistance in plasmid construction, and Helen Ushakov for expert microinjection. This work was supported by NIH Grant 5R01GM135326 to B.D.G and Q.C., and 5R01NS089737 to Q.C.
Funding Statement
The work was supported by the National Institute of Neurological Disorders and Stroke [F31NS117034]; National Institutes of Health [5R01GM135326, 5R01NS089737].
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The data of this study is available from the corresponding author (BG) upon reasonable request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2269028
References
- [1].Yu L, McPhee CK, Zheng L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465(7300):942–946. doi: 10.1038/nature09076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rong Y, Liu M, Ma L, et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat Cell Biol. 2012;14(9):924–934. doi: 10.1038/ncb2557 [DOI] [PubMed] [Google Scholar]
- [3].Schulze RJ, Weller SG, Schroeder B, et al. Lipid droplet breakdown requires dynamin 2 for vesiculation of autolysosomal tubules in hepatocytes. J Cell Bio. 2013;203(2):315–326. doi: 10.1083/jcb.201306140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Du W, Su Q, Chen Y, et al. Kinesin 1 drives autolysosome tubulation. Dev Cell. 2016;37(4):326–336. doi: 10.1016/j.devcel.2016.04.014 [DOI] [PubMed] [Google Scholar]
- [5].Eramo MJ, Gurung R, Mitchell CA, et al. Bidirectional interconversion between PtdIns4P and PtdIns(4,5)P2 is required for autophagic lysosome reformation and protection from skeletal muscle disease. Autophagy. 2021;17(5):1287–1289. doi: 10.1080/15548627.2021.1916195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Munson MJ, Allen GF, Toth R, et al. mTOR activates the VPS 34– UVRAG complex to regulate autolysosomal tubulation and cell survival. EMBO J. 2015;34(17):2272–2290. doi: 10.15252/embj.201590992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Boland B, Kumar A, Lee S, et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in alzheimer’s disease. J Neurosci. 2008;28(27):6926. doi: 10.1523/JNEUROSCI.0800-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Maday S, Holzbaur EL.. Compartment-specific regulation of autophagy in primary neurons. J Neurosci. 2016;36(22):5933–5945. doi: 10.1523/JNEUROSCI.4401-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Takalo M, Salminen A, Soininen H, et al. Protein aggregation and degradation mechanisms in neurodegenerative diseases. Am J Neurodegener Dis. 2013;2(1):1–14. [PMC free article] [PubMed] [Google Scholar]
- [10].Tan C-C, Yu J-T, Tan M-S, et al. Autophagy in aging and neurodegenerative diseases: implications for pathogenesis and therapy. Neurobiol Aging. 2014;35(5):941–957. doi: 10.1016/j.neurobiolaging.2013.11.019 [DOI] [PubMed] [Google Scholar]
- [11].Chang J, Lee S, Blackstone C. Spastic paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J Clin Investig. 2014;124(12):5249–5262. doi: 10.1172/JCI77598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Francis V, Alshafie W, Kumar R, et al. The ARSACS disease protein sacsin controls lysosomal positioning and reformation by regulating microtubule dynamics. J Biol Chem. 2022;298(9):298(9. doi: 10.1016/j.jbc.2022.102320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Vilarino-Guell C, Rajput A, Milnerwood AJ, et al. DNAJC13 mutations in Parkinson disease. Hum Mol Genet. 2014;23(7):1794–1801. doi: 10.1093/hmg/ddt570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Appel-Cresswell S, Rajput AH, Sossi V, et al. Clinical, positron emission tomography, and pathological studies of DNAJC13 p.N855S parkinsonism. Mov Disord. 2014;29(13):1684–1687. doi: 10.1002/mds.26019 [DOI] [PubMed] [Google Scholar]
- [15].Norris A, McManus CT, Wang S, et al. Mutagenesis and structural modeling implicate RME-8 IWN domains as conformational control points. PLoS Genet. 2022;18(10):e1010296. doi: 10.1371/journal.pgen.1010296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Spillantini MG, Schmidt ML, Lee VM-Y, et al. α-synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. doi: 10.1038/42166 [DOI] [PubMed] [Google Scholar]
- [17].Rajput A, Ross JP, Bernales CQ, et al. VPS35 and DNAJC13 disease-causing variants in essential tremor. Eur J Hum Genet. 2015;23(6):887–888. doi: 10.1038/ejhg.2014.164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Grant B, Hirsh D, Kimble J. Receptor-mediated endocytosis in the caenorhabditis elegans oocyte. Mol Biol Cell. 1999;10(12):4311–4326. doi: 10.1091/mbc.10.12.4311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhang Y, Grant B, Hirsh D, et al. RME-8, a conserved J-Domain protein, is required for endocytosis in caenorhabditis elegans. Mol Biol Cell. 2001;12(7):2011–2021. doi: 10.1091/mbc.12.7.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Greener T, Grant B, Zhang Y, et al. Caenorhabditis elegans auxilin: a J-domain protein essential for clathrin-mediated endocytosis in vivo. Nat Cell Biol. 2001;3(2):215–219. doi: 10.1038/35055137 [DOI] [PubMed] [Google Scholar]
- [21].Xhabija B, Taylor GS, Fujibayashi A, et al. Receptor mediated endocytosis 8 is a novel PI(3)P binding protein regulated by myotubularin-related 2. FEBS Lett. 2011;585(12):1722–1728. doi: 10.1016/j.febslet.2011.04.016 [DOI] [PubMed] [Google Scholar]
- [22].Norris A, Grant BD. Endosomal microdomains: formation and function. Curr Opinion Cell Biol. 2020;65:86–95. doi: 10.1016/j.ceb.2020.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Shi A, Sun L, Banerjee R, et al. Regulation of endosomal clathrin and retromer-mediated endosome to Golgi retrograde transport by the J-domain protein RME-8. EMBO J. 2009;28(21):3290–3302. doi: 10.1038/emboj.2009.272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Norris A, Tammineni P, Wang S, et al. SNX-1 and RME-8 oppose the assembly of HGRS-1/ESCRT-0 degradative microdomains on endosomes. Proc Natl Acad Sci U S A. 2017;114(3):E307–E316. doi: 10.1073/pnas.1612730114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Raiborg C, Wesche J, Malerød L, Stenmark H. Flat clathrin coats on endosomes mediate degradative protein sorting by scaffolding Hrs in dynamic microdomains. Journal of Cell Science. 2006;119(12):2414–2424. doi: 10.1242/jcs.02978 [DOI] [PubMed] [Google Scholar]
- [26].Hurley JH. The ESCRT complexes. Crit Rev Biochem Mol Biol. 2010;45(6):463–487. doi: 10.3109/10409238.2010.502516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hurley JH, Hanson PI. Membrane budding and scission by the ESCRT machinery: it’s all in the neck. Nat Rev Mol Cell Biol. 2010;11(8):556–566. doi: 10.1038/nrm2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wollert T, Hurley JH. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature. 2010;464(7290):864–869. doi: 10.1038/nature08849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Popoff V, Mardones GA, Bai SK, et al. Analysis of articulation between Clathrin and retromer in retrograde Sorting on early endosomes. Traffic. 2009;10(12):1868–1880. doi: 10.1111/j.1600-0854.2009.00993.x [DOI] [PubMed] [Google Scholar]
- [30].Gagliardi M, Annesi G, Procopio R, et al. DNAJC13 mutation screening in patients with Parkinson’s disease from South Italy. parkinsonism relat Disord. 2018;55:134–137. doi: 10.1016/j.parkreldis.2018.06.004 [DOI] [PubMed] [Google Scholar]
- [31].Gustavsson EK, Trinh J, Guella I, et al. DNAJC13 genetic variants in parkinsonism. Mov Disord. Mov Disord. 2015;30(2):273–278. doi: 10.1002/mds.26064 [DOI] [PubMed] [Google Scholar]
- [32].Lorenzo-Betancor O, Ogaki K, Soto‐Ortolaza AI, et al. DNAJC13 p.Asn855Ser mutation screening in Parkinson’s disease and pathologically confirmed Lewy body disease patients. Eur J Neurol. 2015;22(9):1323–1325. doi: 10.1111/ene.12770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yoshida S, Hasegawa T, Suzuki M, et al. Parkinson’s disease-linked DNAJC13 mutation aggravates alpha-synuclein-induced neurotoxicity through perturbation of endosomal trafficking. Hum Mol Genet. 2018;27(5):823–836. doi: 10.1093/hmg/ddy003 [DOI] [PubMed] [Google Scholar]
- [34].Schafer WR. Mechanosensory molecules and circuits in C. elegans. Pflugers Arch - Eur J Physiol. 2015;467(1):39–48. doi: 10.1007/s00424-014-1574-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hamelin M, Scott IM, Way JC, et al. The mec-7 beta-tubulin gene of caenorhabditis elegans is expressed primarily in the touch receptor neurons. EMBO J. 1992;11(8):2885–2893. doi: 10.1002/j.1460-2075.1992.tb05357.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lőrincz P, Juhász G. Autophagosome-lysosome fusion. J Mol Biol. 2020;432(8):2462–2482. doi: 10.1016/j.jmb.2019.10.028 [DOI] [PubMed] [Google Scholar]
- [37].Nguyen JA, Yates RM. Better together: Current insights into phagosome-lysosome fusion. Front Immunol. 2021;12:12. doi: 10.3389/fimmu.2021.636078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Malik BR, Maddison DC, Smith GA, et al. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Mol Brain. 2019;12(1):100. doi: 10.1186/s13041-019-0504-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].McGrath MJ, Eramo MJ, Gurung R, et al. Defective lysosome reformation during autophagy causes skeletal muscle disease. J Clin Invest. 2021;131(1). doi: 10.1172/JCI135124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Serra-Vinardell J, Sandler MB, De Pace R, et al. LYST deficiency impairs autophagic lysosome reformation in neurons and alters lysosome number and size. Cell Mol Life Sci. 2023;80(2):53. doi: 10.1007/s00018-023-04695-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Cao Y, Klionsky DJ. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res. 2007;17(10):839–849. doi: 10.1038/cr.2007.78 [DOI] [PubMed] [Google Scholar]
- [42].Kihara A, Noda T, Ishihara N, et al. Two distinct Vps34 phosphatidylinositol 3–kinase complexes function in autophagy and carboxypeptidase Y Sorting inSaccharomyces cerevisiae. J Cell Bio. 2001;152(3):519–530. doi: 10.1083/jcb.152.3.519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ruck A, Attonito J, Garces KT, et al. The Atg6/Vps30/Beclin 1 ortholog BEC-1 mediates endocytic retrograde transport in addition to autophagy in C. elegans. Autophagy. 2011;7(4):386–400. doi: 10.4161/auto.7.4.14391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Clark SG, Shurland D-L, Meyerowitz EM, et al. A dynamin GTPase mutation causes a rapid and reversible temperature-inducible locomotion defect in C. elegans. Proc Natl Acad Sci, USA. 1997;94(19):10438–10443. doi: 10.1073/pnas.94.19.10438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Xhabija B, Vacratsis PO. Receptor-mediated endocytosis 8 utilizes an N-terminal phosphoinositide-binding motif to regulate endosomal clathrin dynamics. J Biol Chem. 2015;290(35):21676–21689. doi: 10.1074/jbc.M115.644757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hu Y, Reggiori F. Molecular regulation of autophagosome formation. Biochem Soc Trans. 2022;50(1):55–69. doi: 10.1042/BST20210819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Nishimura T, Tamura N, Kono N, et al. Autophagosome formation is initiated at phosphatidylinositol synthase-enriched ER subdomains. EMBO J. 2017;36(12):1719–1735. doi: 10.15252/embj.201695189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Tian E, Wang F, Han J, et al. Epg-1 functions in autophagy-regulated processes and may encode a highly divergent Atg13 homolog in C. elegans. Autophagy. 2009;5(5):608–615. doi: 10.4161/auto.5.5.8624 [DOI] [PubMed] [Google Scholar]
- [49].Lu Q, Yang P, Huang X, et al. The WD40 repeat PtdIns(3)P-Binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev Cell. 2011;21(2):343–357. doi: 10.1016/j.devcel.2011.06.024 [DOI] [PubMed] [Google Scholar]
- [50].Yang P, Zhang H. The coiled-coil domain protein EPG-8 plays an essential role in the autophagy pathway in C. elegans. Autophagy. 2011;7(2):159–165. doi: 10.4161/auto.7.2.14223 [DOI] [PubMed] [Google Scholar]
- [51].Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy. 2007;3(4):323–328. doi: 10.4161/auto.4012 [DOI] [PubMed] [Google Scholar]
- [52].Zhang H, Chang JT, Guo B, et al. Guidelines for monitoring autophagy in caenorhabditis elegans. Autophagy. 2015;11(1):9–27. doi: 10.1080/15548627.2014.1003478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Krüger U, Wang Y, Kumar S, et al. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol Aging. 2012;33(10):2291–2305. doi: 10.1016/j.neurobiolaging.2011.11.009 [DOI] [PubMed] [Google Scholar]
- [54].Feng T, Tammineni P, Agrawal C, et al. Autophagy-mediated regulation of BACE1 protein trafficking and degradation. J Biol Chem. 2017;292(5):1679–1690. doi: 10.1074/jbc.M116.766584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Umezawa H, Aoyagi T, Morishima H, et al. Pepstatin, a new pepsin inhibitor produced by Actinomycetes. J Antibiot (Tokyo). 1970;23(5):259–262. doi: 10.7164/antibiotics.23.259 [DOI] [PubMed] [Google Scholar]
- [56].Kleina LG, Grubman MJ. Antiviral effects of a thiol protease inhibitor on foot-and-mouth disease virus. J Virol. 1992;66(12):7168–7175. doi: 10.1128/jvi.66.12.7168-7175.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Stavoe AKH, Holzbaur ELF. Autophagy in neurons. Annu Rev Cell Dev Biol. 2019;35:477–500. doi: 10.1146/annurev-cellbio-100818-125242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ferguson SM. Neuronal lysosomes. Neurosci Lett. 2019;697:1–9. doi: 10.1016/j.neulet.2018.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hirst J, Hesketh GG, Gingras A-C, et al. Rag GTPases and phosphatidylinositol 3-phosphate mediate recruitment of the AP-5/SPG11/SPG15 complex. J Cell Bio. 2021;220(2). doi: 10.1083/jcb.202002075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Eisenberg E, Greene L E. Multiple Roles of Auxilin and Hsc70 in Clathrin‐Mediated Endocytosis. Traffic. 2007;8(6):640–646. doi: 10.1111/j.1600-0854.2007.00568.x [DOI] [PubMed] [Google Scholar]
- [61].Ken Sato AN, Sato M, Grant Barth D.. C. elegans as a model for membrane traffic. WormBook: the online review of C. elegans Biology [internet]. 2018. [DOI] [PMC free article] [PubMed]
- [62].Kirchhausen T, Harrison SC. Protein organization in clathrin trimers. Cell. 1981;23(3):755–761. doi: 10.1016/0092-8674(81)90439-6 [DOI] [PubMed] [Google Scholar]
- [63].Chen Y, Yu L. Scissors for autolysosome tubules. EMBO J. 2015;34(17):2217–2218. doi: 10.15252/embj.201592519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Lenk GM, Meisler MH. Chapter fourteen - mouse models of PI(3,5)P2 deficiency with impaired lysosome function. In: Conn PM, editor. Methods in enzymology. Elsevier: Academic Press; 2014. pp. 245–260. https://www.sciencedirect.com/science/article/pii/B9780123979261000147?casa_token=3fe8fQfZrlcAAAAA:VoWEihZSOe7ynPROpzk2AgI5b5ZkhpLQr4kNXzmRi_zxuMYFM5N5NkJbPRD-kioagemZx2Td [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Rivero-Ríos P, Weisman LS. Roles of PIKfyve in multiple cellular pathways. Curr Opinion Cell Biol. 2022;76:102086. doi: 10.1016/j.ceb.2022.102086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Klein DE, Lee A, Frank DW, et al. The pleckstrin homology domains of dynamin isoforms require oligomerization for high affinity phosphoinositide binding. J Biol Chem. 1998;273(42):27725–27733. doi: 10.1074/jbc.273.42.27725 [DOI] [PubMed] [Google Scholar]
- [67].Yarar D, Surka MC, Leonard MC, et al. SNX9 activities are regulated by multiple phosphoinositides through both PX and BAR domains. Traffic. 2008;9(1):133–146. doi: 10.1111/j.1600-0854.2007.00675.x [DOI] [PubMed] [Google Scholar]
- [68].Lu N, Shen Q, Mahoney TR, et al. Three sorting nexins drive the degradation of apoptotic cells in response to PtdIns(3)P signaling. Mol Biol Cell. 2011;22(3):354–374. doi: 10.1091/mbc.e10-09-0756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lu N, Shen Q, Mahoney TR, et al. Two PI 3-kinases and one PI 3-phosphatase together establish the cyclic waves of phagosomal PtdIns(3)P critical for the degradation of apoptotic cells. PLoS Biol. 2012;10(1):e1001245. doi: 10.1371/journal.pbio.1001245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Cheng S, Wang K, Zou W, et al. PtdIns(4,5)P₂ and PtdIns3P coordinate to regulate phagosomal sealing for apoptotic cell clearance. J Cell Bio. 2015;210(3):485–502. doi: 10.1083/jcb.201501038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Zhou C, Wu Z, Du W, et al. Recycling of autophagosomal components from autolysosomes by the recycler complex. Nat Cell Biol. 2022;24(4):497–512. doi: 10.1038/s41556-022-00861-8 [DOI] [PubMed] [Google Scholar]
- [72].Besemer AS, Maus J, Ax MDA, et al. Receptor-mediated endocytosis 8 (RME-8)/DNAJC13 is a novel positive modulator of autophagy and stabilizes cellular protein homeostasis. Cell Mol Life Sci. 2021;78(2):645–660. doi: 10.1007/s00018-020-03521-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Brenner S. The genetics of caenorhabditis elegans. Genetics. 1974;77(1):71–94. doi: 10.1093/genetics/77.1.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Frokjaer-Jensen C, Wayne Davis M, Hopkins CE, et al. Single-copy insertion of transgenes in caenorhabditis elegans. Nat Genet. 2008;40(11):1375–1383. doi: 10.1038/ng.248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Cai Q, Lu L, Tian J-H, et al. Snapin-regulated late endosomal transport is critical for efficient autophagy-lysosomal function in neurons. Neuron. 2010;68(1):73–86. doi: 10.1016/j.neuron.2010.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Cai Q, Zakaria H, Simone A, et al. Spatial Parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol. 2012;22(6):545–552. doi: 10.1016/j.cub.2012.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ye X, Cai Q. Snapin-mediated BACE1 retrograde transport is essential for its degradation in lysosomes and regulation of APP processing in neurons. Cell Rep. 2014;6(1):24–31. doi: 10.1016/j.celrep.2013.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Ye X, Sun X, Starovoytov V, et al. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum Mol Genet. 2015;24(10):2938–2951. doi: 10.1093/hmg/ddv056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Ye X, Feng T, Tammineni P, et al. Regulation of synaptic Amyloid-β generation through BACE1 retrograde transport in a mouse model of Alzheimer’s disease. J Neurosci. 2017;37(10):2639–2655. doi: 10.1523/JNEUROSCI.2851-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Tammineni P, Jeong YY, Feng T, et al. Impaired axonal retrograde trafficking of the retromer complex augments lysosomal deficits in Alzheimer’s disease neurons. Hum Mol Genet. 2017;26(22):4352–4366. doi: 10.1093/hmg/ddx321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Han S, Zhang M, Jeong YY, et al. The role of mitophagy in the regulation of mitochondrial energetic status in neurons. Autophagy. 2021;17(12):4182–4201. doi: 10.1080/15548627.2021.1907167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Han S, Jeong YY, Sheshadri P, et al. Mitophagy regulates integrity of mitochondria at synapses and is critical for synaptic maintenance. EMBO Rep. 2020;21(9):e49801. doi: 10.15252/embr.201949801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Jeong YY, Han S, Jia N, et al. Broad activation of the Parkin pathway induces synaptic mitochondrial deficits in early tauopathy. Brain. 2022;145(1):305–323. doi: 10.1093/brain/awab243 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data of this study is available from the corresponding author (BG) upon reasonable request.