

Abstract
DUF692 multinuclear iron oxygenases (MNIOs) are an emerging family of tailoring enzymes involved in the biosynthesis of ribosomally synthesized and post-translationally modified peptides (RiPPs). Three members, MbnB, TglH, and ChrH, have been characterized to date and shown to catalyze unusual and complex transformations. Using a co-occurrence-based bioinformatic search strategy, we recently generated a sequence similarity network of MNIO-RiPP operons that encode one or more MNIOs adjacent to a transporter. The network revealed >1000 unique gene clusters, evidence of an unexplored biosynthetic landscape. Herein, we assess an MNIO-RiPP cluster from this network that is encoded in Proteobacteria and Actinobacteria. The cluster, which we have termed mov (for methanobactin-like operon in Vibrio), encodes a 23-residue precursor peptide, two MNIOs, a RiPP recognition element, and a transporter. Using both in vivo and in vitro methods, we show that one MNIO, homologous to MbnB, installs an oxazolone-thioamide at a Thr-Cys dyad in the precursor. Subsequently, the second MNIO catalyzes N–Cα bond cleavage of the penultimate Asn to generate a C-terminally amidated peptide. This transformation expands the reaction scope of the enzyme family, marks the first example of an MNIO-catalyzed modification that does not involve Cys, and sets the stage for future exploration of other MNIO-RiPPs.
Graphical Abstract

INTRODUCTION
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are structurally diverse natural products with a range of biological activities.1–3 Because the biosynthesis of all RiPPs begins with a precursor peptide containing the canonical 20 amino acids, the diversity in this natural product family is generated by tailoring enzymes, which install modifications onto the core region of the precursor, often leading to a variety of alterations that can blur their ribosomal origin.3,4 A convenient means of categorizing RiPPs is, therefore, by the modifications and the corresponding tailoring enzymes involved in their biosynthesis. The ever-growing repository of bacterial genomes has especially benefitted the study of RiPPs through genome mining and prioritization of biosynthetic gene clusters (BGCs) that are predicted to produce novel chemotypes.5–9 Application of genome mining to RiPPs has expanded the compound family and revealed numerous novel transformations, notably those catalyzed by metalloenzymes, such as radical S-adenosylmethionine (SAM),9–15 cytochrome P450,16,17 or copper-dependent enzymes.18
Especially intriguing among RiPPs are those synthesized by the emerging family of multinuclear iron oxygenases (MNIOs), previously known as DUF692 enzymes. Three MNIOs have been studied thus far, MbnB,19–21 TglH,22,23 and ChrH24 from the methanobactin, pearlin, and chryseobasin biosynthetic pathways, respectively. The products of the former two are known, whereas the chryseobasin mature product remains to be characterized. A common theme among the characterized MNIOs is that they modify Cys-rich precursors via four-electron oxidative rearrangements in the absence of reductant, leading to heterocycle/macrocycle formation or β-carbon excision.19,22,24 Specifically, MbnB in conjunction with its partner protein MbnC,20 which acts as a scaffold for peptide binding, installs an oxazolone-thioamide at two Cys residues, thus generating the key motif that chelates copper in methanobactin (Figures 1A and S1A). Genes coding for MbnB, MbnC, and the precursor peptide MbnA have been found in several divergent BGCs, which have been categorized in Groups I–V (Figure S1).25 Those in Group V contain an additional MNIO, MbnX,25 with yet unknown function. The second MNIO to be characterized, TglH, converts a terminal Cys residue to 2-mercaptoglycine, releasing the original Cys-Cβ as formic acid for a net four-electron oxidation (Figure 1A). The pearlin pathway is the first to utilize the precursor peptide catalytically, incorporating a Cys residue to be modified by TglH and then cleaving it to liberate the mature amino acid product.22,23 Finally, ChrH is the first SAM-dependent MNIO and has been shown to introduce a complex set of alterations onto its precursor including thioether macrocyclization, imidazolidinedione heterocycle formation, and thiomethylation.24 Over 10,000 MNIOs can be seen in microbial genomes, with the vast majority uncharacterized, evidence of new chemical space that can be charted by focusing on this enzyme family.
Figure 1.
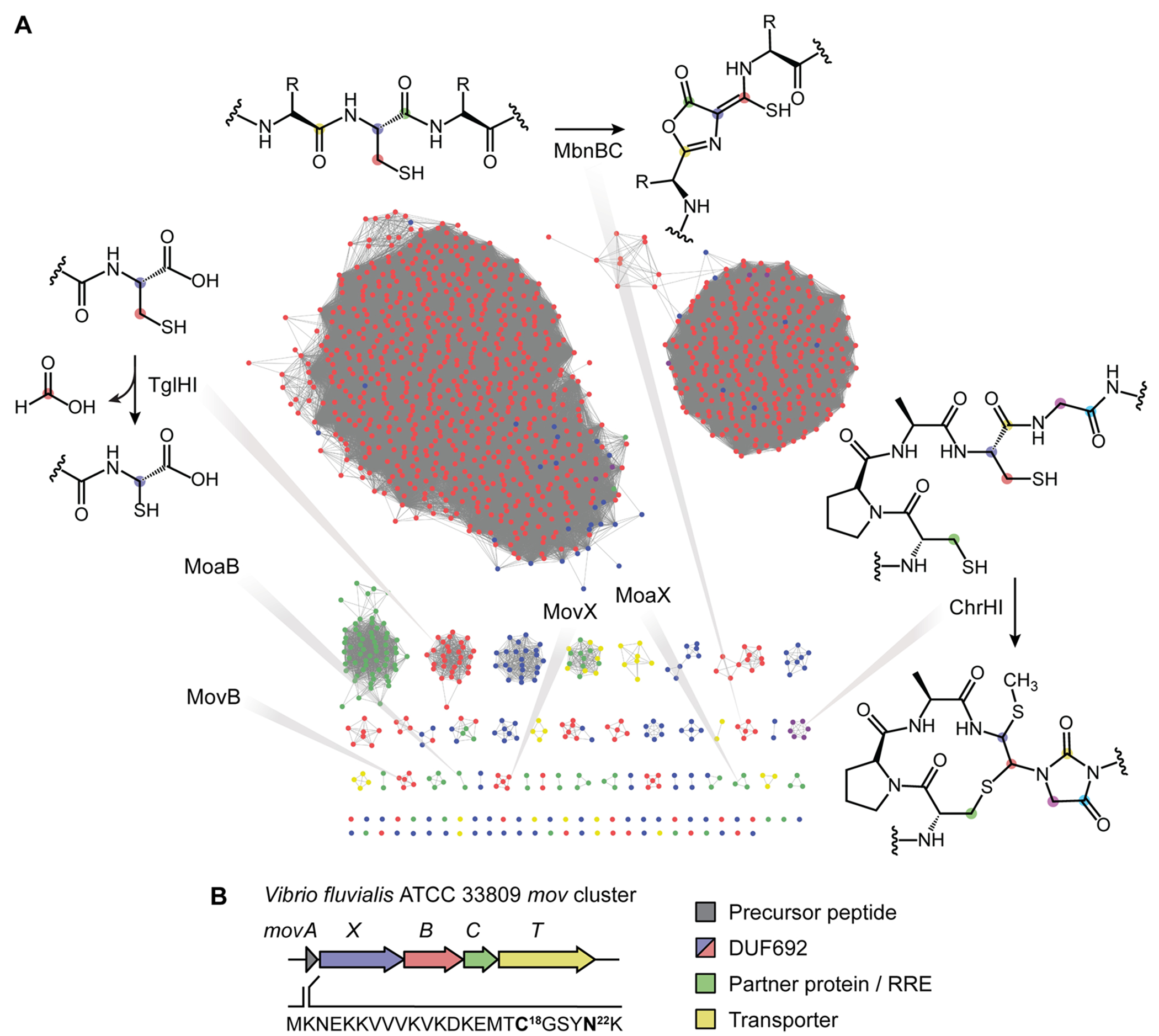
Sequence similarity network (SSN) of MNIO-RiPPs and the mov BGC. (A) SSN of RiPP BGCs that encode an MNIO and an adjacent transporter. Each node marks an individual MNIO enzyme, and lines connecting them indicate sequence similarity. Nodes are colored based on phyla: Proteobacteria (red), Actinobacteria (green), Firmicutes (yellow), Bacteroidetes (purple), and other phyla (blue). MNIO-RiPP families investigated in this work (MovX, MovB), related ones in Actinobacteria (MoaX, MoaB), and previously characterized ones (MbnB, TglH, and ChrH) are highlighted; reactions are shown for the latter. (B) The mov BGC from Vibrio fluvialis encodes a precursor peptide (MovA), two MNIOs (MovX and MovB), a discrete RRE (MovC), and a transporter (MovT). Genes are color-coded; the sequence of the precursor peptide is shown.
Motivated by the biosynthetic promise of MNIOs, we recently generated a sequence similarity network (SSN)26 of DUF692 enzymes in RiPP biosynthesis, using the near-ubiquitous feature of transporters in RiPP BGCs as a bioinformatic hook in a co-occurrence search.27 After accounting for newly deposited sequences, the updated SSN consists of 1382 unique MNIO-RiPP BGCs, arranged in a network of 43 families with at least two homologous members and 57 singletons (Figure 1A). Notably, the three characterized MNIOs, MbnB, TglH, and ChrH, populate distinct families, affirming the validity of the network. Scanning the network also shows that MNIOs co-occur with various other biosynthetic enzymes, suggesting that complex and new modifications remain to be uncovered from MNIO-RiPPs. In the present work, we tackle an MNIO-RiPP BGC that is found in both proteobacterial and actinobacterial genomes and codes for two MNIOs, one that is homologous to MbnB and a divergent one with an extended domain, previously termed MbnX.25 We show that the MbnB homologue installs an oxazolone-thioamide motif, much like the modification in methanobactin, while the other MNIO subsequently cleaves the N–Cα bond of the penultimate Asn to generate a C-terminally amidated peptide, a new reaction for a MNIO enzyme. Our results point to a plausible mechanism for N–Cα bond cleavage and expand the overall scope and catalytic versatility of this emerging enzyme family in RiPP biogenesis.
RESULTS
The mov BGC from Vibrio fluvialis.
To begin exploring the network of MNIO-RiPPs, we focused on an operon that encodes two MNIOs, an MbnB homologue, and a distantly related and uncharacterized MNIO, previously termed MbnX.25 We selected the cluster from the commercially available strain V. fluvialis 33809 (Table S1). As the precursor peptide is divergent from MbnA in Methylosinus trichosporium OB3b and the cluster does not encode methanobactin, we have termed it mov (for methanobactin-like operon in Vibrio). The BGC encodes a precursor peptide (MovA), the two MNIOs (MovX and MovB), a discrete RiPP recognition element (RRE, MovC),28 and a transporter (MovT) (Figure 1B). Compared to MbnAs from Groups I–IV, which contain two conserved Cys residues, Group V precursors contain only one Cys that is conserved in a TCG motif (Figures S1C and S2, Table S2). Assuming that the Cys residue is modified by MovBC, MovX likely performs a new transformation on an unprecedented residue for MNIOs. Sequence alignment of the Group V precursors reveals a conserved Tyr-Asn dipeptide motif that may constitute the site of MovX catalysis (Figure S2).
In Vivo Characterization of the mov BGC.
To characterize the transformations performed by each biosynthetic enzyme, we first followed a heterologous coexpression approach in Escherichia coli. The precursor peptide, MovA, was expressed with an N-terminal hexaHis maltose-binding protein (6HMBP) tag and an intervening HRV3C protease cleavage site. Following coexpression with either no enzyme, MovBC, or MovX, the 6HMBP-tagged MovA was purified, liberated by HRV3C proteolysis, and digested with trypsin. The products were then analyzed via HPLC-coupled high-resolution mass spectrometry (HR-MS) and tandem HR-MS (HR-MS/MS).
As expected, expression of 6HMBP-MovA alone, followed by purification and digestion with trypsin, afforded the unmodified C-terminal 9mer fragment (Figure 2A, Tables S3 and S4). Upon coexpression with MovBC, we observed a mass loss of 4.0313 Da accompanied by a shift in retention time (Figures 2B and S3, Table S3). HR-MS/MS analysis located the modification to the Thr-Cys dyad, consistent with an oxazolone-thioamide modification (Figure 2B, Table S5). We additionally observed side products 13.9793 and 30.0105 Da heavier than the unmodified substrate, reflecting hydrolysis and subsequent decarboxylation of the putative oxazolone ring (Figure S4A,B, Table S6). By contrast, coexpression of 6HMBP-MovA with MovX yielded a C-terminal fragment that was 243.1219 Da lighter than the unmodified 9mer peptide, suggesting cleavage of the C-terminal Asn-Lys dipeptide in addition to a mass loss of 0.9840 Da, consistent with an O-to-NH conversion (Figure 2C, Table S3). Intriguingly, all b-ions generated in the collision-induced dissociation of the MovX product were either unmodified or not observed, whereas all observed y-ions displayed a −0.9840 Da shift relative to the unmodified 7mer peptide, indicating the presence of a C-terminal amide (Figure 2C, Table S7).
Figure 2.
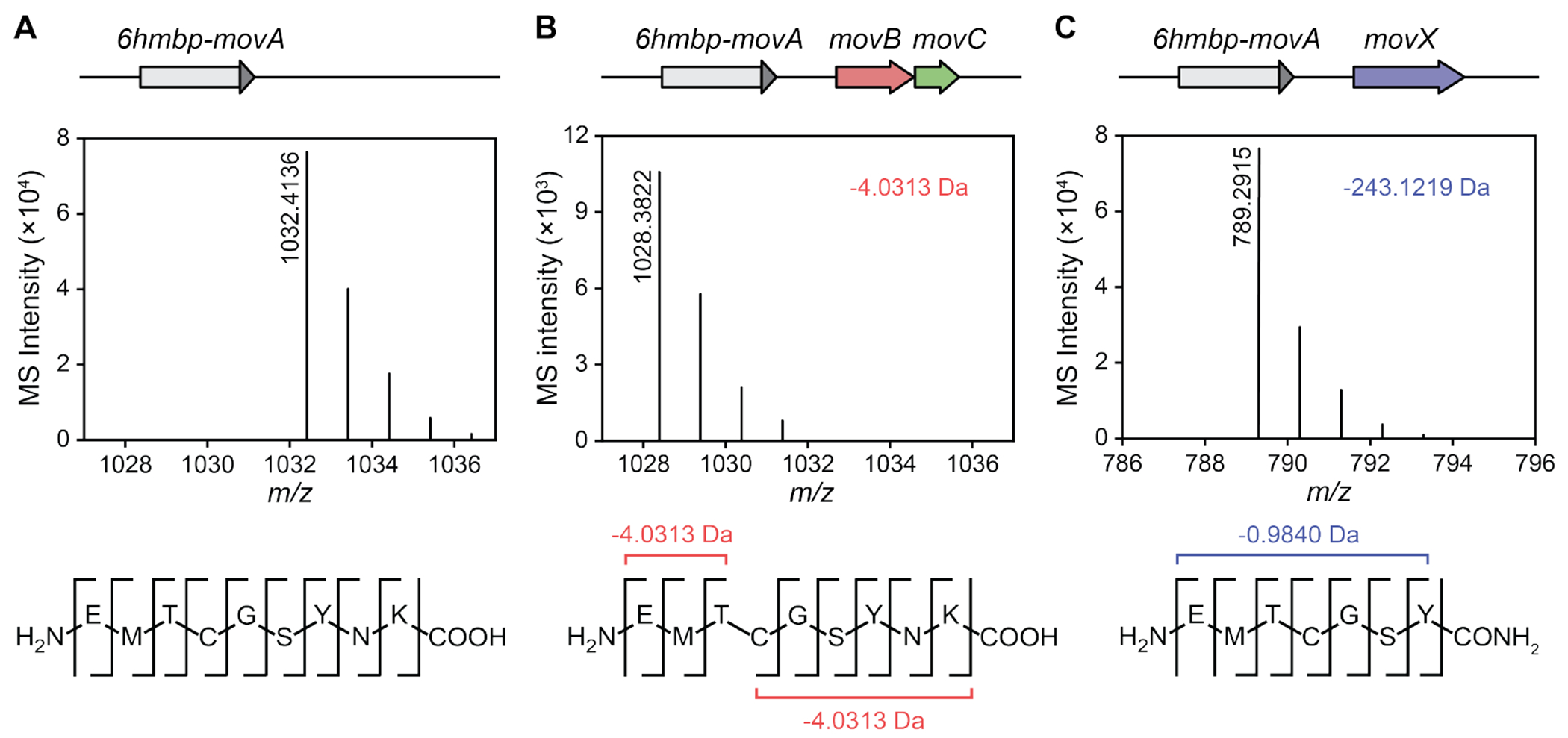
HR-MS and HR-MS/MS analyses of the heterologous expression constructs. Shown are data for trypsin-cleaved MovA after coexpression with no enzymes as control (A), MovBC (B), and MovX (C). The construct is shown above each HR-MS profile, which is zoomed in on the [M+H]+ ion of the trypsin-cleaved product, consisting of 9mer (A,B) or 7mer (C) peptides. Coexpression of MovA with MovBC and MovX gives products that are 4.0313 and 243.1219 Da lighter than the linear 9mer, respectively. The HR-MS/MS fragmentation pattern of the MovBC product is consistent with an oxazolone-thioamide motif. HR-MS/MS analysis of the MovX product reveals that the peptide is devoid of the C-terminal Asn-Lys dipeptide; while b-ions are unaffected, all observed y-ions exhibit a −0.9840 Da shift relative to the unmodified 7mer peptide.
Repeated attempts to characterize the combined MovXBC-modified product failed as E. coli transformants were not viable unless mutations were introduced in the MovA C-terminus, suggesting that the coexpression plasmid, and therefore the MovXBC-modified product, was toxic to E. coli (Table S8). We thus assessed the products of select mutants that emerged during E. coli transformation, including two substitutions, C18S- and C18Y-MovA, as well as a deletion mutant that obliterated the C-terminal Tyr-Asn dyad of MovA. With both C18S- and C18Y-MovA, coexpression with MovXBC delivered C-terminal fragments that were 243.1219 Da lighter than the respective unmodified peptides, indicating that a reaction with MovX had occurred (Tables S9–S11). Meanwhile, coexpression of the deletion mutant with MovXBC yielded a fragment that was 4.0313 Da lighter than the unmodified peptide, indicative of MovBC catalysis (Tables S9 and S12). Overall, the mutations abolished the function of either MovBC or MovX, suggesting that the modifications are mutually exclusive and confirming the discrete sites of post-translational tailoring by MovBC and MovX. The strong evolutionary pressure against the synthesis of MovXBC-modified products possibly provides clues for the biological function of the mature mov-derived RiPP.
In Vitro Characterization of MovBC and MovX.
To access the MovXBC-modified product and further characterize the MovX reaction, we turned to in vitro enzymatic assays. MovX was expressed recombinantly as a hexaHis-tagged construct in E. coli and purified by metal affinity chromatography (Figure S5). Despite previous reports showing that MbnB and MbnC copurify as a heterodimeric complex,19–21 in our hands MovB purified as a discrete protein when coexpressed with MovC. We therefore expressed and purified MovC separately. MovB and MovX were coexpressed with the GroEL/ES chaperone system and purified anaerobically. The use of molecular chaperones was required for obtaining soluble MovX, likely because it harbors an additional domain with a winged helix-turn-helix (wHTH) topology that is predicted to serve as an RRE (Figure S6A).28 Iron quantification revealed that MovB and MovX contain 0.7 ± 0.1 and 1.2 ± 0.1 iron atoms per protomer, respectively, reflecting partial incorporation of the metal cofactor (Table S13). As substrates for enzymatic assays, we generated MovA by solid-phase peptide synthesis (SPPS, Figure S7, Table S14), and acquired the MovBC- and MovX-modified variants via coexpression in E. coli.
Upon incubation of synthetic MovA with MovBC under aerobic conditions, we observed the time-dependent formation of a −4.0313 Da product (Figures 3A and S8A, Table S15). This species was not observed in the absence of MovBC; incubation with ascorbate increased turnover, as reported previously with MbnB.21 HR-MS/MS results located the modification to the Thr-Cys dyad, once again in line with the presence of the oxazolone-thioamide motif (Figure 3D, Table S16). The MovBC product displayed sharp absorption features at 264 and 302 nm, diagnostic of a conjugated system (Figure 3F). We also observed side products that were 13.9793 Da heavier and 30.0105 Da lighter than the unmodified substrate, reflecting hydrolysis and subsequent decarboxylation of the oxazolone ring, in agreement with the heterologous expression data (Figure S4C). Similar results were obtained with MovX-modified MovA (Figure 3C, Table S15), indicating that MovBC can install its modification on both MovA and MovX-reacted MovA. This result is consistent with a recent crystal structure of MbnABC from V. caribbenthicus BAA-2122 (PDB 7DZ9), which shows that the MbnA C-terminus extends out of the MbnB active site and minimally interacts with MbnBC.20
Figure 3.
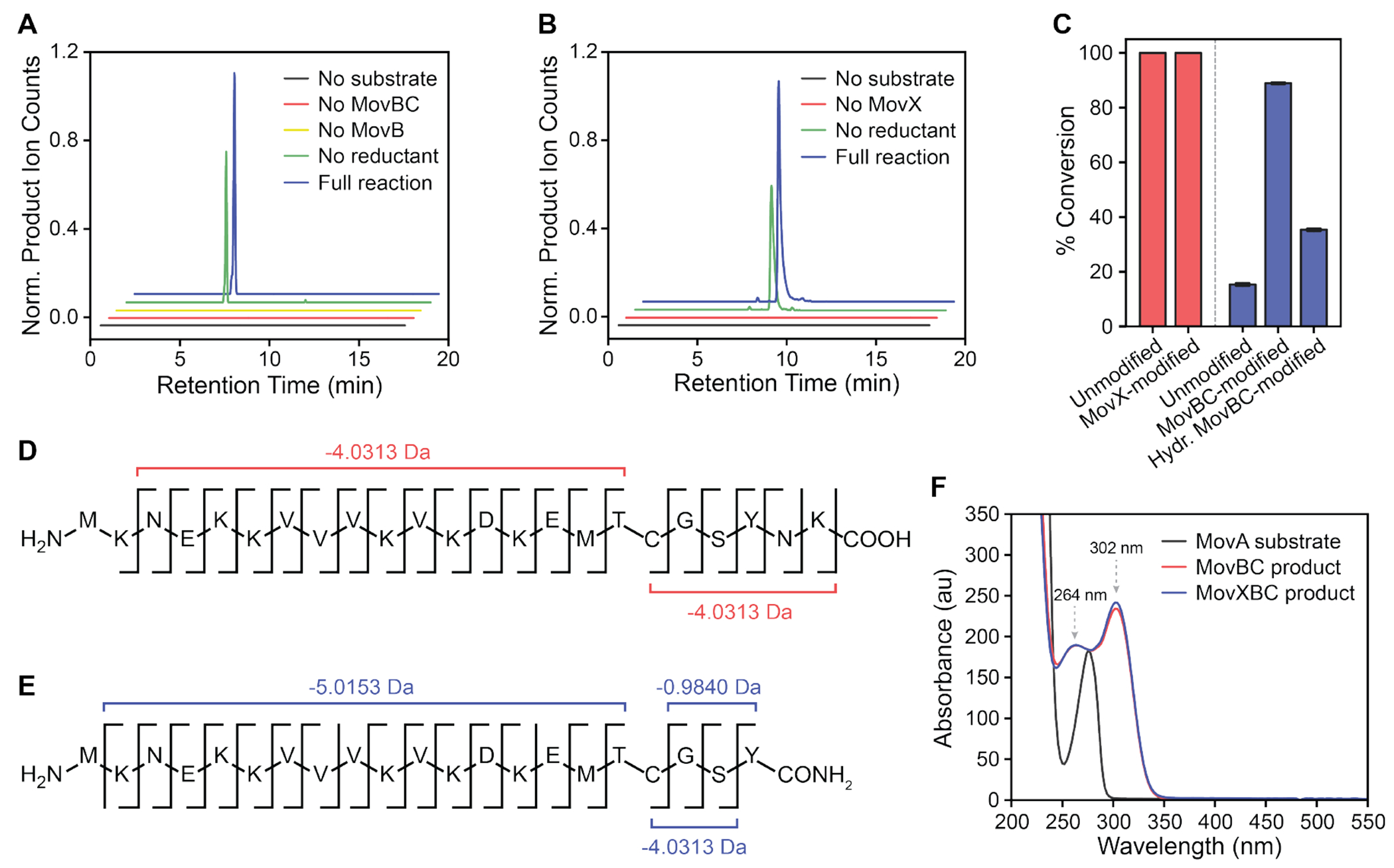
HR-MS and HR-MS/MS analyses of MovBC and MovX products. (A) Enzymatic activity assays of MovBC using MovA as a substrate. Shown are extracted ion chromatograms for the −4.0313 Da product of MovBC relative to the linear 23mer MovA. Product is not observed when MovB, MovBC, or substrate is omitted from the reaction, while the addition of ascorbate increases turnover. (B) Enzymatic activity assays of MovX using MovBC-modified MovA, generated through coexpression of 6HMBP-MovA and MovBC in E. coli, as a substrate. Shown are extracted ion chromatograms for the −247.1532 Da product of MovXBC, which corresponds to the loss of the Asn-Lys dyad and the introduction of a C-terminal amide. Product is not observed when MovX or the substrate is omitted from the reaction, while the addition of ascorbate increases turnover. In panels (A) and (B), traces are offset in both axes for clarity and color-coded as indicated. (C) Substrate preference of MovBC (red) and MovX (blue), as assessed by monitoring conversion of MovA substrate variants by HPLC-coupled HR-MS. MovBC converts both unmodified and MovX-modified MovA to the same extent, while MovX displays a clear preference for MovBC-modified MovA, followed by hydrolyzed MovBC-modified MovA, and unmodified MovA. (D,E) HR-MS/MS analyses of MovBC (D) and MovXBC (E) products. Mass shifts relative to the linear MovA are shown. (F) Ultraviolet–visible spectra of unmodified, MovBC-, and MovXBC-modified MovA. Unmodified MovA solely absorbs at 278 nm, while MovBC- and MovXBC-modified MovA exhibit additional absorption features at 264 and 302 nm.
In contrast to MovB, MovX displayed a clear preference for MovBC-reacted MovA, followed by hydrolyzed MovBC-modified MovA, and finally unmodified MovA (Figure 3C, Tables S15, S17, and S18). Upon incubation of MovX with MovBC-modified MovA under aerobic conditions, we observed time-dependent formation of a −243.1219 Da species relative to the substrate (Figures 3B and S8B), a product that was not observed in the absence of MovX; ascorbate again enhanced product yields. HR-MS/MS analysis indicated an intact oxazolone-thioamide moiety, cleavage of the C-terminal Asn-Lys dipeptide, and presence of a C-terminal amide (Table S17). Finally, one-pot incubation of synthetic MovA with MovXBC under aerobic conditions yielded a product with an overall mass loss of 247.1532 Da and an analogous HR-MS/MS fragmentation pattern as observed for the product of MovX and MovBC-modified MovA (Figure 3E, Tables S15 and S19). Relative to those of the MovBC product, the absorption properties of the MovXBC product were largely unchanged (Figure 3F).
Structural Elucidation of the MovXBC Product.
To definitively elucidate the product of the mov BGC, we conducted a large-scale MovX reaction using MovBC-modified MovA as a substrate. The trypsin-cleaved MovXBC-modified 7mer peptide was isolated and analyzed by 1D/2D nuclear magnetic resonance (NMR) spectroscopy; trypsin-cleaved MovBC-modified and unmodified 9mer peptides were used for comparison (Figures S9–S11, Table S20).1H–13C heteronuclear single quantum coherence (HSQC) spectrum of the MovBC product revealed the disappearance of both Cys-Hα and Cys-Hβ along with a sizeable downfield shift of Thr-Hα from 4.37 to 4.93 ppm (Figure 4A,B). A heteronuclear multiple bond correlation (HMBC) from Gly-Hα to Cys-Cβ, which experienced a significant downshift from 25.7 to 165.6 ppm, provided further evidence for the presence of the oxazolone-thioamide motif (Figure 4C). Equivalent correlations were observed for the MovXBC product, indicating that the modification installed by MovBC remained intact (Figure 4A–C). The HSQC spectrum of the MovXBC product was further devoid of peaks associated with Asn and Lys, indicating the cleavage of these C-terminal residues (Figure 4A,B). An HMBC correlation from Tyr-Hα to the carbonyl carbon of the same residue, which displayed a downshift from 172.8 to 175.5 ppm relative to unmodified and MovBC-modified peptides, marked the new amidated C-terminus (Figure 4D). Collectively, the data are consistent with the structure shown in Figure 4E, and they point to a new reaction for the MNIO MovX.
Figure 4.
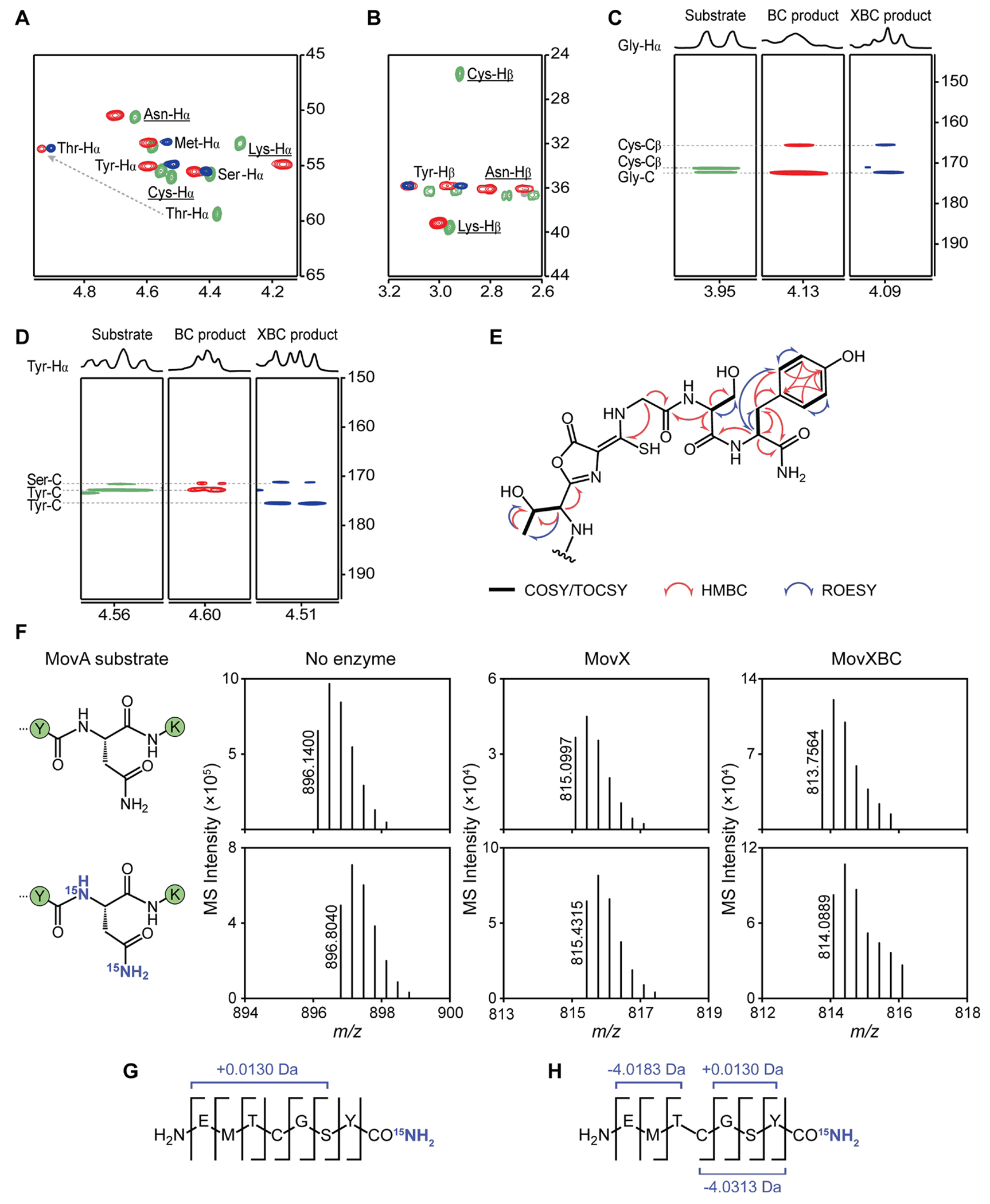
Structural elucidation of the MovBC and MovXBC products by 2D NMR and 15N labeling studies. (A–D) Comparative analysis of 2D NMR spectra of unmodified (green), MovBC-modified (red), and MovXBC-modified (blue) MovA. If present, impurity signals are shown in gray. All observed cross-peaks are labeled, and those that drop out post-modification are underlined. (A) HSQC spectra focusing on the Hα region. Compared to unmodified MovA, Cys-Hα is missing from the MovBC-modified product, and Thr-Hα is significantly downshifted. Moreover, MovXBC-modified MovA is devoid of Asn-Hα and Lys-Hα. (B) HSQC spectra focusing on the Hβ region. Compared to unmodified MovA, Cys-Hβ is missing from the MovBC-modified product. In addition, MovXBC-modified MovA lacks Asn-Hβ and Lys-Hβ. (C) HMBC spectra highlighting cross-peaks between Gly-Hα and Gly-C/Cys-Cβ. Compared to unmodified MovA, Cys-Cβ shifts upfield in both MovBC- and MovXBC-modified products, while Gly-C remains unchanged. (D) HMBC spectra highlighting cross-peaks between Tyr-Hα and Tyr-C and Ser-C. Compared to unmodified and MovBC-modified MovA, Tyr-C is downshifted in the product, while Ser-C remains unchanged. (E) Relevant NMR correlations used to solve the structure of MovXBC-modified MovA. (F) Reactions of MovX and MovXBC with unlabeled MovA and 15N2-Asn-MovA. HR-MS focusing on the [M+3H]3+ ion of each peptide. Reactions with 15N2-Asn-MovA yielded products 0.9970 Da heavier than the respective MovA products, indicating the incorporation of a single 15N label. (G,H) HR-MS/MS analyses of trypsin-cleaved 15N-labeled MovX (G) and MovXBC (H) products after reaction with 15N2-Asn-MovA. The observed fragmentation patterns indicate 15N incorporation at the modified C-terminus.
To obtain further evidence of the presence of a C-terminal amide, we performed 15N labeling studies. Given that Asn contains two amino groups, we generated a MovA isotopolog containing 15N2-Asn at residue 22 by SPPS (Figures S7, S12, and S13) and assessed the fate of the 15N isotopes after reaction with MovX or MovXBC by HR-MS and HR-MS/MS. In both cases, we observed products that were heavier by 0.9970 Da, indicating the incorporation of a single 15N isotope (Figure 4F, Table S21) and demonstrating that Asn is the source of the terminal amide nitrogen. The b-ions generated in the collision-induced dissociation of the 15N-labeled MovX-modified trypsin fragment were either unmodified or not observed, while all observed y-ions displayed a −0.0130 Da shift relative to unmodified 7mer peptide, indicating the presence of a C-terminal 15N-amide (Figure 4G, Table S22). Similarly, HR-MS/MS analysis of the 15N-labeled MovXBC-modified fragment located the 15N isotope at the C-terminus, thus confirming the structure of the MovX-catalyzed modification (Figure 4H, Table S23).
Toward a Mechanism of MovX.
Superposition of an AlphaFold model of MovX onto the crystal structure of MovB from V. caribbenthicus BAA-2122 (PDB 7DZ9)20 reveals all necessary ligands for binding up to three iron atoms (Figure S6B–D). In the MbnB structure, one of these metal ions is coordinated by the Cys residue of MbnA,20 directing H-atom abstraction from the Cys-Cβ, a feature that unites the three MNIOs studied to date.19,22,24 Cys ligation to an active-site iron cofactor is unlikely for MovX, as enzymatic activity was retained after substitution of the only Cys in the precursor peptide with Ser or Tyr (Tables S9–S11). In line with previous work,21,29 MovX may require at least two iron atoms for activity: a ferrous iron responsible for dioxygen binding and catalysis, and a ferric iron that facilitates substrate positioning. Furthermore, MovX catalysis proceeded without the addition of external reductant or cosubstrates, in support of earlier reports in which MNIOs extract all necessary reducing equivalents from their respective substrates.19,22,24 However, the addition of ascorbate did increase turnover (Figure 3B), likely by reducing one of the Fe(III) ions to the catalytically active Fe(II) species.
We explored three plausible mechanisms for the MovX reaction: the first involves heterolytic Lewis acid catalysis, while the second and third entail canonical oxygen activation and radical chemistry (Figure 5). In the heterolytic pathway, succinimide formation via the Asn22 side chain, a reaction with precedent in intein splicing,30 would set up Lewis acid-catalyzed activation of water and subsequent hydrolysis of the succinimide to generate the C-amidated product and an Asp-Lys dipeptide (Figure 5A). This mechanism would be oxygen-independent. Alternatively, oxygen binding to Fe(II) could generate an Fe(III)-O2· species as the oxidant, as previously proposed for the nonheme diiron enzyme myo-inositol oxygenase (MIOX) (Figure 5B).31,32 H-atom abstraction from the Asn-Cα followed by peroxide rebound, as previously proposed for MIOX, would generate a Cα-hydroperoxo Asn intermediate, which upon elimination of H2O2, concomitant with imine formation, and finally hydrolysis would yield product and an Asn-Lys diketone coproduct. Finally, we considered H-atom abstraction from Cβ, as proposed for the three MNIOs studied thus far (Figure 5C).19,22,24 In this case, H-atom abstraction would generate an Asn Cβ-based radical that upon oxygen rebound would give β-OH-Asn and the Fe(IV)-oxo species. Hydroxyl radical formation, mediated by the highly oxidizing Fe(IV)-oxo species, would facilitate β-scission, as observed for 2-hydroxyethylphosphonate dioxygenase (HEPD)33 and proposed for TglH,22 giving rise to a Cα-based radical and 2-oxoacetamide. Another oxygen rebound would set up N–Cα bond cleavage, yielding product and N-glyoxalyl-l-Lys.
Figure 5.
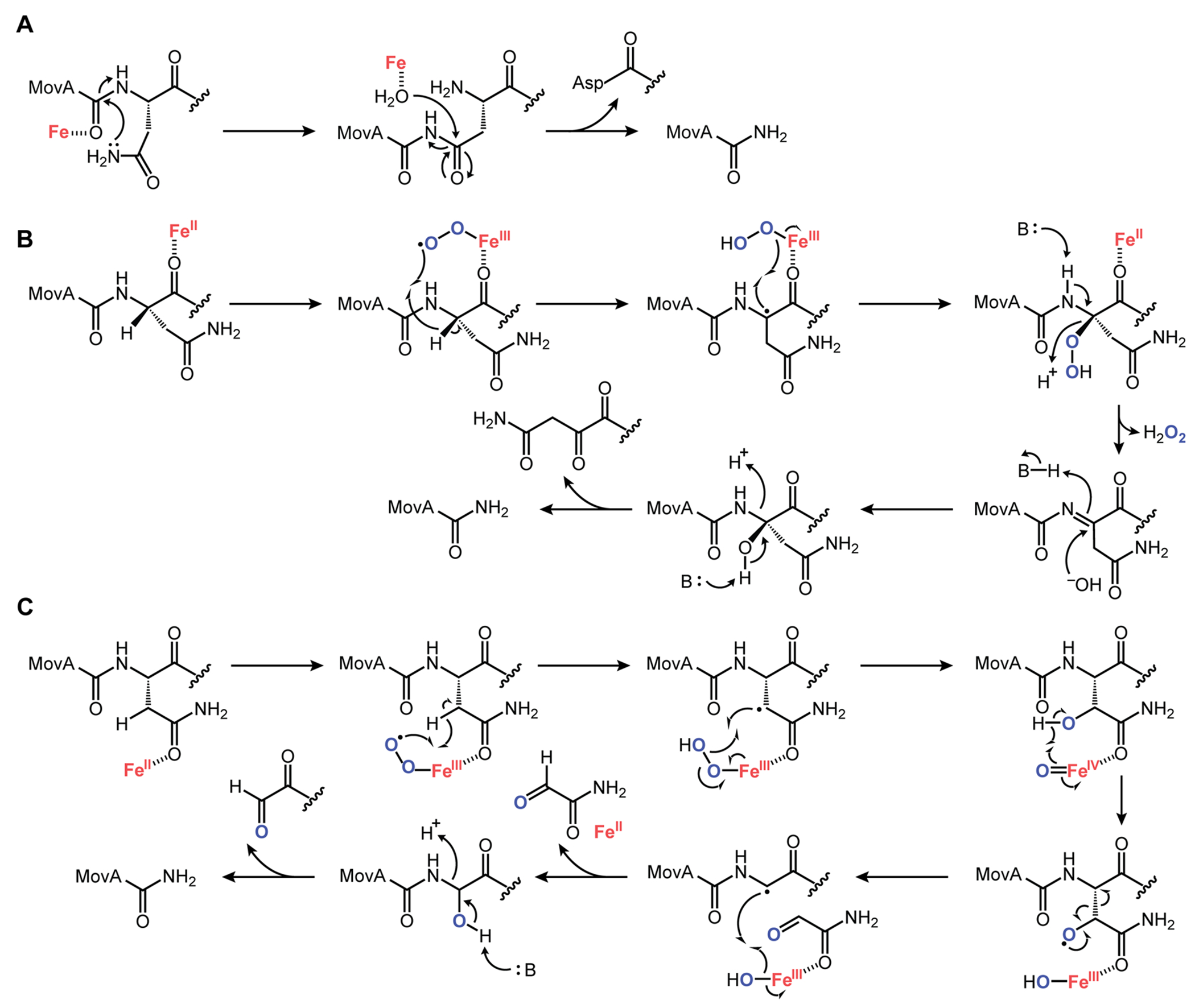
Proposed mechanisms for N–C bond cleavage catalyzed by MovX. (A) Heterolytic mechanism, in which succinimide formation is followed by Lewis acid-catalyzed hydrolysis to give the product. (B) Homolytic mechanism that involves O2 activation, peroxide rebound, and hydrolysis of an imine intermediate. (C) Alternative homolytic mechanism involving O2 activation, hydroxylation at Cβ followed by β-scission, and hydroxylation at Cα followed by cleavage of the N–C– bond. See text for further details.
As a first step toward differentiating between the heterolytic and homolytic mechanisms, we conducted experiments with a MovA isotopolog containing15Nβ-Asn at residue 22, which was accessed by SPPS (Figures S7, S14, and S15). Reaction with MovX or MovXBC, however, only yielded products lacking the 15N isotope (Figure 6A, Table S21). Moreover, no product was observed when the reaction was carried out anaerobically (Figure 6B). Together, these results rule out Mechanism A (Figure 5A) and show that the nitrogen in the product is derived from the α-amino group of Asn (see above). To differentiate between Mechanisms B and C, we attempted to detect the reaction coproduct(s). However, repeated attempts using several detection methods, including HPLC-coupled HR-MS with several columns and elution conditions, as well as 15N isotope tracing, were unsuccessful, likely due to stability issues. Moreover, efforts to trap the coproduct(s) in situ using fluorescein-5-thiosemicarbazide,34 which conjugates to both ketones and aldehydes, were also unsuccessful. A key feature of Mechanism B is the formation of H2O2. Assays with a horseradish peroxidase-based H2O2 detection system indeed showed enzyme concentration-dependent H2O2 formation with kinetics similar to that observed for the product (Figure S16). However, it is possible that the second intermediate shown in Mechanism C (Figure 5C), the Fe(III)-hydroperoxo species, uncouples to deliver H2O2.
Figure 6.
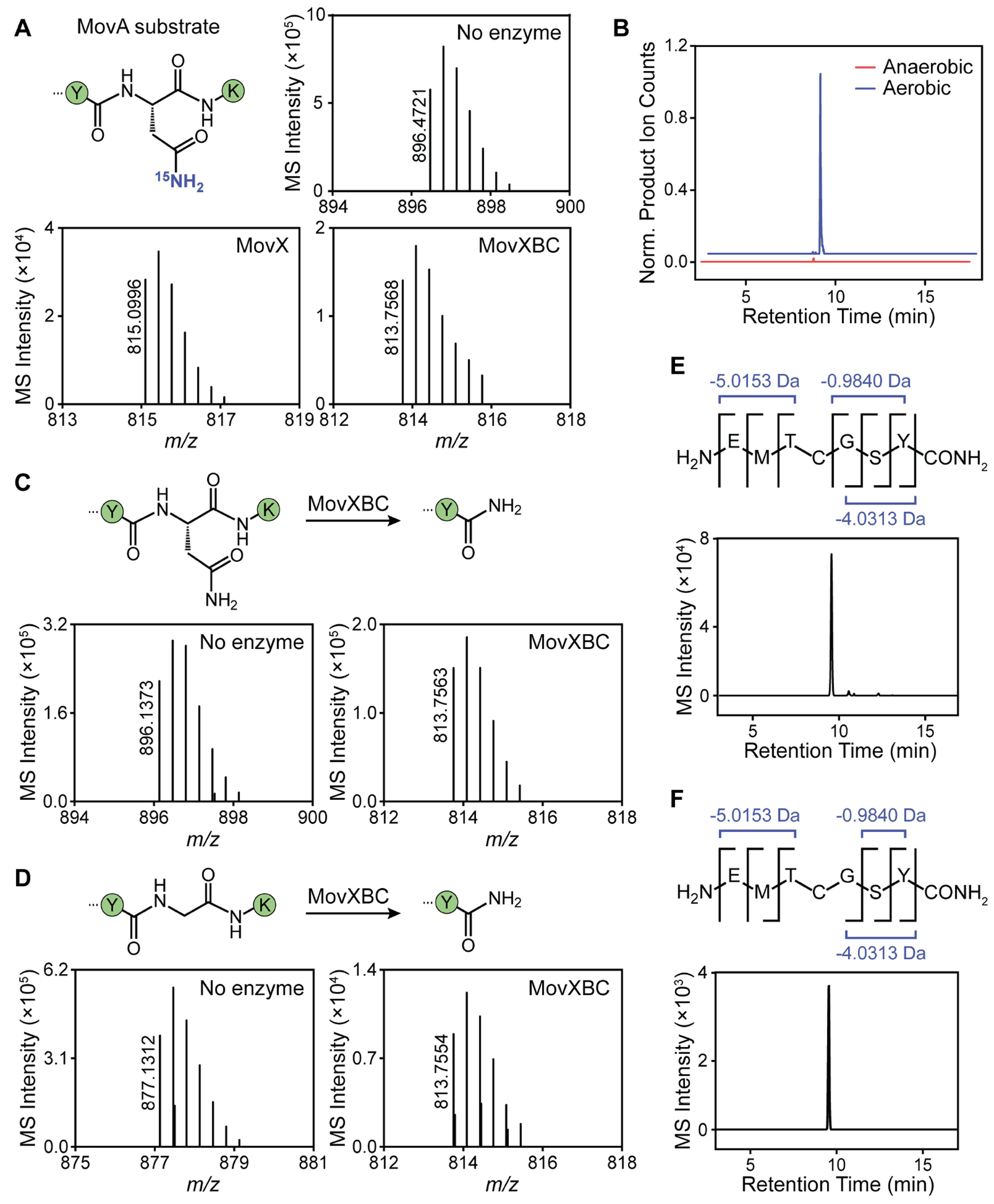
Mechanistic studies performed with MovX. (A) Reactions of MovX and MovXBC with15Nβ-Asn-MovA. HR-MS spectra are zoomed in on the [M+3H]3+ peptide ion. The 15N label is not incorporated in the observed products, indicating that the C-terminal amide originates from the α-amino group of Asn. (B) Enzymatic activity assays of MovX using MovBC-modified MovA as a substrate in the presence or absence of molecular oxygen. Shown are extracted ion chromatograms for MovA after reaction with MovXBC. The MovX reaction proceeds only under aerobic conditions. (C,D) Reaction of MovXBC with MovA (C) and N22G-MovA (D). HR-MS spectra are zoomed in onto the [M+3H]3+ peptide ions. The amidated product is observed for the Gly-substituted substrate, suggesting H-atom abstraction from Cβ is not required for the MovX reaction. (E,F) HR-MS and HR-MS/MS analyses of trypsin-cleaved MovXBC products using MovA (E) and N22G-MovA (F) as substrate. The products are identical, as demonstrated by analogous retention times and fragmentation patterns. The MovBC and MovX modifications, leading to losses of 4.0313 and 0.9840 Da, respectively, are marked.
Another key differentiator between Mechanisms B and C is that the catalytic cycle begins with the abstraction of a β-proton in the latter, as is the case with all MNIOs studied thus far.19,22,24 In Mechanism B, however, H-atom abstraction occurs only at the α-carbon. We therefore conducted assays with an Asn-to-Gly substitution in MovA, reasoning that MovX would be inactive if Mechanism C is operative, while it could retain enzymatic activity if Mechanism B is the pertinent pathway. Upon reaction of N22G-MovA with MovXBC, we detected robust product formation, as determined by retention time analysis, HR-MS, and HR-MS/MS (Figure 6C–F, Table S24). Reaction with MovA and N22G-MovA delivered identical products, as the two C-terminal residues are excised by MovX (Figure 6E,F, Tables S24–S26). These results strongly favor Mechanism B for MovX. Presteady-state spectroscopic assays may be conducted in the future to further corroborate this conclusion.
Attempts To Identify the Mature Product and Bioactivity Assays.
To identify the mature product of the mov cluster, we examined culture supernatants and cell pellets of V. fluvialis by targeted and untargeted HPLC-coupled HR-MS and HR-MS/MS using established methods.35–37 However, we were unable to identify fragments that matched those of the MovA peptide. Supplementing the growth medium with copper, a strategy commonly employed for the isolation of methanobactins,38 also proved unsuccessful, implying that the mov BGC is not responsive to this treatment and perhaps that the mature RiPP lacks copper-chelating properties. We, therefore, resorted to Xenorhabdus beddingii and Kitasatospora viridis, two additional strains that encode orthologous mov clusters, but were similarly unsuccessful in identifying the corresponding products. Mature RiPP products are often difficult to identify due to low expression levels of biosynthetic genes in laboratory culture or inappropriate detection methods.39–42 To explore if this is the case for the mov cluster, we carried out gene expression analysis of V. fluvialis using reverse transcription-quantitative polymerase chain reaction (RT-qPCR) of movX and movM in comparison to the actively expressed nonribosomal peptide synthetase (NRPS) gene in the fluvibactin cluster.43 Our results showed that movX and movM are expressed at significantly lower levels. For example, 5.2 h post inoculation, the fluvibactin nrps copy number was 18-fold and 16-fold higher than those of movX and movM, respectively (Figure S17, Table S27). These results indicate that the mov cluster is transcriptionally silent or sparingly expressed under standard growth conditions and that more sophisticated methods, such as ribosome engineering or high-throughput elicitor screening, will be necessary to enhance its expression.
In the absence of the mature product, we nonetheless explored the biological activity of the modified 7mer (Figure 6E) but did not observe any growth-inhibitory activity against the nine bacteria tested and the HCT116 colon cancer cell line (Table S28). Because C-amidated peptides have been shown to harbor anti-inflammatory activity,44–46 we examined the ability of the modified 7mer to suppress inflammation by monitoring nitric oxide (NO) levels upon addition of lipopolysaccharide, a pro-inflammatory factor. The 7mer reduced NO levels to the background even at low concentrations of 1.25 μg/mL (1.6 μM, Figure S18). While these initial results are intriguing, we emphasize that the assays were conducted with the enzymatic product and need to be repeated with the mature RiPP along with more in-depth bioactivity studies.
DISCUSSION
The increasing availability of microbial genome sequences and bioinformatic tools has brought about a renaissance in natural product research.5 Tapping into the biosynthetic potential of underexplored enzyme families using these tools is paramount to expanding our repertoire of enzyme-catalyzed reactions. With this in mind, we recently generated an SSN of MNIO (DUF692) enzymes present in RiPP BGCs.27 Herein, we have examined a previously uncharacterized MNIO from our network and elucidated a new transformation, thus expanding the reaction scope of this emerging enzyme family and paving the way for future work on other MNIO-RiPP BGCs. It is fair to assume that exciting new reactions will be discovered by exploring MNIO-RiPPs in the future.
All previously studied MNIOs modify Cys residues by oxidative rearrangements that commence with H-atom abstraction from the Cys-Cβ.19,22,24 MovX is the first example of an MNIO that does not modify a Cys residue but instead catalyzes cleavage of the N–Cα bond of an Asn residue to deliver a C-terminally amidated peptide, a reaction that is likely initiated by H-atom abstraction from the Asn-Cα. The MovX reaction resembles that catalyzed by the peptidylglycine α-amidating monooxygenase (PAM), a mammalian bifunctional enzyme involved in the biosynthesis of physiologically important hormones and neurotransmitters, which α-amidates Gly-extended peptides liberating glyoxylate in the process.47–49 While both PAM and MovX require dioxygen, PAM utilizes a copper- and ascorbate-dependent hydroxylating domain and a zinc-dependent lyase domain for catalysis,47,49 whereas MovX harbors a singular catalytic domain that presumably binds only iron. PAM hydroxylates the α-carbon of the terminal Gly residue, setting up N–Cα bond cleavage of the resulting hemiaminal.48 Our proposed pathway involves similar chemistry (Figure 5B), putatively generating the hydroperoxo rebound intermediate, from which H2O2 can be eliminated to deliver a hydrolytically labile imine product. Aside from providing an additional route for metal-catalyzed amidation, future studies could also uncover the coproduct(s), which remained elusive in our work.
Our findings provide a biosynthetic scheme for the putative mature RiPP from the mov BGC. Upon ribosomal synthesis of MovA, MovBC introduces an oxazolone-thioamide motif at Cys18 (Figure 7). Cleavage of the N–Cα bond of the penultimate Asn by MovX then delivers a C-terminally amidated peptide. This modification likely protects from proteolytic degradation and increases the hydrophobicity of the peptide by masking the negative charge of the C-terminus, which could facilitate transport across cellular membranes and/or interaction with a molecular target.50 Possible leader peptide processing completes the pathway, followed by the export of the mature RiPP via MovT. Despite repeated attempts, we were unable to identify the mature product of the mov BGC in three different bacterial strains. In V. fluvialis, the mov cluster is at best sparingly expressed. Future work will be directed toward identifying the mature RiPP through induction of the mov BGC and further exploring its biological activity.
Figure 7.
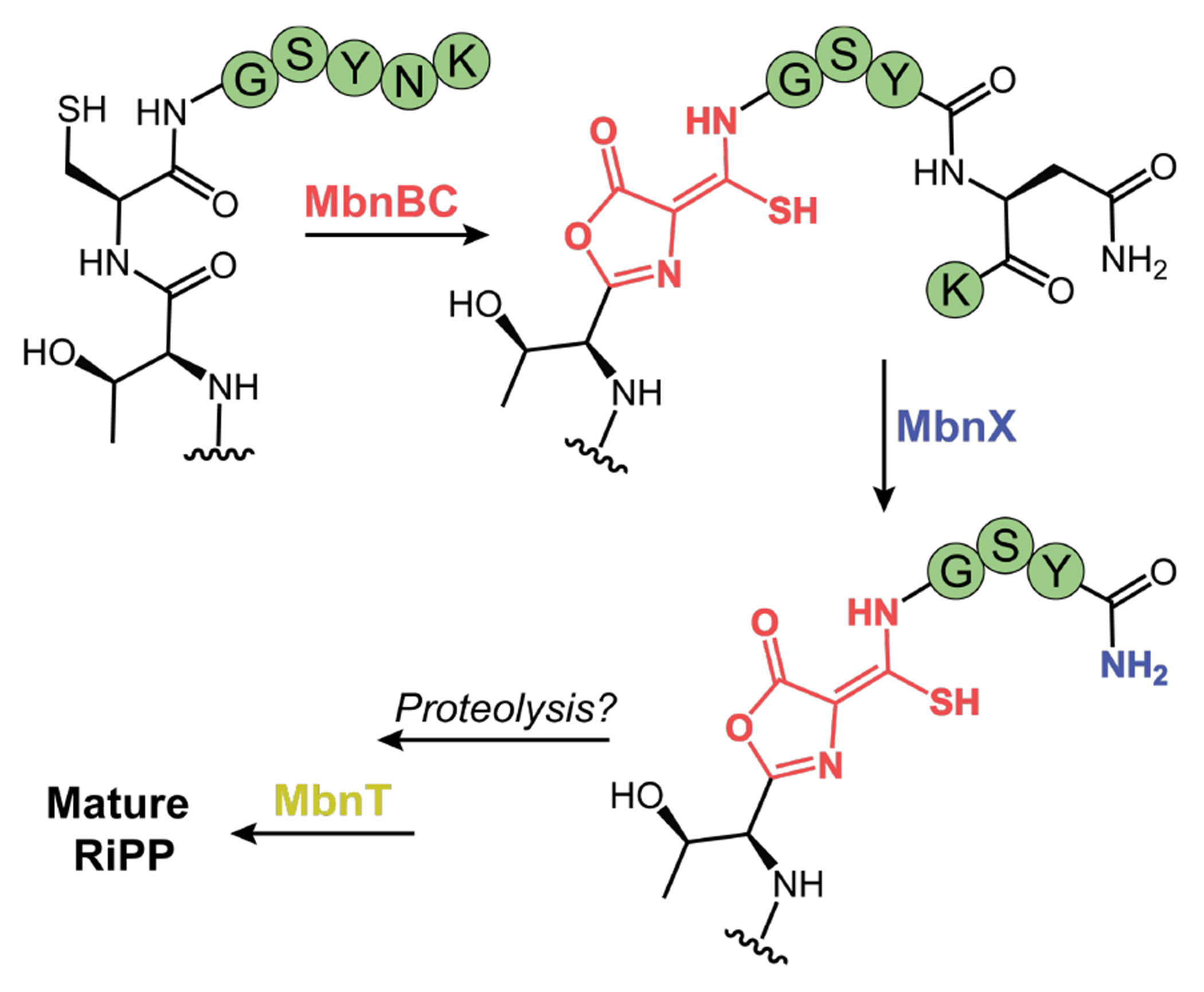
Proposed biosynthesis of RiPP produced by the mov cluster. Upon ribosomal synthesis of the MovA precursor peptide, MovBC catalyzes the formation of the oxazolone-thioamide. MovX then cleaves the N–Cα bond of the penultimate Asn to deliver a C-terminally amidated peptide. Following possible leader peptide processing, MovT likely exports the mature RiPP.
Supplementary Material
ACKNOWLEDGMENTS
We thank Istvan Pelczer for technical support with NMR spectra and Calvin Spolar for helpful conversations.
Funding
We thank the Edward C. Taylor 3rd Year Fellowship in Chemistry (V.T.C.), the Eli Lilly Edward C. Taylor Fellowship in Chemistry (K.A.C.), and the National Science Foundation (NSF CAREER Award 1847932 to M.R.S.) as well as the National Institutes of Health (grant GM129496 and grant GM140034 to M.R.S.) for financial support.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c11740.
Detailed materials and methods, DNA and protein sequences of enzymes examined in this work, tabulated HR-MS and HR-MS/MS data for all products reported, tabulated NMR data for products, catalog of mbn biosynthetic gene clusters, extracted ion chromatograms, mass spectral profiles, UV–visible spectra, and 1D/2D NMR spectra of enzymatic products, AlphaFold structures of MovX, and kinetics of product formation from in vitro enzymatic assays (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.3c11740
The authors declare no competing financial interest.
Contributor Information
Vasiliki T. Chioti, Department of Chemistry, Princeton University, Princeton, New Jersey 08544, United States
Kenzie A. Clark, Department of Chemistry, Princeton University, Princeton, New Jersey 08544, United States
Jack G. Ganley, Department of Chemistry, Princeton University, Princeton, New Jersey 08544, United States
Esther J. Han, Department of Chemistry, Princeton University, Princeton, New Jersey 08544, United States
Mohammad R. Seyedsayamdost, Department of Chemistry and Department of Molecular Biology, Princeton University, Princeton, New Jersey 08544, United States
REFERENCES
- (1).Arnison PG; Bibb MJ; Bierbaum G; Bowers AA; Bugni TS; Bulaj G; Camarero JA; Campopiano DJ; Challis GL; Clardy J; Cotter PD; Craik DJ; Dawson M; Dittmann E; Donadio S; Dorrestein PC; Entian K-D; Fischbach MA; Garavelli JS; Göransson U; Gruber CW; Haft DH; Hemscheidt TK; Hertweck C; Hill C; Horswill AR; Jaspars M; Kelly WL; Klinman JP; Kuipers OP; Link AJ; Liu W; Marahiel MA; Mitchell DA; Moll GN; Moore BS; Müller R; Nair SK; Nes IF; Norris GE; Olivera BM; Onaka H; Patchett ML; Piel J; Reaney MJT; Rebuffat S; Ross RP; Sahl H-G; Schmidt EW; Selsted ME; Severinov K; Shen B; Sivonen K; Smith L; Stein T; Süssmuth RD; Tagg JR; Tang G-L; Truman AW; Vederas JC; Walsh CT; Walton JD; Wenzel SC; Willey JM; Van Der Donk WA Ribosomally Synthesized and Post-Translationally Modified Peptide Natural Products: Overview and Recommendations for a Universal Nomenclature. Nat. Prod. Rep 2013, 30, 108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Montalbán-López M; Scott TA; Ramesh S; Rahman IR; van Heel AJ; Viel JH; Bandarian V; Dittmann E; Genilloud O; Goto Y; Grande Burgos MJ; Hill C; Kim S; Koehnke J; Latham JA; Link AJ; Martínez B; Nair SK; Nicolet Y; Rebuffat S; Sahl H-G; Sareen D; Schmidt EW; Schmitt L; Severinov K; Süssmuth RD; Truman AW; Wang H; Weng J-K; van Wezel GP; Zhang Q; Zhong J; Piel J; Mitchell DA; Kuipers OP; van der Donk WA New Developments in RiPP Discovery, Enzymology and Engineering. Nat. Prod. Rep 2021, 38, 130–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).McIntosh JA; Donia MS; Schmidt EW Ribosomal Peptide Natural Products: Bridging the Ribosomal and Nonribosomal Worlds. Nat. Prod. Rep 2009, 26, 537–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Oman TJ; van der Donk WA Follow the Leader: The Use of Leader Peptides to Guide Natural Product Biosynthesis. Nat. Chem. Biol 2010, 6, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bachmann BO; Van Lanen SG; Baltz RH Microbial Genome Mining for Accelerated Natural Products Discovery: Is a Renaissance in the Making? J. Ind. Microbiol. Biotechnol 2014, 41, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cimermancic P; Medema MH; Claesen J; Kurita K; Wieland Brown LC; Mavrommatis K; Pati A; Godfrey PA; Koehrsen M; Clardy J; Birren BW; Takano E; Sali A; Linington RG; Fischbach MA Insights into Secondary Metabolism from a Global Analysis of Prokaryotic Biosynthetic Gene Clusters. Cell 2014, 158, 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ziemert N; Alanjary M; Weber T The Evolution of Genome Mining in Microbes – a Review. Nat. Prod. Rep 2016, 33, 988–1005. [DOI] [PubMed] [Google Scholar]
- (8).Katz L; Baltz RH Natural Product Discovery: Past, Present, and Future. J. Ind. Microbiol. Biotechnol 2016, 43, 155–176. [DOI] [PubMed] [Google Scholar]
- (9).Scott TA; Piel J The Hidden Enzymology of Bacterial Natural Product Biosynthesis. Nat. Rev. Chem 2019, 3, 404–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Flühe L; Knappe TA; Gattner MJ; Schäfer A; Burghaus O; Linne U; Marahiel MA The Radical SAM Enzyme AlbA Catalyzes Thioether Bond Formation in Subtilosin A. Nat. Chem. Biol 2012, 8, 350–357. [DOI] [PubMed] [Google Scholar]
- (11).Schramma KR; Bushin LB; Seyedsayamdost MR Structure and Biosynthesis of a Macrocyclic Peptide Containing an Unprecedented Lysine-to-Tryptophan Crosslink. Nat. Chem 2015, 7, 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Clark KA; Bushin LB; Seyedsayamdost MR RaS-RiPPs in Streptococci and the Human Microbiome. ACS Bio. Med. Chem. Au 2022, 2, 328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mahanta N; Hudson GA; Mitchell DA Radical S-Adenosylmethionine Enzymes Involved in RiPP Biosynthesis. Biochemistry 2017, 56, 5229–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Latham JA; Barr I; Klinman JP At the Confluence of Ribosomally Synthesized Peptide Modification and Radical S-Adenosylmethionine (SAM) Enzymology. J. Biol. Chem 2017, 292, 16397–16405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yokoyama K; Lilla EA C–C Bond Forming Radical SAM Enzymes Involved in the Construction of Carbon Skeletons of Cofactors and Natural Products. Nat. Prod. Rep 2018, 35, 660–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wyche TP; Ruzzini AC; Schwab L; Currie CR; Clardy J Tryptorubin A: A Polycyclic Peptide from a Fungus-Derived Streptomycete. J. Am. Chem. Soc 2017, 139, 12899–12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Nam H; An JS; Lee J; Yun Y; Lee H; Park H; Jung Y; Oh K-B; Oh D-C; Kim S Exploring the Diverse Landscape of Biaryl-Containing Peptides Generated by Cytochrome P450 Macrocyclases. J. Am. Chem. Soc 2023, 145, 22047–22057. [DOI] [PubMed] [Google Scholar]
- (18).Chigumba DN; Mydy LS; de Waal F; Li W; Shafiq K; Wotring JW; Mohamed OG; Mladenovic T; Tripathi A; Sexton JZ; Kautsar S; Medema MH; Kersten RD Discovery and Biosynthesis of Cyclic Plant Peptides via Autocatalytic Cyclases. Nat. Chem. Biol 2022, 18, 18–28. [DOI] [PubMed] [Google Scholar]
- (19).Kenney GE; Dassama LMK; Pandelia M-E; Gizzi AS; Martinie RJ; Gao P; DeHart CJ; Schachner LF; Skinner OS; Ro SY; Zhu X; Sadek M; Thomas PM; Almo SC; Bollinger JM; Krebs C; Kelleher NL; Rosenzweig AC The Biosynthesis of Methanobactin. Science 2018, 359, 1411–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Dou C; Long Z; Li S; Zhou D; Jin Y; Zhang L; Zhang X; Zheng Y; Li L; Zhu X; Liu Z; He S; Yan W; Yang L; Xiong J; Fu X; Qi S; Ren H; Chen S; Dai L; Wang B; Cheng W Crystal Structure and Catalytic Mechanism of the MbnBC Holoenzyme Required for Methanobactin Biosynthesis. Cell Res. 2022, 32, 302–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Park YJ; Jodts RJ; Slater JW; Reyes RM; Winton VJ; Montaser RA; Thomas PM; Dowdle WB; Ruiz A; Kelleher NL; Bollinger JM; Krebs C; Hoffman BM; Rosenzweig AC A Mixed-Valent Fe(II)Fe(III) Species Converts Cysteine to an Oxazolone/Thioamide Pair in Methanobactin Biosynthesis. Proc. Natl. Acad. Sci. U. S. A 2022, 119, No. e2123566119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ting CP; Funk MA; Halaby SL; Zhang Z; Gonen T; van der Donk WA Use of a Scaffold Peptide in the Biosynthesis of Amino Acid-Derived Natural Products. Science 2019, 365, 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).McLaughlin MI; Yu Y; van der Donk WA Substrate Recognition by the Peptidyl-(S)-2-Mercaptoglycine Synthase TglHI during 3-Thiaglutamate Biosynthesis. ACS Chem. Biol 2022, 17, 930–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ayikpoe RS; Zhu L; Chen JY; Ting CP; van der Donk WA Macrocyclization and Backbone Rearrangement During RiPP Biosynthesis by a SAM-Dependent Domain-of-Unknown-Function 692. ACS Cent. Sci 2023, 9, 1008–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kenney GE; Rosenzweig AC Genome mining for methanobactins. BMC Biol. 2013, 11, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Gerlt JA Genomic Enzymology: Web Tools for Leveraging Protein Family Sequence-Function Space and Genome Context to Discover Novel Functions. Biochemistry 2017, 56, 4293–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Clark KA; Seyedsayamdost MR Bioinformatic Atlas of Radical SAM Enzyme-Modified RiPP Natural Products Reveals an Isoleucine–Tryptophan Crosslink. J. Am. Chem. Soc 2022, 144, 17876–17888. [DOI] [PubMed] [Google Scholar]
- (28).Burkhart BJ; Hudson GA; Dunbar KL; Mitchell DA A Prevalent Peptide-Binding Domain Guides Ribosomal Natural Product Biosynthesis. Nat. Chem. Biol 2015, 11, 564–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wörsdörfer B; Lingaraju M; Yennawar NH; Boal AK; Krebs C; Bollinger JM; Pandelia M-E Organophosphonate-Degrading PhnZ Reveals an Emerging Family of HD Domain Mixed-Valent Diiron Oxygenases. Proc. Natl. Acad. Sci. U.S.A 2013, 110, 18874–18879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Shah NH; Muir TW Inteins: Nature’s Gift to Protein Chemists. Chem. Sci 2014, 5, 446–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Brown PM; Caradoc-Davies TT; Dickson JMJ; Cooper GJS; Loomes KM; Baker EN Crystal Structure of a Substrate Complex of Myo-Inositol Oxygenase, a Di-Iron Oxygenase with a Key Role in Inositol Metabolism. Proc. Natl. Acad. Sci. U.S.A 2006, 103, 15032–15037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Xing G; Diao Y; Hoffart LM; Barr EW; Prabhu KS; Arner RJ; Reddy CC; Krebs C; Bollinger JM Evidence for C–H Cleavage by an Iron–Superoxide Complex in the Glycol Cleavage Reaction Catalyzed by Myo-Inositol Oxygenase. Proc. Natl. Acad. Sci. U.S.A 2006, 103, 6130–6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Peck SC; Wang C; Dassama LMK; Zhang B; Guo Y; Rajakovich LJ; Bollinger JM Jr; Krebs C; van der Donk WA O–H Activation by an Unexpected Ferryl Intermediate during Catalysis by 2-Hydroxyethylphosphonate Dioxygenase. J. Am. Chem. Soc 2017, 139, 2045–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Morinaka BI; Lakis E; Verest M; Helf MJ; Scalvenzi T; Vagstad AL; Sims J; Sunagawa S; Gugger M; Piel J Natural Noncanonical Protein Splicing Yields Products with Diverse β-Amino Acid Residues. Science 2018, 359, 779–782. [DOI] [PubMed] [Google Scholar]
- (35).Covington BC; Seyedsayamdost MR MetEx, a Metabolomics Explorer Application for Natural Product Discovery. ACS Chem. Biol 2021, 16, 2825–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bushin LB; Covington BC; Clark KA; Caruso A; Seyedsayamdost MR Bicyclostreptins are radical SAM enzyme-modified peptides with unique cyclization motifs. Nat. Chem. Biol 2022, 18, 1135–1143. [DOI] [PubMed] [Google Scholar]
- (37).Clark KA; Covington BC; Seyedsayamdost MR Biosynthesis-guided discovery reveals enteropeptins as alternative sactipeptides containing N-methylornithine. Nat. Chem 2022, 14, 1390–1398. [DOI] [PubMed] [Google Scholar]
- (38).Bandow NL; Gallagher WH; Behling L; Choi DW; Semrau JD; Hartsel SC; Gilles VS; DiSpirito AA Chapter Seventeen - Isolation of Methanobactin from the Spent Media of Methane-Oxidizing Bacteria. In Methods in Enzymology; Rosenzweig AC; Ragsdale SW, Eds.; Methods in Methane Metabolism, Part B: Methanotrophy; Academic Press, 2011; Vol. 495, pp 259–269. [DOI] [PubMed] [Google Scholar]
- (39).Nett M; Ikeda H; Moore BS Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat. Prod. Rep 2009, 26, 1362–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Rutledge PJ; Challis GL Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat. Rev. Microbiol 2015, 13, 509–523. [DOI] [PubMed] [Google Scholar]
- (41).Ochi K; Hosaka T New strategies for drug discovery: activation of silent or weakly expressed microbial gene clusters. Appl. Microbiol. Biotechnol 2013, 97, 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Covington BC; Xu F; Seyedsayamdost MR A Natural Product Chemist’s Guide to Unlocking Silent Biosynthetic Gene Clusters. Annu. Rev. Biochem 2021, 90, 763–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Yamamoto S; Okujo N; Fujita Y; Saito M; Yoshida T; Shinoda S Structures of two polyamine-containing catecholate siderophores from Vibrio fluvialis. J. Biochem 1993, 113, 538–544. [DOI] [PubMed] [Google Scholar]
- (44).Varga Z; Gurrola-Briones G; Papp F; Rodríguez de la Vega RC; Pedraza-Alva G; Tajhya RB; Gaspar R; Cardenas L; Rosenstein Y; Beeton C; Possani LD; Panyi G Vm24, a natural immunosuppressive peptide, potently and selectively blocks Kv1.3 potassium channels of human T cells. Mol. Pharmacol 2012, 82, 372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zhang L; Wei X; Zhang R; Koci M; Si D; Ahmad B; Guo H; Hou Y C-Terminal Amination of a Cationic Anti-Inflammatory Peptide Improves Bioavailability and Inhibitory Activity Against LPS-Induced Inflammation. Front Immunol. 2021, 11, No. 618312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Galante P; Campos GAA; Moser JCG; Martins DB; Dos Santos Cabrera MP; Rangel M; Coelho LC; Simon KS; Amado VM; De AI; Muller J; Koehbach J; Lohman RJ; Cabot PJ; Vetter I; Craik DJ; Toffoli-Kadri MC; Monge-Fuentes V; Goulart JT; Schwartz EF; Silva LP; Bocca AL; Mortari MR Exploring the therapeutic potential of an antinociceptive and anti-inflammatory peptide from wasp venom. Sci. Rep 2023, 13, 12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Bradbury AF; Finnie MDA; Smyth DG Mechanism of C-Terminal Amide Formation by Pituitary Enzymes. Nature 1982, 298, 686–688. [DOI] [PubMed] [Google Scholar]
- (48).Young SD; Tamburini PP Enzymatic Peptidyl.Alpha.-Amidation Proceeds through Formation of an.Alpha.-Hydroxyglycine Intermediate. J. Am. Chem. Soc 1989, 111, 1933–1934. [Google Scholar]
- (49).Eipper BA; Milgram SL; Husten EJ; Yun HY; Mains RE Peptidylglycine Alpha-Amidating Monooxygenase: A Multifunctional Protein with Catalytic, Processing, and Routing Domains. Protein Sci. 1993, 2, 489–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Merkler DJ C-Terminal Amidated Peptides: Production by the in Vitro Enzymatic Amidation of Glycine-Extended Peptides and the Importance of the Amide to Bioactivity. Enzyme Microb. Technol 1994, 16, 450–456. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.