

Abstract
Congenital PAX6-aniridia, initially characterized by the absence of the iris, has progressively been shown to be associated with other developmental ocular abnormalities and systemic features making congenital aniridia a complex syndromic disorder rather than a simple isolated disease of the iris. Moreover, foveal hypoplasia is now recognized as a more frequent feature than complete iris hypoplasia and a major visual prognosis determinant, reversing the classical clinical picture of this disease. Conversely, iris malformation is also a feature of various anterior segment dysgenesis disorders caused by PAX6-related developmental genes, adding a level of genetic complexity for accurate molecular diagnosis of aniridia. Therefore, the clinical recognition and differential genetic diagnosis of PAX6-related aniridia has been revealed to be much more challenging than initially thought, and still remains under-investigated.
Here, we update specific clinical features of aniridia, with emphasis on their genotype correlations, as well as provide new knowledge regarding the PAX6 gene and its mutational spectrum, and highlight the beneficial utility of clinically implementing targeted Next-Generation Sequencing combined with Whole-Genome Sequencing to increase the genetic diagnostic yield of aniridia. We also present new molecular mechanisms underlying aniridia and aniridia-like phenotypes. Finally, we discuss the appropriate medical and surgical management of aniridic eyes, as well as innovative therapeutic options.
Altogether, these combined clinical-genetic approaches will help to accelerate time to diagnosis, provide better determination of the disease prognosis and management, and confirm eligibility for future clinical trials or genetic-specific therapies.
Keywords: Congenital aniridia, Foveal hypoplasia, PAX6, Next-generation sequencing, Whole-genome sequencing, Gene therapy
1. Introduction
Congenital aniridia (OMIM# 106210) is a rare panocular malformation classically belonging to the large group of anterior segment dysgenesis (ASD) disorders, and is defined by the absence of iris formation during development. Congenital aniridia is an autosomal dominant condition caused by loss of function variants in the Paired box 6tbox6 gene (PAX6; OMIM 607108) or 11p13 chromosome rearrangements, and more than 700 pathogenic variants have been reported to date. The prevalence of aniridia has been estimated at 1:72.000 (Edén et al., 2008). Diagnosis of congenital aniridia is first made early in infancy as the child’s eyes appear solid black due to the expanded pupil created by the lack of iris tissue. However, classical aniridia resulting from PAX6 mutations is much more than “black eyes” and the term “aniridia” now seems inadequate in the light of recent findings.
As more aniridia patients were ascertained, it became understood that degree of iris hypoplasia can be highly variable between patients, or even between eyes of the same patient, with some aniridia patients having an apparently normal iris. Further, congenital aniridia is usually accompanied by a range of various other ocular anomalies affecting the cornea, iris, lens, fovea, iridocorneal angle, and optic nerve and can include extra-ocular features affecting the brain and the pancreas, making “congenital isolated aniridia” a syndromic panocular congenital disorder. Even more strikingly, was the recent observation that foveal hypoplasia, a congenital malformation of the retina during embryogenesis, is seen more frequently in patients harboring PAX6 mutations than the complete absence of iris. In fact, the degree of foveal hypoplasia is a main determinant factor of visual function in patients with congenital aniridia (Daruich et al., 2021). Notably, congenital iris malformations can also be a feature of other ASDs including Axenfeld-Rieger Syndrome (ARS), Peters anomaly, primary congenital glaucoma, and sclerocornea. These overlapping clinical presentations with aniridia phenotypes due to PAX6 mutations adds to the complexity of the phenotype-genotype correlations in congenital aniridia (Fig. 1).
Fig. 1. Congenital aniridia definition.
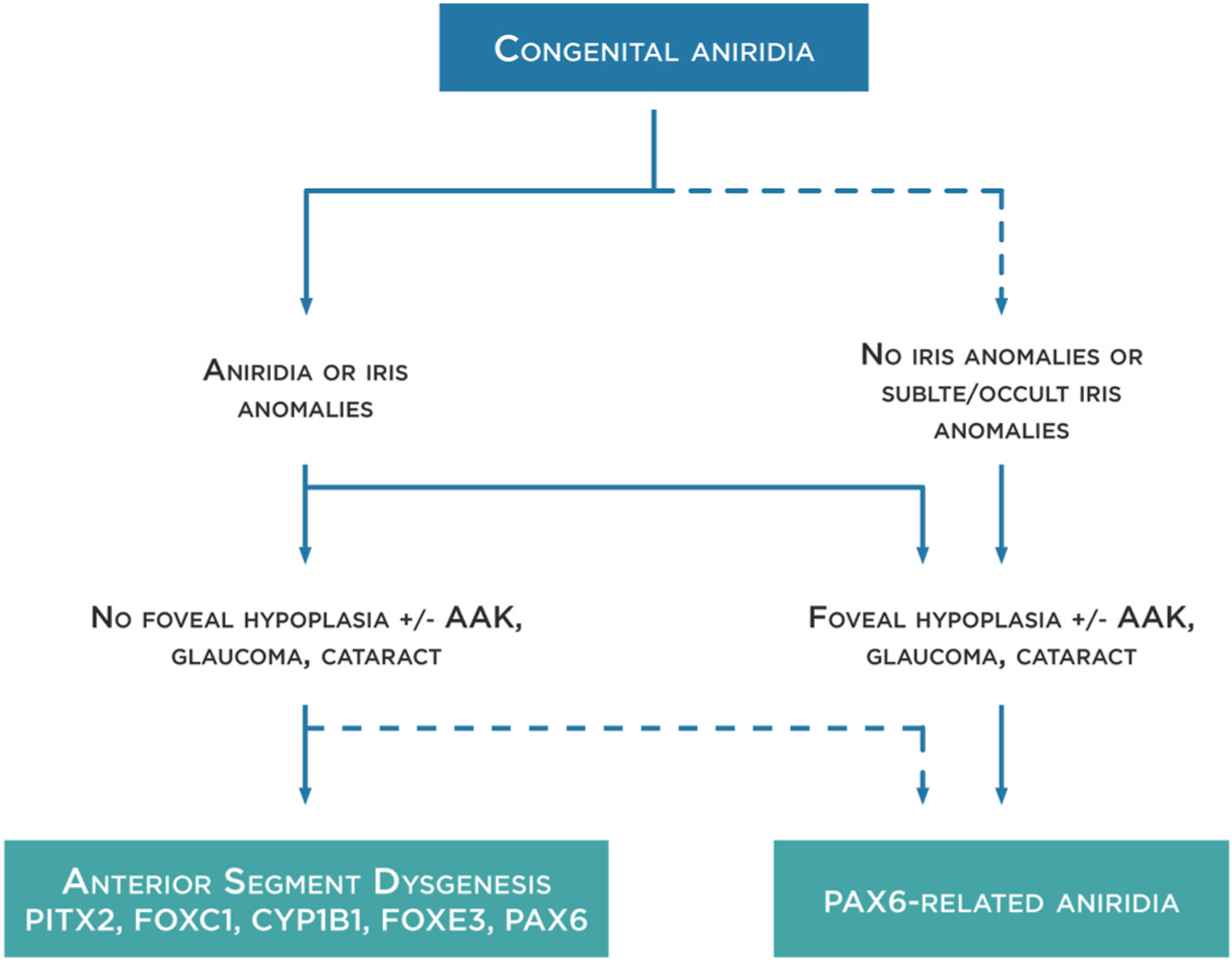
PAX6-related aniridia mostly presents with complete or partial iris hypoplasia (or aniridia) and foveal hypoplasia. However, PAX6-related aniridia could be also observed in the absence of iris anomalies. In these cases, foveal hypoplasia is usually present, as well as other aniridia-related findings such as aniridia-associated keratopathy (AAK), glaucoma and cataracts. Iris anomalies could be also part of an anterior segment dysgenesis phenotype where foveal hypoplasia and other aniridia-related findings are not present. Although PAX6 anomalies could be responsible for these phenotypes, others genes such as PITX2, FOXC1, CYP1B1 and FOXE3 are most frequently detected. Dot lines represent less frequent manifestations.
PAX6 is a highly conserved homeodomain transcriptional factor, that orchestrates the embryonic development of several tissues and organs, including the eyes, pancreas and central nervous system. It functions as a sequence-specific deoxyribonucleic acid (DNA) binding transcription factor that positively or negatively regulates a large spectrum of developmental genes. This explains both the variety of phenotypes resulting from PAX6 mutations, and the overlapping phenotypes caused by mutations in other developmental genes that either regulate PAX6 or are PAX6 regulated. Thus, the number of known genes whose mutation results in aniridia-like phenotypes has expanded over time, even though PAX6 remains the major gene mutated in this condition. Molecular diagnosis of congenital aniridia has long been a challenge due to the heterogeneity of PAX6 variant subtypes and the necessity of screening the entire PAX6 locus from the adjacent downstream ELP4 gene to the upstream WT1 gene, requiring more than one molecular and cytogenetic strategy. Recently, targeted Next-Generation Sequencing (NGS) has become clinically available for the detection of variants at very different genomic scales in one single assay, and has been implemented in molecular laboratories in many countries for the diagnosis of rare disorders. While NGS has the potential to become the one-for-all molecular test, few studies have been performed using this new technology as a single test panel for PAX6, which is desirable as it would allow for the parallel interrogation of other eye developmental genes sharing overlapping phenotypes with PAX6-related aniridia. Here, we highlight the importance of NGS and Whole-Genome Sequencing (WGS) in routine diagnostic practice as a combined platform, offering a high diagnostic yield for congenital aniridia and expanding the mutational spectrum of PAX6 by revealing “cryptic” balanced structural variants.
The purpose of this review is to highlight advances over the past decade in our understanding of the specific clinical features as well as the genetic causes underlying congenital aniridia that are critical for accurate diagnosis and patient follow-up. We also present new molecular mechanisms and genetic causes underlying aniridia and aniridia-like phenotypes, which were discovered using unbiased WGS approaches. Finally, we describe the medical and surgical management of aniridic eyes as some surgery methods routinely used in clinical practice may not be clinically appropriate in aniridia patients, and cover new insights into innovative therapeutic options including gene therapy and pharmacological approaches.
2. Vertebrate eye organogenesis: focus on the function of Pax6
The complexity of the ocular phenotypes observed in humans heterozygous for PAX6 mutations (Lima Cunha et al., 2019), and the anophthalmia observed in both humans (Glaser et al., 1994) and animals (Hill et al., 1991) homozygous for Pax6 mutations, is not surprising in light of the complex functions that Pax6 plays in nearly all aspects of ocular morphogenesis.
2.1. Early eye development
Ocular organogenesis begins at mid-gastrulation and involves intricate coordination between transcription factors, signaling pathways, and morphogenetic movements to form the adult eye (Gunhaga, 2011; Miesfeld and Brown, 2019; Schlosser, 2014).
The first morphological signs of eye development in humans begins around day 22, when the optic sulci appear as shallow grooves in the neural folds. Shh and Six3 are both critical regulators of eye field bifurcation, and their removal results in cyclopia (Jeong et al., 2008). As the eye field is divided, these cells are pushed laterally, resulting in the optic vesicles. Simultaneously, the group of cells of the anterior neuroectoderm that constitute the eye field have already begun to express a set of eye field transcription factors, that are highly evolutionary conserved, including Pax6.
Once Pax6 expression initiates in the anterior portion of the pre-placodal ectoderm, it becomes competent to form the lens, conjunctival epithelium and corneal epithelium when induced to do so by signaling from the optic vesicle (Ashery-Padan et al., 2000; Cvekl and Duncan, 2007). This function explains the complete anophthalmia arising from homozygous mutation of Pax6. The lens placode originates from Pax6-positive surface ectodermal cells (Cvekl and Callaerts, 2017) (Day 27 in humans). Lens formation is controlled by signals emanating from both the optic vesicle (positive) and the periocular mesenchyme (POM) (negative) to the lens placode. The POM inhibits lens placode formation, through Pax6 inhibition by TGFβ/Smad3 and WNT/βcatenin signaling. Once the optic vesicle evaginates from the neural plate, the vesicle physically excludes the POM from the region of ectoderm where the lens placode forms. This process relieves Pax6 repression at the surface ectoderm and brings the optic vesicle close enough to the lens placode to induce lens morphogenesis.
Once the presumptive lens ectoderm is committed, the lens placode thickens due to cell elongation and proliferation, a process that is very sensitive to Pax6 dosage as this process is delayed in mice harboring heterozygous Pax6 mutations contributing to the small lenses seen in the Small eye (Sey) mouse (Favor et al., 2008; van Raamsdonk and Tilghman, 2000). The lens placode subsequently invaginates to form the lens pit, then separates from the overlying ectoderm to form the lens vesicle (Day 36 in humans)(Cvekl and Ashery-Padan, 2014). This process is Pax6 dependent as well. Peter’s anomaly, a stalk between the cornea and lens caused by incomplete separation of the lens vesicle from the head ectoderm, is common in both humans with aniridia and mice heterozygous for Pax6 mutations (Hanson, 2003; Hanson et al., 1994; Prosser and van Heyningen, 1998). The reconstituted Pax6 expressing head ectoderm directly overlying the lens vesicle is then fated to form the Pax6 dependent tissues, the corneal epithelium and conjunctiva (Wolosin et al., 2004). Simultaneously with the commitment of the anterior pre-placodal ectoderm, the optic vesicle begins to extend from the neural tube, eventually contacting the Pax6-expressing head ectoderm where it induces lens placode thickening. Pax6 expression in the optic vesicle is necessary to establish its ability to regulate lens development (Canto-Soler and Adler, 2006; Collinson et al., 2000; Reza and Yasuda, 2004), while Pax6 expression in the head ectoderm is required for optic vesicle morphogenesis into a single retina properly oriented in the eye (Ashery-Padan et al., 2000).This mutually inductive interaction between the lens placode and optic vesicle leads to simultaneous invagination of the lens placode into the lens pit and invagination of the optic vesicle into a two-layered optic cup. The high Pax6 levels detected in the outer leaf of the optic cup cooperate with Mitf to drive the initial expression of the melanin producing enzymes needed for the differentiation of these cells into the retinal pigmented epithelium (RPE) while also blocking inappropriate differentiation of these cells into neural retina (Bäumer et al., 2003; Bharti et al., 2012; Raviv et al., 2014). The inner surface of the optic cup also has high levels of Pax6 expression, but due to the absence of Mitf, these cells instead begin to robustly proliferate forming the neuroectoderm which produces the cells of the neural retina. Pax6 later plays multiple dosage sensitive roles in the time and position dependent production of mature retinal neurons. As the neural retina and RPE are differentiating from the inner and outer layers of the optic cup, high Pax6 levels are also maintained at the hairpin turn found at the anterior of the optic cup that connects the optic cup layers into a continuous neuroectoderm (Bäumer et al., 2002; Nishina et al., 1999). Coincident with the initiation of neural retina development, neural crest cells migrate to surround the anterior optic cup where they begin to form mesenchymal connective tissue that is the first morphological sign of iris development (Fig. 2). This is followed by the extension of the region of the hairpin turn anteriorly into the neural crest derived mesenchyme and it opens up into a loop that proliferates to form the dilator muscles and epithelium of the iris.
Fig. 2.
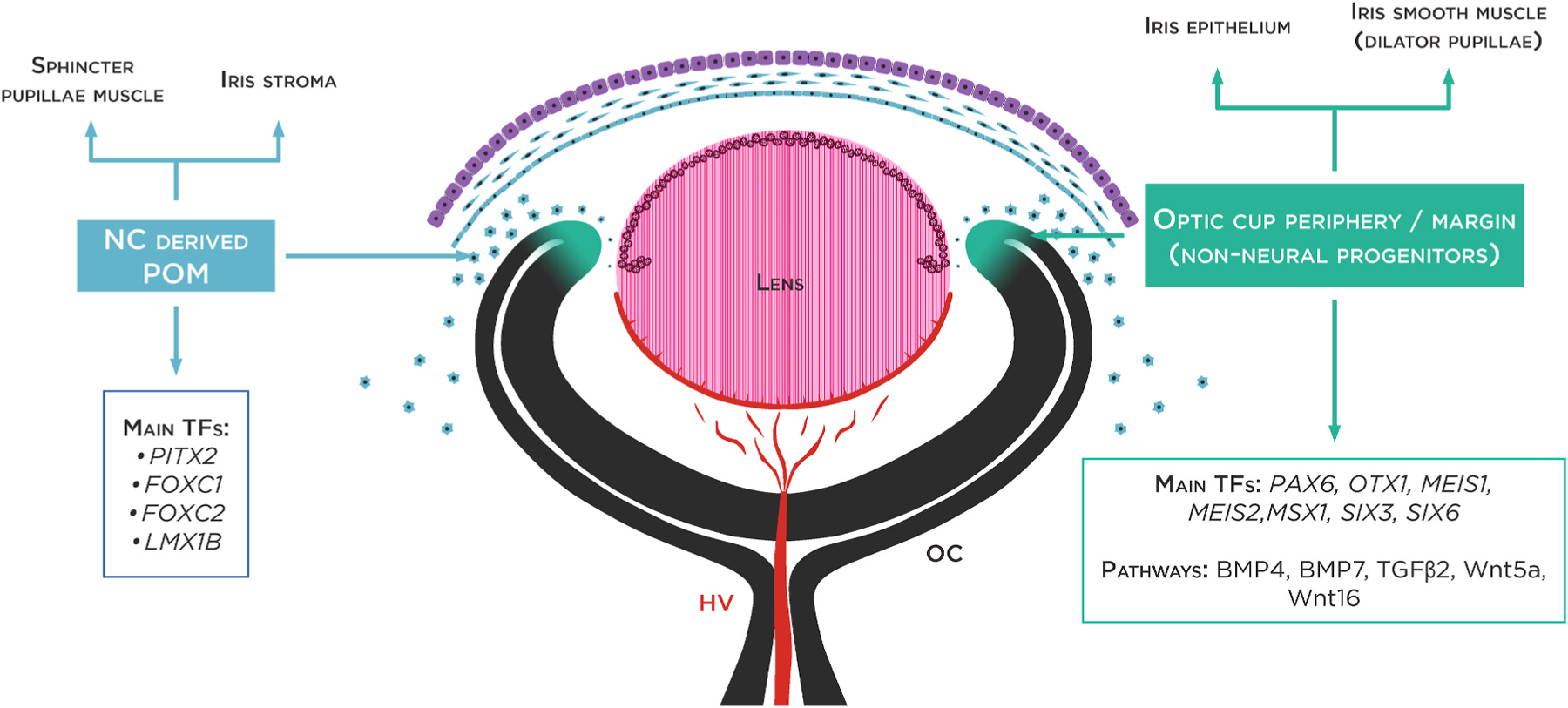
Major genes involved in iris development. Schematic overview of the developing optic cup (8 week old human embryo). Iris development relies on interactions between the periphery of optic cup (OC) or ciliary margin zone (neuroectoderm) and the periocular mesenchyme (POM), which receives contributions from the neural crest (NC) and mesoderm. Main transcription factors (TF) specifically expressed in the optic periphery cup and POM are represented.
Simultaneously, the neural crest derived mesenchyme differentiates into iris nerves and stroma, as well as the pericytes that are associated with the embryonic posterior and anterior fetal vasculatures (including a dense capillary network with hyaloid artery, tunica vasculosa lentis, and pupillary membrane) that nourish the immature retina, lens, and structures of the anterior segment in mammals (Gage et al., 2005; Imaizumi and Kuwabara, 1971; Kanakubo et al., 2006). This process is exquisitely sensitive to Pax6 dosage as the defining phenotype associated with heterozygous loss of PAX6 is aniridia (Lima Cunha et al., 2019) while elevated levels of Pax6 expression also yield iris abnormalities (Schedl et al., 1996). Pax6 expression in the anterior optic cup is necessary for both development of the anterior optic cup into musculature and epithelial layers of the iris (Davis et al., 2009; Davis-Silberman et al., 2005) and the correct recruitment of neural crest cells (which do not seem to express Pax6) to the presumptive iris mesenchyme early in iris development (Kanakubo et al., 2006; Takamiya et al., 2020). Notably, Pax6’s role in iris development may be in part driven by its Pax6(5a) splice variant which has an altered paired domain, and thus distinct DNA interaction properties from canonical Pax6 (Chauhan et al., 2004), as homozygous deletion of the 5a exon in mice results in iris hypoplasia (Singh et al., 2002) although both splice forms are necessary for normal iris development (Davis et al., 2009). The hairpin of the optic cup also gives rise to the ciliary body a structure of the anterior chamber angle that is critical for maintenance of ocular pressure, and are often abnormal in humans with PAX6 mutations (see Section 3.3.7). Specific homozygous deletion of Pax6 in the anterior optic cup profoundly affects ciliary body development demonstrating Pax6 dependence (Davis et al., 2009). However, heterozygous deletion of Pax6 from the anterior optic cup does not affect trabecular meshwork development, while reducing Pax6 expression specifically in the lens and cornea does lead to trabecular meshwork defects, suggesting that Pax6 also regulates chamber angle development non-autonomously, perhaps by dosage sensitive regulation of signaling molecule expression (Kroeber et al., 2010; X. Wang et al., 2017b).
2.2. The lens
The lens is a Pax6 dependent tissue which requires Pax6 function for its initial induction and differentiation from the head ectoderm as Pax6 regulates key downstream transcription factors (Shaham et al., 2012). Pax6 remains important later in lens development with homozygous deletion of the Pax6 gene from the embryonic lens epithelium/secondary fibers resulting in a failure of cell cycle exit at the lens equator, enhanced apoptosis and defects in lens fiber cell differentiation (Cvekl and Zhang, 2017; Shaham et al., 2009). Pax6 also directly regulates the expression of terminal lens differentiation markers, such as crystallins, which are essential for lens function (Cvekl et al., 2017; Cvekl and Zhang, 2017; Duncan et al., 1998; Xie and Cvekl, 2011).
Meis1 and Meis2 homeoproteins are considered as essential upstream regulators of Pax6 during lens morphogenesis based on their interaction with the ectoderm enhancer upstream of the Pax6 transcription start site (Antosova et al., 2016; Conte et al., 2010). The cataract-linked RNA-binding protein (RBP), Celf1, post-transcriptionally regulates Pax6 protein expression in lens development (Aryal et al., 2020). BMP, FGF, and WNT signals within the ectoderm regulate lens induction as well (Faber et al., 2002; Machon et al., 2010; Zhao et al., 2008). BMP activity regulates the specification and formation of the lens placode (Gunhaga, 2011) and is also required for primary lens fiber cell differentiation (Faber et al., 2002).
The lens is very sensitive to Pax6 dosage as heterozygous germ line mutations in mice result in the “small eye (Sey)” phenotype (Hill et al., 1991), which is characterized by congenital reductions in eye size driven by reductions in lens size (Bassnett and Šikić, 2017; Collinson et al., 2001; Voskresenskaya et al., 2021), the extent of which is highly dependent on genetic backgrounds (Hickmott et al., 2018). In some cases these mice exhibit congenital cataracts (Gregory-Evans et al., 2014), in others, Sey mice develop cataracts by middle age with some reports finding a prevalence of anterior subcapsular cataracts due to the fibrotic transition of the lens epithelium (Duncan et al., 2000; Lovicu et al., 2004) and cortical opacities driven by lens fiber cell abnormalities (Duncan et al., 2000). Interestingly, it has been suggested that defects of the iris and cornea may be secondary consequences of primary defects in the lens (Beebe and Coats, 2000; Collinson et al., 2001; Takamiya et al., 2020).
Two other key regulators of lens development are the transcription factors PITX3 and FOXE3. Loss of either gene causes lens aphakia (Blixt et al., 2000; Semina et al., 2000; Valleix et al., 2006).
2.3. The cornea
The cornea develops from the surface ectoderm and POM, which receives contributions from both the neural crest and mesoderm (Johnston et al., 1979; Lwigale, 2015; Trainor et al., 1994; Trainor and Tam, 1995; Walker et al., 2020).
The corneal epithelium, like the lens placode, originates from Pax6 expressing cells of the surface ectoderm. Once the lens placode invaginates and separates from the surface ectoderm to create the lens vesicle, the adjacent Pax6-positive cell population merges to create a contiguous cornea epithelium (Collomb et al., 2013). If proper levels of Pax6 are not present at this stage, improper separation of the lens and cornea leading to Peter’s anomaly can occur.
The laminar structure of the cornea is established by three waves of neural crest cells/POM migration from the dorsal neural tube to populate the periocular space between the lens vesicle and corneal epithelium (day 39 in humans) (Cvekl and Tamm, 2004; Gage et al., 2005; Williams and Bohnsack, 2015). The first set of POM cells differentiate into the corneal endothelium, the second wave into the corneal stroma, and the third one contributes to iris stroma development.
After the laminar structure of the cornea is established, the corneal epithelium begins to differentiate into a stratified epithelium with a basal proliferative cell population that expresses high levels of Pax6. This transient amplifying cell population is maintained by a Pax6-expressing peripheral corneal stem cell niche at the corneal “limbus” which is found in humans in an anatomically distinct region denoted as the “Palisades of Vogt” (Kitazawa et al., 2017; Li et al., 2015; Wolosin et al., 2004). Corneal development completely fails in mouse embryos homozygous for Pax6 mutations due to the failure of early lens development. Later, Pax6 expression is maintained at high levels in the corneal epithelium where it plays critical cell autonomous roles in maintaining corneal epithelial fate (Collinson et al., 2003).
Like the lens, the cornea is exquisitely sensitive to Pax6 gene dosage. Overexpression of Pax6 in the mouse corneal epithelium leads to defects throughout the cornea that become progressively more severe with age (Davis and Piatigorsky, 2011; Mort et al., 2011). Pax6 haploinsufficiency resulting in reduced levels of Pax6 expression in corneal epithelial cells does not appear to affect primary corneal morphogenesis as humans with aniridia and small eye mice are typically born with transparent corneas. However, subtle corneal epithelial defects quickly develop in the aniridic cornea after birth (Lagali et al., 2020a) which often progresses to the potentially blinding condition aniridia-associated keratopathy (AAK, see section 3.3.5).
Moreover, in all species, Pitx2 and Foxc1 are expressed in the POM and are essential for proper anterior segment genesis. Loss of Foxc1 causes abnormal cornea stromal thickening. Both Pitx2 germline and POM-specific mutant mice completely lack the corneal stroma (Evans and Gage, 2005; Gage et al., 1999). Interestingly, TGFβ released from the lens is required for the expression of transcription factors Pitx2 and Foxc1 in the POM (Ittner et al., 2005). Retinoic acid signals also control expression of Pitx2 and Foxc1 in POM (Matt et al., 2005).
2.4. The iris
The iris is made up of several different cell types whose organization during embryogenesis relies on interactions between the optic cup neuroectoderm and the POM (Fig. 2) and include specification of the peripheral optic cup to a non-neuronal fate, migration of cells from the surrounding POM and an atypical formation of smooth muscles from the neuroectoderm (Davis-Silberman and Ashery-Padan, 2008) with signals from the lens serving as important triggers for iris development. Interestingly, the pupillary membrane initially covers the whole surface of the lens and then vacuolates to create the opening in the iris sphincter.
2.4.1. Iris pigment epithelium
The ciliary body, iris pigment epithelium and the dilatator muscle of the iris derive from the non-neural progenitors at the periphery of the optic cup (ciliary margin) as of the third month of gestation in humans. The development of the iris muscle is a rare example of muscles derived ectodermally rather than mesodermally (Davis-Silberman and Ashery-Padan, 2008; Jensen, 2005). Prior to the morphogenesis of the iris, the enhanced expression of transcription factors (Pax6, Otx1, Meis1 and Meis2) and the growth-arrest-specific protein Gas1 at the periphery of the optic cup leads to its compartmentalization into neuronal/central versus non-neuronal/peripheral progenitors (Davis-Silberman and Ashery-Padan, 2008; Dupacova et al., 2021). High levels of Pax6 protein along the optic cup periphery are required for the specification of the iris progenitor cells. The developing iris smooth muscle expresses Pax6 (Jensen, 2005). Pax6 regulation of iris epithelium development is cell autonomous and some of the downstream targets include Foxc1, BMP4 and TGFβ2 (X. Wang et al., 2017b). Otx1 null mice lack a ciliary epithelium and have an underdeveloped iris epithelium (Acampora et al., 1996). Deletion of Tsc1 leads to a premature upregulation of mTORC1 activity within the ciliary margin and a reduction in the number of cells that express Pax6 (Hägglund et al., 2017). In conjunction with transcription factor-mediated regulation, several signaling pathways orchestrate iris epithelium development, such as BMP4, BMP7, canonical WNT signaling (β-catenin mediated) and the Hippo signaling pathway (Davis-Silberman and Ashery-Padan, 2008; Fokina and Frolova, 2006; Gage and Zacharias, 2009). The essential role of PAX6, BMP4 and WNT2b in the specification and morphogenesis of the iris is also suggested by clinical syndromes and functional studies (Davis-Silberman and Ashery-Padan, 2008).
2.4.2. Iris stroma
The pupillary membrane and iris stroma originate from POM, arising from the third wave of migrating neural crest cells (Fig. 2) (Creuzet et al., 2005). As POM also give rise to the cornea endothelium and stroma, the corneal endothelium, corneal stroma, ciliary stroma, and iris stroma all require the same transcription factors and signaling pathways for proper specification and differentiation. For instance, Pitx2, Foxc1, Foxc2 and Lmx1b are all expressed in the POM, and mutations in each gene can cause ciliary body and iris hypoplasia (Smith et al., 2000). The combined loss of Foxc1/2 downregulates Pitx2 expression, and a subset of Pitx2 target genes, Dkk2 and Tfap2b (Seo et al., 2017). Retinoic acid signaling also regulates ciliary body and iris development, by activating Pitx2, Foxc1, and Lmx1b expression (Matt et al., 2005; Miesfeld and Brown, 2019). Interestingly, three retinoic acid (RA) receptors, RARα, RARβ, and RARɣ, directly bind to a retinoic acid response element in the Pitx2 promoter (Kumar and Duester, 2010). Thus, the loss of retinoic acid signaling at this developmental stage results in iris hypoplasia. Pax6 expression in the iris epithelium and musculature then regulates the expression of molecules involved in guidance to, or adhesion of, migratory cells to the developing iris (N-cadherin, R-cadherin) (Davis-Silberman and Ashery-Padan, 2008).
2.5. The iridocorneal angle
The structures of the iridocorneal angle are the last to differentiate during anterior eye development and include the trabecular meshwork and Schlemm’s canal. These Iridocorneal structures originate from POM particularly under the expression of Pitx2 and Foxc1. After the beginning of iris elongation (at gestational age 15th to 20th weeks in human), the chamber angle is occupied by a dense mass of mesenchymal cells. These cells elongate, flatten and separate leaving small spaces between them that are partially occupied by extracellular fibers (Cvekl and Tamm, 2004). At that time, blood vessels appear in the adjacent sclera. Then, the extracellular fibers in the chamber angle form beams, that are then covered by endothelial-like cells. This leads to the mature structure of the trabecular meshwork, which consists of trabecular beams separated by intertrabecular spaces that allow aqueous humor outflow. The scleral vessels next to the chamber angle coalesce into a circumferential Schlemm’s canal that touches the outer side of the trabecular meshwork (Cvekl and Tamm, 2004).
Pax6 activity is required for the differentiation of the iridocorneal angle. However, there are some discrepancies concerning the expression of Pax6 in neural crest-derived cells during the development of the iridocorneal angle (Baulmann et al., 2002; Kanakubo et al., 2006). In Pax6 (lacZ/+) mice, trabecular meshwork cells remained undifferentiated and Schlemm’s canal was absent (Baulmann et al., 2002). However, it is not clear if these changes are caused by cell autonomous or non-autonomous mechanisms. It has been suggested that Pax6 probably controls the expression of signaling molecules in lens and corneal cells that regulate trabecular meshwork and Schlemm’s canal formation (Kroeber et al., 2010).
2.6. The retina and fovea
2.6.1. The retina
Shortly after optic cup formation, Pax6 levels are highest in the anterior most portion, and lowest in the most posterior portion, of the presumptive neural retina, and this regulates the differences in axonal projection from the retinal ganglion cells in the adult neural retina (Bäumer et al., 2002) and the posterior-anterior gradient in the timing of photoreceptor production (Oron-Karni et al., 2008).
Another dosage sensitive role that Pax6 plays is in regulating the correct timing of neurogenesis across the retina. It is now recognized that retinal neurons are formed from the retinal neuroepithelium based on the “age” of the retinal precursors, with the ganglion cells produced first, followed by the horizontal cells, cones, and amacrine cells while the rods, bipolar cells, and Muller glia are produced in a second wave of neurogenesis (Diacou et al., 2022; Marquardt and Gruss, 2002). Optic vesicles from mice homozygous for the Pax6 null allele exhibit reduced neuroepithelial proliferation and precocious differentiation of retinal neurons (Philips et al., 2005) while specific deletion of Pax6 in the inner optic cup leads to precocious differentiation of the neuroepithelium to amacrine cells (Marquardt et al., 2001). Meis1 and Meis2 homeobox genes function to maintain the retinal progenitor cells pool (Dupacova et al., 2021). Later in retinal development, Pax6 also is critical for the production of bipolar neurons, and glycinergic amacrine neurons, while simultaneously inhibiting photoreceptor differentiation (Remez et al., 2017). While Pax6 expression fades in most retina cell types during their differentiation, it is sustained throughout life in humans within retinal ganglion cells and a subset of neurons of the inner nuclear layer (Stanescu et al., 2007). Consistent with this, Pax6 regulates retinal ganglion cell specification (Farhy et al., 2013; Mu and Klein, 2004) and its correct dosage in these cells is critical for the proper elongation and routing of retinal ganglion cell axons to the optic nerve and their final synaptic contacts in the thalmus (Lalitha et al., 2020; Manuel et al., 2008; Sebastián-Serrano et al., 2012). These complex roles of Pax6 in retinal development likely explain the defects observed in the thickness of the plexiform layers in both the Sey mouse (Curto et al., 2007) and humans with aniridia (Pedersen et al., 2020), as well as the defective foveal development that drives a great deal of the primary visual deficits seen in human aniridia (Casas-Llera et al., 2020) (see section 3.3.3, 3.3.4 and 3.3.8).
2.6.2. The fovea
The fovea is a structure found in primate retinas which is responsible for high resolution color vision due to its high concentration of cones (fovea externa) and the exclusion of retinal cell types found in the inner retina to form a pit (fovea interna) (Bringmann et al., 2018). Foveal development has been recently reviewed in detail (He et al., 2022).
The location of the future fovea is determined by the specification of the central rod-free zone during the development of the optic vesicle (fetal week 3–4 in humans), a precondition for foveal pit development (fetal weeks 25–26 in humans) (Bringmann et al., 2018).The development of the foveal pit also requires the absence of retinal blood vessels (Provis, 2001; Provis et al., 2013) with the depth of the foveal pit and the area of the fovea correlating with the size of the foveal avascular zone in humans. This foveal avascularity appears to be induced by ganglion and Müller cells in the foveal center, which express antiangiogenic molecules such as pigment epithelium-derived growth factor and brain natriuretic peptide (Kozulin et al., 2009a, 2009b). In the developing macaque retina, Eph-A6 -A1 and -A4 repellent signaling has a role in retinal vascular patterning, and in the postnatal maintenance of projections from macular and foveal ganglion cells (Kozulin et al., 2009a, 2009b). Another possible factor which was suggested be involved in inhibiting foveal vascularization is the macular pigment (Gariano, 2010).
The fovea externa develops relatively independently from the fovea interna (Bringmann et al., 2018). The fovea externa is formed by a centripetal displacement of photoreceptors, while the fovea interna is formed by a centrifugal displacement of inner retinal layers. The centripetal displacement of photoreceptors to originate the fovea externa, takes place over three phases, from fetal week 8 until the postnatal period in humans. The post-natal displacement of central photoreceptors results in an at least a 4-fold increase in foveolar cone density (Bringmann et al., 2018).
Foveal development is very sensitive to Pax6 dosage as foveal hypoplasia is one of the most consistent phenotypes observed in patients carrying heterozygous PAX6 mutations. However, Pax6’s role in foveal development is not well understood, because mice, the most common vertebrate model for studies on Pax6 function, do not have a foveated retina. However, observations in primates have concluded that Pax6 plays an important role in cell type specification/differentiation and migration of cones toward the foveal center (Bringmann et al., 2018; Pedersen et al., 2020; Shaham et al., 2012). PAX6 may also regulate foveal development via its established roles in retinal ganglion cell development (a cell type excluded from the fovea) and the development of the retinal pigmented epithelium which secretes antiproliferative and antiangiogenic factors, which could be critical to prevent retinal blood vessels from invading the developing fovea (Pedersen et al., 2020). Consistent with this possibility, it was shown that Pax6 activity was required prior to foveal pit formation in anoles, a lizard species that has a foveated retina (Sannan et al., 2018).
3. Aniridia: definition and clinical forms
Congenital aniridia is a rare congenital panocular disorder defined as a complete or partial absence of the iris (Landsend et al., 2021), which is commonly combined with developmental abnormalities of the cornea, lens, optic nerve and fovea. Foveal hypoplasia is a consistent finding resulting in low visual acuity and nystagmus (Fig. 1). The clinical diagnosis of congenital aniridia is often established soon after birth as the iris is missing, causing the baby to appear to have a very dark brown or black eye color, although it can be misdiagnosed as bilateral congenital mydriasis. However, in other cases, the entire iris may be present and apparently intact,or with only subtle changes, making clinical diagnosis more difficult and genetic testing necessary to establish the final diagnosis of aniridia (Kit et al., 2021).
Corneal opacification resulting from limbus insufficiency, cataracts, and glaucoma, usually of later onset, and optic disc hypoplasia further exacerbate visual impairment. Non-ocular anomalies including diabetes, hyposmia, hearing difficulties, sleep disorders and structural brain changes have also been reported in association with aniridia (Dansault et al., 2007; Sisodiya et al., 2001).
About two-thirds of aniridia cases are familial with the remaining cases being sporadic (Hingorani et al., 2012). Human PAX6 is the major gene responsible for autosomal dominant forms of congenital aniridia (Lima Cunha et al., 2019). Aniridia can be either isolated or syndromic, manifesting as part of WAGR (Wilms’ tumor-aniridia-genital anomalies-retardation), WAGRO (WAGR and obesity) or Gillespie syndromes (non-progressive cerebellar ataxia, intellectual disability, iris hypoplasia).
3.1. Clinical diagnosis
There are classically four main ocular phenotypes that lead to a diagnosis of aniridia: black eye color at birth or soon after, nystagmus in infancy, low vision in a child leading to the diagnosis of a foveal hypoplasia, and infantile cataracts. Early diagnosis of congenital aniridia relies on clinical and genetic findings (Fig. 3), and precise phenotypic characterization is important for appropriate management. First of all, it should be determined whether congenital aniridia is isolated or syndromic by assessing the patient for genitourinary and neurological anomalies. Prompt diagnosis of syndromic aniridia is critical to minimize the risk of life threatening cancer as WAGR or WAGRO diagnoses are associated with Wilms’ tumor in half of patients with WT1 deletion (WT1 kidney tumor suppressor gene) (Millar et al., 2017). In Gillespie syndrome, the early diagnosis of cerebellar ataxia is also essential for prompt onset of therapies that improve prognosis. Next, an extensive ophthalmological workups needs to be performed on newly diagnosed patients to fully characterize the ocular phenotype so that vision preserving therapies can be initiated when warranted. Assessing foveal hypoplasia is of special interest in the diagnosis of congenital aniridia, particularly when iris defects are subtle. The grading of foveal hypoplasia will also be predictive of visual prognosis. Anterior segment anomalies should be identified early in life as well as, especially those needing urgent treatment, such as congenital glaucoma.
Fig. 3. Congenital aniridia workup.
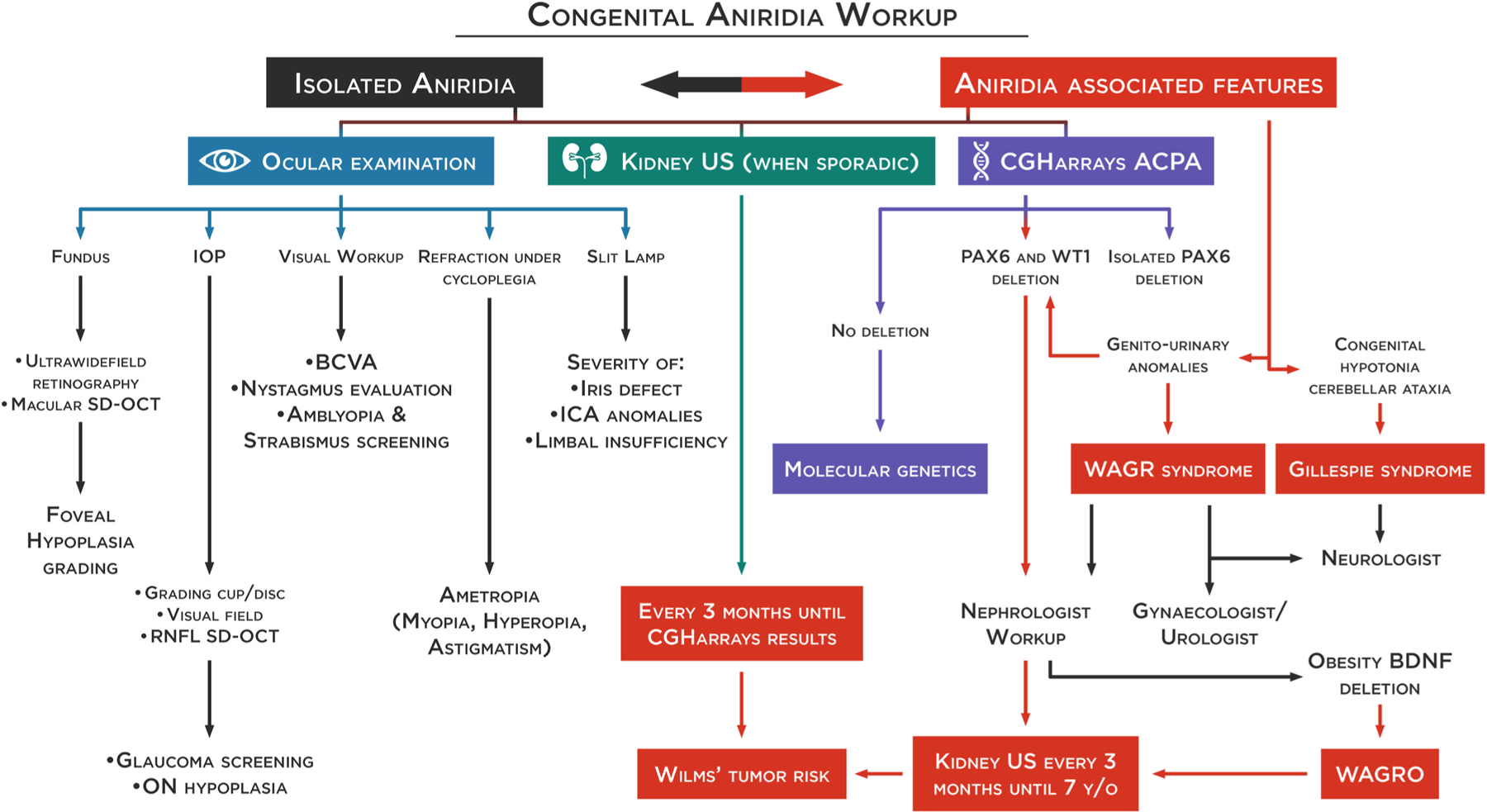
Differentiation of isolated or syndromic congenital aniridia is essential for appropriate management and follow-up. Prompt diagnosis of WAGR or WAGRO is critical to avoid vital risks. An extensive ophthalmological workup needs to be performed to characterize the ocular phenotype, especially foveal hypoplasia through spectral-domain optical coherence tomography (SD-OCT). A genetic workup should first include comparative genomic hybridization arrays to allow for the quick identification of isolated deletions on the PAX6 gene or deletions involving the WT1 gene. In sporadic cases, and until the results are from comparative genomic hybridization arrays, a kidney ultrasound should be performed every 3 months for early detection of a Wilms’ tumor. In case of WT1 deletion, a kidney ultrasound should be continued every 3 months until the age of 7. If no deletion is found, a molecular genetic test (NGS and/or WGS) will be performed to confirm the clinical diagnosis and identify the pathogenic variants allowing phenotype-genotype correlations. It is to be noted that NGS-based protocol has the potential of becoming the one-for-all test for molecular diagnosis of aniridia (see section 4.3 and 4.4), replacing the standard strategy represented here. BCVA: best-corrected visual acuity, ON: optic nerve, ICA: iridocorneal angle, RNFL: retinal nerve fiber layer.
The genetic workup should first include comparative genomic hybridization arrays to enable the quick identification of isolated deletions within PAX6 structural gene or its regulatory elements as well as deletions involving the WT1 gene. If no deletion is found,a molecular genetic testing should be performed to confirm the clinical diagnosis and identify the pathogenic variants allowing phenotype-genotype correlations (and possible eligibility for future clinical trials). It is to be noted that NGS-based diagnostic protocols have the potential to become the one-for-all test for molecular diagnosis of aniridia, replacing the standard strategy represented here (see Section 4.3 and 4.4). This diagnostic algorithm is the currently strategy used because the implementation of NGS is not available in all molecular laboratories worldwide because of the cost involved, the infrastructure required and the bioinformatic analytical challenges. Fig. 4 shows the phenotype and genotype of the congenital aniridia cohort from BaMaRa, the French national database for rare diseases.
Fig. 4. Genetic characteristics of the aniridia cohort from BaMaRa,
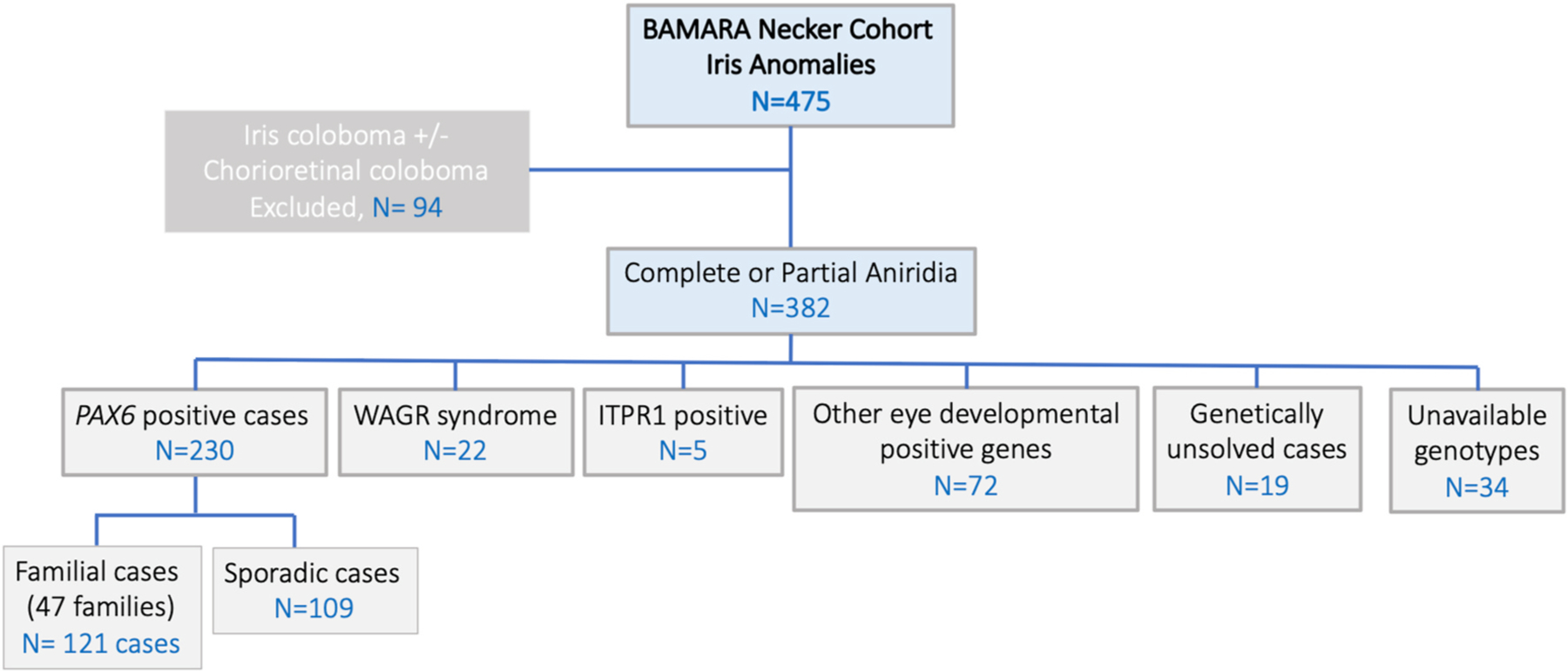
the French national database for rare diseases. Schematic representation of the cohort of 475 patients with iris anomalies. Distribution of genes found to be mutated among 382 patients who presented with aniridia phenotype after excluding patients presenting iris coloboma.
3.2. Syndromic aniridia
3.2.1. WAGR and WAGRO syndrome
Aniridia can be a feature of three syndromes: WAGR, WAGRO and Gillespie syndromes. WAGR syndrome (Wilms’ tumor, Aniridia, Genitourinary anomalies, and mental Retardation) (MIM194072) is a rare contiguous gene deletion syndrome, characterized by a de novo deletion of 11p13 including the WT1 (Wilms’ tumor and genitourinary anomalies) and PAX6 (aniridia) genes. The acronym WAGR is used even in cases where all four conditions are not present. Several non-canonical WAGR features have been also described, including obesity, obstructive sleep apnea, autism spectrum disorders, severe dental malocclusion and musculoskeletal anomalies, among others (Fischbach et al., 2005). Of these non-canonical WAGR features, obesity has generated the greatest clinical interest and has led to the suggestion that WAGR be re-designated as WAGRO (Hall et al., 2019) (MIM 612469). The responsible genes (or gene) for neurodevelopmental problems and obesity in WAGR syndrome have not yet been confirmed. PAX6 has been proposed, given its involvement in central nervous system (CNS) development and the small proportion of cases associating PAX6 mutations and developmental delay or autism (Chien et al., 2009; Davis et al., 2008). Alterations of the brain-derived neurotrophic factor (BDNF) gene have been also implicated in both the neurodevelopmental problems and obesity observed in WAGR (Hall et al., 2019; Han et al., 2008, 2013; Xu et al., 2008). Finally, alterations in the genes SLC1A2 and PRRG4 may also influence the neurodevelopmental component of WAGR syndrome (Xu et al., 2008; Yamamoto et al., 2014).
Early diagnosis of WAGR syndrome is essential to maximize prognosis. A kidney ultrasound for Wilms’ tumor detection should be performed every 3 months when sporadic aniridia (isolated or not) is present until molecular diagnosis rules out WAGR deletion. If WAGR deletion is confirmed, a kidney ultrasound should be performed every 3 months until the age of 7. Additionally, a neurological and urological/gynecological follow-up will be needed for early recognition and management of intellectual disability, autism and genitourinary anomalies.
3.2.2. Gillespie syndrome
Gillespie syndrome (OMIM 206700) is characterized by partial aniridia, presenting as congenital mydriasis, in association with non-progressive cerebellar hypoplasia and ataxia, intellectual disability and congenital hypotonia. The aniridia phenotype in Gillespie syndrome differs from that in classical aniridia. The iris defect is bilateral with a distinct scalloped pupillary margin and iridolenticular strands. This scalloped pupillary margin reveals the persistent pupillary membrane on the rim of the iris sphincter. Interestingly, foveal hypoplasia has rarely been observed in Gillespie syndrome (Dollfus et al., 1998; Hall et al., 2019; Ticho et al., 2006). In our cohort, all patients affected with Gillespie syndrome had a normal foveal structure. Gillespie syndrome is considered a monogenic disorder caused by monoallelic or biallelic pathogenic mutations of the ITPR1 gene, which regulates the formation of anterior eye segment tissues derived from neural crest cells (Kinoshita et al., 2021).
A Gillespie syndrome-like iris defect is also evident in the fixed dilated pupils of individuals with severe multisystemic smooth muscle dysfunction syndrome (OMIM 613834) caused by Arg179 substitutions in the ACTA2 gene (actin, alpha 2, smooth muscle, aorta; OMIM 102620). In these cases iris defects are associated with congenital cardiac features including patent ductus arteriosus (PDA, 91%) or aortopulmonary septal defect (9%) (Hall et al., 2019).
3.3. Isolated aniridia
3.3.1. Nystagmus characteristics
Repetitive, uncontrolled eye movements known as nystagmus usually appear between one and three months of age in aniridia caused by PAX6 mutations; this symptom also lead to the diagnosis of aniridia. Its clinical and oculographic features are those of infantile nystagmus syndrome (INS) (Choi et al., 2021; Lee et al., 2019), and are frequently associated with a vertical component. In a series of 5 patients with PAX6-related aniridia with interpretable oculomotor recordings seen in our clinic, we found specific INS clinical characteristic and waveforms, associated in 4 cases with a vertical, upbeat nystagmus, component.
3.3.2. Iris phenotype
Variable degrees of iris hypoplasia have been described in congenital aniridia. In most cases, there is a complete absence of the iris giving the appearance of a black eye. However, iris hypoplasia can be partial or present as a pseudo-coloboma. In some cases, the entire iris can be present with only subtle changes. In such cases, “aniridia” is a misnomer, but can yet be used, as the phenotype can otherwise be unremarkable. Most congenital aniridia patients display symmetrical changes, but some of them can present with very asymmetrical iris defects (Kit et al., 2021). Variability of iris hypoplasia is often observed among family members (Kit et al., 2021; Landsend et al., 2021).
Missense mutations of the PAX6 gene has been significantly associated with milder grades of iris hypoplasia, compared with deletion or nonsense, frameshift and intronic mutations (Kit et al., 2021). Importantly, for the Gillespie Syndrome the iris defect is bilateral and has a distinct scalloped appearance resulting from the aplasia of the tissue central to the collarette (Hall et al., 2019). Fig. 5 represents the variability of iris hypoplasia found in congenital aniridia and the associated gene mutation.
Fig. 5. Variability of iris hypoplasia in congenital aniridia and genotype correlation.
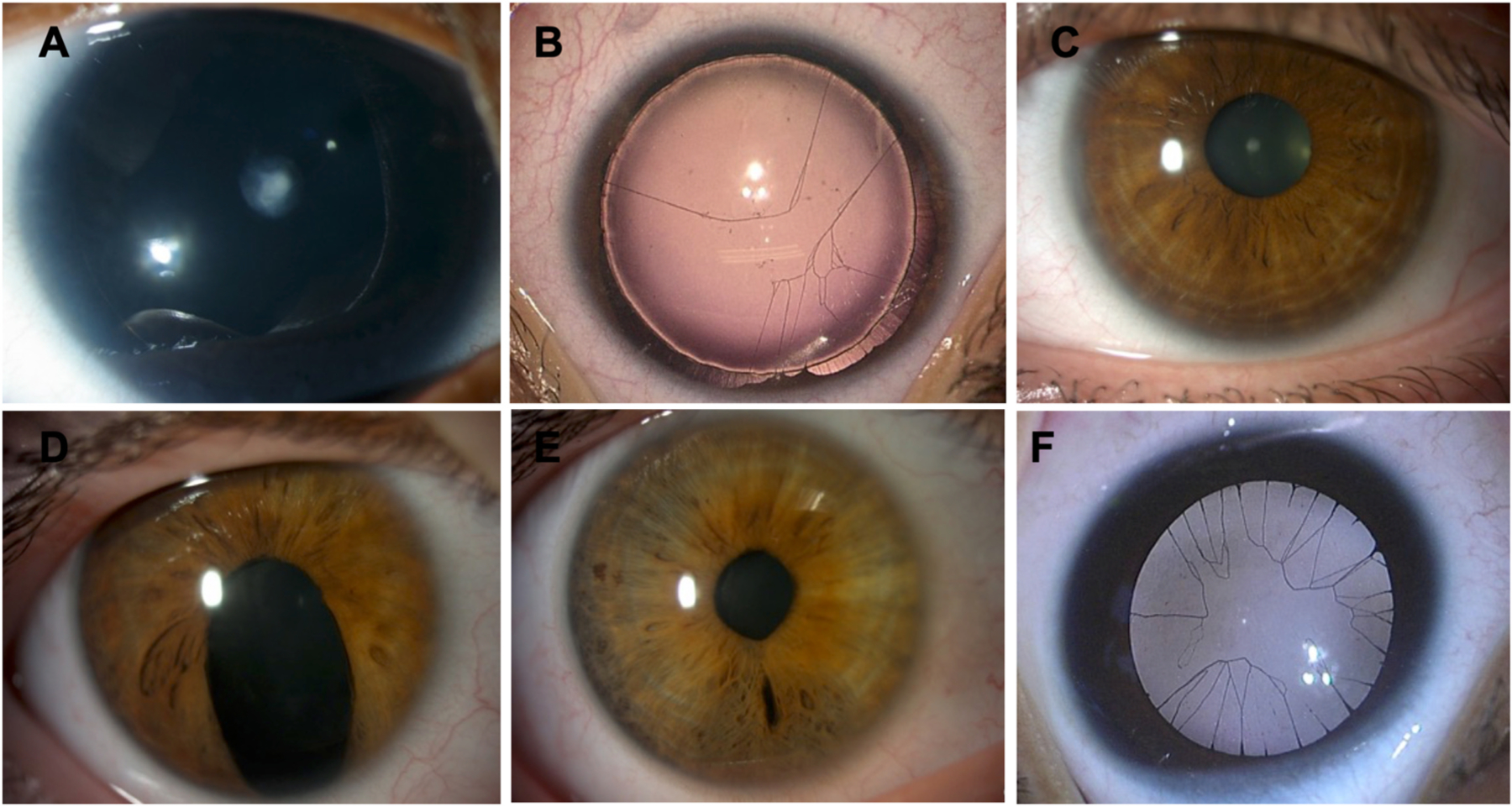
PAX6-related congenital aniridia is associated to various iris phenotypes such as complete aniridia with subcapsular anterior cataracts (A), iris root and iris membrane remnants (B), almost normal iris (C), iris pseudocoloboma (D), discrete iris defects in the contralateral eye of the same patient (E) and congenital aniridia in Gillespie syndrome with iris membrane remnants (F).
3.3.3. Foveal phenotype
More than 90% of patients with congenital aniridia display foveal hypoplasia of variable severity (Daruich et al., 2021; Pedersen et al., 2018) which is likely the reason for nystagmus being common on congenital aniridia. Since foveal hypoplasia is the most reliable sign for aniridia diagnosis, an early Spectral Domain Optical Coherence Tomography (SD-OCT) is recommended, particularly in cases with mild iris anomalies or asymmetrical features. The degree of foveal hypoplasia is most frequently correlated with the severity of iris defects. However, it has been reported that about 70% of patients with a PAX6 mutation and mild iris phenotype presented severe foveal hypoplasia (Daruich et al., 2021). Foveal hypoplasia is symmetrical in most patients, with only a few cases showing different stages of arrest in foveal development between the eyes (Daruich et al., 2021). Foveal hypoplasia variability has been seen within members of the same family (Pedersen et al., 2019).
About 70% of patients diagnosed with congenital aniridia present with severe foveal hypoplasia (grade 3 or 4 in the Thomas classification) (Thomas et al., 2011), suggesting that the stage of arrested foveal development is likely during the centripetal migration of cone photoreceptors from the periphery towards the fovea, which is consistent with the major role of PAX6 in the very early events of foveal morphogenesis (Klimova and Kozmik, 2014; Sannan et al., 2018; Shaham et al., 2012) (see Section 2.6.2). The degree of foveal hypoplasia is usually correlated with best-corrected visual acuity (Daruich et al., 2021). Interestingly, the thickness of foveal outer retinal layers progressively decreases with the severity of foveal hypoplasia. It has been shown that the thickness of the foveal outer retinal layers is the strongest predictor of high visual acuity in congenital aniridia (Pedersen et al., 2020). Aniridia subjects also have reduced maximum cone density in the central fovea compared with normal control individuals, assessed through an adaptive optics scanning light ophthalmoscopy (Pedersen et al., 2019).
The loss of function mutations in PAX6, including prematurely truncating nonsense, frameshift, and whole/partial coding sequence deletions, has been associated with a more severe foveal hypoplasia than mutations that delete 3’ flanking regulatory PAX6 regions without removing any of its coding sequences (Daruich et al., 2021; Pedersen et al., 2020) (See section 4.5). Missense variants showed a variable degree of foveal hypoplasia, including grade 3 and 4 foveal hypoplasia (Daruich et al., 2021; Kit et al., 2021). Interestingly, foveal hypoplasia has rarely been observed in the Gillespie Syndrome (Dollfus et al., 1998; Hall et al., 2019; Ticho et al., 2006) (see section 3.2.2)
3.3.4. Other retinal findings
Patients with aniridia have varying degree of retinal dysfunction, ranging from severely abnormal to almost normal. Rod and cone function are equally affected. The mutations affecting the paired domain appears to have more impact on retinal function than other regions of the PAX6 protein (Tremblay et al., 1998; Wu et al., 1991). It has also been reported that 86% of aniridia patients exhibit a hypopigmented fundus (Erlend C. S. Landsend et al., 2019), that may result from the role of Pax6 in embryonic differentiation of the RPE.
Finally, retinal detachment has been reported in congenital aniridia, specially associated to giant retinal tears (Hama et al., 2010; Landsend et al., 2021; Mirrahimi et al., 2019). Prior to ocular surgery, vitreoretinal abnormalities, and buphthalmos could possibly contribute to these detachments. In our cohort of 230 PAX6-related aniridia patients, 2 children presented with retinal detachment during follow-up.
3.3.5. Corneal phenotype
In recent years, a more detailed phenotypic characterization of the cornea in aniridia has revealed that AAK is not just a prominent central, white opacity in the cornea, but rather that keratopathy in congenital aniridia is a slowly progressive opacification or conjunctivalization of the cornea, which in the earliest stages preserves central corneal transparency (Ihnatko et al., 2016; Lagali et al., 2013, 2018). Although AAK is believed to originate from an insufficiency in limbal stem cells (Latta et al., 2021a), keratopathy distinguishes itself phenotypically from limbal stem cell deficiency (Deng et al., 2019) through the gradual, radial ingrowth of conjunctival tissue in a characteristic pattern, which only affects the central cornea in the later stages, eventually transforming into a thick, whitish opacity. Recognizing the need for careful staging of AAK according to published grading scales (Ihnatko et al., 2016; Lagali et al., 2020b), the prevalence of AAK exceeds 90% in congenital aniridia under these refined definitions. This grading distinguishes a complete absence of limbal involvement (Grade 0), the early encroachment of the conjunctival pannus within 1 mm of the limbal border (Grade 1), the subsequent radial ingrowth of the pannus into the peripheral cornea (Grade 2), central corneal involvement (Grade 3) and late-stage thick, vascularized pannus (Grade 4). This grading scheme characterizes the ‘classic’ form of AAK that progresses with age, and thus it is only meaningful to quote the AAK grade in relation to patient age. Unfortunately, many reports do not include this information, or overlook the early stages of keratopathy and thus erroneously conclude an absence of corneal involvement. Moreover, as classical AAK is progressive and reported to affect the central cornea typically only starting in the second to third decade of life (Lagali et al., 2018, 2020a), studies reporting genotype-phenotype associations in children may not note keratopathy as being a defining phenotypic feature. Moreover, with the more recent and broader definition of AAK including inflammatory, neurotrophic, tear film and meibomian gland changes (Ihnatko et al., 2013; Lagali et al., 2013, 2018, 2020a; E. C. S. E.C.S. Landsend et al., 2018; Erlend Christoffer Sommer Landsend et al., 2019), a reappraisal of the impact of the genotype on AAK is warranted. Nevertheless, despite an incomplete picture of corneal alterations in aniridia, corneal findings in relation to genotypes have been reported in various patient cohorts, with broad classes of genotype-phenotype associations of AAK beginning to emerge. Interestingly in our cohort, terminal forms of AAK may be distinguished into two types: one with classical limbal insufficiency with central opacities and neovascularization and a less common type with dense fibrotic whitish central opacities without neovascularization (Fig. 6). The current progress on AAK genotype-phenotype associations are critically examined in Section 4.6.
Fig. 6. Aniridia-associated keratopathy (AAK).
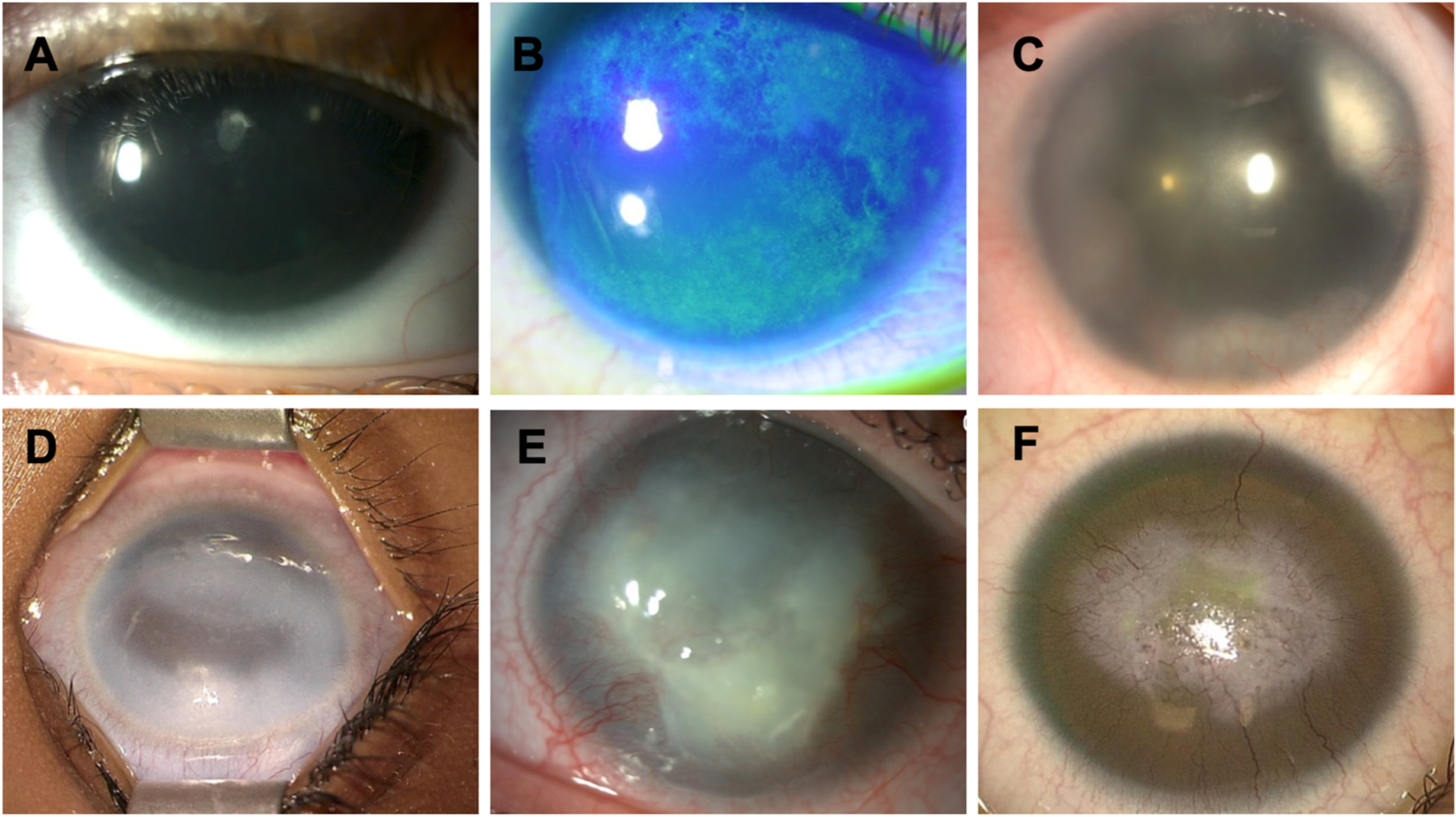
Grade 1 limbal insufficiency with mild anterior subcapsular cataracts (A) or with irregular ocular surface after fluoresceine instillation (B). Grade 2 limbal insufficiency (C). Grade 3 limbal insufficiency with corneal opacity. Note the isolated corneal fibrotic reaction without neovascularization (D). Grade 4 limbal insufficiency with corneal opacity and progressive neovascularization (E). Grade 4 limbal insufficiency with fibrotic reaction and extended corneal neovascularization (F). AAK terminal forms may be distinguished into two types, one with classical limbal insufficiency with central opacities and neovascularization (E, F) and a less common type with dense fibrotic whitish central opacities without neovascularization (D).
3.3.6. Lens anomalies and cataracts
Several alterations of the lens in size, shape, position, or transparency, have been associated with PAX6-related aniridia (Angmo et al., 2021; D’Oria et al., 2021; Kit et al., 2021; Voskresenskaya et al., 2021).
As seen in the Sey mouse (Hill et al., 1991) (see Section 2.2), there is tantalizing evidence that lens size is disturbed in aniridia as microspherophakia (small lens diameter) has been reported in these patients (D’Oria et al., 2021; Voskresenskaya et al., 2021). However, a recent report did find that lens thickness is usually normal in young children with aniridia, although the age dependent increase in lens thickness, which normally occurs during adolescence is less apparent in those with aniridia, leading to reduced lens thickness in adulthood (Voskresenskaya et al., 2021). Defects in lens shape including aniridia-associated lens coloboma can also be a feature of aniridia (Angmo et al., 2021; Singh et al., 2014), suggesting that mice and humans harboring PAX6 mutations may have similar lens phenotypes. Finally, zonular abnormalities have been observed in aniridia, leading to partial or complete lens dislocation (D’Oria et al., 2021; Kusumesh and Ambastha, 2016; Puthalath et al., 2021; Voskresenskaya et al., 2021), although it is not clear whether this is a primary event arising during eye development, a defect in zonular homeostasis or caused by stress to the zonules due to the presence of a small lens in a normal-size eye.
Premature cataracts are a highly prevalent clinical feature in aniridia observed in around 50–70% of patients (Kit et al., 2021; Landsend et al., 2021). Variations in cataract prevalence depending on the type of mutation have been noted, with lower rates of prevalence associated with frameshift and missense mutations (less than 60%) compared to intronic, nonsense or C-terminal extension mutations (>75%) (Kit et al., 2021). Cataracts could be present at birth or occur later during childhood or early adulthood (Landsend et al., 2021). Importantly, clinicians should be aware that mutations in the PAX6 gene can initially present as bilateral congenital cataracts especially when foveal hypoplasia is associated (Nieves-Moreno et al., 2021). Surgery indications should take into account the possibility of an associated, sometimes overlooked, foveal hypoplasia, when the latter – and not the non-surgical associated cataract – is actually responsible for the low vision, in a patient with no obvious iris defects and, therefore, sometimes no diagnosis.
Congenital central anterior subcapsular cataracts are common (D’Oria et al., 2021), and can be associated with metaplastic tissue extending into the anterior chamber (Lagali et al., 2020a). It is possible that this lens anomaly is derived from a delayed separation between the lens vesicle and head ectoderm, consistent with the observation that Peters anomaly, deriving from failed complete lens vesicle separation from the head ectoderm, is seen in a subset of aniridia patients (Hanson et al., 1994). However, studies on Sey mice have revealed that neural crest migration is improperly regulated in Pax6 heterozygotes, leading to neural crest-derived keratolenticular strands connecting the lens to the cornea (Baulmann et al., 2002; Kanakubo et al., 2006). Aniridia patients born with clear lenses develop posterior lens opacities in late childhood/early adulthood at high rates (D’Oria et al., 2021; Voskresenskaya et al., 2021). These observations indicate that the correct dosage of PAX6 is essential for the homeostasis of the adult lens, which is consistent with its sustained expression in both the lens epithelium and newly born fiber cells throughout the lifespan (Faranda et al., 2021; Zhang et al., 2001).
In our cohort, we have observed an equal distribution of two types of cataracts. The most classical: anterior subcapsular congenital cataract, usually present at birth, has very few initial surgical indications, as vision is either not or only slightly impaired by the cataract. The other form, posterior, is a cartwheel cataract. These lens opacities are not present at birth and instead develop progressively in a cartwheel fashion with a posterior subcapsular opacity. The association of congenital aniridia and cartwheel cataracts seems pathognomonic of a PAX6 gene anomaly (Bremond-Gignac, 2019) (Fig. 7). Cataract surgery is required in about 50–85% of aniridia patients at a mean of age of 30 years, with a younger surgery age reported in patients with missense mutations compared to frameshift and intronic mutations (Kit et al., 2021).
Fig. 7. Cataract-related congenital aniridia. A-H. Cartwheel cataract pathognomonic of a PAX6 gene anomaly.
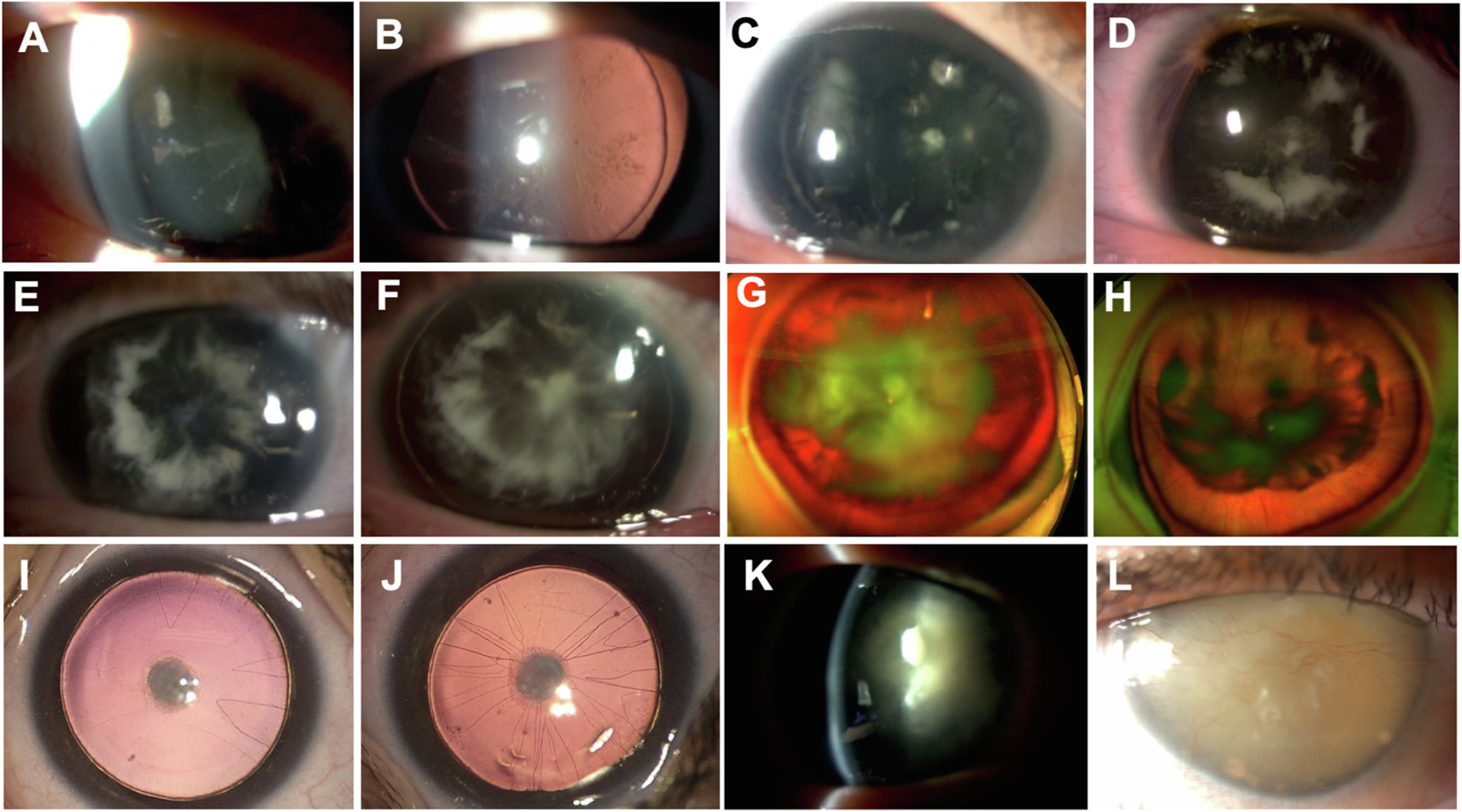
Early onset (A–B) and progressive evolution of cartwheel cataracts with posterior subcapsular opacities (C–H). Note the asymmetrical severity of cartwheel cataracts between the eyes of the same patient (G and H). I-J. Anterior subcapsular congenital cataracts with pupillary membrane remnants. K. Anterior subcapsular cataract with marked posterior cartwheel cataract. L. Total Morgagnian cataract.
3.3.7. Glaucoma
The prevalence of glaucoma in congenital aniridia is estimated at 46–70% of cases, with a prevalence of around 15% in children below 10 year of age (Landsend et al., 2021). The mean age at the diagnosis of glaucoma varies from 8 to 25 years in different reports (Hama et al., 2010; Landsend et al., 2021; Muñoz-Negrete et al., 2021). Whilst congenital glaucoma presenting with buphthalmos is rare in PAX6-related aniridia, glaucoma often occurs later in childhood or adulthood. Among patients presenting with the congenital aniridia phenotype and congenital glaucoma in our cohort (n = 22/329), 50% (n = 11/22) are PAX6-related and 14% (n = 3/22) have WAGR syndrome. Other genes involved in the phenotype associating both congenital aniridia and congenital glaucoma (36%, n = 8) are PITX2, FOXC1 and CYP1B1 (n = 1,3 and 4/22 respectively).
In aniridia patients under the age of 20, an association was found between more pronounced iris hypoplasia and the increased risk of developing glaucoma (Landsend et al., 2021). We have observed a higher frequency of glaucoma in individuals exhibiting the most severe iris phenotype where only a small remnant iris root remains (unpublished data). The prevalence of glaucoma has also been reported to be higher in patients with gene deletions compared to frameshift mutations (Kit et al., 2021). Potential mechanisms of aniridic glaucoma include absence or abnormality of Schlemm’s canal, anomaly of the trabecular meshwork with open angle or development of tissue, resembling synechiae or ridges between the iris stump and the angle wall. This last possibility can lead in some cases to the anterior rotation of the iris stump by traction and closure of the anterior chamber angle (Baulmann et al., 2002; Hall et al., 2019; Erlend C. S. Landsend et al., 2019). Furthermore, some patients undergo cataract, cornea or iris surgery, which carries an independent risk of secondary glaucoma (Kit et al., 2021).
3.3.8. Optic nerve anomalies
PAX6 mutations have been suspected in a large variety of optic disc malformations. The most frequently encountered one, however, is probably optic disk hypoplasia, which is observed in around 10% of aniridia patients, and seems unrelated to the presence of a foveal hypoplasia (McCulley et al., 2005). Other optic disc malformations associated with PAX6 mutations are: papillorenal syndrome, optic disc coloboma, morning glory disc anomaly, optic disc aplasia and persistent hyperplastic primary vitreous (Azuma et al., 2003; Sellheyer and Spitznas, 1987). The fact that PAX6 downregulates the expression of PAX2 (and vice versa) can probably explain for some of these specific clinical pictures (Azuma et al., 2003).
3.3.9. Systemic extra-ocular features
3.3.9.1. Brain anomalies and hearing deficits.
PAX6 is a master regulatory transcription factor regulating neuron proliferation and differentiation; it is key to the establishment of the dorsal-ventral and anterior-posterior axes of the brain. Its functional alterations therefore lead to abnormal embryonic – and postnatal – neurogenesis, abnormal gliogenesis (Osumi et al., 2008) and are responsible for a large spectrum of cerebral malformations (Ochi et al., 2022). These have been studied both in the Sey mouse and aniridia patient populations.
3.3.9.1.1. Structural brain abnormalities in aniridia.
Aniridia associated brain abnormalities affect six structures of the midline: the optic chiasm, the olfactory bulb, the pineal gland, both anterior and posterior commissures and the corpus callosum (Abouzeid et al., 2009; Bamiou et al., 2007; Free et al., 2003; Grant et al., 2017; Sisodiya et al., 2001). They can all be hypoplastic, or even absent – except for the optic chiasm and the corpus callosum. Although the proportion of absent vs. reduced vs. normal structures varies across studies, brain anomalies are likely present in more than half of patients with non-syndromic PAX6 aniridia.
Furthermore, several studies have showed volumetric changes in the grey and white matter of patients with aniridia: an increase in grey matter volume (Ellison-Wright et al., 2004; Free et al., 2003), and a premature age-related cortical thinning (Yogarajah et al., 2016). A greater decline in thickness of the frontoparietal cortex with age and accelerated decline in working memory was also shown in aniridia patients when compared to controls. Subjects with the most severe genotypes showed the most widespread differences compared with controls (Yogarajah et al., 2016).
3.3.9.1.2. Functional brain abnormalities in aniridia.
Decussation patterns in aniridia, unlike in albinism, have been proven normal (Neveu et al., 2005). Hearing difficulties have been demonstrated in patients with aniridia: despite normal audiograms, they exhibit abnormal central auditory processing (difficulty localizing sounds and understanding speech in noise), resulting likely from reduced auditory interhemispheric transfer (Neveu et al., 2005) and reduced functional cortical integration of auditory information (Bobilev et al., 2019).
PAX6 haploinsufficiency causes olfactory dysfunction, which is consistent with olfactory bulb hypoplasia (Grant et al., 2021; Sisodiya et al., 2001). A PAX6 splice mutation in the proline/serine/threonine-rich domain has been reported in an aniridia patient with anosmia (Martha et al., 1995).
The neuropsychological profile of patients with aniridia has not been the subject of systematic studies. Mental retardation is not always present in WAGR syndrome (Van Hare et al., 2007); conversely, variable cognitive dysfunction, possibly overlooked, has been reported in patients and families with PAX6-related aniridia (Dansault et al., 2007; Ellison-Wright et al., 2004; Heyman et al., 1999; Malandrini et al., 2001). Similarly, autism spectrum disorder is a common feature of patients with WAGR syndrome, often attributed to the deletion of PRRG4 (Justice et al., 2017). However, autism spectrum disorder has also been reported in patients with missense mutations or deletions of PAX6. This could possibly be related to the fact that PAX6 also regulates or binds to many autism spectrum disorder-related genes (Kikkawa et al., 2019).
Grant et al. have extensively studied age-related cerebral abnormalities in the Sey mouse, which suggest a global decrease in adult structural plasticity (Grant et al., 2020). By using functional MRI, Pierce et al. demonstrated increased functional connectivity in aniridia patients compared to normal controls, suggesting the compensating recruitment of additional neural regions (Pierce et al., 2014).
3.3.9.2. Sleep disorders.
In mammals, including humans, visual information encoded by the retina is routed to the visual cortex through ganglion cells, but a small number of specialized ganglion cells, called melanopsin ganglion cells, send their messages to the central circadian clock in the suprachiasmatic nuclei of the hypothalamus. Melanopsin ganglion cells constitute only ~2–4% of total ganglion cells, but they play a key role in synchronizing biological rhythms with the environment as the melanopsin makes them directly photosensitive to blue light at around 460–480 nm (Prayag et al., 2019).
Light entering the eye and reaching the retina is proportional to the square of the pupil size (Harley and Sliney, 2018). It is thus expected that patients with aniridia will have highly increased retina exposure. Melanopsin ganglion cells might be over-stimulated permanently, even in very low ambient light levels and even when eyelids are closed. Aniridia patients could thus be more at risk of having circadian rhythm disruption and sleep disorders, particularly insomnia if melanopsin is produced permanently, which has not yet been demonstrated.
On the other hand, several studies have shown sleep disorders associated with aniridia due to the PAX6 mutation. Berntsson et al. described a family with PAX6 haploinsufficiency associated with clinical and biological signs of narcolepsy type 1 and pineal gland hypoplasia (Berntsson et al., 2020). Two other studies showed that patients carrying PAX6 mutations display hypoplasia of the pineal gland and low melatonin secretion (Abouzeid et al., 2009; Hanish et al., 2016), while a recent study in adolescents with PAX6 haploinsufficiency did not report any impairment in the quality of sleep and no typical narcolepsy, but found an increase in their sleep onset latency (Hanish and Han, 2018). Hence, the authors hypothesized that the increase in sleep onset latency in these patients could be explain by the pineal hypoplasia and the reduction of melatonin secretion.
Thus, whether aniridia per se induces sleep disorders, independent of the loss of Pax6 function remains unclear. This question is further complicated when aniridia patients suffer from reduced electroretinographic activity, optic nerve hypoplasia and other severe complications including glaucoma. Such alterations can have a significant impact on their capacity to process light-related information by melanopsin-expressing cells as they are, in part, responsible for sleep and circadian rhythm. Such disturbances have been already reported in AMD and glaucoma patients which exhibit ganglion cell dysfunction (Ahmadi et al., 2020; Maynard et al., 2017; Obara et al., 2016). Overall, the link between aniridia, circadian rhythm disruption and sleep disorders is unclear. Whilst developmental pineal hypoplasia and subsequent melatonin insufficiency is associated with PAX6 mutations, over-stimulation of melanopsin ganglion cells due to high retina exposure, melanopsin ganglion cells damage or dysfunction secondary to glaucoma or optic nerve atrophy could also interfere with the proper synchronization of circadian rhythms through light. Further studies are required to clarify this link in patients with aniridia due to different types of genetic defects and at different ages.
3.3.9.3. Diabetes mellitus.
The pancreas is an organ that originates from the evaginations of pancreatic progenitor cells in the epithelium of the foregut endoderm. The islets of Langerhans constitute the endocrine pancreas, which produces and releases insulin, glucagon, and other endocrine hormones. PAX6 gene is crucial for islet development, whereas the null mutation of PAX6 results in the near absence of glucagon-producing alpha cells underlining its special role in alpha (glucagon secreting) cells (Panneerselvam et al., 2019). The transcription factor Pax6 is required for pancreatic β cell identity, glucose-regulated ATP synthesis, and Ca2+ dynamics in adult mice. Pax6 is a direct activator of β cell genes, thus maintaining mature β cell function and identity. In parallel, Pax6 binds promoters and enhancers to repress alternative islet cell genes including ghrelin, glucagon, and somatostatin (Swisa et al., 2017) (Fig. 8).
Fig. 8. PAX6 role in pancreas organogenesis, particularly in islet development and in mature islets cells.
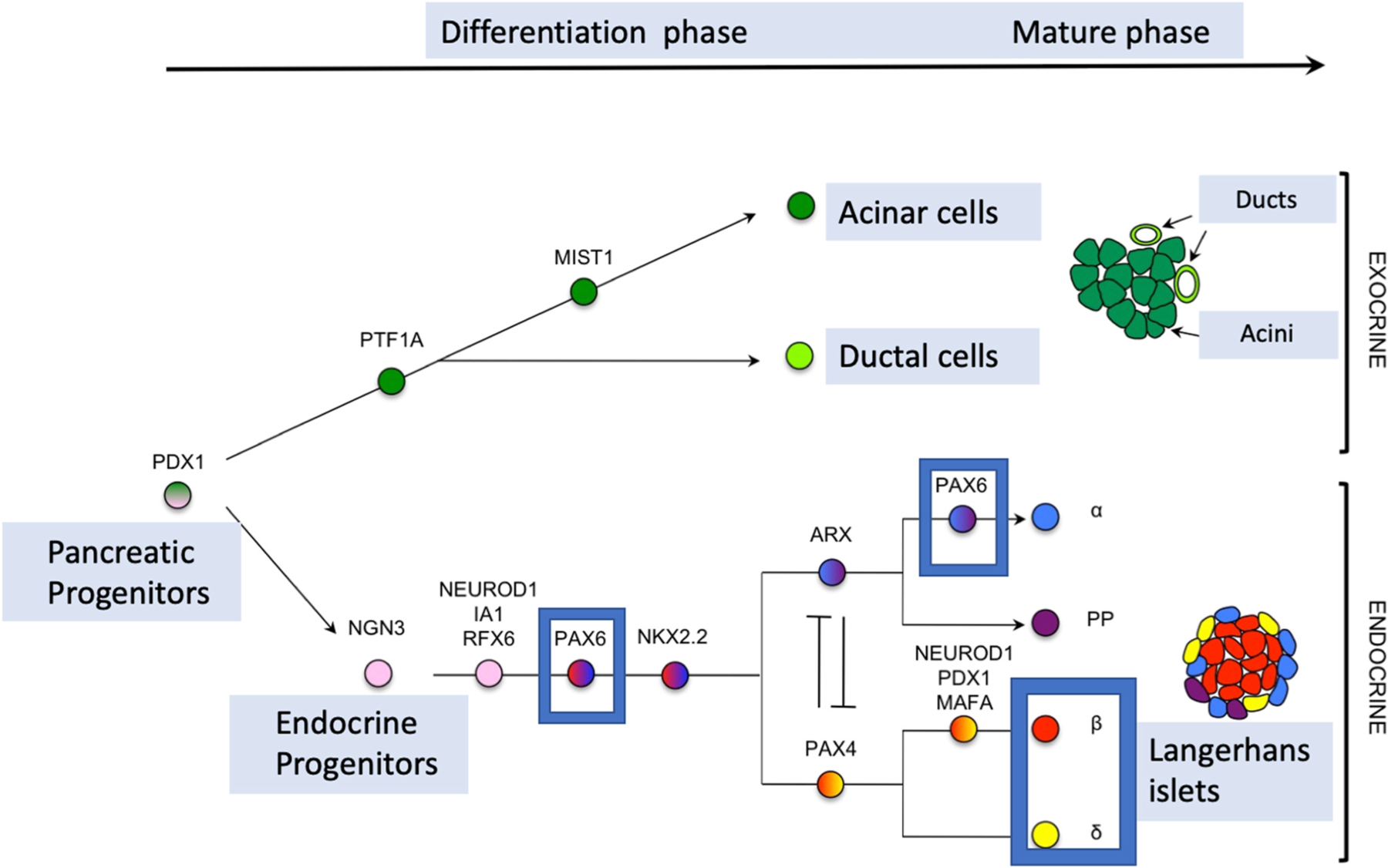
PAX6 is involved in pancreatic endocrine cells development. PAX6 regulates directly expression of insulin genes and of somatostatin in delta-cells in mature cells.
Additionally, PAX6 mutation leads to decreased proinsulin processing (Wen et al., 2009), which may lead, in the long run, to abnormal glucose metabolism as proinsulin, the precursor of insulin, which is less active than insulin. Therefore, one can envision that a mutation in PAX6 results or at least can contribute to impaired glucose homeostasis, or diabetes mellitus (Yasuda et al., 2002).
The most recent informative cohort comprised 86 subjects with PAX6-related aniridia and provided us with a UK-specific view of obesity, glucose intolerance and diabetes mellitus in that setting (Kit et al., 2021). Obesity and type-2 diabetes were the most common systemic associations observed in adult patients: 23.3% (20/86) of the patients were affected with obesity, which is below the estimated UK general population prevalence of 27.8%. Type-2 diabetes was observed in 12.8% (11/86) of the subjects, which is higher than a previously reported 7% prevalence in a survey of patients with aniridia (Netland et al., 2011) and is twice the prevalence of diabetes in the UK general population (4.5%–6%) (“Diabetes Digital Media. Diabetes Prevalence. https://www.diabetes.co.uk/diabetes-prevalence.html. Updated January 15, 2019. Accessed october 10th, 2021.,” n.d.). Cases of glucose intolerance, type-1 and −2 diabetes have been reported among adult patients with aniridia, but rarely in children (Boese et al., 2020; Motoda et al., 2019; Peter et al., 2013), while type-1 diabetes, which can be coincidental and not related to a PAX6 mutation, was not observed.One must remember that the development of type −2 diabetes is multifactorial, with contributions from genetics, epigenetics, life-style factors, and obesity (Zheng et al., 2018). Mice with heterozygous Pax6 variants were shown to have decreased insulin levels (Mitchell et al., 2017). One study reported 5 unrelated patients with aniridia, 4 of which had confirmed PAX6 mutations, all with either glucose intolerance or diabetes (Yasuda et al., 2002); however, the causal relationship for diabetes remains unclear, as most patients with aniridia do not develop it. However, mice with heterozygous Pax6 variants were shown to have decreased insulin levels. (Mitchell et al., 2017; Swisa et al., 2017).
Further studies have shed possible light on the mechanisms linking PAX6 mutation and insulin secretion. Among 10 cases from the five families, two were diagnosed with newly identified diabetes and another two were diagnosed with impaired glucose tolerance. Among controls (n = 12), two had impaired glucose tolerance. Levels of proinsulin in the cases were significantly higher than that of the controls, despite similar levels of total insulin. The areas under the curve of proinsulin in the cases were significantly higher than that of the controls (Tian et al., 2021). This suggests that proinsulin processing is altered; this has been described in another type of diabetes mellitus linked to an insulin gene mutation that increased endoplasmic reticulum stress, ultimately leading the death of insulin-secreting β-cells (Meur et al., 2010).
Furthermore, another study showed that PAX6 can bind to the promoter of the prohormone convertase (PC)1/3 gene, an enzyme essential for the conversion of proinsulin to insulin and directly upregulate the production. Pax6 mutations lead to PC1/3 deficiency, resulting in defective proinsulin processing and abnormal glucose metabolism (Wen et al., 2009).
An interesting case report described a boy with a PAX6 mutation, born with anophthalmia, who experienced hypoglycemic seizures starting at 5 months old due to hyperinsulinemia hypoglycemia, and showed a prediabetic condition at 60 months. This patient provides new evidence that connects PAX6 to glucose homeostasis and highlights that life-threatening hypoglycemia or early onset glucose intolerance may be encountered, albeit exceptionally (Kim et al., 2021). This may be related to a major ER stress leading to increased insulin secretion and then to insulin cell alteration. Taken together, these data suggest that, while obesity may influence the development of diabetes mellitus in some patients with aniridia, the presence of PAX6 mutations, through developmental mechanisms as well as insulin secretion alterations, is a likely factor. Therefore, it is recommended to have a low threshold approach for diabetic assessment in patients with aniridia and a PAX6 mutation to enable clinicians to prospectively monitor and modify risk factors to delay the onset or severity of diabetes mellitus although glucose metabolism alterations remain rare in childhood. Further prospective studies are needed to determine the best screening strategy for impaired glucose tolerance and diabetes mellitus in aniridia.
3.3.10. Associated diseases
In our cohort of 230 patients with PAX6-related congenital aniridia, we have observed various associated diseases (Table 1).
Table 1.
Clinical entities found in patients with PAX6-related congenital aniridia.
| Associated clinical entity, N | PAX6 variant related to congenital aniridia |
|---|---|
| Ehlers Danlos Syndrome, 1 | Exon 9: c.692_693del; p. (Arg231Asnfs*20) |
| Cleft palate, 1 | Exon 13: c.1268A > T; p. (*423Leuext*14) |
| Ankylosing spondylitis, 1 | Exon 5::c.109G > C; p.(Ala37Pro) |
| Microcephaly associated gene mutation, 1 | Exon 8: c.607C > T; p.(Arg203*) |
| Thrombotic and Platelet Disorders, 1 | Exon 8::c.532C > T; p.(Gln178*) |
| Hepatorenal Polycystic Disease, 1 | Exon 12:c.1037del; p.(Pro346Glnfs*19) |
| Bifid uvula, 1 | Exon 8:c.566_576del; p.(Ser189*) |
| Mucoviscidosis, 2 (same family) | Microdeletion PAX6, ELP4 |
4. Genetics of aniridia and aniridia-like disorders
4.1. The human PAX6 gene
The discovery of the aniridia gene 30 years ago was a genetic triumph resulting from the complementary research of animal models and patients leading to the parallel identification of the gene in mouse and humans in 1991 (Ton et al., 1991; Walther and Gruss, 1991). A pivotal role was provided by the Sey mouse strain which was proposed to be the mouse model for human aniridia, suggesting that the small eye phenotype in the mouse and absence of iris in humans could result from mutations in orthologous genes (Glaser et al., 1990; van der Meer-de Jong et al., 1990). The historical backgrounds of PAX6 identification in humans and mice as well as in other numerous vertebrates and invertebrates are documented in details in several major reviews (Cvekl and Callaerts, 2017; Hall et al., 2019; Hanson and Van Heyningen, 1995; Shaham et al., 2012). The human PAX6 gene for congenital aniridia was identified by positional cloning and discovered to be deleted in aniridia patients in 1991 (Ton et al., 1991), 65 years after the disease was first described by Rush in 1926 (Rush, 1926). During the same year, murine Pax6 was cloned (Walther and Gruss, 1991) and then demonstrated to be the causal gene explaining the small eye phenotype (Hill et al., 1991). Very soon after, intragenic mutations in PAX6 were reported in additional aniridia patients, strongly suggesting that mutations in PAX6 might be the most common genetic causes for aniridia in humans (Glaser et al., 1992; Hanson et al., 1993; Jordan et al., 1992). In the following years, hundreds of PAX6 pathogenic variants of various types were reported, and now, at least more than 700 unique variants in PAX6 are listed in PAX6 databases (see section 4.2) (Brown et al., 1998; Kokotas and Petersen, 2010; Tzoulaki et al., 2005).
Ocular development is particularly sensitive to PAX6 dosage, and in most cases, congenital aniridia results from an insufficient quantity of the functional PAX6 protein (PAX6 haploinsufficiency) (Hanson et al., 1993; Prosser and van Heyningen, 1998). Conversely, an increased amount of PAX6 protein may also be deleterious, as exemplified by abnormal phenotypes in mice carrying multiple copies of the human PAX6 gene, or the few cases of PAX6 gene duplication in humans that have been reported (Aalfs et al., 1997; Aradhya et al., 2011; Schedl et al., 1996; Schilter et al., 2013).
PAX6 is a highly conserved sequence-specific DNA binding transcription factor that positively and negatively regulates transcription of a wide range of genes coding various regulatory, signaling, and structural proteins required for eye, brain or pancreas morphogenesis (Callaerts et al., 1997; Cvekl and Callaerts, 2017; Dohrmann et al., 2000; Nomura et al., 2007; Shaham et al., 2012; van Heyningen and Williamson, 2002). The PAX6 gene was the first homeobox gene discovered to play a crucial role in eye development, and it is considered as the ‘‘master regulator gene’’ for oculogenesis in numerous vertebrate and invertebrate species (Cvekl et al., 2004; Gehring, 1996; Glaser et al., 1992; Kokotas and Petersen, 2010; Marquardt et al., 2001).
Human PAX6 is divided into 14 exons, with an additional alternatively spliced exon termed exon 5a, and produces two major isoforms consisting of the canonical 422-amino acid PAX6 isoform and the alternatively spliced PAX6(5a) isoform of 436 amino acids, each having a distinct DNA binding and transactivation ability (Azuma et al., 1999; Glaser et al., 1992; Kammandel et al., 1999; Kim and Lauderdale, 2006).
The human PAX6 spans over 22 kb in length with a large 5′ untranslated region (5′UTR) region, and a 3′ untranslated region (3′UTR) of about 850 bp, very close to the adjacent ubiquitously expressed ELP4 gene, transcribed in opposite orientation to PAX6 (Fig. 9) (Glaser et al., 1992; Kammandel et al., 1999; Kim and Lauderdale, 2006). PAX6 possesses two main P0 and P1 promoters, and a third internal promoter, termed Pα, encoding a truncated PAX6 protein that lack the paired domain. PAX6 translation starts at the first ATG in exon 4 (Fig. 9) (Carrière et al., 1995; Hall et al., 2019; Kim and Lauderdale, 2006; Plaza et al., 1995; Walther and Gruss, 1991; Xu and Saunders, 1997). In fact, the PAX6 genomic locus is more complex than initially considered, and comprises 450 kb in total embracing several highly conserved regulatory elements dispersed both upstream and downstream of PAX6 (Griffin et al., 2002; Kleinjan and van Heyningen, 2005). The transcriptional regulation of PAX6 is highly complex and its tightly spatiotemporal developmental expression during development is coordinated by various enhancer sequences specific to each promoter (Anderson et al., 2002; Plaza et al., 1995; P. X. H.E. Xu et al., 1999; Xu and Saunders, 1997), as well as more distant cis-elements identified at the downstream region of the gene by comprehensive analyses of human aniridia patients carrying deletions and chromosomal rearrangements of those areas (Crolla and van Heyningen, 2002; Fantes et al., 1995; Kleinjan et al., 2001; Kleinjan and van Heyningen, 2005; Lauderdale et al., 2000). These PAX6-specific long-range control elements were collectively termed the “distant downstream regulatory region” (DDR), and were located downstream of the most distal breakpoint reported in human aniridia patient (SIMO), at around 150 kb downstream of the PAX6 poly-A site, within the adjacent ELP4 gene (Fig. 9) (Griffin et al., 2002; Kleinjan et al., 2002, 2001; Lauderdale et al., 2000; Simola et al., 1983, p. 19). Bhatia and co-workers revealed that SIMO is essential strong enhancer sequence required for normal PAX6 expression, by maintaining the activation of its upstream promoters through a positive-feedback loop (Bhatia et al., 2013). Therefore, PAX6 itself is able to regulate its own expression in order to maintain a precise dosage of the protein (Aota et al., 2003; Okladnova et al., 1998). PAX6 expression is also regulated by the long non-coding RNAs (lncRNA), PAUPAR/PAX6AS1, which is located upstream the P0 promoter and in the opposite orientation to PAX6 (Fig. 9) (Vance et al., 2014). The function of this PAX6 lncRNA is as-yet not fully understood, but it was recently shown that it coordinates glucose homeostasis by promoting the alternative splicing of the Pax6(5a) isoform resulting in pancreatic glucagon cell-specific production (Singer et al., 2019).
Fig. 9. Schematic representation of the human PAX6 locus.
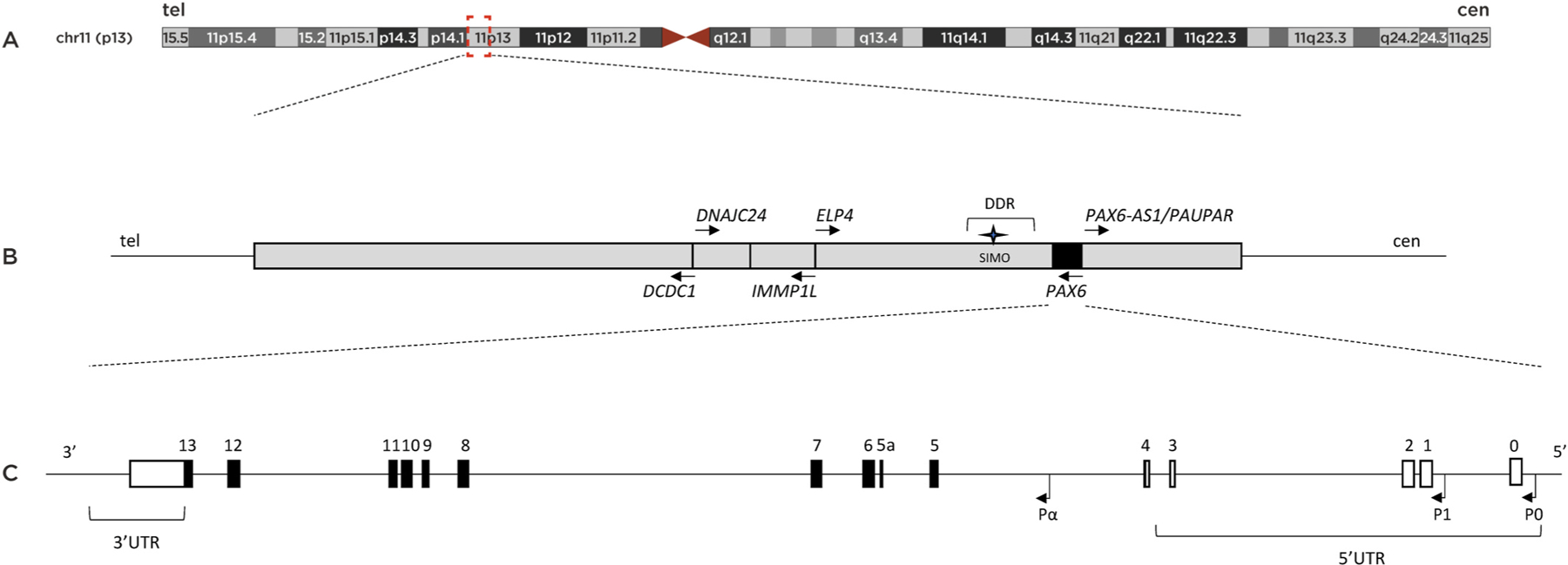
Representation of the PAX6 locus in chromosomal 11p13 region (A). Representation of human PAX6 and its surrounding 3′downstream genes. The downstream regulatory region (DDR) is represented within the neighboring ELP4 gene, with the SIMO enhancer highlighting by a black star. Each grey box represents a gene with its respective DNA strand location, and the arrows above each gene indicate the orientation of transcription. The long non-coding RNA, PAX6-AS1/PAUPAR, is located upstream and in the opposite orientation to PAX6 as indicated by the arrow (B). The human PAX6 gene is represented with its coding exons appearing as black boxes and non-coding exons as white boxes. The coding alternative spliced exon 5a is also represented between exons 5 and 6. The three alternative promoters of PAX6 are indicated by black arrows (C). tel: telomere, cen: centromere, 5′ UTR: 5′ untranslated region, 3′ UTR: 3′ untranslated region.
The canonical PAX6 isoform possesses two DNA-binding domains with a bipartite paired domain, a homeodomain separated by a linker segment, then followed by a C-terminal transactivation domain, enriched in proline-serine-threonine residues that are prone to phosphorylation (Czerny and Busslinger, 1995; Glaser et al., 1994; Kokotas and Petersen, 2010; Mikkola et al., 1999; Tang et al., 1998; Walther and Gruss, 1991). The paired domain and homeodomain can bind to DNA both cooperatively and independently, thereby widening the repertoire of PAX6 target genes (Jun and Desplan, 1996). The paired domain comprises two structurally distinct subdomains, the N-terminal subdomain and the C-terminal subdomain, termed PAI and RED respectively, both folding into three alpha-helices, two of which form a helix-turn-helix motif allowing DNA binding to their respective consensus sequences (H. E. P.X. Xu et al., 1999). The two isoforms have different biological functions, and the ratio between the canonical PAX6 and PAX6(5a) isoforms varies in adult and embryonic tissues with the PAX6(5a) playing a major role in iris formation (Kozmik et al., 1997; Singh et al., 2002). PAX6(5a) isoform is also critical for the structural integrity of the retina with high expression in the fovea (Azuma et al., 2005). More recently, Kiselev et al. observed that a majority of genes were regulated by the canonical PAX6 isoform and that this regulation was mainly negative, with only 50 genes being regulated in common by the two isoforms (Kiselev et al., 2012).
A supplementary layer of complexity of PAX6 regulation emerges from the existence of micro ribonucleic acids (miRNAs) that regulate gene expression post-transcriptionally either through translational suppression and/or degradation by mRNA decay (Bartel, 2009). Several clusters of miRNA recognition elements within the PAX6 3′UTR sequence have been identified to be targets for several miRNAs that collectively play an important role in maintaining a fine-tune expression level of PAX6 at specific spatiotemporal points, defining a functional landscape of cell type-specific miRNA cohorts, as proposed by Ryan et al. (2018). This is well illustrated by the miR-450b-5p that can allow cell-type specific fine tuning of PAX6 expression levels in the corneal epithelium (Selbach et al., 2008; Shalom-Feuerstein et al., 2012), and also by the miR-7 that has been shown to be an important regulator of mouse Pax6 in the pancreas, brain, and eye and is widely co-expressed with Pax6 in these tissues (Arora et al., 2007; de Chevigny et al., 2012; Kredo-Russo et al., 2012; Needhamsen et al., 2014). Also, it was shown that Pax6 may control the down-regulation of multiple genes, notably the neuronal gene Sox1, through direct activation of miR-204 (Conte et al., 2010; Shaham et al., 2013; Shiels, 2020). More importantly, protecting Pax6 3′UTR from miR-7 partially restored protein level in pancreatic islets in an aniridia mouse model, providing a proof of concept for an RNA-based therapy strategy that may be useful for congenital aniridia (Yongblah et al., 2018). Since PAX6 function is particularly sensitive to dosage, boosting the expression of the wild-type copy of PAX6 without excessive overproduction, regardless of the type of loss-of-function mutation, might be promising, since the PAX6 gene is left under its endogenous chromatin environment, allowing a physiological transcriptional regulation, as recently proposed by Ryan and co-workers (Ryan et al., 2018).
Another interesting layer of PAX6 post-transcriptional regulation may be partly mediated by the existence of functional short upstream open reading frames (uORFs) located in the 5ʹUTR of PAX6, as reported by Filatova et al. (2021). It is becoming increasingly clear that post-transcriptional regulation encompassing uORFs can heavily influence the expression and activity of a large number of developmentally relevant genes whose complex and dynamic embryonic patterning require cells to constantly adapt protein levels to orchestrate specific developmental trajectories (Renz et al., 2020).
In conclusion, numerous complex and intricate levels of regulation implicated in PAX6 expression are increasingly discovered. At the transcriptional level, molecular analyses of chromosomal abnormalities whose breakpoints fall within the non-coding 3′ downstream region of PAX6 have greatly contributed to the recognition of specific PAX6 long-range regulatory control elements owing to the phenotypic consequences observed in patients. Similar chromosomal abnormalities are, however, rarely reported in the literature (see section 4.2). In section 4.4 of this review, we show that application of whole-genome sequencing solves the case of a 10-year diagnostic wandering of a five-generation family with typical aniridia by identifying a paracentric PAX6 inversion. This structural variant does not cause direct PAX6 gene disruption, but separates the promoter of PAX6 from its 3′ long range enhancers DDR, likely changing chromatin topology and altering PAX6 expression. Topologically-associated domains raised broad interest in the scientific community with the recently presented evidence that topologically-associated domains are important for maintaining proper enhancer-promoter interactions and ensuring precise spatiotemporal gene expression patterns during development (Ibrahim and Mundlos, 2020; Ren and Dixon, 2015). Therefore, a next challenge will be to determine how structural variants in non-coding regions contribute to alter the 3D chromatin structure of the PAX6 locus, and this could be addressed by Hi-C studies from human ocular tissues and/or ES/iPS cells differentiated into mature ocular cells.
4.2. PAX6 mutational spectrum
Two major PAX6 databases, including the public LOVD PAX6 database (http://lsdb.hgu.mrc.ac.uk/home.php?select_db = PAX6) and the professional “PAX6 Human Gene Mutation Database (HGMD) Pro Mutation Database” catalog all reported PAX6 variants, and more than 700 unique variants have been registered according to the last update in 2022. Here, we provide a summary of PAX6 molecular changes which have been reported in detail in several recent comprehensive reviews (E. S. E.S. Landsend et al., 2018; Lima Cunha et al., 2019; Wawrocka and Krawczynski, 2018). The genetic landscape of PAX6 is characterized by a wide range of molecular defects including single nucleotide variants as well as balanced and unbalanced structural variants. The most common pathogenic variations are single nucleotide variants of various classes, scattered throughout the PAX6 gene, while approximately 15% of the remaining molecular defects are structural variants including large chromosomal aberrations and microdeletions of chromosome 11p13 first reported in 1995, and which are now increasingly reported in the literature (Fantes et al., 1995). These microdeletions are of various sizes and remove a part of or a complete genomic sequence of PAX6 and its neighboring genes located at its 3′ downstream region and are generally associated with a typical aniridia phenotype (Wawrocka et al., 2013). The large majority of pathogenic PAX6 single nucleotide variants introduce a premature termination codon through different mechanisms including nonsense, frameshift small nucleotide deletions, insertions, and indels variants. The reading frame of these premature termination codon variants is shifted, leading to the incorporation of aberrant amino acids into the protein during translation with many stop codons. Consequently, the mutant mRNA produced is usually targeted for nonsense-mediated decay process by which it is rapidly degraded with no production of the protein. This leads to the loss of 50% of PAX6 protein level, resulting in haploinsufficiency mechanism similar to cases with whole PAX6 deletion (Hingorani et al., 2012). Therefore, premature termination codon variants and whole gene deletions are commonly associated with a classical aniridia phenotype (Lima Cunha et al., 2019). Recent functional assays showed that several PAX6 splicing variants, some missense as well as rare synonymous variants may also be classified as premature termination codon variants, and accordingly they are associated with the classical form of aniridia (Filatova et al., 2019). A severe aniridia phenotype is also often associated with “nonstop” or C-terminal extension variants, i.e. variants occurring at the normal stop codon converting the natural stop codon of the C-terminus of PAX6 into a codon, allowing translation to continue into the 3′ untranslated region of PAX6. Nonsense-mediated decay is not predicted for C-terminal extension transcripts and the mechanisms of action for this mutation subtype is still unclear (Tzoulaki et al., 2005). By contrast to premature termination codon, most missense variants lead to the production of a full-length PAX6 protein having only a single amino acid substitution in its coding sequence, and they have been associated with both classical aniridia and non-classical phenotypes (Hanson and Van Heyningen, 1995; Tzoulaki et al., 2005). Functional studies of some PAX6 missense variants have shown that the degree of their structural and functional impairment is highly variable, and is dependent on their location within the protein (Gupta et al., 1998; Tang et al., 1997). However, other functional studies of missense variants located within the PAI subdomain, the linker segment, or the RED subdomain as well as two nonsense mutations have shown that all could reduce, enhance, or leave transcription unchanged, depending on the utilized cell lines, suggesting that the functional outcome of a same mutation is highly dependent, not only on variant location, but also on the specific cellular environment (Chauhan et al., 2004). A largely unexplored area remains the vast non-coding portion of the PAX6 locus (Fig. 9). However, some studies have successfully identified variants affecting regulatory regions of PAX6. The majority of them, correspond to rare chromosomal rearrangements or 11p13 microdeletions whose breakpoints fall within the 3′ downstream region of PAX6 without disrupting the integrity of its genomic sequence (Fantes et al., 1995; Lauderdale et al., 2000; Simola et al., 1983). They result in loss of PAX6 3′ cis-regulatory elements or in their physical separation from the PAX6 promoter located at the 5′ upstream region of PAX6, resulting in decreased PAX6 expression, and ultimately aniridia (Fantes et al., 1995; Lauderdale et al., 2000; Simola et al., 1983). Also, a non-coding single nucleotide change in the SIMO enhancer was reported to cause aniridia by altering a PAX6 consensus binding sequence and thus disrupting its autoregulation loop on PAX6 expression (Bhatia et al., 2013). Finally, about 10%–20% of aniridia cases still remain genetically unexplained depending on the screening methodology used (Ansari et al., 2016). With the advent of next-generation sequencing (NGS) technology, identification of new variants will continue, notably in the context of the increasing number of sporadic cases reported. As illustrated in section 4.4, a significant number of new molecular defects in PAX6 in sporadic cases was identified owing to NGS panel, supporting that PAX6 mutation spectrum is far from being saturated. Therefore, the next step that needs to be addressed is to gain insight into the exact functional consequences of each PAX6 genotype. This challenging knowledge is crucial since in coming years the precision medicine will be based on the pathogenic mechanisms of mutations. As the functional consequences of PAX6 variants depend on both its location and cellular environment, the best way would be to use induced pluripotent stem cells (iPSCs) and 3D organoid-based approaches to explore a variant of interest in its “native” cellular context. Skin fibroblasts of PAX6 patients can be reprogrammed toward iPSCs, which in turn can be differentiated toward specific ocular-like cells, accordingly to the associated-phenotype, modeling most faithfully the human PAX6 disease in its “native” context (Doss and Sachinidis, 2019). In conclusion, centralized PAX6 variant databases, robust genotype-phenotype correlations, and streamlined approaches to functional studies will pave the way for novel treatment opportunities for aniridia.
4.3. Molecular diagnostic strategies: shift towards NGS technologies
It has been challenging to achieve a maximum detection rate in the comprehensive molecular diagnosis of PAX6, due to the large variety of variant subtypes and the necessity of screening the entire PAX6 locus including the 5′- and 3′-regulatory regions and the adjacent downstream ELP4 and upstream WT1 genes, therefore requiring more than one molecular and cytogenetic technology. Consequently, for a long time, molecular diagnostic testing strategies for PAX6 included an initial screening to detect large deletions or chromosomal rearrangements using karyotype analysis, microarray-based comparative genomic hybridization and fluorescence in situ hybridization. This was then followed by PCR-based Sanger sequencing of the entire coding region and intron-exon boundaries of PAX6 for the detection of small intragenic sequence variants. Multiplex ligation-dependent probe amplification was also combined to detect heterozygous PAX6 exon copy number variations that escaped the Sanger sequencing screening. Recently, targeted NGS has become clinically available for the detection of variants at very different genomic scales in one single assay, including the analysis of single nucleotide variants and copy number variations, and has been implemented in molecular laboratories in many countries for the diagnosis of rare disorders. While the NGS-based protocol has the potential of becoming the one-for-all (for detecting all variant types) molecular test, few studies have been performed using targeted NGS technology as a single test panel for PAX6, allowing parallel examinations of other eye developmental genes of interest, which share overlapping phenotypes with PAX6-related aniridia.
4.4. Clinical implementation of combined NGS and WGS as a diagnostic strategy for ophthalmic genetics including aniridia and aniridia-like disorders
Here, we present genetic data from a cohort of 450 cases with diverse ASD disorders including congenital aniridia that were investigated at first-line by using a combination of targeted NGS panel combined with parent-offspring trios short-read WGS (Valleix et al., oral communication, VI European Aniridia Conference, June 2022). This diagnostic genetic testing practice combines a custom-made targeted gene panel on an Illumina NextSeq500 platform with a clinically based WGS approach on an Illumina Novaseq6000 platform owing to the French national plan for widespread implementation of whole genome sequencing (WGS) for rare diseases in the clinical setting (Plan France Médecine Génomique 2025; PFMG2025). The diagnostic NGS panel assay was similar to the panel oculome previously developed by Patel et al. (Patel et al., 2019). A subpanel was designed to cover the full spectrum of ocular ASD. Overall, pathogenic and likely pathogenic variants in PAX6 were identified in 230 unrelated families (Fig. 4). Of the 230 PAX6-positive families, 60% were sporadic cases and 40% had a family history, reversing the classic picture of the mode of presentation of this disease. All PAX6 variants were germline except for two cases with mosaic alleles, showing that NGS makes it possible to accurately evaluate the extent of mosaicism for de novo variants. NGS revealed a majority of single nucleotide variants in PAX6, with a total of new 66 as yet unreported variants. Pathogenic and likely pathogenic variants (excluding those identified for subpanels on congenital cataracts, albinism, and corneal dystrophies) were also found in FOXC1, PITX2, CYP1B1, HCCS, CPAMD8, ADAMTSL4, MAF, ADAMTS17, PXDN, MAB21L1, FOXE3, SLC38A8, PRSS56 but also YAP1, ZEB2, and NDP. Besides single nucleotide variants, a total of thirty nine structural variants were identified in PAX6, PITX2, FOXC1, HCCS, NDP, and VSX2. Among copy number variations in PAX6, whole gene deletions (including WT1 or not) and microdeletions affecting the 3’ cis-regulatory elements of PAX6 of variable sizes were the most frequent copy number variations found in the cohort. For families remaining genetically unexplained after NGS analysis, parent-offspring trios paired-end WGS was performed in the clinical setting (Fig. 4). WGS made it possible to identify and characterize the genomic breakpoints of the three distinct structural variants, one corresponding to an inversion of 12-Mb involving the PAX6 locus on chromosome 11 and the two others corresponding to an inversion of 62-Mb and a complex rearrangement with deletion/inversion of 40-Mb affecting the PITX2 locus on chromosome 4 (Valleix et al., VI European Aniridia Conference, June 2022). The 12-Mb PAX6 and the 62-Mb PITX2 inversions were inherited with complete penetrance in an autosomal dominant manner, whereas the 40-Mb PITX2 deletion/inversion occurred de novo. An important point is that these structural variants did not cause direct gene disruption, but physically separated PAX6 and PITX2 genes from their downstream and upstream regulatory elements, most probably contributing to the transcriptional dysregulation of the PAX6 and PITX2 genes and resulting in the complete extinction of their expression by positional effects. This is the first observation describing PAX6 and PITX2 inversions characterized by WGS, highlighting the clinical importance of structural variants in autosomal dominant congenital aniridia and aniridia-like disorders, and suggesting that these structural variants may not be unique to these families since approximately 5%–10% of aniridia and in up to 40% of ARS cases, the genetic causes remain unexplained. This study supports that a combined NGS and WGS screening strategy improves the diagnostic yield of aniridia, allowing detection of variants in a large diversity of genes as well as the identification of structural variants, not previously covered by standard diagnostic technology platforms. This study on a large patient cohort reinforces the notion that PAX6 is undeniably the major gene for aniridia, but diverse classes of genes are also emerging being implicated in typical aniridia or ASD related-disorders, hence providing a rationale for the parallel screening of ASD genes to increase the diagnostic yield of aniridia (see section 4.7). All these related ASD disorders share overlapping embryonic and genetic mechanisms, and identifying variants in any of these genes may reveal biological pathways important for pathogenesis and innovative treatment opportunities. Since precision medicine will be based on genetic etiology, it is crucial to continue to increase the yield of genetic testing for aniridia and ASD related-disorders by implementing WGS early in the diagnostic clinical process.
4.5. New genotype-phenotype correlations: 3′PAX6 non-coding copy number variations and milder aniridia phenotype
The exact pathophysiology of how PAX6 variants can cause distinct clinical phenotypes remains unclear, and until now, there were no clearly established genotype-phenotype correlations. Congenital aniridia is inherited in a Mendelian fashion as an autosomal-dominant trait, and exceptional cases of biallelic inheritance have been reported, resulting in a stillborn fetus with severe craniofacial anomalies including absence of eyes and nose, a phenotype similar to the homozygous Sey mouse mutant (Hodgson and Saunders, 1980). The disease is considered to be fully penetrant, but the penetrance of each aniridia-associated feature is not complete. Most PAX6 variants show high phenotypic variability, both between different families or within the same family, making well-defined genotype-phenotype correlations difficult (Kokotas and Petersen, 2010). Approximately, two-thirds of aniridia cases are familial, the remainder are sporadic with a negative family history. However, an increasing number of sporadic cases are recorded, reversing the classic picture of this disease’s presentation, progressively making sporadic presentation the norm rather than the exception. This situation can have major clinical consequences depending on the timing at which the de novo mutation arises during embryonic development, because it could be present at different levels in multiple tissues or be eye specific and will be associated with a variability of clinical phenotypes (Acuna-Hidalgo et al., 2016). De novo mutations occurring in a parental gonadal cell or early in the zygote will result in all cells in the fetus carrying the variant or a high-level mosaicism with the variant being present in many different tissues of the fetus, thus making the new variant easily detectable in DNA extracted from peripheral blood. In contrast, a post-zygotic mutation or even a late-somatic mutation will be restricted to a small number of somatic cells or even to a single tissue like the eye. It will then be particularly challenging to detect the new variant in DNA extracted from peripheral blood to confirm the diagnosis of aniridia. Aside from tissue mosaicism, other genetic factors may explain the phenotypic variability, such as the co-inheritance of variations in other developmental genes or miRNA targets that could modulate the fine-tuning of PAX6 expression levels in specific tissues according to the spatial expression of the miRNAs involved. However, it remained difficult to assess all these possibilities until recently, because of technological limitations. The use of next-generation sequencing will now make it possible to address these questions accurately by offering the possibility to better evaluate the extent of mosaicism for de novo variants, as well as the co-inheritance of other variants, and thereby their clinical consequences.
Until now, premature termination codon variants, gene deletions, and C-terminal extension variants were commonly associated with the most classical aniridia phenotype with complete iris aplasia, and profound visual impairment with foveal hypoplasia and nystagmus, while missense variants have been usually associated with a mild degree of iris hypoplasia, and in some cases with no iris defects or associated with non-classical phenotypes, like Peters anomaly, microphthalmia, optic nerve anomalies, coloboma, isolated foveal hypoplasia, keratitis, and anterior segment dysgenesis (Aggarwal et al., 2011; Azuma et al., 2003; Grønskov et al., 1999; Hanson and Van Heyningen, 1995). A major advance in the better understanding of genotype-phenotype correlations is recently provided by Vasilyeva et al. who undertook a first statistical analysis on a large cohort of 110 Russian patients with congenital aniridia (Vasilyeva et al., 2021). The patients were allocated into seven groups according to the type of PAX6 variants, which have been classified using the consensus recommendations from the American College of Medical Genetics. They found that PAX6 mutations explained 96.7% of aniridia phenotypes with only 3 out of 91 probands lacking pathogenic variants in the gene. Missense mutations and splicing mutations often result in a variety of phenotypes with over two-thirds of missense mutations resulting in non-aniridia phenotypes, consistent with previous reports. More importantly, they established a new correlation pattern by revealing the existence of a very distinct and mild phenotype associated with 3′-cis-regulatory region copy number variations (Vasilyeva et al., 2021). Their statistical analysis revealed that patients with 3′-cis-regulatory region copy number variations, outside the coding region of PAX6, usually develop a milder aniridia phenotype without nystagmus, keratopathy or foveal hypoplasia. Their results support an existing view of genotype-phenotype correlations, which concludes that all kinds of loss-of-function variants should result in similar severe aniridic phenotypes, but do not support the idea that PAX6 missense variants are associated with a milder aniridia phenotype. Additionally, several recent studies on foveal hypoplasia grading by SD-OCT in PAX6 aniridic cohorts showed that copy number variations involving the 3′ regulatory regions without removing any of the PAX6 coding sequences were statistically associated with better preservation of the fovea and better visual acuity (Daruich et al., 2021; Pedersen et al., 2020). The study of Daruich and colleagues, including the largest cohort to date in terms of SD-OCT retinal phenotype characterization with a broad spectrum of various types of PAX6 variants representative of the diverse PAX6 mutation categories, showed that all kinds of premature termination codon variants, C-terminal extension variants as well as whole/partial PAX6 coding sequence deletions and some missense variants had a more severe retinal phenotype with the worst outcome in term of visual prognosis than genomic microdeletions restricted to the 3′ flanking regulatory regions of PAX6. The authors also showed that the degree of foveal maldevelopment was most commonly correlated with the severity of iris defects, but among PAX6 eyes with mild iris phenotype or no iris abnormality and unexplained vision loss, 74% presented with grade 4 or 3 foveal hypoplasia, highlighting that the severity of iris phenotype did not always correlate with that of the developmental retinal defect, and therefore could not predict the retinal damage and visual prognosis. These mild cases of iris defects associated with severe foveal hypoplasia were associated with various types of PAX6 mutations including some missense variants. Altogether, these studies reinforce the notion that PAX6 variants are associated with arrested foveal development more frequently than previously thought, even when the iris appears clinically intact. Very recently, Kit et al., reported a longitudinal natural history study of 86 aniridic patients with molecularly confirmed PAX6 mutations over a mean 16.3-year follow-up, and described the phenotypical differences in prevalence, severity, and onset of all ocular comorbidities associated with congenital aniridia (Kit et al., 2021). In this study, of the 86 aniridia patients, macular SD-OCT scans were available from 17 patients for foveal hypoplasia grading, and a significant difference in severity was observed between mutation groups with splice and frameshift subgroups displaying a more severe foveal hypoplasia, while the missense subgroup presented milder grades with no grade 3/4.The discrepancy between Kits’ study and those by the Petersen and Daruich groups concerning the retinal phenotype might be explained, in part, by a difference in the mutational PAX6 spectrum and the mutation subgroup classification resulting in underrepresented categories of variants, notably the absence of deletions restricted to the 3′ regulatory regions and missense variants mimicking premature termination codon loss-of-function variants in Kits’ study, resulting in a sampling bias for retinal phenotype-genotype correlation with all mutation categories representative of those reported in PAX6 databases. In conclusion, ophthalmologists also should be aware that almost all aniridia patients, including those with PAX6 missense variants, remain at high risk for severe foveal hypoplasia, regardless of the severity of the iris phenotype (Daruich et al., 2021).
4.6. Corneal phenotype-genotype correlation
The current progress on AAK genotype-phenotype associations are examined according to mutation type. The phenotypes arising from missense mutations can be variable and have generally been reported to result in either a milder version of AAK with slower progression or a classic progressive AAK phenotype. In a Danish study reporting 5 patients with missense mutations, two had no keratopathy (aged 9 and 25), while the remaining three had mild keratopathy (Grade 1–2) at ages ranging from 41 to 55 years (Grønskov et al., 1999). In a Korean study, 3 patients with missense mutations likely had a classical AAK phenotype, ranging from very mild keratopathy in a 4-month-old, to severe AAK in two patients aged 25 and 30 years (Lim et al., 2012). Nine patients with missense mutations were reported in a British study, for which no keratopathy was noted in four children and only mild keratopathy in one of the remaining adults aged 30 (Hingorani et al., 2009). In another British study, 9 of 19 aniridia patients with missense mutations were reported to have AAK (47%), only one of which required corneal surgery at the age of 34; however, AAK was not graded or well defined in the study and early stages could have been overlooked (Kit et al., 2021). By contrast, in a recent Russian study (Vasilyeva et al., 2021) it was conjectured that missense mutations most likely lead to loss-of-function of the PAX6 protein, thus not supporting a milder general ocular phenotype; however, in the same study, 5 of 7 patients with missense mutations had no detected keratopathy (although AAK was not graded). In a German cohort, 5 patients with missense mutations were reported, four with Grade 1–2 AAK (aged 4, 10, 20, 38) possibly indicating milder keratopathy, while the fifth aged 36 had bilateral Grade 3 AAK, being more consistent with classical AAK progression (Lagali et al., 2020b). To summarize, PAX6 missense mutations lead to variable phenotypic expression of AAK ranging from a mild to classical progressive phenotype. The phenotypic findings should consider the age and careful grading of the limbal involvement as AAK is not a static entity. Further studies are needed to examine whether specific subtypes or loci of missense mutations could explain the variable phenotypic expressivity of these mutations.
Premature termination codon mutations follow the expected classical and progressive AAK phenotype, which is age-dependent. The onset and rate of progression of AAK, however, can be variable in these cases. Notably, intronic mutations in non-coding regions of PAX6 also typically lead to premature termination codon and a classical phenotype (Lima Cunha et al., 2019). The effect of premature termination codon was demonstrated in a German cohort where 24 probands with nonsense-mediated decay -inducing premature termination codon mutations (2 of which were intronic) reflected the classical progression of AAK from ages 1 to 64, with milder grades earlier in life progressing to Grade 3 or 4 in adulthood, followed by corneal transplantation, keratoprosthesis implantation or enucleation (Lagali et al., 2020b). Similarly, in a British study with 17 patients harboring premature termination codon mutations, none to mild keratopathy was observed in childhood and moderate to severe keratopathy in adults (Hingorani et al., 2009). In another British study without AAK grading or distribution according to age, keratopathy was recognized in 72% of premature termination codon cases, with one fifth of cases undergoing some form of corneal transplantation in the fourth to fifth decade of life (Kit et al., 2021). Likewise in a Russian study examining 99 patients with either nonsense, frame-shifting or splicing mutations, the majority (65%) had keratopathy, although the AAK Grade and age of subjects were not reported (Vasilyeva et al., 2021). In two Indian studies where AAK was not graded, the studies reported variable keratopathy expressivity in premature termination codon cases, although subject age and progression of keratopathy were not specifically considered (Dubey et al., 2015; Neethirajan et al., 2009). In summary, premature termination codon mutations in PAX6 are nonsense-mediated decay inducing and strongly support the findings that the resulting PAX6 protein deficit results in the classic progressive AAK phenotype. In future studies a clear documentation of AAK grade in relation to age should be reported; detailed mutation information and longitudinal studies may in the future aid in explaining the variable onset and rate of progression of classical AAK.
C-terminal extension mutations lead to a classic AAK phenotype with either similar severity to premature termination codon mutations (Kit et al., 2021; Lagali et al., 2020b; Lima Cunha et al., 2019) or possibly a more severe phenotype (Hingorani et al., 2012). In a Korean study, a 3-year-old child with C-terminal extension mutation had mild keratopathy noted at this young age, while the keratopathy was moderate in a 26-year-old patient with C-terminal extension mutation (Lim et al., 2012). Of ten patients with C-terminal extension mutations in a British study, 3 children had none to mild keratopathy while 7 adults had mild to severe keratopathy (Hingorani et al., 2009). In another British study with 14 C-terminal extension cases, 12 (86%) had keratopathy, 3 of whom had corneal surgery (sign of severe AAK) at a mean age of 43 years (Kit et al., 2021). Likewise, in a German cohort, 3 patients with C-terminal extension mutations aged 8 to 18 had Grade 2 AAK, while a fourth patient with a C-terminal extension mutation aged 51 had Grade 3 AAK (Lagali et al., 2020b). Summarizing, C-terminal extension mutations are associated with a classic AAK phenotype; however, similar to premature termination codon mutations, more longitudinal data in relation to specific mutation is needed to evaluate whether certain C-terminal extension mutations result in earlier onset or a more severe AAK phenotype.
Multiple exon or whole-gene PAX6 deletions cause a more aggressive AAK phenotype, with onset at a younger age and typically progressing more rapidly than premature termination codon mutations. In a German cohort, 2 patients with multiple exon deletions had a severe AAK phenotype, including a 16-year-old patient with Grade 3 AAK in one eye with the other eye enucleated, and another patient aged 52 having had bilateral corneal transplants (Lagali et al., 2020b). In addition to multiple exons, the entire PAX6 gene may also be deleted with neighboring genes in the 11p3 region of chromosome 11. Among the reported cases of whole-gene deletions in various cohorts are a 41-year-old patient with severe keratopathy (Lim et al., 2012); 4 of 6 patients with gene deletions leading to keratopathy, which all required corneal surgery (Lim et al., 2012); a 42-year-old patient with bilateral Grade 3 AAK (Voskresenskaya et al., 2017), a 7-year-old child with Grade 2 AAK (Zhang et al., 2011) and an 11-year-old child with bilateral Grade 4 AAK (Lagali et al., 2020b).
Owing to its rarity, reports describing the AAK phenotype in WAGR are sporadic and limited in the scientific literature. The available evidence indicates that multiple gene deletions in the context of WAGR result in the most severe form of AAK, emerging at a young age and often rapidly progressing to an advanced stage, even in childhood. Four WAGR patients were reported in a German cohort, including a 4-year-old having Grade 2 AAK, an 11-year-old with Grade 4, a 14-year-old with Grade 3–4 and a 20-year-old with Grade 2–3 (Lagali et al., 2020b). In another report, a 3-year-old child with WAGR had cataract extraction and corneal transplantation, similarly suggesting an early aggressive AAK phenotype (Park et al., 2010). Another child with WAGR aged 4 had corneal opacity at this age, while in another cohort, two children aged 3 and 8 also had bilateral corneal opacities (Yokoi et al., 2016). In a case report from Japan, a one-month-old infant with WAGR had microcornea with a pronounced corneal cyst in one eye, and Grade 3 AAK in the other (Kawase et al., 2001). This population could therefore be considered as a preferred target group for the first new AAK therapies aiming to compensate for PAX6 haploinsufficiency by overexpression of the non-mutant allele (Dorot et al., 2022; Oved et al., 2021; Rabiee et al., 2020). The rationale is that in the non-WAGR population, AAK takes decades to progress, making timing and duration of therapy and follow-up difficult for initial clinical trials designed to demonstrate efficacy in slowing down the progression or reversing the AAK phenotype. By contrast, in WAGR cases AAK onset is earlier in life, and progression occurs over a shorter timeframe. Interestingly, although both premature termination codon mutations and multiple exon or whole-gene PAX6 deletions would result in only one functional gene and a corresponding reduction in the level of the PAX6 protein, the increased severity of the AAK phenotype at a younger age in the case of multiple exon or whole-gene PAX6 deletions would seem to indicate some residual function of the mutant allele and its resulting protein in premature termination codon cases despite its nonsense-mediated decay.
In contrast to chromosomal deletions including the PAX6 gene, patients with gene deletions spanning the downstream 3′ PAX6 regulatory regions, for example encompassing the genes ELP4, IMMP1L, DNAJC24 and DCDC1, have no AAK or show very mild disease. Two patients in a German cohort were reported, one with full deletion of ELP4 and DCDC1 having Grade 1 AAK at age 26 and the other with ELP4, IMMP1L, DNAJC24 and DCDC1 deletion having Grade 0–1 AAK at age 34 (Lagali et al., 2020b). Another patient, aged 54, did not have a detectable PAX6 mutation and had Grade 0 AAK with full limbal palisade structures indicating an intact and morphologically normal limbal stem cell niche (Lagali et al., 2013), and was later determined to carry a deletion of the ELP4 gene (unpublished result). Other reports include a 37-year-old Chinese female with ELP4 and DCDC1 deletion with Grade 1 AAK (You et al., 2020); a Chinese family with ELP4 and DCDC1 deletion where both mother (aged 37) and son (aged 7) had Grade 1 AAK (Zhang et al., 2011); three Russian patients aged 26–45 with ELP4 and DCDC1 deletion having Grade 0 AAK, two of which had full limbal palisade structures (Voskresenskaya et al., 2017); and a 44-year-old mother and 13-year-old son from Cyprus with ELP4, IMMP1L, DNAJC24 and DCDC1 deletion having Grade 0–1 AAK as assessed from published photos (Syrimis et al., 2018). In a Russian study reporting 15 patients with regulatory region deletions, 14 were reported not to have keratopathy, and the authors showed a statistically significant association between the presence of these regulatory mutations and the absence of keratopathy (Vasilyeva et al., 2021).These very mild to nonexistent cases of AAK indicate that the 3’ PAX6 regulatory regions in downstream genes have almost no effect on the AAK phenotype, and that AAK and its progression is almost exclusively linked to mutations within or deletions affecting PAX6.
4.7. Genetic differential diagnosis of classical aniridia and non-classical aniridia
While PAX6 is the major gene for congenital aniridia, in rare cases aniridia may result from variants in the ITPR1 gene causing the Gillespie syndrome (Gerber et al., 2016; McEntagart et al., 2016). Also, a single aniridia pedigree has been shown to have a missense mutation in TRIM44, ~4 Mb away from PAX6 on 11p13 (Zhang et al., 2015). The pathogenicity of this TRIM44 variant was supported by in vitro functional data demonstrating that it causes reduced PAX6 expression, which results in the silencing of PAX6 expression.
Congenital aniridia belongs to the spectrum of ocular anterior segment dysgenesis (ASD). ASD disorders are characterized by a high phenotypic variability and are genetically heterogeneous with many genes as-yet undiscovered, explaining the low genetic diagnostic yield and the difficulties in making a clear clinical diagnosis for these disorders. The overlapping phenotypic presentation of ASD disorders reflects the dynamic interaction of the embryological events required for proper development of the ocular structures of the anterior segment which are under the control of diverse transcription factors playing a role in intricate mechanistic pathways (Chrystal and Walter, 2019; Cvekl and Tamm, 2004; Ito and Walter, 2014). ASD disorders encompass different clinical entities, mainly including congenital aniridia, Axenfeld-Rieger syndrome (ARS), primary congenital glaucoma (PCG), Peters anomaly, and other anterior segment dysgenesis phenotypes. Table 2 gives a brief summary of main phenotypes associated with the major ASD genes which can help to prioritize genetic analysis based on phenotype. Foveal hypoplasia or presence of extra ocular features such as dental and umbilical anomalies may help for guide the clinicians. As shown in Table 2, each gene shows phenotypic pleiotropy and each ASD subtype shows genetic heterogeneity, highlighting that clinical phenotyping is central to establish the pathogenicity of a genetic finding and to establish novel accurate genotype-phenotype correlations. The diagnostic yield in ASD is highly variable, and more than 50% of patients being undiagnosed by conventional sequencing methods. This suggests that novel genes or other mechanisms still need to be elucidated, such as presence of variants in noncoding regions of genes, not covered by standard diagnostic techniques, and thus requiring whole genome sequencing (WGS). Identification of novel genetic causes will not only have a major clinical impact but also provide a biological insight into the pathways involved in the development of the anterior segment of the eye, thus leading to the development of drugs targeting those identified pathways. So far, only a limited number of genetic studies with large cohorts of ASD patients have been completed using high throughput sequencing techniques, but some recently genotype-phenotype associations with PXDN, CPAMD8, CYP1B1, FOXE3, COL4A1, ADAMTS17 have been recently described, but in only a handful of patients (Cheong et al., 2016; Hollmann et al., 2017).
Table 2.
Summary of genetic factors associated with aniridia and aniridia-like diagnosis in humans.
| Gene | Genomic Location | Function | Inheritance | Major Human Phenotype | Common ocular features, associated features | Rare ocular features, associated features | References |
|---|---|---|---|---|---|---|---|
| PAX6 OMIM # 607108 | 11p13 | Transcription factor with a paired domain and a homeodomain | Autosomal dominant | Aniridia | Iris Hypoplasia/aniridia Foveal hypoplasia AAK Glaucoma Cataract |
Optic nerve hypoplasia Peters anomaly Anosmia Diabetes |
Jordan et al. (1992) |
| PITX2 OMIM # 601542 | 4q25 | Transcription factor with a paired-related bicoid-type homeodomain | Autosomal dominant | Axenfeld-Rieger syndrome | Pseudopolycoria, Iris Hypoplasia Irido-corneal adhesions Glaucoma Embryotoxon Extraocular features |
Peters anomaly Aniridia-like phenotypes Congenital glaucoma |
Semina et al. (1996) |
| FOXC1 OMIM # 601090 | 6p25.3 | Forkhead transcription factor | Autosomal dominant | Axenfeld-Rieger syndrome | Pseudopolycoria Iris Hypoplasia Irido-corneal adhesions Congenital glaucoma Embryotoxon Extraocular features |
Peters anomaly Aniridia-like phenotypes Foveal hypoplasia |
(Mears et al., 1998; Nishimura et al., 1998) |
| FOXE3 OMIM # 601094 | 1p33 | Forkhead transcription factor | Autosomal recessive/Autosomal dominant | Cataracts with or without anterior segment dysgenesis, microphthalmia | Cataracts Peters anomaly Primary aphakia Sclerocornea Microphthalmia Extraocular features |
Aniridia-like phenotypes Sclerocornea- Microphthalmia- Aphakia |
(Semina et al., 2001; Valleix et al., 2006) |
| CYP1B1 OMIM # 601771 | 2p22.2 | Cytochrome P450 enzyme | Autosomal recessive | Congenital glaucoma | Congenital glaucoma | Peters anomaly Aniridia-like phenotypes |
Stoilov et al. (1997) |
AAK: Aniridia Associated Keratopathy.
5. Therapeutic approaches
During infancy, children with congenital aniridia present marked visual impairment due to foveal hypoplasia. Furthermore, the evolution of the disease is characterized by severe complications: glaucoma with progressive trabecular dysfunction and AAK with progressive corneal opacification. Cataracts are a curable feature, even if risky surgery accelerates limbal deficit and corneal opacification.
5.1. Iris implants
The loss of iris has not just cosmetic implications, it also has direct implications on visual acuity, photophobia, glare and trabecular meshwork anomalies with glaucoma (Beltrame et al., 2003; Landsend et al., 2021). Prosthetic iris devices with or without intraocular lens may be the only choice to improve patients’ symptoms. However, their implantation is a challenging task with a high risk for complications. Prosthetic iris devices has been mostly used in traumatic aniridia, but also in congenital aniridia. French national guidelines for congenital aniridia management do not recommend prosthetic iris devices. The choice of prosthetic iris devices is also difficult, due to the absence of guidelines and vast data in the literature. There are few prosthetic iris devices available: iris-lens diaphragm prosthesis, customized artificial iris and capsular tension ring-based prosthetic iris device, each of them with their specific pros and cons. The main advantage is that Iris-lens diaphragm can correct both aniridia and aphakia simultaneously without the need of an additional intraocular lens. However, they are large, rigid, and brittle, needing large corneal incisions (>9–10 mm) and hindering the bag insertion. Following their implantation, visual acuity and symptoms improve, such as glare and photophobia, specifically two-thirds of cases show an improvement in visual acuity while most of them report a resolution of optical symptoms. However, quality of evidence is not robust and most reports are retrospective series including congenital and traumatic aniridia (Aslam et al., 2008; Reinhard et al., 2000; Sundmacher et al., 1994). The most frequent post-operative complication is secondary glaucoma, followed by prothesis decentration and corneal endothelial decompensation. Secondary glaucoma has been reported in about 20–30% of eyes after sulcus implantation of iris-lens diaphragm (Qiu et al., 2016; Reinhard et al., 2000). Prothesis mispositioning and progression or development of corneal epithelial disease have been reported in about 20% of cases (Aslam et al., 2008; Reinhard et al., 2000; Sundmacher et al., 1994). Additionally, mild persistent intraocular inflammation or anterior uveitis after prosthetic iris devices has been observed in most of cases, over a mean duration of 8 months (Aslam et al., 2008; Reinhard et al., 2000; Sundmacher et al., 1994). It is important that prosthesis positioning does not interfere with the trabecular meshwork, and post-operative topical treatment keeps intraocular chronic inflammation under control. In case of failure, additional surgery is required to manage glaucoma and corneal endothelial decompensation. Limbal stem cell failure, vitreous hemorrhage, and retinal detachment are even more rare. Apart from vitreous hemorrhaging, which usually occurs in the early follow-up, the onset of other complications is later in the follow-up. A customized artificial iris consists of only the iris diaphragm without a central optic and seems to have lower complications rates than iris-lens diaphragm (Mayer et al., 2020). ArtificialIris (Customflex; HumanOptics), the most commonly used, is a foldable hydrophobic silicone elastomer that can be inserted with a small corneal incision (2.5–3.2 mm) with consequently better visual outcomes. However, it needs to be implanted into the bag. ArtificialIris also has the best cosmetic results, due to its customizable nature with a variety of colors available. For these reasons ArtificalIris implantation is becoming more and more popular in recent years. However, the experience with those devices is still limited, particularly in congenital aniridia (Mayer et al., 2020). Finally, a ring-based prosthetic iris device consists in two rings that need to be implanted and dialed into position to form a closed diaphragm. Implantation could be performed with the same small corneal incision created for phacoemulsification and intraocular lens implant. Surgery can be technically challenging, as the alignment must be accurate and the capsule bag needs to be intact. Further studies are needed to determine the efficacy and complication rate of these devices in congenital aniridia.
5.2. Cataract surgery
The most important component of evaluation is to assess the visual significance of the cataract. This can be difficult given the accompanying foveal and/or optic disc hypoplasia and nystagmus. Baseline vision measurements are essential and, on old enough patients, glare testing can be used to see if the vision drops by 2 lines or more.
Surgical techniques differ between children and adults with aniridia. The following discussion is with regards to pediatric cataract surgery aniridia. There are some points that need consideration prior to surgery depending on the age of the child being operated upon. When there is an absent iris, as in congenital aniridia, the anterior chamber depth is shallower compared to normal controls (Voskresenskaya et al., 2021). This means that any lens power equations using ACD as a predicted constant must be customized. This should also be taken into consideration since the implanted lens may be sitting slightly forward if implanted in the bag without optic capture in the posterior rhexis.
5.2.1. Incision
To avoid any deterioration of the limbal stem cells, a clear corneal wound is advised (Landsend et al., 2021) or alternatively a long sclerocorneal tunnel incision.
Capsulorhexis.
The anterior lens capsule has been shown to be much thinner than that of normal controls, but this work was performed on adults with congenital aniridia (Schneider et al., 2003). Nevertheless, this should be taken into consideration since tears in the anterior capsule have been reported during capsulorhexis, albeit in adults again (Uka et al., 2005). It is important that the anterior capsulorhexis opening is smaller than the optic of the intraocular lens to prevent forward displacement and subsequent corneal endothelial damage (J. D. Wang et al., 2017). It is also important since this acts as a natural light block when fibrosis of the anterior capsule occurs. While YAG capsulotomy has been advocated (J. D. Wang et al., 2017), this is not always easy, especially in the presence of nystagmus, and therefore posterior capsulorhexis should be considered for all pediatric ages, with anterior vitrectomy in children 4 years and under and without for children over 4 years old. Using the two incision push-pull rhexis technique allows for a reliable and repeatable method to size the opening of the anterior capsule (Nischal, 2002).
5.2.2. Lens implantation
Whenever possible an intraocular lens implant should be placed; using aphakic contact lenses can adversely affect the limbal stem cells, which often fail with time in PAX6-related aniridia. Scleral-sutured lenses in children often erode, but have been reported in adults with congenital aniridia. If the anterior capsule opening is too big for the optic diameter or there is a tear in the anterior capsule, posterior optic capture with anterior vitrectomy should be considered (Medsinge and Nischal, 2015). Some authors advocate the use of single-piece blue blocking lenses, but a three-piece foldable hydrophobic acrylic lens gives the best capsular bag stretch. That said, the use of a capsular tension ring may also provide good lens capsule stretch (J. D. Wang et al., 2017). The advantage of using a three-piece lens is that if during implantation there is a tear in the anterior capsule (due to the reported fragility of the capsule in congenital aniridia) then the lens can be fastened via optic capsule capture in the posterior capsulorhexis.
It is worth noting that in optic capsule capture there are fewer lens epithelial cells encroaching the visual axis as compared to the bag lens placement. Allowing fibrosis of the lens capsule may be the safest option in younger children. More importantly there is a report suggesting that postoperative vision is the same with or without iris diaphragm lenses in cases of congenital aniridia (Aslam et al., 2008).
5.2.3. Post-operative considerations
While standard postoperative anti-inflammatory drops such as steroids should be used, a preservative-free version should be considered given the limbal stem cell abnormalities seen in these patients (Landsend et al., 2021), especially since the progression of aniridia-associated keratopathy has been noted in adults as well as anecdotally reported in children (usually teenagers) (Park et al., 2010). Anterior fibrosis syndrome may rarely be seen in children (Moshirfar et al., 2022), but the prognosis is extremely poor.
Cataract surgery in congenital aniridia should be undertaken when vision is affected. This can only be detected by comparing visual function to baseline tests using behavioral tests (central visual acuity with glare testing) and electrophysiological testing (visual evoked potentials). Surgical techniques must be meticulous and undertaken by an experienced cataract surgeon.
5.3. Glaucoma treatments
Treating glaucoma in aniridia remains challenging. Medical treatment can control intraocular pressure in many aniridia patients, at least initially (Sihota et al., 2021). Topical medication containing prostaglandin analogs, beta-blockers or carbonic anhydrase inhibitors are the most frequently employed treatment. It is recommended to use preservative-free formulations, and combined therapy is often required (Muñoz-Negrete et al., 2021). Surgical treatment is required in many cases. A recent study showed that glaucoma surgery was performed in half of eyes with aniridic glaucoma during a mean follow-up period of 16 years. The mean age of glaucoma surgery was 30.7 ± 19.5 years (Kit et al., 2021).
There is no consensus on the nature of first-line surgery for glaucoma in aniridia. Surgical options include goniotomy, trabeculotomy, trabeculectomy with or without antimitotic agents, drainage devices and cyclodestructive procedures.
Prophylactic goniotomy could be considered in aniridia patients with tissue extensions between the iris stump and angle wall, but without glaucoma. Prophylactic goniotomy in eyes with iris extensions covering more than half of the circumference of the trabecular meshwork made it possible to control intraocular pressure (<22 mm Hg) and not increase optic disk cupping in 89% of eyes (49 of 53 eyes) after an average follow-up time of 9 years and 6 months (Chen and Walton, 1999).
Trabeculotomy has been proposed as initial surgery in aniridic uncontrolled glaucoma. Adachi et al. reported controlled intraocular pressure (≤21 mm Hg) in 10 of 12 eyes (83%) after trabeculotomy. The mean patient age upon initial trabeculotomy was 4.7 years old. Six eyes were treated once, and 4 eyes twice during a mean follow-up of 9.5 years (Adachi et al., 1997).
Trabeculectomy may be considered in older children or adults with aniridic glaucoma, preferentially associated with antimetabolites (Sihota et al., 2021). Long-term success of trabeculectomy in aniridia remains variable among rapports (Landsend et al., 2021). Okada et al. reported that, in aniridia patients aged <40 years, intraocular pressure of ≤20 mmHg persisted for a mean 14.6 months after trabeculectomy (Okada et al., 2000). A recent study has found that 96% and 52% of eyes surgically treated with trabeculectomy with mitomycin C achieved intraocular pressure of ≤18 mmHg with or without topical medication, respectively, during a mean follow-up of 7 years (Sihota et al., 2021).
In aniridia patients after infancy or early childhood, the implantation of a drainage device is the choice for many clinicians if eye drops are not sufficient to control the intraocular pressure (Landsend et al., 2021). A drainage device could also be the first choice in closed angle aniridic glaucoma (Muñoz-Negrete et al., 2021). The studies on various drainage devices including Baerveldt, Molteno or Ahmed devices, show relatively high success rates in aniridic glaucoma (Landsend et al., 2021). A success rate of 100% at 6 months and 88% after one year have been reported after drainage device surgery (Almousa and Lake, 2014; Arroyave et al., 2003). A recent study resulted in lower intraocular pressure and a higher rate of surgical success for the Aurolab aqueous drainage implant compared to trabeculectomy with mitomycin C in eyes with aniridia-associated glaucoma (Durai et al., 2021).
Cyclodestructive procedures could control intraocular pressure in about 25–50% of refractory glaucoma (Blake, 1952; Wiggins and Tomey, 1992). It has been suggested that there is a higher rate of complications in aniridia patients compared to other cases of refractory pediatric glaucoma. Authors argued that absent iris tissue possibly reduces the isolating capacity during cyclocryotherapy, increasing the risk of complications (Wagle et al., 1998).
5.4. AAK treatments
5.4.1. Conservative approaches
Optimizing the ocular surface using artificial tears without preservatives is generally recommended as the first step in managing AAK. However, while sufficient in patients exhibiting sub-clinical AAK, in cases of mild to moderate AAK, a second-line treatment is required. Autologous serum eye drops or amniotic membrane transplantation – see below – are therefore used (although temporarily) in such cases. Autologous serum eye drops provide lubrication and contain various growth factors. In a prospective interventional case series, 26 eyes from 13 patients with AAK were treated with autologous serum eye drops 8 times a day for 2 months. All patients showed a subjective improvement in keratopathy symptoms after autologous serum applications. Corneal epithelialization, corneal epithelial cell squamous metaplasia, and tear stability improved significantly with the treatment and showed to be superior to conventional therapy with substitute tears for improving the ocular surface and subjective comfort. However, visual acuity, regression of vascular pannus, and sub-epithelial scarring showed only slight improvement (López-García et al., 2008). In another report, a 17-year--old patient with AAK and a persistent epithelial defect was treated successfully using maternal finger-prick blood, a potential alternative to autologous serum eye drops in the management of refractory persistent epithelial defect (Pujari et al., 2021). Amniotic membrane-derived eye drops might also be a valuable alternative, but no reports are available in the literature. Despite their usefulness, a greater knowledge of the growth factors present in such preparations is needed. This would eventually help the development of more efficient eye drops preparations. Since topical cyclosporine is used for dry eye syndrome, its early use in PAX6-related aniridia may be considered (Periman et al., 2020).
5.4.2. Surgical approaches. Amniotic Membrane Transplantation.
Amniotic membrane transplantation is used alone or in combination with limbal stem cell transplantation as a way to reduce pain and promote the healing of the ocular surface. Tseng et al. performed amniotic membrane transplantation in 3 eyes from 3 aniridia patients with mild limbal stem cell deficiency and visual potential limited by nystagmus and other posterior segment abnormalities. Following amniotic membrane transplantation alone, their corneal surfaces became smooth, stable, and transparent enough to regain their best preoperative visual acuity (Tseng et al., 1998). In another study by Lopez-Garcia et al., 14 eyes in patients with congenital aniridia and moderate limbal stem cell deficiency were evaluated after amniotic membrane transplantation alone. Visual acuity showed a mean improvement of 0.3 at 24 months of follow-up. Corneal re-epithelialization was seen in the first 2 months, but after 9 months, persistent epithelial defect and chronic ulceration appeared in some patients. Corneal clarity and peripheral neovascularization markedly improved after 2 months. Impression cytology showed an improvement in squamous metaplasia at 3 and 6 months, however, after 9 months, a progressive worsening was seen in epithelial cell metaplasia. Most importantly, 9 months after amniotic membrane transplantation, a limbal biopsy showed nearly normal epithelium and limbo-corneal stroma (López-García et al., 2005).
5.4.3. Corneal transplantation
The outcome of penetrating keratoplasty alone for AAK is poor, although it may still be a helpful adjunctive procedure for restoring corneal clarity (De La Paz et al., 2008). This is consistent with underlying limbal stem cell deficiency observed in AAK. Tiller et al. showed no difference in long-term visual acuity between aniridia patients treated with corneal transplantation or not. While transient improvement was seen after keratoplasty, most grafts failed because of the recurrence of AAK (Tiller et al., 2003).
Performing penetrating keratoplasty without taking into account the underlying limbal stem cell deficiency is only going to provide temporary improvement. To help assess the three degrees of limbal stem cell deficiency, some authors advocate using OCT to assess the presence or absence of the Palisades of Vogt since these are the microenvironments in which the limbal stem reside; no Palisades of Vogt and likely no or very little limbal stem (Lagali et al., 2013; Nischal and Lathrop, 2016). Lamellar keratoplasty (either Deep Anterior Lamellar Keratoplasty or Anterior Lamellar Keratoplasty) can be indicated for optical and/or tectonic reasons, for example to remove stromal opacities that appear in severe and advanced stages of AAK. Given the inflamed condition and immunocompromised status of AAK eyes, lamellar keratoplasty is proposed instead of penetrating keratoplasty, as it carries fewer risks of immune rejections. However, if a limbal transplant does not accompany or precede the keratoplasty, it usually ends up in recurrent graft failure, as described for approaches involving penetrating keratoplasty.
5.4.4. Limbal allograft
To treat bilateral limbal stem cell deficiency in aniridia, three options exist for the source of stem cells in limbal stem cell transplantation: keratolimbal allografts, living-related conjunctival-limbal allograft and ex vivo expanded epithelial cells (Cultured limbal epithelial transplantation and cultured oral mucosa epithelial transplantation, discussed below). In 2003, Holland et al. described the results obtained in 31 eyes from 23 patients with aniridia undergoing keratolimbal allografts. Twenty-three eyes achieved a stable ocular surface (median follow-up time = 27 months). Twenty eyes (64.5%) had undergone subsequent penetrating keratoplasty and, for the whole group, visual acuity improved from 20/100 to 20/165 after keratolimbal allografts and subsequent penetrating keratoplasty. Interestingly, patients receiving systemic immunosuppression had a greater likelihood of achieving ocular surface stability and improved visual acuity compared with those who received only topical immunosuppression (Holland et al., 2003). In a recent retrospective case series, Cheung et al. compared the long-term outcomes of 224 eyes (of which 124 were from aniridia patients) that underwent keratolimbal allografts alone and 63 eyes (of which 27 were from aniridia patients) that underwent living-related conjunctival-limbal allograft alone, with a mean follow-up time of 7.2 years for all eyes. Although a specific analysis of aniridia eyes was not performed, overall living-related conjunctival-limbal allograft demonstrated lower rejection rates, improved graft survival, and better best-corrected visual acuity compared with keratolimbal allografts. For living-related conjunctival-limbal allograft eyes, 82.5% maintained a stable ocular surface compared with 64.7% of keratolimbal allografts eyes at the last follow-up. Only 6.3% of living-related conjunctival-limbal allograft eyes demonstrated a failed ocular surface compared with 15.6% of keratolimbal allografts eyes. The mean BCVA was 20/158 for keratolimbal allografts eyes compared with 20/100 for living-related conjunctival-limbal allograft eyes at the last follow-up. A smaller proportion of living-related conjunctival-limbal allograft eyes (30.2% compared with 43.3%) developed an episode of acute rejection, and a higher proportion of these episodes were resolved with treatment in the living-related conjunctival-limbal allograft group (79.0% compared with 53.6%) (Cheung et al., 2020). There is some concern whether or not living-related conjunctival-limbal allograft contains a sufficient number of limbal stem cells, but studies seem to indicate that they are sufficient for effective treatment of AAK. Heavy and prolonged immunosuppression regimens are needed for long-term success. Most importantly, choosing the right mmunosuppressive drugs and doses and length of administration is a delicate balancing act. Too much immunosuppression will save the graft, but carries well known side effects, not enough immunosuppression will cause allograft rejection: something that needs to be addressed carefully.
5.4.5. Cultured limbal epithelial transplantation
Because of the PAX6 mutation, the studies so far reported have used unaffected allogeneic donor cells (allo-cultured limbal epithelial transplantation). In the first cohort of patients undergoing allo-cultured limbal epithelial transplantation, Shortt et al. had 3 patients with AAK. The clinical outcome observed between 10 and 13 months showed that 2 out of 3 experienced improved visual acuity (Shortt et al., 2008). However, in their 3-year follow-up study including a cohort of 10 eyes from 9 patients with AAK treated with allogeneic cultured limbal epithelial transplantation, visual acuity gradually declined over the next 30 months (Shortt et al., 2014). In this latter study, immunosuppression was stopped at 6 months, as longer regimens were not considered to be appropriate given the lack of evidence of long-term donor cell survival on the ocular surface following transplantation (Daya et al., 2005). Comparable outcomes were reported by Behaegel et al., who assessed the efficacy and safety of Human Leukocyte Antigen-matched allogeneic cultured limbal epithelial transplantation in 6 patients with severe AAK. After a mean follow-up of 53.6 months, 4 of 5 eyes were graded as failures and 1 as a partial success (Behaegel et al., 2022), thus suggesting that HLA-matching is not sufficient to prevent allo-cultured limbal epithelial transplantation failure. Cultured limbal epithelial transplantation might not be the appropriate approach for treating AAK, thus begging the question of whether replacing defective LSCs alone is sufficient or if a functioning microenvironment should also be targeted for successful grafting.
5.4.6. Cultured oral mucosa epithelial transplantation
Since oral mucosal cells do not express PAX6 in the mouth, their use as an alternative source of autologous stem cells for corneal epithelium regeneration could be beneficial in order to avoid the disadvantages of immune-mediated rejection of allogeneic cells. Interestingly, three-year follow-up studies in non-aniridia patients with limbal stem cells deficiency showed that visual acuity was improved in 50% of eyes receiving cultured oral mucosa epithelial transplantation (Nakamura et al., 2011). Dobrowolski et al. performed cultured oral mucosa epithelial transplantation in 17 eyes from 13 aniridia patients and showed that 76.4% of the eyes had regular transparent epithelium, with 23.5% developing epithelial defects or central corneal haze. Over an observation period of 12–18 months, visual acuity improved in 88.2% of cases, ranging from hand movement at 0.05 before surgery to hand movement up to 0.1 after surgery (Dobrowolski et al., 2015). Long-term follow-up studies will have to be implemented in order to evaluate the potential influence of the mutated PAX6 during oral mucosal cell transplantation onto the ocular surface and whether these cells could have any therapeutic benefit in AAK.
5.4.6.1. Keratoprosthesis surgery.
Keratoprosthesis surgery is often the last option for the treatment of eyes with end-stage AAK. Shah et al. reported a retrospective review of 46 eyes from 34 patients with AAK who underwent Boston type-1 keratoprosthesis surgery with minimum 2-year follow-up. Thirty-four eyes (74%) had previously failed keratoplasty. Thirty-five eyes (76%) previously underwent ocular surface stem cell transplantation, specifically a keratolimbal allograft. Within the first 6 months post-surgery, 74% (34/46) of patients experienced a gain of ≥2 lines of vision. Overall, there was a gain of ≥2 lines of vision in 43.5% (20/46) of patients at last follow-up, thus confirming that visual improvement in AAK cases occurs at a high rate with the Boston type-1 keratoprosthesis. Complications in aniridia patients include failure of the prosthesis, infections, formation of a retroprosthetic membrane, developing or worsening of glaucoma and choroidal detachment. Nevertheless, keratoprosthetic implantation has become a treatment option for severe conditions that have low probabilities of success, and the Boston type-1 keratoprosthesis is the one that is most commonly used, even as a primary indication (Shah et al., 2018)
5.5. Scleral contact lenses
AAK is one of the main causes of visual impairment with corneal erosions, ulcers, neovascularization and opacities. After the failure of well-conducted ocular surface treatment with regular application of preservative-free artificial tears, a scleral lens appears to be an option of interest (Kojima et al., 2016). Autologous serum is also an interesting option, but it is time-consuming for patients and may not be well tolerated, especially in children. Hence, scleral contact lenses may be proposed. A scleral lens is gas-permeable and corrects ocular surface irregularities. The scleral contact lens with a large base curve allows a tear and fluid reservoir to form between the corneal surface and the scleral lens, maintaining a constantly moisturized cornea. Some adapted patients show visual improvement. Permanent corneal tear hydration results in the corneal healing of recurrent epithelial erosions and improvement in ocular pain. Various types of scleral lens may be fitted, however adaptation remains usually long and complex. Long-term results are encouraging in reducing ocular surface defects and corneal pain as well as maintaining visual acuity (Romero-Rangel et al., 2000; Schornack, 2011).
5.6. Future innovative therapeutic approaches
To overcome the limitations of current therapeutic options for AAK, innovative approaches are under evaluation by different teams, including gene therapy (adeno-associated virus (AAV) delivery, in vivo gene editing through clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) and nonsense mutation suppression) and drug repurposing (Fig. 10).
Fig. 10. Innovative therapeutic approaches for aniridia-associated keratopathy (AAK).
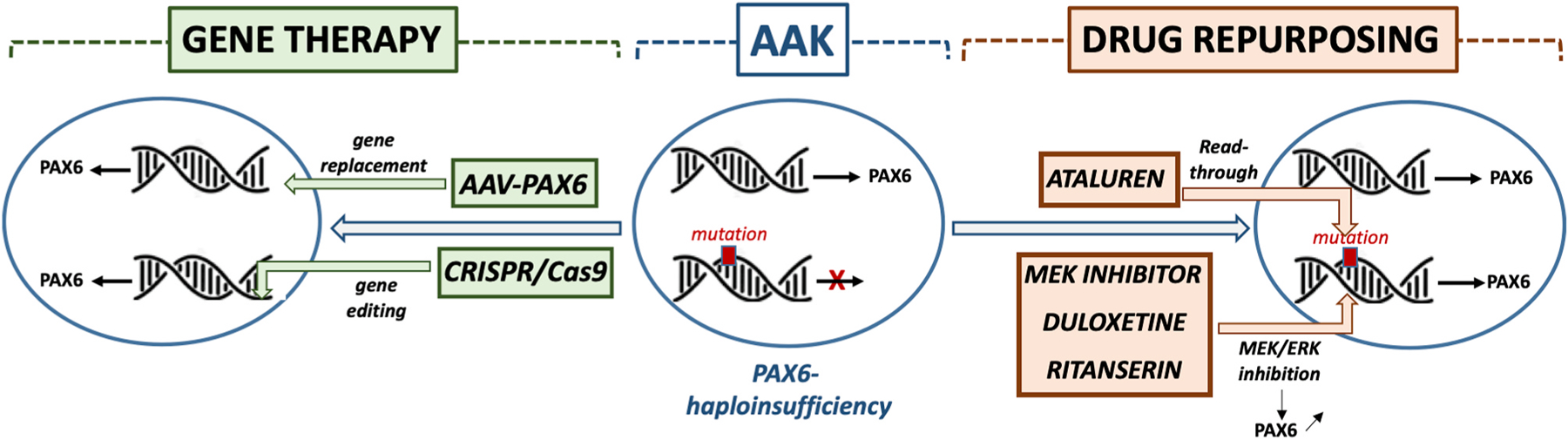
To overcome the limitations of current therapeutic options for AAK, innovative approaches are under evaluation including gene therapy (adeno-associated virus (AAV) delivery, in vivo gene editing through clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) and nonsense mutation suppression) as well as drug repurposing.
5.6.1. Gene therapy
Gene replacement is promising because it can ameliorate the main causes of AAK by directly correcting the genetic information in the cornea. However, because PAX6 dosage is critical for cornea homeostasis, meaning that the overexpression of PAX6 is phenotypically deleterious in mice as is haploinsufficiency (Mort et al., 2011), gene replacement can be effective only if the corrected gene is delivered according to the genuine PAX6 regulatory sequences. Elizabeth Simpson’s team has designed vectors containing small specific regulatory regions of the Pax6 gene called “MiniPromoters” (2). In particular, some are targeting corneal cells (Korecki et al., 2021). These tools can be delivered both topically (eye drop) or intravenously (postnatal day 2). It remains to be demonstrated if these vectors could rescue in vivo aniridia-like mouse models (Mirjalili Mohanna et al., 2020).
In vivo gene editing: the bacterial CRISPR and CRISPR-associated protein 9 (Cas9) nucleases is a highly efficient and precise system that can target dominantly-inherited conditions caused by heterozygous missense mutations through inclusion of the mutated base in the short-guide RNA sequence (Doudna and Charpentier, 2014). It allows for the theoretical permanent treatment of genetic diseases. This technology has been successfully used to produce a new mouse model of aniridia, closer to the human phenotype (Mirjalili Mohanna et al., 2020). In order to be applicable as a potential therapeutic approach for human aniridia, future studies would need to explore direct postnatal eye therapy and demonstrate that no off-target modification of the genome occurred. AAV could also serve as the vehicle for gene correction through genome editing.
Gene silencing: it is generally considered that the nonsense mutated allele does not produce stable mRNA, thus leading to rapid mRNA degradation without producing a mutated protein, such as in PAX6 haploinsufficiency. Therefore, gene silencing using antisense oligonucleotides cannot be applied for aniridia. Similarly, exon-skipping, which could be relevant for diseases in which an exon can be removed without a deleterious phenotype cannot be used for aniridia. In the case of aniridia, clinical evidence indicates that each exon of PAX6 is essential.
miRNA modulation: small non-coding RNAs called miRNAs post-transcriptionally regulate gene expression and protein levels, and many miRNAs have been reported to be important for corneal homeostasis (Mukwaya et al., 2019; Shalom-Feuerstein et al., 2012). In particular, few miRNAs have been reported to be dysregulated in conjunctival cells obtained from aniridia subjects and could repress AAK-specific corneal neovascularization and inflammation (Latta et al., 2021b). This mutation-independent approach could be applicable to the majority of aniridia patients. Encouragingly, several miR-related clinical trials are ongoing for non-ocular conditions.
5.6.2. Pharmacological treatments
Apart from gene therapy, a number of pharmacological approaches potentially relevant for human AAK, either directly or indirectly, have been recently reported. Existing drugs shown to have a therapeutic benefit in animal models of aniridia could be repurposed for use in the eye, potentially providing an even faster route to clinical use.
Nonsense mutation suppression drugs: most PAX6 mutations are nonsense mutations that lead to premature termination codon (Landsend et al., 2021). Ataluren enables ribosomal read-throughs of mRNA containing premature termination codon, essentially bypassing the stop codon UGA (compared to UAA codons), resulting in production of a full-length protein. Ataluren has been approved by the United State Food and Drug Administration to treat congenital aniridia based on promising in vivo data on Pax6Sey mouse (Gregory-Evans et al., 2014), suggesting that developmental eye defects can be efficiently corrected following postnatal treatment (X. Wang et al., 2017a). A Phase-2 clinical trial (STAR Study) was designed to evaluate the safety and efficacy of an oral ataluren suspension (named premature termination codon 124 or Translarna) (Koenig et al., 2009). However, the STAR trial did not meet its primary endpoint, apparently due to patient age and phenotypical discrepancies (https://clinicaltrials.gov/ct2/show/NCT02647359). Inclusion of children, adults and the elderly, presumably with varying grades of AAK, may hinder the assessment of the therapeutic effect on AAK, which may be more sensitive to treatment in earlier stages. Nevertheless, topical administration of the drug could improve availability of the drug for the eye and cornea, in particular (Djayet et al., 2020).
Drugs that reduce neovascularization: in AAK, neovascularization is a major actor for the progressive keratopathy in aniridia patients and driven by the expression of vascular endothelial growth factor A (VEGF-A). Bevacizumab (Avastin), an antibody targeting VEGF-A, has been shown to reduce corneal neovascularization in long term eye diseases (Koenig et al., 2009) and more precisely in a case of aniridia (Lapid-Gortzak et al., 2018). An alternative approach to blocking VEGF-A could be to target the soluble VEGF receptor 1 (called sVEGF-R1 or sflt-1) that binds VEGF-A and prevents the intercellular signaling cascade leading to angiogenesis (Ambati et al., 2006). Notably, the corneal epithelium in aniridia patients is deficient in sflt-1 and injection of recombinant sflt-1 in the cornea of Pax6+/− mice regressed the neovascularization angiogenesis (Ambati et al., 2006).This therapeutic strategy, however, has not to our knowledge been evaluated in humans. Anti-angiogenic treatments in the context of AAK deserve further investigation.
Drugs that enhance Pax6 production: Pax6 represses cell proliferation while promoting differentiation in the corneal epithelium (Ouyang et al., 2006), while the epidermal growth factor-MEK-ERK pathway has a negative correlation with the expression of Pax6 (de la Puente et al., 2016). Thus, MEK inhibition leads to the inhibition of the ERK pathway, which, in turn, increases Pax6 expression. Accordingly, Ali Djalilian’s team found MEK inhibitors can stimulate in vitro Pax6 expression as well as in the eyes of newborn Pax6-deficient mice (Rabiee et al., 2020). Both topical and oral administrations of the drug enhanced Pax6 and partially normalized their eye development.
More recently, an AAK-like cellular model, produced by the gene editing of human limbal cells (Roux et al., 2018) has been used to screen a United State Food and Drug Administration-approved drug library for PAX6 haploinsufficient recovery. Two anti-psychotropic drugs (Duloxetine and Ritanserin) have been identified and validated in vitro for their ability to rescue functional properties of the mutant cells (Dorot et al., 2022; Oved et al., 2021). The effect of these compounds on Pax6 expression is conceivable, as Pax6 is expressed in neural progenitor cells in different regions of the developing CNS and in adult neural stem cells (Brill et al., 2008). Duloxetine is a norepinephrine reuptake inhibitor used (like Cymbalta) against severe depression (Muscatello et al., 2019). Duloxetine enhances dopamine levels within the prefrontal cortex. Of interest, inactivation of Dopamine in the retina induces keratoconus (Bergen et al., 2016) and Pax6 is an important determinant of the dopaminergic phenotype in the olfactory bulb (Ninkovic et al., 2010). Moreover, olfactory bulb dopaminergic neurons are severely reduced in Pax6sey/+ mice, which express reduced amounts of Pax6 (Dellovade et al., 1998), suggesting that Pax6’s function in dopaminergic specification is dose dependent. Ritanserin is a serotonin receptor inhibitor tested for treating schizophrenia and severe sleep disorders (Singh et al., 2010). Of note, aniridia is often accompanied by CNS abnormalities, such as pineal gland atrophy or hypoplasia, leading to disturbed circadian rhythm and sleep disorders (Berntsson et al., 2020). Serotonin, a neurotransmitter known to be involved in nociceptor sensitization, is present in human tears and patients with dry eye symptoms and aqueous tear deficiency had higher tear serotonin levels on their ocular surface, maybe due to enhanced inflammation, like in aniridia (Chhadva et al., 2015). Ritanserin has also been reported to have an antidopaminergic effect in the frontal cortex, which could explain some improvement in negative symptoms of schizophrenia (Ruiu et al., 2000). In addition, serotonin, in cooperation with the autonomic nervous system, regulates tear secretion (Imada et al., 2017). Remarkably, both drugs have been shown to enhance PAX6 production through a specific inhibition of the ERK/MEK pathway (Dorot et al., 2022; Oved et al., 2021).
More than 7000 patients with congenital aniridia in the EU need to be treated. The discovery of small compounds able to enhance PAX6 activity, which could be locally administered using eye drops, associated with drug repurposing is expected to lead to rapid development of applicable drugs for the topical treatment of aniridia.
6. Perspectives and future directions
Further insight in phenotype-genotype correlations based on specific clinical finding using innovative ocular imaging in large cohorts of patients and the implementation of NGS and WGS technologies will help in predicting disease evolution and taking preventive actions against congenital aniridia.
Reference centers with the infrastructure and expertise for precise phenotyping and clinical networks represent the basis of an organizational structure ensuring access to care for patients and phenotype characterization in rare diseases. A multidisciplinary collaboration at the national and international level between clinicians, molecular biologists, clinical geneticists, clinical and scientific researchers should be promising to improve diagnosis, treatment and follow-up of this rare complex disease, as well as to include patients in national registries in order to contact them when mutation-specific therapies will becoming available. For instance, the Clinical Patient Management System (CPMS) was launched by the European Commission in November 2017. It is a web-based application where healthcare professionals from the European Reference Networks (ERN)-Eye can discuss real patient cases. This platform strives to support ERNs by improving the diagnosis and treatment of low prevalence diseases across EU Member States. Additional efforts are necessary to implement international guidelines for congenital aniridia management.
Remarkable progress in exhaustive molecular testing has been made over the last years with the introduction of NGS methods, allowing the discovery of new genes and multiple types of mutations associated with many inherited disorders, including congenital aniridia and the spectrum of ASD disorders. This technological revolution rewires the classical genetic strategies towards new horizons, allowing in the next future to provide an early definitive clinical diagnosis and robust genotype-phenotype information important for optimal care of patients and families. Achieving a molecular diagnosis is of utmost importance in the actual context of emerging therapies because it is a chance for patients to be eligible for gene- or mutation-specific therapies. Consequently, deep-phenotyping and deep-genotyping on large patient cohorts are still highly useful to reveal novel phenotypic associations and novel pathogenic mechanisms and, thus are the first steps towards this ongoing progress. A major barrier is a better knowledge on functional consequences of pathogenic variants discovered in patients. A hope may come directly from themselves with the opportunity to use of iPSCs to explore the specific underlying mechanisms of a mutation of interest in its “native” cellular context accordingly to patient phenotypes. Therefore, knowledge of the full spectrum of phenotypes and genotypes will drive crucially the future targeted therapies, as well as scientific research.
Due to its low prevalence, congenital aniridia, poses a number of specific research challenges, particularly concerning clinical trial design. International multicenter studies could help overcome this point. Moreover, compassionate clinical trials could represent the first strategy to verify safety and some efficacy in this context. Congenital aniridia constitutes a developmental and genetic disease model for gene therapy, especially for AAK and glaucoma are the main causes of blindness in congenital aniridia and should be the target for future innovative research. As a panocular disorder, congenital aniridia represents a model disease for translational research with possible applicability to other ocular malformations.
Acknowledgments
We would like to thank the CRMR (Centre de Reference Maladies Rares en Ophtalmologie) OPHTARA, Gˆeniris association, Aniridia Europe, the European union Cost Action CA18116 ANIRIDIA-NET, as well as the patients and their families for their support. We would also like to thank TG Arch for the graphics. ISM for english editing. DBG received grant support from Gˆeniris Association. MKD received grant support from the National Eye Institute of the USA via EY028597-01 and Aniridia Foundation International.,
Financial Support
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Aniridia-associated keratopathy
- AAV
Adeno-Associated Virus
- ASD
Anterior Segment Dysgenesis
- CNS
Central nervous system
- CRISPR
Clustered regularly interspaced short palindromic repeats
- CRISPR-Cas9
CRISPR-associated protein 9
- DNA
Deoxyribonucleic acid
- FGF
Fibroblast Growth Factor
- miRNAs
MicroRNAs
- NGS
Next-Generation Sequencing
- POM
Periocular Mesenchyme
- SD-OCT
Spectral Domain Optical Coherence Tomography
- RNA
Ribonucleic Acid
- RPE
Retinal Pigment Epithelium
- UTRs
Untranslated Regions
- VEGF-A
Vascular Endothelial Growth Factor A
- WAGR
Wilms’ Tumor-Aniridia-Genital anomalies-Retardation
- WAGRO
WAGR and Obesity
- WGS
Whole-Genome Sequencing
Footnotes
Declarations of competing interest
None.
The three layers of the cornea are defined: the corneal endothelium (CEnd) deriving from the first set of neural crest cells/POM cells, the corneal stroma (CS) deriving from the second wave of NC cells/POM cells and the corneal epithelium (CE) developing from the outer surface ectoderm. The third wave of NC cells/POM cells arrives at the angle of the future cornea and the periphery of the optic cup and contributes to pupillary membrane and iris stroma development. From the periphery of the optic cup will then originate the pigmented epithelium and the smooth muscle of the iris. HV (hyaloid vessels, tunica vasculosa lentis).
Data availability
Data will be made available on request.
References
- Aalfs CM, Fantes JA, Wenniger-Prick LJ, Sluijter S, Hennekam RC, van Heyningen V, Hoovers JM, 1997. Tandem duplication of 11p12-p13 in a child with borderline development delay and eye abnormalities: dose effect of the PAX6 gene product? Am. J. Med. Genet. 73, 267–271. 10.1002/(sici)1096-8628(19971219)73:3<267::aid-ajmg7>3.0.co, 2. [DOI] [PubMed] [Google Scholar]
- Abouzeid H, Youssef MA, ElShakankiri N, Hauser P, Munier FL, Schorderet DF, 2009. PAX6 aniridia and interhemispheric brain anomalies. Mol. Vis. 15, 2074–2083. [PMC free article] [PubMed] [Google Scholar]
- Acampora D, Mazan S, Avantaggiato V, Barone P, Tuorto F, Lallemand Y, Brûlet P, Simeone A, 1996. Epilepsy and brain abnormalities in mice lacking the Otx1 gene. Nat. Genet. 14, 218–222. 10.1038/ng1096-218. [DOI] [PubMed] [Google Scholar]
- Acuna-Hidalgo R, Veltman JA, Hoischen A, 2016. New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 17, 241. 10.1186/s13059-016-1110-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi M, Dickens CJ, Hetherington J, Hoskins HD, Iwach AG, Wong PC, Nguyen N, Ma AS, 1997. Clinical experience of trabeculotomy for the surgical treatment of aniridic glaucoma. Ophthalmology 104, 2121–2125. 10.1016/s0161-6420(97)30041-4. [DOI] [PubMed] [Google Scholar]
- Aggarwal S, Jinda W, Limwongse C, Atchaneeyasakul L, Phadke SR, 2011. Run-on mutation in the PAX6 gene and chorioretinal degeneration in autosomal dominant aniridia. Mol. Vis. 17, 1305–1309. [PMC free article] [PubMed] [Google Scholar]
- Ahmadi H, Lund-Andersen H, Kolko M, Bach-Holm D, Alberti M, Ba-Ali S, 2020. Melanopsin-mediated pupillary light reflex and sleep quality in patients with normal tension glaucoma. Acta Ophthalmol. 98, 65–73. 10.1111/aos.14133. [DOI] [PubMed] [Google Scholar]
- Almousa R, Lake DB, 2014. Intraocular pressure control with Ahmed glaucoma drainage device in patients with cicatricial ocular surface disease-associated or aniridia-related glaucoma. Int. Ophthalmol. 34, 753–760. 10.1007/s10792-013-9868-6. [DOI] [PubMed] [Google Scholar]
- Ambati BK, Nozaki M, Singh N, Takeda A, Jani PD, Suthar T, Albuquerque RJC, Richter E, Sakurai E, Newcomb MT, Kleinman ME, Caldwell RB, Lin Q, Ogura Y, Orecchia A, Samuelson DA, Agnew DW, St Leger J, Green WR, Mahasreshti PJ, Curiel DT, Kwan D, Marsh H, Ikeda S, Leiper LJ, Collinson JM, Bogdanovich S, Khurana TS, Shibuya M, Baldwin ME, Ferrara N, Gerber H-P, De Falco S, Witta J, Baffi JZ, Raisler BJ, Ambati J, 2006. Corneal avascularity is due to soluble VEGF receptor-1. Nature 443, 993–997. 10.1038/nature05249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson TR, Hedlund E, Carpenter EM, 2002. Differential Pax6 promoter activity and transcript expression during forebrain development. Mech. Dev. 114, 171–175. 10.1016/s0925-4773(02)00051-5. [DOI] [PubMed] [Google Scholar]
- Angmo D, Dewan L, Behera A, Gagrani M, 2021. Aniridia with lenticular and choroidal coloboma. Eur. J. Ophthalmol. 31, NP116–NP118. 10.1177/1120672119866106. [DOI] [PubMed] [Google Scholar]
- Ansari M, Rainger J, Hanson IM, Williamson KA, Sharkey F, Harewood L, Sandilands A, Clayton-Smith J, Dollfus H, Bitoun P, Meire F, Fantes J, Franco B, Lorenz B, Taylor DS, Stewart F, Willoughby CE, McEntagart M, Khaw PT, Clericuzio C, Van Maldergem L, Williams D, Newbury-Ecob R, Traboulsi EI, Silva ED, Madlom MM, Goudie DR, Fleck BW, Wieczorek D, Kohlhase J, McTrusty AD, Gardiner C, Yale C, Moore AT, Russell-Eggitt I, Islam L, Lees M, Beales PL, Tuft SJ, Solano JB, Splitt M, Hertz JM, Prescott TE, Shears DJ, Nischal KK, Doco-Fenzy M, Prieur F, Temple IK, Lachlan KL, Damante G, Morrison DA, van Heyningen V, FitzPatrick DR, 2016. Genetic analysis of “PAX6-negative” individuals with aniridia or Gillespie syndrome. PLoS One 11, e0153757. 10.1371/journal.pone.0153757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antosova B, Smolikova J, Klimova L, Lachova J, Bendova M, Kozmikova I, Machon O, Kozmik Z, 2016. The gene regulatory network of lens induction is wired through meis-dependent shadow enhancers of Pax6. PLoS Genet. 12, e1006441 10.1371/journal.pgen.1006441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aota S, Nakajima N, Sakamoto R, Watanabe S, Ibaraki N, Okazaki K, 2003. Pax6 autoregulation mediated by direct interaction of Pax6 protein with the head surface ectoderm-specific enhancer of the mouse Pax6 gene. Dev. Biol. 257, 1–13. 10.1016/s0012-1606(03)00058-7. [DOI] [PubMed] [Google Scholar]
- Aradhya S, Smaoui N, Marble M, Lacassie Y, 2011. De novo duplication 11p13 involving the PAX6 gene in a patient with neonatal seizures, hypotonia, microcephaly, developmental disability and minor ocular manifestations. Am. J. Med. Genet. 155A, 442–444. 10.1002/ajmg.a.33814. [DOI] [PubMed] [Google Scholar]
- Arora A, McKay GJ, Simpson DAC, 2007. Prediction and verification of miRNA expression in human and rat retinas. Invest. Ophthalmol. Vis. Sci. 48, 3962–3967. 10.1167/iovs.06-1221. [DOI] [PubMed] [Google Scholar]
- Arroyave CP, Scott IU, Gedde SJ, Parrish RK, Feuer WJ, 2003. Use of glaucoma drainage devices in the management of glaucoma associated with aniridia. Am. J. Ophthalmol. 135, 155–159. 10.1016/s0002-9394(02)01934-7. [DOI] [PubMed] [Google Scholar]
- Aryal S, Viet J, Weatherbee BAT, Siddam AD, Hernandez FG, Gautier-Courteille C, Paillard L, Lachke SA, 2020. The cataract-linked RNA-binding protein Celf1 post-transcriptionally controls the spatiotemporal expression of the key homeodomain transcription factors Pax6 and Prox1 in lens development. Hum. Genet. 139, 1541–1554. 10.1007/s00439-020-02195-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashery-Padan R, Marquardt T, Zhou X, Gruss P, 2000. Pax6 activity in the lens primordium is required for lens formation and for correct placement of a single retina in the eye. Genes Dev. 14, 2701–2711. 10.1101/gad.184000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslam SA, Wong SC, Ficker LA, MacLaren RE, 2008. Implantation of the black diaphragm intraocular lens in congenital and traumatic aniridia. Ophthalmology 115, 1705–1712. 10.1016/j.ophtha.2008.03.025. [DOI] [PubMed] [Google Scholar]
- Azuma N, Tadokoro K, Asaka A, Yamada M, Yamaguchi Y, Handa H, Matsushima S, Watanabe T, Kohsaka S, Kida Y, Shiraishi T, Ogura T, Shimamura K, Nakafuku M, 2005. The Pax6 isoform bearing an alternative spliced exon promotes the development of the neural retinal structure. Hum. Mol. Genet. 14, 735–745. 10.1093/hmg/ddi069. [DOI] [PubMed] [Google Scholar]
- Azuma N, Yamaguchi Y, Handa H, Hayakawa M, Kanai A, Yamada M, 1999. Missense mutation in the alternative splice region of the PAX6 gene in eye anomalies. Am. J. Hum. Genet. 65, 656–663. 10.1086/302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma N, Yamaguchi Y, Handa H, Tadokoro K, Asaka A, Kawase E, Yamada M, 2003. Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. Am. J. Hum. Genet. 72, 1565–1570. 10.1086/375555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamiou D-E, Free SL, Sisodiya SM, Chong WK, Musiek F, Williamson KA, van Heyningen V, Moore AT, Gadian D, Luxon LM, 2007. Auditory interhemispheric transfer deficits, hearing difficulties, and brain magnetic resonance imaging abnormalities in children with congenital aniridia due to PAX6 mutations. Arch. Pediatr. Adolesc. Med. 161, 463–469. 10.1001/archpedi.161.5.463. [DOI] [PubMed] [Google Scholar]
- Bartel DP, 2009. MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassnett S, Šikić H, 2017. The lens growth process. Prog. Retin. Eye Res. 60, 181–200. 10.1016/j.preteyeres.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulmann DC, Ohlmann A, Flügel-Koch C, Goswami S, Cvekl A, Tamm ER, 2002. Pax6 heterozygous eyes show defects in chamber angle differentiation that are associated with a wide spectrum of other anterior eye segment abnormalities. Mech. Dev. 118, 3–17. 10.1016/s0925-4773(02)00260-5. [DOI] [PubMed] [Google Scholar]
- Bäumer N, Marquardt T, Stoykova A, Ashery-Padan R, Chowdhury K, Gruss P, 2002. Pax6 is required for establishing naso-temporal and dorsal characteristics of the optic vesicle. Development 129, 4535–4545. 10.1242/dev.129.19.4535. [DOI] [PubMed] [Google Scholar]
- Bäumer N, Marquardt T, Stoykova A, Spieler D, Treichel D, Ashery-Padan R, Gruss P, 2003. Retinal pigmented epithelium determination requires the redundant activities of Pax2 and Pax6. Development 130, 2903–2915. 10.1242/dev.00450. [DOI] [PubMed] [Google Scholar]
- Beebe DC, Coats JM, 2000. The lens organizes the anterior segment: specification of neural crest cell differentiation in the avian eye. Dev. Biol. 220, 424–431. 10.1006/dbio.2000.9638. [DOI] [PubMed] [Google Scholar]
- Behaegel J, Tassignon MJ, Lagali N, Consejo A, Koppen C, Dhubhghaill SN, 2022. Outcomes of human Leukocyte antigen-matched allogeneic cultivated limbal epithelial transplantation in aniridia-associated keratopathy-A single-center retrospective analysis. Cornea 41, 69–77. 10.1097/ICO.0000000000002729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrame G, Salvetat ML, Chizzolini M, Driussi GB, Busatto P, Di Giorgio G, Barosco F, Scuderi B, 2003. Implantation of a black diaphragm intraocular lens in ten cases of post-traumatic aniridia. Eur. J. Ophthalmol. 13, 62–68. 10.1177/112067210301300109. [DOI] [PubMed] [Google Scholar]
- Bergen MA, Park HN, Chakraborty R, Landis EG, Sidhu C, He L, Iuvone PM, Pardue MT, 2016. Altered refractive development in mice with reduced levels of retinal dopamine. Invest. Ophthalmol. Vis. Sci. 57, 4412–4419. 10.1167/iovs.15-17784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berntsson SG, Kristoffersson A, Daniilidou M, Dahl N, Ekström C, Semnic R, Markström A, Niemelä V, Partinen M, Hallböök F, Landtblom A-M, 2020. Aniridia with PAX6 mutations and narcolepsy. J. Sleep Res. 29, e12982 10.1111/jsr.12982. [DOI] [PubMed] [Google Scholar]
- Bharti K, Gasper M, Ou J, Brucato M, Clore-Gronenborn K, Pickel J, Arnheiter H, 2012. A regulatory loop involving PAX6, MITF, and WNT signaling controls retinal pigment epithelium development. PLoS Genet. 8, e1002757 10.1371/journal.pgen.1002757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia S, Bengani H, Fish M, Brown A, Divizia MT, de Marco R, Damante G, Grainger R, van Heyningen V, Kleinjan DA, 2013. Disruption of autoregulatory feedback by a mutation in a remote, ultraconserved PAX6 enhancer causes aniridia. Am. J. Hum. Genet. 93, 1126–1134. 10.1016/j.ajhg.2013.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake EM, 1952. The surgical treatment of glaucoma complicating congenital aniridia. Trans. Am. Ophthalmol. Soc. 50, 47–53. [PMC free article] [PubMed] [Google Scholar]
- Blixt A, Mahlapuu M, Aitola M, Pelto-Huikko M, Enerbäck S, Carlsson P, 2000. A forkhead gene, FoxE3, is essential for lens epithelial proliferation and closure of the lens vesicle. Genes Dev. 14, 245–254. [PMC free article] [PubMed] [Google Scholar]
- Bobilev AM, Hudgens-Haney ME, Hamm JP, Oliver WT, McDowell JE, Lauderdale JD, Clementz BA, 2019. Early and late auditory information processing show opposing deviations in aniridia. Brain Res. 1720, 146307 10.1016/j.brainres.2019.146307. [DOI] [PubMed] [Google Scholar]
- Boese EA, Tollefson MR, Schnieders MJ, Darbro BW, Alward WLM, Fingert JH, 2020. Novel intragenic PAX6 deletion in a pedigree with aniridia, morbid obesity, and diabetes. Curr. Eye Res. 45, 91–96. 10.1080/02713683.2019.1649704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremond-Gignac D, 2019. 1st Eye Conference Development and Disease: PAX6, Aniridia and beyond in Charlottesville. [Google Scholar]
- Brill MS, Snapyan M, Wohlfrom H, Ninkovic J, Jawerka M, Mastick GS, Ashery-Padan R, Saghatelyan A, Berninger B, Götz M, 2008. A dlx2- and pax6-dependent transcriptional code for periglomerular neuron specification in the adult olfactory bulb. J. Neurosci. 28, 6439–6452. 10.1523/JNEUROSCI.0700-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringmann A, Syrbe S, Görner K, Kacza J, Francke M, Wiedemann P, Reichenbach A, 2018. The primate fovea: structure, function and development. Prog. Retin. Eye Res. 66, 49–84. 10.1016/j.preteyeres.2018.03.006. [DOI] [PubMed] [Google Scholar]
- Brown A, McKie M, van Heyningen V, Prosser J, 1998. The human PAX6 mutation database. Nucleic Acids Res. 26, 259–264. 10.1093/nar/26.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaerts P, Halder G, Gehring WJ, 1997. PAX-6 in development and evolution. Annu. Rev. Neurosci. 20, 483–532. 10.1146/annurev.neuro.20.1.483. [DOI] [PubMed] [Google Scholar]
- Canto-Soler MV, Adler R, 2006. Optic cup and lens development requires Pax6 expression in the early optic vesicle during a narrow time window. Dev. Biol. 294, 119–132. 10.1016/j.ydbio.2006.02.033. [DOI] [PubMed] [Google Scholar]
- Carrière C, Plaza S, Caboche J, Dozier C, Bailly M, Martin P, Saule S, 1995. Nuclear localization signals, DNA binding, and transactivation properties of quail Pax-6 (Pax-QNR) isoforms. Cell Growth Differ. 6, 1531–1540. [PubMed] [Google Scholar]
- Casas-Llera P, Siverio A, Esquivel G, Bautista C, Alió JL, 2020. Spectral-domain optical coherence tomography foveal morphology as a prognostic factor for vision performance in congenital aniridia. Eur. J. Ophthalmol. 30, 58–65. 10.1177/1120672118818352. [DOI] [PubMed] [Google Scholar]
- Chauhan BK, Yang Y, Cveklová K, Cvekl A, 2004. Functional properties of natural human PAX6 and PAX6(5a) mutants. Invest. Ophthalmol. Vis. Sci. 45, 385–392. 10.1167/iovs.03-0968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TC, Walton DS, 1999. Goniosurgery for prevention of aniridic glaucoma. Arch. Ophthalmol. 117, 1144–1148. 10.1001/archopht.117.9.1144. [DOI] [PubMed] [Google Scholar]
- Cheong S-S, Hentschel L, Davidson AE, Gerrelli D, Davie R, Rizzo R, Pontikos N, Plagnol V, Moore AT, Sowden JC, Michaelides M, Snead M, Tuft SJ, Hardcastle AJ, 2016. Mutations in CPAMD8 cause a unique form of autosomal-recessive anterior segment dysgenesis. Am. J. Hum. Genet. 99, 1338–1352. 10.1016/j.ajhg.2016.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung AY, Eslani M, Kurji KH, Wright E, Sarnicola E, Govil A, Holland EJ, 2020. Long-term outcomes of living-related conjunctival limbal allograft compared with keratolimbal allograft in patients with limbal stem cell deficiency. Cornea 39, 980–985. 10.1097/ICO.0000000000002329. [DOI] [PubMed] [Google Scholar]
- Chhadva P, Lee T, Sarantopoulos CD, Hackam AS, McClellan AL, Felix ER, Levitt RC, Galor A, 2015. Human tear serotonin levels correlate with symptoms and signs of dry eye. Ophthalmology 122, 1675–1680. 10.1016/j.ophtha.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien Yin-Hsuan, Huang H-P, Hwu W-L, Chien Yin-Hsiu, Chang T-C, Lee N-C, 2009. Eye anomalies and neurological manifestations in patients with PAX6 mutations. Mol. Vis. 15, 2139–2145. [PMC free article] [PubMed] [Google Scholar]
- Choi J-H, Kim S-J, Thomas MG, Jung J-H, Oh EH, Shin J-H, Cho JW, Kim H-S, Park J-Y, Choi SY, Choi HY, Choi K-D, 2021. Diagnostic yield of targeted next-generation sequencing in infantile nystagmus syndrome. Ophthalmic Genet. 42, 561–569. 10.1080/13816810.2021.1938138. [DOI] [PubMed] [Google Scholar]
- Chrystal PW, Walter MA, 2019. Aniridia and Axenfeld-Rieger Syndrome: clinical presentations, molecular genetics and current/emerging therapies. Exp. Eye Res. 189, 107815 10.1016/j.exer.2019.107815. [DOI] [PubMed] [Google Scholar]
- Collinson JM, Hill RE, West JD, 2000. Different roles for Pax6 in the optic vesicle and facial epithelium mediate early morphogenesis of the murine eye. Development 127, 945–956. 10.1242/dev.127.5.945. [DOI] [PubMed] [Google Scholar]
- Collinson JM, Quinn JC, Buchanan MA, Kaufman MH, Wedden SE, West JD, Hill RE, 2001. Primary defects in the lens underlie complex anterior segment abnormalities of the Pax6 heterozygous eye. Proc. Natl. Acad. Sci. U. S. A. 98, 9688–9693. 10.1073/pnas.161144098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinson JM, Quinn JC, Hill RE, West JD, 2003. The roles of Pax6 in the cornea, retina, and olfactory epithelium of the developing mouse embryo. Dev. Biol. 255, 303–312. 10.1016/s0012-1606(02)00095-7. [DOI] [PubMed] [Google Scholar]
- Collomb E, Yang Y, Foriel S, Cadau S, Pearton DJ, Dhouailly D, 2013. The corneal epithelium and lens develop independently from a common pool of precursors. Dev. Dynam. 242, 401–413. 10.1002/dvdy.23925. [DOI] [PubMed] [Google Scholar]
- Conte I, Carrella S, Avellino R, Karali M, Marco-Ferreres R, Bovolenta P, Banfi S, 2010. miR-204 is required for lens and retinal development via Meis2 targeting. Proc. Natl. Acad. Sci. U. S. A. 107, 15491–15496. 10.1073/pnas.0914785107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creuzet S, Vincent C, Couly G, 2005. Neural crest derivatives in ocular and periocular structures. Int. J. Dev. Biol. 49, 161–171. 10.1387/ijdb.041937sc. [DOI] [PubMed] [Google Scholar]
- Crolla JA, van Heyningen V, 2002. Frequent chromosome aberrations revealed by molecular cytogenetic studies in patients with aniridia. Am. J. Hum. Genet. 71, 1138–1149. 10.1086/344396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curto GG, Lara JM, Parrilla M, Aijón J, Velasco A, 2007. Modifications of the retina neuronal populations of the heterozygous mutant small eye mouse, the Sey (Dey). Brain Res. 1127, 163–176. 10.1016/j.brainres.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Cvekl A, Ashery-Padan R, 2014. The cellular and molecular mechanisms of vertebrate lens development. Development 141, 4432–4447. 10.1242/dev.107953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvekl A, Callaerts P, 2017. PAX6: 25th anniversary and more to learn. Exp. Eye Res. 156, 10–21. 10.1016/j.exer.2016.04.017. [DOI] [PubMed] [Google Scholar]
- Cvekl A, Duncan MK, 2007. Genetic and epigenetic mechanisms of gene regulation during lens development. Prog. Retin. Eye Res. 26, 555–597. 10.1016/j.preteyeres.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvekl A, Tamm ER, 2004. Anterior eye development and ocular mesenchyme: new insights from mouse models and human diseases. Bioessays 26, 374–386. 10.1002/bies.20009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvekl A, Yang Y, Chauhan BK, Cveklova K, 2004. Regulation of gene expression by Pax6 in ocular cells: a case of tissue-preferred expression of crystallins in lens. Int. J. Dev. Biol. 48, 829–844. 10.1387/ijdb.041866ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvekl A, Zhang X, 2017. Signaling and gene regulatory networks in mammalian lens development. Trends Genet. 33, 677–702. 10.1016/j.tig.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvekl A, Zhao Y, McGreal R, Xie Q, Gu X, Zheng D, 2017. Evolutionary origins of Pax6 control of crystallin genes. Genome Biol Evol 9, 2075–2092. 10.1093/gbe/evx153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czerny T, Busslinger M, 1995. DNA-binding and transactivation properties of Pax-6: three amino acids in the paired domain are responsible for the different sequence recognition of Pax-6 and BSAP (Pax-5). Mol. Cell Biol. 15, 2858–2871. 10.1128/MCB.15.5.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dansault A, David G, Schwartz C, Jaliffa C, Vieira V, de la Houssaye G, Bigot K, Catin F, Tattu L, Chopin C, Halimi P, Roche O, Van Regemorter N, Munier F, Schorderet D, Dufier J-L, Marsac C, Ricquier D, Menasche M, Penfornis A, Abitbol M, 2007. Three new PAX6 mutations including one causing an unusual ophthalmic phenotype associated with neurodevelopmental abnormalities. Mol. Vis. 13, 511–523. [PMC free article] [PubMed] [Google Scholar]
- Daruich A, Robert MP, Leroy C, DE Vergnes N, Beugnet C, Malan V, Valleix S, Bremond-Gignac D, 2021. Foveal hypoplasia grading in 95 cases of congenital aniridia: correlation to phenotype and PAX6 genotype. Am. J. Ophthalmol. 237, 122–129. 10.1016/j.ajo.2021.12.007. [DOI] [PubMed] [Google Scholar]
- Davis J, Piatigorsky J, 2011. Overexpression of Pax6 in mouse cornea directly alters corneal epithelial cells: changes in immune function, vascularization, and differentiation. Invest. Ophthalmol. Vis. Sci. 52, 4158–4168. 10.1167/iovs.10-6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis LK, Meyer KJ, Rudd DS, Librant AL, Epping EA, Sheffield VC, Wassink TH, 2008. Pax6 3’ deletion results in aniridia, autism and mental retardation. Hum. Genet. 123, 371–378. 10.1007/s00439-008-0484-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis N, Yoffe C, Raviv S, Antes R, Berger J, Holzmann S, Stoykova A, Overbeek PA, Tamm ER, Ashery-Padan R, 2009. Pax6 dosage requirements in iris and ciliary body differentiation. Dev. Biol. 333, 132–142. 10.1016/j.ydbio.2009.06.023. [DOI] [PubMed] [Google Scholar]
- Davis-Silberman N, Ashery-Padan R, 2008. Iris development in vertebrates; genetic and molecular considerations. Brain Res. 1192, 17–28. 10.1016/j.brainres.2007.03.043. [DOI] [PubMed] [Google Scholar]
- Davis-Silberman N, Kalich T, Oron-Karni V, Marquardt T, Kroeber M, Tamm ER, Ashery-Padan R, 2005. Genetic dissection of Pax6 dosage requirements in the developing mouse eye. Hum. Mol. Genet. 14, 2265–2276. 10.1093/hmg/ddi231. [DOI] [PubMed] [Google Scholar]
- Daya SM, Watson A, Sharpe JR, Giledi O, Rowe A, Martin R, James SE, 2005. Outcomes and DNA analysis of ex vivo expanded stem cell allograft for ocular surface reconstruction. Ophthalmology 112, 470–477. 10.1016/J.OPHTHA.2004.09.023. [DOI] [PubMed] [Google Scholar]
- de Chevigny A, Coré N, Follert P, Gaudin M, Barbry P, Béclin C, Cremer H, 2012. miR-7a regulation of Pax6 controls spatial origin of forebrain dopaminergic neurons. Nat. Neurosci. 15, 1120–1126. 10.1038/nn.3142. [DOI] [PubMed] [Google Scholar]
- De La Paz MF, Alvarez De Toledo J, Barraquer RI, Barraquer J, 2008. Long-term visual prognosis of corneal and ocular surface surgery in patients with congenital aniridia. Acta Ophthalmol. 86, 735–740. 10.1111/J.1755-3768.2008.01293.X. [DOI] [PubMed] [Google Scholar]
- de la Puente P, Muz B, Jin A, Azab F, Luderer M, Salama NN, Azab AK, 2016. MEK inhibitor, TAK-733 reduces proliferation, affects cell cycle and apoptosis, and synergizes with other targeted therapies in multiple myeloma. Blood Cancer J. 6, e399. 10.1038/bcj.2016.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellovade TL, Pfaff DW, Schwanzel-Fukuda M, 1998. Olfactory bulb development is altered in small-eye (Sey) mice. J. Comp. Neurol. 402, 402–418. [PubMed] [Google Scholar]
- Deng SX, Borderie V, Chan CC, Dana R, Figueiredo FC, Gomes JAP, Pellegrini G, Shimmura S, Kruse FE, 2019. The international limbal stem cell deficiency working group, 2019. Global consensus on definition, classification, diagnosis, and staging of limbal stem cell deficiency. Cornea 38, 364–375. 10.1097/ICO.0000000000001820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diacou R, Nandigrami P, Fiser A, Liu W, Ashery-Padan R, Cvekl A, 2022. Cell fate decisions, transcription factors and signaling during early retinal development. Prog. Retin. Eye Res. 101093 10.1016/j.preteyeres.2022.101093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djayet C, Bremond-Gignac D, Touchard J, Secretan P-H, Vidal F, Robert MP, Daruich A, Cisternino S, Schlatter J, 2020. Formulation and stability of ataluren eye drop oily solution for aniridia. Pharmaceutics 13, E7. 10.3390/pharmaceutics13010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolski D, Orzechowska-Wylegala B, Wowra B, Wroblewska-Czajka E, Grolik M, Szczubialka K, Nowakowska M, Puzzolo D, Wylegala EA, Micali A, Aragona P, 2015. Cultivated oral mucosa epithelium in ocular surface reconstruction in aniridia patients. BioMed Res. Int. 10.1155/2015/281870, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohrmann C, Gruss P, Lemaire L, 2000. Pax genes and the differentiation of hormone-producing endocrine cells in the pancreas. Mech. Dev. 92, 47–54. 10.1016/s0925-4773(99)00324-x. [DOI] [PubMed] [Google Scholar]
- Dollfus H, Joanny-Flinois O, Doco-Fenzy M, Veyre L, Joanny-Flinois L, Khoury M, Jonveaux P, Abitbol M, Dufier JL, 1998. Gillespie syndrome phenotype with a t (X;11)(p22.32;p12) de novo translocation. Am. J. Ophthalmol. 125, 397–399. 10.1016/s0002-9394(99)80157-3. [DOI] [PubMed] [Google Scholar]
- D’Oria F, Barraquer R, Alio JL, 2021. Crystalline lens alterations in congenital aniridia. Arch. Soc. Esp. Oftalmol. 96 (Suppl. 1), 38–51. 10.1016/j.oftale.2020.12.008. [DOI] [PubMed] [Google Scholar]
- Dorot O, Roux LN, Zennaro L, Oved K, Bremond-Gignac D, Pichinuk E, Aberdam D, 2022. The antipsychotropic drug Duloxetine rescues PAX6 haploinsufficiency of mutant limbal stem cells through inhibition of the MEK/ERK signaling pathway. Ocul. Surf. 23, 140–142. 10.1016/j.jtos.2021.12.003. [DOI] [PubMed] [Google Scholar]
- Doss MX, Sachinidis A, 2019. Current challenges of iPSC-based disease modeling and therapeutic implications. Cells 8, E403. 10.3390/cells8050403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E, 2014. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096. 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Dubey SK, Mahalaxmi N, Vijayalakshmi P, Sundaresan P, 2015. Mutational analysis and genotype-phenotype correlations in southern Indian patients with sporadic and familial aniridia. Mol. Vis. 21, 88–97. [PMC free article] [PubMed] [Google Scholar]
- Duncan MK, Cvekl A, Li X, Piatigorsky J, 2000. Truncated forms of Pax-6 disrupt lens morphology in transgenic mice. Invest. Ophthalmol. Vis. Sci. 41, 464–473. [PubMed] [Google Scholar]
- Duncan MK, Haynes JI, Cvekl A, Piatigorsky J, 1998. Dual roles for Pax-6: a transcriptional repressor of lens fiber cell-specific beta-crystallin genes. Mol. Cell Biol. 18, 5579–5586. 10.1128/MCB.18.9.5579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupacova N, Antosova B, Paces J, Kozmik Z, 2021. Meis homeobox genes control progenitor competence in the retina. Proc. Natl. Acad. Sci. U. S. A. 118, e2013136118 10.1073/pnas.2013136118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durai I, Pallamparthy S, Puthuran GV, Wijesinghe HK, Uduman MS, Krishnadas SR, Robin AL, Palmberg P, Gedde SJ, 2021. Outcomes of glaucoma drainage device implantation and trabeculectomy with mitomycin C in glaucoma secondary to aniridia. Am. J. Ophthalmol. 227, 173–181. 10.1016/j.ajo.2021.03.008. [DOI] [PubMed] [Google Scholar]
- Edén U, Iggman D, Riise R, Tornqvist K, 2008. Epidemiology of aniridia in Sweden and Norway. Acta Ophthalmol. 86, 727–729. 10.1111/j.1755-3768.2008.01309.x. [DOI] [PubMed] [Google Scholar]
- Ellison-Wright Z, Heyman I, Frampton I, Rubia K, Chitnis X, Ellison-Wright I, Williams SCR, Suckling J, Simmons A, Bullmore E, 2004. Heterozygous PAX6 mutation, adult brain structure and fronto-striato-thalamic function in a human family. Eur. J. Neurosci. 19, 1505–1512. 10.1111/j.1460-9568.2004.03236.x. [DOI] [PubMed] [Google Scholar]
- Evans AL, Gage PJ, 2005. Expression of the homeobox gene Pitx2 in neural crest is required for optic stalk and ocular anterior segment development. Hum. Mol. Genet. 14, 3347–3359. 10.1093/hmg/ddi365. [DOI] [PubMed] [Google Scholar]
- Faber SC, Robinson ML, Makarenkova HP, Lang RA, 2002. Bmp signaling is required for development of primary lens fiber cells. Development 129, 3727–3737. 10.1242/dev.129.15.3727. [DOI] [PubMed] [Google Scholar]
- Fantes J, Redeker B, Breen M, Boyle S, Brown J, Fletcher J, Jones S, Bickmore W, Fukushima Y, Mannens M, 1995. Aniridia-associated cytogenetic rearrangements suggest that a position effect may cause the mutant phenotype. Hum. Mol. Genet. 4, 415–422. 10.1093/hmg/4.3.415. [DOI] [PubMed] [Google Scholar]
- Faranda AP, Shihan MH, Wang Y, Duncan MK, 2021. The aging mouse lens transcriptome. Exp. Eye Res. 209, 108663 10.1016/j.exer.2021.108663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhy C, Elgart M, Shapira Z, Oron-Karni V, Yaron O, Menuchin Y, Rechavi G, Ashery-Padan R, 2013. Pax6 is required for normal cell-cycle exit and the differentiation kinetics of retinal progenitor cells. PLoS One 8, e76489. 10.1371/journal.pone.0076489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favor J, Gloeckner CJ, Neuhäuser-Klaus A, Pretsch W, Sandulache R, Saule S, Zaus I, 2008. Relationship of Pax6 activity levels to the extent of eye development in the mouse, Mus musculus. Genetics 179, 1345–1355. 10.1534/genetics.108.088591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filatova AY, Vasilyeva TA, Marakhonov AV, Sukhanova NV, Voskresenskaya AA, Zinchenko RA, Skoblov MY, 2021. Upstream ORF frameshift variants in the PAX6 5’UTR cause congenital aniridia. Hum. Mutat. 42, 1053–1065. 10.1002/humu.24248. [DOI] [PubMed] [Google Scholar]
- Filatova AY, Vasilyeva TA, Marakhonov AV, Voskresenskaya AA, Zinchenko RA, Skoblov MY, 2019. Functional reassessment of PAX6 single nucleotide variants by in vitro splicing assay. Eur. J. Hum. Genet. 27, 488–493. 10.1038/s41431-018-0288-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach BV, Trout KL, Lewis J, Luis CA, Sika M, 2005. WAGR syndrome: a clinical review of 54 cases. Pediatrics 116, 984–988. 10.1542/peds.2004-0467. [DOI] [PubMed] [Google Scholar]
- Fokina VM, Frolova EI, 2006. Expression patterns of Wnt genes during development of an anterior part of the chicken eye. Dev. Dynam. 235, 496–505. 10.1002/dvdy.20621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Free SL, Mitchell TN, Williamson KA, Churchill AJ, Shorvon SD, Moore AT, van Heyningen V, Sisodiya SM, 2003. Quantitative MR image analysis in subjects with defects in the PAX6 gene. Neuroimage 20, 2281–2290. 10.1016/j.neuroimage.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Gage PJ, Rhoades W, Prucka SK, Hjalt T, 2005. Fate maps of neural crest and mesoderm in the mammalian eye. Invest. Ophthalmol. Vis. Sci. 46, 4200–4208. 10.1167/iovs.05-0691. [DOI] [PubMed] [Google Scholar]
- Gage PJ, Suh H, Camper SA, 1999. Dosage requirement of Pitx2 for development of multiple organs. Development 126, 4643–4651. [DOI] [PubMed] [Google Scholar]
- Gage PJ, Zacharias AL, 2009. Signaling “cross-talk” is integrated by transcription factors in the development of the anterior segment in the eye. Dev. Dynam. 238, 2149–2162. 10.1002/dvdy.22033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gariano RF, 2010. Special features of human retinal angiogenesis. Eye 24, 401–407. 10.1038/eye.2009.324. [DOI] [PubMed] [Google Scholar]
- Gehring WJ, 1996. The master control gene for morphogenesis and evolution of the eye. Gene Cell. 1, 11–15. 10.1046/j.1365-2443.1996.11011.x. [DOI] [PubMed] [Google Scholar]
- Gerber S, Alzayady KJ, Burglen L, Brémond-Gignac D, Marchesin V, Roche O, Rio M, Funalot B, Calmon R, Durr A, Gil-da-Silva-Lopes VL, Ribeiro Bittar MF, Orssaud C, Héron B, Ayoub E, Berquin P, Bahi-Buisson N, Bole C, Masson C, Munnich A, Simons M, Delous M, Dollfus H, Boddaert N, Lyonnet S, Kaplan J, Calvas P, Yule DI, Rozet J-M, Fares Taie L, 2016. Recessive and dominant de novo ITPR1 mutations cause Gillespie syndrome. Am. J. Hum. Genet. 98, 971–980. 10.1016/j.ajhg.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser T, Jepeal L, Edwards JG, Young SR, Favor J, Maas RL, 1994. PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat. Genet. 7, 463–471. 10.1038/ng0894-463. [DOI] [PubMed] [Google Scholar]
- Glaser T, Lane J, Housman D, 1990. A mouse model of the aniridia-Wilms tumor deletion syndrome. Science 250, 823–827. 10.1126/science.2173141. [DOI] [PubMed] [Google Scholar]
- Glaser T, Walton DS, Maas RL, 1992. Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat. Genet. 2, 232–239. 10.1038/ng1192-232. [DOI] [PubMed] [Google Scholar]
- Grant MK, Bobilev AM, Branch A, Lauderdale JD, 2021. Structural and functional consequences of PAX6 mutations in the brain: implications for aniridia. Brain Res. 1756, 147283 10.1016/j.brainres.2021.147283. [DOI] [PubMed] [Google Scholar]
- Grant MK, Bobilev AM, Pierce JE, DeWitte J, Lauderdale JD, 2017. Structural brain abnormalities in 12 persons with aniridia. F1000Res 6, 255. 10.12688/f1000research.11063.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant MK, Bobilev AM, Rasys AM, Branson Byers J, Schriever HC, Hekmatyar K, Lauderdale JD, 2020. Global and age-related neuroanatomical abnormalities in a Pax6-deficient mouse model of aniridia suggests a role for Pax6 in adult structural neuroplasticity. Brain Res. 1732, 146698 10.1016/j.brainres.2020.146698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory-Evans CY, Wang X, Wasan KM, Zhao J, Metcalfe AL, Gregory-Evans K, 2014. Postnatal manipulation of Pax6 dosage reverses congenital tissue malformation defects. J. Clin. Invest. 124, 111–116. 10.1172/JCI70462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin C, Kleinjan DA, Doe B, van Heyningen V, 2002. New 3’ elements control Pax6 expression in the developing pretectum, neural retina and olfactory region. Mech. Dev. 112, 89–100. 10.1016/s0925-4773(01)00646-3. [DOI] [PubMed] [Google Scholar]
- Grønskov K, Rosenberg T, Sand A, Brøndum-Nielsen K, 1999. Mutational analysis of PAX6: 16 novel mutations including 5 missense mutations with a mild aniridia phenotype. Eur. J. Hum. Genet. 7, 274–286. 10.1038/sj.ejhg.5200308. [DOI] [PubMed] [Google Scholar]
- Gunhaga L, 2011. The lens: a classical model of embryonic induction providing new insights into cell determination in early development. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 1193–1203. 10.1098/rstb.2010.0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta SK, De Becker I, Tremblay F, Guernsey DL, Neumann PE, 1998. Genotype/phenotype correlations in aniridia. Am. J. Ophthalmol. 126, 203–210. 10.1016/s0002-9394(98)00191-3. [DOI] [PubMed] [Google Scholar]
- Hägglund A-C, Jones I, Carlsson L, 2017. A novel mouse model of anterior segment dysgenesis (ASD): conditional deletion of Tsc1 disrupts ciliary body and iris development. Dis Model Mech 10, 245–257. 10.1242/dmm.028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall HN, Williamson KA, FitzPatrick DR, 2019. The genetic architecture of aniridia and Gillespie syndrome. Hum. Genet. 138, 881–898. 10.1007/s00439-018-1934-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hama Y, Hirakata A, Tomita K, Inoue M, 2010. Retinal detachment with giant oral dialysis in an eye with congenital aniridia. Jpn. J. Ophthalmol. 54, 105–107. 10.1007/s10384-009-0746-8. [DOI] [PubMed] [Google Scholar]
- Han JC, Liu Q-R, Jones M, Levinn RL, Menzie CM, Jefferson-George KS, Adler-Wailes DC, Sanford EL, Lacbawan FL, Uhl GR, Rennert OM, Yanovski JA, 2008. Brain-derived neurotrophic factor and obesity in the WAGR syndrome. N. Engl. J. Med. 359, 918–927. 10.1056/NEJMoa0801119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JC, Thurm A, Golden Williams C, Joseph LA, Zein WM, Brooks BP, Butman JA, Brady SM, Fuhr SR, Hicks MD, Huey AE, Hanish AE, Danley KM, Raygada MJ, Rennert OM, Martinowich K, Sharp SJ, Tsao JW, Swedo SE, 2013. Association of brain-derived neurotrophic factor (BDNF) haploinsufficiency with lower adaptive behaviour and reduced cognitive functioning in WAGR/11p13 deletion syndrome. Cortex 49, 2700–2710. 10.1016/j.cortex.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanish AE, Butman JA, Thomas F, Yao J, Han JC, 2016. Pineal hypoplasia, reduced melatonin and sleep disturbance in patients with PAX6 haploinsufficiency. J. Sleep Res. 25, 16–22. 10.1111/jsr.12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanish AE, Han JC, 2018. Delayed onset of sleep in adolescents with PAX6 haploinsufficiency. Biol. Res. Nurs. 20, 237–243. 10.1177/1099800417753670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson I, Van Heyningen V, 1995. Pax6: more than meets the eye. Trends Genet. 11, 268–272. 10.1016/s0168-9525(00)89073-3. [DOI] [PubMed] [Google Scholar]
- Hanson IM, 2003. PAX6 and congenital eye malformations. Pediatr. Res. 54, 791–796. 10.1203/01.PDR.0000096455.00657.98. [DOI] [PubMed] [Google Scholar]
- Hanson IM, Fletcher JM, Jordan T, Brown A, Taylor D, Adams RJ, Punnett HH, van Heyningen V, 1994. Mutations at the PAX6 locus are found in heterogeneous anterior segment malformations including Peters’ anomaly. Nat. Genet. 6, 168–173. 10.1038/ng0294-168. [DOI] [PubMed] [Google Scholar]
- Hanson IM, Seawright A, Hardman K, Hodgson S, Zaletayev D, Fekete G, van Heyningen V, 1993. PAX6 mutations in aniridia. Hum. Mol. Genet. 2, 915–920. 10.1093/hmg/2.7.915. [DOI] [PubMed] [Google Scholar]
- Harley SK, Sliney DH, 2018. Pupil size in outdoor environments. Health Phys. 115, 354–359. 10.1097/HP.0000000000000887. [DOI] [PubMed] [Google Scholar]
- He Y, Chen X, Tsui I, Vajzovic L, Sadda SR, 2022. Insights into the developing fovea revealed by imaging. Prog. Retin. Eye Res. 101067 10.1016/j.preteyeres.2022.101067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyman I, Frampton I, van Heyningen V, Hanson I, Teague P, Taylor A, Simonoff E, 1999. Psychiatric disorder and cognitive function in a family with an inherited novel mutation of the developmental control gene PAX6. Psychiatr. Genet. 9, 85–90. 10.1097/00041444-199906000-00006. [DOI] [PubMed] [Google Scholar]
- Hickmott JW, Gunawardane U, Jensen K, Korecki AJ, Simpson EM, 2018. Epistasis between Pax6Sey and genetic background reinforces the value of defined hybrid mouse models for therapeutic trials. Gene Ther. 25, 524–537. 10.1038/s41434-018-0043-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill RE, Favor J, Hogan BL, Ton CC, Saunders GF, Hanson IM, Prosser J, Jordan T, Hastie ND, van Heyningen V, 1991. Mouse small eye results from mutations in a paired-like homeobox-containing gene. Nature 354, 522–525. 10.1038/354522a0. [DOI] [PubMed] [Google Scholar]
- Hingorani M, Hanson I, van Heyningen V, 2012. Aniridia. Eur. J. Hum. Genet. 20, 1011–1017. 10.1038/ejhg.2012.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani M, Williamson KA, Moore AT, van Heyningen V, 2009. Detailed ophthalmologic evaluation of 43 individuals with PAX6 mutations. Invest. Ophthalmol. Vis. Sci. 50, 2581–2590. 10.1167/iovs.08-2827. [DOI] [PubMed] [Google Scholar]
- Hodgson SV, Saunders KE, 1980. A probable case of the homozygous condition of the aniridia gene. J. Med. Genet. 17, 478–480. 10.1136/jmg.17.6.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland EJ, Djalilian AR, Schwartz GS, 2003. Management of aniridic keratopathy with keratolimbal allograft: a limbal stem cell transplantation technique. Ophthalmology 110, 125–130. 10.1016/S0161-6420(02)01451-3. [DOI] [PubMed] [Google Scholar]
- Hollmann AK, Dammann I, Wemheuer WM, Wemheuer WE, Chilla A, Tipold A, Schulz-Schaeffer WJ, Beck J, Schütz E, Brenig B, 2017. Morgagnian cataract resulting from a naturally occurring nonsense mutation elucidates a role of CPAMD8 in mammalian lens development. PLoS One 12, e0180665. 10.1371/journal.pone.0180665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim DM, Mundlos S, 2020. The role of 3D chromatin domains in gene regulation: a multi-facetted view on genome organization. Curr. Opin. Genet. Dev. 61, 1–8. 10.1016/j.gde.2020.02.015. [DOI] [PubMed] [Google Scholar]
- Ihnatko R, Eden U, Fagerholm P, Lagali N, 2016. Congenital aniridia and the ocular surface. Ocul. Surf. 14, 196–206. 10.1016/j.jtos.2015.10.003. [DOI] [PubMed] [Google Scholar]
- Ihnatko R, Edén U, Lagali N, Dellby A, Fagerholm P, 2013. Analysis of protein composition and protein expression in the tear fluid of patients with congenital aniridia. J. Proteonomics 94, 78–88. 10.1016/j.jprot.2013.09.003. [DOI] [PubMed] [Google Scholar]
- Imada T, Nakamura S, Hisamura R, Izuta Y, Jin K, Ito M, Kitamura N, Tanaka KF, Mimura M, Shibuya I, Tsubota K, 2017. Serotonin hormonally regulates lacrimal gland secretory function via the serotonin type 3a receptor. Sci. Rep. 7, 6965. 10.1038/s41598-017-06022-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi M, Kuwabara T, 1971. Development of the rat iris. Invest. Ophthalmol. 10, 733–744. [PubMed] [Google Scholar]
- Ito YA, Walter MA, 2014. Genomics and anterior segment dysgenesis: a review. Clin. Exp. Ophthalmol. 42, 13–24. 10.1111/ceo.12152. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Wurdak H, Schwerdtfeger K, Kunz T, Ille F, Leveen P, Hjalt TA, Suter U, Karlsson S, Hafezi F, Born W, Sommer L, 2005. Compound developmental eye disorders following inactivation of TGFbeta signaling in neural-crest stem cells. J Biol 4, 11. 10.1186/jbiol29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen AM, 2005. Potential roles for BMP and Pax genes in the development of iris smooth muscle. Dev. Dynam. 232, 385–392. 10.1002/dvdy.20224. [DOI] [PubMed] [Google Scholar]
- Jeong Y, Leskow FC, El-Jaick K, Roessler E, Muenke M, Yocum A, Dubourg C, Li X, Geng X, Oliver G, Epstein DJ, 2008. Regulation of a remote Shh forebrain enhancer by the Six3 homeoprotein. Nat. Genet. 40, 1348–1353. 10.1038/ng.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston MC, Noden DM, Hazelton RD, Coulombre JL, Coulombre AJ, 1979. Origins of avian ocular and periocular tissues. Exp. Eye Res. 29, 27–43. 10.1016/0014-4835(79)90164-7. [DOI] [PubMed] [Google Scholar]
- Jordan T, Hanson I, Zaletayev D, Hodgson S, Prosser J, Seawright A, Hastie N, van Heyningen V, 1992. The human PAX6 gene is mutated in two patients with aniridia. Nat. Genet. 1, 328–332. 10.1038/ng0892-328. [DOI] [PubMed] [Google Scholar]
- Jun S, Desplan C, 1996. Cooperative interactions between paired domain and homeodomain. Development 122, 2639–2650. 10.1242/dev.122.9.2639. [DOI] [PubMed] [Google Scholar]
- Justice ED, Barnum SJ, Kidd T, 2017. The WAGR syndrome gene PRRG4 is a functional homologue of the commissureless axon guidance gene. PLoS Genet. 13, e1006865 10.1371/journal.pgen.1006865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammandel B, Chowdhury K, Stoykova A, Aparicio S, Brenner S, Gruss P, 1999. Distinct cis-essential modules direct the time-space pattern of the Pax6 gene activity. Dev. Biol. 205, 79–97. 10.1006/dbio.1998.9128. [DOI] [PubMed] [Google Scholar]
- Kanakubo S, Nomura T, Yamamura K, Miyazaki J, Tamai M, Osumi N, 2006. Abnormal migration and distribution of neural crest cells in Pax6 heterozygous mutant eye, a model for human eye diseases. Gene Cell. 11, 919–933. 10.1111/j.1365-2443.2006.00992.x. [DOI] [PubMed] [Google Scholar]
- Kawase E, Tanaka K, Honna T, Azuma N, 2001. A case of atypical WAGR syndrome with anterior segment anomaly and microphthalmos. Arch. Ophthalmol. 119, 1855–1856. [PubMed] [Google Scholar]
- Kikkawa T, Casingal CR, Chun SH, Shinohara H, Hiraoka K, Osumi N, 2019. The role of Pax6 in brain development and its impact on pathogenesis of autism spectrum disorder. Brain Res. 1705, 95–103. 10.1016/j.brainres.2018.02.041. [DOI] [PubMed] [Google Scholar]
- Kim J, Lauderdale JD, 2006. Analysis of Pax6 expression using a BAC transgene reveals the presence of a paired-less isoform of Pax6 in the eye and olfactory bulb. Dev. Biol. 292, 486–505. 10.1016/j.ydbio.2005.12.041. [DOI] [PubMed] [Google Scholar]
- Kim J-M, Kim S-K, Kim S-H, Cho W-K, Cho K-S, Jung M-H, Suh B-K, Ahn M-B, 2021. Transient hyperinsulinemic hypoglycemia linked to PAX6 mutation. Medicina 57, 582. 10.3390/medicina57060582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita A, Ohyama K, Tanimura S, Matsuda K, Kishino T, Negishi Y, Asahina N, Shiraishi H, Hosoki K, Tomiwa K, Ishihara N, Mishima H, Mori R, Nakashima M, Saitoh S, Yoshiura K-I, 2021. Itpr1 regulates the formation of anterior eye segment tissues derived from neural crest cells. Development 148, dev188755. 10.1242/dev.188755. [DOI] [PubMed] [Google Scholar]
- Kiselev Y, Eriksen TE, Forsdahl S, Nguyen LHT, Mikkola I, 2012. 3T3 cell lines stably expressing Pax6 or Pax6(5a)–a new tool used for identification of common and isoform specific target genes. PLoS One 7, e31915. 10.1371/journal.pone.0031915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kit V, Lima Cunha D, Hagag AM, Moosajee M, 2021. Longitudinal genotype-phenotype analysis in 86 PAX6-related aniridia patients. JCI Insight. 10.1172/jci.insight.148406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa K, Hikichi T, Nakamura T, Sotozono C, Kinoshita S, Masui S, 2017. PAX6 regulates human corneal epithelium cell identity. Exp. Eye Res. 154, 30–38. 10.1016/j.exer.2016.11.005. [DOI] [PubMed] [Google Scholar]
- Kleinjan DA, Seawright A, Elgar G, van Heyningen V, 2002. Characterization of a novel gene adjacent to PAX6, revealing synteny conservation with functional significance. Mamm. Genome 13, 102–107. 10.1007/s00335-001-3058-y. [DOI] [PubMed] [Google Scholar]
- Kleinjan DA, Seawright A, Schedl A, Quinlan RA, Danes S, van Heyningen V, 2001. Aniridia-associated translocations, DNase hypersensitivity, sequence comparison and transgenic analysis redefine the functional domain of PAX6. Hum. Mol. Genet. 10, 2049–2059. 10.1093/hmg/10.19.2049. [DOI] [PubMed] [Google Scholar]
- Kleinjan DA, van Heyningen V, 2005. Long-range control of gene expression: emerging mechanisms and disruption in disease. Am. J. Hum. Genet. 76, 8–32. 10.1086/426833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimova L, Kozmik Z, 2014. Stage-dependent requirement of neuroretinal Pax6 for lens and retina development. Development 141, 1292–1302. 10.1242/dev.098822. [DOI] [PubMed] [Google Scholar]
- Koenig Y, Bock F, Horn F, Kruse F, Straub K, Cursiefen C, 2009. Short- and long-term safety profile and efficacy of topical bevacizumab (Avastin) eye drops against corneal neovascularization. Graefes Arch. Clin. Exp. Ophthalmol. 247, 1375–1382. 10.1007/s00417-009-1099-1. [DOI] [PubMed] [Google Scholar]
- Kojima T, Hasegawa A, Nakamura T, Isogai N, Kataoka T, Ichikawa K, 2016. Five-year PROSE treatment for aniridic keratopathy. Optom. Vis. Sci. 93, 1328–1332. 10.1097/OPX.0000000000000942. [DOI] [PubMed] [Google Scholar]
- Kokotas H, Petersen MB, 2010. Clinical and molecular aspects of aniridia. Clin. Genet. 77, 409–420. 10.1111/j.1399-0004.2010.01372.x. [DOI] [PubMed] [Google Scholar]
- Korecki AJ, Cueva-Vargas JL, Fornes O, Agostinone J, Farkas RA, Hickmott JW, Lam SL, Mathelier A, Zhou M, Wasserman WW, Di Polo A, Simpson EM, 2021. Human MiniPromoters for ocular-rAAV expression in ON bipolar, cone, corneal, endothelial, Müller glial, and PAX6 cells. Gene Ther. 28, 351–372. 10.1038/s41434-021-00227-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozmik Z, Czerny T, Busslinger M, 1997. Alternatively spliced insertions in the paired domain restrict the DNA sequence specificity of Pax6 and Pax8. EMBO J. 16, 6793–6803. 10.1093/emboj/16.22.6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozulin P, Natoli R, Madigan MC, O’Brien KMB, Provis JM, 2009a. Gradients of Eph-A6 expression in primate retina suggest roles in both vascular and axon guidance. Mol. Vis. 15, 2649–2662. [PMC free article] [PubMed] [Google Scholar]
- Kozulin P, Natoli R, O’Brien KMB, Madigan MC, Provis JM, 2009b. Differential expression of anti-angiogenic factors and guidance genes in the developing macula. Mol. Vis. 15, 45–59. [PMC free article] [PubMed] [Google Scholar]
- Kredo-Russo S, Mandelbaum AD, Ness A, Alon I, Lennox KA, Behlke MA, Hornstein E, 2012. Pancreas-enriched miRNA refines endocrine cell differentiation. Development 139, 3021–3031. 10.1242/dev.080127. [DOI] [PubMed] [Google Scholar]
- Kroeber M, Davis N, Holzmann S, Kritzenberger M, Shelah-Goraly M, Ofri R, Ashery-Padan R, Tamm ER, 2010. Reduced expression of Pax6 in lens and cornea of mutant mice leads to failure of chamber angle development and juvenile glaucoma. Hum. Mol. Genet. 19, 3332–3342. 10.1093/hmg/ddq237. [DOI] [PubMed] [Google Scholar]
- Kumar S, Duester G, 2010. Retinoic acid signaling in perioptic mesenchyme represses Wnt signaling via induction of Pitx2 and Dkk2. Dev. Biol. 340, 67–74. 10.1016/j.ydbio.2010.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusumesh R, Ambastha A, 2016. Congenital aniridia with ectopia lentis. J. Clin. Diagn. Res. 10, NJ01. 10.7860/JCDR/2016/18703.8165, 02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagali N, Edén U, Utheim TP, Chen X, Riise R, Dellby A, Fagerholm P, 2013. In vivo morphology of the limbal palisades of vogt correlates with progressive stem cell deficiency in aniridia-related keratopathy. Invest. Ophthalmol. Vis. Sci. 54, 5333–5342. 10.1167/iovs.13-11780. [DOI] [PubMed] [Google Scholar]
- Lagali N, Wowra B, Dobrowolski D, Utheim TP, Fagerholm P, Wylegala E, 2018. Stage-related central corneal epithelial transformation in congenital aniridia-associated keratopathy. Ocul. Surf. 16, 163–172. 10.1016/j.jtos.2017.11.003. [DOI] [PubMed] [Google Scholar]
- Lagali N, Wowra B, Fries FN, Latta L, Moslemani K, Utheim TP, Wylegala E, Seitz B, Käsmann-Kellner B, 2020a. Early phenotypic features of aniridia-associated keratopathy and association with PAX6 coding mutations. Ocul. Surf. 18, 130–140. 10.1016/j.jtos.2019.11.002. [DOI] [PubMed] [Google Scholar]
- Lagali N, Wowra B, Fries FN, Latta L, Moslemani K, Utheim TP, Wylegala E, Seitz B, Käsmann-Kellner B, 2020b. PAX6 mutational status determines aniridia-associated keratopathy phenotype. Ophthalmology 127, 273–275. 10.1016/j.ophtha.2019.09.034. [DOI] [PubMed] [Google Scholar]
- Lalitha S, Basu B, Surya S, Meera V, Riya PA, Parvathy S, Das AV, Sivakumar KC, Nelson-Sathi S, James J, 2020. Pax6 modulates intra-retinal axon guidance and fasciculation of retinal ganglion cells during retinogenesis. Sci. Rep. 10, 16075 10.1038/s41598-020-72828-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsend ECS, Lagali N, Utheim TP, 2021. Congenital aniridia - a comprehensive review of clinical features and therapeutic approaches. Surv. Ophthalmol. 10.1016/j.survophthal.2021.02.011. [DOI] [PubMed] [Google Scholar]
- Landsend Erlend C.S., Pedersen HR, Utheim ØA, Rueegg CS, Baraas RC, Lagali N, Bragadóttir R, Moe MC, Utheim TP, 2019. Characteristics and utility of fundus autofluorescence in congenital aniridia using scanning laser ophthalmoscopy. Invest. Ophthalmol. Vis. Sci. 60, 4120–4128. 10.1167/iovs.19-26994. [DOI] [PubMed] [Google Scholar]
- Landsend ECS, Utheim ØA, Pedersen HR, Aass HCD, Lagali N, Dartt DA, Baraas RC, Utheim TP, 2018. The level of inflammatory tear cytokines is elevated in congenital aniridia and associated with meibomian gland dysfunction. Invest. Ophthalmol. Vis. Sci. 59, 2197–2204. 10.1167/iovs.18-24027. [DOI] [PubMed] [Google Scholar]
- Landsend ES, Utheim ØA, Pedersen HR, Lagali N, Baraas RC, Utheim TP, 2018. The genetics of congenital aniridia-a guide for the ophthalmologist. Surv. Ophthalmol. 63, 105–113. 10.1016/j.survophthal.2017.09.004. [DOI] [PubMed] [Google Scholar]
- Lapid-Gortzak R, Santana NTY, Nieuwendaal CP, Mourits MP, van der Meulen IJE, 2018. Topical bevacizumab treatment in aniridia. Int. Ophthalmol. 38, 1741–1746. 10.1007/s10792-017-0605-4. [DOI] [PubMed] [Google Scholar]
- Latta L, Figueiredo FC, Ashery-Padan R, Collinson JM, Daniels J, Ferrari S, Szentmáry N, Solá S, Shalom-Feuerstein R, Lako M, Xapelli S, Aberdam D, Lagali N, 2021a. Pathophysiology of aniridia-associated keratopathy: developmental aspects and unanswered questions. Ocul. Surf. 22, 245–266. 10.1016/j.jtos.2021.09.001. [DOI] [PubMed] [Google Scholar]
- Latta L, Ludwig N, Krammes L, Stachon T, Fries FN, Mukwaya A, Szentmáry N, Seitz B, Wowra B, Kahraman M, Keller A, Meese E, Lagali N, Käsmann-Kellner B, 2021b. Abnormal neovascular and proliferative conjunctival phenotype in limbal stem cell deficiency is associated with altered microRNA and gene expression modulated by PAX6 mutational status in congenital aniridia. Ocul. Surf. 19, 115–127. 10.1016/j.jtos.2020.04.014. [DOI] [PubMed] [Google Scholar]
- Lauderdale JD, Wilensky JS, Oliver ER, Walton DS, Glaser T, 2000. 3’ deletions cause aniridia by preventing PAX6 gene expression. Proc. Natl. Acad. Sci. U. S. A. 97, 13755–13759. 10.1073/pnas.240398797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Choi D-G, Chun BY, Oh EH, Lee Y-J, Kim U-K, Park J-S, 2019. A family with a mild form of congenital nystagmus and optic disc coloboma caused by a novel PAX6 mutation. Gene 705, 177–180. 10.1016/j.gene.2019.04.035. [DOI] [PubMed] [Google Scholar]
- Li G, Xu F, Zhu J, Krawczyk M, Zhang Y, Yuan J, Patel S, Wang Y, Lin Y, Zhang Ming, Cai H, Chen D, Zhang Meixia, Cao G, Yeh E, Lin D, Su Q, Li W, Sen GL, Afshari N, Chen S, Maas RL, Fu X-D, Zhang K, Liu Y, Ouyang H., 2015. Transcription factor PAX6 (paired box 6) controls limbal stem cell lineage in development and disease. J. Biol. Chem. 290, 20448–20454. 10.1074/jbc.M115.662940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HT, Seo E-J, Kim G-H, Ahn H, Lee H, Shin KH, Lee J-K, Yoo H-W, 2012. Comparison between aniridia with and without PAX6 mutations: clinical and molecular analysis in 14 Korean patients with aniridia. Ophthalmology 119, 1258–1264. 10.1016/j.ophtha.2011.12.010. [DOI] [PubMed] [Google Scholar]
- Lima Cunha D, Arno G, Corton M, Moosajee M, 2019. The spectrum of PAX6 mutations and genotype-phenotype correlations in the eye. Genes (Basel) 10. 10.3390/genes10121050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-García JS, Rivas L, García-Lozano I, 2005. [Moderate limbal deficiency in patients with congenital aniridia treated with amniotic membrane transplantation]. Arch. Soc. Esp. Oftalmol. 80, 517–523. 10.4321/S0365-66912005000900005. [DOI] [PubMed] [Google Scholar]
- López-García JS, Rivas L, García-Lozano I, Murube J, 2008. Autologous serum eyedrops in the treatment of aniridic keratopathy. Ophthalmology 115, 262–267. 10.1016/J.OPHTHA.2007.03.087. [DOI] [PubMed] [Google Scholar]
- Lovicu FJ, Steven P, Saika S, McAvoy JW, 2004. Aberrant lens fiber differentiation in anterior subcapsular cataract formation: a process dependent on reduced levels of Pax6. Invest. Ophthalmol. Vis. Sci. 45, 1946–1953. 10.1167/iovs.03-1206. [DOI] [PubMed] [Google Scholar]
- Lwigale PY, 2015. Corneal development: different cells from a common progenitor. Prog Mol Biol Transl Sci 134, 43–59. 10.1016/bs.pmbts.2015.04.003. [DOI] [PubMed] [Google Scholar]
- Machon O, Kreslova J, Ruzickova J, Vacik T, Klimova L, Fujimura N, Lachova J, Kozmik Z, 2010. Lens morphogenesis is dependent on Pax6-mediated inhibition of the canonical Wnt/beta-catenin signaling in the lens surface ectoderm. Genesis 48, 86–95. 10.1002/dvg.20583. [DOI] [PubMed] [Google Scholar]
- Malandrini A, Mari F, Palmeri S, Gambelli S, Berti G, Bruttini M, Bardelli AM, Williamson K, van Heyningen V, Renieri A, 2001. PAX6 mutation in a family with aniridia, congenital ptosis, and mental retardation. Clin. Genet. 60, 151–154. 10.1034/j.1399-0004.2001.600210.x. [DOI] [PubMed] [Google Scholar]
- Manuel M, Pratt T, Liu M, Jeffery G, Price DJ, 2008. Overexpression of Pax6 results in microphthalmia, retinal dysplasia and defective retinal ganglion cell axon guidance. BMC Dev. Biol. 8, 59. 10.1186/1471-213X-8-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt T, Ashery-Padan R, Andrejewski N, Scardigli R, Guillemot F, Gruss P, 2001. Pax6 is required for the multipotent state of retinal progenitor cells. Cell 105, 43–55. 10.1016/s0092-8674(01)00295-1. [DOI] [PubMed] [Google Scholar]
- Marquardt T, Gruss P, 2002. Generating neuronal diversity in the retina: one for nearly all. Trends Neurosci. 25, 32–38. 10.1016/s0166-2236(00)02028-2. [DOI] [PubMed] [Google Scholar]
- Martha A, Strong LC, Ferrell RE, Saunders GF, 1995. Three novel aniridia mutations in the human PAX6 gene. Hum. Mutat. 6, 44–49. 10.1002/humu.1380060109. [DOI] [PubMed] [Google Scholar]
- Matt N, Dupé V, Garnier J-M, Dennefeld C, Chambon P, Mark M, Ghyselinck NB, 2005. Retinoic acid-dependent eye morphogenesis is orchestrated by neural crest cells. Development 132, 4789–4800. 10.1242/dev.02031. [DOI] [PubMed] [Google Scholar]
- Mayer CS, Hoffmann AM, Prahs P, Reznicek L, Khoramnia R, 2020. Functional outcomes after combined iris and intraocular lens implantation in various iris and lens defects. BMC Ophthalmol. 20, 370. 10.1186/s12886-020-01621-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard ML, Zele A, Kwan A, Feigl B, 2017. Intrinsically photosensitive retinal ganglion cell function, sleep efficiency and depression in advanced age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 58, 990–996. 10.1167/iovs.16-20659. [DOI] [PubMed] [Google Scholar]
- McCulley TJ, Mayer K, Dahr SS, Simpson J, Holland EJ, 2005. Aniridia and optic nerve hypoplasia. Eye 19, 762–764. 10.1038/sj.eye.6701642. [DOI] [PubMed] [Google Scholar]
- McEntagart M, Williamson KA, Rainger JK, Wheeler A, Seawright A, De Baere E, Verdin H, Bergendahl LT, Quigley A, Rainger J, Dixit A, Sarkar A, López Laso E, Sanchez-Carpintero R, Barrio J, Bitoun P, Prescott T, Riise R, McKee S, Cook J, McKie L, Ceulemans B, Meire F, Temple IK, Prieur F, Williams J, Clouston P, Németh AH, Banka S, Bengani H, Handley M, Freyer E, Ross A, , DDD Study, van Heyningen V, Marsh JA, Elmslie F, FitzPatrick DR, 2016. A restricted repertoire of de novo mutations in ITPR1 cause Gillespie syndrome with evidence for dominant-negative effect. Am. J. Hum. Genet. 98, 981–992. 10.1016/j.ajhg.2016.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears AJ, Jordan T, Mirzayans F, Dubois S, Kume T, Parlee M, Ritch R, Koop B, Kuo WL, Collins C, Marshall J, Gould DB, Pearce W, Carlsson P, Enerbäck S, Morissette J, Bhattacharya S, Hogan B, Raymond V, Walter MA, 1998. Mutations of the forkhead/winged-helix gene, FKHL7, in patients with Axenfeld-Rieger anomaly. Am. J. Hum. Genet. 63, 1316–1328. 10.1086/302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medsinge A, Nischal KK, 2015. Cataract surgery in children with congenital keratolenticular adhesion (Peters anomaly type 2). J AAPOS 19, 24–28. 10.1016/j.jaapos.2014.10.026. [DOI] [PubMed] [Google Scholar]
- Meur G, Simon A, Harun N, Virally M, Dechaume A, Bonnefond A, Fetita S, Tarasov AI, Guillausseau P-J, Boesgaard TW, Pedersen O, Hansen T, Polak M, Gautier J-F, Froguel P, Rutter GA, Vaxillaire M, 2010. Insulin gene mutations resulting in early-onset diabetes: marked differences in clinical presentation, metabolic status, and pathogenic effect through endoplasmic reticulum retention. Diabetes 59, 653–661. 10.2337/db09-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesfeld JB, Brown NL, 2019. Eye organogenesis: a hierarchical view of ocular development. Curr. Top. Dev. Biol. 132, 351–393. 10.1016/bs.ctdb.2018.12.008. [DOI] [PubMed] [Google Scholar]
- Mikkola I, Bruun JA, Bjorkoy G, Holm T, Johansen T, 1999. Phosphorylation of the transactivation domain of Pax6 by extracellular signal-regulated kinase and p38 mitogen-activated protein kinase. J. Biol. Chem. 274, 15115–15126. 10.1074/jbc.274.21.15115. [DOI] [PubMed] [Google Scholar]
- Millar AJW, Cox S, Davidson A, 2017. Management of bilateral Wilms tumours. Pediatr. Surg. Int. 33, 737–745. 10.1007/s00383-017-4091-6. [DOI] [PubMed] [Google Scholar]
- Mirjalili Mohanna SZ, Hickmott JW, Lam SL, Chiu NY, Lengyell TC, Tam BM, Moritz OL, Simpson EM, 2020. Germline CRISPR/Cas9-Mediated gene editing prevents vision loss in a novel mouse model of aniridia. Mol. Ther. Methods Clin. Dev. 17, 478–490. 10.1016/j.omtm.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirrahimi M, Sabbaghi H, Ahmadieh H, Jahanmard M, Hassanpour K, Suri F, 2019. A novel PAX6 mutation causes congenital aniridia with or without retinal detachment. Ophthalmic Genet. 40, 146–149. 10.1080/13816810.2019.1597374. [DOI] [PubMed] [Google Scholar]
- Mitchell RK, Nguyen-Tu M-S, Chabosseau P, Callingham RM, Pullen TJ, Cheung R, Leclerc I, Hodson DJ, Rutter GA, 2017. The transcription factor Pax6 is required for pancreatic β cell identity, glucose-regulated ATP synthesis, and Ca2+ dynamics in adult mice. J. Biol. Chem. 292, 8892–8906. 10.1074/jbc.M117.784629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mort RL, Bentley AJ, Martin FL, Collinson JM, Douvaras P, Hill RE, Morley SD, Fullwood NJ, West JD, 2011. Effects of aberrant Pax6 gene dosage on mouse corneal pathophysiology and corneal epithelial homeostasis. PLoS One 6, e28895. 10.1371/journal.pone.0028895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moshirfar M, Hastings J, Ronquillo Y, 2022. Aniridic fibrosis syndrome. StatPearls. StatPearls Publishing, Treasure Island (FL. [PubMed] [Google Scholar]
- Motoda S, Fujita S, Kozawa J, Kimura T, Fukui K, Ikuno Y, Imagawa A, Iwahashi H, Shimomura I, 2019. Case of a novel PAX6 mutation with aniridia and insulin-dependent diabetes mellitus. J. Diabetes Investig. 10, 552–553. 10.1111/jdi.12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu X, Klein WH, 2004. A gene regulatory hierarchy for retinal ganglion cell specification and differentiation. Semin. Cell Dev. Biol. 15, 115–123. 10.1016/j.semcdb.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Mukwaya A, Jensen L, Peebo B, Lagali N, 2019. MicroRNAs in the cornea: role and implications for treatment of corneal neovascularization. Ocul. Surf. 17, 400–411. 10.1016/j.jtos.2019.04.002. [DOI] [PubMed] [Google Scholar]
- Muñoz-Negrete FJ, Teus MA, García-Feijoó J, Canut MI, Rebolleda G, 2021. Aniridic glaucoma: an update. Arch. Soc. Esp. Oftalmol. 96 (Suppl. 1), 52–59. 10.1016/j.oftale.2020.11.011. [DOI] [PubMed] [Google Scholar]
- Muscatello MRA, Zoccali RA, Pandolfo G, Mangano P, Lorusso S, Cedro C, Battaglia F, Spina E, Bruno A, 2019. Duloxetine in psychiatric disorders: expansions beyond major depression and generalized anxiety disorder. Front. Psychiatr. 10, 772. 10.3389/fpsyt.2019.00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Takeda K, Inatomi T, Sotozono C, Kinoshita S, 2011. Long-term results of autologous cultivated oral mucosal epithelial transplantation in the scar phase of severe ocular surface disorders. Br. J. Ophthalmol. 95, 942–946. 10.1136/BJO.2010.188714. [DOI] [PubMed] [Google Scholar]
- Needhamsen M, White RB, Giles KM, Dunlop SA, Thomas MG, 2014. Regulation of human PAX6 expression by miR-7. Evol Bioinform Online 10, 107–113. 10.4137/EBO.S13739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neethirajan G, Solomon A, Krishnadas SR, Vijayalakshmi P, Sundaresan P, 2009. Genotype/phenotype association in Indian congenital aniridia. Indian J. Pediatr. 76, 513–517. 10.1007/s12098-009-0075-4. [DOI] [PubMed] [Google Scholar]
- Netland PA, Scott ML, Boyle JW, Lauderdale JD, 2011. Ocular and systemic findings in a survey of aniridia subjects. J AAPOS 15, 562–566. 10.1016/j.jaapos.2011.07.009. [DOI] [PubMed] [Google Scholar]
- Neveu MM, Holder GE, Sloper JJ, Jeffery G, 2005. Optic chiasm formation in humans is independent of foveal development. Eur. J. Neurosci. 22, 1825–1829. 10.1111/j.1460-9568.2005.04364.x. [DOI] [PubMed] [Google Scholar]
- Nieves-Moreno M, Noval S, Peralta J, Palomares-Bralo M, Del Pozo A, Garcia-Miñaur S, Santos-Simarro F, Vallespin E, 2021. Expanding the phenotypic spectrum of PAX6 mutations: from congenital cataracts to nystagmus. Genes 12, 707. 10.3390/genes12050707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninkovic J, Pinto L, Petricca S, Lepier A, Sun J, Rieger MA, Schroeder T, Cvekl A, Favor J, Götz M, 2010. The transcription factor Pax6 regulates survival of dopaminergic olfactory bulb neurons via crystallin αA. Neuron 68, 682–694. 10.1016/j.neuron.2010.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nischal KK, 2002. Two-incision push-pull capsulorhexis for pediatric cataract surgery. J. Cataract Refract. Surg. 28, 593–595. 10.1016/s0886-3350(01)01125-7. [DOI] [PubMed] [Google Scholar]
- Nischal KK, Lathrop KL, 2016. The palisades of vogt in congenital corneal opacification (an American ophthalmological society thesis). Trans. Am. Ophthalmol. Soc. 114, T8. [PMC free article] [PubMed] [Google Scholar]
- Nishimura DY, Swiderski RE, Alward WL, Searby CC, Patil SR, Bennet SR, Kanis AB, Gastier JM, Stone EM, Sheffield VC, 1998. The forkhead transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nat. Genet. 19, 140–147. 10.1038/493. [DOI] [PubMed] [Google Scholar]
- Nishina S, Kohsaka S, Yamaguchi Y, Handa H, Kawakami A, Fujisawa H, Azuma N, 1999. PAX6 expression in the developing human eye. Br. J. Ophthalmol. 83, 723–727. 10.1136/bjo.83.6.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura T, Haba H, Osumi N, 2007. Role of a transcription factor Pax6 in the developing vertebrate olfactory system. Dev. Growth Differ. 49, 683–690. 10.1111/j.1440-169X.2007.00965.x. [DOI] [PubMed] [Google Scholar]
- Obara EA, Hannibal J, Heegaard S, Fahrenkrug J, 2016. Loss of melanopsin-expressing retinal ganglion cells in severely staged glaucoma patients. Invest. Ophthalmol. Vis. Sci. 57, 4661–4667. 10.1167/iovs.16-19997. [DOI] [PubMed] [Google Scholar]
- Ochi S, Manabe S, Kikkawa T, Osumi N, 2022. Thirty years’ history since the discovery of Pax6: from central nervous system development to neurodevelopmental disorders. Int. J. Mol. Sci. 23, 6115. 10.3390/ijms23116115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada K, Mishima HK, Masumoto M, Tsumamoto Y, Tsukamoto H, Takamatsu M, 2000. Results of filtering surgery in young patients with aniridia. Hiroshima J. Med. Sci. 49, 135–138. [PubMed] [Google Scholar]
- Okladnova O, Syagailo YV, Mössner R, Riederer P, Lesch KP, 1998. Regulation of PAX-6 gene transcription: alternate promoter usage in human brain. Brain Res Mol Brain Res 60, 177–192. 10.1016/s0169-328x(98)00167-3. [DOI] [PubMed] [Google Scholar]
- Oron-Karni V, Farhy C, Elgart M, Marquardt T, Remizova L, Yaron O, Xie Q, Cvekl A, Ashery-Padan R, 2008. Dual requirement for Pax6 in retinal progenitor cells. Development 135, 4037–4047. 10.1242/dev.028308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osumi N, Shinohara H, Numayama-Tsuruta K, Maekawa M, 2008. Concise review: pax6 transcription factor contributes to both embryonic and adult neurogenesis as a multifunctional regulator. Stem Cell. 26, 1663–1672. 10.1634/stemcells.2007-0884. [DOI] [PubMed] [Google Scholar]
- Ouyang J, Shen Y-C, Yeh L-K, Li W, Coyle BM, Liu C-Y, Fini ME, 2006. Pax6 overexpression suppresses cell proliferation and retards the cell cycle in corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 47, 2397–2407. 10.1167/iovs.05-1083. [DOI] [PubMed] [Google Scholar]
- Oved K, Zennaro L, Dorot O, Zerbib J, Frank E, Roux LN, Bremond-Gignac D, Pichinuk E, Aberdam D, 2021. Ritanserin, a potent serotonin 2A receptor antagonist, represses MEK/ERK signalling pathway to restore PAX6 production and function in aniridia-like cellular model. Biochem. Biophys. Res. Commun. 582, 100–104. 10.1016/j.bbrc.2021.10.036. [DOI] [PubMed] [Google Scholar]
- Panneerselvam A, Kannan A, Mariajoseph-Antony LF, Prahalathan C, 2019. PAX proteins and their role in pancreas. Diabetes Res. Clin. Pract. 155, 107792 10.1016/j.diabres.2019.107792. [DOI] [PubMed] [Google Scholar]
- Park SH, Park YG, Lee MY, Kim MS, 2010. Clinical features of Korean patients with congenital aniridia. Kor. J. Ophthalmol. 24, 291–296. 10.3341/kjo.2010.24.5.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Hayward JD, Tailor V, Nyanhete R, Ahlfors H, Gabriel C, Jannini TB, Abbou-Rayyah Y, Henderson R, Nischal KK, Islam L, Bitner-Glindzicz M, Hurst J, Valdivia LE, Zanolli M, Moosajee M, Brookes J, Papadopoulos M, Khaw PT, Cullup T, Jenkins L, Dahlmann-Noor A, Sowden JC, 2019. The Oculome Panel Test: Next-Generation Sequencing to Diagnose a Diverse Range of Genetic Developmental Eye Disorders. Ophthalmology 126, 888–907. 10.1016/j.ophtha.2018.12.050. [DOI] [PubMed] [Google Scholar]
- Pedersen HR, Baraas RC, Landsend ECS, Utheim ØA, Utheim TP, Gilson SJ, Neitz M, 2020. PAX6 genotypic and retinal phenotypic characterization in congenital aniridia. Invest. Ophthalmol. Vis. Sci. 61, 14. 10.1167/iovs.61.5.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen HR, Hagen LA, Landsend ECS, Gilson SJ, Utheim ØA, Utheim TP, Neitz M, Baraas RC, 2018. Color vision in aniridia. Invest. Ophthalmol. Vis. Sci. 59, 2142–2152. 10.1167/iovs.17-23047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen HR, Neitz M, Gilson SJ, Landsend ECS, Utheim ØA, Utheim TP, Baraas RC, 2019. The cone photoreceptor mosaic in aniridia: within-family phenotype-genotype discordance. Ophthalmol. Retina. 3, 523–534. 10.1016/j.oret.2019.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periman LM, Perez VL, Saban DR, Lin MC, Neri P, 2020. The immunological basis of dry eye disease and current topical treatment options. J. Ocul. Pharmacol. Therapeut. 36, 137–146. 10.1089/jop.2019.0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter NM, Leyland M, Mudhar HS, Lowndes J, Owen KR, Stewart H, 2013. PAX6 mutation in association with ptosis, cataract, iris hypoplasia, corneal opacification and diabetes: a new variant of familial aniridia? Clin. Exp. Ophthalmol. 41, 835–841. 10.1111/ceo.12109. [DOI] [PubMed] [Google Scholar]
- Philips GT, Stair CN, Young Lee H, Wroblewski E, Berberoglu MA, Brown NL, Mastick GS, 2005. Precocious retinal neurons: pax6 controls timing of differentiation and determination of cell type. Dev. Biol. 279, 308–321. 10.1016/j.ydbio.2004.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JE, Krafft CE, Rodrigue AL, Bobilev AM, Lauderdale JD, McDowell JE, 2014. Increased functional connectivity in intrinsic neural networks in individuals with aniridia. Front. Hum. Neurosci. 8, 1013. 10.3389/fnhum.2014.01013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaza S, Dozier C, Langlois MC, Saule S, 1995. Identification and characterization of a neuroretina-specific enhancer element in the quail Pax-6 (Pax-QNR) gene. Mol. Cell Biol. 15, 892–903. 10.1128/MCB.15.2.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prayag AS, Najjar RP, Gronfier C, 2019. Melatonin suppression is exquisitely sensitive to light and primarily driven by melanopsin in humans. J. Pineal Res. 66, e12562. 10.1111/jpi.12562. [DOI] [PubMed] [Google Scholar]
- Prosser J, van Heyningen V, 1998. PAX6 mutations reviewed. Hum. Mutat. 11, 93–108. 10.1002/(SICI)1098-1004, 1998. [DOI] [PubMed] [Google Scholar]
- Provis JM, 2001. Development of the primate retinal vasculature. Prog. Retin. Eye Res. 20, 799–821. 10.1016/s1350-9462(01)00012-x. [DOI] [PubMed] [Google Scholar]
- Provis JM, Dubis AM, Maddess T, Carroll J, 2013. Adaptation of the central retina for high acuity vision: cones, the fovea and the avascular zone. Prog. Retin. Eye Res. 35, 63–81. 10.1016/j.preteyeres.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujari R, Deshmukh R, Sheth C, Rajan MS, 2021. Maternal finger-prick allogenic blood for persistent corneal epithelial defects. BMJ Case Rep. 14 10.1136/BCR-2020-241138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthalath AS, Gupta N, Samanta R, Verma PK, 2021. Congenital aniridia with ectopia lentis and unilateral buphthalmos: an unusual presentation. BMJ Case Rep. 14, e244000 10.1136/bcr-2021-244000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Ji Y, Zheng T, Lu Y, 2016. The efficacy and complications of black diaphragm intra-ocular lens implantation in patients with congenital aniridia. Acta Ophthalmol. 94, e340–e344. 10.1111/aos.12923. [DOI] [PubMed] [Google Scholar]
- Rabiee B, Anwar KN, Shen X, Putra I, Liu M, Jung R, Afsharkhamseh N, Rosenblatt MI, Fishman GA, Liu X, Ghassemi M, Djalilian AR, 2020. Gene dosage manipulation alleviates manifestations of hereditary PAX6 haploinsufficiency in mice. Sci. Transl. Med. 12, eaaz4894. 10.1126/scitranslmed.aaz4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raviv S, Bharti K, Rencus-Lazar S, Cohen-Tayar Y, Schyr R, Evantal N, Meshorer E, Zilberberg A, Idelson M, Reubinoff B, Grebe R, Rosin-Arbesfeld R, Lauderdale J, Lutty G, Arnheiter H, Ashery-Padan R, 2014. PAX6 regulates melanogenesis in the retinal pigmented epithelium through feed-forward regulatory interactions with MITF. PLoS Genet. 10, e1004360 10.1371/journal.pgen.1004360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard T, Engelhardt S, Sundmacher R, 2000. Black diaphragm aniridia intraocular lens for congenital aniridia: long-term follow-up. J. Cataract Refract. Surg. 26, 375–381. 10.1016/s0886-3350(99)00432-0. [DOI] [PubMed] [Google Scholar]
- Remez LA, Onishi A, Menuchin-Lasowski Y, Biran A, Blackshaw S, Wahlin KJ, Zack DJ, Ashery-Padan R, 2017. Pax6 is essential for the generation of late-born retinal neurons and for inhibition of photoreceptor-fate during late stages of retinogenesis. Dev. Biol. 432, 140–150. 10.1016/j.ydbio.2017.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Dixon JR, 2015. A CRISPR connection between chromatin topology and genetic disorders. Cell 161, 955–957. 10.1016/j.cell.2015.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renz PF, Valdivia-Francia F, Sendoel A, 2020. Some like it translated: small ORFs in the 5’UTR. Exp. Cell Res. 396, 112229 10.1016/j.yexcr.2020.112229. [DOI] [PubMed] [Google Scholar]
- Reza HM, Yasuda K, 2004. Lens differentiation and crystallin regulation: a chick model. Int. J. Dev. Biol. 48, 805–817. 10.1387/ijdb.041863hr. [DOI] [PubMed] [Google Scholar]
- Romero-Rangel T, Stavrou P, Cotter J, Rosenthal P, Baltatzis S, Foster CS, 2000. Gas-permeable scleral contact lens therapy in ocular surface disease. Am. J. Ophthalmol. 130, 25–32. 10.1016/s0002-9394(00)00378-0. [DOI] [PubMed] [Google Scholar]
- Roux LN, Petit I, Domart R, Concordet J-P, Qu J, Zhou H, Joliot A, Ferrigno O, Aberdam D, 2018. Modeling of aniridia-related keratopathy by CRISPR/Cas9 genome editing of human limbal epithelial cells and rescue by recombinant PAX6 protein. Stem Cell. 36, 1421–1429. 10.1002/stem.2858. [DOI] [PubMed] [Google Scholar]
- Ruiu S, Marchese G, Saba PL, Gessa GL, Pani L, 2000. The 5-HT2 antagonist ritanserin blocks dopamine re-uptake in the rat frontal cortex. Mol. Psychiatr. 5, 673–677. 10.1038/sj.mp.4000804. [DOI] [PubMed] [Google Scholar]
- Rush CC, 1926. Congenital aniridia. Trans. Am. Ophthalmol. Soc. 24, 332–341. [PMC free article] [PubMed] [Google Scholar]
- Ryan BC, Lowe K, Hanson L, Gil T, Braun L, Howard PL, Chow RL, 2018. Mapping the Pax6 3’ untranslated region microRNA regulatory landscape. BMC Genom. 19, 820. 10.1186/s12864-018-5212-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sannan NS, Shan X, Gregory-Evans K, Kusumi K, Gregory-Evans CY, 2018. Anolis carolinensis as a model to understand the molecular and cellular basis of foveal development. Exp. Eye Res. 173, 138–147. 10.1016/j.exer.2018.05.012. [DOI] [PubMed] [Google Scholar]
- Schedl A, Ross A, Lee M, Engelkamp D, Rashbass P, van Heyningen V, Hastie ND, 1996. Influence of PAX6 gene dosage on development: overexpression causes severe eye abnormalities. Cell 86, 71–82. 10.1016/s0092-8674(00)80078-1. [DOI] [PubMed] [Google Scholar]
- Schilter KF, Reis LM, Schneider A, Bardakjian TM, Abdul-Rahman O, Kozel BA, Zimmerman HH, Broeckel U, Semina EV, 2013. Whole-genome copy number variation analysis in anophthalmia and microphthalmia. Clin. Genet. 84, 473–481. 10.1111/cge.12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosser G, 2014. Early Embryonic Specification of Vertebrate Cranial Placodes, vol. 3. Wiley Interdiscip Rev Dev Biol, pp. 349–363. 10.1002/wdev.142. [DOI] [PubMed] [Google Scholar]
- Schneider S, Osher RH, Burk SE, Lutz TB, Montione R, 2003. Thinning of the anterior capsule associated with congenital aniridia. J. Cataract Refract. Surg. 29, 523–525. 10.1016/s0886-3350(02)01602-4. [DOI] [PubMed] [Google Scholar]
- Schornack MM, 2011. Limbal stem cell disease: management with scleral lenses. Clin. Exp. Optom. 94, 592–594. 10.1111/j.1444-0938.2011.00618.x. [DOI] [PubMed] [Google Scholar]
- Sebastián-Serrano A, Sandonis A, Cardozo M, Rodríguez-Tornos FM, Bovolenta P, Nieto M, 2012. Pαx6 expression in postmitotic neurons mediates the growth of axons in response to SFRP1. PLoS One 7, e31590. 10.1371/journal.pone.0031590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N, 2008. Widespread changes in protein synthesis induced by microRNAs. Nature 455, 58–63. 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- Sellheyer K, Spitznas M, 1987. Ultrastructure of the human posterior tunica vasculosa lentis during early gestation. Graefes Arch. Clin. Exp. Ophthalmol. 225, 377–383. 10.1007/BF02153410. [DOI] [PubMed] [Google Scholar]
- Semina EV, Brownell I, Mintz-Hittner HA, Murray JC, Jamrich M, 2001. Mutations in the human forkhead transcription factor FOXE3 associated with anterior segment ocular dysgenesis and cataracts. Hum. Mol. Genet. 10, 231–236. 10.1093/hmg/10.3.231. [DOI] [PubMed] [Google Scholar]
- Semina EV, Murray JC, Reiter R, Hrstka RF, Graw J, 2000. Deletion in the promoter region and altered expression of Pitx3 homeobox gene in aphakia mice. Hum. Mol. Genet. 9, 1575–1585. 10.1093/hmg/9.11.1575. [DOI] [PubMed] [Google Scholar]
- Semina EV, Reiter R, Leysens NJ, Alward WL, Small KW, Datson NA, Siegel-Bartelt J, Bierke-Nelson D, Bitoun P, Zabel BU, Carey JC, Murray JC, 1996. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat. Genet. 14, 392–399. 10.1038/ng1296-392. [DOI] [PubMed] [Google Scholar]
- Seo S, Chen L, Liu W, Zhao D, Schultz KM, Sasman A, Liu T, Zhang HF, Gage PJ, Kume T, 2017. Foxc1 and Foxc2 in the neural crest are required for ocular anterior segment development. Invest. Ophthalmol. Vis. Sci. 58, 1368–1377. 10.1167/iovs.16-21217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah KJ, Cheung AY, Holland EJ, 2018. Intermediate-term and long-term outcomes with the Boston type 1 keratoprosthesis in aniridia. Cornea 37, 11–14. 10.1097/ICO.0000000000001412. [DOI] [PubMed] [Google Scholar]
- Shaham O, Gueta K, Mor E, Oren-Giladi P, Grinberg D, Xie Q, Cvekl A, Shomron N, Davis N, Keydar-Prizant M, Raviv S, Pasmanik-Chor M, Bell RE, Levy C, Avellino R, Banfi S, Conte I, Ashery-Padan R, 2013. Pax6 regulates gene expression in the vertebrate lens through miR-204. PLoS Genet. 9, e1003357 10.1371/journal.pgen.1003357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham O, Menuchin Y, Farhy C, Ashery-Padan R, 2012. Pax6: a multi-level regulator of ocular development. Prog. Retin. Eye Res. 31, 351–376. 10.1016/j.preteyeres.2012.04.002. [DOI] [PubMed] [Google Scholar]
- Shaham O, Smith AN, Robinson ML, Taketo MM, Lang RA, Ashery-Padan R, 2009. Pax6 is essential for lens fiber cell differentiation. Development 136, 2567–2578. 10.1242/dev.032888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalom-Feuerstein R, Serror L, De La Forest Divonne S, Petit I, Aberdam E, Camargo L, Damour O, Vigouroux C, Solomon A, Gaggioli C, Itskovitz-Eldor J, Ahmad S, Aberdam D, 2012. Pluripotent stem cell model reveals essential roles for miR-450b-5p and miR-184 in embryonic corneal lineage specification. Stem Cell. 30, 898–909. 10.1002/stem.1068. [DOI] [PubMed] [Google Scholar]
- Shiels A, 2020. TRPM3_miR-204: a complex locus for eye development and disease. Hum. Genom. 14, 7. 10.1186/s40246-020-00258-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortt AJ, Bunce C, Levis HJ, Blows P, Doré CJ, Vernon A, Secker GA, Tuft SJ, Daniels JT, 2014. Three-year outcomes of cultured limbal epithelial allografts in aniridia and Stevens-Johnson syndrome evaluated using the Clinical Outcome Assessment in Surgical Trials assessment tool. Stem Cell. Trans. Med. 3, 265–275. 10.5966/SCTM.2013-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortt AJ, Secker GA, Rajan MS, Meligonis G, Dart JK, Tuft SJ, Daniels JT, 2008. Ex vivo expansion and transplantation of limbal epithelial stem cells. Ophthalmology 115, 1989–1997. 10.1016/J.OPHTHA.2008.04.039. [DOI] [PubMed] [Google Scholar]
- Sihota R, Selvan H, Azmira K, Dada T, Sharma A, Gupta A, Upadhyay A, 2021. Improving long-term intraocular pressure and visual outcomes in eyes with aniridic glaucoma. Graefes Arch. Clin. Exp. Ophthalmol. 259, 3749–3755. 10.1007/s00417-021-05335-5. [DOI] [PubMed] [Google Scholar]
- Simola KO, Knuutila S, Kaitila I, Pirkola A, Pohja P, 1983. Familial aniridia and translocation t(4;11)(q22;p13) without Wilms’ tumor. Hum. Genet. 63, 158–161. 10.1007/BF00291536. [DOI] [PubMed] [Google Scholar]
- Singer RA, Arnes L, Cui Y, Wang J, Gao Y, Guney MA, Burnum-Johnson KE, Rabadan R, Ansong C, Orr G, Sussel L, 2019. The long noncoding RNA paupar modulates PAX6 regulatory activities to promote alpha cell development and function. Cell Metabol. 30, 1091–1106. 10.1016/j.cmet.2019.09.013 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, Mohamed A, Chaurasia S, Ramappa M, Mandal AK, Jalali S, Sangwan VS, 2014. Clinical manifestations of congenital aniridia. J. Pediatr. Ophthalmol. Strabismus 51, 59–62. 10.3928/01913913-20131223-01. [DOI] [PubMed] [Google Scholar]
- Singh S, Mishra R, Arango NA, Deng JM, Behringer RR, Saunders GF, 2002. Iris hypoplasia in mice that lack the alternatively spliced Pax6(5a) isoform. Proc. Natl. Acad. Sci. U. S. A. 99, 6812–6815. 10.1073/pnas.102691299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SP, Singh V, Kar N, Chan K, 2010. Efficacy of antidepressants in treating the negative symptoms of chronic schizophrenia: meta-analysis. Br. J. Psychiatry 197, 174–179. 10.1192/bjp.bp.109.067710. [DOI] [PubMed] [Google Scholar]
- Sisodiya SM, Free SL, Williamson KA, Mitchell TN, Willis C, Stevens JM, Kendall BE, Shorvon SD, Hanson IM, Moore AT, van Heyningen V, 2001. PAX6 haploinsufficiency causes cerebral malformation and olfactory dysfunction in humans. Nat. Genet. 28, 214–216. 10.1038/90042. [DOI] [PubMed] [Google Scholar]
- Smith RS, Zabaleta A, Kume T, Savinova OV, Kidson SH, Martin JE, Nishimura DY, Alward WL, Hogan BL, John SW, 2000. Haploinsufficiency of the transcription factors FOXC1 and FOXC2 results in aberrant ocular development. Hum. Mol. Genet. 9, 1021–1032. 10.1093/hmg/9.7.1021. [DOI] [PubMed] [Google Scholar]
- Stanescu D, Iseli HP, Schwerdtfeger K, Ittner LM, Remé CE, Hafezi F, 2007. Continuous expression of the homeobox gene Pax6 in the ageing human retina. Eye 21, 90–93. 10.1038/sj.eye.6702166. [DOI] [PubMed] [Google Scholar]
- Stoilov I, Akarsu AN, Sarfarazi M, 1997. Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (Buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Hum. Mol. Genet. 6, 641–647. 10.1093/hmg/6.4.641. [DOI] [PubMed] [Google Scholar]
- Sundmacher R, Reinhard T, Althaus C, 1994. Black-diaphragm intraocular lens for correction of aniridia. Ophthalmic Surg. 25, 180–185. [PubMed] [Google Scholar]
- Swisa A, Avrahami D, Eden N, Zhang J, Feleke E, Dahan T, Cohen-Tayar Y, Stolovich-Rain M, Kaestner KH, Glaser B, Ashery-Padan R, Dor Y, 2017. PAX6 maintains β cell identity by repressing genes of alternative islet cell types. J. Clin. Invest. 127, 230–243. 10.1172/JCI88015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syrimis A, Nicolaou N, Alexandrou A, Papaevripidou I, Nicolaou M, Loukianou E, Christophidou-Anastasiadou V, Malas S, Sismani C, Tanteles GA, 2018. Aniridia due to a novel microdeletion affecting PAX6 regulatory enhancers: case report and review of the literature. J. Genet. 97, 555–562. [PubMed] [Google Scholar]
- Takamiya M, Stegmaier J, Kobitski AY, Schott B, Weger BD, Margariti D, Cereceda Delgado AR, Gourain V, Scherr T, Yang L, Sorge S, Otte JC, Hartmann V, van Wezel J, Stotzka R, Reinhard T, Schlunck G, Dickmeis T, Rastegar S, Mikut R, Nienhaus GU, Strähle U, 2020. Pax6 organizes the anterior eye segment by guiding two distinct neural crest waves. PLoS Genet. 16, e1008774 10.1371/journal.pgen.1008774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang HK, Chao LY, Saunders GF, 1997. Functional analysis of paired box missense mutations in the PAX6 gene. Hum. Mol. Genet. 6, 381–386. 10.1093/hmg/6.3.381. [DOI] [PubMed] [Google Scholar]
- Tang HK, Singh S, Saunders GF, 1998. Dissection of the transactivation function of the transcription factor encoded by the eye developmental gene PAX6. J. Biol. Chem. 273, 7210–7221. 10.1074/jbc.273.13.7210. [DOI] [PubMed] [Google Scholar]
- Thomas MG, Kumar A, Mohammad S, Proudlock FA, Engle EC, Andrews C, Chan W-M, Thomas S, Gottlob I, 2011. Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography a predictor of visual acuity? Ophthalmology 118, 1653–1660. 10.1016/j.ophtha.2011.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W, Zhu X-R, Qiao C-Y, Ma Y-N, Yang F-Y, Zhou Z, Feng J-P, Sun R, Xie R-R, Lu J, Cao X, Zhou J-B, Yang J-K, 2021. Heterozygous PAX6 mutations may lead to hyper-proinsulinaemia and glucose intolerance: a case-control study in families with congenital aniridia. Diabet. Med. 38, e14456 10.1111/dme.14456. [DOI] [PubMed] [Google Scholar]
- Ticho BH, Hilchie-Schmidt C, Egel RT, Traboulsi EI, Howarth RJ, Robinson D, 2006. Ocular findings in Gillespie-like syndrome: association with a new PAX6 mutation. Ophthalmic Genet. 27, 145–149. 10.1080/13816810600976897. [DOI] [PubMed] [Google Scholar]
- Tiller AM, Odenthal MTP, Verbraak FD, Gortzak-Moorstein N, 2003. The influence of keratoplasty on visual prognosis in aniridia: a historical review of one large family. Cornea 22, 105–110. 10.1097/00003226-200303000-00004. [DOI] [PubMed] [Google Scholar]
- Ton CC, Hirvonen H, Miwa H, Weil MM, Monaghan P, Jordan T, van Heyningen V, Hastie ND, Meijers-Heijboer H, Drechsler M, 1991. Positional cloning and characterization of a paired box- and homeobox-containing gene from the aniridia region. Cell 67, 1059–1074. 10.1016/0092-8674(91)90284-6. [DOI] [PubMed] [Google Scholar]
- Trainor PA, Tam PP, 1995. Cranial paraxial mesoderm and neural crest cells of the mouse embryo: co-distribution in the craniofacial mesenchyme but distinct segregation in branchial arches. Development 121, 2569–2582. 10.1242/dev.121.8.2569. [DOI] [PubMed] [Google Scholar]
- Trainor PA, Tan SS, Tam PP, 1994. Cranial paraxial mesoderm: regionalisation of cell fate and impact on craniofacial development in mouse embryos. Development 120, 2397–2408. 10.1242/dev.120.9.2397. [DOI] [PubMed] [Google Scholar]
- Tremblay F, Gupta SK, De Becker I, Guernsey DL, Neumann PE, 1998. Effects of PAX6 mutations on retinal function: an electroretinographic study. Am. J. Ophthalmol. 126, 211–218. 10.1016/s0002-9394(98)00190-1. [DOI] [PubMed] [Google Scholar]
- Tseng SCG, Prabhasawat P, Barton K, Gray T, Meiler D, 1998. Amniotic membrane transplantation with or without limbal allografts for corneal surface reconstruction in patients with limbal stem cell deficiency. Archieve Opthalmol. 116, 431–441. 10.1001/ARCHOPHT.116.4.431, 1960. [DOI] [PubMed] [Google Scholar]
- Tzoulaki I, White IMS, Hanson IM, 2005. PAX6 mutations: genotype-phenotype correlations. BMC Genet. 6, 27. 10.1186/1471-2156-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uka J, Minamoto A, Hirayama T, Adachi T, Yamane K, Mishima HK, 2005. Endoscope-aided cataract surgery in corneal opacity associated with aniridia. J. Cataract Refract. Surg. 31, 1455–1456. 10.1016/j.jcrs.2005.05.015. [DOI] [PubMed] [Google Scholar]
- Valleix S, Niel F, Nedelec B, Algros M-P, Schwartz C, Delbosc B, Delpech M, Kantelip B, 2006. Homozygous nonsense mutation in the FOXE3 gene as a cause of congenital primary aphakia in humans. Am. J. Hum. Genet. 79, 358–364. 10.1086/505654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer-de Jong R, Dickinson ME, Woychik RP, Stubbs L, Hetherington C, Hogan BL, 1990. Location of the gene involving the small eye mutation on mouse chromosome 2 suggests homology with human aniridia 2 (AN2). Genomics 7, 270–275. 10.1016/0888-7543(90)90550-e. [DOI] [PubMed] [Google Scholar]
- Van Hare GF, Perry J, Berul CI, Triedman JK, 2007. Cost effectiveness of neonatal ECG screening for the long QT syndrome. Eur. Heart J. 28, 137. 10.1093/eurheartj/ehl414 author reply137-139. [DOI] [PubMed] [Google Scholar]
- van Heyningen V, Williamson KA, 2002. PAX6 in sensory development. Hum. Mol. Genet. 11, 1161–1167. 10.1093/hmg/11.10.1161. [DOI] [PubMed] [Google Scholar]
- van Raamsdonk CD, Tilghman SM, 2000. Dosage requirement and allelic expression of PAX6 during lens placode formation. Development 127, 5439–5448. 10.1242/dev.127.24.5439. [DOI] [PubMed] [Google Scholar]
- Vance KW, Sansom SN, Lee S, Chalei V, Kong L, Cooper SE, Oliver PL, Ponting CP, 2014. The long non-coding RNA Paupar regulates the expression of both local and distal genes. EMBO J. 33, 296–311. 10.1002/embj.201386225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilyeva TA, Marakhonov AV, Voskresenskaya AA, Kadyshev VV, Käsmann-Kellner B, Sukhanova NV, Katargina LA, Kutsev SI, Zinchenko RA, 2021. Analysis of genotype-phenotype correlations in PAX6-associated aniridia. J. Med. Genet. 58, 270–274. 10.1136/jmedgenet-2019-106172. [DOI] [PubMed] [Google Scholar]
- Voskresenskaya A, Pozdeyeva N, Batkov Y, Vasilyeva T, Marakhonov A, West RA, Caplan JL, Cvekl A, Wang Y, Duncan MK, 2021. Morphometric analysis of the lens in human aniridia and mouse Small eye. Exp. Eye Res. 203, 108371 10.1016/j.exer.2020.108371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskresenskaya A, Pozdeyeva N, Vasilyeva T, Batkov Y, Shipunov A, Gagloev B, Zinchenko R, 2017. Clinical and morphological manifestations of aniridia-associated keratopathy on anterior segment optical coherence tomography and in vivo confocal microscopy. Ocul. Surf. 15, 759–769. 10.1016/j.jtos.2017.07.001. [DOI] [PubMed] [Google Scholar]
- Wagle NS, Freedman SF, Buckley EG, Davis JS, Biglan AW, 1998. Long-term outcome of cyclocryotherapy for refractory pediatric glaucoma. Ophthalmology 105, 1921. 10.1016/S0161-6420(98)91042-9. –1926; discussion 1926–1927. [DOI] [PubMed] [Google Scholar]
- Walker H, Akula M, West-Mays JA, 2020. Corneal development: role of the periocular mesenchyme and bi-directional signaling. Exp. Eye Res. 201, 108231 10.1016/j.exer.2020.108231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther C, Gruss P, 1991. Pax-6, a murine paired box gene, is expressed in the developing CNS. Development 113, 1435–1449. 10.1242/dev.113.4.1435. [DOI] [PubMed] [Google Scholar]
- Wang JD, Zhang JS, Xiong Y, Li J, Li XX, Liu X, Zhao J, Tsai FF, Vishal J, You QS, Huang Y, Wan XH, 2017. Congenital aniridia with cataract: case series. BMC Ophthalmol. 17, 115. 10.1186/s12886-017-0503-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Gregory-Evans K, Wasan KM, Sivak O, Shan X, Gregory-Evans CY, 2017a. Efficacy of postnatal in vivo nonsense suppression therapy in a Pax6 mouse model of aniridia. Mol. Ther. Nucleic Acids 7, 417–428. 10.1016/j.omtn.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Shan X, Gregory-Evans CY, 2017b. A mouse model of aniridia reveals the in vivo downstream targets of Pax6 driving iris and ciliary body development in the eye. Biochim. Biophys. Acta, Mol. Basis Dis. 1863, 60–67. 10.1016/j.bbadis.2016.10.018. [DOI] [PubMed] [Google Scholar]
- Wawrocka A, Krawczynski MR, 2018. The genetics of aniridia - simple things become complicated. J. Appl. Genet. 59, 151–159. 10.1007/s13353-017-0426-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawrocka A, Sikora A, Kuszel L, Krawczynski MR, 2013. 11p13 deletions can be more frequent than the PAX6 gene point mutations in Polish patients with aniridia. J. Appl. Genet. 54, 345–351. 10.1007/s13353-013-0154-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen JH, Chen YY, Song SJ, Ding J, Gao Y, Hu QK, Feng RP, Liu YZ, Ren GC, Zhang CY, Hong TP, Gao X, Li LS, 2009. Paired box 6 (PAX6) regulates glucose metabolism via proinsulin processing mediated by prohormone convertase 1/3 (PC1/3). Diabetologia 52, 504–513. 10.1007/s00125-008-1210-x. [DOI] [PubMed] [Google Scholar]
- Wiggins RE, Tomey KF, 1992. The results of glaucoma surgery in aniridia. Arch. Ophthalmol. 110, 503–505. 10.1001/archopht.1992.01080160081036. [DOI] [PubMed] [Google Scholar]
- Williams AL, Bohnsack BL, 2015. Neural crest derivatives in ocular development: discerning the eye of the storm. Birth Defects Res C Embryo Today. 105, 87–95. 10.1002/bdrc.21095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolosin JM, Budak MT, Akinci MAM, 2004. Ocular surface epithelial and stem cell development. Int. J. Dev. Biol. 48, 981–991. 10.1387/ijdb.041876jw. [DOI] [PubMed] [Google Scholar]
- Wu L, Ma Q, Chen Y, Wu DZ, Luo T, 1991. Abnormalities of ERG in congenital aniridia. Yan Ke Xue Bao 7, 151–152, 119. [PubMed] [Google Scholar]
- Xie Q, Cvekl A, 2011. The orchestration of mammalian tissue morphogenesis through a series of coherent feed-forward loops. J. Biol. Chem. 286, 43259–43271. 10.1074/jbc.M111.264580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HE, Rould MA, Xu W, Epstein JA, Maas RL, Pabo CO, 1999. Crystal structure of the human Pax6 paired domain-DNA complex reveals specific roles for the linker region and carboxy-terminal subdomain in DNA binding. Genes Dev. 13, 1263–1275. 10.1101/gad.13.10.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu PX, Zhang X, Heaney S, Yoon A, Michelson AM, Maas RL, 1999. Regulation of Pax6 expression is conserved between mice and flies. Development 126, 383–395. 10.1242/dev.126.2.383. [DOI] [PubMed] [Google Scholar]
- Xu S, Han JC, Morales A, Menzie CM, Williams K, Fan Y-S, 2008. Characterization of 11p14-p12 deletion in WAGR syndrome by array CGH for identifying genes contributing to mental retardation and autism. Cytogenet. Genome Res. 122, 181–187. 10.1159/000172086. [DOI] [PubMed] [Google Scholar]
- Xu ZP, Saunders GF, 1997. Transcriptional regulation of the human PAX6 gene promoter. J. Biol. Chem. 272, 3430–3436. 10.1074/jbc.272.6.3430. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Togawa M, Shimada S, Sangu N, Shimojima K, Okamoto N, 2014. Narrowing of the responsible region for severe developmental delay and autistic behaviors in WAGR syndrome down to 1.6 Mb including PAX6, WT1, and PRRG4. Am. J. Med. Genet. 164A, 634–638. 10.1002/ajmg.a.36325. [DOI] [PubMed] [Google Scholar]
- Yasuda T, Kajimoto Y, Fujitani Y, Watada H, Yamamoto S, Watarai T, Umayahara Y, Matsuhisa M, Gorogawa S, Kuwayama Y, Tano Y, Yamasaki Y, Hori M, 2002. PAX6 mutation as a genetic factor common to aniridia and glucose intolerance. Diabetes 51, 224–230. 10.2337/diabetes.51.1.224. [DOI] [PubMed] [Google Scholar]
- Yogarajah M, Matarin M, Vollmar C, Thompson PJ, Duncan JS, Symms M, Moore AT, Liu J, Thom M, van Heyningen V, Sisodiya SM, 2016. PAX6, brain structure and function in human adults: advanced MRI in aniridia. Ann Clin. Transl. Neurol. 3, 314–330. 10.1002/acn3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi T, Nishina S, Fukami M, Ogata T, Hosono K, Hotta Y, Azuma N, 2016. Genotype-phenotype correlation of PAX6 gene mutations in aniridia. Hum. Genome Var. 3, 15052 10.1038/hgv.2015.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yongblah K, Alford SC, Ryan BC, Chow RL, Howard PL, 2018. Protecting pax6 3’ UTR from MicroRNA-7 partially restores PAX6 in islets from an aniridia mouse model. Mol. Ther. Nucleic Acids 13, 144–153. 10.1016/j.omtn.2018.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You B, Zhang X, Xu K, Xie Y, Ye H, Li Y, 2020. Mutation spectrum of PAX6 and clinical findings in 95 Chinese patients with aniridia. Mol. Vis. 26, 226–234. [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Cveklova K, Oppermann B, Kantorow M, Cvekl A, 2001. Quantitation of PAX6 and PAX6(5a) transcript levels in adult human lens, cornea, and monkey retina. Mol. Vis. 7, 1–5. [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Qin G, Chen G, Li T, Gao L, Huang L, Zhang Y, Ouyang K, Wang Y, Pang Y, Zeng B, Yu L, 2015. Variants in TRIM44 cause aniridia by impairing PAX6 expression. Hum. Mutat. 36, 1164–1167. 10.1002/humu.22907. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang Q, Tong Y, Dai H, Zhao X, Bai F, Xu L, Li Y, 2011. Large novel deletions detected in Chinese families with aniridia: correlation between genotype and phenotype. Mol. Vis. 17, 548–557. [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Yang T, Madakashira BP, Thiels CA, Bechtle CA, Garcia CM, Zhang H, Yu K, Ornitz DM, Beebe DC, Robinson ML, 2008. Fibroblast growth factor receptor signaling is essential for lens fiber cell differentiation. Dev. Biol. 318, 276–288. 10.1016/j.ydbio.2008.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Ley SH, Hu FB, 2018. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 14, 88–98. 10.1038/nrendo.2017.151. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available on request.