

Abstract
A growing body of literature suggests that alterations in the human microbiome are causative of disease initiation and progression. Aykut et al1. present data supporting the argument that alterations in the gut fungal microbiome (the “mycobiome”), along with the presence of fungal elements within pancreatic tissue (specifically those of the genus Malassezia), are associated with pancreatic oncogenesis. Upon analyzing the human sequencing data presented in the original manuscript, we found few fungal reads in pancreatic tissue samples and did not identify differences in pancreatic or gut mycobiome composition between healthy and pancreatic ductal adenocarcinoma (PDAC) patients. Our re-analysis of these data does not support an association between an intrinsic pancreatic mycobiome and the development of human PDAC, and illustrates the challenges in analyzing microbiome sequencing data from low biomass samples.
Using sequence analysis of the internal transcribed spacer (ITS) region of fungi, Aykut et al. characterized the mycobiome of 5 normal human pancreatic tissue samples, 13 PDAC tissue samples, and 18 fecal samples from patients with PDAC. They also sequenced 9 fecal samples from healthy patients, although these data were not presented in the manuscript. Focusing only on the analysis of these human samples, we retrieved the raw sequencing data that were publicly available through the National Center for Biotechnology Information’s (NCBI) Sequence Read Archive (SRA) to confirm the findings from the manuscript and to compare the gut and pancreatic mycobiomes of the healthy and PDAC patients.
Over the past decade, a number of bioinformatic pipelines have become available for the analysis of amplicon sequencing data. The pipeline we initially used to analyze the sequencing files employed DADA2, an open-source software package for R2. DADA2 infers differences in sample sequences exactly (down to a single nucleotide) to generate amplicon sequence variants (ASVs). In contrast, the pipeline utilized by Aykut et al. used an open-reference operational taxonomic unit (OTU) picking approach implemented in QIIME13. Prior studies suggest that the QIIME1 OTU picking strategy used in this manuscript has a relatively high rate of false taxonomic classification and tends to overestimate microbial diversity4,5. Recently published data suggest that DADA2 is a more accurate and reproducible method for analyzing amplicon sequencing data than an OTU picking strategy6–8.
When we analyzed the pancreatic tissue samples using our DADA2 pipeline, we found the raw sequencing data from the Aykut experiments to have a mean quality score of ≤ 30 in the majority of reads (Extended Data Fig. 1a), and we did not identify an abundant fungal presence within normal or malignant pancreatic tissues (Extended Data Fig. 1b). In our analysis of these data, we identified only 17 reads assigned to Malasezzia spp., all of which were found in a single PDAC sample (Extended Data Fig. 1c–e).
Due to our inability to replicate the findings from the Aykut et al. manuscript using a DADA2 pipeline, we next sought to analyze the human pancreatic tissue sequencing data as described in the original manuscript. As QIIME1 has been succeeded by QIIME2 and is no longer supported, we utilized QIIME2 for our re-analysis but kept all parameters the same as reported in Aykut et al. for OTU clustering, chimera removal, and OTU filtering. We again found very few fungal reads in the pancreatic tissue samples. Specifically, we identified a total of 25 fungal reads in the 5 normal pancreatic samples, and a total of 116 reads in the 13 pancreatic samples from patients with PDAC (Fig. 1a). Among the fungal reads identified, only 19 reads were assigned to the genus Malassezia (6 reads in 2 of the normal pancreas samples and 13 reads in 7 of the PDAC samples; Fig. 1b). There were no significant differences in mycobiome composition or diversity by patient group (Fig. 1c–d). Moreover, while Malassezia was among the most abundant genera in these samples, this genus was inconsistently present in both normal and PDAC pancreas tissues (Fig. 1e).
Fig. 1 |. QIIME2 re-analysis of human pancreatic ITS sequencing data made publicly available by Aykut et al.
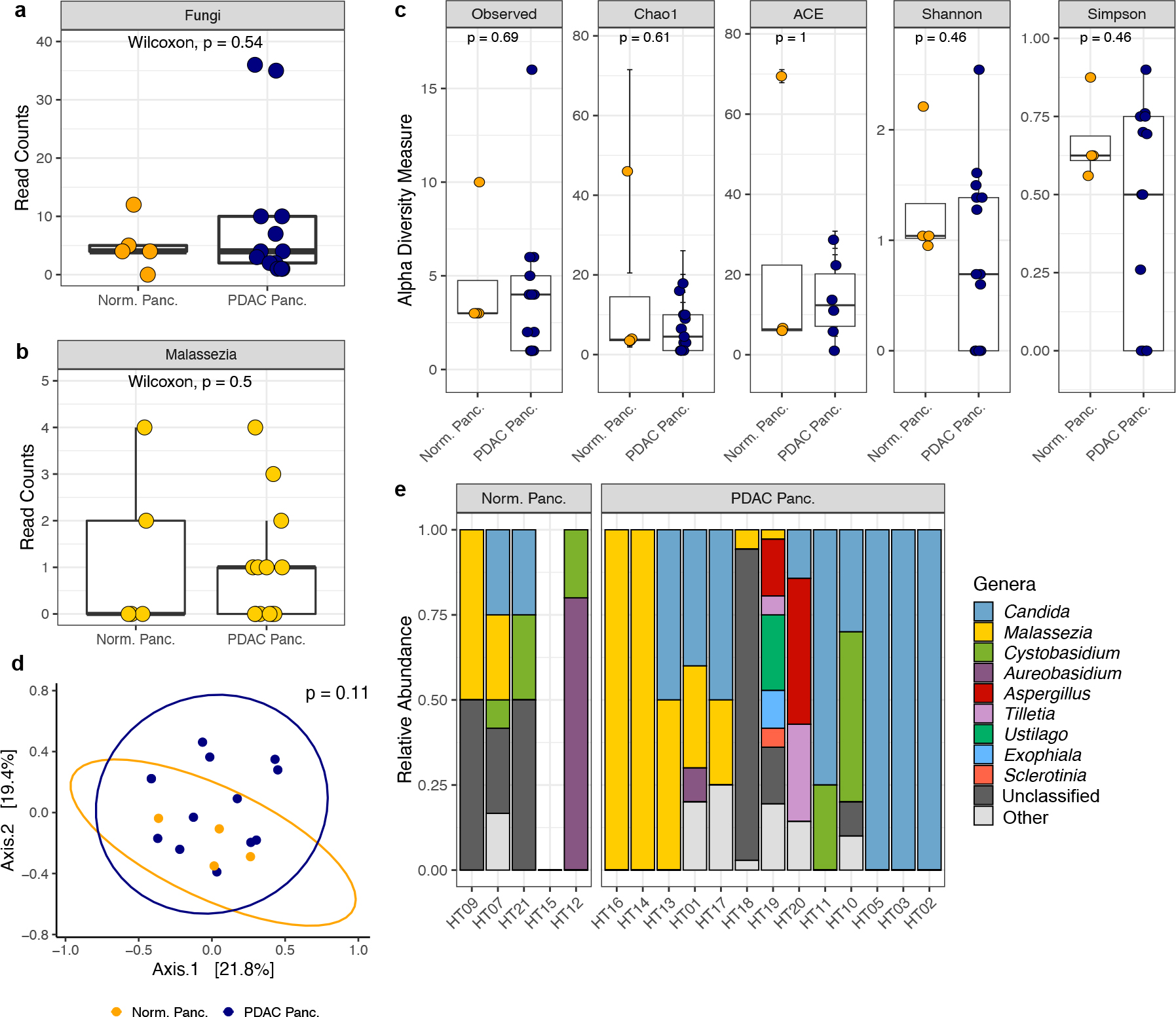
a, Box plots depicting total fungal reads in normal pancreas tissue (n=5 biologically independent samples) and pancreatic ductal adenocarcinoma (PDAC) tissue (n=13 biologically independent samples). b, Box plots depicting sequencing reads assigned to the fungal genus Malassezia in normal pancreas tissue (n=5 biologically independent samples) and PDAC tissue (n=13 biologically independent samples). c, Alpha diversity measures of normal pancreas tissue (n=4 biologically independent samples) and PDAC tissue (n=13 biologically independent samples). Diversity measures shown are the number of observed taxa, the Chao1 index, abundance-based coverage estimates (ACE), and the Shannon and Simpson’s indices. d, Non-metric multi-dimensional scaling plot of normal pancreas tissue (n=4 biologically independent samples) and PDAC tissue (n=13 biologically independent samples) fungal communities, based on Bray–Curtis dissimilarity. e, Relative abundances of the top ten fungal genera identified in normal and PDAC pancreatic tissue samples. Box plot minima and maxima bounds represent the 25th and 75th percentiles, respectively; the centre bound represents the median (a, b, c). Whiskers extend to 1.5 times the interquartile range (IQR) (a, b), and data in c are presented as mean ± SEM after a single sample with zero fungal reads was dropped from the analysis. P values were estimated using two-sided Wilcoxon rank-sum tests (a, b, c) or two-way PERMANOVA (d). Individual data points are shown.
To further explore our findings, we had a separate analysis performed on the sequencing data from the pancreatic tissue samples by the Duke Genomic Analysis and Bioinformatics Core. Using a DADA2 pipeline similar to ours, they also did not identify a significant fungal presence in pancreatic tissues, with only 16 reads assigned to Malasezzia spp., all of which were found in a single PDAC sample (Extended Data Fig. 2a–d). Therefore, given the similar results from multiple approaches to the analysis of these sequencing data, we believe that the fungal sequencing reads generated from the human pancreatic tissues in this study are insufficient for analyses of mycobiome diversity and composition and do not support the conclusion that there is an increased presence of Malasezzia in tissue from human PDAC tumors.
Next, we sought to compare the composition and diversity of the gut mycobiome between healthy and PDAC patients using the sequencing data available from the Aykut et al. study. Fecal samples from both patient populations were sequenced and deposited into the SRA by Aykut et al., although analyses comparing the gut mycobiomes of healthy and PDAC patients were not presented in the original article. However, to maintain consistency with other analyses reported by Aykut et al. in this manuscript, we analyzed these sequencing data using the aforementioned QIIME2 pipeline. We found fewer observed taxa in the gut mycobiomes of PDAC patients compared to healthy individuals (median: 6 vs. 10 OTUs; p = 0.02), but there were no other significant differences in analyses comparing the gut mycobiomes of these patient populations (Fig. 2a–e).
Fig. 2 |. QIIME2 re-analysis of human fecal ITS sequencing data made publicly available by Aykut et al.
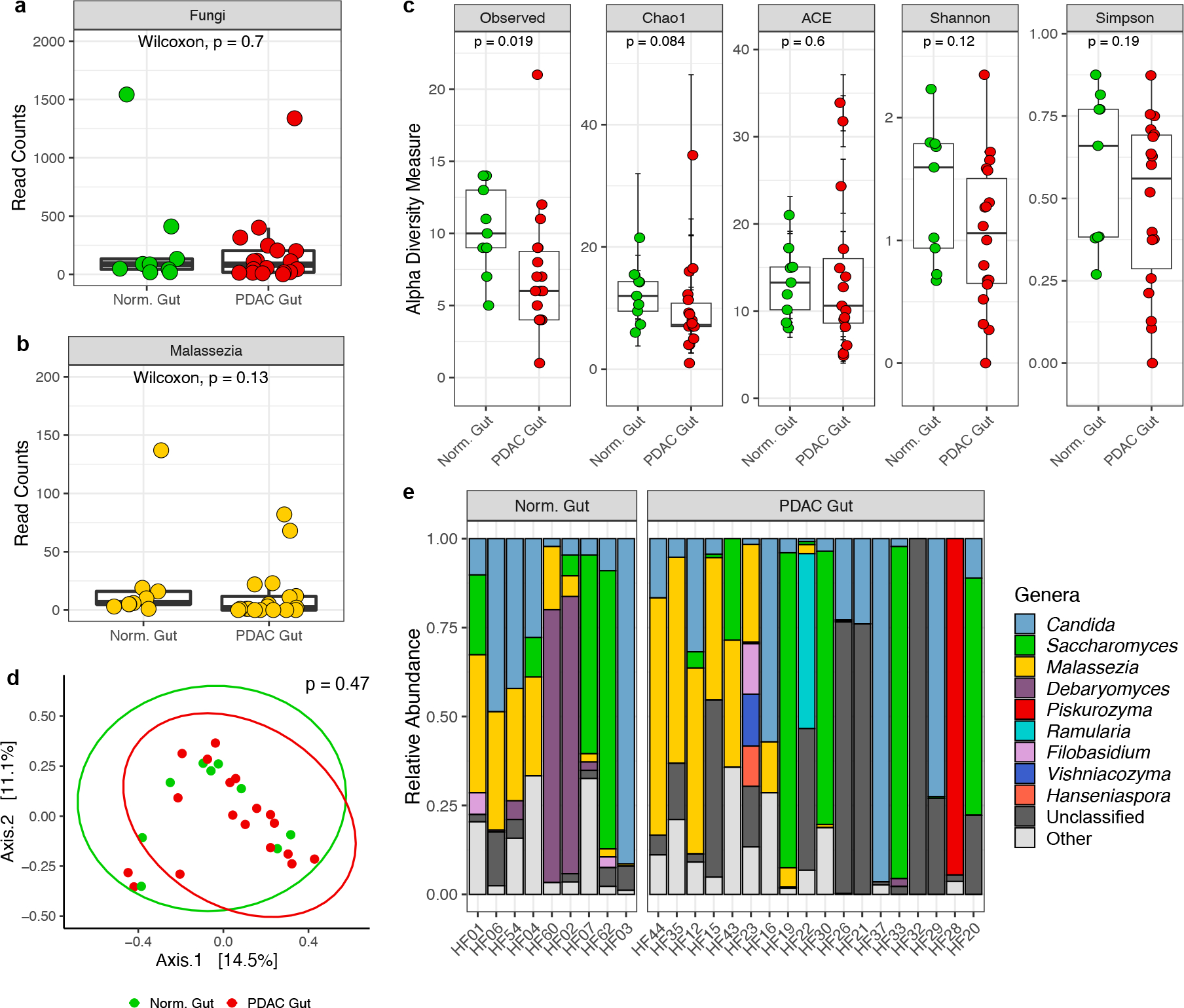
a, Box plots depicting fungal reads in normal gut (n=9 biologically independent samples) and pancreatic ductal adenocarcinoma (PDAC) gut (n=18 biologically independent samples). b, Box plots depicting sequencing reads assigned to the fungal genus Malassezia in normal gut (n=9 biologically independent samples) and PDAC gut (n=18 biologically independent samples). c, Alpha diversity measures of normal gut (n=9 biologically independent samples) and PDAC gut (n=18 biologically independent samples), including the number of observed taxa, the Chao1 index, abundance-based coverage estimates (ACE), and the Shannon and Simpson’s indices. d, Non-metric multi-dimensional scaling plot of normal gut (n=9 biologically independent samples) and PDAC gut (n=18 biologically independent samples), based on Bray–Curtis dissimilarity. e, Relative abundances of the top ten fungal genera in fecal samples from healthy individuals and patients with PDAC. Box plot minima and maxima bounds represent the 25th and 75th percentiles, respectively; the centre bound represents the median. Individual (a, b, c). Whiskers extend to 1.5 times the interquartile range (IQR) (a, b), and data in c are presented as mean ± SEM. P values were estimated using two-sided Wilcoxon rank-sum tests (a, b, c) or two-way PERMANOVA (d). Individual data points are shown.
Finally, we believe our findings highlight the challenges of using low biomass samples for microbiome sequencing studies. Human tissues are known to have a low microbial burden, and investigating the microbiome of these samples creates the challenge of discriminating between low biomass microbial communities and microbial DNA contamination that can be introduced during sample collection and processing, DNA extraction, and library preparation9. The inclusion of appropriate negative controls and efforts to identify and remove sequencing contaminants is critical to the interpretation of microbiome data from low biomass samples10.
In conclusion, although Aykut et al. showed fungal dysbiosis in the pancreatic tissues and guts of wild-type and PDAC mice, our analyses did not identify similar differences within human pancreatic tissues or fecal samples. Thus, we believe there is currently insufficient evidence to support the hypothesis that the pancreatic or gut mycobiome promotes pancreatic oncogenesis in humans. Our findings emphasize the need for standardized methods for generating and analyzing microbiome sequencing data, especially data generated from low biomass samples, to improve the reproducibility of results across studies.
Methods
QIIME2 bioinformatics and data analysis of Aykut et al. ITS sequences
The raw, demultiplexed sequencing reads from this manuscript were downloaded from the NCBI BioProject database (PRJNA557226). Consistent with the original manuscript, the forward (R1) reads were processed using a QIIME2 (v2019.10.0) pipeline based on the following methods and parameters reported by Aykut et al. Using Cutadapt (v2.8), primers were verified and removed and sequences shorter than 100 bases were discarded. These reads were imported into QIIME2 and quality filtered at a Phred score of 20, using qiime quality-filter q-score (--p-min-quality 19). Defaults were used for all other parameters. The 1,836,920 quality-filtered reads (mean reads per sample: 11,339; number of samples: 162) were then de-replicated using qiime vsearch dereplicate-sequences and chimeric sequences were removed using VSEARCH (v2.8) with the UNITE UCHIME reference dataset (v7.2). OTUs were clustered with qiime vsearch cluster-features-open-reference (per QIIME’s workflow, this approach “is synonymous to using split_libraries*.py commands in QIIME1”) at 97% identity match to the UNITE reference database (v7.2). There were 66,928 OTUs, corresponding to 1,773,779 reads (96.6% of the total reads), that did not align to fungi; these OTUs were excluded from downstream analyses. All fungal OTUs that were unidentified were blasted against NCBI’s ITS database (blastn v2.7.1), and taxonomy was reassigned with the best hit producing ≥ 97% identity or query coverage. A total of 59,686 sequence reads were clustered into 558 OTUs for mouse fecal samples (52,312 reads), 69 OTUs for mouse pancreatic tissue samples (1,413 reads), 114 OTUs for human fecal samples (5,790 reads), and 44 OTUs for human pancreatic tissue samples (171 reads). Low-abundance OTUs in fewer than two samples were removed as described in the original manuscript. Statistical analyses were performed in R (v4.0.5). Mycobiome alpha diversity measures were estimated using the R package Phyloseq (v1.34.0). Non-metric multi-dimensional scaling plots were generated based on Bray–Curtis dissimilarity. Wilcoxon rank-sum tests and permutational multivariate analysis of variance (PERMANOVA; Vegan v2.5–7) were used to evaluate for differences across patient groups11,12. P < 0.05 was considered to be statistically significant.
Fletcher et al. DADA2 analysis of Aykut et al. human ITS data
Demultiplexed ITS sequences (forward or R1 reads only) generated by Illumina were processed using a DADA2 (v1.14.0) pipeline. Raw reads were imported into DADA2 and dereplicated. The “plotQualityProfile” tool from DADA2 was used to determine the median base quality for each position of the reads for each region. Based on this profile, reads were truncated after 150 bases. Reads with an expected error rate higher than 5 (maxEE=5) were removed. All other parameters were set to default. Representative sequences from each ITS region were aligned against the UNITE database (v7.2) within DADA2 for taxonomical annotation using assignTaxonomy(). Sequences not found within the UNITE database were blasted against the Blast ITS_RefSeq_Fungi database (blastn v2.7.1, blastdb v5) and reassigned if the percent identity or query coverage was ≥ 97%. Analyses were performed in RStudio (v4.0.5) using the Phyloseq (v1.34.0), Microbiomeutilities (v1.0.16), and Microbiome (v1.14.0) packages.
Duke Genomic Analysis and Bioinformatics Core analysis of Aykut et al. human ITS data
The fastx_quality_stats tool from Fastx-toolkit (v0.0.14) was used to determine median base quality for each position of the forward reads (R1) for each region. Reads were imported into qiime2 (v2019.10.0) and denoised and dereplicated with DADA2 (q2-dada2 v2019.10.0). In DADA2, reads were trimmed at the beginning or truncated at the end if the median base quality fell below a score of 30 as determined by Fastx-toolkit. Representative sequences from the ITS region were blasted against the UNITE database for all eukaryotes (v8.0) with blastn using the megablast algorithm (blastv 2.9.0, blastdb v5). To assign taxonomy to sequences not found within the UNITE database, representative sequences were blasted to the blast nt database (blastn v2.7.1, blastbd v5) using both the megablast and discontinuous megablast algorithm. Demultiplexing and blast results were aggregated in the R statistical programming environment (v4.0.5).
Statistics and Reproducibility
The raw sequences from this manuscript were analyzed by two independent laboratories using QIIME2 and DADA2 using various quality filtering parameters and taxonomy assignment algorithms. Each independent analysis yielded similar results.
Extended Data
Extended Data Fig. 1 |. DADA2 analysis by Fletcher et al. of ITS sequencing data from Aykut et al. human pancreatic tissue samples.
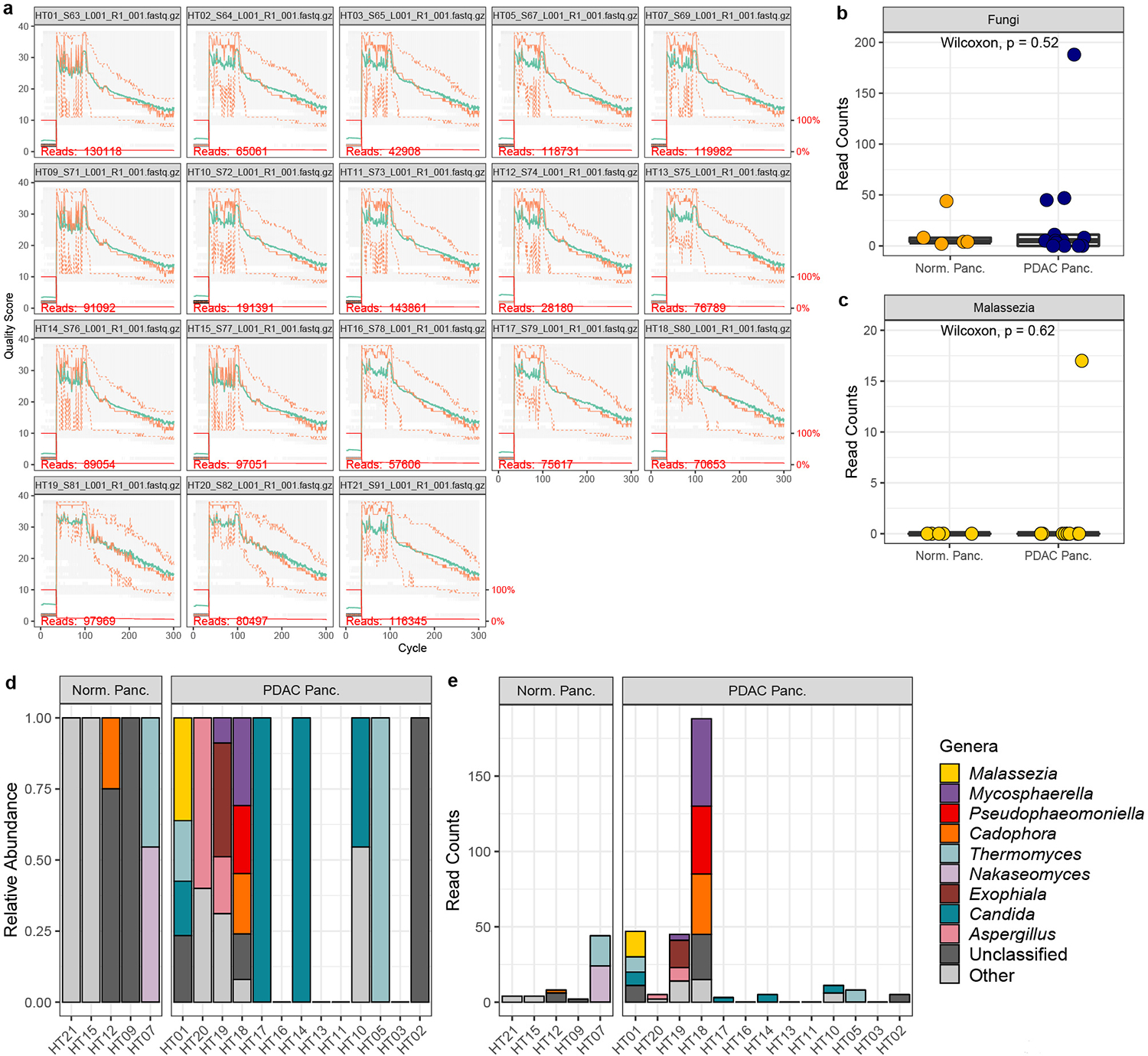
a, Quality plot of raw sequencing reads. The y-axis represents the Phred quality score and the x-axis represents the cycle, which corresponds to the base position of sequencing reads. The mean quality score at each base position is shown by a green line and the quartiles of the quality score distribution are shown by orange lines. The number of sequencing reads in each sample is shown in red font. The red line shows the scaled proportion of reads that extend to at least that position. b, Box plots depicting fungal reads in normal pancreas tissue (n=5 biologically independent samples) and pancreatic ductal adenocarcinoma (PDAC) tissue (n=13 biologically independent samples) c, Box plots depicting sequencing reads assigned to the fungal genus Malassezia in normal pancreas (n=5 biologically independent samples) and PDAC tissue (n=13 biologically independent samples). d, Relative abundances, and e, read counts of the top ten fungal genera in pancreatic tissue samples from healthy individuals and patients with PDAC. Box plot minima and maxima bounds represent the 25th and 75th percentiles, respectively; the centre bound represents the median. Whiskers extend to 1.5 times the interquartile range (IQR). P values were estimated using two-sided Wilcoxon rank-sum tests (b, c). Individual data points are shown.
Extended Data Fig. 2 |. DADA2 analysis by Duke Genomic Analysis and Bioinformatics Core of ITS sequencing data from Aykut et al. human pancreatic tissue samples.
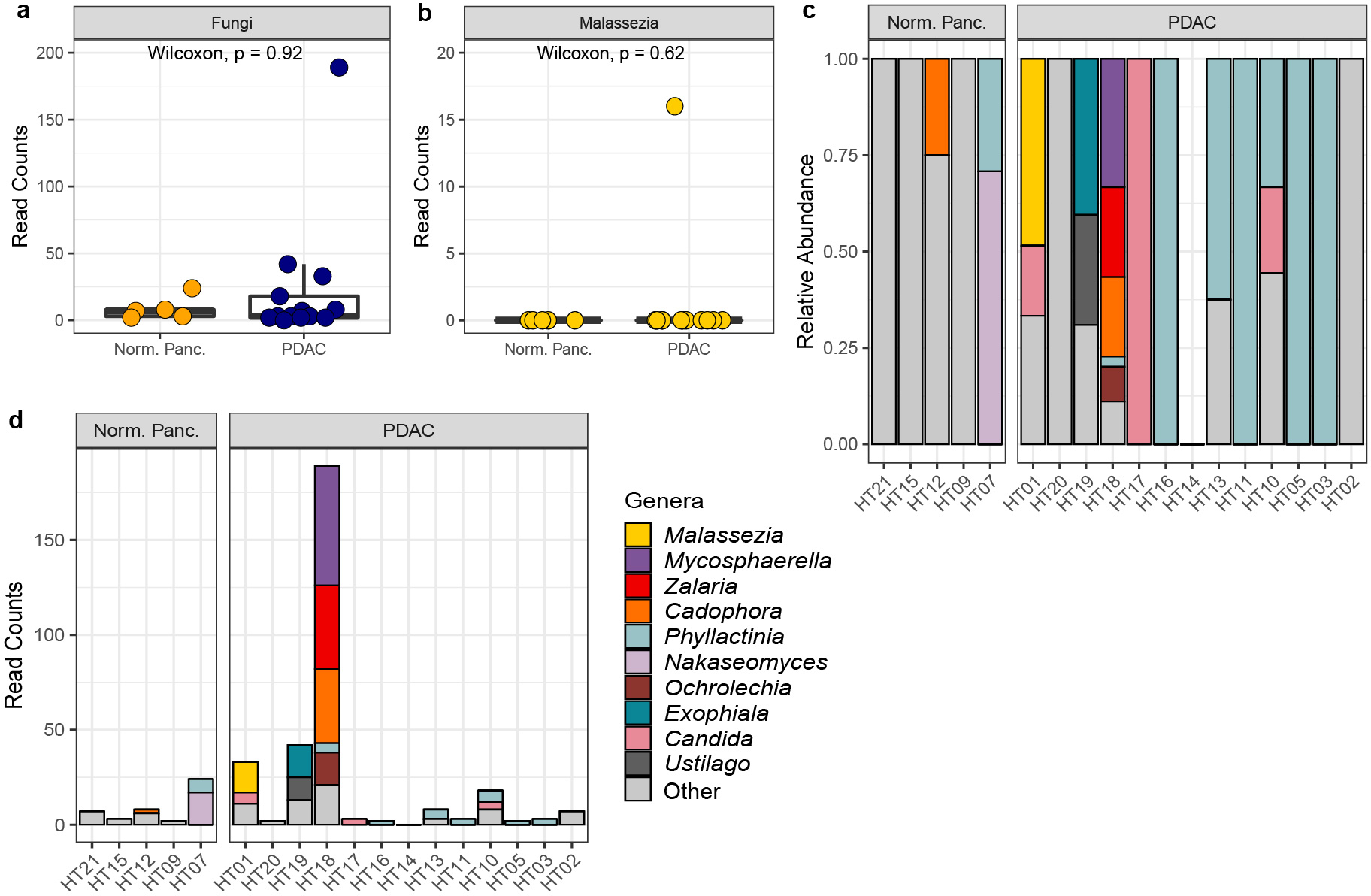
a, Box plots depicting fungal reads in normal pancreas tissue (n=5 biologically independent samples) and pancreatic ductal adenocarcinoma (PDAC) tissue (n=13 biologically independent samples) b, Box plots depicting sequencing reads assigned to the fungal genus Malassezia in normal pancreas tissue (n=5 biologically independent samples) and PDAC tissue (n=13 biologically independent samples). c, Relative abundances, and d, read counts of the top ten fungal genera in pancreatic tissue samples from healthy individuals and patients with PDAC. Box plot minima and maxima bounds represent the 25th and 75th percentiles, respectively; the centre bound represents the median. Whiskers extend to 1.5 times the interquartile range (IQR). P values were estimated using two-sided Wilcoxon rank-sum tests (a, b). Individual data points are shown.
Acknowledgements
We thank the Duke University School of Medicine for use of the Sequencing and Genomic Technologies Shared Resource, which provided services for sequence analysis. We also thank A. Cosentino for providing graphical consultation and color palette development.
Funding
This work was funded by the Duke University School of Medicine through a grant from the Duke Microbiome Center. Dr. Kelly was supported by a National Institutes of Health Career Development Award (K23-AI135090). Dr. Eckhoff was supported by a National Institutes of Health T-32 Grant (T32-CA093245) for translational research in surgical oncology.
Footnotes
Competing interests
The authors declare no competing interests.
Code availability
The scripts for the QIIME2 and DADA2 pipelines and downstream analyses performed in R are available in a public GitHub repository (https://github.com/afletch00/Fungi-Nature-2022).
Reprints and permissions information is available at www.nature.com/reprints
Data availability
The sequencing dataset generated in the experiments conducted by Aykut et al. is available in the NCBI Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra; PRJNA557226). Raw count and taxonomy tables generated by QIIME2 and DADA2 as part of our re-analysis of these original sequencing data are available in a public GitHub repository (https://github.com/afletch00/Fungi-Nature-2022).
References
- 1.Aykut B et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 574, 264–267, doi: 10.1038/s41586-019-1608-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Callahan BJ et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13, 581–583, doi: 10.1038/nmeth.3869 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caporaso JG et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336, doi: 10.1038/nmeth.f.303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prodan A et al. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS One 15, e0227434, doi: 10.1371/journal.pone.0227434 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edgar RC Accuracy of microbial community diversity estimated by closed- and open-reference OTUs. PeerJ 5, e3889, doi: 10.7717/peerj.3889 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Callahan BJ, McMurdie PJ & Holmes SP Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J 11, 2639–2643, doi: 10.1038/ismej.2017.119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caruso V, Song X, Asquith M & Karstens L Performance of Microbiome Sequence Inference Methods in Environments with Varying Biomass. mSystems 4, doi: 10.1128/mSystems.00163-18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Narayan NR et al. Piphillin predicts metagenomic composition and dynamics from DADA2-corrected 16S rDNA sequences. BMC Genomics 21, 56, doi: 10.1186/s12864-019-6427-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Selway CA, Eisenhofer R & Weyrich LS Microbiome applications for pathology: challenges of low microbial biomass samples during diagnostic testing. J Pathol Clin Res 6, 97–106, doi: 10.1002/cjp2.151 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eisenhofer R et al. Contamination in Low Microbial Biomass Microbiome Studies: Issues and Recommendations. Trends Microbiol 27, 105–117, doi: 10.1016/j.tim.2018.11.003 (2019). [DOI] [PubMed] [Google Scholar]
- 11.McMurdie PJ & Holmes S phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217, doi: 10.1371/journal.pone.0061217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dixon P VEGAN, a package of R functions for community ecology. J Veg Sci 14, 927–930, doi:DOI 10.1111/j.1654-1103.2003.tb02228.x (2003). [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The sequencing dataset generated in the experiments conducted by Aykut et al. is available in the NCBI Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra; PRJNA557226). Raw count and taxonomy tables generated by QIIME2 and DADA2 as part of our re-analysis of these original sequencing data are available in a public GitHub repository (https://github.com/afletch00/Fungi-Nature-2022).