

Abstract
INTRODUCTION:
Ulcerative colitis (UC) is a chronic condition that may require long-term treatment. We report the final efficacy and safety results of the UNIFI long-term extension study of ustekinumab in patients with UC through 4 years.
METHODS:
Ustekinumab induction responders who completed 44 weeks of maintenance treatment and agreed to enter the long-term extension continued their subcutaneous maintenance therapy (90 mg ustekinumab every 8 or 12 weeks [q8w or q12w] or placebo). Starting at week 56, randomized patients could receive dose adjustment to 90 mg q8w. Symptoms and adverse events were assessed through the study; endoscopic assessment was conducted at week 200.
RESULTS:
Of the 348 patients randomized to subcutaneous ustekinumab at maintenance baseline (q8w and q12w combined), 55.2% were in symptomatic remission at week 200. A greater proportion of biologic-naive patients (67.2% [117/174]) were in symptomatic remission than those with a history of biologic failure (41.6% [67/161]). Among patients in symptomatic remission at week 200, 96.4% were corticosteroid-free. Of the 171 patients with endoscopic evaluation at week 200, 81.6% (71/87) in the q12w group and 79.8% (67/84) in the q8w group had endoscopic improvement. From weeks 156 to the final safety visit (up to week 220), no deaths, major adverse cardiovascular events, or tuberculosis occurred in patients receiving ustekinumab. Nasopharyngitis, UC worsening, and upper respiratory tract infections were the most frequently reported adverse events.
DISCUSSION:
The long-term efficacy of ustekinumab maintenance in patients with UC was confirmed through 4 years. No new safety signals were observed. ClinicalTrials.gov number NCT02407236.
KEYWORDS: endoscopic improvement, long-term extension, symptomatic remission, ulcerative colitis, ustekinumab

INTRODUCTION
Ulcerative colitis (UC) is a chronic, immune-mediated disorder characterized by inflammation of the colon. Due to the lifelong nature of UC, long-term treatment is often required to induce and maintain remission. Advances in biologic therapies have allowed a paradigm shift in treatment goals from controlling symptoms to endoscopic healing, thus improving long-term outcomes (1).
Ustekinumab, a fully human interleukin-12/23p40 inhibitor, has shown to be safe and effective for maintaining remission through 3 years of subcutaneous (SC) maintenance therapy after intravenous (IV) induction in patients with moderate to severely active UC (2,3). The UNIFI study consisted of an induction study, a maintenance study (4), and a long-term extension (LTE) through 4 years.
In this report, the final efficacy and safety results of the UNIFI LTE study through 4 years are presented. Of note, endoscopic assessments were performed at the final efficacy evaluation, which enables more objective evaluation of long-term efficacy in addition to patient-reported symptoms.
METHODS
Study design
The UNIFI study consisted of 2 randomized, double-blind, placebo-controlled studies: an 8-week induction study and a 44-week maintenance study, previously described (4). The LTE began after week 44 of maintenance (2,3) through week 220 (final safety visit), with a final efficacy visit at week 200. The study design (see Supplementary Figure S1, http://links.lww.com/AJG/D138) has been previously reported (2–4).
Overall, 523 IV ustekinumab induction responders were randomized to SC maintenance therapy (intent-to-treat [ITT] population). Patients not in full Mayo clinical response 8 weeks after IV ustekinumab induction received SC ustekinumab 90 mg at week 8, and those in full Mayo clinical response 16 weeks after the initial IV ustekinumab infusion were followed up in the nonrandomized population of the maintenance study and received ustekinumab 90 mg q8w. All patients completing week 44 of the maintenance study were eligible to continue treatment in the LTE. The maintenance study was unblinded after analysis of the week-44 end points. Patients receiving SC placebo at the time of study unblinding were discontinued.
At week 56, randomized patients whose UC worsened based on investigator's judgment could adjust to ustekinumab 90 mg q8w; dose adjustment was allowed once. The last dose adjustment was at week 188.
Assessments
Symptom and physician global assessments were conducted every 12 weeks until study unblinding and then every 8 or 12 weeks depending on assigned dose regimen. Patients who remained in the trial underwent an endoscopic assessment (either sigmoidoscopy or colonoscopy) at week 200, and the endoscopic evaluation was performed by a local reader (not by central review as during the main study).
C-reactive protein (CRP) and fecal calprotectin were assessed every 3 months in the LTE through week 200. Inflammatory Bowel Disease Questionnaire (IBDQ) was used to assess disease-specific health-related quality of life across 4 dimensional scores: bowel, systemic, social, and emotional (5). Use of the IBDQ was made under license from McMaster University, Hamilton, Canada. The IBDQ was assessed every 6 months in the LTE through week 200.
Efficacy end points
Symptomatic remission was defined as a Mayo stool frequency subscore of 0 or 1 and a rectal bleeding subscore of 0 and was evaluated at each visit through week 200. Corticosteroid-free symptomatic remission was defined as patients in symptomatic remission and not receiving oral corticosteroids (including beclomethasone dipropionate and budesonide) at the time point (visit) of the efficacy assessment.
Efficacy end points related to the Mayo score were evaluated at week 200. Full Mayo clinical remission was defined as a Mayo score ≤2 points, with no individual subscore >1. Full Mayo clinical response was defined as a decrease from induction baseline in the Mayo score by ≥30% and ≥3 points, with either a decrease from induction baseline in the rectal bleeding subscore ≥1 or a rectal bleeding subscore of 0 or 1. Modified Mayo response was defined as a decrease from induction baseline in the modified Mayo score (no physician global assessment subscore) by ≥30% and ≥2 points, with either a decrease from induction baseline in the rectal bleeding subscore ≥1 or a rectal bleeding subscore of 0 or 1. Endoscopic improvement was defined as an endoscopy subscore (of the Mayo score) of 0 or 1. Median CRP and fecal calprotectin concentrations and IBDQ remission (IBDQ score ≥170 points) were assessed through week 200.
Safety
Adverse events (AE), serious AE (SAE), infections, serious infections, and laboratory assessments were evaluated during the LTE (week 44 through the final safety visit, which was week 220 or the discontinuation visit if the patient discontinued before week 220). Patients randomized to placebo could dose adjust to ustekinumab 90 mg q8w in the LTE before study unblinding, and any AE reported after dose adjustment were included in the ustekinumab group.
Immunogenicity
Serum blood samples for immunogenicity assessments were collected every 6 months during the LTE. Antibodies to ustekinumab were detected using a validated, drug-tolerant, electrochemiluminescent immunoassay on the MesoScale Discovery platform. This assay can detect antidrug antibodies (ADA) in the presence of up to 100 mg/mL of ustekinumab in the sample (6,7). Patients were classified as positive if ADA were detected at any time after the first ustekinumab administration of the induction study through the end of the study.
Statistical analysis
The efficacy analysis populations included the following: (i) all patients randomized to ustekinumab in the maintenance study; (ii) randomized patients in the maintenance study who continued to receive ustekinumab in the LTE; and (iii) nonrandomized patients in the maintenance study who received ustekinumab in the LTE (see Supplementary Table S1, http://links.lww.com/AJG/D138). No statistical comparisons were made between treatment groups.
For the ITT analyses of symptomatic remission and corticosteroid-free symptomatic remission, all patients randomized to ustekinumab (q12w or q8w) in the maintenance study were included, regardless of whether patients entered the LTE. Patients who met treatment failure criteria were considered nonresponders (see Supplementary Table S1, http://links.lww.com/AJG/D138). Patients with missing data pertaining to a dichotomous end point at a visit were considered not to have achieved the dichotomous end point, including those who did not enter the LTE.
Randomized patients who entered the LTE were evaluated for symptomatic remission, stool frequency, rectal bleeding, corticosteroid use, outcomes associated with endoscopic evaluation at week 200 (full Mayo clinical remission, full Mayo clinical response, modified Mayo clinical response, and endoscopic improvement), inflammatory biomarkers (CRP and fecal calprotectin), and IBDQ remission. Symptomatic remission among patients who entered the LTE was evaluated using 3 approaches: (i) nonresponder imputation for treatment failure and missing data; (ii) observed case analysis only for patients with available data at an analysis visit; and (iii) modified observed case analysis up to the time of dose adjustment with nonresponder imputation for patients who met treatment failure criteria (see Supplementary Table S1, http://links.lww.com/AJG/D138). Other dichotomous end points, including proportions of patients with ≤3 stools/day, Mayo stool frequency subscores of 0 or 1, rectal bleeding subscores of 0 (see Supplementary Table S2, http://links.lww.com/AJG/D138), and IBDQ remission, were conservatively analyzed using nonresponder imputation for treatment failure and missing data. For continuous end points, including absolute number of stools/day, daily corticosteroid dose, CRP, and fecal calprotectin, patients who met treatment failure criteria had their week-0 (induction) value carried forward to the visits thereafter and patients who had missing data at a visit had their last available value carried forward to that visit. Symptomatic remission was also evaluated for nonrandomized patients who entered the LTE with nonresponder imputation for treatment failure and missing data.
Safety was evaluated using number of AE, SAE, infections, serious infections, AE leading to discontinuation, malignancies, and deaths per 100 patient-years (PY) of follow-up for all patients treated with ustekinumab in the LTE (randomized and nonrandomized). Event rates per 100 PY from week 44 through the final LTE safety visit were summarized. Event rates were also summarized for each year of the maintenance study (1st year: weeks 0–44; 2nd year: LTE weeks 44–96; 3rd year: LTE weeks 96–156; and 4th year: LTE weeks 156–220). All authors had access to the data and have reviewed/approved the final manuscript.
RESULTS
Patient disposition
At maintenance baseline, 523 IV ustekinumab induction responders were randomized to SC maintenance therapy: SC placebo, n = 175; ustekinumab 90 mg q12w, n = 172; and ustekinumab 90 mg q8w, n = 176. Of these patients, 399 (76.3%) continued treatment in the LTE, including 284 patients who continued ustekinumab treatment (n = 141 q12w; n = 143 q8w). LTE baseline clinical disease characteristics were previously described (2), including whether patients were biologic naive or had a history of biologic failure and prior UC treatments. During the LTE, UC-specific concomitant medications were permitted per investigator's discretion.
Of the patients randomized in maintenance who continued ustekinumab treatment in the LTE, 29.8% (42/141) in the 90 mg SC q12w group and 29.4% (42/143) in the 90 mg SC q8w group discontinued treatment (Table 1). Patients with a history of biologic failure were more likely to discontinue treatment (42.7%; 53/124) than biologic-naive patients (18.8%; 28/149).
Table 1.
Study agent discontinuation before scheduled final dosing visit: Randomized patients in maintenance treated in the LTEa

An additional 157 patients who received ustekinumab IV induction were not in full Mayo clinical response at induction week 8, received a SC dose of ustekinumab 90 mg at induction week 8, and were in full Mayo clinical response at induction week 16. These patients continued to receive SC ustekinumab 90 mg q8w as part of the nonrandomized population in the maintenance study. At week 44, 116 of these patients entered the LTE and continued SC ustekinumab 90 mg q8w. Of these patients, 18.1% (n = 21) discontinued study treatment before the final dosing visit. Reasons for discontinuation included AE (n = 9), lack of efficacy (n = 6), and other (n = 6). Results for dose adjustment were previously reported (3).
Efficacy
Symptomatic remission.
Among all patients randomized to ustekinumab at maintenance baseline, proportions of patients in symptomatic remission were maintained through week 200 (Figure 1a–c).
Figure 1.

Symptomatic remissiona and corticosteroid-free symptomatic remissiona from maintenance baseline through week 200 for all ustekinumab IV induction responders randomized in the maintenance study (ITT analysis).b,c,d,e Symptomatic remission for the (a) overall population, (b) biologic-naive patients, and (c) patients with a history of biologic failure. Corticosteroid-free symptomatic remission for the (d) overall population, (e) biologic-naive patients, and (f) patients with a history of biologic failure. aSymptomatic remission was defined as a stool frequency subscore of 0 or 1 and a rectal bleeding subscore of 0. bData are shown by randomized treatment group at maintenance week 0, regardless of whether patients received dose adjustment during the long-term extension. cPatients who had both stool frequency and rectal bleeding subscores missing at a visit were considered not to be in symptomatic remission for that visit. dPatients who had a prohibited change in UC medication, an ostomy or colectomy, or used a rescue medication after clinical flare, or discontinued study agent due to lack of therapeutic effect or due to an AE of worsening of UC before week 44 were considered not to be in symptomatic remission at every visit thereafter up to week 44. ePatients who had an ostomy or colectomy or discontinued study agent due to lack of therapeutic effect or due to an AE of worsening of UC after week 44 were considered not to be in symptomatic remission at every visit thereafter. AE, adverse event; ITT, intent-to-treat; LTE, long-term extension; q8w, every 8 weeks; q12w, every 12 weeks; SC, subcutaneous, UC, ulcerative colitis.
Overall, 55.2% (192/348) of all patients randomized to ustekinumab at maintenance baseline (combined q8w/q12w) were in symptomatic remission at week 200 (Figure 1a). Specifically, 54.5% (96/176) of patients randomized to ustekinumab q8w and 55.8% (96/172) randomized to ustekinumab q12w (including those who dose-adjusted to q8w) were in symptomatic remission at week 200.
A greater proportion of biologic-naive patients (Figure 1b) were in symptomatic remission at each time point than those with a history of biologic failure (Figure 1c). Overall, 67.2% (117/174) of biologic-naive patients randomized to ustekinumab at maintenance baseline (combined q8w/q12w) were in symptomatic remission at week 200, compared with 41.6% (67/161) of patients with a history of biologic failure. The proportion of patients in symptomatic remission was maintained more consistently over time in the biologic-naive patients than in those with a history of biologic failure. In addition, the proportion of patients in symptomatic remission in the biologic-naive q8w subgroup decreased <1% per year between weeks 44 and 200, while the proportion in q8w patients with a history of biologic failure decreased approximately 7% per year during this period.
Corticosteroid tapering was allowed starting at the beginning of the maintenance study. Proportions of patients in corticosteroid-free symptomatic remission increased through maintenance week 16 and were generally sustained thereafter (Figure 1d–f) in a pattern similar to symptomatic remission. Overall, 53.2% (185/348) of patients randomized to ustekinumab (combined q8w/q12w) were in corticosteroid-free symptomatic remission at week 200. A total of 65.5% (114/174) of biologic-naive patients (combined q8w/q12w) and 39.8% (64/161) of patients with a history of biologic failure randomized to ustekinumab (combined q8w/q12w) were in corticosteroid-free symptomatic remission at week 200.
Among patients in symptomatic remission at each time point in the maintenance study and LTE, most of them were not receiving steroids. Of the 96 q8w patients in symptomatic remission at week 200, 91 (94.8%) were corticosteroid-free. Similarly, of the 96 q12w patients in symptomatic remission at week 200, 94 (97.9%) were corticosteroid-free.
In addition, among the 55 q8w biologic-naive patients in symptomatic remission at week 200, 54 (98.2%) were corticosteroid-free, and of the 62 q12w biologic-naive patients, 60 (96.8%) were in corticosteroid-free symptomatic remission at week 200. Among the 37 q8w patients with a history of biologic failure in symptomatic remission at week 200, 34 (91.9%) were corticosteroid-free. Of the 30 q12w patients with a history of biologic failure in symptomatic remission at week 200, 30 (100%) were corticosteroid-free.
Among patients randomized to ustekinumab who entered the LTE, the proportion of patients in symptomatic remission was maintained through week 200 (Figure 2a–f). Symptomatic remission outcomes through week 200 were evaluated in the overall population of randomized patients who entered the LTE and in the subgroups of biologic-naive patients and patients with a history of biologic failure, in which patients with missing data/meeting treatment failure criteria were considered nonresponders (Figure 2a–c). An observed case analysis of symptomatic remission was performed in which only patients with available data at each visit were analyzed (Figure 2d–f). A modified observed case analysis included patients in symptomatic remission up to the time of dose adjustment, and patients who met treatment criteria were considered nonresponders (see Supplementary Figure S2, http://links.lww.com/AJG/D138).
Figure 2.

Symptomatic remissiona from week 44 to week 200 among patients randomized to ustekinumab at maintenance baseline and treated in the LTE using (a-c) nonresponder imputation analysis,b,c,d or (d-f) observed case analysise for the overall population, biologic-naive patients, and patients with a history of biologic failure. aSymptomatic remission was defined as a stool frequency subscore of 0 or 1 and a rectal bleeding subscore of 0. bData are shown by randomized treatment group at maintenance week 0, regardless of whether patients received dose adjustment during the LTE. cPatients who had both stool frequency and rectal bleeding subscores missing at a visit were considered not to be in symptomatic remission for that visit. dPatients who had an ostomy or colectomy or discontinued study agent due to lack of therapeutic effect or due to an AE of worsening of UC after week 44 were considered not to be in symptomatic remission at every visit thereafter. eDenominator is number of patients with available data at each visit. AE, adverse event, LTE, long-term extension; q8w, every 8 weeks; q12w, every 12 weeks; SC, subcutaneous; UC, ulcerative colitis.
Among randomized patients who entered the LTE, absolute stool numbers remained low and proportions of patients with an absolute stool number ≤3/day were maintained through week 200. Proportions of patients with Mayo stool frequency subscores of 0 or 1 or no rectal bleeding (Mayo rectal bleeding subscore of 0) were also generally maintained (see Supplementary Table S2, http://links.lww.com/AJG/D138).
Symptomatic remission outcomes through week 200 in the nonrandomized study population who received ustekinumab in the LTE (see Supplementary Figure S3, http://links.lww.com/AJG/D138) were consistent with randomized patients who entered the LTE (Figure 2a).
Corticosteroid use in the LTE.
Among randomized patients receiving corticosteroids at maintenance baseline who entered the LTE, 77.9% (53/68) in the q12w group and 80.3% (57/71) in the q8w group were not receiving corticosteroids at week 200. The average daily P.Eq corticosteroid dose among patients receiving corticosteroids upon entering the LTE was low and generally maintained through week 200 (see Supplementary Figure S4, http://links.lww.com/AJG/D138).
Clinical and endoscopic outcomes.
Of the 213 patients with observed symptom data at week 200, 171 also had endoscopic assessments at that time point, which enabled Mayo clinical and endoscopic outcome evaluations. Full Mayo clinical remission, full Mayo clinical response, modified Mayo score response, and endoscopic improvement were evaluated for all patients with available data (observed case analysis) and with nonresponder imputation for patients who met treatment failure criteria (modified observed case analysis) (Figure 3).
Figure 3.
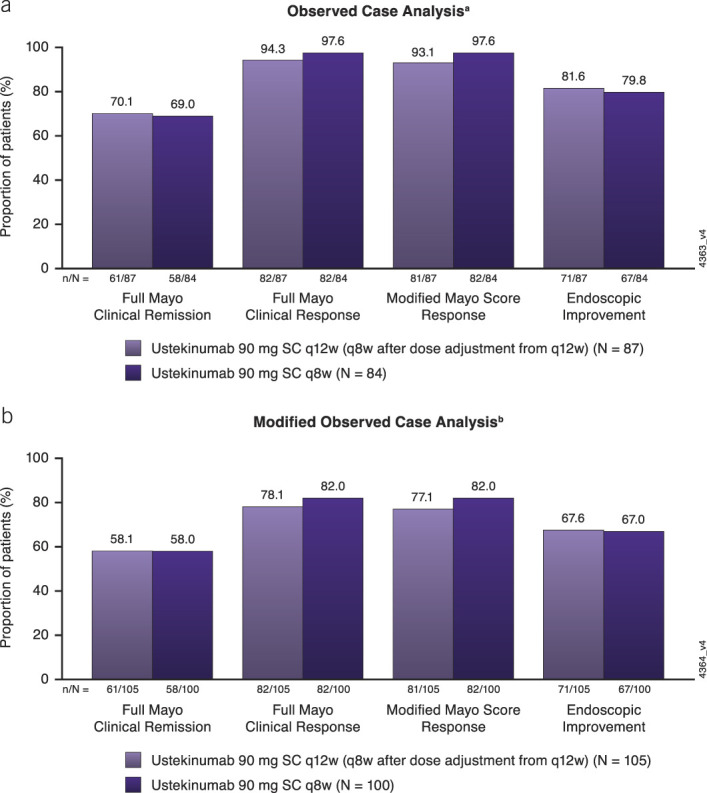
Full Mayo clinical remission, full Mayo clinical response, modified Mayo score response, and endoscopic improvement among patients with endoscopic evaluations at week 200 using (a) observed case analysis and (b) modified observed case analysis. Data are shown by randomized treatment group at maintenance week 0, regardless of whether patients received dose adjustment during the LTE. Full Mayo clinical remission was defined as a Mayo score ≤2 points, with no individual subscore >1. Full Mayo clinical response was defined as a decrease from induction baseline in the total Mayo score by ≥ 30% and ≥3 points, with either a decrease from induction baseline in the rectal bleeding subscore ≥1 or a rectal bleeding subscore of 0 or 1. Modified Mayo score response was defined as a decrease from induction baseline in the modified Mayo score by ≥ 30% and ≥2 points, with either a decrease from induction baseline in the rectal bleeding subscore ≥1 or a rectal bleeding subscore of 0 or 1. Endoscopic improvement was defined as an endoscopy subscore of 0 or 1. aIncludes data for patients who had endoscopic subscores at week 200. No imputation for missing data or treatment failure. bIncludes data for patients who had endoscopic subscores at week 200 or those who met treatment failure criteria. Patients who met treatment failure criteria (i.e., had an ostomy or colectomy or discontinued study agent due to lack of therapeutic effect or due to an AE of worsening of UC before week 200) were not considered to have achieved the end point. AE, adverse event; LTE, long-term extension; q8w, every 8 weeks; q12w, every 12 weeks; SC, subcutaneous; UC, ulcerative colitis.
Biomarkers.
Median serum CRP (see Supplementary Figure S5A, http://links.lww.com/AJG/D138) and fecal calprotectin (see Supplementary Figure S5B, http://links.lww.com/AJG/D138) concentrations were maintained from week 44 to week 200 among randomized patients who continued ustekinumab treatment in the LTE.
The IBDQ.
The proportion of patients who were in IBDQ remission was maintained from week 44 to week 200 (see Supplementary Figure S6, http://links.lww.com/AJG/D138).
Immunogenicity
The incidence of antibodies to ustekinumab was low through the final safety visit of the LTE. Overall, 5.5% (22/400) of randomized and nonrandomized patients who continued ustekinumab in the LTE were positive for ADA through the final safety visit. Overall, 5 of these 22 patients (22.7%) were positive for neutralizing antibodies. ADA were often transient and seemed to have no effect on efficacy.
Safety
Key safety events per 100 PY of follow-up for all patients treated in the LTE are summarized in Table 2. During the LTE (week 44 through the final safety visit [week 220], unless the patient discontinued study participation before week 220), key safety event rates in ustekinumab-treated patients were not greater than those observed in placebo-treated patients. The most frequently reported AE (>5 events per 100 PY) for patients receiving ustekinumab (combined q8w/q12w) during the LTE were nasopharyngitis (16.82 per 100 PY), UC worsening (15.04 per 100 PY), and upper respiratory tract infections (5.35 per 100 PY). Overall, 4 ustekinumab-treated patients in the LTE reported opportunistic infections (cytomegalovirus infection n = 2, Listeria monocytogenes n = 1, and oral herpes with mouth ulceration and concurrent neutropenia n = 1). The number of nonmelanoma skin carcinomas per 100 PY was 0.54 for ustekinumab-treated patients and 0.63 for placebo-treated patients (Table 2). Among ustekinumab-treated patients, 1 case of colorectal cancer and 1 case of rectal cancer were reported.
Table 2.
Summary of key safety findings per 100 patient-years of follow-up from week 44 through the final safety visit: Patients treated in the LTE

In the LTE, one previously reported death due to cardiac arrest occurred in a patient who received 1 dose of ustekinumab 90 mg SC after dose adjustment from placebo between weeks 44 and 96 (2). The patient presented with multiple comorbidities, and the death was considered unrelated to ustekinumab. During the final year of the LTE, no new deaths or major adverse cardiovascular events (MACE) were reported in ustekinumab-treated patients. No cases of active tuberculosis or posterior reversible encephalopathy syndrome were reported throughout the maintenance study and the LTE. Rates of key safety events by year of ustekinumab treatment showed no increase in the fourth year of ustekinumab treatment (Figure 4).
Figure 4.
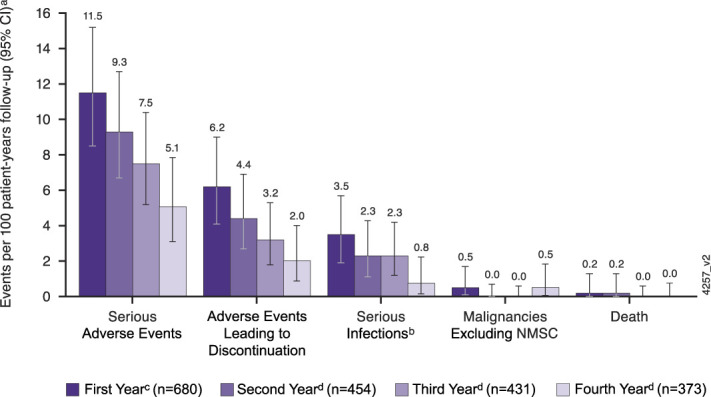
Key safety events per 100 patient-years of follow-up for the first, second, third, and fourth years of ustekinumab maintenance therapy. aNumber of adverse events per 100 patient-years of follow-up and 95% CI (rates by each year of follow-up) in the combined ustekinumab ulcerative colitis safety cohort. CI were based on an exact method assuming that the observed number of events follows a Poisson distribution. bInfection as assessed by the investigator. c1st year includes the following: (i) patients who received ustekinumab SC (q8w or q12w) in the maintenance study with data from maintenance weeks 0 through 44; (ii) patients who were in full Mayo clinical response to ustekinumab IV induction dosing and received placebo SC on entry into this maintenance study with data from maintenance weeks 0 through 8. d2nd, 3rd, and 4th years include the following: (i) patients who were in full Mayo clinical response to ustekinumab IV induction dosing and were randomized to ustekinumab 90 mg SC q12w or q8w on entry into the maintenance study; (ii) patients who were in full Mayo clinical response to ustekinumab IV induction dosing, randomized to receive placebo SC on entry into the maintenance study, and had a dose adjustment to ustekinumab 90 mg SC q8w, with data from the time of dose adjustment onward; and (iii) patients who were not in full Mayo clinical response to ustekinumab IV at induction week 8 but were in full Mayo clinical response at induction week 16 after an SC administration of ustekinumab at induction week 8 and received ustekinumab 90 mg SC q8w on entry into the maintenance study. The 2nd year included data from week 44 through week 96, the 3rd year included data from week 96 through week 156, and the 4th year included data from week 156 through the final safety visit up to week 220. CI, confidence interval; IV, intravenous; LTE, long-term extension; q8w, every 8 weeks; q12w, every 12 weeks; SC, subcutaneous.
DISCUSSION
In this final report from the UNIFI maintenance and LTE study, we present efficacy and safety data through 4 years for ustekinumab in patients with moderately to severely active UC who responded to IV induction and received SC maintenance treatment. The results showed that long-term SC maintenance therapy of q8w and q12w ustekinumab in patients who responded to IV ustekinumab induction was safe and effective at maintaining symptomatic remission. These results are consistent with previously reported findings from this LTE (2,3) and the IM-UNITI LTE study of ustekinumab in patients with Crohn's disease (8). Biologic-naive patients had greater persistence of therapy and efficacy outcomes than those with a history of biologic failure. Ustekinumab maintained efficacy without the need for concomitant corticosteroids. Safety results were consistent with the known safety profile of long-term ustekinumab treatment. Exposure-adjusted analysis showed that ustekinumab AE rates were not greater than placebo.
A unique feature of this LTE was the endoscopic assessment at week 200, which enabled the analysis of full Mayo score–related outcomes 3 years after the primary analyses of the maintenance study at 1 year. The results showed that patients who remained in the study through week 200 (most of whom were doing well symptomatically) had high rates of full Mayo clinical remission, full Mayo clinical response, modified Mayo score response, and endoscopic improvement.
Symptomatic remission data were analyzed and presented using 3 different approaches, each addressing different clinical questions related to long-term data. The ITT analysis evaluated all patients randomized in the maintenance study. Patients who discontinued treatment for any reason at any time through week 200, including those who did not enter the LTE, were considered nonresponders. These results help conservatively estimate the probability that a patient who responded to ustekinumab IV induction may maintain symptomatic remission through up to 4 years of ustekinumab treatment. The observed case analyses included patients with available data at each visit in the LTE; patients who discontinued the study for any reason were excluded from time of discontinuation forward. These results help estimate the probability that a patient continuing ustekinumab treatment will be in symptomatic remission. In long-term studies, patients who continue treatment tend to have better efficacy and safety outcomes than those who discontinue (9,10). The modified observed case analysis included patients with available data in the LTE up to the time of dose adjustment with nonresponder imputation for patients who met prespecified treatment failure criteria. This approach may be more reflective of the real-world long-term efficacy of ustekinumab because it excluded patients who discontinued ustekinumab for reasons other than efficacy.
Although 2 ustekinumab maintenance doses were evaluated throughout the LTE, the study was not powered to compare the efficacy and safety between doses. In the modified observed case analysis, the proportion of patients in symptomatic remission at week 200 in the q12w group was numerically greater than that in the q8w group, particularly among patients with a history of biologic failure. However, this was a result of the analysis approach because data were censored for patients who received dose adjustment. Dose adjustment (q12w to q8w) was conducted at the investigator's discretion starting at week 56. The effects of ustekinumab dose adjustment were previously reported (3).
The safety results during long-term maintenance treatment were consistent with those previously reported in controlled studies across approved indications (2,3,11,12). Exposure-adjusted analysis of key safety events, including AE, SAE, infections, serious infections, AE leading to discontinuations, and malignancies, occurred at rates that were not greater than placebo. Consistent with safety data previously reported during the UNIFI LTE, the most frequently reported AE were nasopharyngitis, upper respiratory tract infection, and UC worsening. No cases of tuberculosis occurred. One death and 3 MACE occurred between weeks 44 and 96, as previously reported (2). No new deaths or MACE occurred between weeks 96 and 200. Two patients had solid tumors (rectal and colon cancers).
Limitations of this study should be considered when interpreting the results. The LTE was double-blinded until all patients completed the 1-year maintenance study and the primary analysis was completed, at which time all patients were unblinded, and patients receiving placebo discontinued. Therefore, the duration of blinded treatment and assessments varied among patients, depending on when they were enrolled in the trial, and efficacy comparisons with placebo were not viable after week 44 (placebo comparison for safety was based on events per 100 PY). Endoscopic evaluations at week 200 were not available for all patients and were not centrally read.
In conclusion, in this population of patients with treatment-refractory moderate-to-severe UC, patients who responded to ustekinumab IV induction and received SC maintenance treatment generally maintained clinical benefit through 4 years. The safety profile of ustekinumab maintenance treatment was consistent with the known long-term safety profile in other approved indications.
Study Highlights.
WHAT IS KNOWN
✓ Ulcerative colitis requires chronic treatment.
✓ Ustekinumab subcutaneous (90 mg) was shown to be safe and effective in patients with moderate-to-severe ulcerative colitis through 3 years.
WHAT IS NEW HERE
✓ This UNIFI long-term extension study demonstrates that ustekinumab treatment can maintain clinical and endoscopic outcomes through 4 years.
✓ No new safety signals were reported through 4 years.
✓ Ustekinumab maintenance provided clinical and endoscopic benefit to patients through 4 years of treatment.
✓ Most patients in symptomatic remission maintained remission without corticosteroids.
CONFLICTS OF INTEREST
Guarantor of the article: Waqqas Afif, MD.
Specific author contributions: W.A., R.P.A., M.T.A., S.D., W.J.S., R.P., T.H., E.J.S., R.W.L., D.S.R., L.P.-B., and B.E.S. participated in the conception and design of the study, analysis and interpretation of the data, and drafted/revised the manuscript for important intellectual content. Y.M., H.Z., and C.M. participated in the conception and design of the study, participated in acquisition/collection of data, analysis and interpretation of data, and drafted/revised the manuscript for important intellectual content. All authors approved the final version of the manuscript for submission.
Financial support: This work was supported by Janssen Research & Development, LLC.
Potential competing interests: W.A. reports having received speaker, advisory board member, and or clinical investigator for AbbVie, Amgen, BMS, Dynacare, Eli-Lilly, Janssen, Merck, Novartis, Pfizer, Prometheus, Sandoz, Sanofi, Takeda, and Theradiag. R.P.A. reports having received grant support and consulting fees from AbbVie, Dr Falk Pharma, and Janssen Research & Development, LLC. M.T.A. reports having received consulting fees from AbbVie, Janssen Research & Development, LLC., Prometheus Biosciences, Takeda, Focus Medical Communications, Pfizer, Boehringer Ingelheim Pharmaceuticals, Gilead, Imedex, Cornerstone Health, Inc., UCB Biopharma SRL, Eli Lilly, Bristol Myers Squibb, and Celsius. S.D. reports having received consultancy fees from AbbVie, Alimentiv, Allergan, Amgen, AstraZeneca, Athos Therapeutics, Biogen, Boehringer Ingelheim, Celgene, Celltrion, Eli Lilly, Enthera, Ferring Pharmaceuticals Inc., Gilead, Hospira, Inotrem, Janssen, Johnson & Johnson, MSD, Mundipharma, Mylan, Pfizer, Roche, Sandoz, Sublimity Therapeutics, Takeda, TiGenix, UCB Inc. and Vifor; and received lecture fees from AbbVie, Amgen, Ferring Pharmaceuticals Inc., Gilead, Janssen, Mylan, Pfizer, and Takeda. W.J.S. reports having received research grants from AbbVie, Abivax, Arena Pharmaceuticals, Boehringer Ingelheim, Celgene, Genentech, Gilead Sciences, Glaxo Smith Kline, Janssen, Eli Lilly, Pfizer, Prometheus Laboratories, Seres Therapeutics, Shire Pharmaceuticals, Takeda, Theravance Biopharma; consulting fees from AbbVie, Abivax, Admirx, Alfasigma, Alimentiv, Alivio Therapeutics, Allakos, Amgen, Arena Pharmaceuticals, AstraZeneca, Atlantic Pharmaceuticals, Bausch Health (Salix), Beigene, Bellatrix Pharmaceuticals, Biora (Progenity), Boehringer Ingelheim, Boston Pharmaceuticals, Bristol Meyers Squibb, Celgene, Celltrion, Clostrabio, Codexis, Equillium, Forbion, Galapagos, Genentech, Gilead Sciences, GlaxoSmithKline, Gossamer Bio, Immunic (Vital Therapies), Index Pharmaceuticals, Inotrem, Intact Therapeutics, Iota Biosciences, Janssen, Kiniksa Pharmaceuticals, Kyverna Therapeutics, Landos Biopharma, Eli Lilly, Morphic Therapeutics, Novartis, Ono Pharmaceuticals, Oppilan Pharma (now Ventyx Biosciences), Otsuka, Pandion Therapeutics, Pfizer, Pharm Olam, Polpharm, Prometheus Biosciences, Protagonist Therapeutics, PTM Therapeutics, Quell Therapeutics, Reistone Biopharma, Seres Therapeutics, Shanghai Pharma Biotherapeutics, Shoreline Biosciences, Sublimity Therapeutics, Surrozen, Takeda, Theravance Biopharma, Thetis Pharmaceuticals, Tillotts Pharma, Vedanta Biosciences, Ventyx Biosciences, Vimalan Biosciences, Vivelix Pharmaceuticals, Vividion Therapeutics, Vivreon Gastrosciences, Xencor, Zealand Pharma; stock or stock options from Allakos, BeiGene, Biora (Progenity), Gossamer Bio, Oppilan Pharma (now Ventyx Biosciences), Prometheus Biosciences, Prometheus Laboratories, Protagnoists Therapeutics, Shoreline Biosciences, Ventyx Biosciences, Vimalan Biosciences, Vivreon Gastrosciences; and employee at Shoreline Biosciences and Ventyx Biosciences. Spouse: Iveric Bio–consultant, stock options; Progenity–stock; Oppilan Pharma (now Ventyx Biosciences)–stock; Prometheus Biosciences–employee, stock, stock options; Prometheus Laboratories–stock, stock options, consultant, Ventyx Biosciences–stock, stock options; Vimalan Biosciences–stock. R.P. reports having received consulting fees from Abbott, AbbVie, Abbivax, Alimentiv (formerly Robarts), Amgen, Arena Pharmaceuticals, AstraZeneca, Biogen, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Celltrion, Cosmos Pharmaceuticals, Eisai, Elan, Eli Lilly, Ferring, Galapagos, Fresenius Kabi, Genentech, Gilead Sciences, Glaxo-Smith Kline, JAMP Bio, Janssen, Merck, Mylan, Novartis, Oppilan Pharma, Organon, Pandion Pharma, Pendopharm, Pfizer, Progenity, Prometheus Biosciences, Protagonist Therapeutics, Roche, Sandoz, Satisfai Health, Shire, Sublimity Therapeutics, Takeda Pharmaceuticals, Theravance Biopharma, Trellus, Viatris, Ventyx, UCB; Speaker's Fees from AbbVie, Amgen, Arena Pharmaceuticals, Bristol Myers Squibb, Celgene, Eli Lilly, Ferring, Fresenius Kabi, Gilead Sciences, Janssen, Merck, Organon, Pfizer, Roche, Sandoz, Shire, Takeda Pharmaceuticals and served on Advisory Boards for: AbbVie, Alimentiv (formerly Robarts), Amgen, Arena Pharmaceuticals, AstraZeneca, Biogen, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Eli Lilly, Ferring, Fresenius Kabi, Genentech, Gilead Sciences, Glaxo-Smith Kline, JAMP Bio, Janssen, Merck, Mylan, Novartis, Oppilan Pharma, Organon, Pandion Pharma, Pfizer, Progenity, Protagonist Therapeutics, Roche, Sandoz, Shire, Sublimity Therapeutics, Takeda Pharmaceuticals, and Ventyx. T.H. reports having received grant support from AbbVie, Daiichi-Sankyo, EA Pharma Co, Ltd. JIMRO, Mitsubishi Tanabe Pharma Corporation, Mochida Pharmaceutical Co., Ltd., Nippon Kayaku Co., Ltd., Pfizer Inc., and Takeda Pharmaceutical Co., Ltd.; received consulting fees from EA Pharma Co, Ltd. and Janssen Research & Development, LLC.; and received lecture fees from AbbVie, EA Pharma Co, Ltd., Mitsubishi Tanabe Pharma Corporation, Eli Lilly, Gilead Sciences, Kissei Pharmaceutical Co, Ltd., and Takeda Pharmaceutical Co., Ltd. E.J.S. reports having received grant support from AbbVie, AstraZeneca, Crohn's and Colitis Foundation, Janssen Research & Development, LLC., New York Crohn's Foundation, Pfizer, UCB, Genentech, Seres Therapeutics, and Celgene; received consulting fees from AbbVie, Abgenomics, Crohn's and Colitis Foundation, Evidera, GI Health Foundation, Janssen, Protagonist, Seres, and Takeda Stock: Gilead; and received speaker fees from GI Health Foundation and Prime Therapeutics. R.W.L. reports having received advisory board fees from AbbVie, Aspen, BMS, Celgene, Celltrion, Chiesi, Ferring, Glutagen, Hospira, Janssen, Lilly, MSD, Novartis, Pfizer, Prometheus Biosciences, and Takeda; received research grants from Celltrion, Shire, Janssen, Takeda, Joanna Tiddy grant from University of Sydney, McCusker Charitable Trust, Gastroenterological Society of Australia, NHMRC, Gutsy Group, and Pfizer. D.S.R. reports having received speaker fees from AbbVie, Janssen Research & Development, LLC., and Emerge Health and served as an advisory board member for AbbVie and Janssen Research & Development, LLC. L.P.-B. reports having received personal fees from AbbVie, Allergan, Alma, Amgen, Applied Molecular Transport, Arena Pharmaceuticals, Biogen, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Celltrion, Eli Lilly, Enterome, Enthera, Ferring, Fresenius, Genentech, Gilead, Hikma, Index Pharmaceuticals, Janssen, MSD, Mylan, Nestlé, Norgine, Oppilan Pharma, OSE Immunotherapeutics, Pfizer, Pharmacosmos, Roche, Samsung Bioepis, Sandoz, Sterna, Sublimity Therapeutics, Takeda, Tillots, and Vifor; received grants from AbbVie, MSD, and Takeda; and received stock options from CTMA. Y.M., H.Z., and C.M. all are employees of Janssen Research & Development, LLC and own stock/stock options in Johnson & Johnson. B.E.S. reports consulting fees from AbbVie, Alimentiv, Amgen, Arena Pharmaceuticals, Artugen Therapeutics, Astra Zeneca, Boehringer Ingelheim, Boston Pharmaceuticals, Calibr, Celgene, Celltrion, ClostraBio, Equillium, Enthera, Evommune, Fresenius Kabi, Galapagos, Genentech (Roche), Gilead Sciences, GlaxoSmithKline, Gossamer Bio, Index Pharmaceuticals, Innovation Pharmaceuticals, Inotrem, Kaleido, Kallyope, Merck, Morphic Therapeutics, MRM Health, Progenity, Prometheus Biosciences, Prometheus Laboratories, Protagonist Therapeutics, Q32 Bio, Sun Pharma, Surrozen, Target RWE, Teva, TLL Pharmaceutical, and Ventyx Biosciences; consulting and speaking fees from Abivax; consulting and speaking fees and other support from Eli Lilly; research grants, consulting and speaking fees and other support from Bristol Myers Squibb, Janssen, Pfizer, and Takeda; research grants and consulting fees from Theravance Biopharma; and stock options from Ventyx Biopharma.
Writing assistance: Medical writing support was provided by Kristin Ruley Sharples, PhD, of Janssen Scientific Affairs, LLC, under the direction of the authors in accordance with Good Publication Practice guidelines (Ann Inter Med. 2015;163:461-464) and was funded by Janssen Scientific Affairs, LLC.
Data transparency statement: The data sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
The authors thank the patients, investigators, and study personnel who made the UNIFI LTE possible. All authors had access to the study data and reviewed and approved the final manuscript.
Supplementary Material
Footnotes
SUPPLEMENTARY MATERIAL accompanies this paper at http://links.lww.com/AJG/D138
Contributor Information
Ramesh P. Arasaradnam, Email: r.arasaradnam@warwick.ac.uk.
Silvio Danese, Email: sdanese@hotmail.com.
William J. Sandborn, Email: wsandborn@health.ucsd.edu.
Ye Miao, Email: ymiao3@its.jnj.com.
Hongyan Zhang, Email: hzhang23@its.jnj.com.
Remo Panaccione, Email: rpanacci@ucalgary.ca.
Tadakazu Hisamatsu, Email: thisamatsu@ks.kyorin-u.ac.jp.
Ellen J. Scherl, Email: ejs2005@med.cornell.edu.
Rupert W. Leong, Email: rupertleong@hotmail.com.
David S. Rowbotham, Email: davidrb@adhb.govt.nz.
Laurent Peyrin-Biroulet, Email: peyrinbiroulet@gmail.com.
Bruce E. Sands, Email: bruce.sands@mssm.edu.
Colleen Marano, Email: cmarano@its.jnj.com.
REFERENCES
- 1.Le Berre C, Ricciuto A, Peyrin-Biroulet L, et al. Evolving short- and long-term goals of management of inflammatory bowel diseases: Getting it right, making it last. Gastroenterology 2022;162(5):1424–38. [DOI] [PubMed] [Google Scholar]
- 2.Panaccione R, Danese S, Sandborn WJ, et al. Ustekinumab is effective and safe for ulcerative colitis through 2 years of maintenance therapy. Aliment Pharmacol Ther 2020;52(11-12):1658–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abreu MT, Rowbotham DS, Danese S, et al. Efficacy and safety of maintenance ustekinumab for ulcerative colitis through 3 years: UNIFI long-term extension. J Crohns Colitis 2022;16(8):1222–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sands BE, Sandborn WJ, Panaccione R, et al. Ustekinumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med 2019;381(13):1201–14. [DOI] [PubMed] [Google Scholar]
- 5.Irvine EJ, Feagan B, Rochon J, et al. Quality of life: A valid and reliable measure of therapeutic efficacy in the treatment of inflammatory bowel disease. Canadian Crohn’s relapse prevention trial study group. Gastroenterology 1994;106(2):287–96. [DOI] [PubMed] [Google Scholar]
- 6.Adedokun OJ, Xu Z, Gasink C, et al. Pharmacokinetics and exposure response relationships of ustekinumab in patients with Crohn’s disease. Gastroenterology 2018;154(6):1660–71. [DOI] [PubMed] [Google Scholar]
- 7.Adedokun OJ, Xu Z, Marano C, et al. Ustekinumab pharmacokinetics and exposure response in a phase 3 randomized trial of patients with ulcerative colitis. Clin Gastroenterol Hepatol 2020;18(10):2244–55.e9. [DOI] [PubMed] [Google Scholar]
- 8.Sandborn WJ, Rebuck R, Wang Y, et al. Five-year efficacy and safety of ustekinumab treatment in Crohn’s disease: The IM-UNITI trial. Clin Gastroenterol Hepatol 2022;20(3):578–90.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casanova MJ, Chaparro M, García-Sánchez V, et al. Evolution after anti-TNF discontinuation in patients with inflammatory bowel disease: A multicenter long-term follow-up study. Am J Gastroenterol 2017;112(1):120–31. [DOI] [PubMed] [Google Scholar]
- 10.Kennedy NA, Warner B, Johnston EL, et al. Relapse after withdrawal from anti-TNF therapy for inflammatory bowel disease: An observational study, plus systematic review and meta-analysis. Aliment Pharmacol Ther 2016;43(8):910–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghosh S, Gensler LS, Yang Z, et al. Ustekinumab safety in psoriasis, psoriatic arthritis, and Crohn’s disease: An integrated analysis of phase II/III clinical development programs. Drug Saf 2019;42(6):751–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sandborn WJ, Feagan BG, Danese S, et al. Safety of ustekinumab in inflammatory bowel disease: Pooled safety analysis of results from phase 2/3 studies. Inflamm Bowel Dis 2021;27(7):994–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]