

Abstract
The field of oncology has been transformed by immune checkpoint inhibitors (ICI) and other immune-based agents, however many patients do not receive a durable benefit. While biomarker assessments from pivotal ICI trials have uncovered certain mechanisms of resistance, results thus far have only scraped the surface. Mechanisms of resistance are as complex as the tumor microenvironment (TME) itself, and the development of effective therapeutic strategies will only be possible by building accurate models of the tumor-immune interface. With advancement of multi-omic technologies, high-resolution characterization of the TME is now possible. In addition to sequencing of bulk tumor, single-cell transcriptomic, proteomic, and epigenomic data as well as T cell receptor profiling can now be simultaneously measured and compared between responders and non-responders to ICI. Spatial sequencing and imaging platforms have further expanded the dimensionality of existing technologies. Rapid advancements in computation and data sharing strategies enables development of biologically interpretable machine learning models to integrate data from high-resolution, multi-omic platforms. These models catalyze the identification of resistance mechanisms and predictors of benefit in ICI-treated patients, providing scientific foundation for novel clinical trials. Moving forward, we propose a framework by which in silico screening, functional validation, and clinical trial biomarker assessment can be used for the advancement of combined immunotherapy strategies.
Keywords: immune checkpoint inhibitors, immunotherapy, multi-omics, single-cell sequencing, spatial omics, imaging, computational biology, machine learning, artificial intelligence
Introduction: The need for advanced immunotherapy combinations
Immune checkpoint inhibitors (ICI) have revolutionized the treatment of many cancer types, though the majority of patients do not receive a durable benefit from monotherapy alone.(1,2) Since the FDA approval of CTLA4 and PD1 antibodies for the treatment of advanced melanoma, ICIs have been studied in nearly every malignancy. These trials provided an early paradigm of response criteria to explain an ever-widening gap between long-term responders and those with primary or secondary resistance.(3) The “immunogenicity” of the tumor is the backbone of this paradigm, with highly mutagenic cancers (e.g. lung and skin) initially believed to derive the most benefit.(4) However, biomarkers aimed at defining a tumor’s immunogenic potential, including tumor mutational burden (TMB), PD-L1 immunohistochemistry (IHC) and tumor infiltrating lymphocyte (TIL) assays, do not always correlate strongly with ICI response.(5) Instead, markers based on static, unidimensional parameters of the tumor microenvironment (TME) only highlight the overall need for integrated, high-dimensional measures that capture the immense complexity of the tumor-immune interface. Dual CTLA4 plus PD1 blockade is used in multiple cancers, with LAG3 inhibition recently approved alongside anti-PD1 therapy in advanced melanoma.(6) However, a lack of advanced biomarkers has limited the identification of populations most likely to receive benefit, leading to high toxicity and limited efficacy of other combination strategies, such as co-stimulatory agonists and intratumoral therapies.(7–9)
Similar to ICI, multi-omics technologies have transformed the field of immuno-oncology (IO) on scientific exploration and clinical translation. Historically, high costs and resource barriers prevented these technologies from being applied broadly toward the discovery of novel IO targets.(10) Nowadays, omics production is no longer a rate-limiting step, with data analytics and computation now charged with harnessing these high-dimensional data to better characterize the TME. As an example, next-generation sequencing (NGS) of the tumor transcriptome allowed the development of inflammatory gene signatures based on interferon-gamma (IFNγ) signaling and cytolytic scores.(11) Though imperfect, these signatures provide a more comprehensive depiction of T cell activity as compared to clinically used biomarkers.(12) Given the relative paucity of ICI-treated cohorts with expression data, these inflammatory signatures also serve as more accurate, predictive surrogates for ICI response.(11,13) Most importantly, by comparing non-T cell-inflamed versus T cell-inflamed phenotypes, a model can be established by which molecular drivers of immune exclusion may be identified.(14) Apart from bulk tissue sequencing, multi-omics now encompasses nearly all aspects of cancer biology, including single-cell RNAseq/CITEseq/ATACseq, T cell/B cell receptor (TCR/BCR) sequencing, microbiome sequencing, metabolomics, spatial transcriptomics/proteomics, pathomics, and radiomics, among others.
In order to expand the efficacy of ICI and other immune-based agents, several critical hurdles need to be addressed. First, both ICI and co-stimulatory agonists require T cell-infiltration of the TME. While current histological analyses can detect rudimentary TIL levels, they do not necessarily qualify the degree of cytotoxicity or exhaustion.(5) Additionally, most analyses and associated biomarkers are focused on immune cell activity, despite strong evidence for both tumor cell-intrinsic and stromal cell-mediated immune evasion.(15,16) Characterizing the interaction and signaling mechanisms among all cells in the TME will be critical for augmenting ICI therapy. Further, the heterogeneity of each cancer type and within individual tumors themselves has been difficult to capture, yet this heterogeneity is a fundamental source of resistance via subclonal evolution.(17) Other sources of ICI resistance include intracellular proteins that can act as bypass mechanisms to extracellular checkpoint blockade; these downstream mediators are difficult to detect and target.(18) Lastly, neoantigenicity has long been heralded as a key marker of immune activity, yet the ability to detect tumor-specific antigens and translate this measurement to T cell cytotoxicity has been limited.(19)
By defining this needs assessment, the field of IO is poised to apply novel, multi-omic platforms to advance combined immunotherapy strategies. Ideally, the overall objective should be to comprehensively and accurately characterize the TME based on the interaction among immune, stromal, and tumor cells. Using these high-dimensional assessments from tumors of IO-treated patients, the interactions between non-responding versus responding cohorts can be compared to identify novel and translationally relevant resistance mechanisms. Advances in multi-omics technology and robust clinical trial tissue banking have led to an exponential growth in data acquisition, highlighting the primary need for clinically-oriented data analytics and integration. By combining these multi-omics data from IO-treated patients using computational methods and AI-driven strategies, we may be able to more accurately identify and target molecular pathways of resistance and augment existing immune-based therapies in future clinical trials (Fig. 1). Finally, an emphasis on equitable data sharing has enabled large scale AI models from published cohorts, catalyzing the discovery of novel IO combinations.(20)
Figure 1.
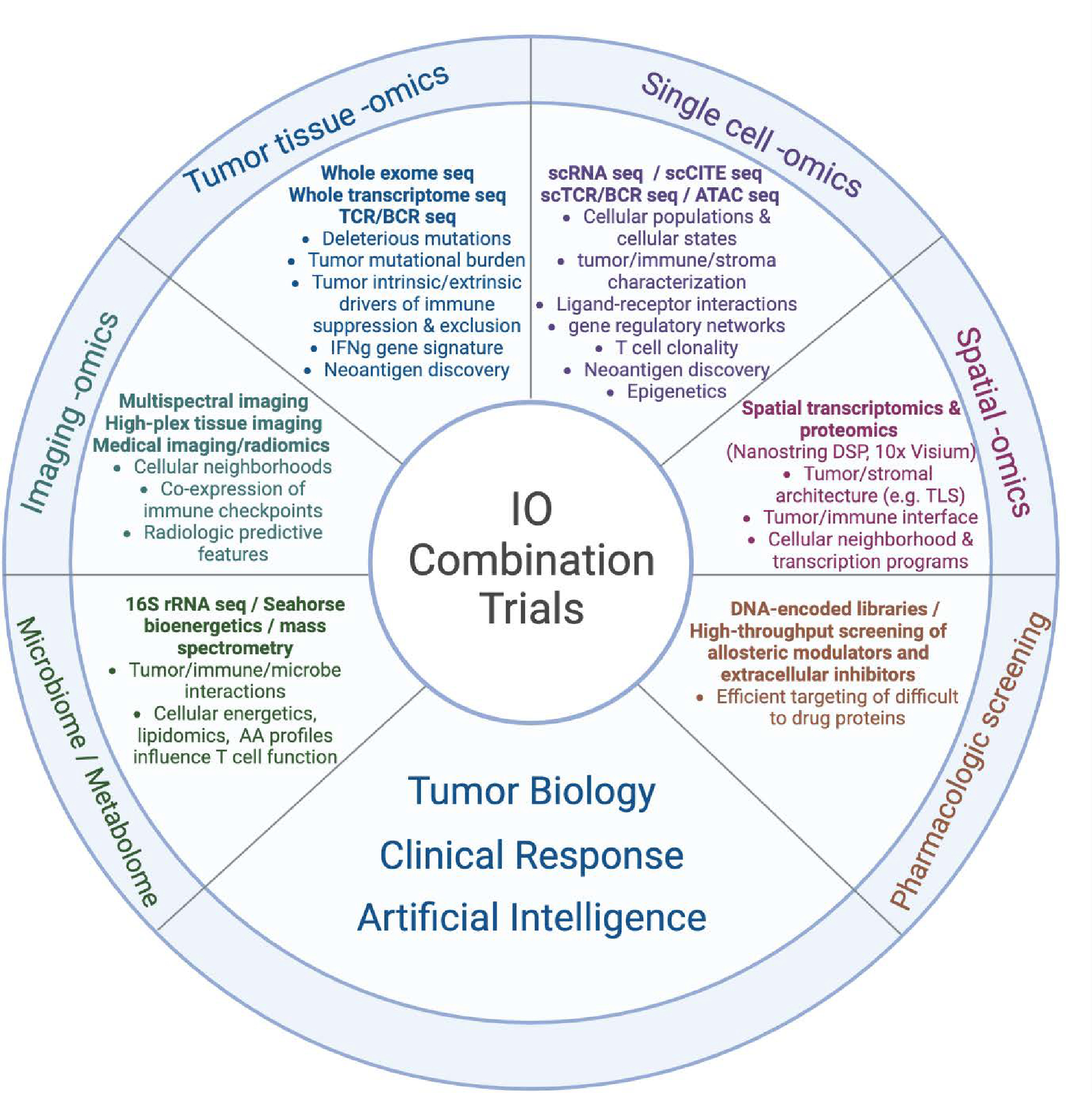
An array of multi-omics technology and AI platforms can be used to characterize the tumor-immune-stromal interface, identify resistance pathways, predict clinical response, and catalyze a new era of combined immunotherapy strategies.
Tumor tissue omics: Includes high-throughput sequencing of the tumor genome, exome, and transcriptome. Specific mutations in the tumor DNA can be detected through whole-genome sequencing (WGS), WES, targeted sequencing (e.g., via companion diagnostic (CDx)), among others. RNAseq of the tumor transcriptome can detect differentially expressed genes in clinically relevant samples that may correspond to targetable pathways. The expression of IFNγ based gene signatures can also be used to categorize tumor samples into T cell-inflamed versus non-T cell-inflamed phenotypes and identify the upregulation of pathways specific to immune exclusion or ICI resistance. Sequencing of the T cell receptor (TCR) or B cell receptor (BCR) may indirectly identify tumor-specific T cell or B cell clonal populations. Neoantigens can be predicted from integration of DNAseq (mutations), RNAseq (expression), and potentially mass spectrometry-based proteogenomics.
Single cell omics: High-throughput profiling of individually isolated cells provides transcript sequence and abundance from specific cell types, providing a high resolution of the tumor microenvironment. Advances in multiplexing technologies can further quantify cell-surface protein expression (CITEseq), TCR/BCR repertoire, and chromatin accessibility (ATACseq) in addition to single cell transcriptomics. By increasing the resolution of gene and protein expression to the cellular level, differentially expressed pathways can be specifically identified within the tumor, immune, and stromal cell populations to further characterize molecular mediators of immune exclusion in the TME. Further, cell surface protein expression via CITEseq can provide additional phenotypic data to determine cell types. TCR sequencing of infiltrating lymphocytes can identify neoantigen-specific TCRs and allow engineering of neoantigen-specific TCRs in vitro.
Imaging omics: An array of technologies including multiplex immunofluorescence, codetection by indexing (CODEX), iterative indirect immunofluorescence imaging (4i), and imaging mass cytometry (among others) now permit dozens of unique protein markers to be digitally measured from fixed tumor tissue. On the macroscopic level, thousands of features from radiologic body imaging can be computationally extracted to build machine learning models in prediction of clinical outcome.
Spatial omics: As another platform to characterize tumor-immune-stromal interactions, both Nanostring digital spatial profiling (DSP) GeoMx and 10X Visium can be used to map gene expression values to fresh frozen or formalin-fixed paraffin embedded (FFPE) tissue, identifying pathways of immune exclusion at a geographical level along with cellular neighborhoods that may be associated with clinical outcomes. Moreover, single-cell resolution has been achieved by Nanostring CosMx single-molecule imaging (SMI), 10x Xenium, and Vizgen MERSCOPE (Multiplexed Error-Robust Fluorescence in situ Hybridization, MERFISH). These technologies profile several hundred to a few thousand targeted genes within individual cells in a spatial context. This advancement allows for a deeper understanding of cell-to-cell communication and interactions between ligands and receptors in cellular neighborhoods of TME.
Metabolomics and the Microbiome: In addition to gene and protein expression, a number of metabolic platforms have been developed to measure parameters of cellular energetics, including oxygen consumption, extracellular acidification, and lipid and amino acid profiling. Comparing the microbiota of ICI responders versus non-responders using 16s rRNA sequencing, metagenomics, and metatranscriptomics has also identified unique taxa correlated with clinical outcomes.
Pharmacologic screening: While multi-omic platforms can be used to identify novel pathways of IO resistance, advances in pharmacologic screening are required to target these pathways in clinical trials. DNA encoded libraries allow high-throughput screening of millions of compounds to efficiently identify candidate agents to target historically “undruggable” proteins.
Artificial intelligence: Machine learning (ML) and deep learning (DL) have been pivotal in driving our understanding and therapeutic approaches to diseases at a personalized level, including but not limited to, multimodal data integration across radiology scans, pathology images, electronic health records (EHR), and multi-omics; predictive modeling on patient stratification/selection, adverse event monitoring, and immunotherapy treatment response; new immunotherapeutic agent discovery or drug repurposing from in vitro screens and/or cell imaging; and foundation models for medical imaging segmentation, single-cell phenotypic annotation, and drug discovery from chemical language representations. As technology and data evolve, it’s anticipated that AI will play an even more significant role in propelling these fields forward.
IO, immuno-oncology; sc, single-cell; seq, sequencing; CITE, cellular indexing of transcriptomes and epitopes; TCR, T cell receptor; BCR, B cell receptor; ATAC, assay for transpose-accessible chromatin; DSP, digital spatial profiler; TLS, tertiary lymphoid structure; IMC, imaging mass cytometry; CODEX, co-detection by indexing; AA, amino acid
Figure made with BioRender
Established multi-omic platforms used in translational immunotherapy
Whole genome/exome & somatic mutation profiling
On the heels of the Human Genome Project, The Cancer Genome Atlas (TCGA) was launched, laying the groundwork for publicly available, cancer-specific genomic and transcriptomic reference data.(21) With the exponential decrease in NGS costs and collaborative efforts across academic institutions and industrial partners, over 20,000 samples across 33 malignancies were sequenced. At the same time, ICIs were reaching widespread clinical use, and the first generation of high-dimensional tumor data have been mined towards ICI response and prediction strategies. For example, whole-exome sequencing (WES) from an anti-CTLA4 cohort revealed an association between TMB and long-term response.(22) As a prelude to future studies, however, this association alone was not found to be predictive of durable benefit. Instead, nonsynonymous mutations were filtered according to MHC I binding potential and categorized into mutant versus nonmutant translated peptides. Using both hierarchical clustering and supervised learning platforms, a set of tetrapeptide sequences defined a “neoepitope signature” that successfully predicted response in a validation cohort.(22) Despite a limited number of tumors and utilization of only one omics platform, this study exemplified the strength of combining high-dimensional data with predictive modeling to more accurately connect tumor biology with treatment outcomes. A similar study of PD1 inhibition in NSCLC also demonstrated an association between TMB and therapeutic response, however neither TMB nor a smoking-specific mutational pattern were specific for a durable benefit.(23) Rather, an exome-wide differential analysis of deleterious mutations revealed an explicit pattern in genes involved in DNA repair, such as POLD1, POLE, and MSH2, the latter tied to mismatch repair deficiency (MMRd). This exome-wide analysis not only identified specific pathway aberrations tied to ICI response, but also foreshadowed a series of studies leading to the tumor-agnostic approval of pembrolizumab for dMMR and microsatellite instability-high (MSI-H) cancers.(24) Though not performed in this study, a computational model combining mutational signatures, total TMB, and histopathological variables may have offered higher predictive power in validation cohorts.
From a clinical standpoint, somatic mutational profiling is now routine practice for most solid tumors. Ten unique oncogenic drivers are now targetable in the treatment of non-small cell lung cancer, for example, and many of these targeted therapies have been approved alongside specific gene panels. In addition to MSI-H, a pan-cancer approval of pembrolizumab was granted for TMB-high (>10 mutations/megabase) solid tumors alongside a companion diagnostic based on the results of KEYNOTE-158.(25,26) While this approval provides an additional therapeutic option in patients without a reasonable alternative, additional biomarkers are still needed to help strengthen the predictive utility of TMB.
Whole transcriptome and gene expression signatures
While genomics can detect deleterious mutations associated with therapeutic response, high-resolution quantification of the transcriptome can provide a more global characterization of tumor biology, part of the overall goal in identifying tumor-intrinsic and extrinsic drivers of immune suppression and immune exclusion. A precursor to RNAseq, researchers first used microarray technologies to perform differential expression analyses between anti-PD1 resistant and wild-type murine models.(27) BMP7, a member of the TGFβ family, was found to be significantly upregulated. Additionally, BMP7 was shown to modulate IFNγ, IL2, MAPK14, and SMAD1; shRNA-mediated BMP7 knockdown restored ICI efficacy, and BMP7 transcript levels were found to negatively correlate with survival in multiple NSCLC cohorts.(27) These results helped build a model by which a biological mechanism of immune suppression was deduced, targeted inhibition was shown to reverse anti-PD1 resistance, and predictive biomarkers were validated from publicly available expression cohorts.
Using banked tumor samples from early phase trials with pembrolizumab, another study developed a gene expression signature after comparing a set of tumor- and immune-related genes in responders versus non-responders.(11) Nearly all top-ranked genes were related to IFNγ signaling; unsupervised clustering, regression modeling, and validation among nine additional PD1-treated cohorts produced a final 18-gene T cell-inflamed signature. In comparing this signature to the clinical outcomes of a multi-tumor, anti-PD1 clinical trial (KEYNOTE-012), both response and progression-free survival were associated with higher gene expression. This study also confirmed a seemingly paradoxical but homeostatic upregulation of inhibitory molecules (IDO1, PDL1, TIGIT, LAG3) in T cell-inflamed tumors,(28) a finding with important implications for therapeutic combination strategies. Lastly, a similar but separate study took advantage of transcriptome-wide expression data from TCGA, identifying a 160-gene, pan-cancer signature that clustered tightly with established T cell effector genes.(12) This signature, denoting an IFNγ-enriched, inflamed phenotype, was used to stratify tumors into T cell-inflamed and non-T cell-inflamed subgroups.(14) Over 9000 tumors across 30 cancer types were analyzed from publicly available expression cohorts; and as opposed to other studies focused on specific immune cell processes driving T cell suppression, this analysis focused on tumor-intrinsic drivers of immune evasion and identified distinct pathways shared by non-T cell-inflamed tumors across cancer types. For example, the CTNNB1 (Wnt/b-catenin), KLF4, and MYC were found to be upregulated transcriptional programs from non-T cell-inflamed tumors across multiple cancers, with expression significantly higher in PD1-responding patients from a separate melanoma cohort.(14)
Single-cell transcriptomics, TCR sequencing, metabolomics, and the microbiome
Based on these foundational studies and with further advances in technology, a second generation of multi-omic platforms has provided additional insight into the biology of immune excluded tumors. Perhaps of greatest impact, single-cell RNA sequencing (scRNAseq) has enabled high-resolution characterization of cellular heterogeneity in the TME and provided a foundation for additional multiplexing methods.(29) That being said, scRNA sequencing depth is limited for each cell whereas bulk RNAseq can provide an entire transcriptome with fewer logistical barriers. Utilizing both bulk and single-cell technologies, researchers identified a significantly higher proportion of TREM2hi macrophages in non-responding versus responding melanoma cohorts treated with PD1 blockade.(30) These PD1-resistant tumors also comprised a significantly higher subset of γδ T cells, an identifiable target with therapeutic implications. Using predictive modeling and the feature genes from these differentially represented cell types, an ‘ICI outcome signature’ was developed and subsequently validated in multiple bulk expression datasets.(30) In separate studies, however, another subset of γδ T cells was shown to strongly correlate with ICI response in a cohort of dMMR colon cancer patients, and increasing evidence now points to a TCF7+CD8+ T cell population highly correlating with improved clinical outcomes to IO therapy.(31,32) Thus, even with the enhanced resolution of scRNAseq, the results from these studies continue to emphasize the immense complexity of the TME, heterogeneity among cancer types, and need for continued advancement of high-resolution platforms. One of these advancements included sequencing of the T cell receptor (TCRseq) itself as a step towards neoantigen prediction. In a proof of concept study, targeted TCRseq showed a significant correlation between TCR clonality and response, with TCR diversity more prognostic of overall survival in an anti-PD1 treated melanoma cohort.(33) With important implications for biomarker development, additional work demonstrated a greater proportion of shared clonotypes between intratumoral and peripheral T cells among NSCLC tumors achieving major pathologic response with neoadjuvant ICI.(34) Moving forward, however, more personalized assays would be needed to identify specific sequences tied to response.
Apart from nucleic acid sequencing, an array of other multi-omic platforms have been developed to further characterize the immune microenvironment and identify targetable mediators of ICI response, including metabolomic profiling and microbiome sequencing. Numerous metabolic processes have been tied to T cell dysfunction and ICI resistance, including macrophage-specific arginine catabolism(35) and tumor-intrinsic oxidative phosphorylation.(36) With advances in liquid chromatography–mass spectrometry (LC-MS) and nuclear magnetic resonance (NMR), however, high-dimensional metabolic profiles can be compared between ICI-resistant and -responding populations. For instance, NMR spectroscopy identified a significant enrichment of pyruvate from anti-PD1-resistant patients with NSCLC,(37) LC-MS detected significantly higher levels of kynurenine in a multi-cohort study of anti-PD1 treated patients with decreased survival,(38) and a specific lipid profile was identified in antigen-stimulated type 1 dendritic cells via untargeted mass spectrometry.(39) Lastly, metagenomic profiles of patient stool samples have revealed significant associations between microbial organisms and ICI efficacy.(40) An intratumoral analysis of melanoma samples similarly revealed distinct bacterial taxa correlating with ICI response.(41) Using a murine tumor model, fecal microbial transplant (FMT) with specific bacterial strains from responding patients was shown to reverse ICI resistance,(42) findings recently validated in early phase clinical trials.(43,44)
Novel multi-omics technology and artificial intelligence (AI)
Multiplexing technologies at the single-cell level
The next generation of omics technology will augment the dimensionality of existing platforms, add spatial resolution to expression data, integrate pharmacologic screening programs, and utilize machine learning (ML) and deep learning (DL) algorithms for optimized neoantigen discovery. With the increased accessibility of scRNAseq, multiplexing technologies now provide tiered levels of biological data using the same high-throughput platform. Examples include CITEseq (cellular indexing of transcriptomes and epitopes) and ATACseq (assay for transposase-accessible chromatin) which can tag specific proteins and epigenetic modifications, respectively, alongside single cell transcript expression. Applying this multiplex technology to models of immune resistance, researchers targeted hundreds of immune-related genes and compared high-resolution expression data using a patient-derived, tumor-TIL co-culture platform.(45) This Perturb-CITE-seq screening assay identified known drivers of immune exclusion, including defects in the IFNγ and antigen presentation pathways, but also novel immune modulators such as CD58, an adhesion protein binding macrophages and T cells, whose knockout decreased both T and NK cell effector activity.(45) In another experiment utilizing single-cell ATACseq, specific regulatory networks were identified and shown to promote the exhaustion of cytotoxic T cells from anti-PD1 treated tumors.(46) While perhaps less applicable to ICI augmentation, the results from these studies carry important implications for the design of adoptive cellular therapy in future trials.
Spatial profiling
As NGS technologies continue to evolve, it has become increasingly clear that the spatial context of these data are needed to fully characterize the tumor-immune interface.(47) For example, factors harboring the metastatic potential of different tumors have remained elusive, but advances in digital spatial profiling (DSP) may provide key insights necessary for therapeutic intervention.(48,49) Given the unique biology and high morbidity associated with brain metastases (BrM), researchers used DSP to compare spatial transcriptomics landscape between primary tumor and BrM tissue from a cohort of NSCLC patients.(50) Multiple “environments” were collected for each patient, including primary tumor, tumor-immune interface, brain metastasis, and brain tumor-immune interface, providing a transcriptome landscape across disease states. Compared to primary tumors, BrMs were enriched in cancer associated fibroblasts (CAFs) and M2-type macrophages, while the brain tumor-immune interface showed decreased lymphocyte infiltration and expression of antigen presentation genes. Additionally, transcripts from the primary tumor-immune interface were compared between fast and slow metastasis cohorts, revealing a ‘metastasis gene signature’ which included S100A11 (associated with the pro-tumor cytokine IL-8) and genes encoding adhesion molecules. By localizing transcript data within the tumor-immune interface, specific pathways can be identified and targeted to potentially decrease the risk of BrM development.(50) Further, with PD1 inhibitors used as first-line treatment for advanced NSCLC lacking driver mutations, these results carry important implications for combination strategies in high-risk patients. An emerging biomarker for ICI response, tertiary lymphoid structures have also been identified with DSP technology, with increasing evidence pointing to a critical immunomodulatory role of oligoclonal B cell populations in the peritumoral region.(51)
Multispectral immunofluorescence (mIF) and other imaging modalities also offer high-resolution spatial information,(52–54) yet establishing meaningful patterns within the exorbitant amount of three-dimensional output data has been problematic. Adapting imaging algorithms from the field of astronomy, researchers established relational databases linking single-cell mIF markers with annotated pathology slides of the tumor-host interface.(55) With this impressive breadth of histopathologic variables, predictive modeling identified biomarkers associated with anti-PD1 response from a cohort of melanoma patients. CD8+FoxP3+ T cells were strongly tied to response, despite prior studies showing suppressive activity, while CD163+PD-L1– myeloid cells, a common phenotypic marker of M2-type macrophages, were associated with therapeutic resistance.(55) While this study accumulated an overwhelming amount of data (>40 TB), it provided a framework by which single-cell proteomic data can characterize tumor-immune interactions and identify drivers of immune exclusion—a critical step forward as computational power continues to grow.
Pharmacologic screening
Though high-dimensional proteomic data can now be used to identify drivers of ICI resistance, pharmacologic targeting remains another challenge for effective combination strategies.(56) Transmembrane proteins can be targeted with cell-surface antibodies and tyrosine kinase inhibitors, but many intracellular proteins (e.g. KRAS) have been deemed “undruggable” for decades.(57) Cbl-b, a ubiquitin-ligase and downstream mediator of CD28 and CTLA4 signaling, is an intracellular protein strongly tied to immune resistance in preclinical models.(58,59) Until recently, pharmacologic inhibition was not possible, but with the advent of DNA encoded libraries (DEL), billions of small molecules can be screened for inhibitory activity against proteins of interest.(60) Using this platform, specific allosteric inhibitors against Cbl-b have been identified for use in both monotherapy and ICI combination trials.(61) Overall, DEL screening helps translate the proteomic data from IO-treated cohorts to novel therapeutic strategies.
Neoantigen identification
While multiple attempts have been made to predict the neoantigenicity of a tumor, specific neoantigen detection has remained challenging. DNAseq and RNAseq can identify thousands of mutations and unique sequences between tumor and host tissue, yet selecting peptides that are both recognized by autologous T cells and elicit a tumor-specific cytotoxic response require additional tools.(62) To develop a predictive model, researchers screened thousands of mutant peptides from individuals with metastatic cancer and identified a subset of 185 neoantigens upon recognition by MHC class I-restricted T cells.(63) Numerous features were profiled and compared between these verified neoantigens and negative candidates, including MHC binding potential, relative expression, variant allele frequency, subcellular localization, and microbial similarity, among others. The resulting ML model was used to calculate an ‘immunogenicity score’ for candidate neoantigen sequences with the goal of more rapid therapeutic targeting.(63) The TCR repertoire can also be used to identify immunogenic antigens in the TME, providing direct access to neoepitope plus HLA data while bypassing the numerous filtering steps between WES to final peptide presentation. Further, the predictive accuracy of HLA genotyping using NGS data alone has been limited.(64) Based on this principle, researchers used a DL algorithm, denoted DeepTCR, to compare the TCR repertoire between ICI responders and non-responders.(65) Notably, antigen-specific signatures were identified in both cohorts, but non-responders were found to exhibit a high turnover of tumor-specific T cells while responders demonstrated a maintenance pool of effector and memory T cells following ICI therapy. Thus, even with a high rate of antigen-specific T cell infiltration, the functional/exhaustive state of these T cells were more predictive of ICI response as compared to the engagement of any one specific neoantigen.(65) As personalized vaccines against tumor associated antigens enter clinical trials,(66) combination strategies may be needed to prevent T cell exhaustion and maintain a neoantigen-specific effector response.
Artificial intelligence
Lastly, ML and DL algorithms can be used to reliably extract features from high-dimensional data and predict treatment response among various anti-neoplastic and immunotherapeutic agents. Starting with a syngeneic murine model, researchers developed a joint dimension reduction framework to incorporate whole transcriptome data, phenotypic variables, and ICI response outcomes across hundreds of tumors.(67) Upon training and validation, this model accurately predicted ICI response in 88% (SE = 1.1%) of treatment-naïve murine samples and also achieved meaningful predictive power with a clinical ICI dataset (AUC = 0.74).(67) Another computational pipeline, ENLIGHT, evolved from an algorithm utilizing synthetic lethality and synthetic rescue genetic interactions to predict treatment outcomes.(68) Again utilizing transcriptomic data, the ENLIGHT model predicted response (OR > 1) in 19 of 21 cohorts treated with ICI or targeted therapies. While NGS technologies provide the historic standard of biological features, digital imaging has emerged as a more cost-efficient platform for high-throughput, high-resolution data extraction.(69) With a vast increase in feature quantity, however, DL has been utilized as a more sophisticated and efficient route for feature transformation and dimensionality reduction. This form of “self-supervision” permits a substantial number of cell-based features (with or without genetic or drug perturbations) to be used for image-based profiling in a new era of response prediction.(69) In addition to cell-based imaging, DL has been used to build CT-based imaging signatures, extracting radiomic features as an independent biomarker for IO response.(70,71) Moving forward, these tools could inform clinical trial design by eliminating patients least likely to benefit from novel IO agents, increasing the likelihood of reaching a statistical benefit in early phase IO trials.
Conclusions: Data integration and framework for developing immunotherapy combinations
While each advancement in omics technology adds another dimension in characterizing the TME, the greatest benefit towards combined immunotherapy strategies involves the integration of these platforms. A computational, multi-omics assessment of genomic mutations, RNAseq, and proteomic data in classically non-IO responsive tumors (e.g. pancreas, breast) found that lymphocyte infiltration and neoantigen predictions did not correlate with immune invasion.(72) Rather, a DNA damage response protein, ATM, was the primary predictor of immunogenic potential, providing rationale for ongoing trials with innate immune agonists in combination with ICI.(72,73) Another study of 80,000 TIL from pancreatic tumors integrated scRNAseq, scTCRseq, and in vivo assays to identify specific features of dysfunctional T cell phenotypes, potentially targetable features to augment adoptive TIL therapy.(74) Importantly, certain aspects of IO resistance remain difficult to measure, including T cell exhaustion and anti-inflammatory immune cell phenotypes—i.e. omic platforms are only useful if there are identifiable data points to collect. By integrating multiple, indirect features associated with various immune phenotypes, however, a more global and accurate characterization can be gathered. Even key clinical variables (e.g. sex) have become controversial predictors of IO response, requiring a more comprehensive analysis of molecular and demographic profiles using multi-omic and AI platforms, with critical implications for trial design.(75)
Moving forward, we propose a framework (Fig. 2) by which in silico screening of integrated, multi-omics data can nominate resistance pathways from existing IO-treated cohorts. Next, genetic knockdown assays using specialized organoids and even organ-on-a-chip models can provide functional validation.(76) Clinical trials utilizing these novel targets in combination with existing IO therapy will also collect multi-omics data in addition to safety and response assessment. Finally, in repeating this cycle, computational models can be built using prospective outcomes data and multi-omic variables to identify biomarkers of response and resistance to these novel combination therapies.
Figure 2.
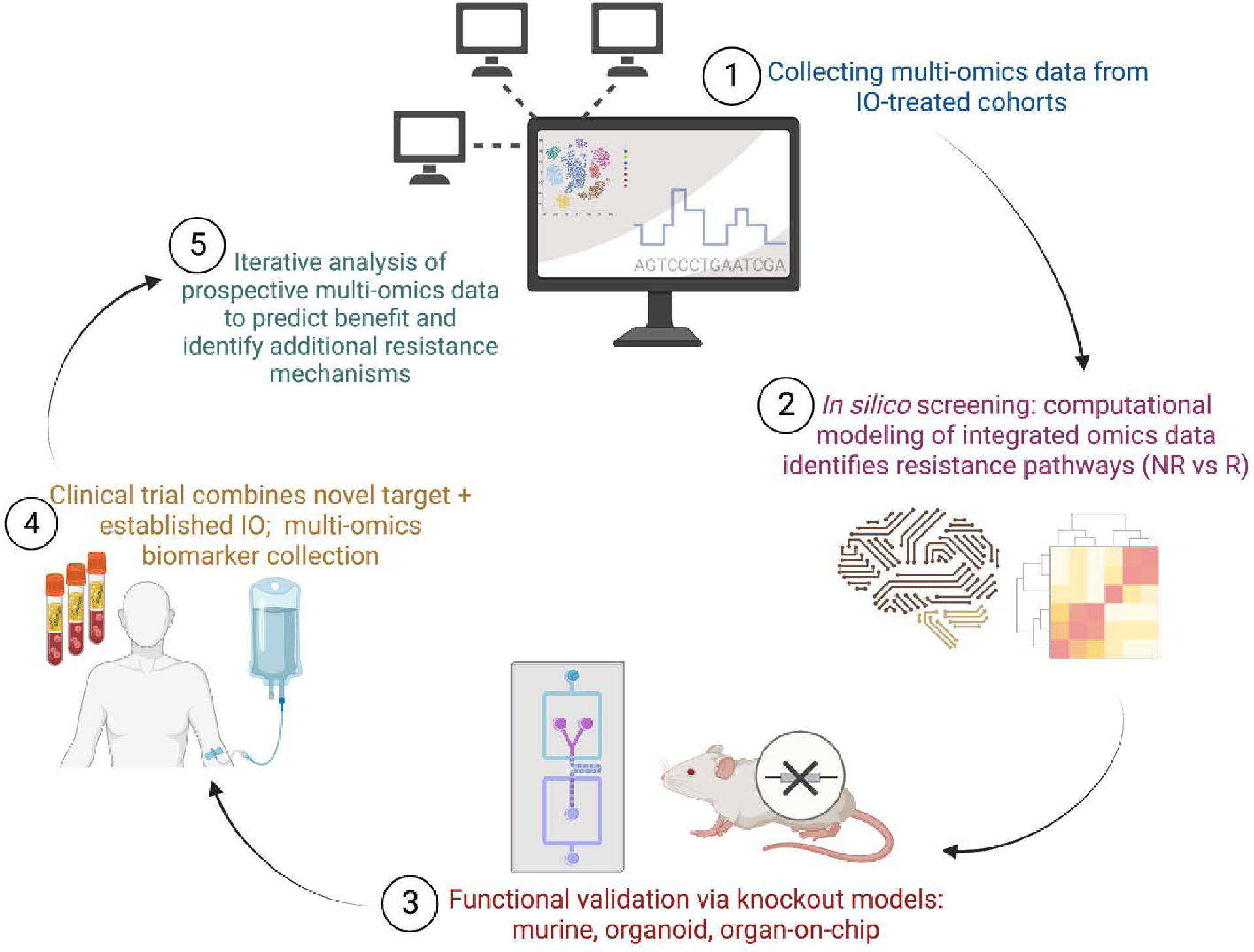
Framework for utilizing multi-omics data to advance immunotherapy combination strategies. An iterative cycle of in silico screening, functional validation, and clinical trial biomarker collection can be used to identify novel resistance pathways, develop predictive models of benefit, and highlight additional resistance mechanisms.
In step 1, multi-omics and/or image data is collected from clinically annotated or biologically distinct patient cohorts, using bulk RNAseq, single-cell RNAseq, multispectral imaging, spatial profiling, among others. These data are computationally integrated and compared between IO-non-responders and IO-responders along with non-T cell-inflamed versus T cell-inflamed tumors samples. This in silico screening identifies candidate genes and molecular pathways that may be associated with IO-resistance and/or immune exclusion for therapeutic target nomination (step 2). In step 3, functional assays are used to experimentally validate the immune-exclusive role of these candidate genes/pathways and assess novel inhibitors and other agents. Based on these preclinical data, early phase clinical trials combine novel pathway agents with established IO therapies; clinical outcome along with translational biomarker data is collected (step 4). In returning to the start of the cycle (step 5), multi-omics biomarker data from these novel clinical trials are used to identify predictors of response as well as additional genes/pathways tied to therapy resistance.
IO, immuno-oncology; NR, non-responder; R, responder
Figure made with BioRender
Support:
This work was supported by National Institutes of Health (NIH) Grant R01DE031729 (R.B., J.J.L.), P50CA097190 (R.B.), UM1CA186690 (J.J.L.), P50CA254865 (R.B., J.J.L.), in part by National Cancer Institute through the UPMC Hillman Cancer Center CCSG award (P30CA047904).
Footnotes
Disclosures: R.C.A. and W.C. have no declartions. J.J.L. declares DSMB: Abbvie, Agenus, Immutep, Evaxion; Scientific Advisory Board: (no stock) 7 Hills, Affivant, BioCytics, Bright Peak, Exo, Fstar, Inzen, RefleXion, Xilio (stock) Actym, Alphamab Oncology, Arch Oncology, Duke Street Bio, Kanaph, Mavu, NeoTx, Onc.AI, OncoNano, physIQ, Pyxis, Saros, STipe, Tempest; Consultancy with compensation: Abbvie, Agenus, Alnylam, AstraZeneca, Atomwise, Bayer, Bristol-Myers Squibb, Castle, Checkmate, Codiak, Crown, Cugene, Curadev, Day One, Eisai, EMD Serono, Endeavor, Flame, G1 Therapeutics, Genentech, Gilead, Glenmark, HotSpot, Kadmon, Ko Bio Labs, Krystal, KSQ, Janssen, Ikena, Inzen, Immatics, Immunocore, Incyte, Instil, IO Biotech, LegoChem, Macrogenics, Merck, Mersana, Nektar, Novartis, Partner, Pfizer, Pioneering Medicines, PsiOxus, Regeneron, Replimmune, Ribon, Roivant, Servier, STINGthera, Sumoitomo, Synlogic, Synthekine, Teva; Research Support: (all to institution for clinical trials unless noted) AbbVie, Astellas, Astrazeneca, Bristol-Myers Squibb, Corvus, Day One, EMD Serono, Fstar, Genmab, Hot Spot, Ikena, Immatics, Incyte, Kadmon, KAHR, Macrogenics, Merck, Moderna, Nektar, Next Cure, Novartis, Numab, Palleon, Pfizer, Replimmune, Rubius, Servier, Scholar Rock, Synlogic, Takeda, Trishula, Tizona, Xencor; Patents: US-11638728 (Microbiome Biomarkers for Anti-PD-1/PD-L1 Responsiveness: Diagnostic, Prognostic and Therapeutic Uses Thereof); R.B. declares PCT/US15/612657 (Cancer Immunotherapy), PCT/US18/36052 (Microbiome Biomarkers for Anti-PD-1/PD-L1 Responsiveness: Diagnostic, Prognostic and Therapeutic Uses Thereof), PCT/US63/055227 (Methods and Compositions for Treating Autoimmune and Allergic Disorders).
References
- 1.de Castro G, Kudaba I, Wu YL, Lopes G, Kowalski DM, Turna HZ, et al. Five-Year Outcomes With Pembrolizumab Versus Chemotherapy as First-Line Therapy in Patients With Non–Small-Cell Lung Cancer and Programmed Death Ligand-1 Tumor Proportion Score ≥ 1% in the KEYNOTE-042 Study. J Clin Oncol. 2022. Oct 28;JCO.21.02885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. CheckMate 067: 6.5-year outcomes in patients (pts) with advanced melanoma. J Clin Oncol. 2021. May 20;39(15_suppl):9506–9506. [Google Scholar]
- 3.Robert C A decade of immune-checkpoint inhibitors in cancer therapy. Nat Commun. 2020. Jul 30;11(1):3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGrail DJ, Pilié PG, Rashid NU, Voorwerk L, Slagter M, Kok M, et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann Oncol. 2021. May 1;32(5):661–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li H, van der Merwe PA, Sivakumar S. Biomarkers of response to PD-1 pathway blockade. Br J Cancer. 2022. Jun;126(12):1663–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Research C for DE and. FDA approves Opdualag for unresectable or metastatic melanoma. FDA; [Internet]. 2022. Mar 21 [cited 2022 Sep 15]; Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-opdualag-unresectable-or-metastatic-melanoma [Google Scholar]
- 7.Kim TW, Burris HA III, de Miguel Luken MJ, Pishvaian MJ, Bang YJ, Gordon M, et al. First-In-Human Phase I Study of the OX40 Agonist MOXR0916 in Patients with Advanced Solid Tumors. Clin Cancer Res. 2022. Aug 15;28(16):3452–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.GSK provides update on feladilimab, an investigational inducible T cell co-stimulatory (ICOS) agonist | GSK; [Internet]. 2021. [cited 2023 Feb 18]. Available from: https://www.gsk.com/en-gb/media/press-releases/gsk-provides-update-on-feladilimab-an-investigational-inducible-t-cell-co-stimulatory-icos-agonist/ [Google Scholar]
- 9.Idera Pharmaceuticals Announces Results From ILLUMINATE-301 Trial of Tilsotolimod + Ipilimumab in anti-PD-1 Refractory Advanced Melanoma | Idera Pharmaceuticals, Inc. [Internet]. [cited 2022 Sep 15]. Available from: https://ir.iderapharma.com/news-releases/news-release-details/idera-pharmaceuticals-announces-results-illuminate-301-trial [Google Scholar]
- 10.Ma A, Xin G, Ma Q. The use of single-cell multi-omics in immuno-oncology. Nat Commun. 2022. May 18;13(1):2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017. Aug 1;127(8):2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spranger S, Luke JJ, Bao R, Zha Y, Hernandez KM, Li Y, et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci U S A. 2016. Nov 29;113(48):E7759–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Danaher P, Warren S, Lu R, Samayoa J, Sullivan A, Pekker I, et al. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): results from The Cancer Genome Atlas (TCGA). J Immunother Cancer. 2018. Dec 1;6(1):63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bao R, Stapor D, Luke JJ. Molecular correlates and therapeutic targets in T cell-inflamed versus non-T cell-inflamed tumors across cancer types. Genome Med. 2020. Oct 27;12(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spranger S, Gajewski TF. Mechanisms of Tumor Cell–Intrinsic Immune Evasion. Annu Rev Cancer Biol. 2018;2(1):213–28. [Google Scholar]
- 16.Du W, Pasca di Magliano M, Zhang Y. Therapeutic Potential of Targeting Stromal Crosstalk-Mediated Immune Suppression in Pancreatic Cancer. Front Oncol. 2021;11:682217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brady SW, McQuerry JA, Qiao Y, Piccolo SR, Shrestha G, Jenkins DF, et al. Combating subclonal evolution of resistant cancer phenotypes. Nat Commun. 2017. Nov 1;8(1):1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Augustin RC, Bao R, Luke JJ. Targeting Cbl-b in cancer immunotherapy. J Immunother Cancer. 2023. Feb 7;11(2):e006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li S, Simoni Y, Zhuang S, Gabel A, Ma S, Chee J, et al. Characterization of neoantigen-specific T cells in cancer resistant to immune checkpoint therapies. Proc Natl Acad Sci. 2021. Jul 27;118(30):e2025570118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rehm HL, Page AJH, Smith L, Adams JB, Alterovitz G, Babb LJ, et al. GA4GH: International policies and standards for data sharing across genomic research and healthcare. Cell Genomics. 2021. Nov 10;1(2):100029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The Cancer Genome Atlas Program - NCI [Internet]. 2018. [cited 2023 Feb 18]. Available from: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga
- 22.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N Engl J Med. 2014. Dec 4;371(23):2189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science. 2015. Apr 3;348(6230):124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Research C for DE and. FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication. FDA; [Internet]. 2019. Feb 9 [cited 2023 Feb 9]; Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pembrolizumab-first-tissuesite-agnostic-indication [Google Scholar]
- 25.Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020. Oct 1;21(10):1353–65. [DOI] [PubMed] [Google Scholar]
- 26.Research C for DE and. FDA approves pembrolizumab for adults and children with TMB-H solid tumors. FDA; [Internet]. 2020. Jun 17 [cited 2023 Aug 27]; Available from: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-pembrolizumab-adults-and-children-tmb-h-solid-tumors [Google Scholar]
- 27.Cortez MA, Masrorpour F, Ivan C, Zhang J, Younes A, Lu Y, et al. Bone morphogenetic protein 7 promotes resistance to immunotherapy. Nat Commun. 2020. Sep 24;11:4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013. Aug 28;5(200):200ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Erfanian N, Derakhshani A, Nasseri S, Fereidouni M, Baradaran B, Jalili Tabrizi N, et al. Immunotherapy of cancer in single-cell RNA sequencing era: A precision medicine perspective. Biomed Pharmacother Biomedecine Pharmacother. 2022. Feb;146:112558. [DOI] [PubMed] [Google Scholar]
- 30.Xiong D, Wang Y, You M. A gene expression signature of TREM2hi macrophages and γδ T cells predicts immunotherapy response. Nat Commun. 2020. Oct 8;11:5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Vries NL, van de Haar J, Veninga V, Chalabi M, Ijsselsteijn ME, van der Ploeg M, et al. γδ T cells are effectors of immunotherapy in cancers with HLA class I defects. Nature. 2023;613(7945):743–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell. 2018. Nov 1;175(4):998–1013.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valpione S, Mundra PA, Galvani E, Campana LG, Lorigan P, De Rosa F, et al. The T cell receptor repertoire of tumor infiltrating T cells is predictive and prognostic for cancer survival. Nat Commun. 2021. Jul 2;12(1):4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang J, Ji Z, Caushi JX, El Asmar M, Anagnostou V, Cottrell TR, et al. Compartmental Analysis of T-cell Clonal Dynamics as a Function of Pathologic Response to Neoadjuvant PD-1 Blockade in Resectable Non-Small Cell Lung Cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2020. Mar 15;26(6):1327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Menjivar RE, Nwosu ZC, Du W, Donahue KL, Hong HS, Espinoza C, et al. Arginase 1 is a key driver of immune suppression in pancreatic cancer. DeNicola GM, editor. eLife. 2023. Feb 2;12:e80721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol Res. 2017. Jan;5(1):9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghini V, Laera L, Fantechi B, del Monte F, Benelli M, McCartney A, et al. Metabolomics to Assess Response to Immune Checkpoint Inhibitors in Patients with Non-Small-Cell Lung Cancer. Cancers. 2020. Nov 30;12(12):3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, Bullock K, Gurjao C, Braun D, Shukla SA, Bossé D, et al. Metabolomic adaptations and correlates of survival to immune checkpoint blockade. Nat Commun. 2019. Sep 25;10:4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Basit F, van Oorschot T, van Buggenum J, Derks RJE, Kostidis S, Giera M, et al. Metabolomic and lipidomic signatures associated with activation of human cDC1 (BDCA3+/CD141+) dendritic cells. Immunology. 2022;165(1):99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCulloch JA, Davar D, Rodrigues RR, Badger JH, Fang JR, Cole AM, et al. Intestinal microbiota signatures of clinical response and immune-related adverse events in melanoma patients treated with anti-PD-1. Nat Med. 2022. Mar;28(3):545–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type–specific intracellular bacteria. Science. 2020. May 29;368(6494):973–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science. 2018. Jan 5;359(6371):91–7. [DOI] [PubMed] [Google Scholar]
- 43.Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin JM, Morrison RM, et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science. 2021. Feb 5;371(6529):595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Routy B, Lenehan JG, Miller WH, Jamal R, Messaoudene M, Daisley BA, et al. Fecal microbiota transplantation plus anti-PD-1 immunotherapy in advanced melanoma: a phase I trial. Nat Med. 2023. Jul 6;1–12. [DOI] [PubMed] [Google Scholar]
- 45.Frangieh CJ, Melms JC, Thakore PI, Geiger-Schuller KR, Ho P, Luoma AM, et al. Multimodal pooled Perturb-CITE-seq screens in patient models define mechanisms of cancer immune evasion. Nat Genet. 2021. Mar;53(3):332–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat Biotechnol. 2019. Aug;37(8):925–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bassiouni R, Gibbs LD, Craig DW, Carpten JD, McEachron TA. Applicability of spatial transcriptional profiling to cancer research. Mol Cell. 2021. Apr 15;81(8):1631–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu Q, Jiang M, Wu L. Spatial transcriptomics technology in cancer research. Front Oncol. 2022. Oct 13;12:1019111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hunter MV, Moncada R, Weiss JM, Yanai I, White RM. Spatially resolved transcriptomics reveals the architecture of the tumor-microenvironment interface. Nat Commun. 2021. Nov 1;12(1):6278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Q, Abdo R, Iosef C, Kaneko T, Cecchini M, Han VK, et al. The spatial transcriptomic landscape of non-small cell lung cancer brain metastasis. Nat Commun. 2022. Oct 10;13(1):5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020. Jan;577(7791):549–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goltsev Y, Nolan G. CODEX multiplexed tissue imaging. Nat Rev Immunol. 2023. Aug 31;1–1. [DOI] [PubMed] [Google Scholar]
- 53.He S, Bhatt R, Brown C, Brown EA, Buhr DL, Chantranuvatana K, et al. High-plex imaging of RNA and proteins at subcellular resolution in fixed tissue by spatial molecular imaging. Nat Biotechnol. 2022. Dec;40(12):1794–806. [DOI] [PubMed] [Google Scholar]
- 54.Bosisio FM, Van Herck Y, Messiaen J, Bolognesi MM, Marcelis L, Van Haele M, et al. Next-Generation Pathology Using Multiplexed Immunohistochemistry: Mapping Tissue Architecture at Single-Cell Level. Front Oncol. 2022. Jul 29;12:918900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berry S, Giraldo NA, Green BF, Cottrell TR, Stein JE, Engle EL, et al. Analysis of multispectral imaging with the AstroPath platform informs efficacy of PD-1 blockade. Science. 2021. Jun 11;372(6547):eaba2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dang CV, Reddy EP, Shokat KM, Soucek L. Drugging the “undruggable” cancer targets. Nat Rev Cancer. 2017. Aug;17(8):502–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Asimgil H, Ertetik U, Çevik NC, Ekizce M, Doğruöz A, Gökalp M, et al. Targeting the undruggable oncogenic KRAS: the dawn of hope. JCI Insight. 2022. Jan 11;7(1):e153688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Q, Zhou H, Langdon WY, Zhang J. E3 ubiquitin ligase Cbl-b in innate and adaptive immunity. Cell Cycle. 2014. Jun 15;13(12):1875–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wallner S, Gruber T, Baier G, Wolf D. Releasing the brake: targeting Cbl-b to enhance lymphocyte effector functions. Clin Dev Immunol. 2012;2012:692639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Satz AL, Brunschweiger A, Flanagan ME, Gloger A, Hansen NJV, Kuai L, et al. DNA-encoded chemical libraries. Nat Rev Methods Primer. 2022. Jan 17;2(1):1–17. [Google Scholar]
- 61.Sharp A, Williams A, Blagden SP, Plummer ER, Hochhauser D, Krebs M, et al. A first-in-human phase 1 trial of nx-1607, a first-in-class oral CBL-B inhibitor, in patients with advanced solid tumor malignancies. J Clin Oncol. 2022. Jun;40(16_suppl):TPS2691–TPS2691. [Google Scholar]
- 62.Yarchoan M, Johnson BA, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017. Apr;17(4):209–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gartner JJ, Parkhurst MR, Gros A, Tran E, Jafferji MS, Copeland A, et al. A machine learning model for ranking candidate HLA class I neoantigens based on known neoepitopes from multiple human tumor types. Nat Cancer. 2021. May;2(5):563–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bauer DC, Zadoorian A, Wilson LOW, Melbourne Genomics Health Alliance, Thorne NP. Evaluation of computational programs to predict HLA genotypes from genomic sequencing data. Brief Bioinform. 2018. Mar 1;19(2):179–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sidhom JW, Oliveira G, Ross-MacDonald P, Wind-Rotolo M, Wu CJ, Pardoll DM, et al. Deep learning reveals predictive sequence concepts within immune repertoires to immunotherapy. Sci Adv. 8(37):eabq5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ott PA, Hu-Lieskovan S, Chmielowski B, Govindan R, Naing A, Bhardwaj N, et al. A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-small Cell Lung Cancer, or Bladder Cancer. Cell. 2020. Oct 15;183(2):347–362.e24. [DOI] [PubMed] [Google Scholar]
- 67.Zeng Z, Gu SS, Wong CJ, Yang L, Ouardaoui N, Li D, et al. Machine learning on syngeneic mouse tumor profiles to model clinical immunotherapy response. Sci Adv. 8(41):eabm8564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dinstag G, Shulman ED, Elis E, Ben-Zvi DS, Tirosh O, Maimon E, et al. Clinically oriented prediction of patient response to targeted and immunotherapies from the tumor transcriptome. Med N Y N. 2023. Jan 13;4(1):15–30.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chandrasekaran SN, Ceulemans H, Boyd JD, Carpenter AE. Image-based profiling for drug discovery: due for a machine-learning upgrade? Nat Rev Drug Discov. 2021;20(2):145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saad MB, Hong L, Aminu M, Vokes NI, Chen P, Salehjahromi M, et al. Predicting benefit from immune checkpoint inhibitors in patients with non-small-cell lung cancer by CT-based ensemble deep learning: a retrospective study. Lancet Digit Health. 2023. Jul 1;5(7):e404–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Colen RR, Rolfo C, Ak M, Ayoub M, Ahmed S, Elshafeey N, et al. Radiomics analysis for predicting pembrolizumab response in patients with advanced rare cancers. J Immunother Cancer. 2021. Apr 13;9(4):e001752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McGrail DJ, Federico L, Li Y, Dai H, Lu Y, Mills GB, et al. Multi-omics analysis reveals neoantigen-independent immune cell infiltration in copy-number driven cancers. Nat Commun. 2018. Apr 3;9(1):1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rothlin CV, Ghosh S. Lifting the innate immune barriers to antitumor immunity. J Immunother Cancer. 2020. Apr 1;8(1):e000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schalck A, Sakellariou-Thompson D, Forget MA, Sei E, Hughes TG, Reuben A, et al. Single-Cell Sequencing Reveals Trajectory of Tumor-Infiltrating Lymphocyte States in Pancreatic Cancer. Cancer Discov. 2022. Oct 5;12(10):2330–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ye Y, Jing Y, Li L, Mills GB, Diao L, Liu H, et al. Sex-associated molecular differences for cancer immunotherapy. Nat Commun. 2020. Apr 14;11(1):1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ingber DE. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat Rev Genet. 2022. Aug;23(8):467–91. [DOI] [PMC free article] [PubMed] [Google Scholar]