

Summary
Hypoxia-inducible factor-1α (HIF1α) attenuates mitochondrial activity while promoting glycolysis. However, lower glycolysis is compromised in human clear cell renal cell carcinomas, in which HIF1α acts as a tumor suppressor by inhibiting cell-autonomous proliferation. Here, we find that, unexpectedly, HIF1α suppresses lower glycolysis after the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) step, leading to reduced lactate secretion in different tumor cell types when cells encounter a limited pyruvate supply such as that typically found in the tumor microenvironment in vivo. This is because HIF1α-dependent attenuation of mitochondrial oxygen consumption increases the NADH/NAD+ ratio that suppresses the activity of the NADH-sensitive GAPDH glycolytic enzyme. This is manifested when pyruvate supply is limited, since pyruvate acts as an electron acceptor that prevents the increment of the NADH/NAD+ ratio. Furthermore, this anti-glycolytic function provides a molecular basis to explain how HIF1α can suppress tumor cell proliferation by increasing the NADH/NAD+ ratio.
Keywords: HIF1α, glycolysis, oxygen, renal cell carcinoma, mitochondria, NADH, hypoxia, tumor
Graphical abstract
Highlights
-
•
HIF1α suppresses lower glycolysis when pyruvate supply is limited
-
•
Insufficient pyruvate facilitates HIF1α-dependent elevation of NADH/NAD+ ratio
-
•
Inhibition of NADH/NAD+ elevation restores lower glycolysis upon HIF1α activation
-
•
HIF1α-dependent increase in NADH/NAD+ ratio reduces cell proliferation
In this article, Urrutia et al. explain the metabolic pattern of ccRCC tumors characterized by the suppression of lower glycolysis and constitutive HIF1α activation. Urrutia et al. show that HIF1α elevates the NADH/NAD+ ratio, which attenuates lower glycolysis when pyruvate supply is limited, as occurs in the solid tumor microenvironment.
Introduction
Hypoxia-inducible factors (HIFs) are transcription factors central to cellular adaptation to insufficient oxygen supply. HIFs are heterodimeric transcription factors composed of an α (HIF1α, HIF2α, or HIF3α) and a β subunit (HIFβ) also named the aryl hydrocarbon receptor nuclear translocator. Whereas the HIFβ subunit is constitutively expressed, the stability of the HIFα subunits is controlled by the prolyl-4-hydroxylase (PHD) domain proteins (PHD1, PHD2, and PHD3), which are 2-oxoglutarate-dependent Fe2+-dioxygenases.1,2 When oxygen is available (i.e., in normoxic conditions), PHDs can use this oxygen to hydroxylate two conserved proline residues in the HIFα subunits, which can then be recognized by the von Hippel Lindau (VHL)/E3 ubiquitin ligase complex, which targets the HIFα subunits for proteasomal degradation.3,4,5,6,7 By contrast, in hypoxic conditions, there is insufficient oxygen for the PHDs to hydroxylate the HIFα subunits, which prevents their recognition by VHL/E3 and results in their stabilization. HIFα subunits can also be stabilized in normoxic conditions by treatment with pharmacological inhibitors of PHDs, such as dimethyloxalylglycine (DMOG).5,6 Moreover, HIFα subunits are also constitutively stabilized in human clear cell renal cell carcinomas (ccRCCs) due to the loss of VHL.8,9,10 When HIFα subunits are stabilized, they can shuttle to the nucleus, where they heterodimerize with HIFβ subunits and bind to DNA at hypoxia response elements of target genes, thereby driving a HIF-dependent transcriptional program.11,12,13
A key role of the HIF pathway is to rewire cell metabolism to save oxygen, inducing an anaerobic switch whereby cellular oxidative pathways are attenuated while glycolysis is potentiated. HIF1α attenuates glucose and glutamine oxidation through the tricarboxylic acid (TCA) cycle14,15,16,17,18,19 and directly reduces the activity of different mitochondrial respiratory chain complexes as well as the mitochondrial content. Indeed, HIF1α induces key genes involved in the suppression of mitochondrial complex I activity and assembly,20,21,22,23 while also reducing the expression of complex II subunits, and rewires the activity and expression of mitochondrial complex IV subunits.24,25 In sharp contrast, HIF1α can potentiate glycolysis to assure ATP generation in an oxygen- and mitochondrial-independent manner. In this context, HIF1α induces the transcription of genes encoding glycolytic enzymes and glucose transporters.26,27,28 Moreover, the expression of glycolytic enzymes is elevated in ccRCC, which is characterized by the loss of VHL activity and, hence, constitutive activation of HIF1α isoforms.29 However, the abundance of lower glycolysis intermediates is reduced in ccRCC, despite the constitutive HIF1α activation in these carcinomas.30,31 These metabolomics data are difficult to reconcile with the aforementioned potential of HIF1α to enhance the expression of glycolytic enzymes, and the molecular basis underlying this effect still has to be explored.
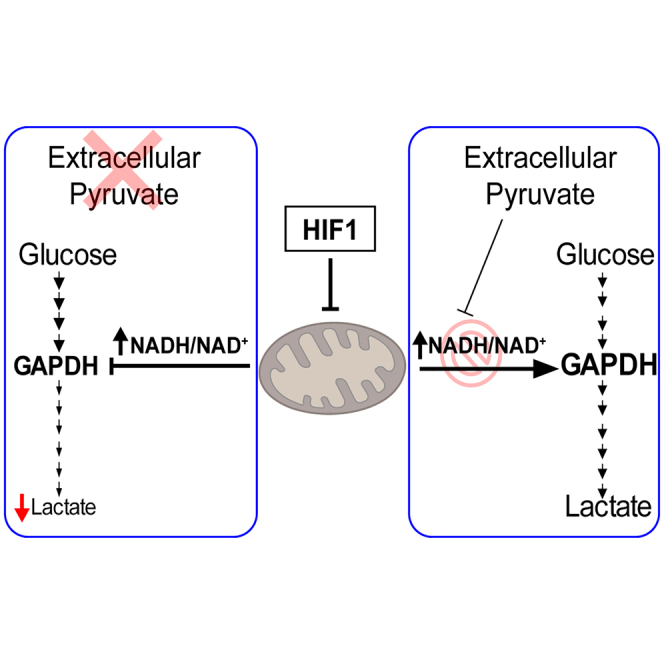
Here, we show that HIF1α not only reduces mitochondrial oxygen consumption but also suppresses lower glycolysis leading to reduced lactate secretion in different cell models including VHL-deficient ccRCC, glioblastoma, and breast cancer cells. This is a consequence of HIF1α-dependent suppression of mitochondrial activity, which leads to an elevation of the cellular NADH/NAD+ ratio that reduces glycolytic flux and the abundance of lower glycolysis metabolites after the reaction catalyzed by the NADH-sensitive glyceraldehyde 3-phosphate dehydrogenase (GAPDH). This unanticipated HIF1α-dependent suppression of glycolytic flux is operative in conditions of limited extracellular pyruvate supply typically found in the solid tumor microenvironment. This is because exogenous pyruvate reduces the excess of NADH into NAD+ after its conversion to lactate through potentiation lactate dehydrogenase (LDH) activity, impeding the HIF1α-dependent increment of the NADH/NAD+ ratio. Therefore, it is likely that this HIF1α-dependent suppression of lower glycolysis was not observed previously due to the high concentrations of pyruvate normally used in culture media, in which the role of HIF1α on glycolysis has generally been studied. Along a similar line, we also found that HIF1α-dependent inhibition of lower glycolysis provides a metabolic basis for the anti-proliferative potential of HIF1α, which is especially manifested in ccRCC, where HIF1α acts as a tumor suppressor by inhibiting ccRCC cell proliferation, as mentioned above.
Results
HIF1α-dependent reduction of lower glycolysis
A central role of the HIF1α isoform is the transcriptional upregulation of genes encoding glycolytic enzymes,26,27 which would suggest that VHL-deficient ccRCC should display a marked elevation in glycolytic metabolites, as these tumors are characterized by constitutive HIF1α activation. However, metabolomic profiling of ccRCC showed a reduction in lower glycolytic metabolites in these tumors when compared to the adjacent healthy renal tissue.30,31 Indeed, a more detailed analysis of available data corresponding to individual ccRCC samples shown in the study of Hakimi et al. revealed that many of these tumor samples had reduced levels of the lower glycolytic intermediates relative to the healthy renal tissue, such as phosphoglycerates (2-PG and 3-PG) and phosphoenolpyruvate (PEP) (Figure 1A), whereas the levels of the upper glycolytic metabolites like glucose 6-phosphate and fructose 6-phosphate were elevated in the vast majority of ccRCC samples analyzed (Figure 1A). Moreover, the mean value for lactate was much lower than the upper glycolytic intermediates, and in some of these ccRCC samples, lactate levels were surprisingly lower than the adjacent healthy renal tissue (Figure 1A) (see also discussion).
Figure 1.

HIF1α-dependent attenuation of glucose-dependent biosynthesis of glycolytic intermediates and lactate release in VHL-deficient renal cell carcinoma cells
(A) Relative content of glycolytic intermediates in 138 human VHL-deficient renal cell carcinomas compared to their adjacent kidney tissue. The graph shows those samples in which metabolite induction is positive (red) and negative (blue).
(B) Scheme of glycolysis indicating those that are upper and lower glycolytic intermediates.
(C) Total metabolite abundance of intracellular glyceraldehyde-3-phosphate (GAP), dihydroxyacetone phosphate (DHAP), phosphoglycerates (2/3-PGs), and phosphoenolpyruvate (PEP) in 786-O-HIF1α (n = 3) and control cells (n = 3).
(D) Total metabolite abundance of intracellular lactate in 786-O-HIF1α cells (n = 3) and control cells (n = 3).
(E) Relative amount of extracellular lactate released by 786-O-HIF1α cells, 786-O-HIF1α-bHLH∗ cells, and control cells cultured in media at glucose (Glc) concentrations of 25 and 5 mM (n = 16–24).
In the bar graphs, the values represent the mean ± SEM, and statistical analysis was performed using a two-tailed unpaired t test when comparing 2 groups and two-way ANOVA followed by Tukey’s post hoc test for (E): ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 represent p values.
Then, we evaluated whether this unexpected glycolytic profile could be driven by HIF1α activation in VHL-deficient ccRCC cells. First, we used VHL-deficient 786-O cells, which lack HIF1α expression. This is a widely recognized cellular model to assess the biological activity of HIF1α in ccRCC cells by restoring HIF1α expression (Figure S1A).18,32 HIF1α activity was confirmed by measuring the expression of carbonic anhydrase IX (CAIX), a HIF1α-dependent gene in ccRCC,32 showing that CAIX was more strongly expressed in HIF1α-transduced cells than in the control 786-O cells (Figure S1A). Next, we assessed the content of upper and lower glycolytic intermediates (Figure 1B). We observed that the total content of upper glycolytic intermediates like glyceraldehyde-3-phosphate (GAP) and dihydroxyacetone phosphate (DHAP) was elevated in 786-O-HIF1α cells (Figure 1C). Conversely, there was less content of lower glycolytic intermediates like 2/3-PGs and PEP in 786-O-HIF1α cells (Figure 1C), in line with the metabolic data from human ccRCC samples. These data suggest that the suppression of lower glycolysis observed in human ccRCC samples is a consequence of HIF1α activation in these tumors. Moreover, our data also suggest that reduction of lower glycolysis occurs at the GAPDH reaction because the levels of GAP (the substrate of GAPDH) were elevated upon HIF1α activation, whereas there was a reduction in the lower glycolytic intermediates downstream of the GAPDH reaction.
Reduced lactate release upon HIF1α activation
In line with the HIF1α-dependent suppression of lower glycolysis, intracellular lactate levels were reduced in 786-O-HIF1α cells (Figure 1D). This was confirmed when 786-O cells were cultured in different glucose concentrations, including a concentration of 5 mM that is closer to physiological extracellular glucose concentrations (Figure 1E). Moreover, lactate release was not affected when we expressed in 786-O cells a mutant inactive HIF1α DNA-binding domain (HIF1α basic-helix-loop-helix [HIF1α-bHLH]∗) version that is not able to bind the DNA (Figures 1E and S1A), confirming that the attenuation of lactate release depends on HIF1α transcriptional activity. Then, we assessed whether endogenous HIF1α could also reduce lactate release in other cellular models. Hence, we exposed G55 glioblastoma or MDA-MB-231 breast cancer cells to hypoxia or treated them with DMOG, a PHD oxygen sensor inhibitor, which leads to endogenous HIF1α stabilization. First, we confirmed that hypoxia and DMOG-treated cells induced an increase of HIF1α protein expression as well as of its target gene CAIX (Figure S1B). Exposure to either hypoxia or DMOG provoked a remarkable decline in lactate release in G55 glioblastoma cells (Figure 2A). Similar results were found in MDA-MB-231 breast cancer cells exposed to DMOG (Figures 2B and S1B). Moreover, silencing endogenous HIF1α in G55 and MDA-MB-231 cells largely attenuated the decline in lactate release upon DMOG treatment (Figures 2C, 2D, and S1C). Hence, lactate release is attenuated not only by restoring HIF1α in 786-O renal cells but also by activating endogenous HIF1α in other cell types, such as G55 and MDA-MB-231 cells. Notably, this effect was not evident in cell cultures of A549 lung carcinoma cells or murine embryonic fibroblasts (MEFs) at doses of DMOG that enhanced HIF1α-dependent target genes and HIF1α protein levels (Figures 2E and S1D).
Figure 2.

Suppression of lactate release upon endogenous HIF1α activation in breast cancer and glioblastoma cells
(A) Relative amount of extracellular lactate released by G55 control cells (n = 9) and G55 cells exposed to hypoxia (n = 3) or treated with DMOG (0.1 mM, n = 6 or 0.2 mM, n = 4).
(B) Relative amount of extracellular lactate released by MDA-MB-231 control cells or those treated with 0.5 mM DMOG (n = 6).
(C) Relative amount of extracellular lactate released by G55 control cells, HIF1α-silenced G55 cells, G55 cells treated with 0.2 mM DMOG, or HIF1α-silenced G55 cells treated with 0.2 mM DMOG (n = 6).
(D) Relative amount of extracellular lactate released by MDA-MB-231 control cells, HIF1α-silenced MDA-MB-231 cells, MDA-MB-231 cells treated with 0.5 mM DMOG, and HIF1α-silenced MDA-MB-231 cells treated with 0.5 mM DMOG (n = 5).
(E) Relative amount of extracellular lactate released by A549 control cells and A549 cells treated with 1 mM DMOG (n = 9), control murine embryonic fibroblasts (MEFs), and MEFs treated with 0.2 mM DMOG (n = 6).
In the bar graphs, the values represent the mean ± SEM, and statistical analysis was performed using a two-tailed unpaired t test when comparing 2 groups, with Welch’s correction when needed, or one-way or two-way ANOVA followed by Tukey’s post hoc test for (A) and (C) and for (D), respectively: ∗p < 0.05 and ∗∗∗p < 0.001 represent significant p values.
Collectively, these data show that HIF1α has the unexpected potential to compromise the abundance of lower glycolysis metabolites resulting in reduced lactate release in some biological settings including ccRCCs.
Extracellular pyruvate restores glycolytic flux in HIF1α-expressing cells
It is generally accepted that glycolysis is a metabolic pathway in which glucose is simply converted to lactate in a cell-autonomous manner without the requirement of extracellular metabolites different than glucose. For this reason, the glycolytic metabolite measurements shown above were performed in the absence of exogenous pyruvate. Of note, although we did not add pyruvate, and this might be considered an extreme condition, it should be noted that it does not mean that there is not pyruvate in the media. This is because cells secrete endogenous pyruvate into the medium reaching concentrations we found to range from 5.8 ± 0.91 to 24.86 ± 1.36 μM (n = 4), which also resembles the levels of pyruvate found in the tumor microenvironment.33,34,35,36 We therefore wondered whether this unexpected HIF1α-dependent suppression of glycolysis could be recovered by extracellular pyruvate, which is a lower glycolysis metabolite that can enter into the cells using specific carriers such as monocarboxylate transporters.37,38,39 Indeed, we found that exogenous pyruvate fully restored the reduced lactate release in 786-O-HIF1α cells (Figure 3A). Moreover, the reduced lactate secretion in G55 or MDA-MB-231 cells exposed to hypoxia or treated with DMOG was also fully prevented in the presence of extracellular pyruvate (Figures 3B–3D). Then, we wondered whether the restoration of lactate release was a consequence of the recovery of glycolytic flux in HIF1α-expressing cells. To assess glycolytic flux, we incubated cells with D-[5-3H]-glucose and measured the 3H2O production during the lower glycolysis reaction catalyzed by enolase, which converts [2-3H]-2-PG to PEP (Figure 3E). Importantly, we found that glycolytic flux was significantly reduced in G55 cells treated with DMOG and 786-O-HIF1α cells and that this effect was reversed in the presence of exogenous pyruvate (Figures 3F and 3G). In agreement with these data, we also found that the accumulation of upper glycolytic intermediates like DHAP/GAP (the pool of DHAP and GAP), previously noted in 786-O-HIF1α cells, was also completely prevented by extracellular pyruvate in G55 cells treated with DMOG, as well as in 786-O-HIF1α cells (Figures 3H and 3J). Moreover, accumulation of other upper glycolytic intermediates such as fructose bisphosphate (FBP; pool of F1,6-BP and F2,6-BP),was also markedly attenuated in the presence of extracellular pyruvate in DMOG-treated G55 cells (Figure 3I). Similar results were found in 786-O-HIF1α cells when in the presence of extracellular pyruvate (Figure 3K). Next, we evaluated whether HIF1α also promotes the accumulation of upper-glycolysis-derived metabolites, such as those of the pentose phosphate pathway (Figure S2A). First, we confirmed that pentose phosphate pathway metabolites were elevated in a large number of those individual human ccRCC samples analyzed above (Figure S2B). Similar results were obtained in 786-O and G55 cells, where we found that pentose 5-phosphate metabolites (the pool of ribose-5-phosphate + ribulose-5-phosphate) accumulate upon HIF1α activation conditions. This accumulation was markedly reduced when glycolytic flux was reactivated by exogenous pyruvate (Figures S2C and S2D). These data suggest that the accumulation of upper glycolytic intermediates is a consequence of the HIF1α-dependent suppression of lower glycolysis at the level of GAPDH. Along these lines, extracellular pyruvate relieves glycolytic flux, impeding the increase in upper glycolysis metabolites and the potentiation of the pentose phosphate pathway upon HIF1α activation.
Figure 3.

HIF1α-dependent attenuation of glycolytic flux and lactate release is recovered in the presence of extracellular pyruvate
(A) Relative amount of extracellular lactate released by 786-O-HIF1α cells, 786-O-HIF1α-bHLH∗ cells, and their corresponding control cells in the presence or absence of 1 mM extracellular pyruvate (n = 6).
(B) Relative amount of extracellular lactate released by control G55 cells and G55 cells exposed to 0.5% hypoxia in the presence or absence of 1 mM extracellular pyruvate (n = 6).
(C) Relative amount of extracellular lactate released by control G55 cells and G55 treated with 0.2 mM DMOG in the presence or absence of 1 mM extracellular pyruvate (n = 6).
(D) Relative amount of extracellular lactate released by control MDA-MB-231 cells and MDA-MB-231 cells treated with 0.5 mM DMOG in the presence or absence of 1 mM extracellular pyruvate (n = 4).
(E) The scheme represents the evaluation of glycolytic flux measured through the 3H2O production during the reaction catalyzed by enolase that converts 2-PG to PEP in cells cultured with D-[5-3H]-glucose. The position of the 3H (green) is indicated in the labeled glucose and all the glycolytic metabolites until 2-PG.
(F) Glycolytic flux in control G55 cells and G55 treated with 0.2 mM DMOG in the presence (n = 10) or absence (n = 15) of 1 mM extracellular pyruvate.
(G) Glycolytic flux in 786-O-HIF1α and control cells in either the presence (n = 4) or absence (n = 4) of extracellular pyruvate (1 mM).
(H) Relative amount of DHAP/GAP (the pool of DHAP and GAP) in G55 control cells and G55 cells treated with DMOG (0.2 mM) in the presence or absence of extracellular pyruvate (1 mM, n = 3).
(I) Relative amount of FBP (the pool of F1,6-BP and F2,6-BP) in G55 control cells and G55 cells treated with DMOG (0.2 mM) in either the presence or absence of extracellular pyruvate (1 mM, n = 3).
(J) Relative amount of DHAP/GAP in 786-O-HIF1α cells and their corresponding control cells in the presence or absence of extracellular pyruvate (1 mM, n = 3).
(K) Relative amount of FBP in 786-O-HIF1α cells and their corresponding control cells in the presence or absence of extracellular pyruvate (1 mM, n = 3).
In the bar graphs, the values represent the mean ± SEM, and statistical analysis was performed using a two-way ANOVA followed by a Tukey’s post hoc test: ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 represent significant p values.
Collectively, our data suggest that the ability of HIF1α to reduce lactate release is hidden in conditions containing excessive pyruvate content frequently found in those culture media. Thus, this HIF1α-dependent anti-glycolytic effect can emerge under conditions of insufficient pyruvate supply as the solid tumor microenvironment, which thereby explains why ccRCCs show a simultaneous suppression of lower glycolysis and constitutive HIF1α activation (see also the discussion).
Minimal contribution of glutamine to pyruvate biosynthesis
Because pyruvate is essential to recover the HIF1α-dependent suppression of glycolysis and it can potentially be generated from glutamine, we evaluated whether glutamine-derived pyruvate biosynthesis was compromised in HIF1α-expressing cells, leading to insufficient pyruvate synthesis to sustain glycolytic flux. Pyruvate can potentially be derived from glutamine via two different pathways. After its initial conversion to malate or oxaloacetate (Figure 4A), the latter is converted into PEP by PEP carboxykinase (PEPCK) and finally to pyruvate through pyruvate kinase (Figure 4A).40 Alternatively, malate can be decarboxylated and converted to pyruvate by malic enzyme (ME) (Figure 4A).41 It has previously been shown that HIF1α attenuates glutamine oxidation,18,19 which might limit glutamine-dependent pyruvate biosynthesis. Therefore, we asked whether glutamine-dependent biosynthesis of pyruvate and lactate was operative in our cells. To assess whether pyruvate and lactate can be generated from glutamine in 786-O and G55 control cells, we labeled the cells with 5-13C-glutamine and measured the level of 1-13C-lactate generated by PEPCK or ME activity (Figure 4A). Our data showed that 5-13C-glutamine barely contributed any labeling to lactate (M+1) in 786-O and G55 cells (Figures 4B–4E). These data show that reduced lactate release in HIF1α-expressing cells cannot be attributed to a reduced glutamine-dependent pyruvate biosynthesis because glutamine-derived pyruvate is barely detectable in control 786-O and G55 cells.
Figure 4.

Analysis of the contribution of glutamine to lactate biosynthesis in renal cancer and glioblastoma cells
(A) Biosynthesis of 1-13C-labeled lactate from 13C-labeled glutamine at the fifth position (5-13C-glutamine). Blue circles represent the 13C-labeled carbons and white circles the unlabeled carbons.
(B) Isotopologue distribution of extracellular glutamine in 5-13C-glutamine-labeled 786-O cells (n = 2).
(C) Isotopologue distribution of extracellular glutamine in 5-13C-glutamine-labeled G55 cells (n = 3).
(D) Relative extracellular levels of 1-13C-lactate (M+1) compared to 12C-lactate (M+0) in 5-13C-glutamine-labeled 786-O cells (n = 2).
(E) Relative extracellular levels of 1-13C-lactate (M+1) compared to 12C-lactate (M+0) in 5-13C-glutamine-labeled G55 cells (n = 3).
In the bar graph, the values represent the mean ± SEM.
HIF1α-dependent elevation of the NADH/NAD+ ratio leads to reduced glycolytic activity
Then, we asked why conditions of limited supply of extracellular pyruvate resembling those in the tumor microenvironment enable the HIF1α-dependent suppression of glycolytic flux. Along this line, we consider that a central metabolic action executed by HIF1α is the suppression of mitochondrial oxygen consumption to save oxygen in hypoxic conditions.14,15,42 Suppression of mitochondrial activity reduces the potential of the mitochondria to oxidize NADH to NAD+, thereby leading to accumulation of NADH and elevation of the NADH/NAD+ ratio.43 This can be prevented by extracellular pyruvate, which together with the accumulated NADH are converted to lactate and NAD+, respectively, through LDH activity.44,45,46 Indeed, we first show that HIF1α activation in 786-O cells as well as DMOG-treated G55 cells leads to a marked decline in cellular oxygen consumption rate (Figure 5B), accompanied by an increased NADH/NAD+ ratio (Figures 5C and 5D), when extracellular pyruvate supply is limited. However, this increase of the NADH/NAD+ ratio in 786-O-HIF1α or DMOG-treated G55 cells was markedly reduced when cells were cultured in the presence of extracellular pyruvate (Figures 5C and 5D). In this context, the glycolytic reaction driven by GAPDH converts NAD+ to NADH, and therefore it has been considered sensitive to elevation in the NADH/NAD+ ratio when mitochondria activity is attenuated.47 Since HIF1α attenuates lower glycolysis after the GAPDH step, our data suggest that a HIF1α-dependent increase of the NADH/NAD+ ratio as a consequence of mitochondrial inhibition—when extracellular pyruvate supply is limited—results in the inhibition of glycolytic flux after the GAPDH step, leading to reduced lactate release. To further assess whether the increased NADH/NAD+ ratio was responsible for the suppression of glycolysis, we used another LDH substrate, α-ketobutyrate (αKB), which can also preclude the increment of the NADH/NAD+ ratio upon its conversion to α-hydroxybutyrate by LDH activity45 (Figure 5A). Indeed, αKB firstly impedes the increased NADH/NAD+ ratio in 786-O-HIF1α cells, as well as in G55 cells exposed to DMOG (Figures 5C and 5D), and secondly fully restores lactate release in 786-O-HIF1α cells and G55 cells exposed to DMOG (Figures 5E and 5F). To further support these data, we reasoned that if a HIF1α-dependent increase of the NADH/NAD+ ratio (as a consequence of mitochondrial inhibition) leads to inhibition of lactate release, then the sole mitochondria inhibition should be enough to reduce lactate secretion because of the elevation of the NADH/NAD+ ratio. To address this question, we used metformin as an inhibitor of mitochondrial respiration that suppresses the NADH oxidation to NAD+ leading to an elevation of the intracellular NADH/NAD+ ratio.44,48 Indeed, inhibition of mitochondrial oxygen consumption using metformin—to a similar extent to the HIF1α activator DMOG (Figure 5B)—leads to an increase of the NADH/NAD+ ratio that is, importantly, accompanied by a parallel inhibition of lactate release (Figures 5G and 5H). Moreover, and like HIF1α-expressing cells, αKB reduces the elevation of the NADH/NAD+ ratio and fully restore cellular lactate secretion in cells in which mitochondrial activity is inhibited by metformin (Figures 5G and 5H). Finally, we also wondered how HIF1α can drive cytosolic NADH-dependent suppression of lower glycolysis. The cytosolic NADH content depends on its rate of consumption in the cytosolic side of the malate-aspartate shuttle, which primarily depends on mitochondrial TCA cycle activity.49,50 Therefore, the HIF1α-dependent suppression of mitochondrial TCA might suppress the malate-aspartate shuttle and the ensuing accumulation of cytosolic NADH. Indeed, we previously found that HIF1α reduces not only TCA cycle activity but also key enzymes in the malate-aspartate shuttle like GOT1 and GOT2.18 Hence, we wondered whether suppression of the malate-aspartate shuttle could result in NADH-dependent suppression of lactate release. Indeed, silencing of GOT1 and GOT2 elevates the NADH/NAD+ ratio in parallel with an inhibition of lactate release, which was completely restored when the elevation of the NADH/NAD+ ratio was prevented with αKB (Figures S3A–S3C). As such, HIF1α-dependent suppression of key players in the mitochondrial/cytosolic malate-aspartate shuttle appears to induce the cytosolic NADH-evoked suppression of lactate release.
Figure 5.

HIF1α-dependent elevation of the intracellular NADH/NAD+ ratio leads to reduced lactate secretion
(A) The reaction shows the regeneration of NAD+ by the conversion of pyruvate or αKB to lactate and α-hydroxybutyrate (αHB), respectively, through LDH activity.
(B) Oxygen consumption rate (OCR) in 786-O-HIF1α cells (n = 9) and 786-O control cells (n = 10) (left), as well as in G55 control cells (n = 5) and those treated with 0.2 mM DMOG (n = 5) or 2 mM metformin (n = 5) (right).
(C) NADH/NAD+ ratio in 786-O-HIF1α cells, 786-O-HIF1α-bHLH∗ cells, and their corresponding control cells in the presence (n = 3) or absence (n = 6) of extracellular pyruvate or αKB (1 mM).
(D) NADH/NAD+ ratio in G55 treated with 0.2 mM DMOG and control G55 cells in the presence (n = 3) or absence (n = 6) of extracellular pyruvate or αKB (1 mM).
(E) The relative amount of extracellular lactate released by 786-O-HIF1α cells, 786-O-HIF1α-bHLH∗ cells, and their corresponding control cells in the presence or absence of extracellular αKB (1 mM) (n = 6).
(F) The relative amount of extracellular lactate released by control G55 cells and G55 treated with 0.2 mM DMOG in the presence or absence of extracellular αKB (1 mM) (n = 9).
(G) NADH/NAD+ ratio in G55 treated with 2 mM metformin and control G55 cells in the presence (n = 3–6) or absence (n = 6) of αKB (1 mM).
(H) Relative amount of extracellular lactate released by control G55 cells and G55 treated with 2 mM metformin in the presence (n = 6) or absence (n = 12) of αKB (1 mM).
In the bar graphs, the values represent the mean ± SEM, and statistical analysis was performed using a two-tailed unpaired t test when comparing 2 groups or a one-way (B, right) or two-way (C–H) ANOVA followed by Tukey’s post hoc test: ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 represent significant p values.
Collectively, these data show that mitochondrial inhibition (upon HIF1α activation or pharmacologically) leads to an elevation of the NADH/NAD+ ratio that suppresses cellular glycolytic activity. Importantly, this unexpected action of HIF1α takes place when there is a limited extracellular supply of pyruvate (as in tumor microenvironment) because a large enough supply of pyruvate—or αKB—can act as electron acceptors to oxidize the excess of NADH upon mitochondrial inhibition.
Suppression of cell proliferation through the HIF1α-dependent attenuation of lower glycolysis
A central action of HIF1α is the inhibition of cell proliferation, which has been explored in numerous biological settings including ccRCCs, in which HIF1α functions as a tumor suppressor.18,32,51 786-O-HIF1α or DMOG-treated G55 and MDA-MB-231 cells show a remarkable decline in cell proliferation when compared to 786-O or untreated G55 and MDA-MB-231 control cells (Figure 6A). We therefore investigated whether HIF1α-dependent glycolysis slowdown might contribute to the anti-proliferative potential of HIF1α. Indeed, extracellular pyruvate or αKB recovered the reduced proliferation upon HIF1α activation in 786-O cells as well as in DMOG-treated G55 and MDA-MB-231 cells (Figure 6A), which shows that NADH accumulation underlies the ability of HIF1α to act as suppressor of tumor cell proliferation. In order to assess whether the NADH-dependent inhibition of lower glycolysis at the level of GAPDH was involved in this anti-proliferative effect, we silenced GAPDH expression. Importantly, silencing of GAPDH was enough to decrease cell proliferation at conditions that reduce lactate release to a similar extent to HIF1α activation (Figure 6B).
Figure 6.

Reduced cell proliferation as consequence of HIF1α-dependent lower glycolysis inhibition
(A) Cell proliferation assays in 786-O-HIF1α cells and their corresponding control cells in the presence or absence of extracellular pyruvate or αKB (1 mM) (left; n = 9–10) in G55 control cells and G55 cells treated with 0.2 mM DMOG in the presence or absence of extracellular pyruvate or αKB (1 mM) (middle; n = 9) and MDA-MB-231 control cells and MDA-MB-231 cells treated with 0.5 mM DMOG in the presence or absence of extracellular pyruvate or αKB (1 mM) (right; n = 3).
(B) On the left, representative western blots of the GAPDH and β-ACTIN protein in G55 control cells and GAPDH-silenced G55 cells. In the middle, cell proliferation assay in G55 control cells and GAPDH-silenced G55 cells (n = 9). On the right, the relative amount of extracellular lactate released by G55 control cells and GAPDH-silenced G55 cells, (n = 6).
In the bar graphs, the values represent the mean ± SEM, and statistical analysis was performed using a two-tailed unpaired t test when comparing 2 groups, with Welch’s correction when needed, or two-way ANOVA followed by Tukey’s post hoc test: ∗∗∗p < 0.001 represent significant p values.
Collectively, all these data show that HIF1α-dependent mitochondrial inhibition leads to an elevated NADH/NAD+ ratio when extracellular pyruvate supply is limited. This results in the attenuation of glycolytic flux, providing a molecular explanation for the reduced lower glycolysis manifested in ccRCCs. Moreover, these data show that HIF1α does not promote glycolysis in all cell types because some cells require an external source of pyruvate to sustain their glycolytic flux.
Discussion
The established role of HIF1α as an activator of glycolysis is based on its ability to induce the transcription of the genes encoding glycolytic enzymes as well as glucose and lactate transporters.26,27,28,39,52 Moreover, previous studies have shown that hypoxia increases lactate release in a HIF1α-dependent manner in some cell models.53,54 However, the metabolomic profile of human ccRCCs—characterized by constitutive HIF1α activation—showed a diminished content of lower glycolysis intermediates like the PGs (2-PG and 3-PG) and PEP.30,31 Lactate content was also reduced in some, but not all, ccRCCs. It has to be considered that lactate is released into the extracellular milieu and is present in the blood, and its levels can therefore be heavily influenced by its accumulation in the microenvironment of poorly vascularized tumors.55 Therefore, lactate levels in the ccRCC tumor mass do not accurately reflect glycolysis like other lower glycolytic intermediates, such as PEP or 2-PG. Importantly, this limitation was overcome here by assessing the role of HIF1α on the uncoupling of ccRCC glycolysis and lactate release, specifically in ccRCC cells in the absence of other lactate sources. Our different experimental approaches, such as the measurement of intracellular glycolytic intermediates and glycolytic flux analysis, suggest that the weaker lower glycolysis seen in nephrectomies of human ccRCCs is provoked by HIF1α. The data show that some tumor cell types cannot sustain the expected HIF1α-dependent glycolysis under conditions of low extracellular pyruvate such as those typically found in the tumor microenvironment. In this context, pyruvate concentration in serum—in principle, the maximum exogenous pyruvate that a tumor cell in general can receive—can be as low as 40 μM.36,56,57,58 Moreover, pericellular pyruvate concentrations in solid tumors can be lower than 40 μM in the serum because of their deficient perfusion.34,35,36 Therefore, our data suggest that extracellular pyruvate levels in the ccRCC tumor mass are low enough to enable the HIF1α-dependent anti-glycolytic effect observed in these tumors. Moreover, we also found that G55 glioblastoma and MDA-MB-231 breast cancer cells also required a sufficient exogenous pyruvate supply to sustain glycolysis upon HIF1α activation. Therefore, it is conceivable that the HIF1α-dependent suppression of glycolytic flux might be also operative in the areas of glioblastoma or breast solid tumors with a simultaneous poor supply of oxygen and pyruvate. Since regular pyruvate concentrations in cell culture media do not reflect the extracellular pyruvate levels encountered in vivo, it seems necessary to consider the extracellular pyruvate levels when assessing the impact of HIF1α on glycolysis and possibly on other aspects of the metabolic rewiring controlled by HIF1α. Indeed, together with recent metabolomics data, our results suggest that extracellular pyruvate exerts a pivotal influence on glycolysis, despite the ability of HIF1α to induce the expression of glycolytic enzymes in ccRCCs.26,29,53,59 This dominant role of extracellular pyruvate may be operative under other conditions, such as a limited supply of vitamin B1 that induces GAPDH gene expression.60
NADH oxidation to NAD+ is coupled to mitochondrial complex I activity, and therefore pharmacological inhibition of mitochondrial activity leads to an increased intracellular NADH/NAD+ ratio.44 A central function of HIF1α is to slow down the mitochondrial activity.20,21,22,23 Therefore, our study shows that, indeed, HIF1α activation also elevates the NADH/NAD+ ratio in parallel to its ability to reduce mitochondrial oxygen consumption. Our data show that the HIF1α-dependent suppression of lower glycolysis after the GAPDH step and the subsequent reduced lactate release are consequences of this HIF1α-dependent increment of the NADH/NAD+ ratio when extracellular pyruvate supply is limited. Along this line, recent studies have suggested that NADH accumulation leads to “NADH reductive stress” that compromises the activity of NADH-producing enzymes including, potentially, GAPDH.47,61 Indeed, lactate release was fully restored when the increment of the NADH/NAD+ ratio was prevented by two independent electron acceptors such as pyruvate and αKB, which oxidize the accumulated NADH to NAD+, preventing the accumulation of the NADH/NAD+ ratio upon HIF1α-dependent mitochondrial inhibition. In support of these data, we also found that the sole pharmacological inhibition of mitochondrial activity using metformin—mimicking the action of HIF1α on mitochondrial activity—also results in reduced lactate release, which was completely restored when the cellular increment of the NADH/NAD+ ratio upon mitochondrial inhibition was reduced. It is generally accepted that impairment of mitochondrial activity (as HIF1α does) potentiates glycolysis, but our data show that reduced mitochondrial activity does not necessarily drive glycolysis. Here, we show that this expected response is not executed in different biological settings including human ccRCCs, in which mitochondria activity and lower glycolysis are simultaneously compromised. This is because, in biological settings such as the tumor microenvironment, the extracellular pyruvate supply is low enough to prevent NADH reductive stress, which ultimately attenuates glycolytic flux after the GAPDH step.
Furthermore, our data show that the repression of lower glycolysis occurs in parallel with the accumulation of upper glycolytic intermediates upstream of the GAPDH reaction, such as DHAP, GAP, and FBP. Therefore, our data strongly suggest that HIF1α can uncouple upper and lower glycolysis by compromising the glycolytic step catalyzed by GADPH. In agreement with our data, specific inhibition of GAPDH with the selective inhibitor koningic acid (KA) leads to a disconnection of upper and lower glycolysis,62 similar to what is manifested by HIF1α-expressing cells. As such, KA reduces the intracellular content of lower glycolytic intermediates like 3-PG or PEP, while metabolites of upper glycolysis such as DHAP or F1,6-P are accumulated. In line with this, HIF1α-dependent accumulation of upper glycolysis metabolites is relevant to explain some in vivo biological manifestations executed by HIF1α in tumors, such as ccRCCs. Indeed, potentiation of the pentose phosphate pathway as a molecular feature of ccRCC is a direct consequence of the HIF1α-dependent uncoupling of upper and lower glycolysis we described in this study. Moreover, we also found that HIF1α-dependent attenuation of GAPDH-dependent lower glycolysis contributes to the anti-proliferative potential of HIF1α activation, which has been also largely explored in ccRCCs, where HIF1α acts as a tumor suppressor.18,32,51 In this context, previous studies have shown that suppression of cell proliferation upon mitochondrial inhibition can be attributed to the elevation of the NADH/NAD+ ratio, which compromises the biosynthesis of aspartate, which is essential for DNA synthesis and cell proliferation.44,45,46 Importantly, our data unveil that the anti-proliferative effect of accumulated NADH reductive stress upon HIF1α-dependent mitochondrial inhibition can be also attributed to the attenuation of other NADH vulnerable pathways such as lower glycolysis.
In summary, we found an unexpected role for HIF1α as a suppressor of glycolysis in some cells under conditions of a limited supply of extracellular pyruvate, which enables the HIF1α-dependent cellular increase of the NADH/NAD+ ratio that ultimately compromises lower glycolysis. Our data also imply firstly that unexpectedly extracellular pyruvate can be essential to control glycolytic flux and secondly that mitochondrial inhibition is not necessarily accompanied by the expected increase on glycolysis when cells encounter in vivo conditions of extracellular pyruvate supply resembling those in the tumor microenvironment, such as ccRCCs, characterized by constitutive HIF1α activation and a simultaneous suppression of lower glycolysis and mitochondrial activity.
Limitations of the study
In this study, we have shown that HIF1α compromises lower glycolysis as a consequence of HIF1α-dependent suppression of mitochondrial activity and subsequent NADH reductive stress. This HIF1α-dependent glycolysis uncoupling occurs when cellular pyruvate supply is limited because pyruvate can attenuate NADH reductive stress. Since HIF1α compromises lower glycolysis in human ccRCCs, it can be also presumed that there is a limited supply of extracellular pyruvate in these tumors as has been previously found in some tumor models.36,56,57,58 Therefore, further experimentation is necessary to ascertain whether interstitial pyruvate content in human ccRCCs is also reduced. In addition, we have found that this HIF1α-dependent metabolic response occurs not only in ccRCC cells but also in other tumor cell types. However, we found that there are some other cell models in which HIF1α activation did not result in reduced lactate release, although the molecular mechanism of this differential response remains elusive. Finally, our study has been focused on HIF1α-dependent glycolysis uncoupling in tumor cells, which can preferentially induce this response taking into consideration the limited pyruvate supply in solid tumors. However, it will be relevant to investigate which other non-cancer biological settings might be characterized by limited pyruvate supply to permit this HIF1α-dependent glycolysis uncoupling.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-HIF1α | BD Transduction Laboratories | Cat#610959; RRID: AB_398272 |
| Mouse monoclonal anti-CAIX | Santa Cruz Biotechnology | Cat#sc-365900; RRID: AB_10846466 |
| Monoclonal anti-β-actin-peroxidase | Sigma-Aldrich | Cat#A3854; RRID: AB_262011 |
| Rabbit polyclonal anti-HIF-1α (C-Term) | Cayman Chemical | Cat#10006421; RRID: AB_409037 |
| Rabbit polyclonal anti- PDK1 | Enzo Life Sciences, Inc. | Cat#ADI-KAP-PK112; RRID: AB_10618932 |
| Goat polyclonal anti-HSP 60 | Santa Cruz Biotechnology | Cat#sc-1722; RRID: AB_2233354 |
| Rabbit polyclonal anti-GAPDH | Abcam | Cat#ab9485; RRID: AB_307275 |
| Rabbit polyclonal anti-GOT1 | Proteintech | Cat#14886-1-AP; RRID AB_2113630 |
| Rabbit polyclonal anti-GOT2 | Sigma-Aldrich | Cat#HPA018139; RRID AB_1849903 |
| Chemicals, peptides, and recombinant proteins | ||
| Doxycycline | Sigma-Aldrich | Cat#D9891 |
| Dimethyloxalylglycine (DMOG) | Enzo Life Sciences, Inc. | Cat#BML-EI347 |
| Metformin | Sigma-Aldrich | Cat#D150959 |
| Sodium pyruvate | Lonza | Cat#BE13-115E |
| Sodium α-ketobutyrate (αKB) | Sigma-Aldrich | Cat#K0875 |
| L-Glutamine-5-13 C | Cambridge Isotope Laboratories, Inc. | Cat#CLM-1166 |
| Protease inhibitors | Sigma-Aldrich | Cat#P8340 |
| Phosphatase inhibitors | Sigma-Aldrich | Cat#P5726 |
| Pyridine | Sigma-Aldrich | Cat#270970 |
| Methoxyamine hydrochloride | Sigma-Aldrich | Cat#226904 |
| 5-3H-Glucose | Perkin Elmer | Cat#NET531001MC |
| Dodecyltrimethylammonium bromide (DTAB) | Sigma-Aldrich | Cat#D8638 |
| Critical commercial assays | ||
| L-Lactate Assay Kit (Colorimetric/Fluorometric) | Abcam | Cat#ab65330 |
| DC protein Assay Kit | Bio-Rad | Cat#5000112 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | Cat#23225 |
| NAD/NADH-Glo™ Assay Kit | Promega | Cat#G9071 |
| Clarity Western ECL Substrate | Bio-Rad | Cat#1705061 |
| Super-Signal West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | Cat#34096 |
| Experimental models: Cell lines | ||
| 786-O | Provided by Dr. María J. Calzada | N/A |
| G55 | Provided by Dr. Till Acker | N/A |
| MDA-MB 231 | Provided by Dr. Massimiliano Mazzone | N/A |
| A549 | Provided by Dr. Massimiliano Mazzone | N/A |
| Murine embryonic fibroblasts (MEFs) | This study | N/A |
| Oligonucleotides | ||
| Control siRNA | Santa Cruz Biotechnology | Cat#sc-37007 |
| HIF1α siRNA | Santa Cruz Biotechnology | Cat#sc-35561 |
| GAPDH siRNA | Santa Cruz Biotechnology | Cat#sc-35448 |
| GOT1 siRNA | Dharmacon | Cat#J-011673 |
| GOT2 siRNA | Dharmacon | Cat#J-011674 |
| Recombinant DNA | ||
| pLVX-TetOne-Puro-Luc | Takara | Cat#631849 |
| pLVX-TetOne-HIF1α | Meléndez-Rodríguez et al.18 | N/A |
| pLVX-TetOne-HIF1α bHLH∗ | Meléndez-Rodríguez et al.18 | N/A |
| Other | ||
| Pyruvate-free Dulbecco’s high glucose modified Eagle’s Medium (DMEM) | Cytiva HyClone™ | Cat#SH30022 |
| Pyruvate- and glucose-free Dulbecco’s high glucose modified Eagle′s Medium (DMEM) | Gibco™ | Cat#11966025 |
| Pyruvate- and glutamine-free Dulbecco’s high glucose modified Eagle’s Medium (DMEM) | Gibco™ | Cat#11960044 |
| Pyruvate-, glutamine- and glucose-free Dulbecco’s high glucose modified Eagle’s Medium (DMEM) | Gibco™ | Cat# A1443001 |
| Lipofectamine 2000 Transfection Reagent | Invitrogen | Cat#11668019 |
| Dialyzed fetal bovine serum (dFBS) | Sigma-Aldrich | Cat#F0392 |
| Metabolic data from Human ccRCC samples | Hakimi et al.30 | N/A |
| Mass spectrometry data | This study | EMBL-EBI MetaboLights: MTBLS8901 https://www.ebi.ac.uk/metabolights/MTBLS8901 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Julián Aragonés (julian.aragones@uam.es).
Materials availability
Cell lines generated in this study are available upon request to the lead contact.
Data and code availability
-
•
All original data reported in this study is available from the lead contact upon request. Mass spectrometry data have been deposited to the EMBL-EBI MetaboLights database63 with the identifier MTBLS8901. The dataset can be accessed in the following link https://www.ebi.ac.uk/metabolights/MTBLS8901.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Method details
Cell models
The 786-O-LUC/HIF1α/HIF1α-bHLH∗,18 G55, MDA-MB-231 and A549 cell lines, and the murine embryonic fibroblasts (MEFs) were all maintained in pyruvate-free Dulbecco’s high glucose modified Eagle’s Medium (DMEM: SH30022.01, Cytiva HyClone) supplemented with 100 units/mL of penicillin and 100 μg/mL streptomycin (P/S: DE17-602E, Lonza), 20 mM HEPES buffer (17-737E, Lonza) and 10% fetal bovine serum (FBS: SV30160.03, Cytiva HyClone). In the experiments, 786-O-LUC/HIF1α/HIF1α-bHLH∗ cells were treated with doxycycline (1 μg/mL: D9891, Sigma-Aldrich) to induce vector expression. Cells were kept at 37°C in an atmosphere with 5% of CO2 and 95% air for normoxic conditions. To induce hypoxia the cells were maintained at 0.5% O2 in an Invivo2 400 workstation (Ruskinn) or exposed to dimethyloxalylglycine (DMOG, 0.1–1 mM: BML-EI347, Enzo Life Sciences, Inc.), a pharmacological inhibitor of prolyl-4-hydroxylase. All experiments were carried out in 5 mM glucose unless specifically stated.
Gene silencing by siRNA
Temporary siRNA gene silencing was achieved using a specific HIF1α siRNA (sc-35561, Santa Cruz Biotechnology), GAPDH siRNA (sc-35448, Santa Cruz Biotechnology), GOT1 siRNA (J-011673, Dharmacon), GOT2 siRNA (J-011674) and a control siRNA (sc-37007, Santa Cruz Biotechnology). The G55 and MDA-MB-231 cell lines were seeded in p60 plates and then transfected with the respective siRNA using Lipofectamine 2000 (11668019, Invitrogen). The transfected cells were plated and recovered 72 h after transfection for extracellular lactate measurement and western blot analysis.
Cell treatments
To measure metabolites, cells were plated in pyruvate-free high glucose DMEM supplemented with P/S, HEPES and 10% dialyzed FBS (dFBS, 10,000 Mw cut-off: F0392, Sigma-Aldrich). Then cells were subjected to 0.5% hypoxia or treated with DMOG (0.1 mM–1 mM) or metformin (2 mM: D150959, Sigma-Aldrich), and supplemented as necessary with sodium pyruvate (1 mM: BE13-115E, Lonza) or sodium α-ketobutyrate (αKB, 1 mM: K0875, Sigma-Aldrich) for 24 h. Subsequently, the cells were maintained for 2 h in a 5mM glucose pyruvate-free DMEM medium (SH30022.01, Cytiva HyClone and 11966025, Gibco) with the corresponding treatments. Finally, this media was changed to the same 5mM glucose pyruvate-free DMEM media - with the corresponding treatments - and cells and supernatants were collected after 60 min, or 15 min when specifically stated, to measure intracellular metabolites and extracellular lactate. For L-Glutamine-5-13C tracer analysis, the 786-O-LUC and G55 cells were incubated as above but with pyruvate- and glutamine-free DMEM (11960044, Gibco) supplemented with 4 mM L-Glutamine-5-13C (CLM-1166, Cambridge Isotope Laboratories, Inc.), after which the medium was collected. All the samples collected were quenched in liquid nitrogen and stored at −80°C until they were processed.
Extracellular lactate measurement
Extracellular lactate measurements were assayed with the colorimetric L-Lactate Assay Kit (ab65330, Abcam), according to manufacturer’s instructions. Lactate measurements were normalized to protein levels. Cell samples were homogenized in RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS and 50 mM Tris-HCL [pH 8]) with 5% protease inhibitors (P8340, Sigma-Aldrich) and 1% phosphatase inhibitors (P5726, Sigma-Aldrich), and the protein concentration was quantified with the DC protein Assay Kit (5000112, Bio-Rad).
Mass spectrometry metabolite analysis
A cold two-phase methanol:water:chloroform (5:3:5) solution was used for metabolite extraction. The polar (methanol-water) phase and protein interphase was collected after centrifugation at 4°C and dried using a vacuum concentrator. Dried polar and protein fractions were stored at −80°C until use. The protein fraction was dissolved in 200 μL sodium hydroxide (0.2 M), and the protein concentration was then quantified using the Pierce BCA Protein Assay Kit (23225, Thermo Fisher Scientific) for further normalization.
The pentose-phosphate intermediates, fructose bisphosphate and dihydroxy acetone phosphate/glyceraldehyde 3-phosphate were analyzed by liquid chromatography mass spectrometry (LC-MS). A 1290 Infinity II LC System (Agilent Technologies), with a thermal autosampler set at 4°C, coupled to a 6546 Quadrupole-Time of Flight (Q-TOF, Agilent Technologies) was used for the measurement. Dried polar metabolites were resuspended in 50 μL of 80% methanol and a volume of 2 μL or 10 μL of the sample was injected on a Poroshell HILIC column (Agilent InifinityLab Poroshell 120 HILIC-Z, 2.1 mm × 150 mm, 2.7 μm, PEEK-lined, Agilent Technologies). The separation of metabolites was achieved with a column temperature of 50°C and a flow rate of 0.25 mL/min. A gradient was applied for 32 min (solvent A: 10mM ammonium acetate in water with 2.5 μM InfinityLab Deactivator Additive, pH = 9; solvent B: 10mM ammonium acetate in water/acetonitrile 15:85 (v/v) with 2.5 μM InfinityLab Deactivator Additive, pH = 9) to separate the metabolites (0 min: 96% B, 2 min: 96% min, 5.5min: 88% B, 8.5 min: 88% B, 9min: 86% B, 14 min: 86% B, 19 min: 82% B, 25 min: 65% B, 27 min: 65% B, 28 min: 96%, 32 min: 96% B). The MS operated in negative full scan mode (m/z range: 50–1100) using a gas temperature of 225°C, sheath gas flow of 12 L/min, sheath gas temperature of 350°C and capillary voltage of 3.5 kV. Data were collected using the MassHunter Workstation LC/MS Data Acquisition Version 10.1 (Agilent Technologies) and were analyzed using the Agilent MassHunter Workstation Profinder version 10.0.2.
Intracellular glycolytic intermediates (GAP, DHAP, 2/3-PG, PEP), and lactate as well as extracelular lactate were detected using a 7890 A GC system (Agilent Technologies) combined with 5975C Inert MS system (Agilent Technologies) as described previously.64 Polar metabolites were derivatized with 20 μL methoxyamine (20 mg/mL in pyridine: 270970 and 226904, Sigma-Aldrich) for 90 min at 37°C. Subsequently, 15 μL of N-(tert-butyldimethylsilyl)-N-methyl-trifluoroacetamide, with 1% tert-butyldimethylchlorosilane (TBDMS: 375934Sigma-Aldrich) were added to 7.5 μL of each derivative and incubated for 60 min at 60°C. 1 μL of each sample was injected into a DB35MS column in splitless or in split mode (ratio 1 to 9) using an inlet temperature of 270°C. The carrier gas was helium with a flow rate of 1 mL/min. Upon injection, the GC oven was kept at 100°C for 1 min, increased up to 105°C with a gradient of 2.5 °C/min followed by 2 min at 105°C, then ramped to 240°C with a gradient of 3.5 °C/min, and after that ramped up to 320°C with a gradient of 22 °C/min, which was followed by 2 min at 320°C.The measurement of metabolites was performed under electron impact ionization at 70 eV. Isotopologue distributions were extracted from the raw ion chromatograms using a custom MATLAB M-file, which applies consistent integration bounds and baseline correction to each ion.65 In addition, we corrected for naturally occurring isotopes.66 All labeling fractions were transformed into natural abundance-corrected mass distribution vectors.67
Glycolytic flux
G55 cells, 786-O-LUC and 786-O-HIF1 cells were plated in pyruvate-free high glucose DMEM supplemented with P/S, HEPES and 10% dialyzed FBS (dFBS, 10,000 Mw cut-off: F0392, Sigma-Aldrich). Then cells were treated with DMOG (0.2 mM) and supplemented as necessary with sodium pyruvate (1 mM: BE13-115E, Lonza) for 24 h. Subsequently, the cells were maintained for 2 h in a 5 mM glucose pyruvate-free DMEM medium (SH30022.01, Cytiva HyClone and 11966025, Gibco) with the corresponding treatments. Then, cell media was replaced with labeling solution (pyruvate-free 5 mM glucose DMEM, without FBS, supplemented with 80 μCi/mmol of 5-3H-Glucose (NET531001MC, PerkinElmer) and the corresponding treatments. Cells were incubated in labeling solution at 37°C for 2 h. Then, the supernatant was transferred to glass vials sealed with rubber stoppers. 3H2O was captured over 48 h at 37°C in hanging wells containing Whatman paper soaked with H2O. Radioactivity was quantified by liquid scintillation counting.
NADH/NAD+ measurements
To measure the NADH/NAD+, the cells seeded in 6-well plates were first washed with PBS and then lysed with a 500 μL of dodecyltrimethylammonium bromide (DTAB) Buffer (1% DTAB in 0.2 M NaOH diluted 1:1 with PBS: D8638, Sigma-Aldrich), and assayed using the NAD/NADH-Glo Assay Kit (G9071, Promega), according to the manufacturer’s instructions. Luminescence was recorded on a CLARIOstar Plus apparatus (BMG LABTECH).
Cell proliferation analysis
For cell proliferation assay G55 control, GAPDH-silenced G55 cells, 786-O-LUC, HIF1α and MDA-MB-231 were seeded in DMEM dFBS. After 24 h, media was changed with the corresponding treatments. After 72 h, cells were fixed with 4% paraformaldehyde for at least 24 h, and then cells were stained with 0.025% crystal violet solution (11435027, Fisher Scientific) for 20 min. After the incubation, cells were washed several times with distilled water and finally for the measurement, methanol was added to the stained cells and the results were measured with spectrophotometer at 540 nm.
Oxygen consumption
The 786-O-LUC and HIF1α cells or G55 cells were seeded in XF-24 plates. The G55 cells were treated with DMOG (0.2 mM), 24 h before the experiment was performed. The following day the cells were maintained for 2 h with pyruvate-free DMEM containing 5 mM glucose, and supplemented with P/S, HEPES and dFBS and the necessary treatments. Subsequently, the cells were maintained and equilibrated for another hour at 37°C without CO2 in 500 μL of the same medium without the P/S, HEPES and dFBS but with the treatments. The basal cellular oxygen consumption rate (OCR) was measured using an XF24 Extracellular Flux Analyzer (Seahorse Bioscience). After the experiments the cells were homogenized in RIPA buffer supplemented with 5% protease inhibitors and 1% phosphatase inhibitors, and the protein recovered was quantified with a DC protein Assay kit for normalization.
Western blotting
Cells from the extracellular lactate experiments were lysed in RIPA buffer containing 5% protease inhibitors and 1% phosphatase inhibitors, and the protein concentration was quantified as indicated above. The samples were then denatured in Laemmli buffer, resolved on SDS-polyacrylamide gels and transferred to 0.45 μm pore size nitrocellulose membranes (10600007, Cytiva). The membranes were then blocked and the human cell lines were probed with antibodies against HIF1α (610959, BD Transduction Laboratories), CAIX (sc-365900, Santa Cruz Biotechnology), GAPDH (ab9485, abcam), GOT1 (14886-1-AP, Proteintech), GOT2 (HPA018139, Sigma-Aldrich), HSP60 (ab82520, abcam) and β-actin (A3854, Sigma-Aldrich), while the mouse cells were probed with antibodies against HIF1α (10006421, Cayman Chemical), PDK1 (ADI-KAP-PK112-F, Enzo Life Sciences, Inc.) and HSP60 (sc-1722, Santa Cruz Biotechnology). Antibody binding was detected with the Clarity Western ECL Substrate (1705061, Bio-Rad) or Super-Signal West Femto Maximum Sensitivity Substrate (34096, Thermo Fisher Scientific), and visualized on a digital luminescent image analyzer (Image Quant LAS4000 Mini, GE Healthcare).
Metabolic data from human ccRCC samples
The median normalized values of the metabolites of interest were obtained from the database generated by Hakimi et al.30 from 138 patients with matched RCC tumor and adjacent healthy tissue. The relative increase or decrease in the metabolites within a tumor/healthy adjacent tissue pair was calculated by subtracting the median normalized value of the healthy adjacent tissue sample from the tumor sample. When the result was positive, it was considered enriched in the tumor and when the result was negative it was considered reduced in the tumor.
Quantification and statistical analysis
Statistical analyses were performed using the GraphPad Prism 9.0.0 software. The data were analyzed using a two-tailed Student’s test or two-tailed t test with Welch’s correction when needed, one-way ANOVA and two-way ANOVA, both followed by Tukey’s post hoc analysis. p values < 0.05 were considered statistically significant.
Acknowledgments
This work was supported by grants from the Ministerio de Economía y Competitividad (SAF2016-76815 and SAF2017-90794-REDT), the Ministerio de Ciencia, Innovación y Universidades (PID2019-106371RB-I00), and the Ministerio de Ciencia e Innovación (PID2022-138418OB-I00). A.A.U. was supported by the CAM “Atracción de Talento” program. C.M.-C. was supported by the Ministerio de Economía y Competitividad (BES-2017-082320). A.G.-G. was supported by an FPI-UAM fellowship from the Autonomous University of Madrid. L.F.-A. was supported by the Ministerio de Ciencia e Innovación (PRE2020-095326). H.F.A. was funded by the King Baudouin Foundation and Stichting Tegen Kanker, and A.V. and G.R. received fellowships from FWO. S.-M.F. acknowledges funding from the European Research Council under the ERC Consolidator Grant Agreement no. 771486–MetaRegulation, FWO – Research Projects, KU Leuven – FTBO, the Beug Foundation, and Fonds Baillet Latour. K.D.B. is endowed by the Schulthess Foundation. The mitochondria icon of the graphical abstract was made by jaiganesh (https://github.com/jaiganeshjg) and is licensed under CC0 (https://creativecommons.org/publicdomain/zero/1.0/). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
A.A.U., K.D.B., S.-M.F., and J.A. were involved in the design of the experiments, data analysis, and writing of the manuscript. A.A.U., C.M.-C. and A.G.-G. were involved in the extracellular lactate and NADH/NAD+ measurements and cell treatments. A.A.U., C.M.-C., and A.G.-G. were involved in obtaining oxygen consumption measurements. A.A.U. was involved in analyzing the metabolic data comparing ccRCC and the adjacent healthy renal tissue. H.F.A., G.R., D.L., and A.V. helped with the glucose and glutamine dynamic tracer experiments. H.F.A., G.R., D.L., A.V., A.M.F.C., and S.I. helped with metabolite measurements by mass spectroscopy. I.S.-A. was involved in glycolytic flux analysis. A.A.U. and L.F.-A. were involved in the western blot analysis.
Declaration of interests
S.-M.F. has received funding from BlackBelt Therapeutics, Gilead, and Alesta Therapeutics, is on the advisory board of Alesta Therapeutics, and has consulted for Fund+ and Droia Ventures. Moreover, S.-M.F. is on the editorial board of several journals including Cell Reports.
Published: April 11, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114103.
Supplemental information
References
- 1.Bruick R.K., McKnight S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 2.Epstein A.C., Gleadle J.M., McNeill L.A., Hewitson K.S., O’Rourke J., Mole D.R., Mukherji M., Metzen E., Wilson M.I., Dhanda A., et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 3.Safran M., Kaelin W.G. HIF hydroxylation and the mammalian oxygen-sensing pathway. J. Clin. Invest. 2003;111:779–783. doi: 10.1172/JCI18181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schofield C.J., Ratcliffe P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 5.Ivan M., Kondo K., Yang H., Kim W., Valiando J., Ohh M., Salic A., Asara J.M., Lane W.S., Kaelin W.G. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 6.Jaakkola P., Mole D.R., Tian Y.M., Wilson M.I., Gielbert J., Gaskell S.J., von Kriegsheim A., Hebestreit H.F., Mukherji M., Schofield C.J., et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 7.Yu F., White S.B., Zhao Q., Lee F.S. HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc. Natl. Acad. Sci. USA. 2001;98:9630–9635. doi: 10.1073/pnas.181341498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohh M., Park C.W., Ivan M., Hoffman M.A., Kim T.Y., Huang L.E., Pavletich N., Chau V., Kaelin W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat. Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- 9.Maxwell P.H., Wiesener M.S., Chang G.W., Clifford S.C., Vaux E.C., Cockman M.E., Wykoff C.C., Pugh C.W., Maher E.R., Ratcliffe P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 10.Mandriota S.J., Turner K.J., Davies D.R., Murray P.G., Morgan N.V., Sowter H.M., Wykoff C.C., Maher E.R., Harris A.L., Ratcliffe P.J., Maxwell P.H. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell. 2002;1:459–468. doi: 10.1016/s1535-6108(02)00071-5. [DOI] [PubMed] [Google Scholar]
- 11.Jiang B.H., Rue E., Wang G.L., Roe R., Semenza G.L. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J. Biol. Chem. 1996;271:17771–17778. doi: 10.1074/jbc.271.30.17771. [DOI] [PubMed] [Google Scholar]
- 12.Ratcliffe P.J., O'Rourke J.F., Maxwell P.H., Pugh C.W. Oxygen sensing, hypoxia-inducible factor-1 and the regulation of mammalian gene expression. J. Exp. Biol. 1998;201:1153–1162. doi: 10.1242/jeb.201.8.1153. [DOI] [PubMed] [Google Scholar]
- 13.Ortiz-Barahona A., Villar D., Pescador N., Amigo J., del Peso L. Genome-wide identification of hypoxia-inducible factor binding sites and target genes by a probabilistic model integrating transcription-profiling data and in silico binding site prediction. Nucleic Acids Res. 2010;38:2332–2345. doi: 10.1093/nar/gkp1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim J.W., Tchernyshyov I., Semenza G.L., Dang C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 15.Papandreou I., Cairns R.A., Fontana L., Lim A.L., Denko N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 16.Semenza G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Invest. 2013;123:3664–3671. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Samanta D., Semenza G.L. Metabolic adaptation of cancer and immune cells mediated by hypoxia-inducible factors. Biochim. Biophys. Acta. Rev. Cancer. 2018;1870:15–22. doi: 10.1016/j.bbcan.2018.07.002. [DOI] [PubMed] [Google Scholar]
- 18.Meléndez-Rodríguez F., Urrutia A.A., Lorendeau D., Rinaldi G., Roche O., Böğürcü-Seidel N., Ortega Muelas M., Mesa-Ciller C., Turiel G., Bouthelier A., et al. HIF1α Suppresses Tumor Cell Proliferation through Inhibition of Aspartate Biosynthesis. Cell Rep. 2019;26:2257–2265.e2254. doi: 10.1016/j.celrep.2019.01.106. [DOI] [PubMed] [Google Scholar]
- 19.Sun R.C., Denko N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014;19:285–292. doi: 10.1016/j.cmet.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tello D., Balsa E., Acosta-Iborra B., Fuertes-Yebra E., Elorza A., Ordóñez Á., Corral-Escariz M., Soro I., López-Bernardo E., Perales-Clemente E., et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1α decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 2011;14:768–779. doi: 10.1016/j.cmet.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 21.Chan S.Y., Zhang Y.Y., Hemann C., Mahoney C.E., Zweier J.L., Loscalzo J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009;10:273–284. doi: 10.1016/j.cmet.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Z., Li Y., Zhang H., Huang P., Luthra R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene. 2010;29:4362–4368. doi: 10.1038/onc.2010.193. [DOI] [PubMed] [Google Scholar]
- 23.Favaro E., Ramachandran A., McCormick R., Gee H., Blancher C., Crosby M., Devlin C., Blick C., Buffa F., Li J.L., et al. MicroRNA-210 regulates mitochondrial free radical response to hypoxia and krebs cycle in cancer cells by targeting iron sulfur cluster protein ISCU. PLoS One. 2010;5 doi: 10.1371/journal.pone.0010345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fukuda R., Zhang H., Kim J.W., Shimoda L., Dang C.V., Semenza G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 25.Soro-Arnaiz I., Li Q.O.Y., Torres-Capelli M., Meléndez-Rodríguez F., Veiga S., Veys K., Sebastian D., Elorza A., Tello D., Hernansanz-Agustín P., et al. Role of Mitochondrial Complex IV in Age-Dependent Obesity. Cell Rep. 2016;16:2991–3002. doi: 10.1016/j.celrep.2016.08.041. [DOI] [PubMed] [Google Scholar]
- 26.Semenza G.L., Roth P.H., Fang H.M., Wang G.L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994;269:23757–23763. [PubMed] [Google Scholar]
- 27.Iyer N.V., Kotch L.E., Agani F., Leung S.W., Laughner E., Wenger R.H., Gassmann M., Gearhart J.D., Lawler A.M., Yu A.Y., Semenza G.L. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gordan J.D., Thompson C.B., Simon M.C. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. 2007;12:108–113. doi: 10.1016/j.ccr.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clark D.J., Dhanasekaran S.M., Petralia F., Pan J., Song X., Hu Y., da Veiga Leprevost F., Reva B., Lih T.S.M., Chang H.Y., et al. Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell. 2019;179:964–983.e31. doi: 10.1016/j.cell.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hakimi A.A., Reznik E., Lee C.H., Creighton C.J., Brannon A.R., Luna A., Aksoy B.A., Liu E.M., Shen R., Lee W., et al. An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell. 2016;29:104–116. doi: 10.1016/j.ccell.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lucarelli G., Galleggiante V., Rutigliano M., Sanguedolce F., Cagiano S., Bufo P., Lastilla G., Maiorano E., Ribatti D., Giglio A., et al. Metabolomic profile of glycolysis and the pentose phosphate pathway identifies the central role of glucose-6-phosphate dehydrogenase in clear cell-renal cell carcinoma. Oncotarget. 2015;6:13371–13386. doi: 10.18632/oncotarget.3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raval R.R., Lau K.W., Tran M.G.B., Sowter H.M., Mandriota S.J., Li J.L., Pugh C.W., Maxwell P.H., Harris A.L., Ratcliffe P.J. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol. Cell Biol. 2005;25:5675–5686. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Serrao E.M., Kettunen M.I., Rodrigues T.B., Lewis D.Y., Gallagher F.A., Hu D.E., Brindle K.M. Analysis of 13 C and 14 C labeling in pyruvate and lactate in tumor and blood of lymphoma-bearing mice injected with 13 C- and 14 C-labeled pyruvate. NMR Biomed. 2018;31 doi: 10.1002/nbm.3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Altea-Manzano P., Cuadros A.M., Broadfield L.A., Fendt S.M. Nutrient metabolism and cancer in the in vivo context: a metabolic game of give and take. EMBO Rep. 2020;21 doi: 10.15252/embr.202050635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sattler U.G.A., Walenta S., Mueller-Klieser W. A bioluminescence technique for quantitative and structure-associated imaging of pyruvate. Lab. Invest. 2007;87:84–92. doi: 10.1038/labinvest.3700493. [DOI] [PubMed] [Google Scholar]
- 36.Sullivan M.R., Danai L.V., Lewis C.A., Chan S.H., Gui D.Y., Kunchok T., Dennstedt E.A., Vander Heiden M.G., Muir A. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife. 2019;8 doi: 10.7554/eLife.44235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Halestrap A.P., Price N.T. The proton-linked monocarboxylate transporter (MCT) family: structure, function and regulation. Biochem. J. 1999;343:281–299. [PMC free article] [PubMed] [Google Scholar]
- 38.Halestrap A.P., Wilson M.C. The monocarboxylate transporter family--role and regulation. IUBMB Life. 2012;64:109–119. doi: 10.1002/iub.572. [DOI] [PubMed] [Google Scholar]
- 39.Le Floch R., Chiche J., Marchiq I., Naiken T., Naïken T., Murray C.M., Critchlow S.E., Roux D., Simon M.P., Pouysségur J., et al. CD147 subunit of lactate/H+ symporters MCT1 and hypoxia-inducible MCT4 is critical for energetics and growth of glycolytic tumors. Proc. Natl. Acad. Sci. USA. 2011;108:16663–16668. doi: 10.1073/pnas.1106123108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Montal E.D., Dewi R., Bhalla K., Ou L., Hwang B.J., Ropell A.E., Gordon C., Liu W.J., DeBerardinis R.J., Sudderth J., et al. PEPCK Coordinates the Regulation of Central Carbon Metabolism to Promote Cancer Cell Growth. Mol. Cell. 2015;60:571–583. doi: 10.1016/j.molcel.2015.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang C., Ko B., Hensley C.T., Jiang L., Wasti A.T., Kim J., Sudderth J., Calvaruso M.A., Lumata L., Mitsche M., et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol. Cell. 2014;56:414–424. doi: 10.1016/j.molcel.2014.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aragonés J., Fraisl P., Baes M., Carmeliet P. Oxygen sensors at the crossroad of metabolism. Cell Metab. 2009;9:11–22. doi: 10.1016/j.cmet.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 43.Diebold L.P., Gil H.J., Gao P., Martinez C.A., Weinberg S.E., Chandel N.S. Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat. Metab. 2019;1:158–171. doi: 10.1038/s42255-018-0011-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gui D.Y., Sullivan L.B., Luengo A., Hosios A.M., Bush L.N., Gitego N., Davidson S.M., Freinkman E., Thomas C.J., Vander Heiden M.G. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab. 2016;24:716–727. doi: 10.1016/j.cmet.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sullivan L.B., Gui D.Y., Hosios A.M., Bush L.N., Freinkman E., Vander Heiden M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell. 2015;162:552–563. doi: 10.1016/j.cell.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Birsoy K., Wang T., Chen W.W., Freinkman E., Abu-Remaileh M., Sabatini D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell. 2015;162:540–551. doi: 10.1016/j.cell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patgiri A., Skinner O.S., Miyazaki Y., Schleifer G., Marutani E., Shah H., Sharma R., Goodman R.P., To T.L., Robert Bao X., et al. An engineered enzyme that targets circulating lactate to alleviate intracellular NADH:NAD. Nat. Biotechnol. 2020;38:309–313. doi: 10.1038/s41587-019-0377-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vander Heiden M.G., DeBerardinis R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell. 2017;168:657–669. doi: 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen W.W., Freinkman E., Wang T., Birsoy K., Sabatini D.M. Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell. 2016;166:1324–1337.e11. doi: 10.1016/j.cell.2016.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mick E., Titov D.V., Skinner O.S., Sharma R., Jourdain A.A., Mootha V.K. Distinct mitochondrial defects trigger the integrated stress response depending on the metabolic state of the cell. Elife. 2020;9 doi: 10.7554/eLife.49178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shen C., Beroukhim R., Schumacher S.E., Zhou J., Chang M., Signoretti S., Kaelin W.G. Genetic and functional studies implicate HIF1α as a 14q kidney cancer suppressor gene. Cancer Discov. 2011;1:222–235. doi: 10.1158/2159-8290.CD-11-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ullah M.S., Davies A.J., Halestrap A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006;281:9030–9037. doi: 10.1074/jbc.M511397200. [DOI] [PubMed] [Google Scholar]
- 53.Seagroves T.N., Ryan H.E., Lu H., Wouters B.G., Knapp M., Thibault P., Laderoute K., Johnson R.S. Transcription factor HIF-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol. Cell Biol. 2001;21:3436–3444. doi: 10.1128/MCB.21.10.3436-3444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cramer T., Yamanishi Y., Clausen B.E., Förster I., Pawlinski R., Mackman N., Haase V.H., Jaenisch R., Corr M., Nizet V., et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brooks G.A. Lactate as a fulcrum of metabolism. Redox Biol. 2020;35 doi: 10.1016/j.redox.2020.101454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cantor J.R., Abu-Remaileh M., Kanarek N., Freinkman E., Gao X., Louissaint A., Lewis C.A., Sabatini D.M. Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell. 2017;169:258–272.e17. doi: 10.1016/j.cell.2017.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Psychogios N., Hau D.D., Peng J., Guo A.C., Mandal R., Bouatra S., Sinelnikov I., Krishnamurthy R., Eisner R., Gautam B., et al. The human serum metabolome. PLoS One. 2011;6 doi: 10.1371/journal.pone.0016957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mayers J.R., Vander Heiden M.G. Famine versus feast: understanding the metabolism of tumors in vivo. Trends Biochem. Sci. 2015;40:130–140. doi: 10.1016/j.tibs.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Metallo C.M., Vander Heiden M.G. Understanding metabolic regulation and its influence on cell physiology. Mol. Cell. 2013;49:388–398. doi: 10.1016/j.molcel.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jeong H., Vacanti N.M. Systemic vitamin intake impacting tissue proteomes. Nutr. Metab. 2020;17:73. doi: 10.1186/s12986-020-00491-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goodman R.P., Markhard A.L., Shah H., Sharma R., Skinner O.S., Clish C.B., Deik A., Patgiri A., Hsu Y.H.H., Masia R., et al. Hepatic NADH reductive stress underlies common variation in metabolic traits. Nature. 2020;583:122–126. doi: 10.1038/s41586-020-2337-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liberti M.V., Dai Z., Wardell S.E., Baccile J.A., Liu X., Gao X., Baldi R., Mehrmohamadi M., Johnson M.O., Madhukar N.S., et al. A Predictive Model for Selective Targeting of the Warburg Effect through GAPDH Inhibition with a Natural Product. Cell Metab. 2017;26:648–659.e8. doi: 10.1016/j.cmet.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haug K., Cochrane K., Nainala V.C., Williams M., Chang J., Jayaseelan K.V., O'Donovan C. MetaboLights: a resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2020;48:D440–D444. doi: 10.1093/nar/gkz1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rossi M., Altea-Manzano P., Demicco M., Doglioni G., Bornes L., Fukano M., Vandekeere A., Cuadros A.M., Fernández-García J., Riera-Domingo C., et al. PHGDH heterogeneity potentiates cancer cell dissemination and metastasis. Nature. 2022;605:747–753. doi: 10.1038/s41586-022-04758-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Young J.D., Walther J.L., Antoniewicz M.R., Yoo H., Stephanopoulos G. An elementary metabolite unit (EMU) based method of isotopically nonstationary flux analysis. Biotechnol. Bioeng. 2008;99:686–699. doi: 10.1002/bit.21632. [DOI] [PubMed] [Google Scholar]
- 66.Brand W.A. High precision isotope ratio monitoring techniques in mass spectrometry. J. Mass Spectrom. 1996;31:225–235. doi: 10.1002/(SICI)1096-9888(199603)31:3<225::AID-JMS319>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 67.Buescher J.M., Antoniewicz M.R., Boros L.G., Burgess S.C., Brunengraber H., Clish C.B., DeBerardinis R.J., Feron O., Frezza C., Ghesquiere B., et al. A roadmap for interpreting (13)C metabolite labeling patterns from cells. Curr. Opin. Biotechnol. 2015;34:189–201. doi: 10.1016/j.copbio.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All original data reported in this study is available from the lead contact upon request. Mass spectrometry data have been deposited to the EMBL-EBI MetaboLights database63 with the identifier MTBLS8901. The dataset can be accessed in the following link https://www.ebi.ac.uk/metabolights/MTBLS8901.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.