

Loss of induction of immunogenic cell death in response to T-DM1 leads to resistance that can be overcome by targeting TACC3, providing an attractive strategy to improve the efficacy of T-DM1.
Abstract
Trastuzumab emtansine (T-DM1) was the first and one of the most successful antibody–drug conjugates (ADC) approved for treating refractory HER2-positive breast cancer. Despite its initial clinical efficacy, resistance is unfortunately common, necessitating approaches to improve response. Here, we found that in sensitive cells, T-DM1 induced spindle assembly checkpoint (SAC)-dependent immunogenic cell death (ICD), an immune-priming form of cell death. The payload of T-DM1 mediated ICD by inducing eIF2α phosphorylation, surface exposure of calreticulin, ATP and HMGB1 release, and secretion of ICD-related cytokines, all of which were lost in resistance. Accordingly, ICD-related gene signatures in pretreatment samples correlated with clinical response to T-DM1–containing therapy, and increased infiltration of antitumor CD8+ T cells in posttreatment samples was correlated with better T-DM1 response. Transforming acidic coiled-coil containing 3 (TACC3) was overexpressed in T-DM1–resistant cells, and T-DM1 responsive patients had reduced TACC3 protein expression whereas nonresponders exhibited increased TACC3 expression during T-DM1 treatment. Notably, genetic or pharmacologic inhibition of TACC3 restored T-DM1–induced SAC activation and induction of ICD markers in vitro. Finally, TACC3 inhibition in vivo elicited ICD in a vaccination assay and potentiated the antitumor efficacy of T-DM1 by inducing dendritic cell maturation and enhancing intratumoral infiltration of cytotoxic T cells. Together, these results illustrate that ICD is a key mechanism of action of T-DM1 that is lost in resistance and that targeting TACC3 can restore T-DM1–mediated ICD and overcome resistance.
Significance:
Loss of induction of immunogenic cell death in response to T-DM1 leads to resistance that can be overcome by targeting TACC3, providing an attractive strategy to improve the efficacy of T-DM1.
Graphical Abstract
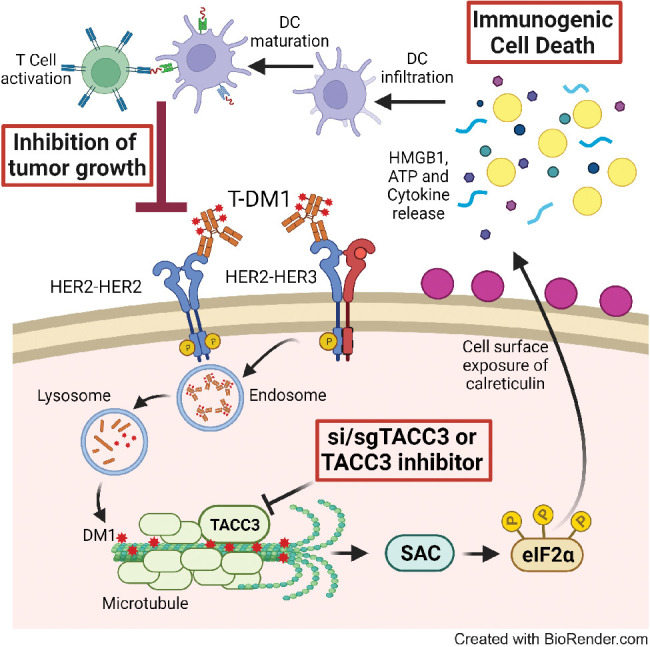
Introduction
Immunogenic cell death (ICD) is a specific form of cell death that is characterized by the release of danger-associated molecular patterns (DAMP), such as cell-surface exposure of calreticulin, ATP, and HMGB1 release. This, in turn, leads to secretion of proinflammatory cytokines that altogether evoke antitumor immune responses upon dendritic cell (DC) maturation and cytotoxic T-cell activation (1, 2). Although ICD has been shown to be a key mechanism of cell death for several different chemotherapies (3, 4) or targeted therapy agents (5), little is known if loss of ICD can trigger resistance to anticancer therapies and if there are therapeutic opportunities to revive ICD and restore drug sensitivity.
Antibody–drug conjugates (ADC) are immunoconjugates containing a monoclonal antibody that is bound to a cytotoxic drug, a so-called payload, with a chemical linker (6). ADCs are highly effective agents and are successfully used in treating many different cancers, including breast cancer. Ado-trastuzumab emtansine (T-DM1) is an FDA-approved iconic ADC of a HER2-targeting antibody (trastuzumab) conjugated to a maytansine derivative (DM1), which is a microtubule depolymerizing agent. It is the first and one of the most successful ADCs approved for the treatment of aggressive HER2+ breast cancer with refractory disease (7, 8). Despite its initial clinical success, resistance is common. Resistance to ADCs may involve alterations in the internalization or recycling of the targeted tumor antigen, e.g., HER2, in the case of T-DM1, changes in payload efficacy, such as enhanced drug efflux or activation of specific survival mechanisms, as demonstrated by us and others (9–11). However, it is still not fully uncovered (i) if response to ADCs may involve activation of ICD, (ii) if resistance to ADCs may be driven by loss of ICD, and (iii) if there are specific molecular targets that may be inhibited to reinstate ICD and thus achieve a much stronger and more durable response.
Transforming acidic coiled-coil containing 3 (TACC3) is upregulated in solid tumors and hematologic malignancies and is strongly associated with worse prognosis in several different cancers (12–14). It is localized to centrosomes as well as microtubules to control spindle stability and microtubule nucleation (15, 16). We previously showed that TACC3 forms distinct functional interactomes regulating different processes in mitosis and interphase to ensure the proliferation and survival of cancer cells with centrosome amplification (13). Despite its roles in tumor growth of highly aggressive cancers, little is known about the role of TACC3 in regulating the immunogenicity of tumors or in driving resistance to anticancer therapies.
In this study, we demonstrated, for the first time, that the T-DM1–induced activation of spindle assembly checkpoint (SAC) triggers ICD, mediated by its payload, DM1, and contributes to T-DM1 cytotoxicity, whereas T-DM1 resistance is characterized by lack of ICD induction. Notably, an ICD-related gene signature correlates with clinical response to T-DM1–containing therapy. We further demonstrated that TACC3 is overexpressed in T-DM1 resistant models and the increase in TACC3 protein after T-DM1 therapy is associated with clinical T-DM1 resistance. Targeting TACC3 not only restores T-DM1–induced SAC activation and mitotic cell death, but it also activates ICD hallmarks and leads to secretion of proinflammatory cytokines in T-DM1 resistant cells. This ultimately results in DC maturation and infiltration of cytotoxic T cells enhancing T-DM1 response in vivo.
Materials and Methods
Cell lines, drugs, and culture conditions
Human HER2+ breast cancer cell lines, SK-BR-3, BT-474, and AU-565, human HER2− breast cancer cell lines, MDA-MB-231 and MDA-MB-157, and murine mammary tumor cell line, EMT6 were obtained from ATCC. T-DM1–resistant (T-DM1R) SK-BR-3 and BT-474 cells were generated previously (11). Meantime, wild-type (WT) cells with no drug incubation were cultured side by side. EMT6.huHER2 cells were generated from the EMT6 cell line by stable overexpression of human HER2. All the cells, except AU-565, were cultured in Dulbecco Modified Eagle Medium supplemented with 50 U/mL penicillin/streptomycin, 1% nonessential amino acids, and 10% fetal bovine serum (Gibco). AU-565 cell line is cultured in RPMI-1640 medium (Corning) supplemented with 50 U/mL penicillin/streptomycin, 1% nonessential amino acids, and 10% fetal bovine serum (Gibco). Cells were routinely tested for Mycoplasma contamination using MycoAlert detection kit (Lonza) and were authenticated by STR sequencing. Except for WT and resistant cell lines, other cells were cultured for less than 20 passages.
HER2+ human tumor samples
To analyze the association between TACC3 protein expression, levels of immune cell infiltration, and response to T-DM1, IHC staining of TACC3, CD3, CD8, and CD4 was performed in 71 HER2+ tumor samples collected before or after T-DM1 therapy from 57 HER2+ breast cancer patients. To separate patients as sensitive versus resistant, we utilized the overall survival status following T-DM1 treatment, and assigned patients who survived upon T-DM1 treatment as sensitive, and those who died as resistant (17). The patients were diagnosed between 2010 and 2019 at Hacettepe University School of Medicine, Ankara, Turkey. The primary tumor specimens were most commonly diagnostic primary breast biopsies or surgical specimens. Post-T-DM1 samples were collected from patients who progressed on T-DM1 treatment or after T-DM1 treatment. These specimens were collected mostly from metastasectomy or rebiopsies to confirm metastases, hormone receptor status or HER2 status. The available patient characteristics are provided in Supplementary Table S1. Written informed consent was obtained from all patients. The use of human tissues from Hacettepe University was conducted in accordance with the Declaration of Helsinki and approved by the Non‐Interventional Clinical Research Ethics Committee of Hacettepe University (approval no: 2020/02-40). The animal experiments were approved by the Institutional Animal Care and Use Committee of the University of South Carolina and the Medical University of South Carolina.
Transient transfection with siRNAs and overexpression vectors
siRNAs were purchased from Dharmacon, and the sequences are provided in Supplementary Table S2. For cell viability, BT-474 T-DM1R (8×103 cells/well) and SK-BR-3 T-DM1R (6×103 cells/well) cells were seeded in 96-well plates with their growth medium without P/S. Twenty-four hours later, cells were transfected with three different siRNAs targeting TACC3 (Dharmacon) at a final concentration of 20 nmol/L (siTACC3#1: D-004155-03, siTACC3#2: D-004155-02, and siTACC3#3: D-004155-04) or two different siRNAs targeting CDC20 (Dharmacon) at a final concentration of 40 nmol/L (siCDC20#1: D-003225-10, siCDC20#2: D-003225-12) using Lipofectamine 2000 (Invitrogen) transfection reagent as described previously (18). Twenty-four hours after transfections, BT-474 and SK-BR-3 T-DM1R cells were treated with 15 and 0.06 μg/mL of T-DM1, respectively. Cell viability was measured with sulforhodamine B (SRB) assay 72 hours after T-DM1 treatment. Empty or TACC3 overexpression vectors were given 12 hours before T-DM1 treatment with an amount of 100 ng/well. 72 hours following transfection, cell viability was measured with SRB assay.
Isolation of bone marrow–derived cells
To obtain bone marrow–derived cells (BMDC), 6- to 8-week-old BALB/c mice were euthanized, and the femur and tibia were dissected, cleaned, and placed in a 6-well dish containing IMDM culture media. A mortar was used to crush the bones, releasing the bone marrow contents into the culture media. The bone marrow cells were plated in a 6-well plate at a seeding density of 1 million cells/mL (5 mL total per well). The cells were cultured for 7 days with 10 ng/mL GM-CSF and 10 ng/mL IL4. Dendritic cell phenotype was confirmed by flow cytometry using CD11c positivity.
DC maturation and T-cell activation assays
The DCs were cultured in conditioned media from EMT6.huHER2 cells treated with T-DM1 alone or in combination with BO-264 for a week, and the expression of maturation markers CD80 and CD86 were analyzed by flow cytometry. For the coculture experiments, DCs were cultured in the conditioned media from EMT6.huHER2 cells treated with T-DM1 alone or in combination with BO-264 overnight in the presence of gp100 peptide (1 μg/mL). The next day, CD3+ T cells isolated from gp100 T-cell receptor bearing Pmel-1 transgenic mice were labeled with CellTrace Violet (Thermo Fisher) and added to the wells containing gp100-loaded DCs at a ratio of 1:10 (DC:T-cell). After 5 days in culture, T cells were collected and analyzed for markers of activation and proliferation. Supernatant from the coculture was collected and sent for multiplex cytokine analysis (Eve Technologies).
Flow cytometry
To analyze the expression of DC maturation and T-cell markers, the DCs and T cells were trypsinized and fixed with 4% PFA. Staining for cell-surface markers was performed by incubating cells with conjugated primary antibodies (Supplementary Table S3) diluted at a 1:200 ratio in FACS buffer (0.1% BSA in PBS) for 30 minutes at 4°C. Samples were then processed using LSRFortessa and analyzed with FlowJo software (v10.8.1; Tree Star).
ATP secretion and HMGB1 release assays
To measure ATP secretion and HMGB1 release, ENLITEN ATP assay system and LUMIT HMGB1 immunoassay (Promega) were utilized, respectively. Cells were seeded in a 96-well plate and treated with drugs for 48 hours. The supernatant was transferred into opaque 96-well plates and analyzed according to the manufacturer's instructions. The luminescence signal was measured using a SpectraMax i3x (Molecular Devices) microplate reader.
Cytokine array
Human Cytokine Array C5 (RayBiotech) was performed according to the manufacturer's instructions. Briefly, BT-474 and SK-BR-3 WT cells or SK-BR-3 T-DM1R cells were treated with T-DM1 only or in combination with BO-264, respectively for 48 hours. Then, supernatants were collected and incubated with the antibody-coated membranes overnight. The next day, they were incubated with biotinylated Ab and labeled with streptavidin. The chemiluminescence imaging was performed using the iBright Imaging system (Thermo Fisher), and iBright Analysis software [v(5.1.0)] was used for data analysis.
In vivo studies
Six- to 8-week-old female BALB/c or FVB mice were housed in a temperature-controlled and 12-hour light/12-hour dark cycle environment. All the in vivo studies were carried out in accordance with the Institutional Animal Care and Use Committee of the University of South Carolina and the Medical University of South Carolina. The human HER2-expressing MMTV.f.huHER2#5 (Fo5) transgenic model was obtained from Genentech under a material transfer agreement (OM-217137 and OR-224086B). Tumor pieces of 2×2 mm in size were transplanted near the MFP of female 6- to 8-week-old immunocompetent FVB mice. After the mean tumor volume reached 100 mm3, mice were andomly allocated to treatment groups. T-DM1 was given once at a dose of 5 mg/kg, by intravenous (i.v.) injection, whereas BO-264 was given at a dose of 50 mg/kg, daily, via oral gavage. For testing the effects of combination therapy on tumor growth, mice were treated for 2 weeks, and all mice were sacrificed, and tumors were collected for Western blotting. To analyze immune cell infiltration and serum cytokine profiling, a separate cohort of mice were treated for a week, and then sacrificed and serum samples and tumors were collected. Tumors were processed for multiplex IHC staining, whereas serum samples were sent for multiplex cytokine analysis (Eve Technologies). There was no blinding during in vivo experiments. Sample sizes were determined based on previous studies (19, 20).
The vaccination assay was done by using combination-treated EMT6.huHER2 cells. Briefly, cells were treated with T-DM1 (5 μg/mL), BO-264 (500 nmol/L) or their combination for 72 hours. During the vaccination assay, 3×105 cells treated with the single agent or combination therapies were collected as a mixture of attached (corresponding to living) and floating (corresponding to dying) cells, washed twice with PBS, and injected into the flank region of BALB/c mice (with 5–7 mice per group) after assessing the apoptotic fraction by Annexin V/PI staining, which showed around 60% apoptosis in the combination-treated group. One week later, the same mice were rechallenged by injecting 1×106 alive (i.e., growing) EMT6.huHER2 cells to the opposite flank as previously described (21).
Bioinformatics and statistical analyses
The microarray data set, GSE194040 (22), was downloaded from the GEO database (23). The metagene and breast cancer-specific ICD gene signatures that were preclinically validated, and shown to be prognostic were retrieved from (24). The expression levels of the ICD-related genes in the T-DM1 + pertuzumab-treated patients from GSE194040 were represented as a heat map using Morpheus software from Broad Institute, https://software.broadinstitute.org/morpheus. The patients were categorized as low versus high ICD score expressers by performing K-means clustering with Euclidean distance metric using the Morpheus tool from Broad Institute (https://software.broadinstitute.org/morpheus/). The significance between the number of responders versus nonresponders among low versus high ICD score expressers was determined using Chi-square testing. To analyze the association of T-DM1 response with the presence of immune cell types in GSE194040, we utilized the CIBERSORTx tool by running the cell fractions module (https://cibersortx.stanford.edu/). This tool deconvolutes the bulk RNA-seq data using signature genes derived from either single-cell transcriptomes or sorted cell populations. We utilized three different immune cell signatures, one derived from microarray data (25) and two of them derived from single-cell RNA-seq (scRNA-seq) data (26, 27), and compared their levels in low versus high ICD-expressers using the Mann–Whitney test. The significance of the percentage of tumors expressing different markers in different groups was calculated using Chi-square testing. All the results are represented as mean ± SD or mean ± SEM, as indicated in the figure legends. All statistical analyses were performed in GraphPad Prism Software [v10.0.0 (153)]. Comparisons between the two groups were done using unpaired two-sided Student t test. Tumor volumes between combination groups versus single agent or vehicle-treated groups were compared using two-way ANOVA with the integration of Dunnett multiple comparison test. Survival curves for the vaccination assay were generated using the Kaplan–Meier method, and significance between groups was calculated by the log-rank test. Experiments were repeated two to three times independently with similar results.
All other methods, including Western blotting, inhibitor treatments, CRISPR/Cas9-mediated gene knockout and stable overexpression, immunofluorescence staining, IHC staining, multiplex IHC staining, and multiplex cytokine array are provided in Supplementary Methods.
Ethical approval
The use of human tissues from Hacettepe University was conducted in accordance with the Declaration of Helsinki and approved by the Non‐Interventional Clinical Research Ethics Committee of Hacettepe University (approval no: 2020/02-40). Written informed consent was obtained from all patients. The animal experiments were approved by the Institutional Animal Care and Use Committee of the University of South Carolina and the Medical University of South Carolina.
Data availability
The RNA-seq data of WT and T-DM1R cells were uploaded to the Sequence Read Archive (SRA) database under the BioProject accession number PRJNA1048320 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1048320). Gene expression data were downloaded from the NCBI Gene Expression Omnibus database under GSE194040 (22). All other raw data generated in this study are available upon request from the corresponding author.
Results
T-DM1 induces ICD markers in T-DM1–sensitive breast cancer cells and ICD signature correlates with T-DM1 sensitivity in patients
To test if ICD is a mechanism of T-DM1 sensitivity, we first analyzed the phosphorylation of eIF2α, one of the major hallmarks of ICD (28) upon T-DM1 treatment, and observed a prominent increase in p-eIF2α (S51) along with microtubule disassembly, mitotic arrest [as shown by p-Histone H3 (S10)] and apoptosis (as shown by cleaved caspase-3 and PARP) in two different HER2+ cell lines, SK-BR-3 and BT-474 (Fig. 1A; Supplementary Fig. S1A). This prompted us to test if T-DM1 would activate also other ICD-related DAMPs that are indispensable for induction of ICD. We observed that upon T-DM1 treatment, there was a significant increase in ATP secretion (Fig. 1B), HMGB1 release (Fig. 1C), and calreticulin cell-surface exposure (Fig. 1D; Supplementary Fig. S1B) in both cell lines. We then assessed the cytokine profiles secreted from sensitive cells upon T-DM1 treatment and observed significant induction of IFNγ, IL12-p40, RANTES (CCL5), IL15, MCP-1 (CCL2), and MCP-2 (CCL8; Fig. 1E; Supplementary Fig. S1C), which are well-established cytokines involved in ICD (29–32). These results show that T-DM1–induced mitotic arrest and apoptosis are accompanied by increased eIF2α phosphorylation, induction of DAMPs and cytokines, indicative of ICD induction.
Figure 1.
![Figure 1. T-DM1 induces ICD markers in T-DM1–sensitive breast cancer cells and ICD correlates with T-DM1 sensitivity in patients. A, Western blot analysis of mitotic arrest [p-H3 (S10)], apoptosis (cleaved caspase-3 and PARP), and ICD marker [p-eIF2α (S51)] in T-DM1–treated SK-BR-3 WT (left) and BT-474 WT (right) cells. B and C, Relative ATP release (B) and HMGB1 release (C) from T-DM1–treated SK-BR-3 WT and BT-474 WT cells (n = 3, 4). D, Immunofluorescence cell-surface staining of calreticulin (green) in T-DM1–treated SK-BR-3 WT cells. Scale bar, 10 μm. DAPI was used to stain the nucleus. Its quantification is provided on the right. E, Cytokine array blot analysis showing the differentially secreted cytokines in T-DM1–treated SK-BR-3 WT cells. F, Schematic summary of the treatment scheme and the sample collection timeline in GSE194040 (22). G, Heatmap of ICD-related genes found in the ICD gene signature score (33) and their correlation with pCR in T-DM1 + pertuzumab-treated patients from GSE194040. pCR: 1, sensitive; pCR: 0, resistant. Chi-square analysis of sensitive vs. resistant tumors expressing low vs. high ICD score is provided below. H, Percentage of CD8+ cells in sensitive (sens) vs. resistant (res) tumors collected pre- (n = 40) and post-T-DM1 (n = 18) treatment. Tables of the percentages of CD8-low or CD8-high tumors (based on average levels of CD8+ cells in each group) are given below and significance was calculated using Chi-square test. I, The representative images from H. Scale bar, 150 μm. Data correspond to mean values ± SD. P values for the bar graphs were calculated with the unpaired, two-tailed Student t test. Significance for the Chi-square analysis was calculated with the Chi-square test. **, P < 0.01. (F, Created with BioRender.com.)](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=11063689_1475fig1.jpg)
T-DM1 induces ICD markers in T-DM1–sensitive breast cancer cells and ICD correlates with T-DM1 sensitivity in patients. A, Western blot analysis of mitotic arrest [p-H3 (S10)], apoptosis (cleaved caspase-3 and PARP), and ICD marker [p-eIF2α (S51)] in T-DM1–treated SK-BR-3 WT (left) and BT-474 WT (right) cells. B and C, Relative ATP release (B) and HMGB1 release (C) from T-DM1–treated SK-BR-3 WT and BT-474 WT cells (n = 3, 4). D, Immunofluorescence cell-surface staining of calreticulin (green) in T-DM1–treated SK-BR-3 WT cells. Scale bar, 10 μm. DAPI was used to stain the nucleus. Its quantification is provided on the right. E, Cytokine array blot analysis showing the differentially secreted cytokines in T-DM1–treated SK-BR-3 WT cells. F, Schematic summary of the treatment scheme and the sample collection timeline in GSE194040 (22). G, Heatmap of ICD-related genes found in the ICD gene signature score (33) and their correlation with pCR in T-DM1 + pertuzumab-treated patients from GSE194040. pCR: 1, sensitive; pCR: 0, resistant. Chi-square analysis of sensitive vs. resistant tumors expressing low vs. high ICD score is provided below. H, Percentage of CD8+ cells in sensitive (sens) vs. resistant (res) tumors collected pre- (n = 40) and post-T-DM1 (n = 18) treatment. Tables of the percentages of CD8-low or CD8-high tumors (based on average levels of CD8+ cells in each group) are given below and significance was calculated using Chi-square test. I, The representative images from H. Scale bar, 150 μm. Data correspond to mean values ± SD. P values for the bar graphs were calculated with the unpaired, two-tailed Student t test. Significance for the Chi-square analysis was calculated with the Chi-square test. **, P < 0.01. (F, Created with BioRender.com.)
To further test the clinical relevance of ICD in determining response to T-DM1, we analyzed the GSE194040 (22) patient data set, containing gene expression profiling data from pretreated HER2+ tumors of patients with clinical information on the status of pathologic complete response (pCR) or non-pCR after T-DM1 + pertuzumab therapy (Fig. 1F). We generated a heatmap of genes found in a preclinically validated, and prognostic ICD-related gene signature (Fig. 1G, top; ref. 33) and correlated its levels with pCR. We showed that genes of the ICD score were expressed at higher levels in sensitive patients, whereas resistant patients were enriched in low ICD score expressers (Fig. 1G, bottom). We further validated these data using a breast cancer-specific ICD signature (Supplementary Fig. S2A and S2B). Notably, the high ICD score-expressing patients showed no enrichment of three different sets of tissue-derived immune cell signatures generated using microarray (25) or scRNA-seq (26, 27) data (Supplementary Fig. S2C, as determined by Mann–Whitney test). These data suggest that higher pretreatment levels of ICD-related genes, but not immune cell types, may predict response to T-DM1 therapy. To further support these findings, we analyzed our own cohort of 57 HER2+ breast cancer patients with variable clinical outcomes, and with available tumor samples having been collected before and after T-DM1 therapy (Hacettepe cohort, Supplementary Table S1). Although the percentage of CD8-high tumors was similar between sensitive versus resistant patients in the pretreatment settings, it was significantly higher in sensitive patients compared with resistant ones after treatment with T-DM1 (75% vs. 43%; Fig. 1H and I). Of note, there was no change in the levels of CD3+ and CD4+ cells based on T-DM1 sensitivity in either pretreatment or posttreatment settings (Supplementary Fig. S2D–S2F). Overall, these data demonstrate that ICD is a novel mechanism of action of T-DM1, expression of ICD-related genes correlates with T-DM1 response in pretreatment samples, and increased infiltration of antitumor CD8+ T cells is correlated with better T-DM1 response in posttreatment samples.
T-DM1–induced ICD is driven by SAC-dependent mitotic arrest induced by the payload, DM1
To test if the T-DM1–induced release of DAMPs is dependent on mitotic arrest and activation of SAC, which are known to induce apoptosis under T-DM1 treatment (9, 10), we inhibited Mps1, one of the major regulators of SAC activation (34), with TC Mps1 in T-DM1–treated SK-BR-3 and BT-474 cells. We observed that T-DM1 induced growth inhibition (Fig. 2A; Supplementary Fig. S3A), eIF2α phosphorylation (Fig. 2B), ATP release (Fig. 2C), HMGB1 release (Fig. 2D; Supplementary Fig. S3B) as well as cell-surface exposure of calreticulin (Fig. 2E and F) were all reversed upon SAC inhibition. We also validated these results using a third HER2+ cell line, AU-565, having high sensitivity to T-DM1 in terms of growth inhibition, and SAC-dependent ICD induction (Supplementary Figs. S3C–S3G). To test if the observed induction of ICD markers is due to the antibody or the payload component of T-DM1, we tested the effect of trastuzumab, the antibody component of T-DM1, and showed that trastuzumab used at the same dose and duration as T-DM1 was not able to elicit the induction of eIF2α phosphorylation, ATP release or calreticulin surface exposure in none of the HER2+ cell lines (Supplementary Fig. S4A–S4F), suggesting that T-DM1 induced ICD markers are due to its payload. Indeed, treatment of both HER2+ and HER2− breast cancer cell lines (Supplementary Fig. S3C) with the payload, DM1 at the corresponding dose caused a SAC-dependent activation of ICD markers (Fig. 2G–L; Supplementary Fig. S5). Overall, our data show that T-DM1 induces mitotic cell death and activation of ICD markers in a SAC-dependent manner via its payload DM1.
Figure 2.

T-DM1–induced ICD is driven by SAC-dependent mitotic arrest induced by the payload, DM1. A, Percent growth inhibition in SK-BR-3 WT cells treated with T-DM1 alone or in combination with 1 μmol/L TC Mps1 (Mps1 inhibitor; n = 4). B, Western blot analysis of p-H3 and p-eIF2α in SK-BR-3 WT cells treated with T-DM1 alone or in combination with 1 μmol/L TC Mps1. Actin was used as a loading control. C and D, Relative ATP (C) and HMGB1 (D) release in SK-BR-3 WT cells treated with T-DM1 alone or in combination with 1 μmol/L TC Mps1 (n = 3). E, Immunofluorescence cell-surface staining of calreticulin (green) in SK-BR-3 WT cells treated with T-DM1 alone or in combination with 1 μmol/L TC Mps1. Scale bar, 10 μm. F, The quantification graph of E. G, Western blot analysis of mitotic arrest, apoptosis, and the ICD marker, p-eIF2α (S51), in BT-474 cells treated with two different doses of DM1 (150 and 300 nmol/L) with or without TC Mps1. Actin was used as the loading control. H and I, Relative ATP (H) and HMGB1 (I) release in SK-BR-3 cells treated with DM1 (15 nmol/L) with or without TC Mps1 (n = 3). J and K, Relative ATP (J) and HMGB1 (K) release in BT-474 cells treated with DM1 (150 nmol/L) with or without TC Mps1 (n = 3). L, Surface calreticulin staining of SK-BR-3 and BT-474 cells treated with DM1 (15 nmol/L for SK-BR-3 and 150 nmol/L for BT-474) with or without TC Mps1. Data correspond to mean values ± SD. P values were calculated with the unpaired, two-tailed Student t test. **, P < 0.01.
ICD-related factors are lost in T-DM1 resistance upon TACC3 overexpression, TACC3 correlates with clinical T-DM1 resistance, and its inhibition overcomes T-DM1 resistance and restores ICD markers in vitro
It is unclear if the loss of ICD is a mechanism of drug resistance. Therefore, we first examined the markers of mitotic arrest, apoptosis, and ICD in our previously published acquired resistant models of T-DM1 (BT-474 T-DM1R and SK-BR-3 T-DM1R; ref. 11). We observed that whereas the induction of p-eIF2α (an indispensable marker of ICD induction) upon T-DM1 treatment was accompanied by the induction of mitotic arrest and apoptosis in sensitive cells, none of these markers were induced in T-DM1R cells (Fig. 3A), suggesting that genes involved in mitotic progression likely control ICD. Supporting these data, T-DM1 was not able to elicit the induction of DAMPs (e.g., ATP release and HMGB1 release) in T-DM1R cell lines (Supplementary Fig. S6A and S6B). To identify the mediators of T-DM1 resistance that suppress the prolonged mitosis and ICD-related markers, which are induced in sensitive cells upon T-DM1 treatment, we examined the changes in mitosis genes (using the gene set “mitotic cell-cycle transition” from Gene Ontology) upon T-DM1 resistance by reanalyzing our RNA-seq data of BT-474 T-DM1R and SK-BR-3 T-DM1R cell lines compared with their WT counterparts (Fig. 3B). 21 genes were differentially expressed only in BT-474 T-DM1R cells whereas 35 genes were differentially expressed only in SK-BR-3 T-DM1R cells. 11 genes were deregulated in both cell lines in the same direction (CDKN1A, UBE2S, TACC3, UBE2C, PLK1, CHMP4B, CCNB2, ZPR1, NFIA, CCNO, and ID4). Among those, TACC3 had the third highest fold change (upregulated in the T-DM1R cells). It is a spindle and centrosome-associated protein, maintaining microtubule dynamics and driving mitotic progression, and it is also druggable (Fig. 3B). We validated the increased expression of TACC3 in T-DM1–resistant cells also at protein level (Fig. 3C). To further test the clinical relevance of TACC3 protein expression in terms of its correlation with T-DM1 response, we stained TACC3 protein in our Hacettepe cohort. We observed that there is downregulation of the percentage of TACC3-high tumors in posttreatment setting as compared with pretreatment within sensitive patients (from 53% to 17%), whereas there is upregulation of the percentage of TACC3-high tumors in posttreatment setting as compared with pretreatment within resistant patients (from 48% to 64%; Fig. 3D and E). Notably, in posttreatment settings, where we observed higher TACC3 levels in resistant patients, the percentage of Ki67-high cells was higher in resistant tumors compared with sensitive ones (57% vs. 33%; Supplementary Fig. S6C and S6D), similar to TACC3 levels (Fig. 3D). This demonstrates the association between TACC3 and proliferative capacity under T-DM1 therapy, and it also confirms the resistance status of the tumors. Overall, these data suggest that higher TACC3 protein expression upon T-DM1 treatment may be associated with increased proliferative capacity and resistance to T-DM1 in clinical samples. Interestingly, the downregulation of TACC3 protein upon T-DM1 or DM1 treatment in a sensitive setting was also observed in vitro in sensitive cell lines (Supplementary Fig. S7A and S7B). To determine the mechanisms of TACC3 protein downregulation, we tested whether the mitotic ubiquitin ligase, the anaphase-promoting complex (APC/C) could trigger TACC3 degradation. Silencing the activator of the APC/C complex, CDC20 reversed the T-DM1–induced TACC3 downregulation, suggesting that APC/C complex in part mediates TACC3 degradation (Supplementary Fig. S7C–S7E).
Figure 3.

ICD-related factors are lost in T-DM1 resistance upon TACC3 overexpression, TACC3 correlates with clinical T-DM1 resistance, and its inhibition overcomes T-DM1 resistance and restores ICD markers in vitro. A, Western blot analysis of mitotic arrest, apoptosis, and ICD markers in SK-BR-3 WT and T-DM1–resistant (T-DM1R) cells treated with 0.05 μg/mL T-DM1 in a time-dependent manner. B, The log-fold change of the mitotic genes differentially expressed only in BT-474, SK-BR-3, or both T-DM1R cells compared with WT counterparts in RNA-seq analysis. C, Western blot analysis of TACC3 protein expression in BT-474 and SK-BR-3 WT vs. T-DM1R cells. Actin was used as a loading control. D, TACC3 IHC score in Hacettepe cohort patients before and after treatment with T-DM1 who are sensitive (left) vs. resistant (right) to T-DM1. The percentages of patients who have TACC3-low or -high tumors in the pre- and posttreatment groups are given below, and significance was calculated using the Chi-square test. E, Representative TACC3 IHC and hematoxylin and eosin (H&E) staining in the tumor tissues of patients from D. Scale bar, 100 μm. F, Percent growth inhibition in SK-BR-3 T-DM1R cells transfected with siTACC3 and treated with 0.03 μmol/L T-DM1 (n = 4–6). G, Percent growth inhibition in SK-BR-3 T-DM1R cells treated with T-DM1 alone or in combination with 1 μmol/L TACC3 inhibitor (BO-264; n = 4–6). H, Western blot analysis of mitotic arrest, apoptosis, and ICD markers in BT-474 T-DM1R cells transfected with siTACC3 and treated with T-DM1. Actin was used as a loading control. I, Western blot analysis of mitotic arrest, apoptosis, and ICD markers in SK-BR-3 T-DM1R cells treated with T-DM1 alone or in combination with BO-264. Actin was used as a loading control. J, Relative ATP release from SK-BR-3 T-DM1R cells treated with T-DM1 alone or in combination with BO-264 (n = 3, 4). K, Relative HMGB1 release from SK-BR-3 T-DM1R cells treated with T-DM1 alone or in combination with BO-264 (n = 3). L, Western blot analysis of mitotic arrest, apoptosis, and ICD markers in SK-BR-3 WT cells overexpressing TACC3 and treated with T-DM1. Actin was used as a loading control. M, Relative ATP release from SK-BR-3 WT cells overexpressing TACC3 and treated with T-DM1 (n = 3). N, Percent growth inhibition in SK-BR-3 WT cells overexpressing TACC3 and treated with T-DM1 (n = 3). Data correspond to mean values ± SD. Significance for D was calculated with one-way Wilcoxon signed-rank test. P values for other subfigures were calculated with the unpaired, two-tailed Student t test. *, P < 0.05; **, P < 0.01.
To determine the causal role of TACC3 overexpression in mediating T-DM1 resistance, we tested if targeting TACC3 overcomes T-DM1 resistance in our acquired T-DM1–resistant models. Inhibition of TACC3 either with three different siRNAs (Fig. 3F; Supplementary Fig. S8A; ref. 19) or with pharmacologic inhibitors BO-264 (Fig. 3G; Supplementary Fig. S8B) or SPL-B (Supplementary Fig. S8C; ref. 35) overcame resistance in both T-DM1R cell lines. TACC3 inhibition in combination with T-DM1 disrupted microtubule dynamics (Supplementary Fig. S8D), inhibited microtubule polymerization (Supplementary Fig. S8E) and caused mitotic arrest and apoptosis (Fig. 3H and I; Supplementary Fig. S8F). Notably inhibiting the SAC kinase, Mps1 using TC Mps1 reversed the TACC3 inhibition-mediated T-DM1 sensitization (Supplementary Fig. S8G), suggesting that SAC activation is also crucial for restoration of T-DM1 sensitivity by TACC3 targeting. Furthermore, combination treatment increased eIF2α phosphorylation (Fig. 3H and I; Supplementary Fig. S8F), ATP release (Fig. 3J; Supplementary Fig. S9A–S9C), HMGB1 release (Fig. 3K; Supplementary Fig. S9D) and calreticulin surface exposure in these T-DM1R cell lines (Supplementary Fig. S9E–S9K). It also induced the secretion of proinflammatory cytokines (Supplementary Fig. S9L). Notably, overexpression of TACC3 in T-DM1 sensitive cells abrogated T-DM1–induced mitotic arrest, apoptosis, eIF2α phosphorylation (Fig. 3L), and decreased ATP release (Fig. 3M), thus conferring T-DM1 resistance (Fig. 3N). Overall, our data show that T-DM1 resistance is characterized by loss of ICD markers, and targeting TACC3 overcomes T-DM1 resistance in a SAC-dependent manner and restores the induction of T-DM1–induced ICD markers in vitro.
Targeting TACC3 in combination with T-DM1 in human HER2-expressing murine cells induces ICD markers and leads to ex vivo DC maturation and T-cell activation
To test the effects of TACC3 inhibition in combination with T-DM1 in a syngeneic T-DM1–resistant setting that will allow us to assess the changes in the immunogenicity of the cells, we first developed the human HER2-overexpressing derivative of the murine EMT6 mammary tumor cells; EMT6.huHER2. We demonstrated that these cells are resistant to T-DM1, in line with the literature (36), and inhibiting TACC3 with BO-264 reversed resistance by reducing cell viability in a dose-dependent manner (Fig. 4A). Furthermore, we validated our results by knocking out TACC3 using CRISPR/Cas9 system. Both sgTACC3 constructs effectively reduced TACC3 expression (Fig. 4B) and mediated T-DM1 sensitization (Fig. 4C), similar to TACC3 inhibitors. Notably, combination of TACC3 knockout or its pharmacologic inhibition with T-DM1 induced mitotic arrest, eIF2α phosphorylation, and apoptosis (Fig. 4D and E; Supplementary Fig. S10A and S10B) and activated the ICD markers; ATP release (Fig. 4F and G) and calreticulin cell-surface exposure (Fig. 4H and I) in the T-DM1–resistant EMT6.huHER2 cells.
Figure 4.

Targeting TACC3 sensitizes the human HER2-expressing EMT6.huHER2 cells to T-DM1 and induces ICD markers. A, Cell viability assay in EMT6.huHER2 cells treated with increasing doses of T-DM1 alone or combination with different doses of BO-264 for 3 days (n = 4). B, Validation of TACC3 knockout in EMT6.huHER2 cells obtained using CRISPR/Cas9 system. C, Cell viability assay in EMT6.huHER2.sgTACC3 vs. sgControl cells treated with increasing doses of T-DM1 for 3 days (n = 4). D, Western blot analysis of mitotic arrest, apoptosis, and ICD markers in EMT6.huHER2 cells treated with T-DM1 alone or in combination with BO-264. Actin was used as a loading control. E, Western blot analysis of TACC3, mitotic arrest, apoptosis, and ICD markers in EMT6.huHER2.sgTACC3 vs. sgControl cells treated with T-DM1. Actin was used as a loading control. F, Relative ATP release from EMT6.huHER2 cells treated with T-DM1 alone or in combination with BO-264 (n = 3). G, Relative ATP release from EMT6.huHER2.sgTACC3 vs. sgControl cells treated with T-DM1 (n = 3, 4). H, Immunofluorescence cell-surface staining of calreticulin (green) in EMT6.huHER2 cells treated with T-DM1 alone or in combination with BO-264. Its quantification is provided on the right. I, Immunofluorescence cell-surface staining of calreticulin (green) in EMT6.huHER2.sgTACC3 vs. sgControl cells treated with T-DM1. Its quantification is provided on the right. Data correspond to mean values ± SD. P values were calculated with the unpaired, two-tailed Student t test. **, P < 0.01.
It has been shown that ICD induction is followed by DC maturation that further results in T-cell activation (37, 38). To test whether the combination of TACC3 inhibition with T-DM1 induces DC maturation and T-cell activation, we isolated bone marrow–derived DCs from BALB/c mice (the strain EMT6 cells were originated from; Fig. 5A). Incubation of DCs with conditioned media (CM) collected from T-DM1 + BO-264–treated EMT6.huHER2 cells resulted in increased expression of the DC maturation markers, CD80 and CD86 (Fig. 5B; Supplementary Fig. S10C). These results were also recapitulated using CRISPR-Cas9-mediated knockout of TACC3 (Fig. 5C and D). Furthermore, coculturing these maturated DCs with T cells increased T-cell activation as shown by CD8 and CD25 staining (Fig. 5E-G; Supplementary Fig. S10D). We profiled the secreted cytokines/chemokines using multiplex cytokine analysis, and observed that proinflammatory markers IL1β, IL2, IL6, IL17, MIP-1α, MIP-1β, and TNFα were all increased in combination-treated samples (Fig. 5H). Overall, these data suggest that inhibition of TACC3 in combination with T-DM1 induces ICD markers and leads to ex vivo DC maturation and T-cell activation in the EMT6.huHER2 murine mammary tumor model.
Figure 5.

Inhibition of TACC3 in combination with T-DM1 leads to ex vivo DC maturation, T-cell activation, and release of ICD-related proinflammatory cytokines. A, Schematic representation of the experimental workflow for DC maturation, T-cell activation, and cytokine profiling experiments. B and C, Flow cytometry analysis of DC maturation markers in DC cells incubated with the CM collected from EMT6.huHER2 cells treated with 7.5 μg/mL T-DM1 and 500 nmol/L BO-264, alone or in combination (B) or in EMT6.huHER2.sgControl vs. sgTACC3 cells treated with 7.5 μg/mL T-DM1 (C). D, Quantification of CD80+/CD86+ cells from B and C (n = 2). E and F, Flow cytometry analysis of T-cell activation marker, CD25 in CD8+ T cells cocultured with DCs from B and C. G, Quantification of the CD25 mean fluorescence intensity (MFI) from E and F (n = 2). H, Levels of proinflammatory cytokines in the media collected from DC-T-cell cocultures from E. Data correspond to mean values ± SD. P values were calculated with the unpaired, two-tailed Student t test. **, P < 0.01. (A, Created with BioRender.com.)
Targeting TACC3 induces ICD markers, leading to immune cell infiltration and potentiating T-DM1 response in vivo
To assess the effects of the combination of T-DM1 with TACC3 inhibition on ICD induction in vivo, we performed the so-called vaccination assay using the T-DM1–resistant EMT6.huHER2 cells (21). Cells were treated with T-DM1 or BO-264 alone or in combination for 48 hours, and the treated cell population (which harbors around 60% apoptotic cells, Supplementary Fig. S10A and S10B) were injected into the flank of BALB/c mice, followed by injection of living cells to the opposite flank after a week, and monitoring of tumor formation (Fig. 6A). Vaccinating mice with combination-treated and mostly apoptotic cells significantly improved tumor-free survival compared with control or single-agent treated groups, demonstrating that our combination therapy elicits ICD in vivo to inhibit tumor formation (Fig. 6B).
Figure 6.

TACC3 inhibition elicits ICD in vivo and potentiates TDM1 response via increasing the infiltration of antitumor immune cells in vivo. A, Schematic representation of the in vivo vaccination assay. B, Tumor-free survival curves of BALB/c mice vaccinated with PBS or single agent or combination-treated EMT6.huHER2 cells (n = 5–7). C, Tumor growth of the MMTV.f.huHER2#5 model under low dose T-DM1 (5 mg/kg, once) in combination with BO-264 (50 mg/kg, daily; n = 6, 7). D, Tumor weights of the mice in C after 14 days of treatment. E and F, Representative resected tumor pictures (E) and body weights (F) from mice in C. G, Western blot analysis of p-eIF2α and eIF2α protein expression levels in tumors from C. Actin was used as a loading control. H, Relative band density graphs for p-eIF2α normalized to eIF2α from G (n = 3). I and J, Multiplex immunofluorescence staining of CD11c/CD86 in short-term-treated MMTV.f.huHER2#5 tumors and its quantification (n = 3). K and L, Multiplex immunofluorescence staining of CD25/CD8 in short-term-treated MMTV.f.huHER2#5 tumors and its quantification (n = 3). Scale bar, 50 μm. M, Levels of the cytokines in the serums of the mice with short-term–treated MMTV.f.huHER2#5 tumors (n = 3). Data for the bar graphs and box plots correspond to mean values ± SD, whereas data for the tumor volume and body weight graphs correspond to mean values ± SEM. Endpoint criteria for mice in C and F are treatment for 14 days or until reaching ethical tumor size cutoff. P values for the bar graphs and box plots were calculated with the unpaired, two-tailed Student t test. The significance for the tumor volume graph and multiplex IHC quantification was calculated with two-way and one-way ANOVA, respectively. *, P < 0.05; **, P < 0.01. (A, Created with BioRender.com.)
To test the effects of TACC3 inhibition on enhancing T-DM1 response in the first-line settings, we utilized the relatively T-DM1 responsive human HER2-overexpressing MMTV.f.huHER2#5 (Fo5) transgenic model (8, 39). Combination treatment completely blocked tumor growth as compared with single-agent T-DM1 (5 mg/kg, i.v., once) or BO-264 (50 mg/kg, oral gavage, daily) treatments (Fig. 6C–E), without affecting body weight (Fig. 6F). Importantly, combination-treated Fo5 tumors exhibited the highest levels of p-eIF2α, the canonical marker of ICD (Fig. 6G and H). To analyze changes in tumor-infiltrated lymphocytes (TIL) and tumor-infiltrated DCs (TIDC) upon combination treatment, we treated the Fo5 tumor-bearing mice with T-DM1 alone or in combination with BO-264 for a week and performed multiplex IHC staining of DCs and T cells in the collected tumors. As a result, we observed that the infiltration of CD11c+CD86+ mature DCs (Fig. 6I and J) and CD8+CD25+ activated cytotoxic T cells (Fig. 6K and L) was significantly increased in tumor samples treated with the combination therapy. Notably, at the sites of DC and T-cell infiltrates, the levels of the ICD markers, p-eIF2α and HMGB1 were also higher (Supplementary Fig. S11A–S11C), suggesting that the induction of ICD following treatment with T-DM1/BO-264 combination is likely involved in the recruitment of antitumor immune cells. Furthermore, there was a trend toward a decrease in T-DM1–induced infiltration of tumor-promoting Foxp3+/CD4+ regulatory T (Treg) cells, whereas NK1.1+/CD27+ NK cells that are responsible for trastuzumab-mediated antibody-dependent cellular cytotoxicity (ADCC), underwent a slight, albeit not significant, decrease in combination therapy (Supplementary Fig. S11D and S11E). We further analyzed the serum levels of cytokines collected from treated mice and observed an increase in the proinflammatory cytokines [IL1α, IL2, RANTES (CCL5), MIP-3α, and LIX (CXCL5)] under combination therapy (Fig. 6M). Altogether, these data demonstrate that TACC3 inhibition restores T-DM1–induced ICD and increases the infiltration of antitumor DCs and cytotoxic T cells without triggering the infiltration of protumorigenic Tregs, thus leading to stronger growth inhibition of human HER2-expressing tumors.
Discussion
ICD is a unique form of cell death that can activate antitumor immune response and has been shown to be induced by several different anticancer therapies. Despite being the first and one of the most successful ADCs approved for refractory HER2-positive breast cancer, little is known if response and resistance to T-DM1 involves ICD modulation that can be leveraged to enhance T-DM1 response. Here, we demonstrate, for the first time, that the iconic ADC, T-DM1 can elicit all the hallmarks of ICD, i.e., eIF2α phosphorylation, ATP secretion, HMGB1 release, and calreticulin surface exposure via its payload DM1, and in a SAC-dependent manner in drug sensitive models (Fig. 7A). In T-DM1 resistance, TACC3 is upregulated and inhibits T-DM1–induced SAC activation, thus blocking eIF2α phosphorylation and DAMPs, which ultimately results in cell survival (Fig. 7B). Inhibiting TACC3 in T-DM1–resistant tumors restores T-DM1–induced SAC activation, mitotic cell death, eIF2α phosphorylation and elevated levels of DAMPs, i.e., the hallmarks of ICD. TACC3 inhibition in combination with T-DM1 further induces ICD markers, proinflammatory cytokine secretion, DC maturation, and T-cell activation, eventually causing inhibition of tumor growth (Fig. 7C).
Figure 7.

Schematic summary of the proposed model of T-DM1 sensitivity, resistance, and targeting T-DM1 resistance. A, In T-DM1–sensitive tumors, the activation of SAC and mitotic arrest lead to apoptosis and activation of ICD markers, e.g., eIF2α phosphorylation, ATP secretion, calreticulin surface exposure, and HMGB1 release, leading to DC maturation and cytotoxic T-cell, culminating in tumor growth inhibition. B, In T-DM1–resistant tumors, overexpression of TACC3 prevents activation of SAC, mitotic cell death, and ICD, thus promoting cell survival. C, Inhibition of TACC3 in combination with T-DM1 in the resistant tumors restores SAC activation and mitotic arrest, leading to apoptosis, induction of ICD hallmarks, and secretion of proinflammatory cytokines, thereby increasing the infiltration of DCs and T cells, thus restoring T-DM1 sensitivity. (Created with BioRender.com.)
In recent years, it has been increasingly recognized that antitumor immune activation is an integral part of response to anticancer therapies. To evoke a cytotoxic immune response, these therapies trigger an immunogenic form of cell death that is characterized by secretion of the so-called eat-me signals, i.e., DAMPs from the dying cancer cells. It has been postulated that levels of DAMPs have prognostic and predictive value in different types of cancers (40). Along these lines, monitoring the levels of DAMPs in the pretreated tumor samples was proposed to have a predictive value by enabling the identification of patients who are intrinsically capable or incapable of responding to ICD-inducing treatment, and for providing novel combinatorial strategies aiming to restore ICD induction in resistant tumors (41). Upon treatment with ICD-inducing single/combination therapies, these DAMPs then lead to infiltration and activation of antitumor immune cells, leading to tumor shrinkage. Along these lines, a higher level of TILs usually indicates a better response to therapy (42). In the context of T-DM1, there were two trials that support our findings that showed an association of T-DM1 response with higher immune cell infiltration, only after T-DM1 therapy. A recent study published the results from the KATHERINE trial showed that levels of predefined immune signatures before T-DM1 therapy showed no association with T-DM1 response in adjuvant settings (43). On the other hand, in an open-label, phase III study (KRISTINE), it was reported that higher levels of immune markers after neoadjuvant T-DM1–containing therapy are associated with higher pCR rates (44). These studies along with our data suggest that expression of ICD-related genes may predict better response to T-DM1, which may further induce antitumor immune activation in sensitive patients. Nevertheless, our study still has limitations due to the lack of availability of a cohort of a larger number of patients with available pre- and posttherapy samples and gene expression profiling.
Association of TILs with clinical response has also been tested for other ADCs, e.g., trastuzumab deruxtecan, and a positive correlation between number of TILs and drug sensitivity was observed (45, 46). In line with the emerging preclinical and clinical data on the immune modulatory effects of ADCs, promising data have been gathered in recent years that support the potential benefits of combining ADCs with immune-oncology drugs. It has been demonstrated that combining ADCs with immune checkpoint inhibitors enhances the homing and activation of immune effector cells, thus leading to stronger therapy responses (47). Therefore, a combination of ADCs with immune-checkpoint blockers, and other immune-oncology drugs are currently in clinical trials in both hematologic malignancies and solid tumors. For instance, Enfortumab vedotin, an ADC of a Nectin-4–directed antibody conjugated to MMAE, a microtubule inhibitor blocking tubulin polymerization, belonging to the same class as DM1, has recently been approved by the FDA as the first ADC to be combined with a PD-1 inhibitor (48). Furthermore, the preliminary results of the combination of Dato-DXd (TROP2-directed deruxtecan) and the PD-L1 antibody durvalumab at the first-line settings for advanced or metastatic TNBC (NCT03742102) demonstrated that 79% of patients had confirmed objective responses, with 10% having complete response, and 69% having partial response (49). Furthermore, the combination therapy was well-tolerated with manageable side effects (49). Here, we demonstrated, for the first time, that ICD induction is one of the mechanisms of T-DM1 sensitivity in vitro and in vivo. Importantly, we showed that tumors from sensitive patients have higher CD8+ T-cell infiltration upon T-DM1 treatment, and combination of T-DM1 with TACC3 inhibition potentiates the induction of ICD markers to induce antitumor immune cell infiltration in vivo in the HER2+ Fo5 transgenic mouse model. These results further encourage testing of the potential of ADCs other than T-DM1 to induce ICD, and investigating the association between sensitivity to ADCs, including T-DM1 or their combination with immune-oncology drugs and ICD hallmarks.
Antimicrotubule agents, such as taxanes, are among the most commonly used chemotherapeutics in cancer and their major mechanism of action involves mitotic cell death via disruption of microtubule dynamics. Intriguingly, recent studies have suggested that the clinical success of microtubule-targeting agents is not only a result of mitotic cell death but potentially involves novel mechanisms that lead to the activation of a strong antitumor immune response (50–52). For instance, paclitaxel, one of the most widely used taxanes was shown to promote a proinflammatory response by activation of innate immunity (51, 53). Paclitaxel may also improve the efficacy of PD-L1 blockade therapy in animal models by causing tumor eradication, metastasis suppression, and preventing recurrence (54). Furthermore, paclitaxel has recently been shown to activate ICD in ovarian cancer cells (55). Despite this preliminary evidence on the potential roles of microtubule-targeting agents on activating antitumor immunity, a mechanistic connection between mitotic arrest, SAC activation, and the induction of ICD markers has not been tested before. In this study, we demonstrated that the T-DM1–induced mitotic arrest and SAC activation are required for the induction of ICD hallmarks, identifying SAC activation as a potentially novel way to induce ICD. However, given that ICD induction is, in part, dictated by the structure of the drug in addition to its molecular mechanisms of action, it has yet to be determined whether other inducers of SAC activation or mitotic arrest could also activate the hallmarks of ICD and elicit immune responses.
TACC3 is a microtubule and centrosome-associated protein playing key roles in mitotic progression (56, 57). TACC3 inhibition was shown to cause formation of multipolar spindles, mitotic arrest, and apoptosis (13, 19, 35). In recent years, novel noncanonical roles of TACC3 in tumor progression are also emerging. Here, we showed that TACC3 inhibition in combination with T-DM1 revives ICD to activate antitumor immune cells. The lack of a profound increase in T-cell activation markers, CD25/CD8 in T-DM1+BO-264/sgTACC3 groups compared with T-DM1 alone (Fig. 5G) might potentially be due to reaching the saturation limit of detecting T-cell activity, which showed certain level of increase in single-agent T-DM1 group as well, due to slightly higher CD80/CD86 (Fig. 5D). Of note, although results of sgRNA-mediated knockout of TACC3 was highly similar to BO-264 treatment, we do not rule out any potential off-target effects of BO-264. An in silico analysis in kidney renal cell carcinoma demonstrated that TACC3 expression correlates with several different types of immune cells, including follicular helper T cells and Tregs (58). Infiltration of the immune-suppressive Tregs into tumors may promote tumor growth via blocking antitumor immune responses (59). For instance, combination of T-DM1 with immune-checkpoint blockers showed efficacy in animal models; however, the infiltration of tumor-promoting Foxp3+/CD4+ Tregs was shown to be increased as well (29). Along these lines, although a trend is observed toward higher progression-free survival in PDL1+ patients upon a combination of T-DM1 with the immune-checkpoint inhibitor, atezolizumab, the difference in survival was not clinically meaningful with more adverse events (60). These findings underlie the necessity to identify novel therapeutic strategies to achieve potent and durable immunogenic responses without activating tumor-promoting immune subsets in larger patient subpopulations. Our data demonstrating the secretion of proinflammatory cytokines, DC maturation, and increased infiltration of cytotoxic effector T cells upon inhibiting TACC3 in combination with T-DM1 without an increase in the infiltration of Foxp3+ Tregs suggest that TACC3 inhibition could be a superior therapeutic strategy to boost the immunogenicity of the tumors without activating tumor-promoting factors.
It has been demonstrated that T-DM1 treatment increases tumor-infiltrating NK cells that are responsible for trastuzumab-induced ADCC (61, 62) in the absence or presence of immunotherapy (29). Interestingly, we did not observe a significant change in the infiltration of NK cells upon T-DM1 treatment or upon treatment with the combination of T-DM1 and TACC3 inhibitors. The lack of NK cell infiltration even in the T-DM1 monotherapy group is probably due to the lower T-DM1 dose used in our study, i.e., 5 mg/kg compared with 15 mg/kg dose used in Muller and colleagues (29). Nonetheless, the strong tumor growth inhibition that we observed upon combination therapy with no NK cell infiltration suggests that TACC3 inhibition-mediated T-DM1 potentiation does not likely involve trastuzumab-mediated ADCC. Overall, our data encourages testing the combination of TACC3 inhibitors with other ADCs beyond T-DM1 or even with immune-checkpoint blockers to achieve superior and durable responses.
Overall, we showed that ICD induction upon SAC-induced mitotic cell death is a novel mechanism of T-DM1 sensitivity and activates T-cell–mediated antitumor immunity, whereas T-DM1 resistance is characterized by loss of ICD. We further identified TACC3 as a novel resistance mediator whose inhibition restores the induction of ICD hallmarks and increases the infiltration of cytotoxic effector T cells into tumors. These data provide preclinical evidence for targeting TACC3 to revive tumor immunogenicity driven by ICD-related DAMPs in T-DM1 refractory HER2+ breast cancer that may ultimately result in improved clinical outcomes.
Supplementary Material
This file contains supplementary methods, 11 supplementary figures and 2 supplementary tables
This file contains Supplementary Table S1
Acknowledgments
We are thankful to the members of the Ozgur Sahin laboratory for their invaluable discussion and advice. We thank the Translational Science Laboratory and the Flow Cytometry and Cell Sorting Shared Resource of the Medical University of South Carolina. We thank Dr. Stephen Royle (University of Warwick) for providing TACC3 ORF-expressing vector. This work was supported by research funding from Mary Kay Ash Foundation Grant MK-07–21 (O. Sahin), and in part, from the NIH (R01CA251374 to O. Sahin) and previously by TUBITAK-BMBF Bilateral Grants (TUBITAK, 214Z130 to O. Sahin and BMBF WTZ, 01DL16003 to S. Wiemann). The core facilities utilized are supported by NIH (C06 RR015455), Hollings Cancer Center Support Grant (P30 CA138313), or Center of Biomedical Research Excellence (COBRE) in Lipidomics and Pathobiology Grant (P30 GM103339). The Zeiss 880 microscope was funded by a Shared Instrumentation grant (S10 OD018113).
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
B. Caliskan reports grants and personal fees from OncoCube Therapeutics outside the submitted work; in addition, B. Akbulut Caliskan has a patent for TR 2018 07646 issued, a patent for EP 3 801 529 B1 issued, and a patent for US 11,622,966 B2 issued. E. Banoglu reports grants and personal fees from OncoCube Therapeutics LLC and personal fees from A2A Pharmaceuticals outside the submitted work; in addition, E. Banoglu has a patent for TR2018 07464 issued, a patent for EP 3 801 529 B1 issued, and a patent for US 11,622,966 B2 issued. S. Mehrotra reports grants from Lipo-Immuno Tech, LLC outside the submitted work. O. Sahin reports grants from Mary Kay Ash Foundation (MK-07-21), National Institutes of Health (R01CA251374, C06 RR015455, P30 CA138313, P30 GM103339, and S10 OD018113), TUBITAK-BMBF Bilateral Program (214Z130), and nonfinancial support from Genentech, Inc. during the conduct of the study; grants, personal fees, and nonfinancial support from OncoCube Therapeutics LLC, grants from LoxiGen, Inc., and personal fees from A2A Pharmaceuticals outside the submitted work; in addition, O. Sahin has a patent for TR2018 07646 issued, a patent for EP 3 801 529 B1 issued, and a patent for US 11,622,966 B2 issued. No disclosures were reported by the other authors.
Authors' Contributions
M.E. Gedik: Conceptualization, resources, data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. O. Saatci: Conceptualization, resources, data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. N. Oberholtzer: Formal analysis, investigation, writing–review and editing. M. Uner: Formal analysis, investigation, writing–review and editing. O. Akbulut Caliskan: Formal analysis, investigation, writing–review and editing. M. Cetin: Formal analysis, investigation, writing–review and editing. M. Aras: Investigation, writing–review and editing. K. Ibis: Investigation, writing–review and editing. B. Caliskan: Data curation, investigation, writing–review and editing. E. Banoglu: Data curation, investigation, writing–review and editing. S. Wiemann: Data curation, investigation, writing–review and editing. A. Üner: Data curation, investigation, writing–review and editing. S. Aksoy: Data curation, investigation, writing–review and editing. S. Mehrotra: Resources, data curation, investigation, methodology, writing–review and editing. O. Sahin: Conceptualization, resources, formal analysis, supervision, funding acquisition, validation, visualization, methodology, project administration, writing–review and editing.
References
- 1. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 2017;17:97–111. [DOI] [PubMed] [Google Scholar]
- 2. Kroemer G, Galassi C, Zitvogel L, Galluzzi L. Immunogenic cell stress and death. Nat Immunol 2022;23:487–500. [DOI] [PubMed] [Google Scholar]
- 3. Galluzzi L, Humeau J, Buque A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol 2020;17:725–41. [DOI] [PubMed] [Google Scholar]
- 4. Pol J, Vacchelli E, Aranda F, Castoldi F, Eggermont A, Cremer I, et al. Trial watch: immunogenic cell death inducers for anticancer chemotherapy. Oncoimmunology 2015;4:e1008866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu P, Zhao L, Pol J, Levesque S, Petrazzuolo A, Pfirschke C, et al. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nat Commun 2019;10:1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chau CH, Steeg PS, Figg WD. Antibody-drug conjugates for cancer. Lancet 2019;394:793–804. [DOI] [PubMed] [Google Scholar]
- 7. Lambert JM, Chari RV. Ado-trastuzumab Emtansine (T-DM1): an antibody-drug conjugate (ADC) for HER2-positive breast cancer. J Med Chem 2014;57:6949–64. [DOI] [PubMed] [Google Scholar]
- 8. Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res 2008;68:9280–90. [DOI] [PubMed] [Google Scholar]
- 9. Garcia-Alonso S, Ocana A, Pandiella A. Trastuzumab emtansine: mechanisms of action and resistance, clinical progress, and beyond. Trends Cancer 2020;6:130–46. [DOI] [PubMed] [Google Scholar]
- 10. Hunter FW, Barker HR, Lipert B, Rothe F, Gebhart G, Piccart-Gebhart MJ, et al. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br J Cancer 2020;122:603–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saatci O, Borgoni S, Akbulut O, Durmus S, Raza U, Eyupoglu E, et al. Targeting PLK1 overcomes T-DM1 resistance via CDK1-dependent phosphorylation and inactivation of Bcl-2/xL in HER2-positive breast cancer. Oncogene 2018;37:2251–69. [DOI] [PubMed] [Google Scholar]
- 12. Du Y, Liu L, Wang C, Kuang B, Yan S, Zhou A, et al. TACC3 promotes colorectal cancer tumourigenesis and correlates with poor prognosis. Oncotarget 2016;7:41885–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saatci O, Akbulut O, Cetin M, Sikirzhytski V, Uner M, Lengerli D, et al. Targeting TACC3 represents a novel vulnerability in highly aggressive breast cancers with centrosome amplification. Cell Death Differ 2023;30:1305–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yun M, Rong J, Lin ZR, He YL, Zhang JX, Peng ZW, et al. High expression of transforming acidic coiled coil-containing protein 3 strongly correlates with aggressive characteristics and poor prognosis of gastric cancer. Oncol Rep 2015;34:1397–405. [DOI] [PubMed] [Google Scholar]
- 15. Booth DG, Hood FE, Prior IA, Royle SJ. A TACC3/ch-TOG/clathrin complex stabilises kinetochore fibres by inter-microtubule bridging. EMBO J 2011;30:906–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gergely F, Karlsson C, Still I, Cowell J, Kilmartin J, Raff JW. The TACC domain identifies a family of centrosomal proteins that can interact with microtubules. Proc Natl Acad Sci USA 2000;97:14352–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Korn EL, Freidlin B, Abrams JS. Overall survival as the outcome for randomized clinical trials with effective subsequent therapies. J Clin Oncol 2011;29:2439–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mutlu M, Saatci O, Ansari SA, Yurdusev E, Shehwana H, Konu O, et al. miR-564 acts as a dual inhibitor of PI3K and MAPK signaling networks and inhibits proliferation and invasion in breast cancer. Sci Rep 2016;6:32541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Akbulut O, Lengerli D, Saatci O, Duman E, Seker UOS, Isik A, et al. A highly potent TACC3 inhibitor as a novel anticancer drug candidate. Mol Cancer Ther 2020;19:1243–54. [DOI] [PubMed] [Google Scholar]
- 20. Saatci O, Kaymak A, Raza U, Ersan PG, Akbulut O, Banister CE, et al. Targeting lysyl oxidase (LOX) overcomes chemotherapy resistance in triple negative breast cancer. Nat Commun 2020;11:2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Humeau J, Levesque S, Kroemer G, Pol JG. Gold standard assessment of immunogenic cell death in oncological mouse models. Methods Mol Biol 2019;1884:297–315. [DOI] [PubMed] [Google Scholar]
- 22. Wolf DM, Yau C, Wulfkuhle J, Brown-Swigart L, Gallagher RI, Lee PRE, et al. Redefining breast cancer subtypes to guide treatment prioritization and maximize response: predictive biomarkers across 10 cancer therapies. Cancer Cell 2022;40:609–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res 2013;41:D991–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Garg AD, De Ruysscher D, Agostinis P. Immunological metagene signatures derived from immunogenic cancer cell death associate with improved survival of patients with lung, breast or ovarian malignancies: a large-scale meta-analysis. Oncoimmunology 2016;5:e1069938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 2019;37:773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016;352:189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bezu L, Sauvat A, Humeau J, Gomes-da-Silva LC, Iribarren K, Forveille S, et al. eIF2alpha phosphorylation is pathognomonic for immunogenic cell death. Cell Death Differ 2018;25:1375–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Muller P, Kreuzaler M, Khan T, Thommen DS, Martin K, Glatz K, et al. Trastuzumab emtansine (T-DM1) renders HER2+ breast cancer highly susceptible to CTLA-4/PD-1 blockade. Sci Transl Med 2015;7:315ra188. [DOI] [PubMed] [Google Scholar]
- 30. Nersesian S, Shakfa N, Peterson N, Vidotto T, AfriyieAsante A, Lightbody E, et al. Chemotherapy induced immunogenic cell death alters response to exogenous activation of STING pathway and PD-L1 immune checkpoint blockade in a syngeneic murine model of ovarian cancer. Biorxiv 2019;824094. [Google Scholar]
- 31. Roussot N, Ghiringhelli F, Rebe C. Tumor immunogenic cell death as a mediator of intratumor CD8 T-cell recruitment. Cells 2022;11:3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li Z, Lai X, Fu S, Ren L, Cai H, Zhang H, et al. Immunogenic cell death activates the tumor immune microenvironment to boost the immunotherapy efficiency. Adv Sci (Weinh) 2022;9:e2201734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen L, Wen Y, Xiong J, Chen Y, Chen CB. An immunogenic cell death-related gene signature reflects immune landscape and predicts prognosis in melanoma independently of BRAF V600E status. Biomed Res Int 2023;2023:1189022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pachis ST, Kops G. Leader of the SAC: molecular mechanisms of Mps1/TTK regulation in mitosis. Open Biol 2018;8:180109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yao R, Kondoh Y, Natsume Y, Yamanaka H, Inoue M, Toki H, et al. A small compound targeting TACC3 revealed its different spatiotemporal contributions for spindle assembly in cancer cells. Oncogene 2014;33:4242–52. [DOI] [PubMed] [Google Scholar]
- 36. D'Amico L, Menzel U, Prummer M, Muller P, Buchi M, Kashyap A, et al. A novel anti-HER2 anthracycline-based antibody-drug conjugate induces adaptive anti-tumor immunity and potentiates PD-1 blockade in breast cancer. J Immunother Cancer 2019;7:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou J, Wang G, Chen Y, Wang H, Hua Y, Cai Z. Immunogenic cell death in cancer therapy: present and emerging inducers. J Cell Mol Med 2019;23:4854–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Moriya T, Kitagawa K, Hayakawa Y, Hemmi H, Kaisho T, Ueha S, et al. Immunogenic tumor cell death promotes dendritic cell migration and inhibits tumor growth via enhanced T cell immunity. iScience 2021;24:102424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Finkle D, Quan ZR, Asghari V, Kloss J, Ghaboosi N, Mai E, et al. HER2-targeted therapy reduces incidence and progression of midlife mammary tumors in female murine mammary tumor virus huHER2-transgenic mice. Clin Cancer Res 2004;10:2499–511. [DOI] [PubMed] [Google Scholar]
- 40. Fucikova J, Kepp O, Kasikova L, Petroni G, Yamazaki T, Liu P, et al. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis 2020;11:1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fucikova J, Moserova I, Urbanova L, Bezu L, Kepp O, Cremer I, et al. Prognostic and predictive value of DAMPs and DAMP-associated processes in cancer. Front Immunol 2015;6:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang K, Xu J, Zhang T, Xue D. Tumor-infiltrating lymphocytes in breast cancer predict the response to chemotherapy and survival outcome: a meta-analysis. Oncotarget 2016;7:44288–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Denkert C, Lambertini C, Fasching PA, Pogue-Geile KL, Mano MS, Untch M, et al. Biomarker data from the phase III KATHERINE study of adjuvant T-DM1 versus trastuzumab for residual invasive disease after neoadjuvant therapy for HER2-positive breast cancer. Clin Cancer Res 2023;29:1569–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. de Haas SL, Slamon DJ, Martin M, Press MF, Lewis GD, Lambertini C, et al. Tumor biomarkers and efficacy in patients treated with trastuzumab emtansine + pertuzumab versus standard of care in HER2-positive early breast cancer: an open-label, phase III study (KRISTINE). Breast Cancer Res 2023;25:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Aoki M, Iwasa S, Boku N. Trastuzumab deruxtecan for the treatment of HER2-positive advanced gastric cancer: a clinical perspective. Gastric Cancer 2021;24:567–76. [DOI] [PubMed] [Google Scholar]
- 46. Iwata TN, Ishii C, Ishida S, Ogitani Y, Wada T, Agatsuma T. A HER2-targeting antibody-drug conjugate, trastuzumab deruxtecan (DS-8201a), enhances antitumor immunity in a mouse model. Mol Cancer Ther 2018;17:1494–503. [DOI] [PubMed] [Google Scholar]
- 47. Nicolo E, Giugliano F, Ascione L, Tarantino P, Corti C, Tolaney SM, et al. Combining antibody-drug conjugates with immunotherapy in solid tumors: current landscape and future perspectives. Cancer Treat Rev 2022;106:102395. [DOI] [PubMed] [Google Scholar]
- 48. Hoimes CJ, Flaig TW, Milowsky MI, Friedlander TW, Bilen MA, Gupta S, et al. Enfortumab vedotin plus pembrolizumab in previously untreated advanced urothelial cancer. J Clin Oncol 2023;41:22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schmid P, Wysocki PJ, Ma CX, Park YH, Fernandes R, Lord S, et al. 379MO. Datopotamab deruxtecan (Dato-DXd) + durvalumab (D) as first-line (1L) treatment for unresectable locally advanced/metastatic triple-negative breast cancer (a/mTNBC): updated results from BEGONIA, a phase Ib/II study. Ann Oncol 2023;34 (suppl_2):S334–90. [Google Scholar]
- 50. Bates D, Eastman A. Microtubule destabilising agents: far more than just antimitotic anticancer drugs. Br J Clin Pharmacol 2017;83:255–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Serpico AF, Visconti R, Grieco D. Exploiting immune-dependent effects of microtubule-targeting agents to improve efficacy and tolerability of cancer treatment. Cell Death Dis 2020;11:361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wordeman L, Vicente JJ. Microtubule targeting agents in disease: classic drugs, novel roles. Cancers (Basel) 2021;13:5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fong A, Durkin A, Lee H. The potential of combining tubulin-targeting anticancer therapeutics and immune therapy. Int J Mol Sci 2019;20:586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Feng B, Niu Z, Hou B, Zhou L, Li Y, Yu H. Enhancing triple negative breast cancer immunotherapy by ICG-templated self-assembly of paclitaxel nanoparticles. Adv Funct Mater 2020;30:1906605. [Google Scholar]
- 55. Lau TS, Chan LKY, Man GCW, Wong CH, Lee JHS, Yim SF, et al. Paclitaxel induces immunogenic cell death in ovarian cancer via TLR4/IKK2/SNARE-dependent exocytosis. Cancer Immunol Res 2020;8:1099–111. [DOI] [PubMed] [Google Scholar]
- 56. Ding ZM, Huang CJ, Jiao XF, Wu D, Huo LJ. The role of TACC3 in mitotic spindle organization. Cytoskeleton (Hoboken) 2017;74:369–78. [DOI] [PubMed] [Google Scholar]
- 57. Singh P, Thomas GE, Gireesh KK, Manna TK. TACC3 protein regulates microtubule nucleation by affecting gamma-tubulin ring complexes. J Biol Chem 2014;289:31719–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fan X, Liu B, Wang Z, He D. TACC3 is a prognostic biomarker for kidney renal clear cell carcinoma and correlates with immune cell infiltration and T cell exhaustion. Aging (Albany NY) 2021;13:8541–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Scott EN, Gocher AM, Workman CJ, Vignali DAA. Regulatory T cells: barriers of immune infiltration into the tumor microenvironment. Front Immunol 2021;12:702726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Emens LA, Esteva FJ, Beresford M, Saura C, De Laurentiis M, Kim SB, et al. Trastuzumab emtansine plus atezolizumab versus trastuzumab emtansine plus placebo in previously treated, HER2-positive advanced breast cancer (KATE2): a phase 2, multicentre, randomised, double-blind trial. Lancet Oncol 2020;21:1283–95. [DOI] [PubMed] [Google Scholar]
- 61. Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front Immunol 2015;6:368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li F, Liu S. Focusing on NK cells and ADCC: a promising immunotherapy approach in targeted therapy for HER2-positive breast cancer. Front Immunol 2022;13:1083462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file contains supplementary methods, 11 supplementary figures and 2 supplementary tables
This file contains Supplementary Table S1
Data Availability Statement
The RNA-seq data of WT and T-DM1R cells were uploaded to the Sequence Read Archive (SRA) database under the BioProject accession number PRJNA1048320 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1048320). Gene expression data were downloaded from the NCBI Gene Expression Omnibus database under GSE194040 (22). All other raw data generated in this study are available upon request from the corresponding author.