

Abstract
Immune checkpoint inhibitors (ICI) have displayed impressive clinical efficacy in the context of non–small cell lung cancer (NSCLC). However, most patients do not achieve long-term survival. Minimally invasive collected samples are attracting significant interest as new fields of biomarker study, and metabolomics is one of these growing fields. We concentrated on the augmented value of the metabolomic profile in differentiating long-term survival from short-term survival in patients with NSCLC subjected to ICIs. We prospectively recruited 97 patients with stage IV NSCLC who were treated with anti–PD-1 inhibitor, including patients treated with monoimmunotherapy as second-line treatment (Cohort 1), and patients treated with combination immunotherapy as first-line treatment (Cohort 2). Each cohort was divided into long-term and short-term survival groups. All blood samples were collected before beginning immunotherapy. Serum metabolomic profiling was performed by UHPLC-Q-TOF MS analysis. Pareto-scaled principal component analysis (PCA) and orthogonal partial least-squares discriminant analysis were performed. In Cohort 1, the mPFS and mOS of long-survival patients are 27.05 and NR months, respectively, and those of short-survival patients are 2.79 and 10.59 months. In Cohort 2, the mPFS and mOS of long-survival patients are 27.35 and NR months, respectively, and those of short-survival patients are 3.77 and 12.17 months. A total of 41 unique metabolites in Cohort 1 and 47 in Cohort 2 were screened. In Cohorts 1 and 2, there are 6 differential metabolites each that are significantly associated with both progression-free survival and overall survival. The AUC values for all groups ranged from 0.73 to 0.95. In cohort 1, the top 3 enriched KEGG pathways, as determined through significant different metabolic pathway analysis, were primary bile acid biosynthesis, African trypanosomiasis, and choline metabolism in cancer. In Cohort 2, the top 3 enriched KEGG pathways were the citrate cycle (TCA cycle), PPAR signaling pathway, and primary bile acid biosynthesis. The primary bile acid synthesis pathway had significant differences in the long-term and short-term survival groups in both Cohorts 1 and 2. Our study suggests that peripheral blood metabolomic analysis is critical for identifying metabolic biomarkers and pathways responsible for the patients with NSCLC treated with ICIs.
Introduction
Lung cancer still has a significant disease burden worldwide. Over 61% of patients are diagnosed in advanced stages with limited therapeutic options and a lower five-year survival rate (1). The search for biomarkers to aid in choosing from the available treatment strategies, including immunotherapy, is ongoing. Immune checkpoint inhibitors (ICI) are one of the most significant improvements in non–small cell lung cancer (NSCLC) treatment and have demonstrated durable clinical activity in patients with advanced NSCLC. However, responses are observed in only 20%–25% of unselected patients. Thus, the search for predictive biomarkers for durable activity and long-term survival is still a topic of interest (2). Within this context, there has been interest in liquid biopsy, including any tumor-derived circulating material through blood or non-blood body fluids (3).
Circulation biomarkers can facilitate the understanding of effectiveness, prognosis, and tumor recurrence with minimum invasion (4). The identification of serum molecules, such as proteins, microRNAs, lncRNAs, exosomes, and metabolites, has become a great concern in the clinic (5, 6). Metabolic dysregulation is one of a complex network of events in tumor development, including NSCLC diagnosis, tumor characterization, and progression (7). The metabolic changes in cancer cells have been well recorded, such as upregulated glycolysis, glutaminolysis and amino acid, and fatty acid synthesis pathways (8). An increasing number of studies have screened candidate biomarkers from serum or cancer tissues, and over 150 metabolites have been found to be associated with lung cancer. However, most metabolites were screened by comparing patients with lung cancer with healthy subjects (9). Few studies have focused on the relationship between serum metabolic markers and the clinical outcomes of patients with advanced NSCLC, which could be useful in guiding personalized ICI treatment. Thus, there remains a critical need to further harness the power of circulating metabolites for differentiating ICI treatment options.
Metabolite concentrations at a certain time can be used as a metabolic “fingerprint” representative of the state of the organism. LC/MS-based metabolomics is a powerful tool that has been used to discover novel circulating biomarkers for many cancers. Our study focused specifically on long-term and short-term survivors of advanced NSCLC treated with ICI and examined the role of baseline metabolic markers in differentiating survival.
Materials and Methods
Study design and participants
Between April 2017 and December 2021, at Zhejiang Cancer Hospital, patients of age older than 18 years with NSCLC who received anti–PD-1 therapeutic agents (including Pembrolizumab, Tislelizumab, and Sintilimab) were prospectively screened. Serum samples of patients were collected at baseline, with inclusion criteria comprising advanced NSCLC, received anti–PD-1 agents as first-line (Cohort 2, combination immunotherapy) or second-line therapy (Cohort 1, monoimmunotherapy), and radiological evaluability based on RECIST version 1.1, had complete survival data. Because immunotherapy is not standard of care for patients with NSCLC with mutant EGFR/BRAF/ALK/ROS1, the major exclusion criteria were harboring EGFR or BRAF mutations or ALK/ROS1 rearrangements and incomplete follow-up data, post-prandial blood samples, stage I–III NSCLC, patients who experienced thoracic radiotherapy. A total of 97 patients were enrolled. Patients in this study received anti–PD-1 therapeutic agents that were approved for marketing. Progression-free survival (PFS) is the time from initiation of anti–PD-1 therapy to disease progression. Overall survival (OS) was calculated from the date of initiation of anti–PD-1 therapy until death from any cause or the date of last follow-up. Durable clinical benefit (DCB) was defined as the absence of proven disease progression for at least 6 months after anti–PD-1 therapy; whereas cases with disease progression or disease stabilization lasting ≤6 months were considered to have no durable benefit (10). Thus, the long-term survival group was defined as PFS for at least 6 months after ICI treatment, whereas PFS less than 6 months was considered the short-term survival group. The last follow-up date was September 1, 2023 with 100% follow-up rate. This study was conducted in accordance with the Declaration of Helsinki and was approved by the ethics committee of Zhejiang Cancer Hospital. Written informed consent was obtained from each patient.
Sample preparation
Blood samples from all patients were collected before the first administration of the anti–PD-1 agent according to normal first- and second-line clinical practice. 5-mL Vacutainer tubes containing the chelating agent ethylene diamine tetraacetic acid (EDTA) were used to collect fasting blood samples, followed by centrifugation (15 minutes, 1,500 × g, 4°C). Serum aliquots (150 μL) were stored at –80°C until UPLC-Q-TOF/MS analysis. Thawed samples (4°C) underwent centrifugation (15 minutes, 14,000 × g, 4°C) after mixing with cold methanol/acetonitrile (1:1, v/v). The dried supernatant was reconstituted for LC/MS analysis in positive (ESI+) and negative (ESI−) electrospray ionization modes.
LC/MS-MS analysis
Analysis was performed using a UHPLC (1290 Infinity LC, Agilent Technologies) coupled to a quadrupole time-of-flight (AB Sciex TripleTOF 6600) at Shanghai Applied Protein Technology Co., Ltd. To achieve HILIC separation, we used a 2.1 × 100 mm ACQUITY UPLC BEH 1.7 μm column (Waters, Ireland). The mobile phases for both ESI+ and ESI− modes consisted of A, which was 25 mmol/L ammonium acetate and 25 mmol/L ammonium hydroxide in water, and B, which was acetonitrile. To obtain RPLC separation, we used a 2.1 × 100 mm ACQUITY UPLC HSS T3 1.8-μm column (Waters, Ireland). For ESI+ mode, the mobile phases included A, which was water with 0.1% formic acid, and B, which was acetonitrile with 0.1% formic acid. On the other hand, for ESI− mode, the mobile phases comprised A, which was 0.5 mmol/L ammonium fluoride in water, and B, which was acetonitrile. During the MS-only acquisition, the instrument was set to capture data within the m/z range of 60–1,000 Da, with an accumulation time of 0.20 s/spectra for the TOF MS scan. Meanwhile, in auto MS-MS acquisition, the instrument captured data within the m/z range of 25–1,000 Da, with an accumulation time of 0.05 s/spectra for the product ion scan. To conduct the product ion scan, we used information-dependent acquisition, specifically selecting the high-sensitivity mode (11).
Data processing
Raw MS data (wiff.scan files) underwent conversion to MzXML files before importing into freely available XCMS software. CAMERA (Collection of Algorithms of MEtabolite pRofile Annotation) facilitated the annotation of isotopes and adducts. Only the variables having >50% of the nonzero measurement values in at least one group were kept in the extracted ion features. Compound identification relied on comparing accuracy m/z values (<25 ppm) and MS/MS spectra with an in-house database of authentic standards.
Statistical analysis
Following sum normalization, processed data underwent analysis using the R package ropls. Multivariate data analysis, including Pareto-scaled principal component analysis (PCA) and orthogonal partial least-squares discriminant analysis (OPLS-DA), was conducted. Robustness was evaluated through 7-fold cross-validation and response permutation testing. The OPLS-DA model's variable importance in the projection (VIP) value and Student t test identified significantly changed metabolites (VIP > 1; P < 0.05). Pearson's correlation analysis explored variable relationships. To conduct survival analyses, we used the log-rank test to compare the Kaplan–Meier curves. In addition, the Cox proportional hazards model was used to calculate the hazard ratio (HR) and determine the corresponding 95% confidence interval (CI). For all tests performed, a two-sided P value of less than 0.05 was regarded as statistically significant, unless stated otherwise. The statistical software used for all analyses was R, specifically version 3.6.0. Moreover, the Receiver Operating Characteristic (ROC) analysis was carried out using the “pROC” package within the R software package (R version 4.2.3; R: The R-Project for Statistical Computing, Vienna, Austria).
Bioinformatic Analysis
KEGG pathway annotation
We conducted a blast analysis of the metabolites using the online Kyoto Encyclopedia of Genes and Genomes (KEGG) database (available at http://geneontology.org/). This allowed us to retrieve the compound identifiers and further link them to pathways within KEGG11. Hence, we extracted the relevant KEGG pathways (12).
Functional enrichment analysis
To delve deeper into the influence of metabolites that are expressed differentially, we conducted an enrichment analysis. The application of KEGG pathway enrichment analyses relied on the Fisher's exact test, with consideration of the entire set of metabolites in each pathway as the background dataset. Only pathways possessing P values below a threshold of 0.05 were deemed significant.
Hierarchical clustering
We used hierarchical clustering analysis using the data on relative expression of the metabolites under investigation. To this end, we used Cluster3.0 (http://bonsai.hgc.jp/∼mdehoon/software/cluster/software.htm) and the Java Treeview software (http://jtreeview.sourceforge.net). During the execution of hierarchical clustering, we opted for the Euclidean distance algorithm to measure similarity and the average linkage clustering algorithm (which uses centroids of observations) for clustering. Alongside the dendrogram, a heat map is commonly used as a visual aid.
Ethics approval and consent to participate
This study was conducted in accordance with the Declaration of Helsinki and was approved by the ethics committee of Zhejiang Cancer Hospital. Written informed consent was obtained from each patient.
Consent for publication
All authors gave their consent for publication.
Data availability
Data were generated by the authors and included in the article.
Results
Demographics of the study cohort
The workflow of the study is presented in Fig. 1. To define potential metabolic profiles, 55 serum samples of patients with advanced NSCLC treated with anti–PD-1 antibody as second-line monoimmunotherapy (Cohort 1) were collected. Patients in Cohort 1 were required to be immunotherapy-naïve in their first-line treatment. Among the patients in Cohort 1, 30 were evaluated as having long-term survival with mPFS 27.05 months (95% CI, 21.26–31.68) and mOS NR months, and 25 had short-term survival with mPFS 2.79 months (95% CI, 1.62–3.97) and mOS 10.59 months (95% CI, 8.06–13.11; Fig. 2A and B). Cohort 2 comprised 42 patients with advanced NSCLC treated with an anti–PD-1 inhibitor as first-line chemo-immunotherapy. Among the patients in Cohort 2, 22 were evaluated as having long-term survival with mPFS 27.35 months (95% CI, 16.29–38.42) and mOS NR months, and 20 had short-term survival with mPFS 3.77 months (95% CI, l 0.94–5.84) and mOS 12.17months (95% CI, 10.81–13.12; Fig. 2C and D). Clinical characteristics are detailed in Supplementary Table S1.
Figure 1.

Design and workflow of the study. By FigDraw tool (http://www.figdraw.com)
Figure 2.

The progression-free survival (PFS) and overall survival (OS) in Cohorts 1 and 2. In Cohort 1, long-term survival patients have mPFS 27.05 months and mOS NR months, and short-term survival patients have mPFS 2.79 months and mOS 10.59 months (A and B). Among the patients in Cohort 2, long-term survival patients have mPFS 27.35 months and mOS NR months, and short-term survival patients have mPFS 3.77 months and mOS 12.17months (C and D).
Quality evaluation of the experimental data
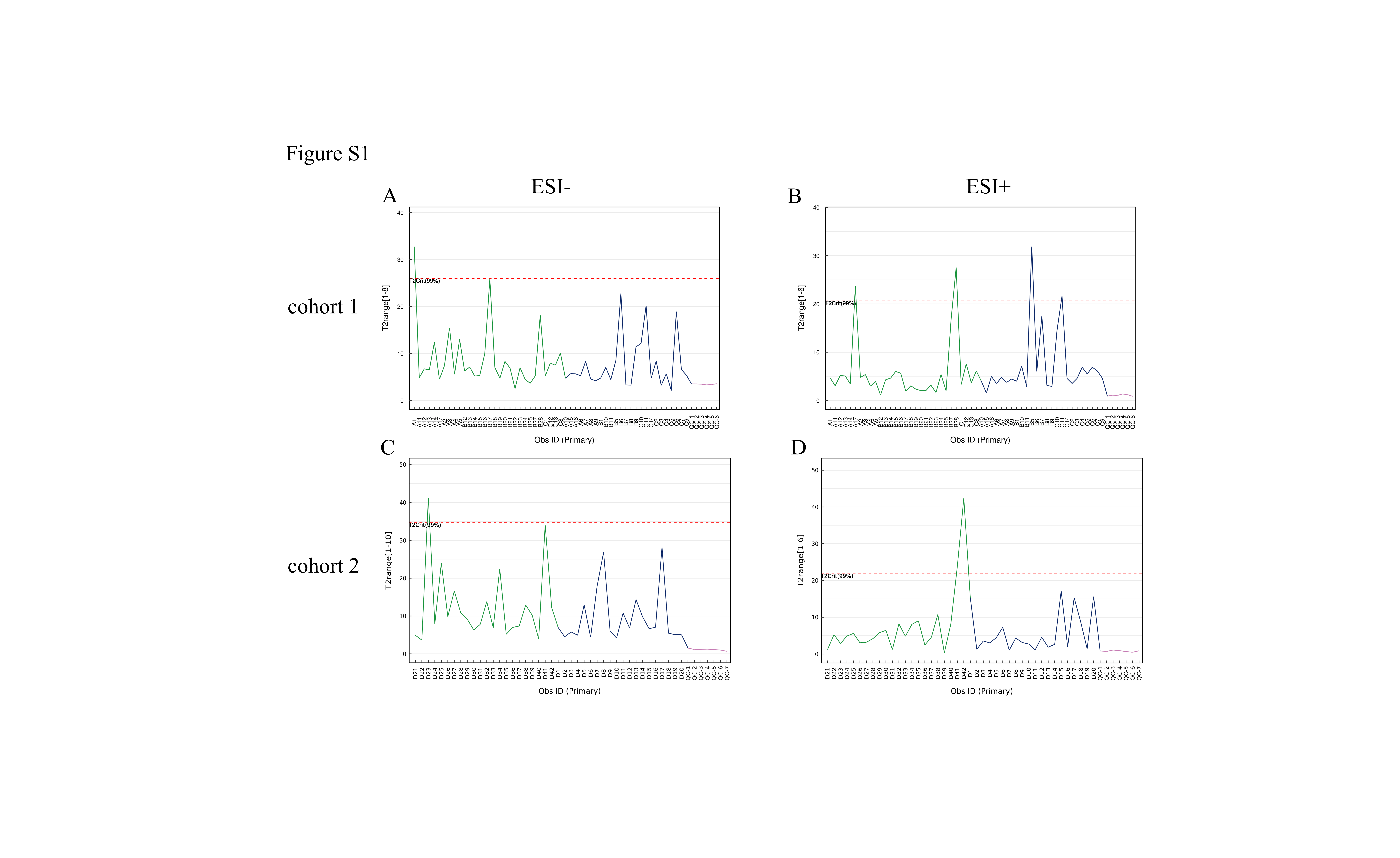
In Cohort 1, a total of 1,067 metabolite peaks were identified, with 494 detected using ESI− ionization and 573 using ESI+ ionization. Similarly, in Cohort 2, a total of 696 metabolite peaks were detected, with 268 detected using ESI− and 428 using ESI+ ionization. Typical extracted ion chromatograms from the two ESI modes are displayed in Supplementary Fig. S1. The findings from the experiment demonstrate that the response intensity and retention time of each chromatographic peak exhibit considerable overlap, indicating minimal variation attributed to instrument error throughout the entire procedure. Furthermore, the quality control samples exhibited close clustering in the score plot generated by PCA, affirming the excellent reproducibility of the metabolomics analysis (Supplementary Fig. S2).
Metabolic profiling of serum
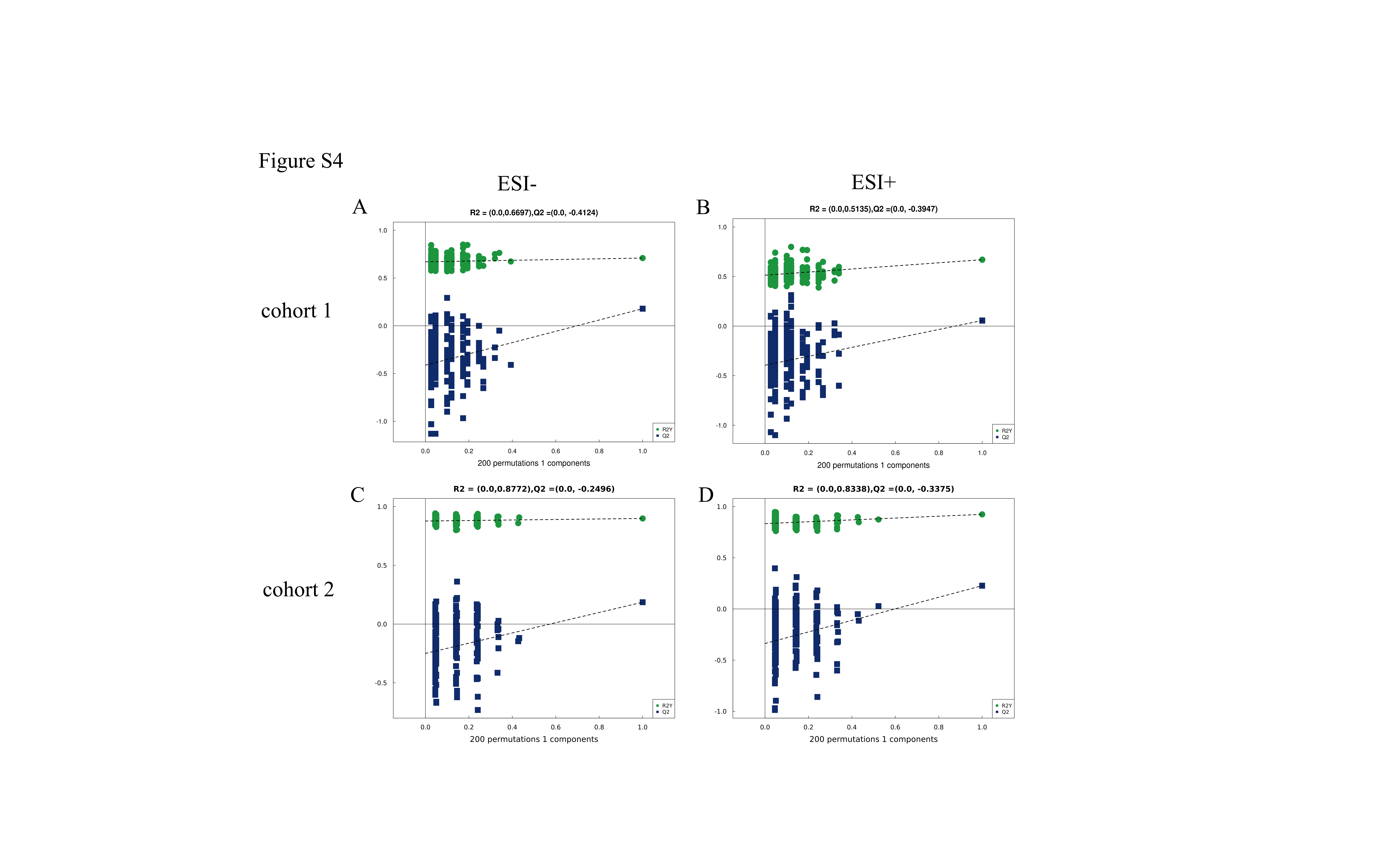
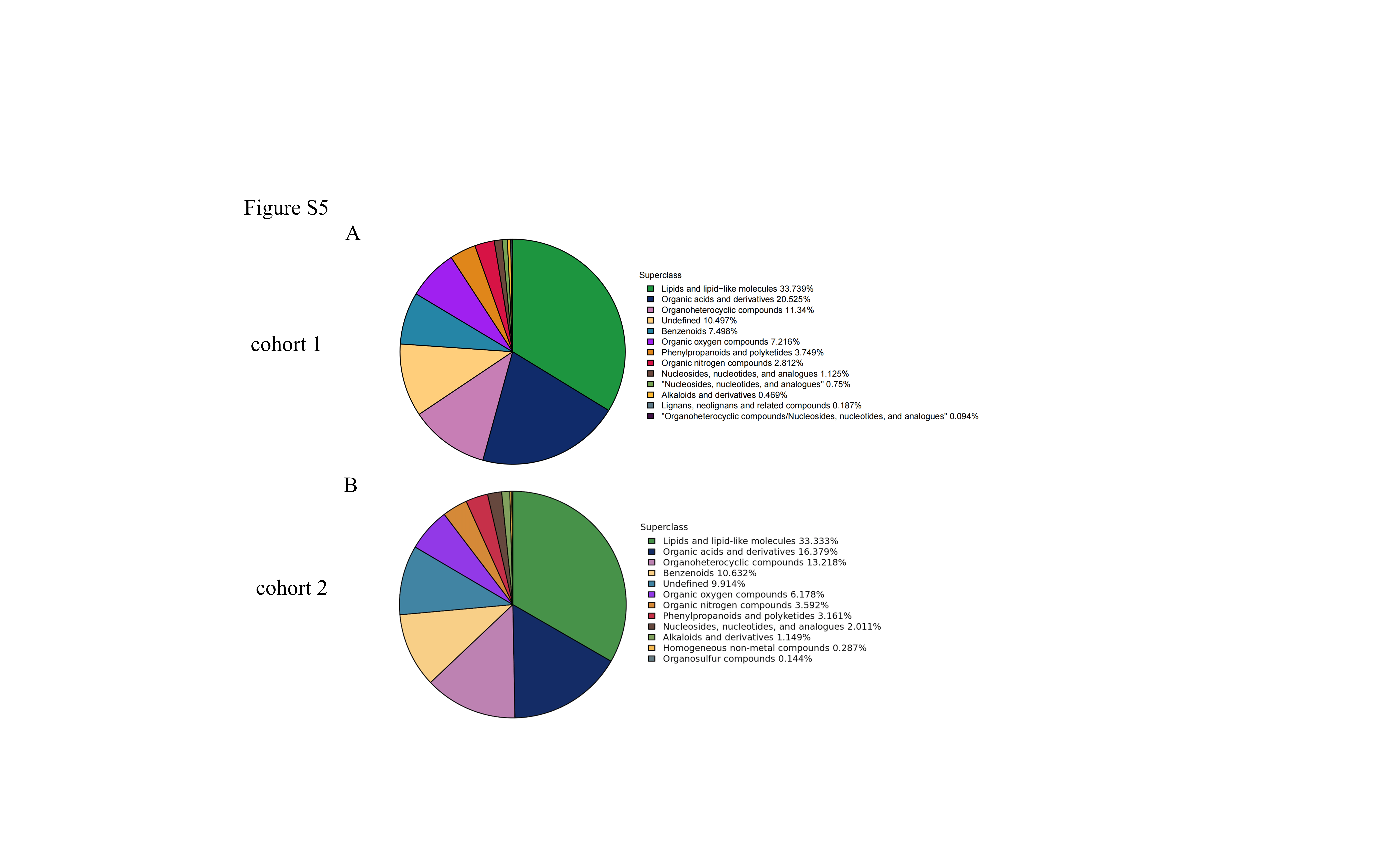
To screen for potential labeled metabolites, OPLS-DA was used. The OPLS-DA score plots of both Cohorts 1 and 2 revealed a clear separation between long-term and short-term survival groups without over-fitting (Supplementary Fig. S3). To avoid over-fitting of the supervised model in the modeling process, the permutation test is used to ensure the validity of the model. Supplementary Fig. S4 shows the permutation test map of the OPLS-DA model in the example comparison group. As the retention of displacement gradually decreased, both R2 (ESI+: R2, 0.0–0.5315; ESI−: R2, 0.0–0.6697) and Q2 (ESI+: Q2, 0.0 to −0.3947; ESI−: Q2, 0.0 to −0.4124) of the random model gradually decreased, indicating that the original model does not over-fitting and the model has good robustness and predictive power. The identified metabolites proportions in each chemical classification were similar in both Cohorts and are shown in Supplementary Fig. S5.
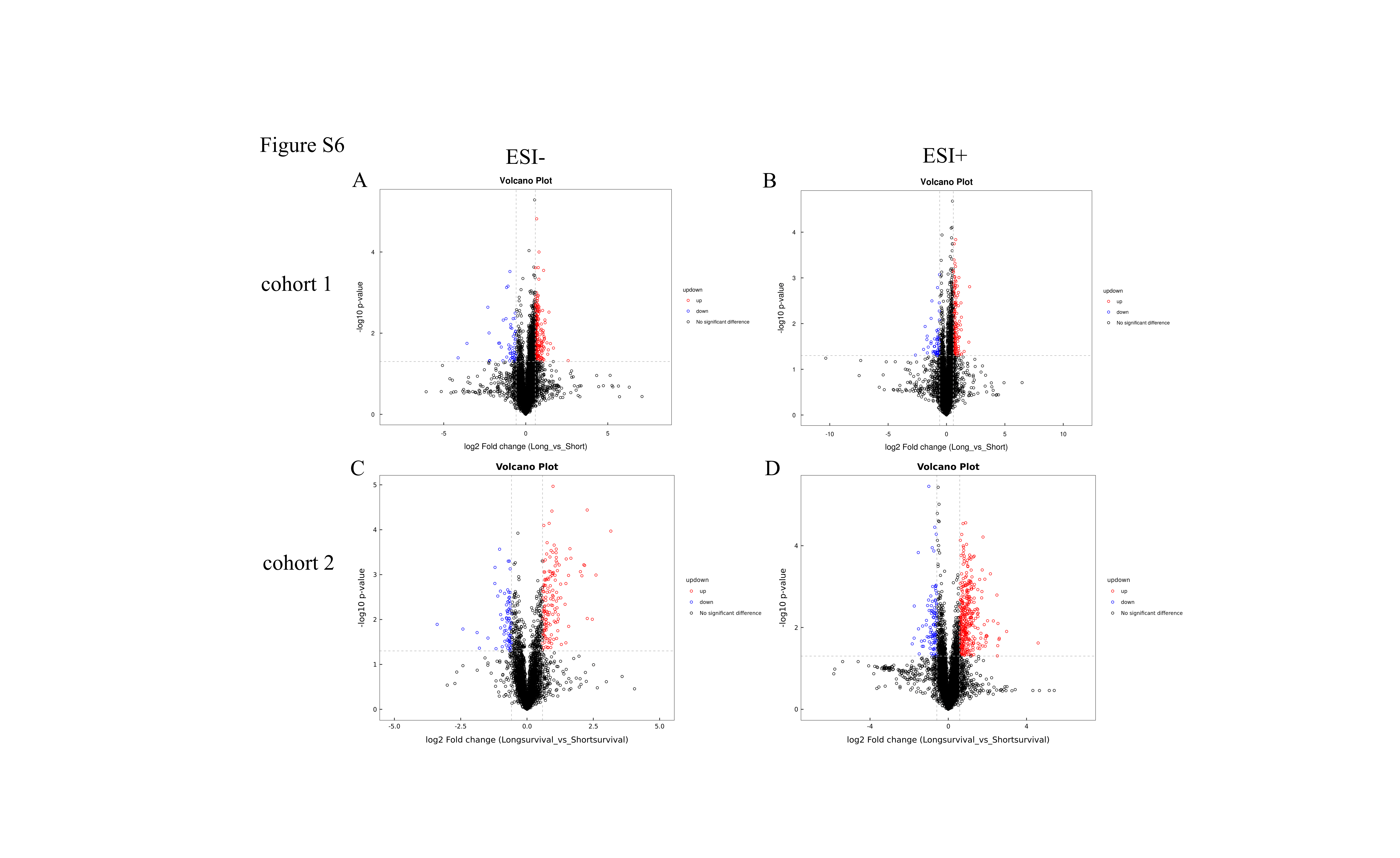
All metabolites detected in the ESI+ and ESI− modes were used for univariate analysis at first. Differentially expressed metabolites with FC > 1.5 or FC < 0.67 and P < 0.05 are represented by volcano plots (Supplementary Fig. S6). As shown in Supplementary Fig. S6, black color indicates no difference in metabolite expression between the long-term and short-term survival groups. Pink color indicates upregulated endogenous metabolites with FC > 1.5 and P < 0.05. Blue color indicates downregulated metabolites with FC < 0.67 and P < 0.05.
Defining potential metabolic biomarkers for NSCLC
Next, we used OPLS-DA VIP > 1 and P < 0.05 as stringent screening criteria for significant differential metabolites in metabolomics analysis. A total of 41 differential metabolites in Cohort 1 and 47 differential metabolites in Cohort 2 were screened (data were shown as Supplementary Material cohort1–41 and cohort2–47). We further performed bio-credit analysis of the selected significantly different metabolites, including cluster analysis, correlation analysis, and pathway analysis.
First, the significant up- or downregulated metabolic fold changes with FC > 1.5 or FC < 0.67, OPLS-DA VIP > 1 and P < 0.05 were screened further and the TOP 10 were visually displayed in histograms, as shown in Fig. 3, Supplementary Table S2. In Cohort 1, 4 common metabolites in ESI− mode (Fig. 3A; Supplementary Table S2) and 8 metabolites in ESI+ mode (Fig. 3B; Supplementary Table S2) were significantly upregulated in the long-term survival group when compared with the short-term survival group. And 1 metabolite in ESI− mode (Fig. 3A; Supplementary Table S2) and 2 metabolites in ESI+ mode (Fig. 3B; Supplementary Table S2) were significantly downregulated. In cohort 2, 8 common metabolites in ESI− mode (Fig. 3C; Supplementary Table S3) and 16 metabolites in ESI+ mode (Fig. 3D; Supplementary Table S3; data showed the Top 10) were significantly upregulated in the long-term survival group when compared with the short-term survival group. And 5 metabolites in ESI− mode (Fig. 3C; Supplementary Table S3) and 5 in ESI+ mode (Fig. 3D; Supplementary Table S3) were significantly downregulated.
Figure 3.

Differential fold analysis of significant different metabolite expressions in Cohorts 1 and 2. The abscissa shows the multiple different expressions; red indicates greater than 1 and green indicates less than 1. The ordinate indicates the significant different metabolites
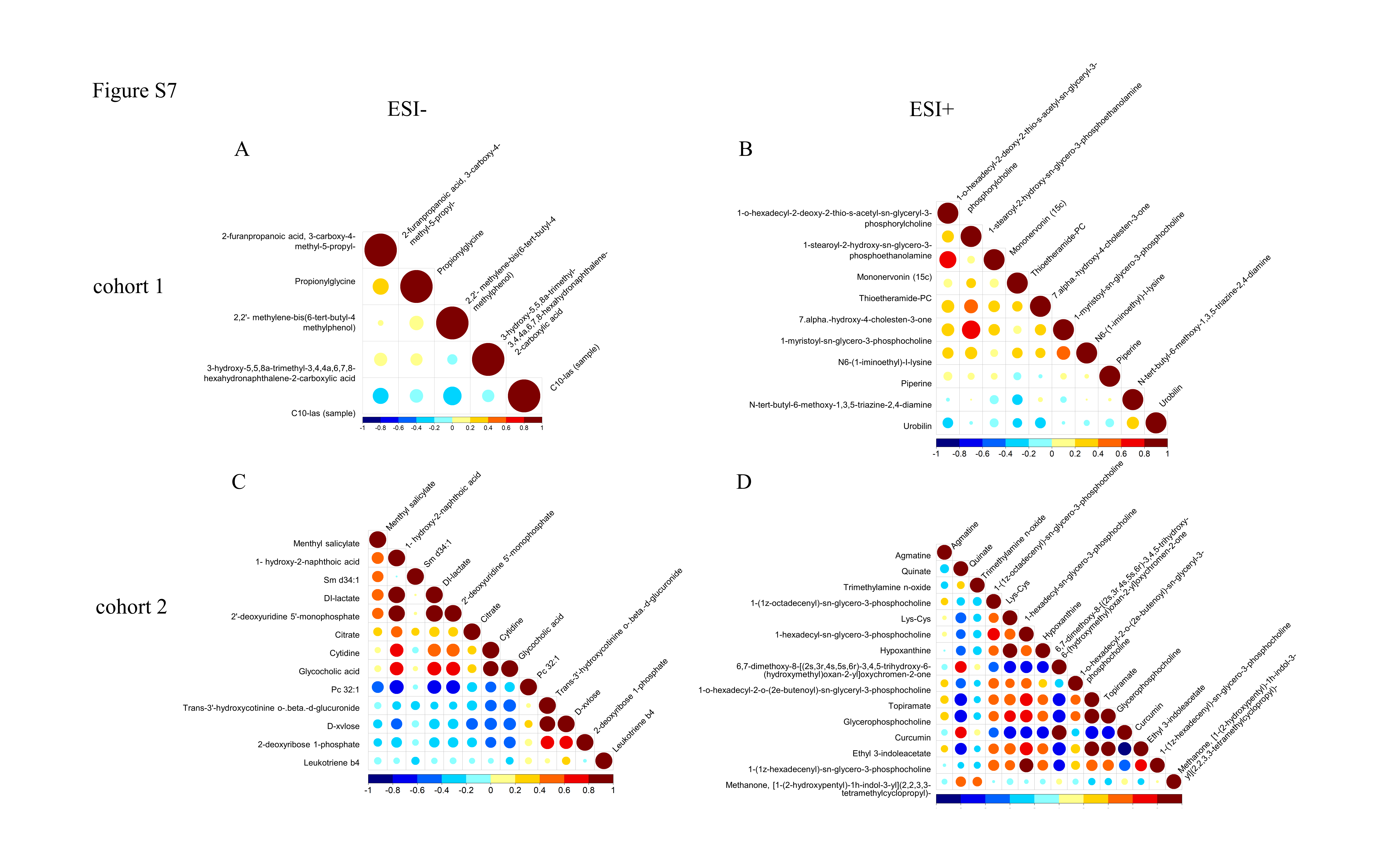
Correlation analysis can measure the closeness of metabolism between significantly different metabolites with sufficient fold change (VIP > 1, P < 0.05, FC > 1.5 or < 0.67; metabolic proximities), conducive to further understanding the mutual regulatory relationship between metabolites during changing biological states. In Cohort 1, correlation analysis indicated that C10-las (sample) had a high negative correlation with 2-furanpropanoic acid, 3-carboxy-4-methyl-5-propyl- and 2,2′-methylene-bis (6-tert-butyl-4 methylphenol) in ESI− mode (Supplementary Fig. S7A). In ESI+ mode, Mononervonin (15c) and 1-o-hexadecyl-2-deoxy-2-thio-s-acetyl-sn-glyceryl-3-phosphorylcholine had a high positive correlation; 1-myristoyl-sn-glycero-3-phosphocholine and 1-stearoyl-2-hydroxy-sn-glycero-3-phosphoethanolamine had a high positive correlation (Supplementary Fig. S7B). Correlation analysis of Cohort 2 is shown in Supplementary Fig. S7C–S7D.
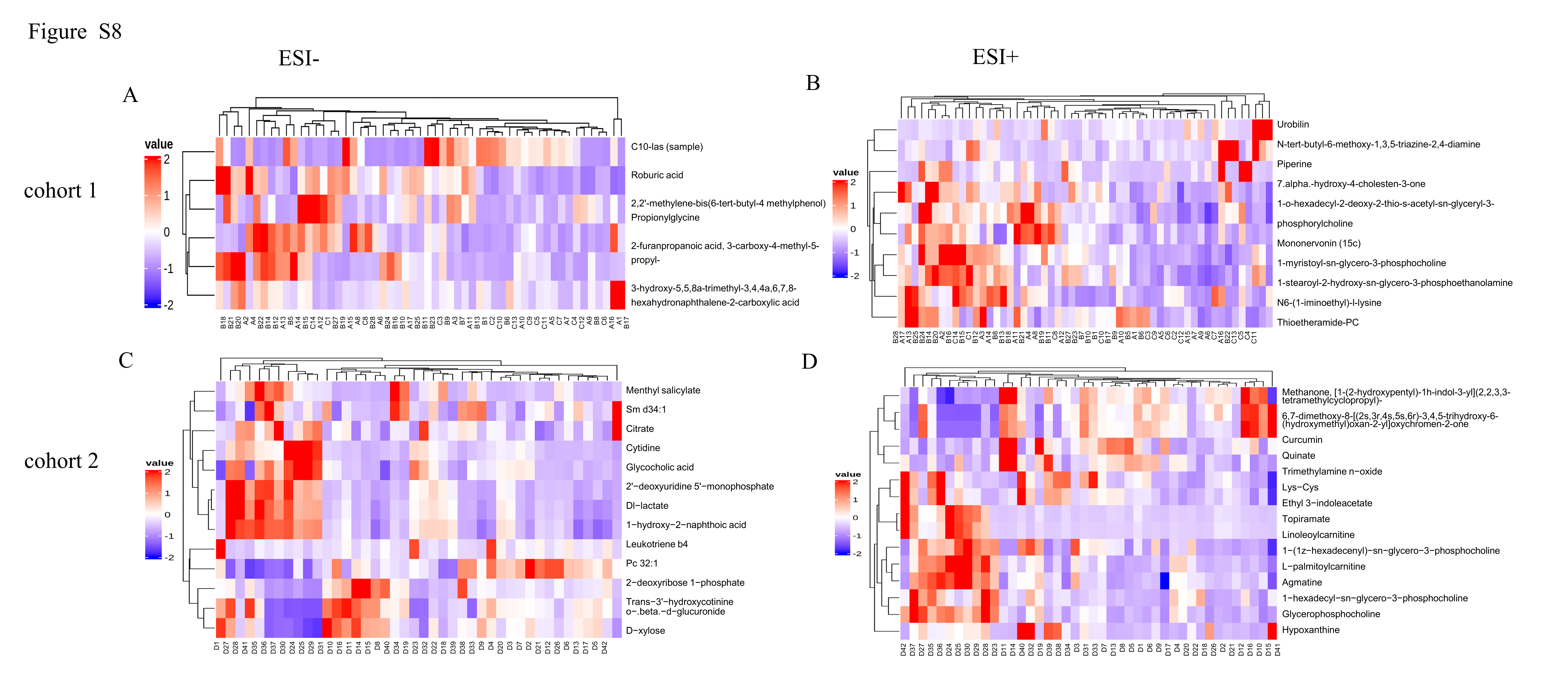
To analyze the metabolic pattern in different groups, metabolite heat maps were also generated (Supplementary Fig. S8). Each row in the graph represents one differential metabolite, and each column represents a set of samples. Red represents significant upregulation, blue represents significant downregulation, color shades indicate the degree of up- and downregulation, and metabolites with close expression patterns were clustered under the same cluster on the left.
Defining potential metabolic signal pathways for NSCLC
In Cohort 1, the top 3 enriched KEGG pathways, as determined by significant different metabolic pathway analysis, were primary bile acid biosynthesis (P = 0.006), African trypanosomiasis (P = 0.02), and choline metabolism in cancer (P = 0.0287). Among them, the primary bile acid synthesis pathway had the most significant differences in the long-term and short-term survival groups. Two related metabolites of the primary bile acid synthesis pathway metabolites differed significantly with a P value less than 0.05, 3 beta, 7 alpha-dihydroxy-5-cholestenoic acid and 7 alpha-hydroxy-4-cholesten-3-one (Fig. 4A and B). In Cohort 2, the top 3 enriched KEGG pathways, as determined by significant different metabolic pathway analysis, were the citrate cycle (TCA cycle; P = 0.0057), PPAR signaling pathway (P = 0.0285), and primary bile acid biosynthesis (P = 0.0294; Fig. 4C and D). Among them, the citrate cycle (TCA cycle) had the most significant differences in the long-term and short-term survival groups. Two related metabolites, citrate and cis-aconitate, of the citrate cycle (TCA cycle) differed significantly, with a P value less than 0.05. Moreover, the primary bile acid synthesis pathway had significant differences in the long-term and short-term survival groups in both Cohorts 1 and 2.
Figure 4.

The KEGG pathways in which the significantly different metabolites take part between Cohorts 1 and 2. In Cohort 1, the top 3 enriched KEGG pathways were primary bile acid biosynthesis, African trypanosomiasis, and choline metabolism in cancer (A and B). In Cohort 2, the top 3 enriched KEGG pathways were the citrate cycle (TCA cycle), PPAR signaling pathway, and primary bile acid biosynthesis (C and D). KEGG, Kyoto Encyclopedia of Genes and Genomes.
Clinical outcomes based on the baseline metabolics
Univariate Cox analysis of PFS or OS by the levels of different metabolics in Cohorts 1and 2 showed that pretreatment-circulating metabolics levels could predict the outcomes of immunotherapy. The differential metabolites simultaneously associated with PFS and OS are displayed in Supplementary Table S4 (6 metabolites in Cohort 1) and Supplementary Table S5 (6 metabolites in Cohort 2). In cohort 1, these 6 differential metabolites showed high expression in the long-term survival group patients, as compared with short-term survival patients. In cohort 2, 3 differential metabolites were highly expressed in the long-term survival group patients, whereas three were lowly expressed. Representative images of the most significant differential metabolites related to PFS and OS in Cohorts 1 and 2 are shown in Fig. 5. The expression levels of metabolites were divided by the median and ≥ median was identified as upregulated. In Cohort 1, patients with 7.alpha. -hydroxy-4-cholesten-3-one expression level ≥ median have a longer mPFS (19.10 vs. 4.62 months, P = 0.002, Fig. 5A) and mOS (NR vs. 15.93 months, P < 0.001, Fig. 5B) when compared with those with expression levels < median. In Cohort 2, patients with Agmatine expression level ≥ median have a longer mPFS (NR vs. 4.27 months, P < 0.001, Fig. 5C) and mOS (NR vs. 14.01 months, P < 0.001, Fig. 5D) when compared with those with expression levels < median.
Figure 5.

Representative images of Univariate Cox analysis of progression-free survival (PFS) or overall survival (OS) by the levels of metabolics in Cohorts 1and 2. A, PFS and (B) OS curves for patients with 7.alpha. -hydroxy-4-cholesten-3-one expression level ≥ median and < median in Cohort 1. C, PFS and (D) OS curves for patients with Agmatine expression level ≥ median and < median in Cohort 2.
Identified metabolics can differentiate patients with long-term or short-term survival
Finally, in both Cohorts 1 and 2, we selected those 6 metabolites that were significantly correlated with both PFS and OS, and normalized their signal values to obtain z-scores. Subsequently, we assessed the enrichment of metabolite signals in the long-term and short-term survival groups and categorized metabolite signals as positively correlated when they were expressed highly in the long-term survival group and negatively correlated when they were expressed highly in the short-term survival group. In Cohort 1, the positively correlated metabolites included all the 6 metabolites (Supplementary Table S4), whereas in Cohort 2, the positively correlated metabolites included Agmatine, Linoleoyl carnitine, and Menthyl salicylate. The negatively correlated metabolites in Cohort 2 included Curcumin, 2-deoxyribose 1-phosphate, and Leukotriene b4. For both positively and negatively correlated metabolites, we calculated the sum of z-scores. The threshold values for the sum of z-scores of metabolites with the same features were determined through ROC analysis, which provided the best discrimination between long-term and short-term survival. We observed that the AUC values for all groups ranged from 0.73 to 0.95.
The model based on the 6 differential upregulated metabolites in distinguishing patients with and without long-term PFS in cohort 1, the ROC curve yielded an AUC of 0.81, with a sensitivity and specificity of 76.9% and 75.9%, respectively (Fig. 6A). The same model in distinguishing patients with and without long-term OS in cohort 1, the ROC curve yielded an AUC of 0.734, with a sensitivity and specificity of 75.0% and 71.8%, respectively (Fig. 6B). The model based on the 3 differential upregulated metabolites (Agmatine, Linoleoyl carnitine, Menthyl salicylate) in distinguishing patients with and without long-term PFS in cohort 2, the ROC curve yielded an AUC of 0.948, with a sensitivity and specificity of 100% and 81.8%, respectively (Fig. 6C). The same model in distinguishing patients with and without long-term OS in cohort 2, the ROC curve yielded an AUC of 0.83, with a sensitivity and specificity of 90.9% and 67.7%, respectively (Fig. 6D). The model based on the 3 differential down-regulated metabolites (Curcumin, 2-deoxyribose 1-phosphateLeukotriene b4) in distinguishing patients with and without long-term PFS in cohort 2, the ROC curve yielded an AUC of 0.816, with a sensitivity and specificity of 59.1% and 100%, respectively (Fig. 6E). The same model in distinguishing patients with and without long-term OS in cohort 2, the ROC curve yielded an AUC of 0.789, with a sensitivity and specificity of 64.5% and 90.9%, respectively (Fig. 6F). Taken together, we identified differential upregulated metabolites from pretreatment serum samples that can potentially distinguish patients with and with long-term survival.
Figure 6.

Receiver Operating Characteristic curves were generated to assess the performance of the predictive model using the 6 differential metabolites to distinguish between patients with and without long-term survival; AUC, area under curve.
Discussion
Metabolomics is considered as a powerful approach to obtain reliable and reproducible biomarkers for detecting temporal physiological changes in certain time for disease diagnosis, disease progression, and for predicting drug response, including cancer field (13–15). The concentrations of the metabolites at a given time in a given sample were thought of as a metabolic “fingerprint,” which were representative of the state of the human at a certain time (16). In this study, we assessed baseline serum metabolomics in Chinese patients with advanced NSCLC following ICI treatment, either monoimmunotherapy as second-line treatment (Cohort 1), or combination immunotherapy as first-line treatment (Cohort 2), and found that there are different metabolics and different metabolic pathways between long-term and short-term survivors. More interestingly, the discovered serum metabolite biomarker panel may help separate patients into long-term and short-term survival groups. The enriched KEGG pathways analysis indicated that primary bile acid biosynthesis was significantly different between long-term and short-term survivors in both the monoimmunotherapy and combination immunotherapy groups. However, it is a bit concern that the extent of concordance between the two cohorts is limited. As mentioned above, the most advantage of the metabolome is that it is much more dynamic than the genome or proteome, because metabolomics is influenced by many factors, for example, dietary intake, medication, lifestyle, variability in sample handling and preparation, co-morbidities such as diabetes, and genetic variations. Thus, using metabolomics to identify biomarkers requires conducting studies in specific populations at particular times, within standardized criteria for participant selection.
First, our results found that more significant differential metabolites could be detected in the baseline serum of patients receiving first-line immune-combination chemotherapy (Cohort 2), when compared with Cohort 1. The main reason for this may be because the patients in Cohort 1 were second-line patients receiving monoimmunotherapy, who had previously received first-line chemotherapy and some other adjuvant drugs, the use of which affects the metabolomic performance of the patients to a certain extent. This result suggests that if we are to apply metabolomics as a predictor of immunotherapy efficacy and survival in the clinic, it may be more appropriate to use it in patient groups that have not received any treatment, mainly due to that there are some exogenous exposures like dietary intake and medication can affect metabolomics (17).
We further used the Univariate Cox analysis method to screen out differential metabolites significantly associated with both PFS and OS. Subsequently, we performed ROC curve analysis on these metabolites to assess their signal enrichment in long-survival and short-survival patient groups. We classified the metabolite signals into positive and negative correlation groups. The cutoff value for the sum of z-scores of metabolites with the same features was determined through ROC analysis, which best discriminates between long-term and short-term survival. We observed that the AUC values for all groups ranged from 0.73 to 0.95, indicating that the featured metabolites can effectively predict survival outcomes (PFS/OS).
Moreover, in our study, the enriched KEGG pathways analysis indicated that the primary bile acid biosynthesis might be important biomarker. There are two synthetic pathways of bile acids, divided into classical and alternative routes. 3 beta, 7 alpha-dihydroxy-5-cholestenoic acid (3β, 7α-dihydroxy-5 cholenoic acid) is an intermediate in the alternative bile acid synthesis pathway. Primary bile acids are produced first through a classical or alternative pathway and then converted into secondary bile acids in the gut by the gut microbiota. Bile acids directly synthesized from cholesterol in liver cells are called primary bile acids, including bile and goose deoxycholic acids. Our study preliminarily suggests that the primary bile acid metabolism pathway may be closely related to long-term survival after immunotherapy. Bile acids are known to affect host metabolism, cancer progression, and innate immunity. Data have also suggested mechanisms through which bile acid metabolites control host immune responses by directly modulating the balance of TH17 and Treg cells (18). These results also further confirm the importance of our results.
Metabolic pathways, including African trypanosomiasis, choline metabolism in cancer, citrate cycle (TCA cycle) and PPAR signaling pathway, were also found to be significantly different between long-term and short-term survivors in our study. Among them, a study showed that an increase in serum choline level was significantly associated with better PFS (aHR, 0.48; 95% CI, 0.28–0.83; P = 0.009) and a trend toward better OS (aHR, 0.64; 95% CI, 0.37–1.12, P = 0.119) in patients receiving pembrolizumab (19). The consistent results obtained by the metabolomics analysis of peripheral blood and the current study reflects the credibility of the analytic method and suggests that bile acid metabolites and choline may be potential biomarkers for ICIs in peripheral blood.
There have been some studies of metabolomics in NSCLC diagnosis, characterization, and progression (20). However, the shortcomings of previous studies are the lack of standardized patient selection and few studies have focused on the value of metabolomics in predicting response or long-term survival in patients with lung cancer receiving immunotherapy. The current study mainly focused on the population of patients with NSCLC receiving chemotherapy, and the results suggest that the baseline blood metabolite markers could suggest better survival. Shen and colleagues, Tian and colleagues, and Hao and colleagues (21–23) examined the role of serum metabolites in predicting benefit among patients with metastatic NSCLC who underwent platinum-based chemotherapy and found altered metabolite levels. These small-molecule metabolites may be potentially useful biomarkers for identifying patients who benefit from platinum-based chemotherapy.
More recently, Ghini and colleagues (24) used metabolomics to analyze serum samples from 50 patients with NSCLC treated with ICIs and showed that the metabolomic fingerprint before treatment acted as a predictive biomarker of treatment response. Another study, which enrolled 74 Chinese patients with stage IIIB/IV NSCLC, showed that hypoxanthine and histidine in serum samples early in treatment were biomarkers that predicted response to PD-1 blockade therapy (25). The strength of our study is that the sample size is larger than previous studies. We divided the patients into Cohorts 1 and 2 for analysis and screened out the targets after cross-comparison, making the study results more credible.
There are limitations in our study. First, there was no dynamic detection. One of the advantages of metabolome was much more dynamic when compared with proteome or genome, which allows detecting, alterations in metabolites in shorter times for predicting cancer risk or drug response (26). Second, no healthy controls were available. Third, there is no validation set. Furthermore, although metabolomics is an emerging field when compared with genomics, transcriptomics, and proteomics, there is heterogeneity across studies. This may be attributed to some endogenous and exogenous factors such as race, smoking status, dietary intake, and sample collection and preparation. Analytic platforms and methods used to identify and quantify metabolites should be further harmonized to standardize the workflow used in metabolomics studies.
Conclusion and future directions
The search for new biomarkers allows for personalized management of patients. The same is true in the era of immunotherapy, which has become the standard of treatment for advanced NSCLC. Liquid biopsy has become a useful procedure in biomarker studies mainly due to its minimal invasiveness. Compared with genomics, proteomics, and transcriptomics, metabolomics is an emerging field and has been newly applied to the study of lung cancer. Our study suggests that peripheral blood metabolomic analysis is important for discovering peripheral blood biomarkers, and there is an urgent need for large-scale clinical validations. Regarding future research directions, we plan to investigate the following aspects: (i) Further screening of key regulatory enzymes in the differential metabolic pathways identified in this study. We will prospectively collect baseline plasma samples from patients with lung cancer receiving frontline immunotherapy and validate them using ELISA. (ii) Our center is currently conducting several clinical research projects, including studies on new immunotherapeutic drugs and novel immune combination therapy strategies. We have already collected baseline blood samples from patients before treatment for future validation and analysis of metabolites or metabolic enzymes. (iii) We will engage in translational medical research and basic research to further explore the mechanisms of metabolites/metabolic enzymes in regulating the efficacy of immunotherapy, such as their impact on the immune microenvironment.
Supplementary Material
Volcano plot of serum metabolomics in Cohorts 1 and 2. The left figures (A, C) were obtained in the negative polarity mode, and the right figures (B, D) were obtained in the positive polarity mode. The abscissa is the log value of log2 of multiple differential expression (fold change), and the ordinate is the log value of -log10 of significant p values. Significant different metabolites: those with an FC> 1.5 and p value <0.05 are shown in rose red, and those with an FC <0.67, and p value <0.05 are colored in blue. Nonsignificantly different metabolites are indicated in black.
{kind=link}
Total ion chromatograms (TICs) of the sample metabolites of Cohort 1 and Cohort 2 detected by UHPLC under negative (A, C) and positive (B, D) ion modes.
{kind=link}
Principal component analysis (PCA) of the metabolites of Cohort 1 and Cohort 2 under negative (A, C) and positive (B, D) ion modes. Green indicates samples from the long-term survival group, blue indicates samples from the short-term survival group and red indicates QC samples.
{kind=link}
Score plot from OPLS-DA of the two groups in Cohort 1 (A, B) and Cohort 2 (C, D). Each data point represents a function of the entire spectral profile of each subject. Partial least-squares discriminant analysis showed a clear separation between the groups with acceptable goodness of fit. Cross-validation plot with a permutation test repeated 200 times. The intercepts of R2 and Q2 under negative (A, C) and positive (B, D) ion modes illustrate that the OPLS-DA model is not overfitted.
{kind=link}
The proportion of the identified metabolites in each chemical classification in Cohort 1 (A) and Cohort 2 (B). Colors in the figure express different chemical classification attribution entries, and the percentage represents the number of metabolites in the chemical classification entry as a proportion of all identified metabolites. Metabolites without chemical classification attribution were defined as undefined.
{kind=link}
Correlation heatmap in Cohorts 1 and 2. Correlation analysis of metabolites with significant differences between positive and negative polarity modes. Red indicates a positive correlation, blue indicates a negative correlation, and white indicates a nonsignificant correlation. The depth of color is related to the absolute value of the correlation coefficient, that is, the higher the degree of positive or negative correlation, the darker the color. The point size is related to the significance of the correlation: more significant, smaller p values and larger points.
{kind=link}
Metabolite heatmap between Cohorts 1 and 2. The left panels (A, C) were obtained in the negative polarity mode, and the right panels (B, D) were obtained in the positive polarity mode. Each row in the graph represents one differential metabolite, and each column represents a set of samples. Red represents significant upregulation, blue represents significant downregulation, color shades indicate the degree of upper downregulation, and metabolites with close expression patterns are clustered under the same cluster on the left.
{kind=link}
Table S1: Patient demographics and baseline clinicopathological characteristics.
Table S2: The TOP10 up- or-down* regulated metabolics in Cohort 1.
Table S3: The TOP10 up- or-down* regulated metabolics in Cohort 2.
Table S4: The differential metabolites simultaneously associated with PFS and OS in cohort 1.
Table S5: The metabolites for significant correlation to both PFS and OS in cohort 2.
original data
original data
Supplementary Figure Legend
O-partial least-squares discrimination analysis (OPLS-DA) of metabolites of Cohort 1 and Cohort 2 under negative (A, C) and positive (B, D) ion modes. Identified compounds could be separated well with optimal goodness of fit. Green indicates samples from the long-term survival group; blue indicates samples from the short-term survival group.
{kind=link}
Acknowledgments
This study was supported by the Natural Scientific Foundation of Zhejiang Province, China (Grant no. LTGY23H160007) and the Science and Technology Program for Health and Medicine in Zhejiang Provice, China (Grant no. 2021KY541). We are grateful to all patients who participated in this study.
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
Authors' Disclosures
No disclosures were reported.
Authors' Contributions
Y. Xu: Conceptualization. K. Ding: Data curation. Z. Peng: Data curation. L. Ding: Conceptualization. H. Li: Resources. Y. Fan: Conceptualization.
References
- 1. Haznadar M, Cai Q, Krausz KW, Bowman ED, Margono E, Noro R, et al. Urinary metabolite risk biomarkers of lung cancer: a prospective cohort study. Cancer Epidemiol Biomarkers Prev 2016;25:978–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sanchez de Cos Escuin J. New immunotherapy and lung cancer. Arch Bronconeumol. 2017;53:682–7. [DOI] [PubMed] [Google Scholar]
- 3. Muinelo-Romay L, Garcia-Gonzalez J, Leon-Mateos L. Lung cancer and liquid biopsy: realities and challenges in routine clinical practice. Arch Bronconeumol 2019;55:289–90. [DOI] [PubMed] [Google Scholar]
- 4. Nandagopal L, Sonpavde G. Circulating biomarkers in bladder cancer. Bladder Cancer 2016;2:369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oberg K, Modlin IM, De Herder W, Pavel M, Klimstra D, Frilling A, et al. Consensus on biomarkers for neuroendocrine tumour disease. Lancet Oncol 2015;16:e435–e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li C, Li C, Zhi C, Liang W, Wang X, Chen X, et al. Clinical significance of PD-L1 expression in serum-derived exosomes in NSCLC patients. J Transl Med 2019;17:355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 8. Miyamoto S, Taylor SL, Barupal DK, Taguchi A, Wohlgemuth G, Wikoff WR, et al. Systemic metabolomic changes in blood samples of lung cancer patients identified by gas chromatography time-of-flight mass spectrometry. Metabolites 2015;5:192–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moreno P, Jimenez-Jimenez C, Garrido-Rodriguez M, Calderon-Santiago M, Molina S, Lara-Chica M, et al. Metabolomic profiling of human lung tumor tissues—nucleotide metabolism as a candidate for therapeutic interventions and biomarkers. Mol Oncol 2018;12:1778–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non–small cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol 2018;36:633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Luo P, Yin P, Hua R, Tan Y, Li Z, Qiu G, et al. A Large-scale, multicenter serum metabolite biomarker identification study for the early detection of hepatocellular carcinoma. Hepatology 2018;67:662–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cheng L, Zhang K, Qing Y, Li D, Cui M, Jin P, et al. Proteomic and lipidomic analysis of exosomes derived from ovarian cancer cells and ovarian surface epithelial cells. J Ovarian Res 2020;13:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stringer KA, McKay RT, Karnovsky A, Quemerais B, Lacy P. Metabolomics and its application to acute lung diseases. Front Immunol 2016;7:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Beuchel C, Becker S, Dittrich J, Kirsten H, Toenjes A, Stumvoll M, et al. Clinical and lifestyle related factors influencing whole blood metabolite levels—a comparative analysis of three large cohorts. Mol Metab 2019;29:76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Playdon MC, Sampson JN, Cross AJ, Sinha R, Guertin KA, Moy KA, et al. Comparing metabolite profiles of habitual diet in serum and urine. Am J Clin Nutr 2016;104:776–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kosmides AKK, Kamisoglu K, Calvano SE; Corbett SA; Androulakis IP. Metabolomic fingerprinting: challenges and opportunities. Crit Rev Biomed Eng 2013;41:205-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stevens VL, Hoover E, Wang Y, Zanetti KA. Pre-analytical factors that affect metabolite stability in human urine, plasma, and serum: a review. Metabolites. 2019;9:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, et al. Bile acid metabolites control T(H)17 and T(reg) cell differentiation. Nature 2019;576:143–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Watson GA, Sanz-Garcia E, Zhang WJ, Liu ZA, Yang SC, Wang B, et al. Increase in serum choline levels predicts for improved progression-free survival (PFS) in patients with advanced cancers receiving pembrolizumab. J Immunother Cancer 2022;10:e004378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shestakova KM, Moskaleva NE, Boldin AA, Rezvanov PM, Shestopalov AV, Rumyantsev SA, et al. Targeted metabolomic profiling as a tool for diagnostics of patients with non–small cell lung cancer. Sci Rep 2023;13:11072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shen J, Ye Y, Chang DW, Huang M, Heymach JV, Roth JA, et al. Circulating metabolite profiles to predict overall survival in advanced non–small cell lung cancer patients receiving first-line chemotherapy. Lung Cancer 2017;114:70–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tian Y, Wang Z, Liu X, Duan J, Feng G, Yin Y, et al. Prediction of chemotherapeutic efficacy in non–small cell lung cancer by serum metabolomic profiling. Clin Cancer Res 2018;24:2100–9. [DOI] [PubMed] [Google Scholar]
- 23. Hao D, Sengupta A, Ding K, Ubeydullah ER, Krishnaiah S, Leighl NB, et al. Metabolites as prognostic markers for metastatic non–small cell lung cancer (NSCLC) patients treated with first-line platinum-doublet chemotherapy. Cancers 2020;12:1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ghini V, Laera L, Fantechi B, Monte FD, Benelli M, McCartney A, et al. Metabolomics to assess response to immune checkpoint inhibitors in patients with non–small cell lung cancer. Cancers 2020;12: 3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nie X, Xia L, Gao F, Liu L, Yang Y, Chen Y, et al. Serum metabolite biomarkers predictive of response to PD-1 blockade therapy in non–small cell lung cancer. Front Mol Biosci 2021;8:678753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wishart DS. Computational approaches to metabolomics. Methods Mol Biol 2010;593:283–313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Volcano plot of serum metabolomics in Cohorts 1 and 2. The left figures (A, C) were obtained in the negative polarity mode, and the right figures (B, D) were obtained in the positive polarity mode. The abscissa is the log value of log2 of multiple differential expression (fold change), and the ordinate is the log value of -log10 of significant p values. Significant different metabolites: those with an FC> 1.5 and p value <0.05 are shown in rose red, and those with an FC <0.67, and p value <0.05 are colored in blue. Nonsignificantly different metabolites are indicated in black.
Total ion chromatograms (TICs) of the sample metabolites of Cohort 1 and Cohort 2 detected by UHPLC under negative (A, C) and positive (B, D) ion modes.
Principal component analysis (PCA) of the metabolites of Cohort 1 and Cohort 2 under negative (A, C) and positive (B, D) ion modes. Green indicates samples from the long-term survival group, blue indicates samples from the short-term survival group and red indicates QC samples.
Score plot from OPLS-DA of the two groups in Cohort 1 (A, B) and Cohort 2 (C, D). Each data point represents a function of the entire spectral profile of each subject. Partial least-squares discriminant analysis showed a clear separation between the groups with acceptable goodness of fit. Cross-validation plot with a permutation test repeated 200 times. The intercepts of R2 and Q2 under negative (A, C) and positive (B, D) ion modes illustrate that the OPLS-DA model is not overfitted.
The proportion of the identified metabolites in each chemical classification in Cohort 1 (A) and Cohort 2 (B). Colors in the figure express different chemical classification attribution entries, and the percentage represents the number of metabolites in the chemical classification entry as a proportion of all identified metabolites. Metabolites without chemical classification attribution were defined as undefined.
Correlation heatmap in Cohorts 1 and 2. Correlation analysis of metabolites with significant differences between positive and negative polarity modes. Red indicates a positive correlation, blue indicates a negative correlation, and white indicates a nonsignificant correlation. The depth of color is related to the absolute value of the correlation coefficient, that is, the higher the degree of positive or negative correlation, the darker the color. The point size is related to the significance of the correlation: more significant, smaller p values and larger points.
Metabolite heatmap between Cohorts 1 and 2. The left panels (A, C) were obtained in the negative polarity mode, and the right panels (B, D) were obtained in the positive polarity mode. Each row in the graph represents one differential metabolite, and each column represents a set of samples. Red represents significant upregulation, blue represents significant downregulation, color shades indicate the degree of upper downregulation, and metabolites with close expression patterns are clustered under the same cluster on the left.
Table S1: Patient demographics and baseline clinicopathological characteristics.
Table S2: The TOP10 up- or-down* regulated metabolics in Cohort 1.
Table S3: The TOP10 up- or-down* regulated metabolics in Cohort 2.
Table S4: The differential metabolites simultaneously associated with PFS and OS in cohort 1.
Table S5: The metabolites for significant correlation to both PFS and OS in cohort 2.
original data
original data
Supplementary Figure Legend
O-partial least-squares discrimination analysis (OPLS-DA) of metabolites of Cohort 1 and Cohort 2 under negative (A, C) and positive (B, D) ion modes. Identified compounds could be separated well with optimal goodness of fit. Green indicates samples from the long-term survival group; blue indicates samples from the short-term survival group.
Data Availability Statement
Data were generated by the authors and included in the article.