

Summary:
Leukocyte recruitment from the vasculature into tissues is a crucial component of the immune system but is also key to inflammatory disease. Chemokines are central to this process but have yet to be therapeutically targeted during inflammation, due to a lack of mechanistic understanding. Specifically, CXCL4 (Platelet Factor 4, PF4) has no established receptor that explains its function. Here we use biophysical, in vitro and in vivo techniques to determine the mechanism underlying CXCL4 mediated leukocyte recruitment. We demonstrate that CXCL4 binds to glycosaminoglycan (GAG) sugars on proteoglycans within the endothelial extracellular matrix resulting in increased adhesion of leukocytes to the vasculature, increased vascular permeability and non-specific recruitment of a range of leukocytes. Furthermore, GAG sulphation confers selectivity onto chemokine localisation. These findings represent a new understanding of chemokine biology, providing novel mechanisms for future therapeutic targeting.
Keywords: CXCL4, PF4, Chemokine, Proteoglycan, Glycosaminoglycan, Extracellular Matrix, Leukocyte, Recruitment, Trafficking
Introduction:
Leukocyte recruitment is central to fighting infection, coordinating the immune response to injury, and driving inflammatory disease1. Chemokines are critical during leukocyte recruitment, facilitating firm adhesion of leukocytes to the vascular endothelium2. Due to the central role of chemokines in many diseases, they are prime therapeutic targets3, but drugs that successfully target them are lacking4. This is, in part, due to an under-developed understanding of the mechanisms underlying chemokine biology.
The dogma of the chemokine field states that they bind to G protein-coupled receptors (GPCRs) on leukocytes, producing integrin activation and firm adhesion to the vascular endothelium2. However, this does not hold for CXCL4 (platelet factor 4, PF4)5. CXCL4 is produced by platelets and macrophages, regulates leukocyte recruitment and is implicated in a range of diseases6. However, it does not have a clearly defined receptor that explains its ability to recruit leukocytes, limiting drug discovery efforts5.
Extracellular matrix proteoglycans are present on most cells and are particularly prominent on the vascular endothelium lining blood vessels where they form the endothelial glycocalyx7. This barrier on the apical side of the endothelium regulates vascular permeability, interactions between circulating leukocytes and the vascular endothelium and is thus a central component of leukocyte recruitment into tissues8. Chemokine interactions with key glycocalyx components, glycosaminoglycan (GAG) sugar side chains on proteoglycans, are critical for chemokine function in vivo but do not directly affect chemokine:receptor interactions9.
The functional role of chemokine:GAG interactions is proposed to be localisation of chemokines to the endothelial surface to ensure that chemokine:receptor interactions occur at a specific site10. This does not explain why chemokines have a wide range of GAG-binding affinities, where some, like CXCL4, have very high-affinity GAG interactions and also crosslink GAG chains10–12. Thus, the biological consequences of CXCL4:GAG interactions have yet to be understood. GAGs and their larger proteoglycan structures can directly mediate cell signalling in other contexts13, but the relevance of this to chemokine function has been largely overlooked. Currently we do not understand the mechanisms that drive CXCL4 mediated leukocyte recruitment and associated disease6. Interactions with extracellular matrix GAGs may provide an “atypical” mechanistic explanation for CXCL4 function.
Herein, we demonstrate that CXCL4 promotes leukocyte adhesion to the endothelium, increases endothelial permeability and recruits a wide range of leukocytes driven by the CXCL4:GAG interaction. This demonstrates a chemokine functioning primarily through GAGs and not via a receptor. We also show that GAG fine structure (sulphation pattern) confers specificity to chemokine:GAG interactions, which illustrates how GAGs can impose selectivity onto the supposedly “redundant” chemokine system.
Results:
CXCL4 atypically recruits a broad spectrum of different leukocytes
To understand the mechanism underlying CXCL4 mediated leukocyte recruitment, we used an in vivo leukocyte recruitment assay to dissect this process (Fig. 1A). This involves injecting air under the mouse dorsum to create a pouch that can be analysed for immune cell recruitment in response to chemokine injection. CXCL4 increased the number of CD45+ (general leukocyte marker) cells, compared to vehicle controls (Fig. 1B). Surprisingly, CXCL4 recruited a wide range of different leukocyte types (Fig. 1C, gating strategy in Fig. S1A); specifically, neutrophils, eosinophils, monocytes, dendritic cells, and T cells (TCR beta+, NK1.1+ and Gamma delta+). In contrast, CCL2, which serves as a comparator for CXCL4, given its well established classical recruitment mechanism via a leukocyte chemokine receptor (CCR2)2, recruited only monocytes (Fig. S1B). Furthermore, using a transcranial window imaging approach (Fig. 1D) we also determined that CXCL4 increases adhesion of leukocytes to the vascular endothelium (Fig. 1E).
Figure 1. CXCL4 recruits a wide range of different leukocytes in vivo.
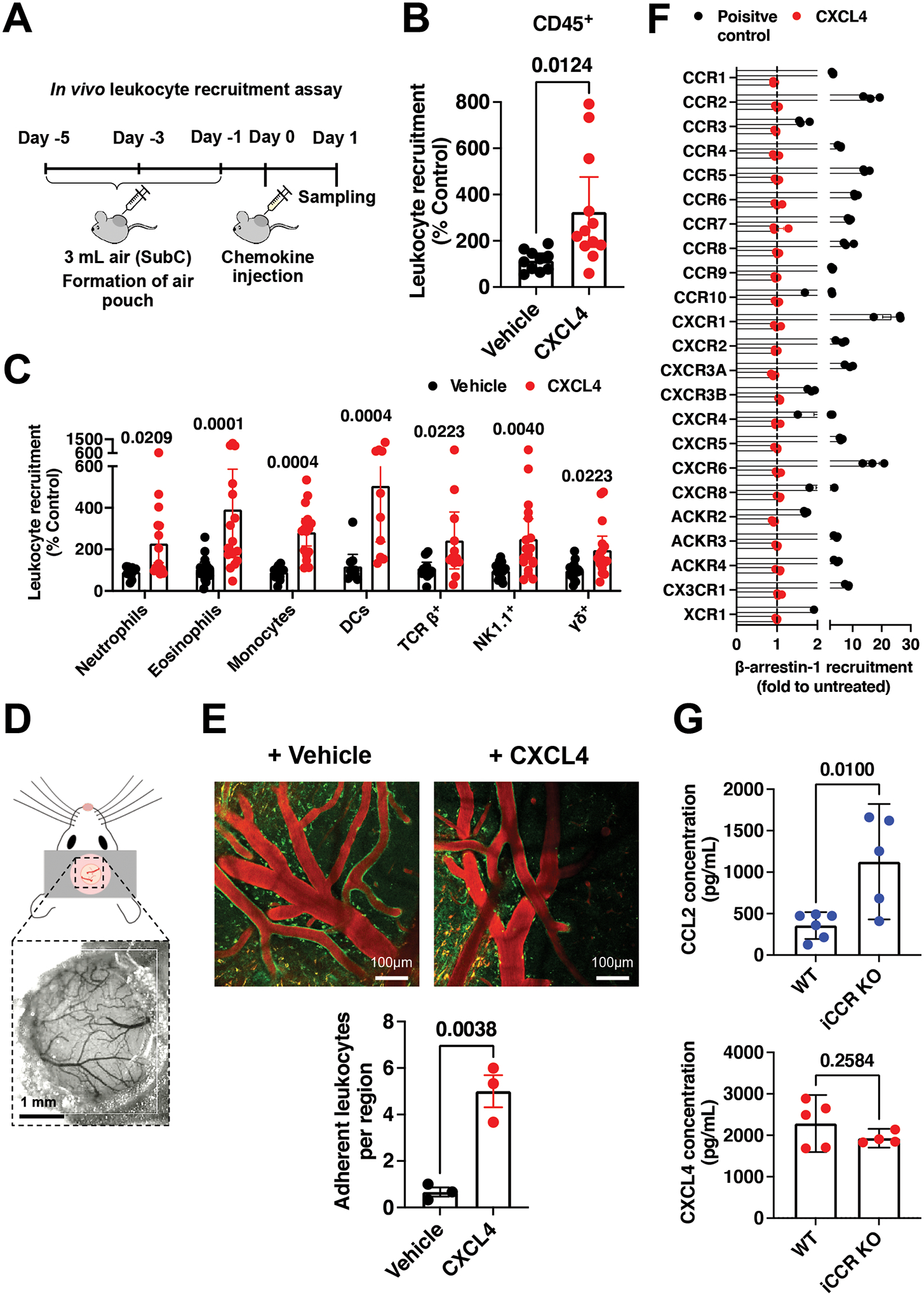
(A) Schematic of the in vivo leukocyte recruitment assay. (B) CD45+ cell counts 24 hr after CXCL4 injection. (C) Quantification of different leukocytes recruited by CXCL4. (D) Schematic of cranial window implantation for in vivo leukocyte adhesion analysis. (E) In vivo analysis of leukocyte (labelled green) adhesion to the walls of the vasculature (labelled red) following injection of CXCL4 or vehicle control. (F) Agonist activity of CXCL4 (100 nM) towards 19 classical and 3 atypical chemokine receptors evaluated in a β-arrestin-1 recruitment assay. For all receptors, one known agonist chemokine (100 nM) listed in the IUPHAR repository of chemokine receptor ligands was added as positive control. (G) CCL2 and CXCL4 quantification in the serum of wild type or iCCR (CCR1, 2, 3 and 5) KO inflamed mice.
All plots are mean with 95% confidence intervals and represent at least two separate experiments, data have been pooled and each dot in B, C, E and G represents an individual mouse and each dot in F represents the mean of an individual experiment. B and C are normalised to vehicle controls. Results in F are expressed as fold change relative to untreated controls and are presented as mean of three independent experiments. Individual p values are shown, B, E and G analysed using an unpaired t-test and C analysed using a one-way ANOVA with a post-hoc Sidak analysis of log-transformed data.
Recruitment of human monocytes by CXCL4 has been reported to be mediated by CCR114. However, CXCL4-mediated recruitment of CD45+ cells or monocytes remained unchanged in the CCR1 KO mouse compared to wild type controls (Fig. S2A). CXCR3 has also been associated with CXCL4 function, albeit at high concentrations6. Analysis of cells recruited by CXCL4 into the air pouch demonstrated that only T cells expressed CXCR3 (Fig. S2B). Furthermore, the Immgen database demonstrates that of the leukocytes recruited by CXCL4, only T cells express CXCR315. Thus, our data suggests that there was no chemokine receptor expression pattern across the leukocyte types recruited by CXCL4 that would explain their recruitment. Further analysis indicates that CXCR3 is also not present on endothelial cells sourced from the air pouch lining (Fig. S2C).
In order to comprehensively determine which chemokine receptors may be responsible for CXCL4 function we examined the ability of CXCL4 to activate all known human chemokine and atypical chemokine receptors in a global screen using β-arrestin-1 recruitment as a functional read-out16. The resulting data indicate that CXCL4 does not activate any known chemokine receptor (Fig. 1F). Therefore, the mechanism underlying widespread CXCL4-mediated recruitment remained unclear.
We next sought to determine whether leukocytes can directly bind and use CXCL4 to facilitate their migration. We exploited previous observations that when chemokine receptors are knocked out, their ligands are increased in the serum due to reduced uptake and degradation17.
For example, iCCR (CCR1, 2, 3 and 5 combined) knock out (KO) mice fail to recruit monocytes to the carrageenan inflamed air pouch17. As a result of the ablated monocyte migration the chemoattractant CCL2 is no longer being used (bound and degraded) during monocyte egress from the blood into the inflamed air pouch of iCCR KO mice, and is found at elevated levels in the serum (Fig. 1G). In contrast, CXCL4 levels remained unchanged under the same conditions. This suggests that monocytes do not directly utilise CXCL4 during trafficking to the inflamed air pouch, even though CXCL4 facilitated their recruitment.
Together these data suggest that CXCL4-mediated recruitment is not driven by binding to chemokine receptors on the leukocyte surface. We hypothesised that CXCL4 would have direct effects on the vascular endothelium regulating processes underlying immune cell recruitment.
CXCL4 increases endothelial permeability
Given that our data suggests a direct effect of CXCL4 on endothelial cells we hypothesised that CXCL4 may regulate endothelial, and thus vascular, permeability. In a Transwell cell migration system (Fig. 2A), purified human monocytes produced a classic chemotaxis response to CCL2 (Fig. S3A). Similarly, CCL5 (monocytes), CXCL12 (Jurkats), CCL21 (L1.2 cells) and CXCL8 (neutrophils) mediated recruitment of their concomitant receptor-expressing cells at 10 nM concentrations (Fig. 2A). In contrast, no increase in monocyte recruitment was produced by CXCL4, even at a relatively high concentration of 10 nM, and despite the ability of CXCL4 to mediate monocyte recruitment in vivo (Fig. 1).
Figure 2. CXCL4 increases endothelial permeability in a receptor independent manner and mediates endothelial intracellular signalling.
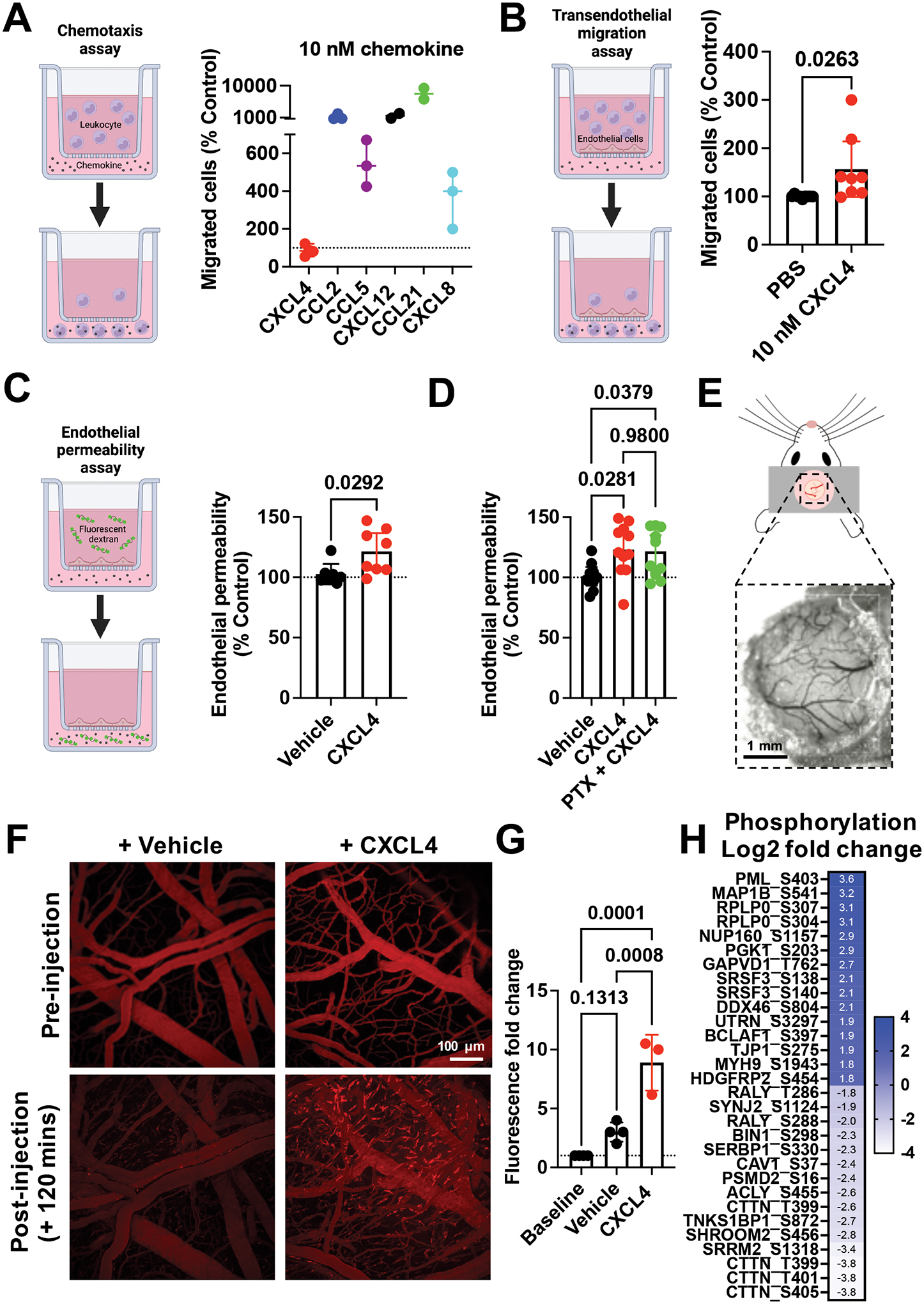
(A) Chemokine mediated chemotaxis of relevant receptor expressing cells; CXCL4, CCL2 or CCL5 (monocytes), CXCL12 (CXCR4+ Jurkat cells), CCL21 (CCR7+ L1.2 cells) and CXCL8 (CXCR2+ neutrophils). (B) Transendothelial migration of human monocytes towards CXCL4. (C) Transwell endothelial permeability in the absence and presence of CXCL4, (D) CXCL4 alone or in combination with pertussis toxin. (E) Schematic of cranial window implantation for in vivo vascular permeability analysis. (F) In vivo analysis of leakage of intravenously injected fluorescent dextran from the vasculature into the meninges following intravenous injection of CXCL4 or vehicle control. (G) Quantification of (F). (H) Heat map of the Log2 fold change of indicated protein phosphorylation sites from endothelial cells stimulated with CXCL4, relative to vehicle controls.
All plots are mean with 95% confidence intervals, represent at least two separate experiments where data have been pooled. Each dot in A-D represents an individual transwell and each dot in G represents an individual mouse. Data in A-D and G are normalised to vehicle controls. Individual p values are shown, and C analysed using an unpaired t-test, D and G using a one-way ANOVA with a post-hoc Tukey analysis.
To better replicate the in vivo environment we added an endothelial cell monolayer to the upper Transwell chamber to analyse transendothelial migration (Fig. 2B). Addition of CXCL4 produced a significant increase in movement of monocytes to the lower Transwell chamber, demonstrating that CXCL4 acts on the endothelium not the leukocyte.
Next, we showed that CXCL4 produced a significant increase in endothelial permeability, CCL5 had a similar effect whereas CCL2 and CXCL8 did not (Fig. 2C and Fig. S3B and C). To determine whether this effect of CXCL4 was mediated by a classical chemokine receptor (Gαi-coupled), we added pertussis toxin (PTx), but observed no effect at concentrations where PTx inhibits GPCR-mediated signalling (Fig. 2D)18. Crucially, CXCL4 increased vascular permeability in vivo as shown by leakage of fluorescent dextran from the brain meningeal vasculature after CXCL4 administration (Fig. 2E–G). These findings suggest that CXCL4 plays a role in regulating the permeability of the vascular endothelium.
CXCL4 regulates protein phosphorylation within endothelial cells
In order to determine how CXCL4 may facilitate leukocyte recruitment we firstly analysed the air pouch for changes in the concentration of a range of different cytokines and chemokines that play a key role in this process following CXCL4 injection (Fig. S4A). No changes, relative to vehicle controls, were observed for any of the proteins analysed suggesting no displacement/release of GAG-bound molecules by CXCL4.
To test whether CXCL4 has a direct effect on endothelial cells to facilitate leukocyte trafficking we utilised phosphorylation enriched mass-spectrometry (Fig. 2H). CXCL4 had limited effects on endothelial protein levels after 2 hours of stimulation (Fig. S4B) but had wide-spread effects on the phosphorylation state of proteins, relative to vehicle controls, indicative of endothelial signalling responses to CXCL4 (Fig. 2H). WebGestalt analysis indicated a number of pathways that were dysregulated following CXCL4 stimulation of endothelial cells19. The phosphorylation state of proteins associated with cell adhesion molecule binding was changed following CXCL4 endothelial stimulation. Specifically PML, GAPVD1, MYH9, and UTRN phosphorylation was increased and CTTN, SERBP1 and TNKS1BP1 phosphorylation was decreased by CXCL4. Dysregulated phosphorylation of proteins associated with adherens junctions and vascular permeability was also observed. Specifically increased phosphorylation of TJP1, MYH9, RPLP0 and decreased phosphorylation of CAV1, CTTN, SHROOM2 and TNKS1BP1 was detected in response to CXCL4 signalling (Fig. 2H). These data suggest that CXCL4 can signal to endothelial cells regulating phosphorylation of proteins that mediate leukocyte adhesion to endothelial cells and also endothelial permeability, supporting our in vivo and in vitro findings of CXCL4 mediated leukocyte recruitment, adhesion and vascular permeability (Fig. 1 and 2).
CXCL4 function is directly mediated via its interaction with the endothelial GAG heparan sulphate (HS)
Given the independence of CXCL4 mediated leukocyte recruitment from a classic chemokine receptor (Fig. 1 and 2), its high affinity for GAG side chains of proteoglycans and the presence of these proteoglycans within the glycocalyx on the blood exposed surface of the endothelium (Fig. 3A)8, we hypothesised that CXCL4 interaction with GAGs on these proteoglycans would directly facilitate leukocyte recruitment. To determine this, we added chemokine to Chinese hamster ovary (CHO) cells with and without genetic KO of the key GAG synthesis gene (B4galt7)20. CXCL4 accumulation was GAG-dependent and much higher than with CCL2 (Fig. 3B and Fig. S5A), despite their similar molecular weight and oligomerisation propensity11,21. HS is thought to be the dominant GAG within the endothelial glycocalyx8. We, therefore, used a biophysical bio-layer interferometry (BLI) approach (Fig. 3C) and determined that the binding of CXCL4 to an HS GAG surrogate, heparin octasaccharide (dp8), was much higher than for CCL2 (Fig. 3D and Fig. S5B). This difference in cellular accumulation and GAG binding indicated that CXCL4 function could be directly dependent on its GAG interaction, unlike CCL2.
Figure 3. CXCL4 function is mediated by its interaction with glycosaminoglycans (GAGs).
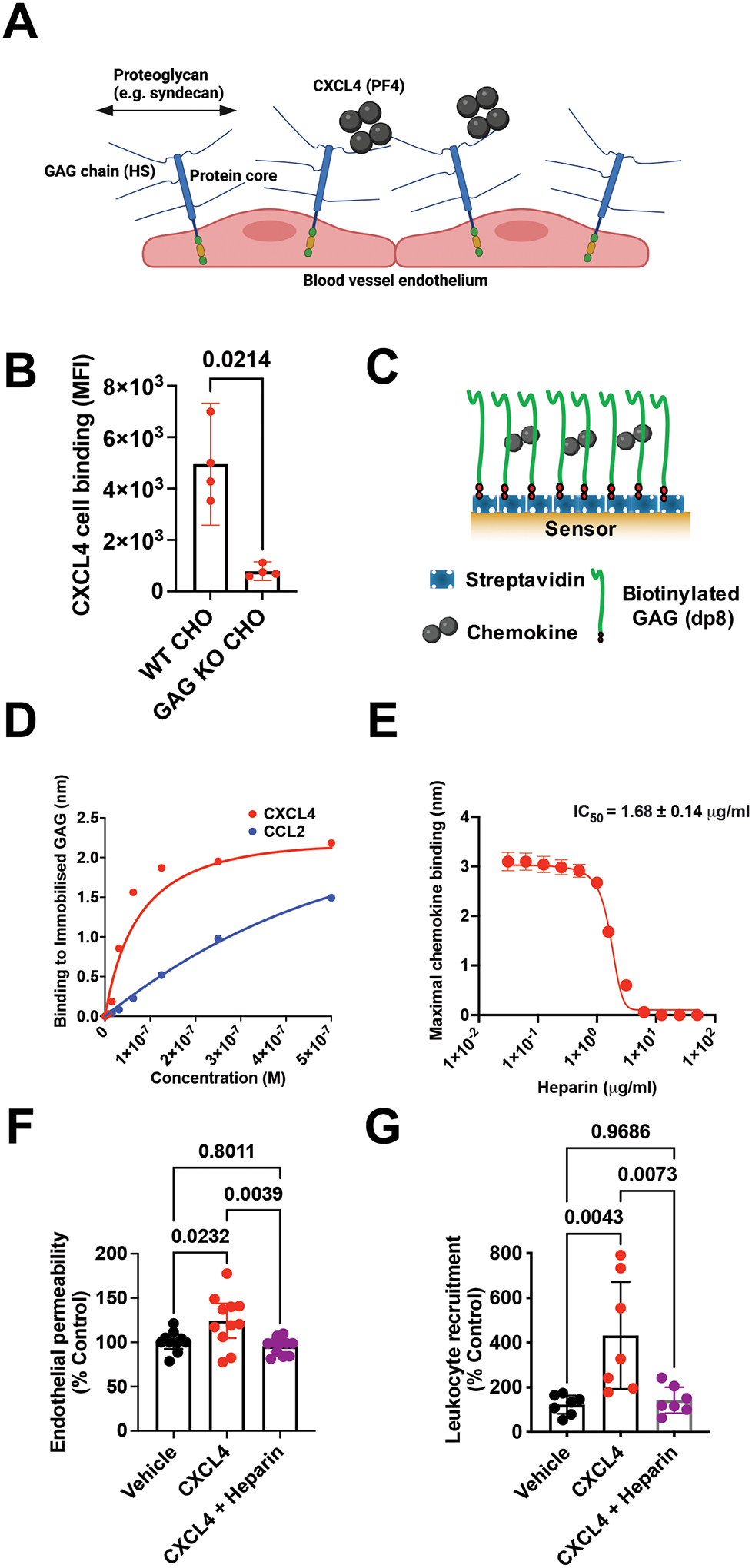
(A) Schematic of proteoglycans forming the luminal endothelial glycocalyx. (B) CXCL4 binding to CHO cells with and without surface GAGs. (C) Schematic of the biophysical BLI GAG-binding assay. (D) Maximum signal of chemokine:dp8 binding at different chemokine concentrations in the BLI assay. (E) Maximum CXCL4 (100 nM) binding to dp8 signal following pre-incubation with increasing concentrations of heparin. (F) Endothelial permeability (Transwell) assay with and without CXCL4 and exogenous heparin. (G) In vivo leukocyte (CD45+) recruitment (air pouch) to CXCL4.
B, F and G are mean with 95% confidence intervals and represent at least two separate experiments where data have been pooled. D and E are representative of at least two separate experiments. Each dot in B and F represents a technical replicate and each dot in G represents an individual mouse. F and G are normalised to vehicle controls. Individual p values are shown, B analysed using an unpaired t-test, F and G analysed using a one-way ANOVA with a post-hoc Tukey analysis.
To test this hypothesis, we firstly showed that CXCL4 binding to GAG was inhibited by pre-incubation with exogenous purified GAG (heparin) in the BLI assay (Fig. 3E and Fig. S5C) and on the cell surface (Fig. S5D), demonstrating specificity of the interaction. Subsequently we determined that pre-incubation with heparin reversed CXCL4 mediated increases in endothelial permeability in vitro (Fig. 3F) and ablated CXCL4 mediated leukocyte recruitment in vivo (Fig. 3G). In contrast, CCL2 mediated recruitment of monocytes to the air pouch was only reduced by a third following pre-incubation with exogenous heparin (Fig. S5E).
These experiments demonstrate that the interaction of CXCL4 with GAGs is responsible for CXCL4 mediated recruitment, likely by modulating endothelial cell functions that regulate leukocyte recruitment. In contrast, the CCL2:GAG interaction has an important but less direct role in facilitating CCL2 mediated leukocyte recruitment; presumably via localisation within the endothelial glycocalyx to facilitate interaction with leukocyte receptors.
CXCL4 oligomerisation mediates GAG binding and leukocyte recruitment
We have previously shown that high-affinity chemokine:GAG interactions are mediated by chemokine oligomerisation and cross-linking of GAG chains11,12. Therefore, we next tested the hypothesis that CXCL4 oligomerisation is a critical driver of leukocyte recruitment via crosslinking of GAGs (Fig. 4).
Figure 4. CXCL4 oligomerisation drives leukocyte recruitment.
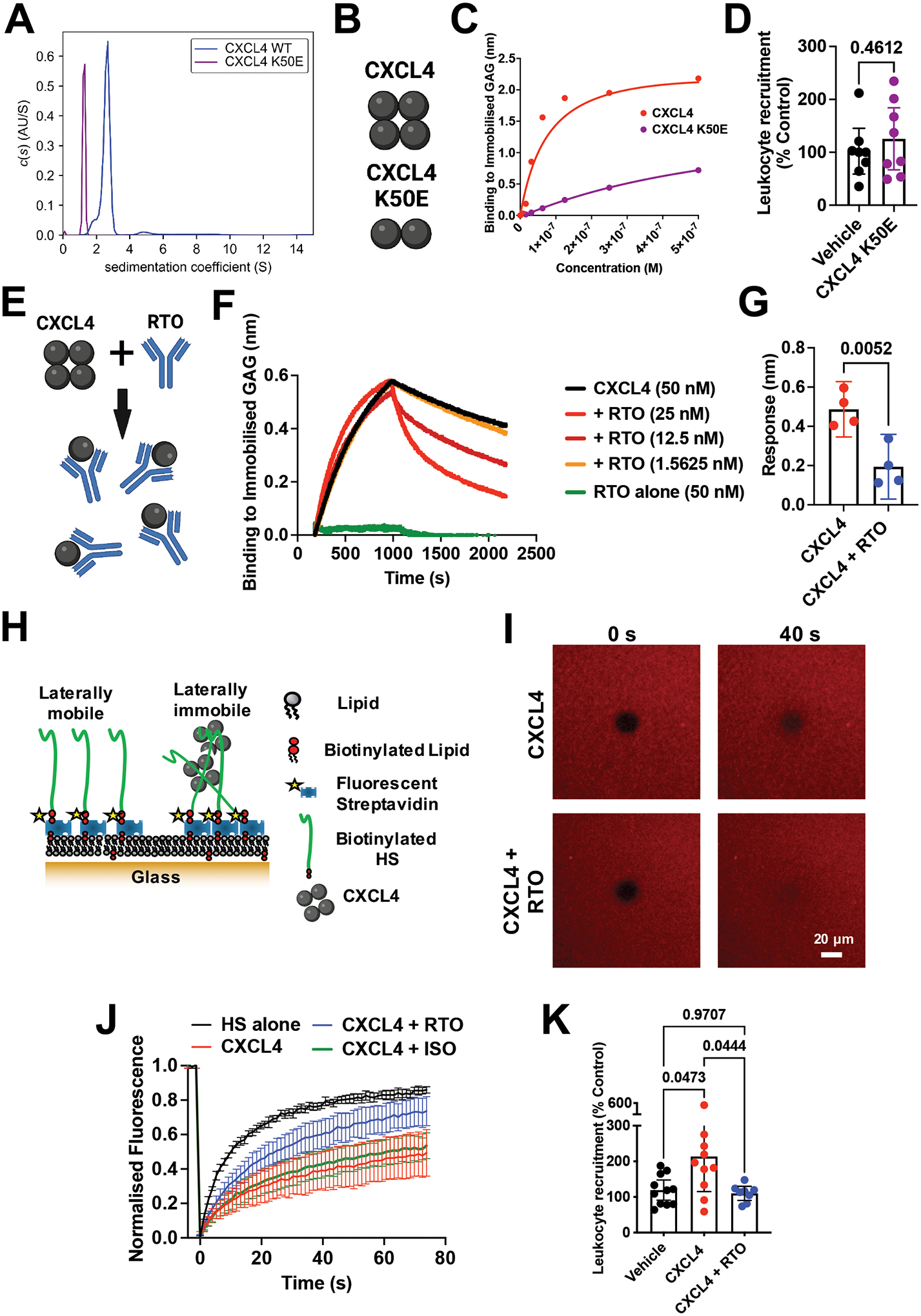
(A) AUC analysis of CXCL4 and the mutant K50E to show sedimentation coefficients (indicative of size). (B) Schematic of oligomerisation states of CXCL4 or the CXCL4 K50E mutant. (C) BLI analysis of CXCL4 K50E binding to immobilised heparin dp8. (D) In vivo leukocyte (CD45+) recruitment (air pouch) to CXCL4 K50E mutant. (E) Schematic of RTO antibody binding to CXCL4 monomer to inhibit oligomerisation. (F) CXCL4 (50 nM) binding to dp8 was monitored in the absence and presence (at a range of concentrations) of RTO antibody. (G) Final CXCL4:dp8 signal (after washing) from (F) is plotted with and without pre-incubation with RTO antibody (25 nM). (H) Schematic of FRAP assay to analyse HS cross-linking. (I) FRAP analysis of GAG in-plane mobility with CXCL4 alone or in combination with the RTO antibody, images post bleaching at indicated times. (J) Fluorescence recovery over time. (K) CXCL4 mediated leukocyte recruitment (CD45+) with and without RTO antibody.
D, G, J and K are mean with 95% confidence intervals and represent at least two separate experiments where data have been pooled. Each dot in D and K represents an individual mouse and each dot in G represents a technical replicate. A, C, F, I and J are representative of at least two separate experiments. Data in D and K are normalised to vehicle controls. Individual p values are shown, G analysed using an unpaired t-test and K analysed using a one-way ANOVA with a post-hoc Tukey analysis.
To analyse chemokine oligomerisation we used sedimentation velocity analytical ultra-centrifugation (SV-AUC). SV-AUC showed that CXCL4 has a sedimentation coefficient of 2.7S, with an estimated frictional ratio of 1.33 and a mass estimate using the Svedberg equation of 28.9 kDa, consistent with its formation of tetramers in solution (Fig. 4A)22,23. A mutant of CXCL4 has previously been generated (K50E) to inhibit oligomerisation of CXCL424. The CXCL4 K50E mutant had a much lower sedimentation coefficient of 1.26S, suggestive of either a compact monomer or an elongated dimer; confirming that the K50E mutation inhibits CXCL4 oligomerisation (Fig. 4A and B)23. CXCL4 K50E also exhibited a large reduction in binding to GAG in the BLI assay, compared to WT CXCL4 (Fig. 4C) and did not recruit leukocytes (CD45+) in vivo (Fig. 4D). We therefore hypothesised that inhibition of CXCL4 oligomerisation is a potential avenue to target CXCL4 driven leukocyte recruitment via inhibition of GAG binding.
Incubation of CXCL4 with an anti-CXCL4 antibody (clone: RTO) that binds monomeric CXCL4 and inhibits CXCL4 oligomerisation (Fig. 4E)25 produced a concentration-dependent inhibition of the CXCL4:GAG interaction in BLI (Fig. 4F–G and Fig. S6A); further demonstrating specificity of the CXCL4:GAG interaction. The inhibitory effect of RTO produced an increased off-rate of CXCL4 from GAG, re-creating that observed for CXCL4 K50E (Fig. S6B). RTO mediated inhibition was specific to CXCL4 since it did not affect CCL2 binding to GAG (Fig. S6C) or bind to the GAG directly (Fig. 4F).
Since the RTO antibody affected the off-rate of the CXCL4:GAG interaction (Fig. 4F), we next sought to determine whether RTO could affect CXCL4 mediated cross-linking of GAG chains. To test this, we used a biophysical model of the glycocalyx formed by HS GAG chains anchored on a fluid supported lipid bilayer, enabling analysis of GAG in-plane mobility and cross-linking using fluorescence recovery after photo-bleaching (FRAP) (Fig. 4H–J and Fig. S7). CXCL4 inhibited recovery 40 seconds after bleaching (Fig. 4I) and over time (Fig. 4J), indicating cross-linking of GAG chains. Pre-incubation of CXCL4 with RTO returned the bleaching recovery close to that seen in the absence of CXCL4 (HS alone) (Fig. 4I–J and Fig. S7). These data demonstrate that RTO mediated inhibition of CXCL4 oligomerisation prevents cross-linking of GAGs. Together, the AUC, BLI and FRAP data demonstrate that chemokine oligomerisation and cross-linking of GAGs are related events that drive the interaction with GAGs.
To determine the importance of CXCL4 oligomerisation, GAG-binding and cross-linking in leukocyte recruitment we utilised RTO in our in vivo leukocyte recruitment model (Fig. 4K). RTO resulted in ablation of CXCL4 mediated CD45+ leukocyte recruitment, confirming the importance of oligomerisation in vivo. This further supports the direct role of GAGs in CXCL4 driven leukocyte recruitment since monomeric chemokines promote chemokine receptor signalling through their GPCRs10,26. Indeed, recent structural studies suggest that chemokine oligomerisation and receptor binding are likely to be mutually exclusive due to overlap in binding sites, at least for many receptor:chemokine pairs9.
GAG sulphation confers selectivity on chemokine localisation
The above data show that CXCL4 function is driven by its interaction with GAG side chains of proteoglycans on the endothelial cell surface. To further characterise chemokine:GAG interactions, we probed the role of specific GAG sulphation positions (Fig. 5A). We hypothesised that changes in GAG sulphation would alter which chemokines are bound and presented on cells.
Figure 5. GAG sulphation mediates chemokine interaction selectivity and cellular localisation.
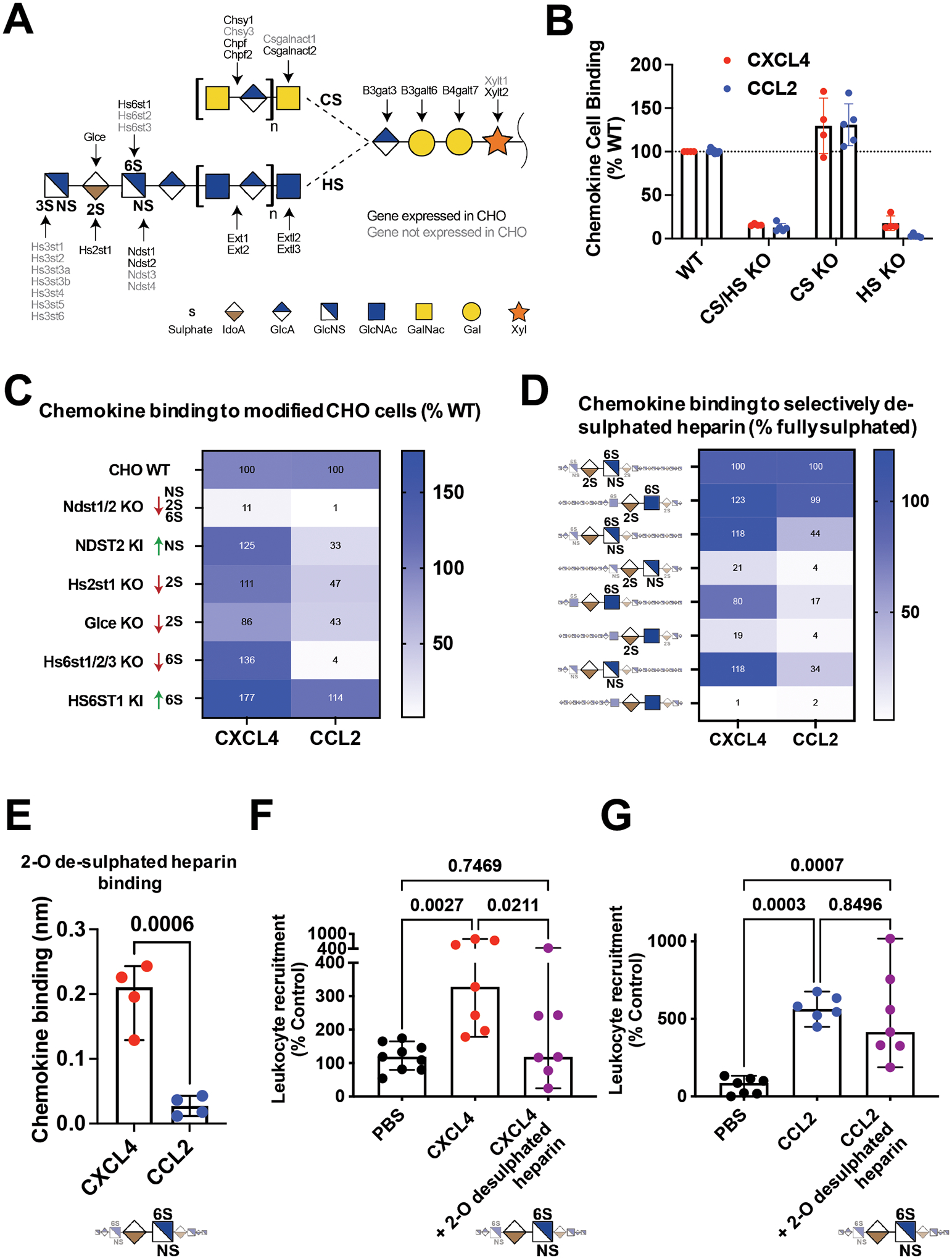
(A) Overview of enzymes involved in the biosynthesis of HS and CS GAGs. Elongation proceeds from right to left, a tetrasaccharide linker is elongated with either HS or CS disaccharide repeats into long linear polysaccharides. HS is further modified by addition of sulphate groups at the N-, 2-O, 6-O or 3-O (rarely) positions, this process is catalysed by the indicated sulphotransferases. (B) Chemokine binding to WT CHO cells or CHO cells with no GAGs (KO B4galt7), no CS (KO Csgalnact1/2/Chsy1) or no HS (KO Extl3). (C) Heat map analysis of chemokine binding to CHO cells with KO/KI of sulfotransferases acting on HS, where data is normalised to WT cells. (D) Heatmap analysis of maximum BLI signal of chemokine (500 nM) binding to differentially de-sulphated heparin fragments, data normalised to binding on fully-sulphated heparin. (E) Raw signal of chemokine binding to 2-O de-sulphated heparin in the BLI assay. Chemokine mediated in vivo leukocyte recruitment with and without 2-O de-sulphated heparin (F) CD45+ cell counts (CXCL4) or (G) monocyte (Ly6C+) cell counts (CCL2).
B, E, F and G are mean with 95% confidence intervals and represent at least two separate experiments where data have been pooled; each dot in B and E represents a technical replicate and each dot in F and G represents an individual mouse. Data in F and G are normalised to vehicle controls. Individual p values are shown, E analysed using an unpaired t test. F and G analysed using a one-way ANOVA with a post-hoc Tukey analysis.
Earlier experiments (Fig. 3B) demonstrated that GAG deficiency in CHO cells resulted in reduced binding to CXCL4. To determine which GAG types are responsible for chemokine binding we used CHO and human embryonic kidney (HEK) cells with gene KOs to remove, individually or in combination, HS and/or chondroitin sulphate (CS) (Fig. 5A and Fig. S8)20,27. We determined that HS bound to CCL2 and CXCL4 (Fig. 5B) whilst CS GAG only bound to CXCL4 on HEK cells (Fig. S8A).
HS GAG chains are 20–200 repeating disaccharides in length and are selectively sulphated on the N-, 2-O, 6-O and 3-O positions, which mediate chemokine:GAG binding (Fig. 5A)28. The enzymes that produce these different sulphation points were systematically knocked out in CHO cells, facilitating analysis of their contribution to chemokine:HS interactions (Fig. 5C). KO of NDST1/2, which reduces N-, 2-O and 6-O sulphation on HS20, largely ablated CHO cell binding to CXCL4 and CCL2. CXCL4 binding was not reduced in any of the other cell lines. CCL2 binding was reduced in the absence of HS2ST1 (2-O sulphation), GLCE (required for 2-O sulphation) and combined HS6ST1, 2 and 3 (6-O Sulphation). Since CHO cells do not make 3-O sulphated HS, the genes that produce it were knocked in29. 3-O sulphation generally increased CHO cell interaction with CXCL4 and CCL2 (Fig. S8B), however, HS3ST1 KI significantly enhanced binding to CCL2 but not CXCL4. These data demonstrate that differential sulphation of GAG chains provides considerable selectivity in binding to chemokines and presenting them on the cell surface.
Preferential targeting of CXCL4 function using GAG mimetics
Disruption of chemokine:GAG interactions is a possible avenue for therapeutic intervention in disease30. Heparin GAG and its derivatives are used as anti-coagulants and are well tolerated in the clinic31. Given the selective interactions mediated by GAG sulphation (Fig. 5A–C), we hypothesised that modified heparin derivatives, with specific sulphation sites removed, may allow preferential inhibition of chemokine driven leukocyte recruitment.
To determine binding selectivity of differentially sulphated heparins we used BLI to analyse binding to CXCL4 and CCL2 (Fig. 5D). This confirmed higher accumulation of CXCL4, over CCL2, on fully sulphated (2-O, 6-O and N- sulphated) heparin (Fig. S8C). Again CCL2 was more sensitive to selective removal of specific sulphate groups than CXCL4 (Fig. 5D). The CXCL4 K50E mutant demonstrated that oligomerisation is important for overall GAG binding and not sulphation specificity (Fig. S8C and D). These data further demonstrated chemokine:GAG selectivity32.
This BLI approach identified a heparin form (2-O desulphated) with full binding activity for CXCL4 and reduced binding to CCL2 (Fig. 5E). Pre-incubation with the 2-O desulphated fragment abolished CXCL4-, but not CCL2-, mediated leukocyte recruitment in vivo (Fig. 5F and G). These data demonstrate sulphation-driven selectivity of chemokine:GAG interactions and suggest that this can be exploited to use GAG mimetics to selectively inhibit certain chemokines.
Discussion:
Here we show that the chemokine CXCL4 mediates recruitment of a range of different leukocytes by binding to the GAG chains of proteoglycans within the endothelial extracellular matrix and not chemokine receptors on leukocytes or the endothelium (Fig. 6). We hypothesise that CXCL4 binding and cross-linking of GAG chains on endothelial proteoglycans triggers signalling mechanisms within the endothelial cells that mediate the effects observed here (Fig. 6)13 and elsewhere33. Whilst we present extensive data demonstrating the importance of GAG binding for CXCL4 function it remains difficult to exclude a role for a non-defined alternative receptor in this system. Future studies will be needed to fully dissect the underlying signalling pathways and integration with known endothelial signalling mechanisms that regulate leukocyte recruitment.
Figure 6. CXCL4 binds to endothelial GAG sugars, resulting in an increase of vascular permeability and non-specific leukocyte recruitment.
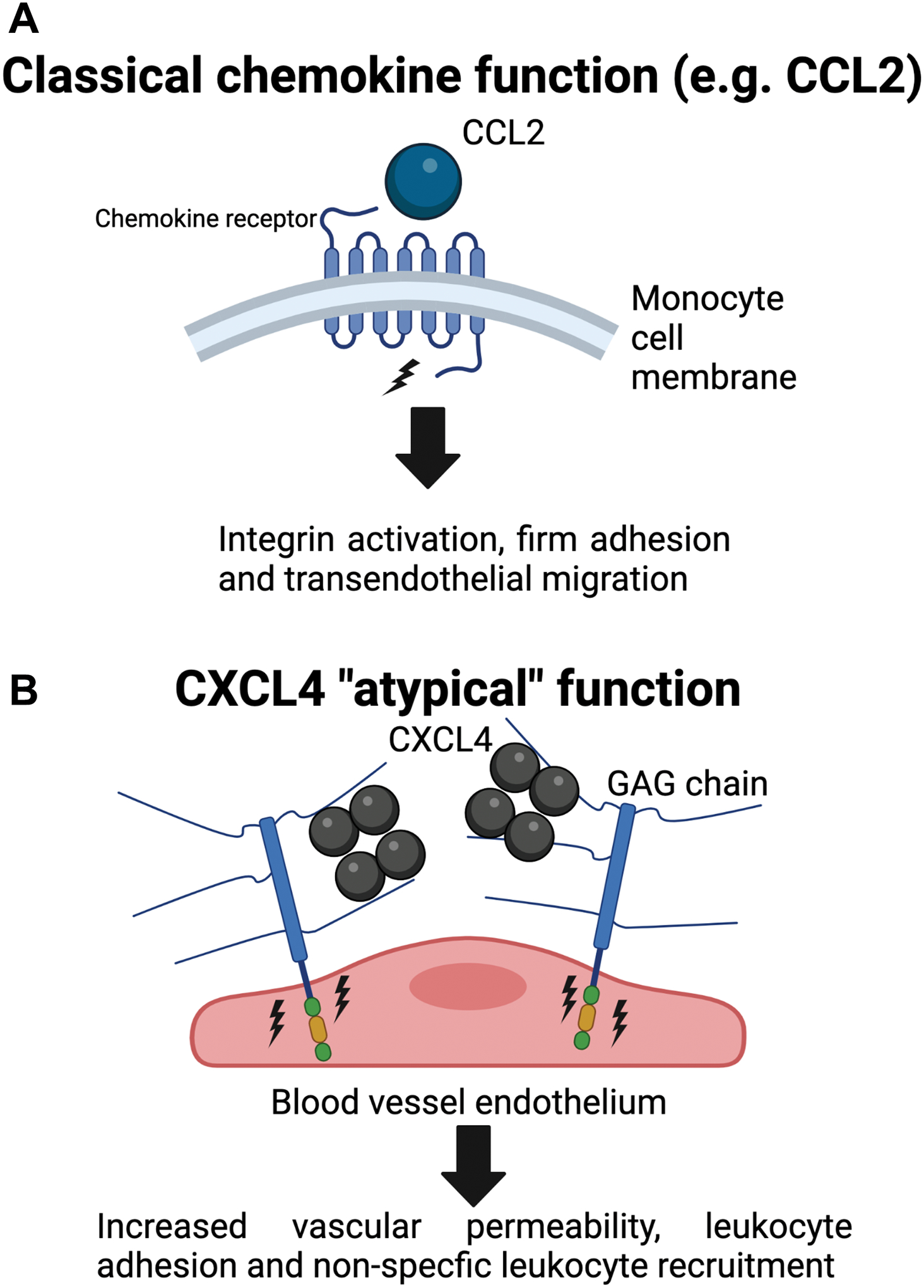
(A) Classical chemokines, e.g. CCL2, facilitate leukocyte recruitment by binding to seven transmembrane receptors on circulating leukocytes leading to signalling, integrin activation and firm adhesion of the leukocyte to the endothelium. (B) We propose that CXCL4 binds and re-models (as a tetramer) endothelial GAG sugars within the glycocalyx. This produces increased vascular permeability and leukocyte adhesion to endothelial cells possibly via signalling through the proteoglycan, facilitating “non-specific” recruitment of a range of different leukocytes from the vasculature and into inflamed tissues.
This generalised recruitment of different leukocytes is supported by previous observations that CXCL4 recruits neutrophils and monocytes in response to ischaemia reperfusion injury34, neutrophils to influenza or Pseudomonas aeruginosa infected lungs35,36, monocytes to atherosclerotic plaques37 and T cells into the malaria infected brain38. Our study may also provide a mechanism explaining previous observations that CXCL4 increases vascular permeability in the brain during cerebral malaria and in the lung following acute injury38,39. CXCL4 mediated increase of vascular permeability and leukocyte adhesion also fits with the role of platelets in facilitating rapid recruitment of leukocytes in response to inflammation40. Thus we propose that CXCL4 production/release is induced during inflammation enabling subsequent CXCL4 mediated vascular permeability, leukocyte adhesion to the endothelium and leukocyte recruitment via binding to GAG chains on endothelial proteoglycans. CCL5, which can also cross-link GAGs12, increased endothelial permeability in vitro whereas CCL2 and CXCL8, which cannot cross-link GAGs, did not. These data further associate GAG cross-linking with proteoglycan mediated effects and suggest other chemokines, cytokines and proteins may function, at least in part, through proteoglycans similarly to CXCL4.
The chemokine system has long been described as redundant, whereby multiple ligands can bind to the same receptor and multiple receptors can bind to the same ligand. However, studies designed to address whether chemokine redundancy exists have found limited evidence for it and in fact suggest specificity17,41–43. Specificity of receptor function is largely explained by co-ordinated cellular expression44. Contrastingly, there are many examples where chemokine ligands that recruit the same cell types are present at comparable concentrations in the same context32,45. Our data suggest that chemokine:GAG interactions, co-ordinated by the differential GAG sulphation patterns found across cells and tissues46, will produce specific localisation of chemokines at the cellular and tissue level.
This study demonstrates ways to target CXCL4 in inflammatory disease by inhibiting oligomerisation and GAG binding using GAG mimetics or specific antibodies. Blocking CXCL4-mediated recruitment of a range of leukocytes and increased vascular permeability is particularly promising in acute hyper-inflamed disease, e.g. sepsis39. Such approaches may also be relevant to targeting CXCL4 in the rare side effects of adenovirus vaccines against SARS-CoV-2 infection47. Future work will be important to address the potential role of CXCL4 as a master regulator of leukocyte recruitment in health and disease.
Limitations of the study
Whilst we present extensive data demonstrating the importance of GAG binding for CXCL4 function it remains difficult to exclude a role for a non-defined alternative receptor in this system. In addition future studies will be needed to fully dissect the underlying signalling pathways and integration with known endothelial signalling mechanisms that regulate leukocyte recruitment. We also demonstrate that CXCL4 mediates signalling in endothelial cells, however, it remains possible that CXCL4 driven cross-linking of GAG chains within the endothelial glycocalyx may play a more “physical” role in facilitating leukocyte recruitment. Furthermore, we present data demonstrating that specific sulphation of GAG chains mediates specificity of binding to chemokines. It remains to be seen whether this is a key mechanism during tissue specific leukocyte recruitment in vivo. It is also possible that the mechanism we describe may be important in the function of other chemokines and related proteins. These issues need to be addressed in future studies.
STAR METHODS
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr Douglas Dyer, (douglas.dyer@manchester.ac.uk)
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Mice
Adult male C57/Bl6 mice (8–12 weeks) were housed in cages of up to four on a 12-hour light/dark cycle, with ad libitum access to food and water. All experiments were carried out following ethical approval from The University of Manchester and University of Glasgow and under licence from the UK Home Office (Scientific Procedures Act 1986).
Cell lines
HEK-293T (ATCC), immortalised human umbilical vein endothelial (Ea.Hy 926 cells) (ATCC), Jurkat and L1.2+ (kind gift from Brian Zabel) cell lines were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS, penicillin (100 Units/ml) and streptomycin (100 μg/ml).
Method details
Materials
All chemokines were purchased from Protein Foundry unless otherwise specified. The dp8 and heparin GAGs were purchased (Iduron) and the de-sulphated heparin fractions were generated as detailed in48.
In vivo leukocyte recruitment
The air pouch was created by injecting 3 mL of sterile air subcutaneously under the dorsal skin on 3 occasions 48 hours apart as described previously17. The indicated quantity of chemokine was re-suspended in 100 μl of PBS (Sigma) and injected into the air pouch. Mice were culled 24 hours later and the air pouch flushed twice with 3 mL of air pouch buffer (PBS containing 1 mM EDTA (Sigma) and 1% (w/v) foetal bovine serum (FBS, Sigma)) to retrieve recruited cells. Retrieved cells were washed in air pouch buffer and stained with fixable cell viability dye (eBioscience, 1:1000 in PBS) for 20 minutes at 4 °C before being washed in flow cytometry buffer and re-suspended in 100 μl antibody staining cocktail (antibodies and FcR blocking reagent (Miltenyi Biotec) diluted 1:200. Cells were incubated for 30 minutes at 4 °C to stain before being washed in flow cytometry buffer and fixed for 10 minutes using 100 μl fixation buffer (BioLegend) at room temperature. CXCR3 staining was performed at 37 °C for 15 minutes prior to surface antibody staining. Cells were then re-suspended in flow cytometry buffer before addition of counting beads (ThermoFisher Scientific) and analysed using a Fortessa flow cytometer (BD Biosciences).
Flow cytometry data was analysed to quantify absolute cell counts or as the percentage of live cells before being normalised relative to vehicle controls to facilitate comparison across experiments.
Chemokine activity analysis (β-arrestin recruitment)
Cells and proteins
HEK-293T cell line was purchased from ATCC and were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS, penicillin (100 Units/ml) and streptomycin (100 μg/ml). Chemokines were purchased from PeproTech with the exception of CCL1 and CXCL17 that were purchased from R&D Systems.
β-arrestin recruitment assay
β-arrestin recruitment to chemokine receptors in response to CXCL4 or positive control chemokines (listed in the IUPHAR repository of chemokine receptors) was monitored by NanoLuc complementation assay [NanoBiT, Promega; Madison, USA, as previously described16,49–51. In brief, 8 × 105 HEK293T cells were plated per well of a 6-well dish and 24 hours later co-transfected with pNBe vectors encoding chemokine receptors C-terminally tagged with SmBiT and human β-arrestin-1 N-terminally tagged with LgBiT. 24 hours after transfection cells were harvested, incubated for 15 minutes at 37 °C with Nano-Glo Live Cell substrate diluted 200-fold and distributed into white 96-well plates (1 × 105 cells per well). CXCL4 or one known agonist chemokine listed in the IUPHAR repository of chemokine receptor ligands was added as positive control to each receptor16 at a final concentration of 100 nM. Ligand-induced β-arrestin recruitment to chemokine receptors was monitored with a GloMax® Discover plate reader (Promega, Madison, USA) for 20 minutes.
ELISA and Luminex analysis of air pouch fluid
Specific concentrations of CXCL4 and CCL2 in mouse serum samples were measured by enzyme-linked immunosorbent assay (ELISA), using Mouse CXCL4/PF4 and Mouse CCL2/JE/MCP-1 DuoSet ELISA kits (R&D Systems, Minneapolis, MN) as per the manufacturer’s instruction. Optical densities were read using a Versamax Microplate Reader (Marshall Scientific, Hampton, NH) at 450 nm wavelength. Air pouch fluid samples were analysed using a Bio-Plex Pro Mouse Chemokine Panel, 31- Plex Assay (Bio-Rad, UK), and the data was acquired on a Bio-Plex Manager™ (Software 6.2)
In vitro chemotaxis, endothelial transmigration and endothelial permeability
Chemotaxis
Monocyte and neutrophil purification was performed as described previously21, briefly blood was obtained from the San Diego Blood Banks and peripheral blood mononuclear cells (PBMCs) were separated by using a Ficoll-Paque gradient and centrifugation. Monocytes were then purified from freshly isolated PBMCs using a CD14+ selection MACS purification (Miltenyi Biotec) followed by final re-suspension in chemotaxis buffer (DMEM (Sigma) + 10% (v/v) FBS (Sigma)). Neutrophils were removed from the bottom of the Ficoll-Paque gradient and red blood cells were removed using red blood cell lysis buffer followed by washing and final re-suspension in chemotaxis buffer.
Chemokine or vehicle control was added in the bottom well of endothelial transwells (5 μm pores (Corning)) at the indicated concentration in 600 μl of chemotaxis buffer. 100 μl of 2 × 106 (2 × 104 total) cell suspension monocytes (CCL2 and CCL5), Jurkats (CXCL12), CCR7+ L1.2 cells (CCL21) and neutrophils (CXCL8) were added to the top well for the indicated chemokine. Transwells were then incubated for 2 hours at 37 °C/5 % CO2, removed and faux adherent cells from the lower membrane washed into the bottom well before migrated cells were counted using a Guava EasyCyte 8HT flow cytometer (Merck Millipore). Data was then presented as the total number of migrated cells or relative to vehicle controls (% control).
Transendothelial migration and permeability
Transendothelial migration experiments were undertaken as above but prior to the addition of migrating cells, endothelial cells were coated onto the upper side of the transwell insert as follows. Transwells with 5 μm pores (Corning) were coated with 50 μl per well of 10 μg/ml of fibronectin (Sigma) and incubated for 1 hour at 37 °C/5% CO2 and washed three times with 50 μl per well of PBS (Sigma). Immortalised human umbilical vein endothelial cells (Ea.Hy 926 cells) (ATCC) were seeded into each well, 100,000 cells per well in 50 μl of culture medium (DMEM (Sigma) containing 1% (v/v) penicillin-streptomycin solution (Thermo Fisher Scientific), 10% (v/v) FBS (Sigma) and 1% (v/v) L-Glutamine (Sigma)) and incubated for 2 days at 37 °C/5% CO2. Transwells were then washed with PBS (three times with 50 μl) and finally chemotaxis buffer prior to endothelial transmigration experiments. In the case of endothelial permeability experiments, 100 μl of chemotaxis buffer containing 25 μM 70,000 MW Texas Red dextran (Thermo Fisher Scientific) was added to the upper side of the endothelial monolayer, following incubation (2 hours 37 °C/5% CO2) fluorescence of the bottom well was determined using a FlexStation 3 fluorescent plate reader (Molecular Devices). Data were expressed and plotted relative to vehicle controls.
In vivo vascular permeability and leukocyte adhesion
Animals
Cranial window implantation surgery was performed on male C57BL/6J mice at 20–25 grams and multiphoton imaging was conducted two weeks later.
Cranial window implantation
Cranial window implantation was conducted as previously described52. In brief, animals were anaesthetised with 2.5% isoflurane in room air and positioned within a stereotactic frame with a heating blanket and temperature probe to ensure maintenance of body temperature at 37.5 °C. The cranium was exposed by removal of the scalp on top of both brain hemispheres. A metal head plate (Narishige CP-2, Japan) was mounted using dental cement (Sun Dental, Japan), to allow later fixation within the multiphoton microscope setup. A 3 mm diameter circular piece of bone was then carefully removed using a dental drill and a circular coverslip (Warner Instruments, USA) was secured in place of the removed bone using dental cement.
Multiphoton imaging
Depth of anaesthesia was maintained throughout the experiment using 1.5–2% isoflurane in 100% oxygen. Animals were fixed into the setup via the head plate and a heating blanket and temperature probe were used to maintain body temperature at 37.5 °C. Images were collected on a Leica SP8 Upright Multiphoton microscope using a Leica 25x / 0.95 L HC Fluotar dipping objective. All images equated to a physical size of 620 × 620 μm but image format and thus pixel sizes were varied between different experiments.
Permeability studies
Animals underwent intravenous injection of 50 μl of 10 mg/ml neutral 70,000 MW Texas Red dextran (Thermo Fisher Scientific) in sterile saline following induction of anaesthesia. Images were captured at 1024 × 1024 pixel resolution and the z planes were spaced 2 μm apart. Samples were imaged using two-photon excitation from a MaiTai MP laser (Spectra Physics) tuned to 880 nm and signal collected using the Leica external non-descanned hybrid detectors (NDHyD) through a BP624/40 filter for the Texas Red dextran signal and simultaneously onto a second NDHyD through a BP440/20 filter to image the dura via second harmonic generation (SHG) imaging. This approach ensured that the equivalent cranial depth was recorded in each experiment. Baseline images were taken for all regions of interest (ROIs). Animals were then intravenously injected with 5 μg of unlabelled CXCL4 diluted in saline or vehicle control (saline alone). Two or three ROIs were acquired for each animal and three or four animals were used per group. Texas Red channel maximum-intensity projection images of the first 60 μm of tissue including the dura were generated and the fluorescence intensities (mode) for each ROI were calculated. These values were taken for each image at 120 minutes following CXCL4/vehicle injection and plotted as fold change from the baseline maximum-intensity projection images.
Leukocyte adhesion studies
Intravenous injection of 100 μl of Wheat Germ Agglutinin Alexa Fluor 488 conjugate (Thermo Fisher Scientific) was performed on awake animals two hours prior to imaging, to label the leukocytes, as previously described53,54. Following induction of anaesthesia 5 μg of unlabelled CXCL4 diluted in saline or vehicle control (saline alone) mixed with 50 μl of 10 mg/ml 70,000 MW Texas Red dextran (Thermo Fisher Scientific) was injected intravenously. Two-photon excitation was used with the MaiTai MP laser tuned to 800 nm and imaged simultaneously on to external NDHyD’s through BP525/50 (for Alexa Fluor 488) and BP624/40 (for Texas Red) filters. To monitor leukocyte activity in each ROI, a 1-minute recording was acquired as a single plane over time (xyt mode) at 256 × 256 pixel resolution at 4 frames per second. Furthermore, images were also acquired at 1024 × 1024 pixel resolution for high resolution representations of each ROI. This imaging regime was repeated 24 hours after CXCL4/vehicle injection under terminal anaesthesia following an additional injection of 50 μl of 10 mg/ml Texas Red dextran to re-label the bloodstream. Leukocyte adhesion was assessed within the venular system 24 hours following CXCL4 or vehicle injection via manual counting. Leukocytes were classed as adhered if they remained in position for the 1 minute duration of the xyt recording and data was recorded as average number of adhered leukocytes across the 3 ROIs for each animal. Three ROIs were recorded per animal and 3 animals were used per group.
Phosphorylation enriched mass-spectrometry analysis of CXCL4 stimulated endothelial cells
Endothelial stimulation
Wells of a 12-well plate (Sigma-Aldrich) were treated with 1 mL fibronectin bovine plasma (10 μg/mL in PBS, Sigma-Aldrich) and incubated for 1 hour at 37°C. Fibronectin was removed and wells were washed with 1 mL PBS. EA.HY 926 endothelial cells were seeded (200,000 cells per well) in culture medium and left to incubate at 37°C for 24 hours. Non-adherent cells were removed by washing with 1 mL DPBS twice. Endothelial cells were then stimulated with vehicle control or CXCL4 (7 μg per well) in culture medium for 2 hours. Media was then removed from wells, cells washed with PBS and were then lysed with RIPA buffer (Merckmillipore, 1:10 in double distilled H2O) supplemented with protease inhibitor (cOmplete tablets, Sigma-Aldrich) and phosphatase inhibitor (Sodium orthovanadate, 5 mM, Sigma-Aldrich) and incubated for 30 mins at 4°C. Lysates were then frozen at −20°C until needed.
Proteolytic digestion and phosphopeptide enrichment
Protein samples were reduced with 15 mM dithiothreitol for 45 min at 56°C. Samples were then cooled and alkylated with 50 mM iodoacetamide (IAM) (45 min, RT, in the dark). −20°C acetone was added at 4 times volume of sample and incubated at −20°C for 1 hour to precipitate. Following centrifugation (16,000 × g, 20 mins, 4°C) the pellet was washed with −20°C acetone and centrifuged again as stated. The pellet was left to dry in a fume hood for 20 min. Pellets were resuspended in 50 mM ammonium bicarbonate supplemented with 0.2% (w/v) Rapigest Surfactant (Waters). Samples were digested with sequencing-grade trypsin (Promega) at an enzyme-to-protein ratio of 1:50 and incubated overnight at 37°C whilst shaking (450 rpm). Digested samples were acidified with trifluoroacetic acid (TFA) to a final concentration of 1% (v/v), pH 3–5. Samples were incubated at 37°C for 45 min, then centrifuged (20,000 × g, 10 min) to precipitate out Rapigest. Desalting was performed with Oasis HLB Cartridges (Waters) which were wet with 50% acetonitrile (ACN) and equilibrated with 0.1% formic acid (FA). Samples were eluted in binding solution (80% v/v ACN, 5% v/v TFA, 1M glycolic acid). Enrichment of phosphopeptides was performed via incubation with MagReSyn® titanium dioxide beads (resynbio). A fraction of digested samples was retained for proteomic analysis. Phosphoproteomic analysis preparation involved washing of beads with 1% ammonium hydroxide and binding solution. Automated phosphopeptide enrichment was performed on the KingFisher Flex robot (ThermoFisherScientific) with a homologous set up to previously described55.
LC-MS/MS Analysis
Digested samples were made up in 12 μL (phospho samples) and 20 μL (protein samples) of 3% ACN and 0.1% FA. Samples were analysed via tandem mass spectrometry (LC-MS/MS) using an UltiMate® 3000 Rapid Separation LC (RSLC, Dionex Corporation, Sunnyvale, CA) coupled to a QE HF (Thermo Fisher Scientific, Waltham, MA) mass spectrometer. Mobile phase A was 0.1% FA in water and B was 0.1% FA in ACN. The column used was 75 mm × 250 μm i.d. 1.7 mM CSH C18, analytical column (Waters). For proteomic analysis 1 μL aliquot of sample was transferred to 5 μL loop and loaded on the column at a flow of 300 nL/min for 5 min at 5% B. For phospho samples, 3 μL was injected and loaded for 13 min. The loop was taken out of line and flow reduced from 300 nL/min to 200 nL/min (in 30 secs). Peptides were separated on a gradient starting from 5% to 18% B (in 63 min 30 sec), then from 18% to 27% B (in 8 min) and from 27% B to 60% B (in 1 min). At 85 min flow is increased back to 300 nL/min until 90 min (end of run).
Data was acquired in data directed manner for 90 min in positive mode. Peptides were automatically selected for fragmentation by data dependent analysis of the top 12 peptides with mass/charge (m/z) between 300 to 1750Th, a charge state of 2/3/4 and dynamic exclusion set at 15 sec. MS resolution was set at 120,000, ACG target of 3e6 and maximum fill time at 20ms. MS2 resolution was set to 30,000, ACG target of 2e5, maximum fill time of 45 ms, isolation window of 1.3Th and collision energy of 28.
Data processing and analysis
Raw data for both proteome and phosphoproteome were processed together using MaxQuant (v1.6.17.0)56 against the human proteome obtained from Uniprot (May 2021)57. Fixed modification of carbamidomethylation of cysteine and variable modifications of methionine oxidation, protein N-terminal acetylation were set for both proteome and phosphoproteome data with phosphorylation of serine, tyrosine and threonine set for the phosphoproteome data only. Precursor tolerance was set at 20ppm and 4.5pm for the first and main searches, with MS/MS tolerance set at 20ppm. A false discovery rate (FDR) of 0.01 was set for PSM and protein level, up to two missed cleavages were permitted and “match-between-runs” was selected.
Processed data were analysed using the Perseus software (1.6.15.0)58. Proteome data were filtered for reverse hits, potential contaminants and proteins only identified by modified peptides. Proteins were retained that were present in 50% of samples and ratios between stimulated/un-stimulated were calculated. Phosphorylation site data were filtered for reverse hits, potential contaminants and localisation probability greater than 0.75 (class I). Phosphorylation sites were retained that were present in 50% of samples and ratios between stimulated/un-stimulated were calculated.
Analysis of chemokine binding to cell surface GAGs
This analysis was adapted from a previous approach20. Briefly, 1 × 105 genetically engineered CHO GS−/− or HEK293 6e cells were harvested from suspension culture and washed in 1 × PBS before being incubated with biotinylated recombinant human CXCL4 or CCL2 (Protein Foundry LLC) at 10 μg/mL in 1 × PBS with 1% FBS for 30 minutes at 4 °C. Cells were washed with 1 × PBS with 1% FBS and incubated with 1:2000 Alexa Fluor 488-streptavidin (S32354, Invitrogen) in 1 × PBS with 1% FBS for 30 min at 4 °C. After washing with 1 × PBS with 1% FBS, cells were resuspended in 1 × PBS with 1% FBS and fluorescence intensity was analysed on a SA3800 spectral cell analyzer (SONY). For the heparin inhibition assays, 10 μg/mL CXCL4 or CCL2 was pre-incubated with varying concentrations of porcine mucosal heparin in 1 × PBS with 1% FBS for 30 min at RT prior to incubation with cells. All experiments were performed a minimum of three times using triplicate samples.
Biophysical chemokine:GAG interaction analysis (Bio-layer interferometry)
Bio-layer interferometry was performed on an Octet Red96 system (Sartorius AG, Goettingen, Germany) using a methodology adapted from11. Firstly, GAGs were biotinylated at their reducing end using an approach described elsewhere59 and immobilised onto High Precision Streptavidin (SAX) biosensors (Sartorius AG, Goettingen, Germany). To immobilise, SAX biosensors were hydrated for 10 minutes in BLI buffer (10 mM Hepes, 150 mM NaCl, 3 mM EDTA, 0.05% Tween-20, pH 7.4). For the heparin dp8 GAG immobilisation was undertaken in BLI buffer at 0.078 μg/ml to achieve an immobilisation level of approx. 1 nm. For the differentially de-sulphated heparin fractions immobilisation was undertaken at 5 μg/ml in BLI buffer until all surfaces were saturated. Sensors were then washed in regeneration buffer (0.1 M Glycine, 1 M NaCl, 0.1% Tween, pH 9.5) and re-equilibrated in assay buffer. Blank reference or GAG coated sensors were then submerged into wells of black-walled 96 well plates containing 200 μL of BLI buffer containing chemokines at the indicated concentrations for the indicated time (association) before being transferred to assay buffer containing wells (dissociation) before finally being washed in regeneration buffer. The binding signal was recorded throughout and signal from binding of chemokine to blank (no immobilised GAG) sensors and by GAG immobilised sensors in assay buffer alone was subtracted from that produced by chemokine binding to GAG immobilised sensors. As well as signal over time the maximum signal during the association phase of the interaction was recorded and is plotted. Data were acquired at 5 Hz and analysed using the Octet HT 10.0 analysis programme. For inhibition analysis signal was plotted against inhibitor concentration and fitted for IC50 values using non-linear regression in the Prism software package (GraphPad).
Analytical ultra-centrifugation
CXCL4 and CXCL4 K50E were re-suspended in water to a concentration of 1 mg/ml and further diluted 1:10 in PBS. Samples were loaded into 2-sector cells with PBS as a reference and centrifuged at 50,000 rpm in a 4-hole An60Ti rotor monitoring the absorbance at 230 nm until sedimentation was reached. The time-resolved sedimenting boundaries were analysed using Sedfit60. The resulting profiles are shown in Gussi61.
Fluorescence recovery after photo-bleaching (FRAP) of films of in-plane mobile GAGs
Heparan sulphate (HS) used in FRAP assays was biotinylated at the reducing end (HS-b), as described previously59. HS was sourced from porcine intestinal mucosa, had a molecular weight of 12 kDa, a polydispersity of 1.6 and an average of 1.4 sulfates per disaccharide62. Dioleoylphosphatidylcholine (DOPC) and dioleoylphosphatidylethanolamine-CAP-biotin (DOPE-CAP-b) lipids were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Streptavidin was labelled with Atto 565 fluorophores (Sav Atto 565), and all chemicals for buffers were supplied by Sigma. Working buffer used for all experimental steps consisted of 10 mM HEPES, pH 7.4, and 150 mM NaCl in ultrapure water.
Preparation of model glycocalyces (films of in-plane mobile GAGs)
Films of one-end anchored HS polysaccharides on supported lipid bilayers were formed as previously described63,64. Briefly, pre-conditioned glass coverslips (24 × 24 mm2, type 1.5; Menzel Gläser, Braunschweig, Germany) were attached, using a bi-component glue (Picodent, Wipperfürth, Germany), to a custom-built teflon holder, thus forming the bottom of four identical wells with a volume of 50 μL each. Supported lipid bilayers (SLBs) were formed by the method of vesicle spreading65, through exposure of the glass surface to small unilamellar vesicles (100 μg/mL for 30 minutes) made from DOPE-CAP-b in a DOPC background (molar ratio 0.5:99.5). SAv Atto 565 (40 μg/mL for 20 min) and HS-b (10 μg/mL for 20 min) were then sequentially applied to the SLBs, to anchor HS via SAv to the biotin-presenting SLB. After each incubation step, the surface was washed with a working buffer to remove excess molecules from the solution phase.
Fluorescence recovery after photobleaching (FRAP) assays
With HS-b anchored to SAv atto 565, FRAP of the fluorescently labelled SAv was used as a reporter of GAG in-plane mobility. Fluorescence images were taken with a confocal laser scanning microscope (Zeiss LSM880, Zeiss, Germany) using ZEN software. The objective was a Plan-Apochromat 40×/1.4 Oil DIC M27, and the light source a 561 nm laser. The pinhole size was 198 μm (5 Airy units). In each FRAP experiment, 3 pre-bleach images of the surface (177.12 × 177.12 μm2; 256 × 256 pixels) were acquired before a circle in the centre of the image (10 μm radius) was bleached, and 57 images were acquired post bleach to monitor the fluorescence recovery.
To quantify fluorescence recovery, all images were analysed in Fiji66. Mean fluorescence intensities were extracted from two regions of equal radius (6.92 μm) in each image in the sequence: the bleach region was located at the image centre and the reference region at the periphery. Fluorescence intensities were corrected for background fluorescence and intensity fluctuations, and normalised using the following equation: . Here, lbg is the background intensity (measured in the absence of fluorophores, and constant across the image), linit. and l(t) are the intensities in the bleach region prior to bleaching (t < 0) and at post-bleach time t (l(0) = l(t = 0)), respectively, and lref,init. and lref(t) are the corresponding intensities in the reference region. With this normalisation, the unbleached (or fully recovered) intensity is 1, and the intensity in the bleach region immediately after bleaching is 0. Recovery curves were plotted as normalised intensity against time, and the area under each curve from t = 0 to t = 40 s was calculated to represent an effective measure of the in-plane mobility of SAv (and the attached HS) on the SLB.
To assess the impact of CXCL4 on HS mobility, the HS film was incubated with CXCL4 alone (250 nM), with a mix of CXCL4 and RTO (at 2:1 molar ratio), or with a mix of CXCL4 and a control IgG (at 2:1 molar ratio) for 1.5 h; the HS film was then analysed by FRAP (within 10 minutes, and with excess proteins in the solution phase). Prior to use, the mixtures of CXCL4 and RTO (or control IgG) were incubated for 1 hour to allow complexes to form. Three independent experiments were performed for each condition, and in each experiment three FRAP series were acquired at different positions on the surface. Data shown in Fig. 4E and Suppl. Fig. 7 represent the mean ± 95% confidence intervals across all 9 data sets per condition.
Immgen
The Immgen data base was probed using the MyGeneSet data browser15.
Quantification and statistical analysis
All statistics were performed using the Prism (GraphPad) software package. Experiments containing two groups were analysed using an unpaired t-test and data containing more than two groups were analysed using a one-way ANOVA with a post-hoc multiple comparison test. p < 0.05 was considered to be statistically significant.
Supplementary Material
Acknowledgements:
The authors would like to acknowledge numerous colleagues for reading and helping in drafting the manuscript. The Bioimaging Facility microscopes used in this study were purchased with grants from BBSRC, Wellcome Trust and the University of Manchester Strategic Fund. Special thanks to Peter March, Roger Meadows and Steven Marsden for their help with the microscopy. FRAP analyses was performed on a microscope within the Bioimaging Facilities at University of Leeds, supported by the Wellcome Trust (WT104818MA). Schematics have been drawn in BioRender.
DPD was supported by a Sir Henry Dale fellowship jointly funded by the Wellcome Trust and Royal Society (Grant Number 218570/Z/19/Z) and a Wellcome Trust centre grant (Grant Number 203128/A/16/Z). RLM was supported by The Carlsberg Foundation CF20-0412. JT and RLM were supported by The European Union’s Horizon 2020 Research and Innovation Programme 899687, HS-SEQ. RL, MS and AC were supported by the Luxembourg Institute of Health (LIH), Luxembourg National Research Fund (INTER/FNRS grants 20/15084569), F.R.S.-FNRS-Télévie (grants 7.4529.19, 7.8504.20 and 7.8508.22). RL is a Luxembourg National Research Fund PhD fellow (PRIDE-14254520 “I2TRON”). TMH was supported by NIH grants 1R01AI161880 and 1R01CA254402.
Footnotes
Declaration of interests: The authors declare no competing interests
References:
- 1.Nourshargh S, and Alon R (2014). Leukocyte migration into inflamed tissues. Immunity 41, 694–707. 10.1016/j.immuni.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 2.Bachelerie F, Ben-Baruch A, Burkhardt AM, Combadière C, Farber JM, Graham GJ, Horuk R, Sparre-Ulrich AH, Locati M, Luster AD, et al. (2014). International Union of Basic and Clinical Pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacological Reviews 66, 1–79. 10.1124/pr.113.007724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schall TJ, and Proudfoot AEI (2011). Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat Rev Immunol 11, 355–363. 10.1038/nri2972. [DOI] [PubMed] [Google Scholar]
- 4.Proudfoot AEI, Bonvin P, and Power CA (2015). Targeting chemokines: Pathogens can, why can’t we? Cytokine 74, 259–267. 10.1016/j.cyto.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 5.von Hundelshausen P, and Schmitt MMN (2014). Platelets and their chemokines in atherosclerosis-clinical applications. Front Physiol 5, 294. 10.3389/fphys.2014.00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vandercappellen J, Damme JV, and Struyf S (2011). The role of the CXC chemokines platelet factor-4 (CXCL4/PF-4) and its variant (CXCL4L1/PF-4var) in inflammation, angiogenesis and cancer. Cytokine Growth Factor Rev 22, 1–18. 10.1016/j.cytogfr.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 7.Gray AL, Pun N, Ridley AJL, and Dyer DP (2022). Role of extracellular matrix proteoglycans in immune cell recruitment. Int J Exp Pathol. 10.1111/iep.12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marki A, Esko JD, Pries AR, and Ley K (2015). Role of the endothelial surface layer in neutrophil recruitment. J Leukocyte Biol 98, 503–515. 10.1189/jlb.3mr0115-011r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graham GJ, Handel TM, and Proudfoot AEI (2019). Leukocyte Adhesion: Reconceptualizing Chemokine Presentation by Glycosaminoglycans. Trends Immunol. 10.1016/j.it.2019.03.009. [DOI] [PubMed] [Google Scholar]
- 10.Handel TM, and Dyer DP (2020). Perspectives on the Biological Role of Chemokine:Glycosaminoglycan Interactions. J Histochem Cytochem, 22155420977971. 10.1369/0022155420977971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dyer DP, Salanga CL, Volkman BF, Kawamura T, and Handel TM (2016). The dependence of chemokine-glycosaminoglycan interactions on chemokine oligomerization. Glycobiology 26, 312–326. 10.1093/glycob/cwv100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dyer DP, Migliorini E, Salanga CL, Thakar D, Handel TM, and Richter RP (2017). Differential structural remodelling of heparan sulfate by chemokines: the role of chemokine oligomerization. Open Biol 7, 160286. 10.1098/rsob.160286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lambaerts K, Wilcox-Adelman SA, and Zimmermann P (2009). The signaling mechanisms of syndecan heparan sulfate proteoglycans. Curr Opin Cell Biol 21, 662–669. 10.1016/j.ceb.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fox JM, Kausar F, Day A, Osborne M, Hussain K, Mueller A, Lin J, Tsuchiya T, Kanegasaki S, and Pease JE (2018). CXCL4/Platelet Factor 4 is an agonist of CCR1 and drives human monocyte migration. Sci Rep 8, 9466. 10.1038/s41598-018-27710-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heng TSP, Painter MW, and Consortium IGP (2008). The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 9, 1091–1094. 10.1038/ni1008-1091. [DOI] [PubMed] [Google Scholar]
- 16.Kouzeli A, Collins PJ, Metzemaekers M, Meyrath M, Szpakowska M, Artinger M, Struyf S, Proost P, Chevigne A, Legler DF, et al. (2020). CXCL14 Preferentially Synergizes With Homeostatic Chemokine Receptor Systems. Front Immunol 11, 561404. 10.3389/fimmu.2020.561404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dyer DP, Medina-Ruiz L, Bartolini R, Schuette F, Hughes CE, Pallas K, Vidler F, Macleod MKL, Kelly CJ, Lee KM, et al. (2019). Chemokine Receptor Redundancy and Specificity Are Context Dependent. Immunity 50, 378–389.e5. 10.1016/j.immuni.2019.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilliland CT, Salanga CL, Kawamura T, Trejo J, and Handel TM (2013). The chemokine receptor CCR1 is constitutively active, which leads to G protein-independent, β-arrestin-mediated internalization. J Biol Chem. 288, 32194–32210. 10.1074/jbc.m113.503797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liao Y, Wang J, Jaehnig EJ, Shi Z, and Zhang B (2019). WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res 47, W199–W205. 10.1093/nar/gkz401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Y-H, Narimatsu Y, Clausen TM, Gomes C, Karlsson R, Steentoft C, Spliid CB, Gustavsson T, Salanti A, Persson A, et al. (2018). The GAGOme: a cell-based library of displayed glycosaminoglycans. Nat Methods 15, 881–888. 10.1038/s41592-018-0086-z. [DOI] [PubMed] [Google Scholar]
- 21.Salanga CL, Dyer DP, Kiselar JG, Gupta S, Chance MR, and Handel TM (2014). Multiple glycosaminoglycan-binding epitopes of monocyte chemoattractant protein-3/CCL7 enable it to function as a non-oligomerizing chemokine. J Biol Chem 289, 14896–14912. 10.1074/jbc.m114.547737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang XX, Chen LL, Bancroft DPD, Lai CKC, and Maione TET (1994). Crystal structure of recombinant human platelet factor 4. Biochemistry 33, 8361–8366. 10.1021/bi00193a025. [DOI] [PubMed] [Google Scholar]
- 23.Rauova L, Zhai L, Kowalska MA, Arepally GM, Cines DB, and Poncz M (2006). Role of platelet surface PF4 antigenic complexes in heparin-induced thrombocytopenia pathogenesis: diagnostic and therapeutic implications. Blood 107, 2346–2353. 10.1182/blood-2005-08-3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rauova L, Poncz M, McKenzie SE, Reilly MP, Arepally G, Weisel JW, Nagaswami C, Cines DB, and Sachais BS (2005). Ultralarge complexes of PF4 and heparin are central to the pathogenesis of heparin-induced thrombocytopenia. Blood 105, 131–138. 10.1182/blood-2004-04-1544. [DOI] [PubMed] [Google Scholar]
- 25.Cai Z, Yarovoi SV, Zhu Z, Rauova L, Hayes V, Lebedeva T, Liu Q, Poncz M, Arepally G, Cines DB, et al. (2015). Atomic description of the immune complex involved in heparin-induced thrombocytopenia. Nat Commun 6, 8277. 10.1038/ncomms9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salanga CL, and Handel TM (2011). Chemokine oligomerization and interactions with receptors and glycosaminoglycans: the role of structural dynamics in function. Exp Cell Res 317, 590–601. 10.1016/j.yexcr.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narimatsu Y, Joshi HJ, Nason R, Coillie JV, Karlsson R, Sun L, Ye Z, Chen Y-H, Schjoldager KT, Steentoft C, et al. (2019). An Atlas of Human Glycosylation Pathways Enables Display of the Human Glycome by Gene Engineered Cells. Mol Cell 75, 394–407.e5. 10.1016/j.molcel.2019.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu D, and Esko JD (2014). Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem 83, 129–157. 10.1146/annurev-biochem-060713-035314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karlsson R, Chopra P, Joshi A, Yang Z, Vakhrushev SY, Clausen TM, Painter CD, Szekeres GP, Chen Y-H, Sandoval DR, et al. (2021). Dissecting structure-function of 3-O-sulfated heparin and engineered heparan sulfates. Sci Adv 7, eabl6026. 10.1126/sciadv.abl6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crijns H, Vanheule V, and Proost P (2020). Targeting Chemokine—Glycosaminoglycan Interactions to Inhibit Inflammation. Front Immunol 11, 907. 10.3389/fimmu.2020.00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Glass CA (2018). Recombinant Heparin-New Opportunities. Front Med (Lausanne) 5, 341. 10.3389/fmed.2018.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dyer DP (2020). Understanding the mechanisms that facilitate specificity, not redundancy, of chemokine mediated leukocyte recruitment. Immunology 290, 21292. 10.1111/imm.13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Affandi AJ, Carvalheiro T, Ottria A, de Haan JJ, Brans MAD, Brandt MM, Tieland RG, Lopes AP, Fernández BM, Bekker CPJ, et al. (2022). CXCL4 drives fibrosis by promoting several key cellular and molecular processes. Cell Reports 38, 110189. 10.1016/j.celrep.2021.110189. [DOI] [PubMed] [Google Scholar]
- 34.Lapchak PH, Ioannou A, Rani P, Lieberman LA, Yoshiya K, Kannan L, Lucca JJD, Kowalska MA, and Tsokos GC (2012). The role of platelet factor 4 in local and remote tissue damage in a mouse model of mesenteric ischemia/reperfusion injury. PLoS ONE 7, e39934. 10.1371/journal.pone.0039934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo L, Feng K, Wang YC, Mei JJ, Ning RT, Zheng HW, Wang JJ, Worthen GS, Wang X, Song J, et al. (2017). Critical role of CXCL4 in the lung pathogenesis of influenza (H1N1) respiratory infection. Mucosal Immunol 10, 1529–1541. 10.1038/mi.2017.1. [DOI] [PubMed] [Google Scholar]
- 36.Yue L, Pang Z, Li H, Yang T, Guo L, Liu L, Mei J, Song X, Xie T, Zhang Y, et al. (2018). CXCL4 contributes to host defense against acute Pseudomonas aeruginosa lung infection. PLoS ONE 13, e0205521. 10.1371/journal.pone.0205521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sachais BS, Turrentine T, McKenna JMD, Rux AH, Rader D, and Kowalska MA (2007). Elimination of platelet factor 4 (PF4) from platelets reduces atherosclerosis in C57Bl/6 and apoE−/− mice. Thromb Haemost 98, 1108–1113. 10.1160/th07-04-0271. [DOI] [PubMed] [Google Scholar]
- 38.Srivastava K, Cockburn IA, Swaim A, Thompson LE, Tripathi A, Fletcher CA, Shirk EM, Sun H, Kowalska MA, Fox-Talbot K, et al. (2008). Platelet Factor 4 Mediates Inflammation in Experimental Cerebral Malaria. Cell Host Microbe 4, 179–187. 10.1016/j.chom.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bdeir K, Gollomp K, Stasiak M, Mei J, Papiewska-Pajak I, Zhao G, Worthen GS, Cines DB, Poncz M, and Kowalska MA (2016). Platelet-specific Chemokines Contribute to the Pathogenesis of Acute Lung Injury. Am J Respir Cell Mol Biol 56, rcmb.2015–0245OC-270. 10.1165/rcmb.2015-0245oc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rainger GE, Chimen M, Harrison MJ, Yates CM, Harrison P, Watson SP, Lordkipanidzé M, and Nash GB (2015). The role of platelets in the recruitment of leukocytes during vascular disease. Platelets 26, 507–520. 10.3109/09537104.2015.1064881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Groom JR, Richmond J, Murooka TT, Sorensen EW, Sung JH, Bankert K, von Andrian UH, Moon JJ, Mempel TR, and Luster AD (2012). CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity 37, 1091–1103. 10.1016/j.immuni.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Girbl T, Lenn T, Perez L, Rolas L, Barkaway A, Thiriot A, del Fresno C, Lynam E, Hub E, Thelen M, et al. (2018). Distinct Compartmentalization of the Chemokines CXCL1 and CXCL2 and the Atypical Receptor ACKR1 Determine Discrete Stages of Neutrophil Diapedesis. Immunity. 10.1016/j.immuni.2018.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coombs C, Georgantzoglou A, Walker HA, Patt J, Merten N, Poplimont H, Busch-Nentwich EM, Williams S, Kotsi C, Kostenis E, et al. (2019). Chemokine receptor trafficking coordinates neutrophil clustering and dispersal at wounds in zebrafish. Nat Commun 10, 5166–17. 10.1038/s41467-019-13107-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Medina-Ruiz L, Bartolini R, Wilson GJ, Dyer DP, Vidler F, Hughes CE, Schuette F, Love S, Pingen M, Hayes AJ, et al. (2022). Analysis of combinatorial chemokine receptor expression dynamics using multi-receptor reporter mice. Elife 11, e72418. 10.7554/elife.72418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ridley AJ, Ou Y, Karlsson R, Pun N, Birchenough HL, Jowitt TA, Lawless C, Miller RL, and Dyer DP (2022). Chemokines form complex signals during inflammation and disease that can be decoded by extracellular matrix proteoglycans. Biorxiv, 2022.09.20.508420. 10.1101/2022.09.20.508420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Warda M, Toida T, Zhang F, Sun P, Munoz E, Xie J, and Linhardt RJ (2006). Isolation and characterization of heparan sulfate from various murine tissues. Glycoconj J 23, 555–563. 10.1007/s10719-006-7668-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baker AT, Boyd RJ, Sarkar D, Teijeira-Crespo A, Chan CK, Bates E, Waraich K, Vant J, Wilson E, Truong CD, et al. (2021). ChAdOx1 interacts with CAR and PF4 with implications for thrombosis with thrombocytopenia syndrome. Sci Adv 7, eabl8213. 10.1126/sciadv.abl8213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yates EA, Santini F, Guerrini M, Naggi A, Torri G, and Casu B (1996). 1H and 13C NMR spectral assignments of the major sequences of twelve systematically modified heparin derivatives. CARBOHYDRATE RESEARCH 294, 15–27. 10.1016/s0008-6215(96)90611-4. [DOI] [PubMed] [Google Scholar]
- 49.Dixon AS, Schwinn MK, Hall MP, Zimmerman K, Otto P, Lubben TH, Butler BL, Binkowski BF, Machleidt T, Kirkland TA, et al. (2016). NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. Acs Chem Biol 11, 400–408. 10.1021/acschembio.5b00753. [DOI] [PubMed] [Google Scholar]
- 50.D’Uonnolo G, Reynders N, Meyrath M, Abboud D, Uchański T, Laeremans T, Volkman BF, Janji B, Hanson J, Szpakowska M, et al. (2022). The Extended N-Terminal Domain Confers Atypical Chemokine Receptor Properties to CXCR3-B. Front Immunol 13, 868579. 10.3389/fimmu.2022.868579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chevigné A, Janji B, Meyrath M, Reynders N, D’Uonnolo G, Uchański T, Xiao M, Berchem G, Ollert M, Kwon Y-J, et al. (2021). CXCL10 Is an Agonist of the CC Family Chemokine Scavenger Receptor ACKR2/D6. Cancers 13, 1054. 10.3390/cancers13051054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goldey GJ, Roumis DK, Glickfeld LL, Kerlin AM, Reid RC, Bonin V, Schafer DP, and Andermann ML (2014). Removable cranial windows for long-term imaging in awake mice. Nature Protocols 9, 2515–2538. 10.1038/nprot.2014.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoon J-H, Shin P, Joo J, Kim GS, Oh W-Y, and Jeong Y (2022). Increased capillary stalling is associated with endothelial glycocalyx loss in subcortical vascular dementia. J Cereb Blood Flow Metabolism, 0271678X2210765. 10.1177/0271678x221076568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pai S, Qin J, Cavanagh L, Mitchell A, El-Assaad F, Jain R, Combes V, Hunt NH, Grau GER, and Weninger W (2014). Real-time imaging reveals the dynamics of leukocyte behaviour during experimental cerebral malaria pathogenesis. PLoS Pathog 10, e1004236. 10.1371/journal.ppat.1004236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tape CJ, Worboys JD, Sinclair J, Gourlay R, Vogt J, McMahon KM, Trost M, Lauffenburger DA, Lamont DJ, and Jørgensen C (2014). Reproducible Automated Phosphopeptide Enrichment Using Magnetic TiO2 and Ti-IMAC. Anal Chem 86, 10296–10302. 10.1021/ac5025842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tyanova S, Temu T, and Cox J (2016). The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc 11, 2301–2319. 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 57.Consortium TU, Bateman A, Martin M-J, Orchard S, Magrane M, Agivetova R, Ahmad S, Alpi E, Bowler-Barnett EH, Britto R, et al. (2020). UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49, D480–D489. 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13, 731–740. 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 59.Thakar D, Migliorini E, Coche-Guerente L, Sadir R, Lortat-Jacob H, Boturyn D, Renaudet O, Labbe P, and Richter RP (2014). A quartz crystal microbalance method to study the terminal functionalization of glycosaminoglycans. Chem. Commun. (Camb.) 50, 15148–15151. 10.1039/c4cc06905f. [DOI] [PubMed] [Google Scholar]
- 60.Schuck P (2000). Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78, 1606–1619. 10.1016/s0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brautigam CA (2015). Calculations and Publication-Quality Illustrations for Analytical Ultracentrifugation Data. Meth Enzymol 562, 109–133. 10.1016/bs.mie.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 62.Jasnin M, van Eijck L, Koza MM, Peters J, Laguri C, Lortat-Jacob H, and Zaccai G (2010). Dynamics of heparan sulfate explored by neutron scattering. Phys Chem Chem Phys 12, 3360–3362. 10.1039/b923878f. [DOI] [PubMed] [Google Scholar]
- 63.Migliorini E, Thakar D, Sadir R, Pleiner T, Baleux F, Lortat-Jacob H, Coche-Guerente L, and Richter RP (2014). Well-defined biomimetic surfaces to characterize glycosaminoglycan-mediated interactions on the molecular, supramolecular and cellular levels. Biomaterials 35, 8903–8915. 10.1016/j.biomaterials.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 64.Migliorini E, Thakar D, Kühnle J, Sadir R, Dyer DP, Li Y, Sun C, Volkman BF, Handel TM, Coche-Guerente L, et al. (2015). Cytokines and growth factors cross-link heparan sulfate. Open Biol 5, 150046. 10.1098/rsob.150046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Richter RP, Bérat R, and Brisson AR (2006). Formation of Solid-Supported Lipid Bilayers: An Integrated View. Langmuir 22, 3497–3505. 10.1021/la052687c. [DOI] [PubMed] [Google Scholar]
- 66.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.