

Abstract
Suppression of the extensor H-reflex by flexor afferent conditioning is thought to be produced by a long-lasting inhibition of extensor Ia-afferent terminals via GABAA receptor-activated primary afferent depolarization (PAD). Considering the recent finding that PAD does not produce presynaptic inhibition of Ia-afferent terminals, we examined in 28 participants if H-reflex suppression is instead mediated by post-activation depression of the extensor Ia-afferents triggered by PAD-evoked spikes and/or by a long-lasting inhibition of the extensor motoneurons. A brief conditioning vibration of the flexor tendon suppressed both the extensor soleus H-reflex and the tonic discharge of soleus motor units out to 150 ms following the vibration, suggesting that part of the H-reflex suppression during this time was mediated by postsynaptic inhibition of the extensor motoneurons. When activating the flexor afferents electrically to produce conditioning, the soleus H-reflex was also suppressed but only when a short-latency reflex was evoked in the soleus muscle by the conditioning input itself. In mice, a similar short-latency reflex was evoked when optogenetic or afferent activation of GABAergic (GAD2+) neurons produced a large enough PAD to evoke orthodromic spikes in the test Ia-afferents, causing post-activation depression of subsequent monosynaptic excitatory postsynaptic potentials. The long duration of this post-activation depression and related H-reflex suppression (seconds) was similar to rate-dependent depression that is also due to post-activation depression. We conclude that extensor H-reflex inhibition by brief flexor afferent conditioning is produced by both post-activation depression of extensor Ia-afferents and long-lasting inhibition of extensor motoneurons, rather than from PAD inhibiting Ia-afferent terminals.
Keywords: presynaptic inhibition, GABA, Ia afferents
Graphical Abstract
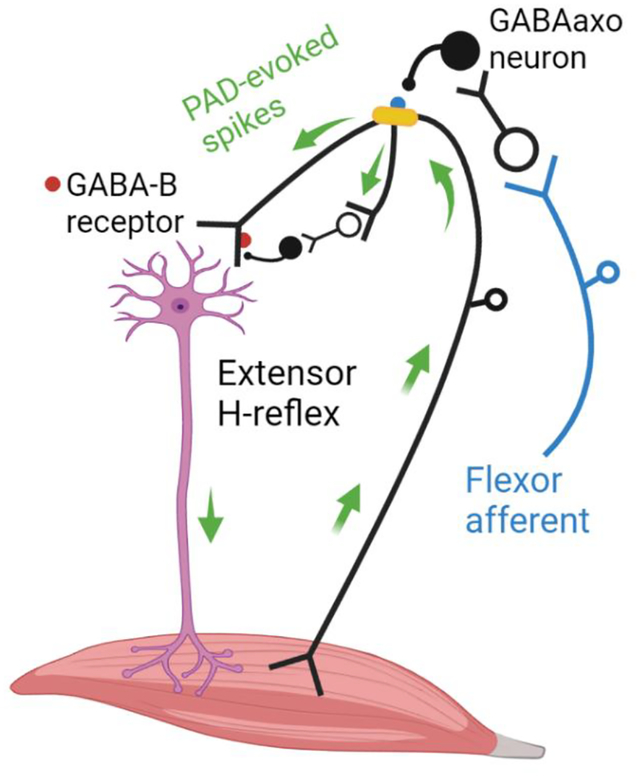
We propose that stimulation of flexor afferents activate GABAergic interneurons (GABAaxo neuron) with axoaxonic connections to or near dorsal nodes of Ranvier (yellow) in extensor Ia afferents. If large enough this nodal PAD, mediated by GABAA receptors (blue), will activate spikes in the extensor Ia afferent (top green arrow) that travel to the extensor motoneuron to produce an excitatory postsynaptic potential and post-activation depression of subsequent H-reflexes evoked by direct extensor Ia afferents. We also hypothesize that PAD-evoked spikes in other branches of the extensor Ia afferent (bottom green arrow) activate more ventrally located GABAaxo interneurons with projections to the Ia afferent terminal where GABAB receptors (red) are activated to also produce post-activation depression of subsequent extensor H-reflexes.
Introduction
The suppression of the H-reflex following a conditioning stimulation to an antagonist nerve was until recently thought to be mediated by the activation of a primary afferent depolarization (PAD) in the Ia afferent terminal (Hultborn et al., 1987a; Stein, 1995; Misiaszek, 2003; Hultborn, 2006). Since the 1960’s, PAD was thought to be activated at the Ia afferent terminal by GABAA receptors, resulting in a shunting of terminal currents, depression of neurotransmitter release and ultimately, a reduction in the excitatory postsynaptic potential (EPSP) evoked in the motoneuron, as reviewed in (Willis, 2006). However, the role of PAD and GABAA receptors in producing presynaptic inhibition in the Ia afferent terminal has recently been disproven by multiple lines of evidence (Hari et al., 2022). For example, there is a sparsity of GABAA receptors on the Ia afferent terminal (Alvarez et al., 1996; Hari et al., 2022) and correspondingly, PAD measured at the unmyelinated Ia afferent terminal is small-to-nonexistent and brief, lasting for only 20 ms compared to more proximal, myelinated regions of the Ia afferent that contain nodes of Ranvier where PAD lasts for 100–200 ms (Lucas-Osma et al., 2018). Further, computer simulations confirm that PAD at the Ia afferent terminal is too weak and brief to produce a physiologically relevant decrease in action potential size to reduce EPSP activation in the motoneuron (Hari et al., 2022). In contrast, Ia afferent terminals are densely covered by GABAB receptors that contribute to terminal presynaptic inhibition (Curtis & Lacey, 1994; Fink, 2013; Hari et al., 2022).
Instead of presynaptic inhibition, the activation of PAD by GABAA receptors facilitate sodium channels at the nodes of Ranvier in Ia afferents, helping spike propagation through branch points to facilitate motoneuron EPSPs (Hari et al., 2022; Metz et al., 2022). This is mediated by axoaxonic connections from GABAergic neurons (GABAaxo neurons) onto GABAA receptors at or near nodes producing a PAD that brings sodium spikes closers to threshold (Hari et al., 2022). Despite its minor influence at the Ia afferent terminal, GABAA receptors have repeatedly been shown to contribute to the inhibition of the monosynaptic reflex in extensor motoneurons following conditioning of an antagonist flexor nerve to evoke PAD, since this inhibition is partly decreased by GABAA receptor blockers (Eccles et al., 1963; Stuart & Redman, 1992; Curtis & Lacey, 1994; Curtis, 1998). This raises the question of how can the activation of GABAA receptors be inhibitory in some instances (i.e., reduce reflexes) and excitatory in other (i.e., facilitate afferent nodal conduction to facilitate reflexes), and how is this related to the observations that the extensor H-reflex is commonly inhibited by an antagonist afferent conditioning stimulation (Mizuno et al., 1971; El-Tohamy & Sedgwick, 1983; Berardelli et al., 1987; Hultborn et al., 1987a; Nakashima et al., 1990; Burke et al., 1992; Capaday et al., 1995; Faist et al., 1996; Iles, 1996; Aymard et al., 2000; Howells et al., 2020) and at other times, the same H-reflex is facilitated by this conditioning stimulation (Hari et al., 2022; Metz et al., 2022)?
One intriguing possibility recently proposed is that PAD can induce post-activation depression in the Ia afferent terminals mediating the H-reflex (Hari et al., 2022) as another type of presynaptic inhibition. Here, we broadly define post-activation depression as a reduction in a monosynaptic EPSP caused by a prior activation of the same EPSP, where the first and second EPSPs are evoked by the same Ia afferent population each time, regardless of how the afferents are activated. Post-activation depression can be caused by numerous mechanisms, including the depletion of transmitter packaging and release in afferent terminals following the first EPSP [proposed by (Curtis & Eccles, 1960a) and measured in the calyx of Held (Neher & Sakaba, 2001)], decreased afferent excitability from the collision of dorsal root reflexes and the second test Ia volley (Eccles et al., 1961a), and even inhibition of the afferent terminals by GABAB receptors indirectly activated by the test afferents that activated the first EPSP (i.e., GABAB receptor-mediated presynaptic inhibition) (Curtis & Lacey, 1994, 1998).
But how would PAD evoked from a conditioning stimulation to an antagonist afferent produce an EPSP in the agonist motoneurons and subsequent post-activation depression of Ia-EPSPs and related H-reflexes? Superficially this seems contrary to the conventional understanding that antagonist afferents generally produce reciprocal postsynaptic inhibition of agonist motoneurons (Sherrington, 1908). However, it is well known that PAD can produce antidromic action potentials in sensory axons that are recorded as dorsal root reflexes in the proximal afferent (termed dorsal root reflexes, DRRs), and PAD is strongest when evoked in extensor Ia afferents by antagonist flexor afferent conditioning (Eccles et al., 1961b; Rudomin & Schmidt, 1999). Further, since the early work of Barron and Matthews, it was recognized that PAD in Ia afferents can also evoke orthodromic action potentials, as evidenced by the generation of motoneuron EPSPs from these PAD-evoked spikes (Barron & Matthews, 1938; Frank, 1959; Eccles et al., 1961a; Duchen, 1986; Willis, 1999). Theoretically, this orthodromic action potential evoked in the Ia afferent terminal should reduce subsequent neurotransmitter release for seconds via post-activation depression and thus, may result in a depression of subsequent EPSPs evoked by directly stimulating this same Ia afferent, just as the motoneuron EPSP is reduced by direct, repetitive activation of the Ia afferent. The latter presynaptic EPSP depression with repetitive activation of the Ia afferent is termed rate dependent depression (RDD) and is caused by post-activation depression of the Ia afferent (Hultborn et al., 1996). This led us to speculate that H-reflexes following a conditioning stimulation of an antagonist nerve may be suppressed if the antagonist afferents activate PAD and orthodromic action potentials in the Ia afferents mediating the H-reflex, as suggested for the suppression of motoneuron EPSPs in the rodent when followed by PAD-evoked spikes [(Duchen, 1986; Hari et al., 2022), see Abstract figure]. To examine this, we first sought confirmation in mice that when orthodromic action potentials are activated in the Ia afferent from PAD circuits (PAD-evoked spikes) that then activate an EPSP in the motoneuron, subsequent direct stimulation of the same Ia afferent produces a smaller EPSP in the motoneuron when tested within the time window of post-activation depression and related RDD (between 20 ms to 2 s), which can last up to 5–10 s in both humans and rodents (Schindler-Ivens & Shields, 2000; Boulenguez et al., 2010). For this, we activated PAD either through optogenetic activation of specific GABA neurons with axoaxonic connections to Ia afferents and their nodes (i.e., light activation of GAD2+ neurons in GAD2-cre//ChR2 mice), or from a separate, heteronymous sensory pathway (Hari et al., 2022). The former optogenetic method is especially powerful because GAD2+ interneurons have both monosynaptic axoaxonic connections onto Ia afferents (termed GABAaxo interneurons) and monosynaptic contacts onto the dendrites of the motoneurons (Pierce & Mendell, 1993; Hughes et al., 2005). Thus, one would expect two events from an isolated activation of the triadic GAD2+ interneurons with light, a monosynaptic IPSP from the direct connection onto the motoneuron and/or a disynaptic EPSP in the motoneuron from the axoaxonic activation of PAD and Ia spikes that then orthodromically activate the motoneuron at a monosynaptic latency. The fact we observe short latency Ia-EPSPs that are evoked by light activation of the GAD2+ interneurons unequivocally demonstrates that PAD-evoked Ia spikes drive these EPSPs.
We also examined in humans if a conditioning stimulation of the antagonist flexor nerve to the extensor soleus muscle, the common peroneal nerve (CPN, also known as the common fibular nerve), could likewise produce a short latency response in the soleus muscle consistent with the activation of an orthodromic, PAD-evoked spike in the soleus Ia afferent, as directly shown in sural and median afferents with near-nerve, extra-axonal recordings (Shefner et al., 1992). We then examined if the suppression of subsequently activated H-reflexes was related to the presence or size of this putative PAD-evoked reflex at stimulation delays previously attributed to the time-course of PAD-mediated presynaptic inhibition (30 to 400 ms). We also examined this antagonist-evoked H-reflex suppression at much longer delays within the post-activation depression window (500 ms to 2500 ms). The profile of the soleus H-reflex suppression by the flexor afferent conditioning was compared to the profile of RDD, and associated post-activation depression, produced from repeated and direct activation of the soleus Ia afferents mediating the H-reflex at similar delays. We hypothesized that a similar long-lasting profile of H-reflex suppression from CPN conditioning or RDD would be consistent with both being mediated by post-activation depression of the Ia afferents.
A more obvious, but rarely measured, mechanism of long-lasting (300 – 400 ms) H-reflex suppression by antagonist conditioning stimulation is postsynaptic inhibition of the motoneuron given the well-known (but brief) glycinergic, Ia-reciprocal postsynaptic inhibition on the motoneuron and the less well-known inhibition caused by GABA (Stuart & Redman, 1992), where ~70% of the GABAergic interneurons with projections to afferent terminals (i.e., GABAaxo) also have projections onto the postsynaptic motoneuron (Pierce & Mendell, 1993; Hughes et al., 2005). These triadic GABAaxo interneurons may produce long lasting postsynaptic inhibition as they are activated for ~ 200 ms by conditioning inputs, as evidenced by the long duration of PAD they evoke in the Ia afferent (Hari et al., 2022; Lin et al., 2022). Thus, we first examined if a brief conditioning stimulation of antagonist flexor afferents directly inhibits the motoneuron with a time course similar to the long-lasting suppression of the H-reflex previously attributed to PAD. Inhibition of the soleus H-reflex from a brief vibration to the antagonist tibialis anterior (TA) tendon, or from percutaneous electrical stimulation to the common peroneal nerve (CPN), was measured at interstimulus intervals (ISIs) between 0 and 500 ms. Direct effects of the conditioning stimulation onto the test motoneuron(s) were measured from changes in the firing rate of tonically discharging single motor units in response to the conditioning stimulation alone [see also (Metz et al., 2022)]. Changes in the firing rate of the tonically discharging motor units were taken as evidence of a postsynaptic effect on the motoneuron (Powers & Binder, 2001). We hypothesized that if the profile of suppression in the firing rate of the motor unit was similar to the profile of inhibition of the H-reflex from the same conditioning stimulation, then postsynaptic inhibition of the test motoneuron mediated a part of the H-reflex inhibition.
Parts of the data from the human vibration experiments have been published (Hari et al., 2022) and are expanded upon here.
Methods
Ethical approvals
Human experiments were approved by the Human Research Ethics Board at the University of Alberta (Protocol 00078057), conformed to the Declaration of Helsinki, and conducted with informed consent of the participants. All experimental procedures were approved by the University of Alberta Animal Care and Use Committee, Health Sciences division (ACUC protocol nos. AUP00000224 and AUP00002891) in accordance with Canadian Council on Animal Care guidelines. Animals were caged in groups of 2–4 and maintained on a 12-hour light/dark cycle, in stable conditions of temperature and humidity, with food and water ad libitum. We understand the ethical principles under which the Journal of Physiology operates, and this work complies with the animal ethics standards outlined in their website.
Human participants and animals
Our sample comprised of 28 participants (11 male) with no known neurological injury or disease, ranging in age from 20 to 58 years [31.6 (13.9), mean (standard deviation]. Thirteen of the participants in this study also participated in the related (Metz et al., 2022) study. In vitro recordings were made from adult mice (2.5 – 6.0 months old, both female and male equally) without (control) and with Cre expressed under the endogenous Gad2 promotor region (Gad2tm1(cre/ERT2)Zjh mice abbreviated to Gad2-CreER mice, The Jackson Laboratory, Stock # 010702) as per Hari et al., 2022. These GAD2-CreER mice were crossed with mice containing flx-stop-flx-ChR2 expressed under the ubiquitous promotor Rosa, to yield mice with GAD2+ neurons expressing light sensitive ChR2 after they were injected with tamoxofin [abbreviated Gad2//ChR2 mice, as detailed in (Hari et al., 2022)]. In vitro and vivo recordings were made from adult rats (3 – 8 months old, female only, Sprague-Dawley) as per (Hari et al., 2022).
Animal experimental setup
In vitro recordings in mice and rats
Rodents were anaesthetized with urethane (for mice 0.11 g/100 g, with a maximum dose of 0.065 g and for rats, 0.18 g per 100 g, with a maximum dose of 0.45 g), a laminectomy was performed, and then the entire sacrocaudal spinal cord was rapidly removed and immersed in oxygenated modified artificial cerebrospinal fluid (mACSF), as detailed previously (Lin et al., 2019). After the spinal cord is extracted, animals are euthanized with an overdose of Euthansol® (sodium pentobarbital, 340mg/ml mice, 240mg/ml rats, i.p. injection). Dorsal and ventral roots (DR and VR) were mounted on silver-silver chloride wires above the nASCF of the recording chamber and covered with grease for monopolar stimulation and recording [see (Hari et al., 2022) for details]. Dorsal roots were stimulated with a constant current stimulator (Isoflex, Israel) with short pulses (0.1 ms) at 1.1 – 1.5 × threshold (T) to specifically activate proprioceptive afferents to evoke both PAD in Ia afferents and monosynaptic EPSPs in motoneurons. This grease gap method was also used to record the composite intracellular response of many sensory axons or motoneurons where the high impedance seal on the dorsal or ventral roots reduces extracellular currents, allowing the recording to reflect intracellular potentials (Lüscher et al., 1979; Leppanen & Stys, 1997; Lucas-Osma et al., 2018). Return and ground wires were placed in the bath and likewise made of silver-silver chloride. Specifically for sensory axons, we recorded from the central ends of dorsal roots cut within about 2 – 4 mm of their entry into the spinal cord, to give the compound potential from all afferents in the root (dorsal root potential, DRP), which has previously been shown to correspond to PAD (Lucas-Osma et al., 2018). The dorsal root recordings were amplified (2,000 times), high-pass filtered at 0.1 Hz to remove drift, low-pass filtered at 10 kHz, and sampled at 30 kHz (Axoscope 8; Axon Instruments/Molecular Devices, Burlingame, CA). These grease gap recordings of PAD on sensory afferents reflect only the response of the largest diameter axons in the dorsal root, mainly group I proprioceptive afferents, as detailed previously (Hari et al., 2022). The composite EPSPs in many motoneurons were likewise recorded from the central cut end of ventral roots mounted in the grease gap, which has also previously been shown to yield reliable estimates of the EPSPs (Fedirchuk et al., 1999). The EPSPs were identified as monosynaptic by their rapid onset (first component, ~1 ms after afferent volley arrives in the ventral horn), lack of variability in latency (< 1 ms jitter), persistence at high rates (10 Hz) and appearance in isolation at the threshold for DR stimulation (< 1.1xT), unlike polysynaptic reflexes which vary in latency, disappear at high rates, and mostly need stronger DR stimulation to activate.
Light was used to evoke PAD in the GAD2//ChR2 mice and dorsal root stimulation was used to evoke PAD in control mice as described previously (Lin et al., 2019). Light was derived from a laser with a 447 nm wavelength (D442001FX lasers from Laserglow Technologies, Toronto) and was passed through a fibre optic cable (MFP_200/220/900–0.22_2m_FC-ZF1.25, Doric Lenses, Quebec City). A half cylindrical prism, the length of about two spinal segments (8 mm; 3.9 mm focal length, Thor Labs, Newton, USA,) collimated the light into a narrow long beam (200 μm wide and 8 mm long). This narrow beam was focused longitudinally on the left side of the spinal cord roughly at the level of the dorsal horn, to target the epicentre of GABAaxo neurons, which are dorsally located. ChR2 rapidly depolarizes neurons (Zhang et al., 2011), and thus we used 5 – 10 ms light pulses to activate GABAaxo neurons, as confirmed by direct recordings from these neurons (Hari et al., 2022). Light was always kept at a minimal intensity, 1.1x T, where T is the threshold to evoke a light response in sensory axons, which made local heating from light unlikely.
Intracellular recordings of Ia afferent branches in the dorsal horn of rats were performed as in (Hari et al., 2022). Briefly, glass capillary tubes (1.5 mm and 0.86 mm outer and inner diameters, respectively; with filament; 603000 A-M Systems; Sequim, USA) with a bevelled hypodermic-shaped point of < 100 nm, were filled through their tips with 1 M K-acetate and 1 M KCl. Intracellular recording and current injection were performed with an Axoclamp2B amplifier (Axon Inst. and Molecular Devices, San Jose, USA). Recordings were low pass filtered at 10 kHz and sampled at 30 kHz (Clampex and Clampfit; Molecular Devices, San Jose, USA). Electrodes were advanced into myelinated afferents of the sacrocaudal spinal cord with a stepper motor (Model 2662, Kopf, USA, 10 μm steps at maximal speed, 4 mm/s), usually at the boundary between the dorsal columns and dorsal horn gray matter. Upon penetration, afferents were identified with direct orthodromic spikes evoked from DR stimulation. The lowest threshold proprioceptive group Ia afferents were identified by their direct response to DR stimulation, very low threshold (< 1.5 × T, T: afferent volley threshold), short latency (group Ia latency, coincident with onset of afferent volley), and antidromic response to micro stimulation of the afferent terminal in the ventral horn [~ 10 μA stimulation via tungsten microelectrode as per (Lucas-Osma et al., 2018)]. Post hoc these were confirmed to be large proprioceptive Ia afferents by their unique extensive terminal branching around motoneurons, unlike large cutaneous Aβ afferents that do not project to the ventral horn. Clean axon penetrations without injury occurred abruptly with a sharp pop detected on speakers attached to the recorded signal, the membrane potential settling rapidly to near - 70 mV, and > 70 mV spikes usually readily evoked by DR stimulation or brief current injection pulses (1 – 3 nA, 20 ms, 1 Hz). Sensory axons also had a characteristic >100 ms long depolarization following stimulation of a dorsal root (primary afferent depolarization, PAD, at 4 – 5 ms latency) and short spike afterhyperpolarization (AHP ~ 10 ms), which further distinguished them from other axons or neurons. Injured axons had higher resting potentials (> - 60 mV), poor spikes (< 60 mV) and low resistance (to current pulse; Rm < 10 MΩ) and were discarded.
In vivo recordings in rats
The influence of evoking PAD in Ia afferents on the monosynaptic reflex (MSR) was performed in awake rats with percutaneous tail EMG recording and nerve stimulation as per (Hari et al., 2022). On the day of experimentation, the rat was housed in a plexiglass tube with the tail protruding out one end and was briefly sedated with isoflurane (1.5%). Fine stainless steel wires (AS 613, Cooner Wire) were percutaneously implanted in the tail for recording EMG and stimulating the caudal tail trunk nerve (wires de-insulated by 2 mm at their tip and inserted in the core of a 27-gauge needle that was removed after insertion. Here, PAD was evoked by a cutaneous conditioning stimulation of the tip of the tail (0.2 ms pulses, 3xT, 40 – 120 ms prior to MSR testing) using an additional pair of fine Cooner wires implanted at the tip of the tail (separated by 8 mm). In rats the MSR latency is later than in mice due to the larger peripheral conduction time, ~12 ms (as again confirmed by a similar latency to the F wave). This MSR was thus quantified by averaging the rectified EMG over a 12 – 20 ms window. Also, to confirm the GABAA receptor involvement in regulating the MSR, the inverse agonist (blocker) L655708 was injected systemically (1 mg/kg i.p., dissolved in 50 μl DMSO and diluted in 900 μl saline). Again, the MSR was tested at matched background EMG levels before and after conditioning (or L655708 application) to rule out changes in postsynaptic inhibition. Following reflex recordings, all percutaneous wires were removed, and animals were brought back to their home cage.
Human experimental setup
Participants were seated in a reclined, supine position on a padded table. The right leg was bent slightly to access the popliteal fossa and padded supports were added to facilitate complete relaxation of all leg muscles because descending activation could potentially activate GABAaxo circuits within the spinal cord (Jankowska et al., 1981b; Rudomin, 1990; Eguibar et al., 1997; Ueno et al., 2018). During H-reflex recordings measured at rest, participants were asked to relax completely with no talking, hand or arm movements.
Surface EMG recordings
A pair of Ag-AgCl electrodes (Kendall; Chicopee, MA, USA, 3.2 cm by 2.2 cm) was used to record surface EMG from the soleus and tibialis anterior (TA) muscles with a ground electrode placed just below the knee. The EMG signals were amplified by 1000 and band-pass filtered from 10 to 1000 Hz (Octopus, Bortec Technologies; Calgary, AB, Canada) and then digitized at a rate of 5000 Hz using Axoscope 10 hardware and software (Digidata 1400 Series, Axon Instruments, Union City, CA). To examine the postsynaptic effects of the conditioning inputs alone on the soleus motoneurons, surface EMG electrodes were also used to record single motor units during weak contractions by placing the surface electrodes on the lateral border of the muscle as done previously (Matthews, 1996). Single motor units were also recorded using a high-density surface EMG electrode (HDsEMG, OT Bioelettronica, Torino, Italy, Semi-disposable adhesive matrix, 64 electrodes, 5×13, 8 mm inter-electrode distance) with ground and differential electrodes wrapped around the ankle and/or below the knee. Signals were amplified (150 times), filtered (10 to 900 Hz) and digitized (16 bit at 5120 Hz) using the Quattrocento Bioelectrical signal amplifier and OTBioLab+ v.1.2.3.0 software (OT Bioelettronica, Torino, Italy). The EMG signal was decomposed into single motor units using custom MatLab software as per (Negro et al., 2016; Afsharipour et al., 2020).
Nerve stimulation to evoke an H-reflex
The tibial nerve (TN) was stimulated in a bipolar arrangement using a constant current stimulator (1-ms rectangular pulse width, Digitimer DS7A, Hertfordshire, UK) to evoke an H-reflex in the soleus muscle. After searching for the TN with a probe to evoke a pure plantarflexion, an Ag-AgCl electrode (cathode: Kendall; Chicopee, MA, USA, 2.2 cm by 2.2 cm) was placed in the popliteal fossa, with the anode (Axelgaard; Fallbrook, CA, USA, 5 cm by 10 cm) placed on the patella. Stimulation intensity was set to evoke a test H-reflex of approximately half maximum on the ascending phase of the H-reflex recruitment curve to allow for both facilitatory and inhibitory effects of the conditioning input to be revealed (Crone et al., 1990). H-reflexes recorded at rest were evoked every 5 seconds to minimize post-activation depression from RDD (Hultborn et al., 1996) and at least 10 test H-reflexes were evoked before conditioning to establish a steady baseline. All H-reflexes were recorded at rest.
Antagonist TA tendon vibration
In 8 participants, the soleus H-reflex was conditioned by a prior vibration of the TA tendon (3 pulses, 200 Hz) to preferentially activate Ia afferents as done previously (Hultborn et al., 1987a). A 7 mm diameter probe attached to an audio amplifier was pressed gently against the TA tendon at the base of the leg. We ensured that the amplitude of the vibration and the amount of pressure applied to the TA tendon was below the threshold to facilitate the H-reflexes over the tested 500 ms ISI intervals (Hultborn et al., 1987a). Approximately 10 baseline soleus H-reflexes were elicited to ensure the H-reflex was stable, followed by 7 conditioned H-reflexes at one of the following ISIs (0, 30, 60, 100, 150, 200, 300, 400 or 500 ms). Following this, 3 to 4 unconditioned H-reflexes were evoked to reestablish baseline and another run of 7 conditioned H-reflexes was applied at a randomly chosen interval. This was repeated until all ISI intervals were applied (Fig. 1A).
Figure 1: TA tendon vibration.
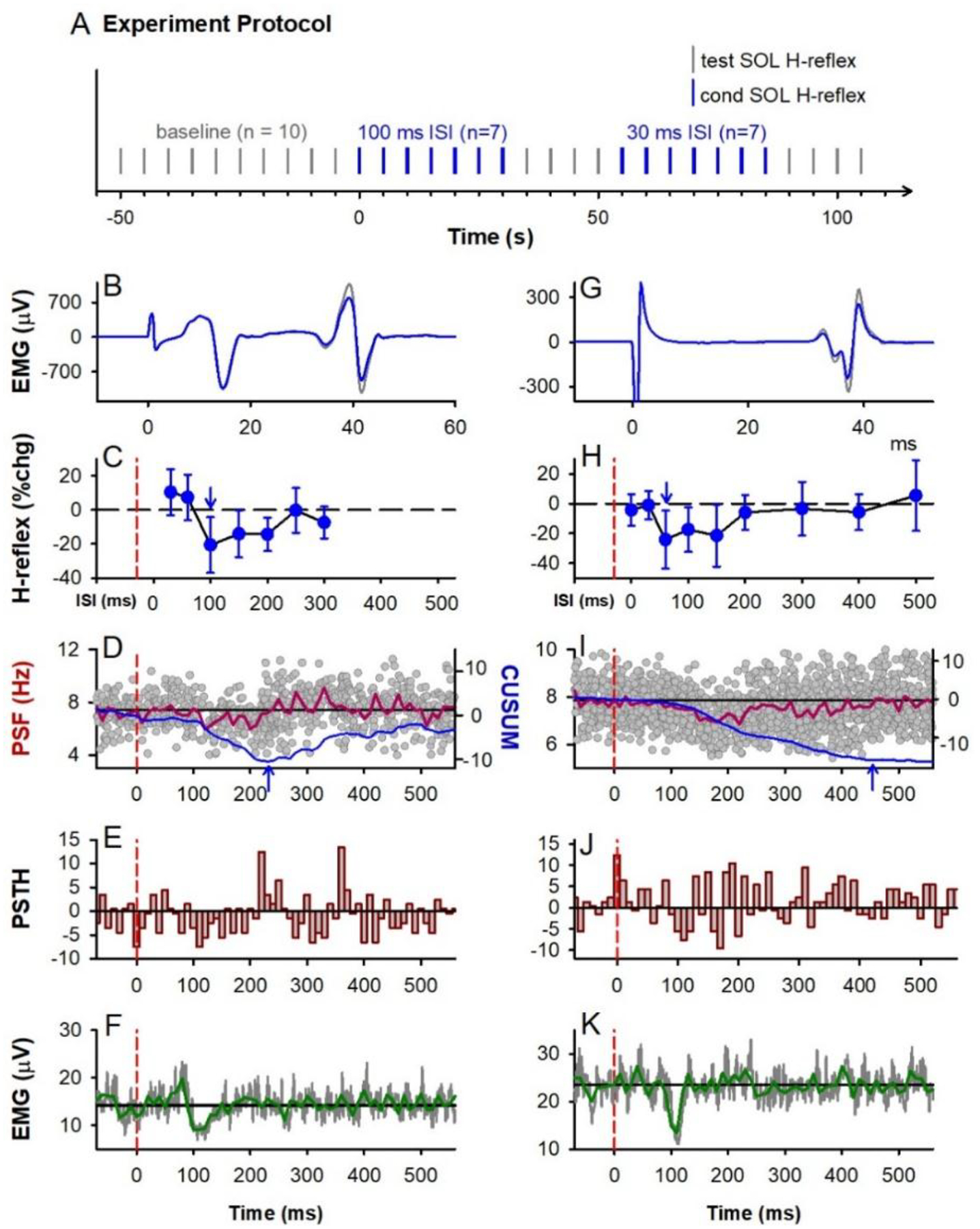
A) General experiment schematic: vertical bars mark application of unconditioned test (grey) and conditioned (blue) H-reflexes to the soleus (SOL) muscle measured at rest, evoked every 5 s with random application of the various conditioning ISIs interposed with a run of test H-reflexes. B-F & G-K: Representative data from 2 participants. B,G) Average of 7 test (grey) and 7 conditioned (blue) soleus H-reflexes (unrectified EMG) at the 100 ms (B) and 60 ms (G) ISI respectively. TN stimulation to evoke H-reflex applied at 0 ms. C,H) % change (chg) of the soleus H-reflex (mean ± SD) plotted at each ISI with peak % change indicated by the downward arrow. Time of vibration onset indicated by red dashed line, occurring to the left of the 0 ms ISI by the latency of H-reflex. D,I) Superimposed firing rates (grey dots, PSF) of soleus motor units in response to TA tendon vibration alone (185 and 340 sweeps respectively), with a 10-ms binned average rate (red line, mean PSF) and mean PSF CUSUM (blue line). E,J) Post-stimulus time histogram (PSTH) showing motor unit count per each 10 ms bin with mean pre-vibration count subtracted. F,K) Rectified soleus EMG (grey lines, 31 and 68 sweeps respectively), with 10-ms binned average (green line). Vertical dashed red lines in C to K mark onset of TA tendon vibration (at time 0). Horizontal black lines mark the average pre-vibration values (from −100 to 0 ms). Parts of G, H, I and K were adapted from Hari et al., 2022.
Modulation in the tonic firing rate of single soleus motor units was used to determine if the TA tendon vibration had any postsynaptic actions on the soleus motoneurons. A small voluntary isometric contraction (~5% of maximum) was used to produce a steady discharge of the single unit(s) using both auditory and visual feedback, while the vibration was delivered every 3 seconds. Seven units from 5 participants were isolated from the surface EMG and 12 units from 3 participants were decomposed from the HDsEMG. Modulation of the whole-muscle surface EMG was also measured while participants held a stronger isometric voluntary contraction at approximately 10% of maximum.
Antagonist CPN stimulation
In a separate experiment on a different day in 13 participants (2 participants from the vibration experiment), the CPN supplying the antagonist TA muscle was stimulated to condition the ipsilateral soleus H-reflex as done previously (Mizuno et al., 1971; El-Tohamy & Sedgwick, 1983; Capaday et al., 1995). The CPN was stimulated using a bipolar arrangement (Ag-AgCl electrodes, Kendall; Chicopee, MA, USA, 2.2 cm by 2.2 cm) with the anode placed anterior and slightly distal to the fibular head on the right leg and the cathode 2–3 cm more proximal. Care was taken to elicit a pure dorsiflexion response without foot eversion. Three pulses (200 Hz, 1-ms pulse width) were applied to the CPN at an intensity of 1.0 and 1.5 × motor threshold (MT) at the 3, 15, 30, 60, 80, 100, 150, 200, 300 and 400 ms ISIs using a similar protocol as in the vibration experiment (Fig. 1A). Motor threshold was determined by the lowest-intensity, single-pulse stimulation of the CPN that produced a small but reproducible direct motor response (M wave, > 10 μV and < 50 μV peak-to-peak) in the TA muscle at rest. In a separate trial, CPN stimulation was applied at longer ISI intervals (500, 1000, 1500, 2000 and 2500 ms) at both the 1.0 and 1.5 × MT stimulation intensities with the same protocol as for the shorter ISIs.
The effect of the 1.0 and 1.5 × MT conditioning CPN stimuli on the firing rate of tonically discharging soleus motor units and whole muscle EMG were also measured as done for the vibration stimuli described above. In 12 participants, 44 units were isolated from the surface EMG and in 1 participant, 2 units were decomposed from the HDsEMG.
Post-activation depression of the soleus H-reflex during RDD
In 7 of the 13 participants from the CPN-conditioning experiment, we examined the suppression of the soleus H-reflex in response to repetitive stimulation of the TN to directly assess post-activation depression and compare it to the long-lasting depression evoked from a conditioning heteronymous CPN stimulation. To measure post-activation depression during RDD, the first H-reflex (H1) of a stimulation trial was evoked at approximately 50% of maximum on the ascending part of the H-reflex recruitment curve at rest. A run of at least 10 H-reflexes were repetitively evoked at the same conditioning-test intervals as for the CPN stimulation at 500, 1000, 1500, 2000 and 2500 ms. Three trials were performed for each stimulation frequency with at least 30 to 40 s in between each trial.
Data analysis
EPSP modulation:
Similar to (Hari et al., 2022), the amplitude of the EPSP recorded in the ventral root evoked from dorsal root stimulation was compared with and without action potentials evoked in the Ia afferent from optogenetic or sensory activated PAD. The average EPSP from ~10 trials evoked every 10 s just before conditioning and 10 trials during conditioning was used to compute the change in the peak size of the monosynaptic EPSP with conditioning. The latency of the direct EPSPs evoked by dorsal root stimulation was also compared to the latency of the EPSPs from PAD-evoked spikes. Along with the ventral root recordings, PAD was simultaneously recorded from the dorsal roots by a similar averaging method (10 trials of conditioning), to establish the relation of changes in EPSPs with associated PAD.
H-reflex modulation:
For a given trial run, the average, unrectified peak-to-peak amplitude of all test (unconditioned) soleus H-reflexes was compared to the average peak-peak amplitude of the 7 conditioned soleus H-reflexes (vibration and CPN stimulation) for each of the ISIs tested. The conditioned H-reflexes were expressed as a % change from the test reflex using the formula: % change H-reflex = ([(condition H - test H)/test H]*100%). Data was also analyzed by averaging the amplitude of the test H-reflexes immediately preceding the 7 conditioned H-reflexes and this provided similar results throughout so only calculations of % change using all test H-reflexes in a trial are reported here. The mean % change of the soleus H-reflex at each ISI was then averaged across participants.
Effect of conditioning stimulation on soleus motoneurons:
The firing rate profile of soleus motor units in response to the conditioning TA tendon vibration or CPN stimulation from multiple trials were superimposed and time-locked to the time of the stimulation (set to 0 ms) to produce a peri-stimulus frequencygram (PSF) in each participant, as done previously (Norton et al., 2008). The PSF in each participant was divided into 10-ms bins and the mean of each bin was expressed as a percentage of the mean, pre-stimulus firing rate measured from a 100-ms window before the conditioning stimulation using the formula: % change PSF = ([(post-stimulus rate - pre-stimulus rate)/pre-stimulus rate]*100%). The mean % change in each corresponding bin was then averaged across participants to produce the group mean PSF and standard deviation. The cumulative sum (CUSUM) of the mean PSF was measured by subtracting the mean pre-stimulus values and integrating over time. A significant increase or decrease in the mean PSF CUSUM was considered when it fell above or below, respectively, 2 standard deviations (SD) of the mean pre-stimulus value. For group changes in EMG activity, a similar bin analysis was done for the rectified surface EMG. The mean pre-stimulus EMG measured between −300 to 0 ms was subtracted from each of the mean bin values to measure the net increase or decrease in EMG activity. The artifact from the CPN stimulation (typically from 0 to 20 ms) was manually removed from the soleus EMG. Similar to (Metz et al., 2022), the H-reflex data plotted at the various ISIs was shifted to the right of the PSF and EMG traces by ~ 30 ms, the average latency of the H-reflex, to estimate the excitability of the spinal motoneurons at the time they were activated by Ia afferents in the H-reflex. Aligning the PSF and EMG data based on each participant’s H-reflex onset latency did not affect the results because H-reflex onset latencies between participants varied by less than 4 ms (30–34 ms), which was smaller than the 10-ms PSF and EMG bin widths.
Post-activation depression of H-reflexes:
To quantify the amount of post-activation depression during the RDD trials, the average peak-to-peak amplitude of the second to eighth H-reflex (H2 to H8) was compared to the peak-to-peak amplitude of the first H-reflex (H1) in each trial run using the formula (for H2 as an example): % change H-reflex = ([(H1− H2)/H1]*100%). In each participant, the % change in H2 to H8 for the three trials at each stimulation frequency was averaged together. The corresponding H2 to H8 values were then averaged across the 7 participants to examine the profile of suppression as a percentage of the first (H1) H-reflex. In addition, the mean % change H-reflex across H2 to H8 for a given stimulation frequency was measured in each participant and then averaged across the group to compare to the profile of % change from the CPN-conditioned soleus H-reflex trials at the corresponding ISIs.
Statistical Analysis
All statistics were performed with SigmaPlot 11 software. To determine if the suppression of the H-reflex across the various conditioning stimulation intervals was different from a 0% change, a One-Way Repeated Measure ANOVA was used because the data was normally distributed. The F value was reported and post hoc Tukey Tests were used to determine which ISIs were significantly different from a 0% change. A Two-Way Repeated Measure ANOVA was used to compare if the H-reflex suppression using antagonist CPN stimulation was different across the various ISIs compared to the repetitive TN conditioning stimulation during RDD. Student t-tests and Pearson Product-Moment Correlation were used to compare the two groups of normally distributed data. Data are presented in the figures and in the text as means and standard deviation (SD). Significance was set as P < 0.05.
Results
Antagonist TA tendon vibration.
We started by examining the action of a conditioning tendon vibration on the soleus H-reflex, as this was the classic method of examining presynaptic inhibition of Ia afferents in humans (Morin et al., 1984; Hultborn et al., 1987a; Hultborn et al., 1987b; Iles & Roberts, 1987; Rossi et al., 1999). The suppression of the soleus H-reflex from a prior vibration (3 cycles at 200 Hz) to the antagonist TA tendon was examined in 8 participants (schematic of experimental protocol in Fig. 1A). Similar to earlier studies, the soleus H-reflex measured at rest was suppressed between 60 to 400 ms following the first pulse of the brief tendon vibration, as shown for participant 1 (Fig. 1B–C) and participant 2 (Fig. 1G–H). Across the 8 participants, the amplitude of the conditioned soleus H-reflex was modulated across the various ISIs, with the H-reflex being significantly suppressed at the 100, 150 and 200 ms ISIs (marked by large white circles, Fig. 2A, see statistics in legend). Note, if the inhibitory effects from the vibration occurred with the third pulse, then all ISIs should be shortened by 10 ms.
Figure 2: TA tendon vibration, group data.
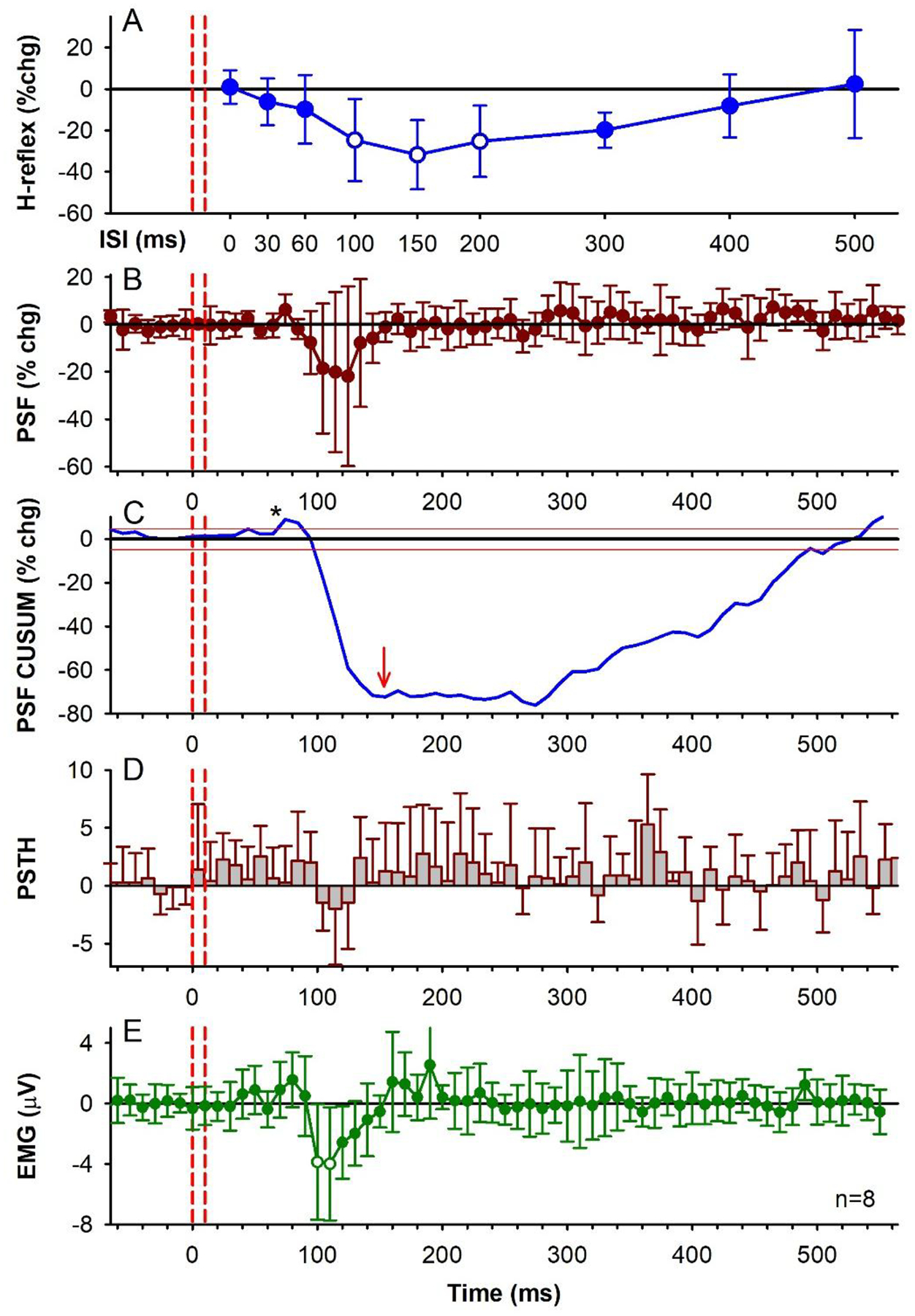
A) % change (chg) of the soleus H-reflex from conditioning TA tendon vibration measured at rest at each ISI averaged across the group (n=8 participants). The average size of the unconditioned test soleus H-reflex was 1,581 (648) μV [50.3 (15.5)% of H-Max]. There was an effect of ISI on the conditioned H-reflexes [F(7, 9)=45.2, P < 0.001, one-way RM ANOVA] with post-hoc analysis showing the 100 ms (P = 0.015), 150 ms (P = 0.005) and 200 ms (P = 0.012) ISIs significantly different from a 0% change (white symbols, Tukey Tests). The 0 ms ISI is shifted to the right of the vibration onset (first dashed red line) by the average latency of the H-reflex. Small open circles are individual participant data points. B) % change of the PSF averaged across the group (10-ms bins) in response to TA tendon vibration alone (as in C-E) during a weak contraction. There was an effect of time on the % change PSF (F[71,64] = 2.02, P < 0.001, one-way RM ANOVA), but no single value was significantly different than a 0% change (post hoc Tukey Test, all P > 0.05). In each participant, 2.13 (1.81) units (range 1–5) were used, on average, to construct the vibration PSF with an average of 164.88 (102.22) stimulation trials (or sweeps, range 63–339), resulting in 9.45 (6.28) frequency values in each 10-ms bin (range 3.8–22.7), and a mean pre-vibration firing rate of 6.54 (0.82) Hz (range 5.33–7.84 Hz) having a firing rate coefficient of variation of 6.24 (1.82) (range 4.44–10.25). C) CUSUM of mean PSF in B, plotted with the mean pre-vibration value (horizontal black line) and 2 SD values above and below the mean line (red lines). D) Number of soleus motor unit action potentials averaged across the group in each 10-ms bin (PSTH) with the mean pre-vibration number subtracted. The count per bin did not significantly change across the time bins (F[71,64] = 1.10, P = 0.288, one-way RM ANOVA). E) Rectified soleus EMG (mean EMG - pre-stimulus EMG) in each 10-ms bin averaged across the group. The EMG was significantly different across the tested time bins (F[7,56] = 2.85, P < 0.001, one-way RM ANOVA), being significantly different from 0 mV at the 100 ms (P = 0.015) and 110 ms bins (P = 0.009, Tukey Test, white symbols). Vertical red dashed lines indicate the onset and offset of TA tendon vibration. Error bars indicate ± SD. * = small increases in motor unit or EMG activity shortly after the vibration.
To determine if the profile of H-reflex suppression was mediated, in part, from a postsynaptic inhibition of the soleus motoneurons, the firing rate of a voluntarily activated soleus motor unit was measured in response to the conditioning tendon vibration applied alone. Following the conditioning stimulus, the mean firing rate of the soleus motor unit measured from the peristimulus frequencygram (PSF) decreased below the mean pre-stimulus rate, with a time course similar to the soleus H-reflex inhibition (the latter between the 80 and 400 ms ISIs in Figs. 1D & I, red trace). Note that the H-reflex data plotted at the various ISIs are shifted to the right of the PSF by ~ 30 ms, the latency of the H-reflex, to estimate the excitability of the spinal motoneurons at the time they were activated by Ia afferents in the H-reflex [see also (Metz et al., 2022)]. The decrease in the PSF is an indication of a prolonged inhibitory postsynaptic potential (IPSP) (Türker & Powers, 2005), with the duration of the IPSP marked by the lowest point in the blue PSF CUSUM line (Figs 1D and I). On average, the PSF was suppressed out to 154 ms (red arrow in PSF CUSUM, Fig. 2C) at time points when the H-reflex was also suppressed (e.g., at the 100 ms ISI). There was also a decrease in the number of motor unit action potentials compared to the mean pre-stimulus count during the early decrease in the PSF, as assessed from the PSTH (Figs. 1E&J, Fig. 2D), that was also reflected in the decreased surface EMG during this period (Figs. 1 F&K, 2E). In some cases, the IPSP was large enough to produce a synchronous resetting of the tonic motor unit discharge, as marked by repetitive clusters of increased firing in the PSTH following the reduced firing periods (near 230 and 360 ms in Fig. 1E). Taken together, our results provide evidence for prolonged inhibition of the soleus motoneurons from the conditioning TA tendon vibration, which likely contributed to the H-reflex suppression.
Unexpectedly, in some participants there was a clear early excitation of the extensor soleus EMG (abbreviated early soleus reflex) following the vibration to the antagonist TA tendon applied alone (e.g., Fig. 1F). This excitation was evident in the group PSF CUSUM, becoming more prominent and consistent across participants starting at 66 ms after the tendon vibration (at * Fig. 2C when the PSF CUSUM crosses the upper 2-SD red line). The relevance of this early soleus excitation evoked by the antagonist afferent conditioning in suppressing the H-reflex was investigated further below in mice and rats.
Post-activation depression of afferent transmission in mice and rats
The early soleus reflex following the TA tendon vibration raised the possibility that the antagonist TA afferents evoked a PAD in the soleus Ia afferents that produced orthodromic action potentials (PAD-evoked spikes) to subsequently activate a monosynaptic reflex (MSR), causing post-activation depression of soleus Ia afferent transmission, as we detail in the Introduction. Thus, we examined if GABAergic neurons with axoaxonic connections to afferents (GABAaxo neurons) could, in principle, evoke PAD and orthodromic spikes in Ia afferents to produce such post-activation depression and a reduction of subsequent motoneuron EPSPs. To do this, grease gap recordings from sacral dorsal and ventral roots were made in adult spinal cords from GAD2//ChR2 positive mice (schematic in Fig. 3A). These recordings allowed simultaneous assessment of afferent PAD and motoneuron EPSPs, with PAD directly evoked by GABAaxo (GAD2+) neurons, which cannot themselves directly evoke monosynaptic EPSPs in motoneurons (Hari et al., 2022). When light activation of GABAaxo neurons produced a PAD without generating action potentials in the afferents (top light blue trace, Fig. 3A), the monosynaptic EPSP evoked from a dorsal root (DR 1) stimulation during this PAD was facilitated (bottom light blue trace) compared to when PAD was not present (pink trace, DR 1 stim alone). As we have recently shown, this EPSP facilitation is due to increased conduction in Ia afferents evoking the EPSP, via PAD facilitating spike generation at the nodes [nodal facilitation (Hari et al., 2022; Metz et al., 2022)]. However, when activation of the GABAaxo neurons produced a larger and faster rising PAD that also generated an action potential in the Ia afferent (PAD-evoked spike, DRR; top black trace Fig. 3A), a fast transient EPSP was evoked in the motoneurons (middle black trace, Fig. 3A), indicating that the PAD-evoked spike traveled orthodromically to the Ia afferent terminal and evoked a monosynaptic EPSP, as has previously demonstrated (Frank, 1959; Duchen, 1986). Indeed, this PAD-evoked spike in the Ia axon activates the motoneuron synchronously at a monosynaptic latency that is similar to the latency of EPSPs generated by direct activation of the Ia afferent by dorsal root stimulation (DR 1, Fig. 3F,G). Following these PAD-evoked spikes and EPSPs, subsequently tested monosynaptic EPSPs evoked by direct DR 1 stimulation were always depressed, regardless of whether the test EPSP was evoked during or after PAD and the related PAD-evoked EPSPs (Figs. 3A and B respectively, black traces), demonstrating that PAD-evoked spikes cause post-activation depression of the Ia-mediated EPSP (see Fig. 3C for group results, n = 5 mice).
Figure 3: GABAA mediated PAD in Ia afferents with and without spiking and subsequent EPSPs.
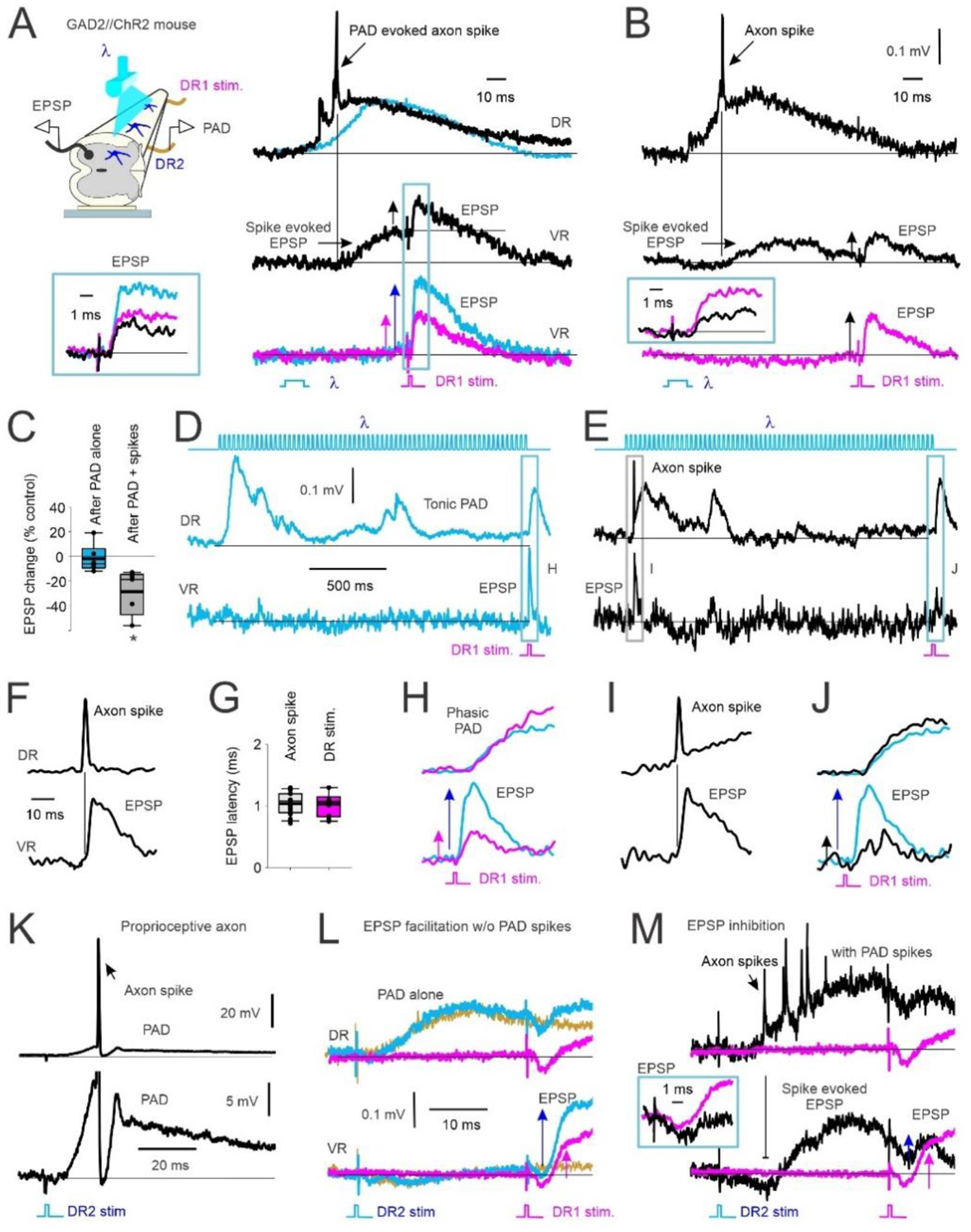
A) Left, In vitro experimental set up showing PAD recorded from dorsal root 2 (DR2) activated by light (λ) in a GAD2//ChR2 positive mouse. Monosynaptic reflex (MSR) activated by stimulation of DR1, subsequent EPSPs recorded from ventral root (VR). Right, top: PAD recorded from DR2 with (black) and without (blue) PAD-evoked axon spike. Middle: VR recording of evoked EPSP from the PAD-evoked axon spike in top trace. Subsequent test EPSP activation (DR1 stimulation) is reduced (vertical arrows indicate EPSP amplitude). Bottom: EPSP from DR1 stimulation alone (pink) compared to EPSP following the light-evoked, non-spiking PAD shown in top trace (blue). Box: Overlay of EPSPs from middle and bottom traces in A. B) Same as in A but test EPSP from DR1 stimulation delivered at a longer interval after the PAD spike-evoked EPSP returned to baseline. Box: Overlay of conditioned (black) and control (pink, DR1 stimulation only) EPSPs. C) Change in amplitude of light-conditioned EPSPs (100 – 150 ms ISI) after a non-spiking PAD (PAD alone) and after a PAD with spike(s) as a percentage of an EPSP evoked from DR1 stimulation alone (control). Box plots: median, thin line; mean, thick line; interquartile range IQR, box bounds; most extreme data points within 1.5 × IQR, standard error bars. EPSPs after a PAD-evoked spike were smaller than a 0% change (* p = 0.021) but not after a non-spiking PAD ended (After PAD alone) (p = 0.151, Mann-Whitney U test, n=5 mice). D-E) Top: Light-evoked tonic PAD recorded from DR2 (middle trace) without (D, blue) and with (E, black) a PAD-evoked spike. Bottom: VR recordings of associated of monosynaptic EPSP from DR1 stimulation and PAD spike-evoked EPSP. F) Example DR and VR recording of a PAD-evoked axon spike and resulting EPSP at a monosynaptic latency on expanded time scale. G) Comparison of EPSP latency following PAD evoked axon spike and DR 1 stimulation (not significantly different, p = 0.683, Student’s t test, n = 6 mice). H-J) Expanded view of PAD (top) and EPSPs (bottom) evoked in D and E. Similar results observed in in n=6/6 mice. K-M) Same as in A and B but DR2 stimulation (1.5 × T) used to evoke PAD instead of light. Similar results in n = 5/5 rats. K) Top: Rat intracellular (IC) recording of proprioceptive (Ia) afferent with PAD-evoked axon spike from DR2 stimulation. Bottom: expanded vertical axis of top trace. L-M) PAD evoked with DR2 stimulation. Top: DR recording. Bottom: VR recording. L) PAD evoked without axon spikes. Pink: EPSP alone from DR1 stim. Blue: Non-spiking PAD evoked from DR2 stimulation and facilitated EPSP from DR1 stimulation. Yellow: PAD from DR2 stimulation applied alone. M) PAD, axon spikes and motoneuron EPSPs evoked from DR2 stimulation and test EPSP from DR1 stimulation without (pink) and with (black) spiking PAD (EPSPs expanded in box).
If the suppression of the monosynaptic EPSP following the PAD-evoked spike is indeed due to post-activation depression of the Ia afferent terminal, this suppression should last for many seconds (Curtis & Eccles, 1960a; Hultborn et al., 1996). In support of this, we evaluated a very long interval between the PAD-evoked spikes and the test EPSP. When an EPSP was evoked during a tonic PAD produced by a long train of light pulses, the EPSP was facilitated by the PAD when no PAD-evoked spikes were produced, as previously reported (Hari et al., 2022) (Fig. 3D and expanded in H, pink trace EPSP alone, blue trace EPSP during tonic PAD). In contrast, when the same long train of light pulses evoked a spike at the start of the tonic PAD (Fig. 3E and expanded in I), the test EPSP that was evoked 1500 ms later was instead depressed (Fig. 3E and expanded in J, black trace), especially compared to the expected facilitation at this time (blue trace overlayed from Fig. 3H). This is broadly similar to Fig. 3 of Duchen 1986, though in that case PAD was evoked indirectly by a DR stimulation, as we detail next.
A similar suppression of the monosynaptic EPSP occurred when PAD-evoked spikes in the Ia afferent (within dorsal root DR1) were activated by sensory-evoked PAD, rather than light-evoked PAD, the former produced by stimulating a separate dorsal root (termed DR2) to that used to evoke the test EPSP from DR1. As shown from an intracellular (IC) axon recording of a proprioceptive Ia afferent from DR1 (top, Fig. 3K), a single pulse stimulation to the dorsal root DR2 sometimes produced a spike in the Ia afferents of DR1 on the rising phase of the PAD evoked by the DR2 stimulation (expanded view in bottom). The same DR2 stimulation produced multiple axon spikes in the afferent population recorded from DR1 (top, Fig. 3M; 1.5xT, black trace), that in turn evoked multiple compounded motoneuron EPSPs recorded in the ventral root (VR, bottom), as with the optogenetically-evoked PAD (Fig 3A). Following these PAD-evoked EPSPs, the EPSP evoked by a direct DR1 stimulation was suppressed (inset in Fig. 3M), even though it was evoked when the membrane potential of the motoneuron returned to near baseline. Thus, an orthodromic action potential in the DR1 Ia afferents was activated twice, once from the PAD produced by the DR2 stimulation, and a second time from the direct stimulation of DR1 to reveal the phenomenon of post-activation depression of the Ia-EPSP. Similar to light activation of GABAaxo neurons, when DR2 stimulation did not produce axon spikes in the DR1 Ia afferent or associated depolarizations (EPSPs) of the motoneurons (DR2 stimulation only: yellow trace, Fig. 3L), the test EPSP evoked by a subsequent DR 1 stimulation was facilitated by the non-spiking PAD (1.5xT; blue trace, Fig. 3L) compared to the EPSP tested alone without PAD (pink trace, Fig. 3L). This absence or presence of PAD-evoked spikes occurred randomly at a fixed DR2 stimulation intensity, as in Figures 3L and M, respectively, though these spikes could also be reduced in occurrence by reducing the stimulation intensity and again, the EPSP was inhibited only when they occurred (not shown). In summary, the same sensory conditioning stimulation can produce either suppression or facilitation of subsequent EPSPs depending on whether the associated PAD activates orthodromic spikes in the Ia afferent. Additionally, afferents from one nerve (e.g., DR 2) can produce widespread activation of PAD-evoked spikes in afferents from other nerves (DR 1) (Lucas-Osma et al., 2018), which may lead to short-latency reflex activation of motoneurons reminiscent of heteronymous polysynaptic reflexes and reflex irradiation (Baldissera et al., 1981).
To explore whether PAD-evoked spikes can produce motoneuron reflexes in vivo, we first examined the special characteristics of these spikes and the reflexes they evoke in vitro so they can be distinguished from polysynaptic reflexes activated from other sources and ultimately, be detected in vivo. We started by determining the origin of the PAD-evoked spikes by recording intracellularly from single group Ia afferents in the rat spinal cord near the dorsal root entry zone (Fig. 4A). A brief DR stimulation that was subthreshold to the Ia afferent being recorded (as indicated by a lack of an orthodromic spike at the green arrowhead in Fig. 4B) evoked a PAD in the recorded Ia afferent. In some of the trials, the DR stimulation produced one or more spikes starting on the rising phase of the PAD (dark blue Fig. 4B), which propagated antidromically toward the recording electrode and out the dorsal root and was recorded as a dorsal root reflex [DRR; (Eccles et al., 1961b)]. In trials where the DR stimulation did not produce these antidromic spikes, there were underlying oscillations on top of the PAD at times where the spikes occurred in other trials (light blue, Fig. 4B). Previously, we have demonstrated that these oscillations represent spikes that are generated by PAD deep in the spinal cord (distal to the recording electrode) that fail to propagate antidromically out the dorsal root, but likely propagate orthodromically toward the motoneurons (Lucas-Osma et al., 2018), and so have functional importance. One key characteristic of these PAD-evoked spikes and oscillations is that they are blocked by hyperpolarization, with a small hyperpolarization of the Ia afferent consistently eliminating the spikes (pink, Fig 4B) and a stronger hyperpolarization eliminating the underlying oscillations (pink, Fig. 4D). Consistent with the recorded oscillations being mediated by distal sodium spikes, we found that inducing sodium spike inactivation by evoking a direct orthodromic spike in the Ia afferent via a supra-threshold stimulation to its DR (at green arrowhead, Fig. 4C) reduced the subsequent PAD-triggered oscillations measured in the DR (dark blue, Fig. 4C). Furthermore, after blocking these oscillations with a strong intracellular hyperpolarization, they resumed when the hyperpolarization was removed (pink, Fig. 4D), the latter showing that they are mediated intrinsically to the axon and not by synaptic inputs. Another characteristic of the PAD-evoked spikes and oscillations is their unique timing, starting at about a 5–7 ms latency on the rising portion of a phasic PAD evoked by sensory stimulation (Fig. 4E, top left), and sometimes repeating at about 7–10 ms intervals, driving a characteristic oscillation in the population recording of afferents in the DRs, with a frequency (100 to 140 Hz) that is likely determined by the axon’s refractory period and associated AHP (seen in Fig. 4B). Finally, as with direct hyperpolarization of the Ia afferent, indirect afferent hyperpolarization induced by blocking spontaneous tonic PAD on the afferents with either a low dose of the non-selective GABAA receptor antagonist bicuculline (Fig. 4E, top traces) or the selective extra-synaptic α5 GABAA receptor blocker L655708 (with inverse agonist properties) [Fig. 4F, top traces; see (Lucas-Osma et al., 2018) for details] reduced PAD-evoked spikes and oscillations. These same activation characteristics appear in the motoneurons (VR recordings, Fig. 4E&F, bottom traces) when simultaneously recorded with the PAD, consistent with these PAD-evoked spikes driving the motoneuron EPSPs. That is, the population of motoneuron EPSPs in the VR recording followed the oscillatory profile of the population of Ia PAD measured on the DR recording with the same timing and were eliminated by hyperpolarizing the afferents with bicuculline (Fig. 4E, bottom) or L655708 (Fig. 4F, bottom), consistent with previous observations of Duchen (1986) in mice, also studied in vitro.
Figure 4. GABAA mediated synchronous PAD and motoneuron oscillations.
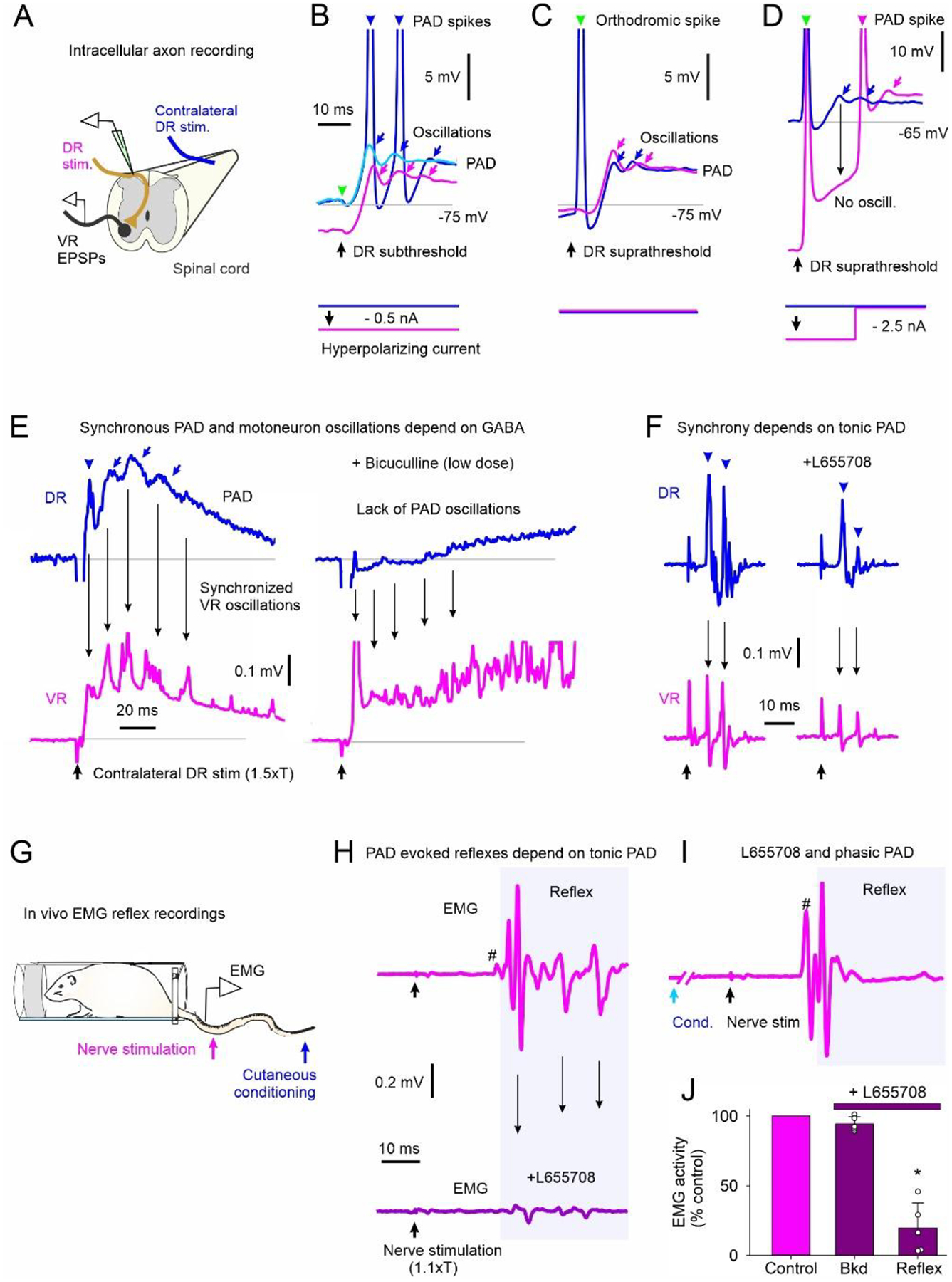
A-D) PAD recorded intra-axonally from a rat Ia afferent. A) In vitro experimental set up. B) Stimulation of DR that is subthreshold to orthodromic spike for recorded Ia afferent without (blue & light blue traces) and with (pink) a small, applied hyperpolarization. PAD-evoked spikes and oscillations marked by arrowheads and arrows, respectively. Predicted timing of non-activated orthodromic spike indicated by green arrowhead. C) Suprathreshold DR stimulation producing orthodromic spike (green arrow and blue trace) compared to PAD oscillations produced with subthreshold DR stimulation in A (pink). D) Suprathreshold DR stimulation with strong hyperpolarization of Ia afferent to reduce PAD oscillations from distal spikes, n = 10/10 axons similar to results in B-D. E-F) Left: Synchronized PAD (DR recording, top) and motoneuron EPSP (VR recording, bottom) oscillations from PAD evoked by contralateral DR stimulation in rat. Right: Application of the wide spectrum GABAA receptor blocker bicuculine (E) or the a5 GABAA receptor blocker L655708 (both inverse agonists) (F) to the recording bath, similar findings in 7/7 rats. G) PAD evoked by stimulating cutaneous afferents in tip of tail in awake rat and MSR EMG recordings from more proximal tail nerve stimulation. H) Top: Small MSR (#) and oscillating EMG reflex responses from tail nerve stimulation (1.1xT) to recruit proprioceptive afferents. Bottom: EMG response to tail nerve stimulation following i.p. application of L655708. I) Facilitation of MSR and later reflex from prior (60 ms ISI) conditioning cutaneous stimulation of tip of tail. J) Pre-stimulus background (Bkd) EMG and post-MSR reflex activity (shaded window in I) following application of L655708 as a percentage of pre-drug (control). * = reflex significantly different from control (Mann Whitney U test, p < 0.001, n = 6 rats).
In the awake rat (Fig. 4G), cutaneous nerve stimulation known to evoke PAD caused EMG reflexes with repeated bursts of activity (Fig. 4H, top), again separated by about 7–10 ms (140 to 100 Hz), similar to that seen in vitro (Fig. 4E, bottom left), suggesting that they may be due to the oscillating PAD-evoked spikes driving the oscillating reflexes. Indeed, hyperpolarizing the afferents with L655708 reduced these reflex oscillations (Fig. 4H, bottom) without changing the postsynaptic activity of the motoneurons assessed by the background EMG (Bkg, Fig. 4J), consistent with an action on the Ia afferents rather than on the motoneurons. L655708 reduced both the small monosynaptic reflex (# in Fig. 4H), as previously reported (Hari et al., 2022), and the repeated bursts of EMG at longer intervals. This is consistent with these long-latency reflexes being mediated by monosynaptic EPSPs triggered by PAD-evoked spikes, rather than polysynaptic reflexes from other sources, the latter which should be increased, rather than decreased, by reducing GABA action. Note that even with L655708, the monosynaptic reflex (#) and some of the later putative PAD-evoked reflexes were facilitated with a prior brief cutaneous conditioning stimulation known to activate phasic PAD circuits and facilitate the monosynaptic EPSP by depolarizing the afferents (Fig. 4I), showing that the L655708 simply eliminated the EPSPs by hyperpolarizing the sensory axons in Fig 4H, rather than indirectly acting to eliminate the EPSP by other pre- or postsynaptic pathways (e.g. via GABAB receptors).
Soleus H-reflex inhibition following an antagonist-evoked early soleus reflex associated with post-activation depression.
Similar to mice and rats, a conditioning electrical stimulation to the human flexor CPN supplying the TA muscle often produced an early excitatory reflex response in the extensor soleus muscle (early soleus reflex), which is not expected from our classical understanding of the reciprocal inhibitory organization of reflexes, but is similar to what we observed with vibration, as detailed above. As described below, this early soleus reflex is consistent with a CPN-evoked PAD causing extensor afferent spikes and associated reflexes based on its latency and frequency of oscillation. Moreover, when we observed an early soleus reflex from antagonist CPN stimulation, subsequent soleus H-reflexes were suppressed (Figs. 5 & 6). In contrast, soleus H-reflexes were facilitated when we did not observe an early soleus reflex from the CPN stimulation (Fig. 7), consistent with a non-spiking PAD that facilitated Ia conduction and motoneuron EPSPs, similar to that seen in rodents detailed above.
Figure 5: CPN stimulation: early H-reflex suppression.
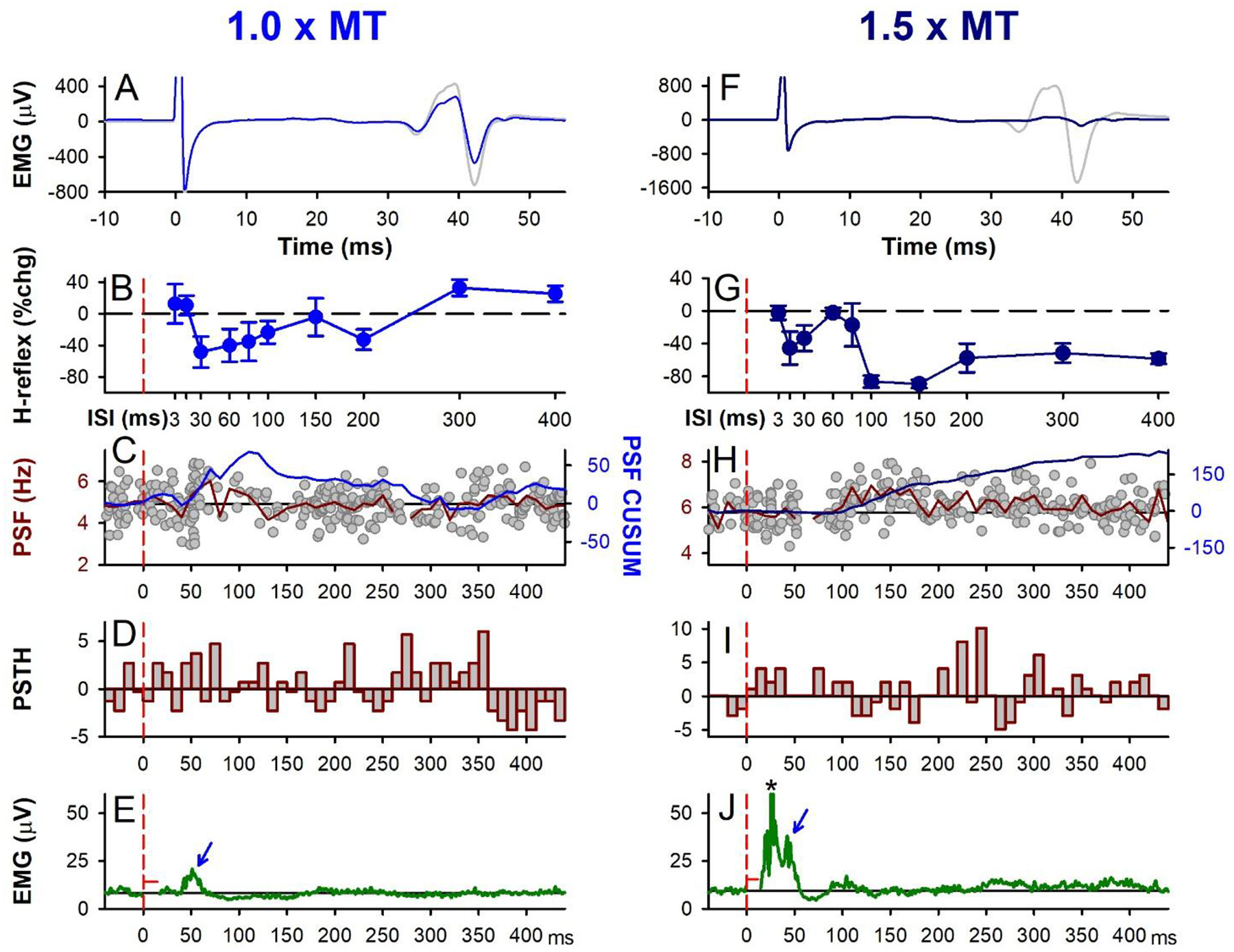
Same presentation as Figure 1 but for 1.0 × MT (A-E) and 1.5 × MT (F-J) electrical CPN conditioning stimulation (3 pulses at 200 Hz), representative data from a single participant. A,F) Unrectified EMG of test (grey) and conditioned H-reflex (blue: 1.0 × MT at 30 ms ISI and dark blue: 1.5 × MT at 100 ms ISI) measured at rest. B,G) % change soleus H-reflex (Mean ± SD) plotted at each ISI tested. C,H) PSF (grey dots) with 108 sweeps in C and 98 sweeps in H, mean PSF (red line) measured from CPN stimulation alone during weak contraction (as in D-J) and CUSUM of mean PSF (blue line). D,I) Number of motor unit counts per 10-ms time bin with average pre-stimulation count subtracted. E,J) Average rectified soleus EMG with 131 sweeps in E and 100 sweeps in J. Stimulation artifact has been removed (horizontal red line near 0 ms). B-J) Vertical dashed red lines indicate the onset of the CPN conditioning stimulation train (at time 0). * in J marks crosstalk from TA M-wave.
Figure 6: CPN stimulation: early H-reflex suppression, group data.
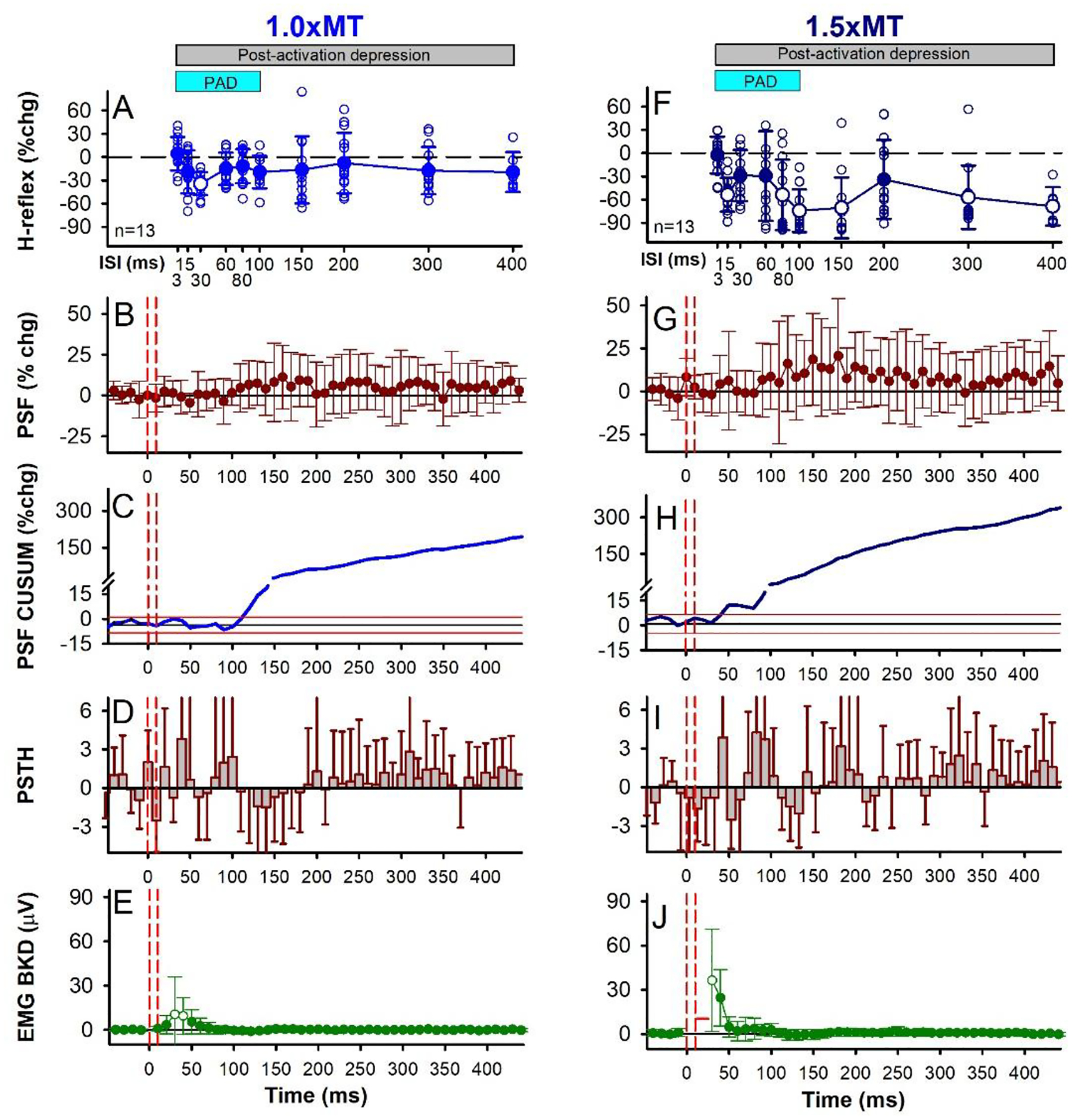
Same presentation as Figure 2 but for the 1.0 × MT (A-E) and 1.5 × MT (F-J) CPN conditioning stimulation (3 pulses at 200 Hz) with data averaged across the 13 participants. The average size of the unconditioned H-reflex measured at rest was 961.2 (454.1) μV [30.9 (13.8)% of Hmax; 16.6 (6.6)% of Mmax]. A&F) There was an effect of ISI on the % change of the H-reflex for both the 1.0 × MT (F[12,10] =2.392, P = 0.030) and 1.5 × MT (F[12,10] =7.801, P < 0.001, one-way RM ANOVAs) stimulus intensities, with the 30 ms ISI (P = 0.017, 1.0 × MT) and 15, 80, 100, 300 and 400 ms ISIs (P’s < 0.01, 1.5 × MT) significantly different from a 0% change (Tukey Test, white symbols). Light blue bar indicates predicted duration of PAD and grey bar the predicted window of post-activation depression. Small open circles are individual participant data points. B,G) There was no effect of time on the % change PSF for the 1.0 × MT (F[12,61] = 1.094, P = 0.298) and 1.5 × MT (F[12,61] = 1.270, P = 0.087, CPN conditioning stimulation applied alone (one-way RM ANOVAs). In each participant, 1.18 (0.35) units (range 1–2) were used, on average, to construct the 1.0 × MT CPN PSF with 113.23 (36.37) sweeps (range 36–167), 6.26 (2.49) frequency points in each 10-ms bin (range 2.2–12.3), and a 6.49 (1.38) Hz mean pre-stimulus firing rate (range 4.88–8.80 Hz) having a firing rate coefficient of variation of 6.16 (1.56) (range 3.90–8.90). Likewise, 1.14 (0.30) units (range 1–2) were used, on average, to construct the 1.5 × MT CPN PSF with 100.69 (34.40) sweeps (range 37–168), 5.10 (2.09) frequency points in each 10-ms bin (range 2.8–10.2), and a 6.91 (1.29) Hz mean pre-stimulus firing rate (range 4.83–10.06 Hz) having a firing rate coefficient of variation of 5.88 (1.80) (range 3.54–10.20). C,H) % change CUSUM of the mean PSF averaged across participants. Note the expanded y-axis below 15% to better visualize the early part of the PSF CUSUM. D,I) There was an effect of time on the count per bin (PSTH) for the 1.0 × MT (F[116,47] = 1.579, P = 0.010), but not the 1.5 × MT (F[116,47] = 1.352, P = 0.064), CPN conditioning stimulation (one-way RM ANOVAs). E,J) There was an effect of time on the EMG with the 1.0 × MT (F[12,46] = 3.361, P < 0.001) and 1.5 × MT (F[12,59] = 10.708, P < 0.001) CPN stimulation (one-way RM ANOVAs), with the 30 and 40 ms (P < 0.001, 1.0 × MT) and 30 ms (P < 0.001, 1.5 × MT) time bins significantly greater than 0 mV (Tukey Test, white symbols). Error bars ± SD.
Figure 7: CPN stimulation: early H-reflex facilitation, individual and group data.
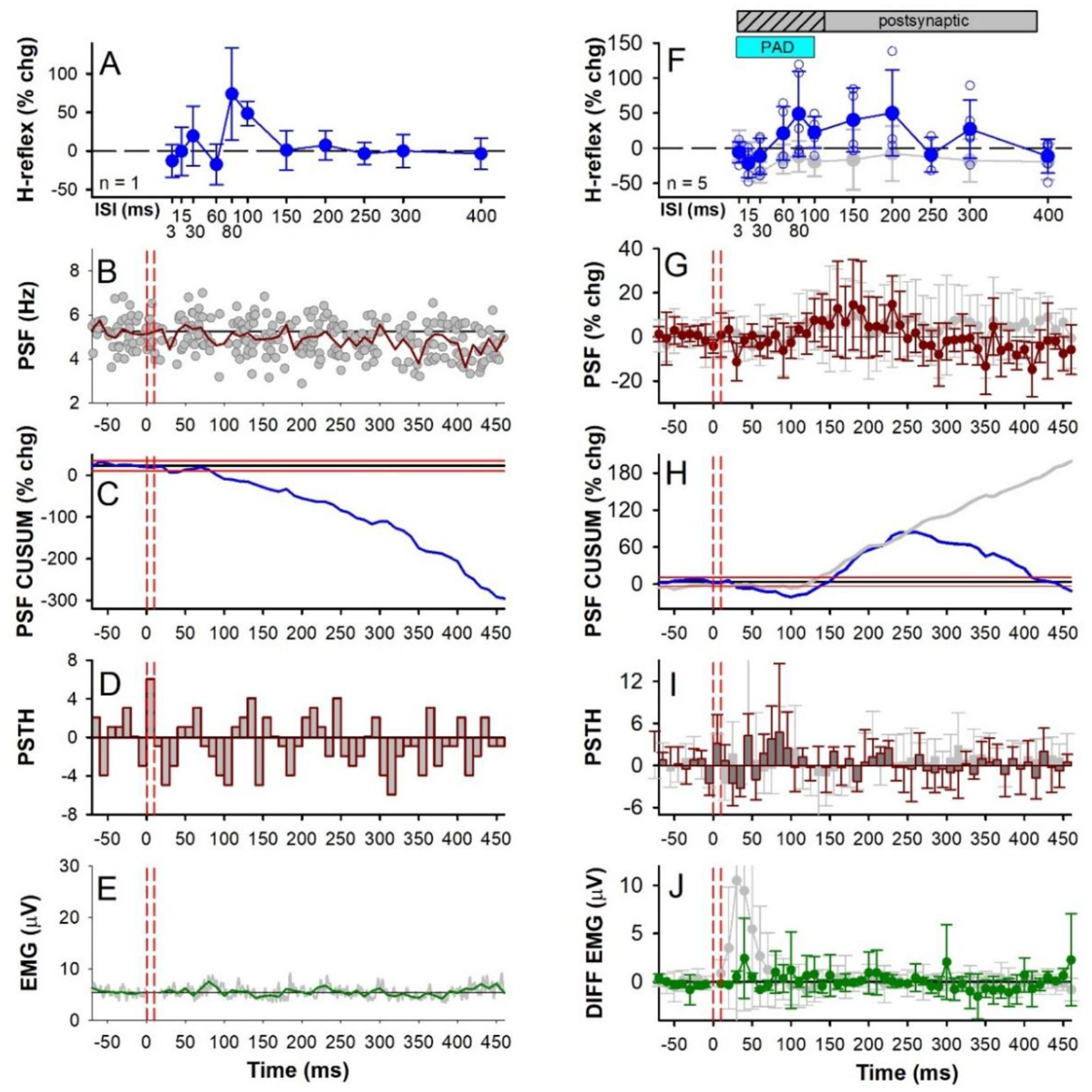
Similar presentation as Figure 6 but with 1.0 × MT CPN conditioning stimulation that did not evoke an early reflex response in the soleus muscle (3 pulses at 200 Hz). A-E data from an individual participant and F-J group data (n = 5 participants). Grey data points in F-J are replotted from Figs. 6A–E where 1.0 × MT CPN stimulation produced an early soleus reflex response. Note however the more compressed y-axis scales compared to Fig. 6 and the removal of the axis break in H. There was an effect of ISI on the % change H-reflex (F) (F[4,11] =2.113, P = 0.043) and an effect of time on the % change PSF (G) (F[3,61] = 1.673, P = 0.005) but no effect of time on the count per bin in the PSTH (I) (F[3,61] = 1.120, P = 0.281) or EMG (J) (F[3, 60] = 0.994, P = 0.498), all one-way RM ANOVAs. In each participant, an average of 1.70 (0.96) units (range 1–3) were used to construct the PSF that formed the group data in (G), with an average of 118.00 (44.64) sweeps (range 83–180), resulting in 7.00 (2.83) frequency points in each 10-ms bin (range 4.4–11.0), and with a pre-stimulus firing rate of 6.56 (1.54) Hz (range 5.31–8.75 Hz) having a firing rate coefficient of variation of 7.62 (1.22) (range 6.21–8.90). All H-reflexes measured at rest. Small open circles in F are individual participant data points. Error bars ± SD.
In participants where CPN conditioning produced an early soleus reflex consistent with PAD-evoked spikes (in 13/18 participants tested, Figs. 5 & 6), there was an associated inhibition of the H-reflex. As shown for two of these participants, an early soleus reflex was visible in the surface EMG near 40 ms at both the 1.0 and 1.5 × MT CPN stimulation intensities (see arrows in Figs. 5E and J) that was more readily observed at 1.0 × MT in this participant since there was no crosstalk from the TA M-wave between 10 to 30 ms (* in Fig. 5J). At the 1.5 × MT CPN intensity, the average latency of the early soleus reflex was 39.71 ms (3.36) and occurred earlier than the TA H-reflex at 41.6 ms (3.50), consistent with the TA H-reflex being activated by slower conducting motor axons. Although partly overlapping in time, the TA H-reflex (not shown) and the early soleus reflex waveforms were not correlated to rule out crosstalk between the two signals. Overall, the average amplitude of the early reflex response was larger for the 1.5 × MT stimulation (Fig. 6J) compared to the 1.0 × MT stimulation (Fig. 6E). This consistent reflex response produced marked synchronization in the firing probability of the soleus motor units, as shown in the PSTH (Figs. 6D, I; see also Fig. 5I) that was not readily apparent in the surface EMG. Interestingly, power spectrum analysis of the early soleus reflex EMG during the predicted early PAD window (i.e., 30–80 ms after the first CPN stimulation) revealed a frequency spike at 161.9 (27.4) Hz (not shown, 1.5 × MT), similar to the frequency of the PAD oscillations recorded in the VR in the rodent (140 Hz, Fig. 4 E).
The early soleus reflex was also reflected in the firing rate profiles of the soleus motor units in response to the CPN stimulation applied alone (PSF and PSF CUSUM), which reflect the profile of the underlying motoneuron membrane potential (Türker & Powers, 2005). The PSF increased near 40 ms, as shown for the 1.0 × MT stimulation in Figure 5C, and in the group data where, at the 1.5 × MT intensity, the PSF increased above 2 SD near 40 ms (Fig. 6H), which was around 7 ms later than the onset latency of the soleus H-reflex [32.3 (2.0) ms] evoked from direct TN stimulation. Although similar in onset, the PSF and PSF CUSUM displayed a longer-lasting increase following the CPN stimulation compared to the surface EMG, lasting out to 400 ms as illustrated by the PSF remaining above the baseline rate (Fig. 6B,G) and by the sustained increase in the PSF CUSUM (Figs. 6C,H). This is consistent with multiple PAD-evoked Ia-EPSPs or a polysynaptic reflex response (see multiple Ia spikes in Figs. 3M and 4B–F). Similar effects are seen in cat motoneurons, where the flexor-evoked EPSPs appear in antagonist extensor motoneurons, sometimes alone (Frank, 1959) and sometimes riding on an IPSP (Stuart & Redman, 1992), and these too likely resulted from PAD-evoked spikes and caused post-activation depression of the Ia afferents (Hari et al., 2022).
Following the early soleus reflex, the inhibition of the H-reflex had two phases, an early phase (D1) starting at the 15–30 ms ISI and a later, more sustained phase of inhibition (D2) starting at the 80 ms ISI that was larger with the higher intensity 1.5 × MT CPN stimulation (Fig. 5F,G) compared to the lower intensity 1.0 × MT CPN stimulation (Fig. 5A,B), similar to previous studies (Mizuno et al., 1971; El-Tohamy & Sedgwick, 1983; Capaday et al., 1995). When averaged across the 13 participants, the soleus H-reflex was suppressed at the 30 ms ISI when conditioned with the 1.0 × MT CPN stimulation (Fig. 6A, white circle) and at several ISIs starting at 15 ms when conditioned with the 1.5 × MT CPN stimulation (Fig. 6F, see legend for statistics). In summary, the CPN conditioning stimulation produced an early reflex in the soleus muscle consistent with PAD-evoked spikes in the soleus Ia afferent producing soleus monosynaptic EPSPs, as we and others have seen in rodents and cats. This early reflex response is associated with an inhibition of the soleus H-reflex lasting seconds (detailed later), consistent with post-activation depression of the H-reflex.
Soleus H-reflex facilitation in the absence of an antagonist-evoked early soleus reflex.
Based on the findings in the mice, if a flexor conditioning stimulation does not activate a PAD-evoked spike in the extensor Ia afferents and thus, does not pre-activate the monosynaptic extensor Ia-EPSPs (as evident by a lack of early soleus reflex), then the H-reflex should not be inhibited by post- activation depression, but if anything facilitated by PAD [Fig. 3A&D and (Hari et al., 2022; Metz et al., 2022)]. Indeed, we found that when there was a lack of an early soleus reflex evoked by conditioning, consistent with a lack of PAD-evoked spikes in the soleus Ia afferents, the soleus H-reflex was not inhibited but instead, facilitated by conditioning (seen in 5/18 participants in response to the 1.0 × MT CPN conditioning) [see also (Metz et al., 2022)]. This is shown for one participant in Figure 7 where the CPN stimulation does not increase the PSF or PSF CUSUM (Fig. 7B and C respectively) or the rectified surface EMG (Fig. 7E), whereas the H-reflex was facilitated at the 80 and 100 ms ISIs within the predicted PAD window. When averaged across these 5 participants, the H-reflex was facilitated between the 30 and 250 ms ISIs (blue trace, Fig. 7F) compared to the H-reflex suppression that occurred in the other group of 13 participants where an early reflex response was present (the latter replotted from Fig. 6A, grey trace). The amplitude of the H-reflex was significantly modulated across the different ISIs but post hoc analysis revealed that no H-reflex at a given ISI was different than a 0% change, likely due to the small number of participants [Fig. 7F, see legend for statistics]. However, this is consistent with our main conclusion that the lack of an early soleus reflex and associated PAD-evoked spikes prevents an inhibition of the H-reflex, due to a lack of post-activation depression. Further, when comparing the peak H-reflex facilitation that occurred at the 60 or 80 ms ISIs in each participant (during the predicted PAD window), the conditioned H-reflex was significantly greater than a 0% change [see Fig. 3C in the accompanying (Metz et al., 2022)]. Note that on average, there was a brief period of motoneuron inhibition from 30 to 100 ms after the 1.0 × MT CPN stimulation, as reflected in the PSF (Fig. 7G), PSF CUSUM (Fig. 7H) and clustering of motor unit firing probability in the PSTH starting at 60 ms (Fig. 7I). This early motoneuron inhibition may have reduced the early facilitation of the conditioned H-reflex between the 15 to 60 ms ISIs within the PAD window. Following this early inhibition was a period of motoneuron facilitation as shown in the PSF (Fig. 7G, red) and PSF CUSUM (Fig. 7H, blue) that may have shunted the Ia-EPSPs at the 150 to 200 ms ISIs (as in Fig. 3A), but it was much briefer compared to when there was an early soleus reflex response in the other group (the latter again plotted in grey from Fig. 6). Like in the PSF, the amplitude of the early excitatory reflex response in the soleus EMG was much smaller or absent in these 5 participants compared to when H-reflexes were suppressed by the CPN conditioning (Fig. 7J, compare green and grey traces respectively).
As described above, our observed early excitatory reflex in the soleus EMG or PSF from the CPN conditioning stimulation may be indicative of PAD-evoked spiking in the soleus Ia afferents that produces long-lasting post-activation depression and subsequent H-reflex suppression. In contrast, when we observed no or very little early reflex activity, PAD evoked from the conditioning afferent input likely facilitates Ia afferent conduction and thus, increases subsequent H-reflexes. In further support of this, we observed that the amount of H-reflex inhibition at the 100 ms ISI was greater when the amplitude of the early soleus reflex measured between 30 – 50 ms following a CPN stimulation alone was also greater, as demonstrated by the negative correlation between these two measures (Fig. 8). Facilitation of the soleus H-reflex at the 100 ms ISI was typically produced only when there was no or very little (< 2 μV) early soleus reflex activity in the soleus muscle compared to background.
Figure 8: CPN stimulation: % change soleus H-reflex at the 100 ms ISI vs early soleus reflex EMG.
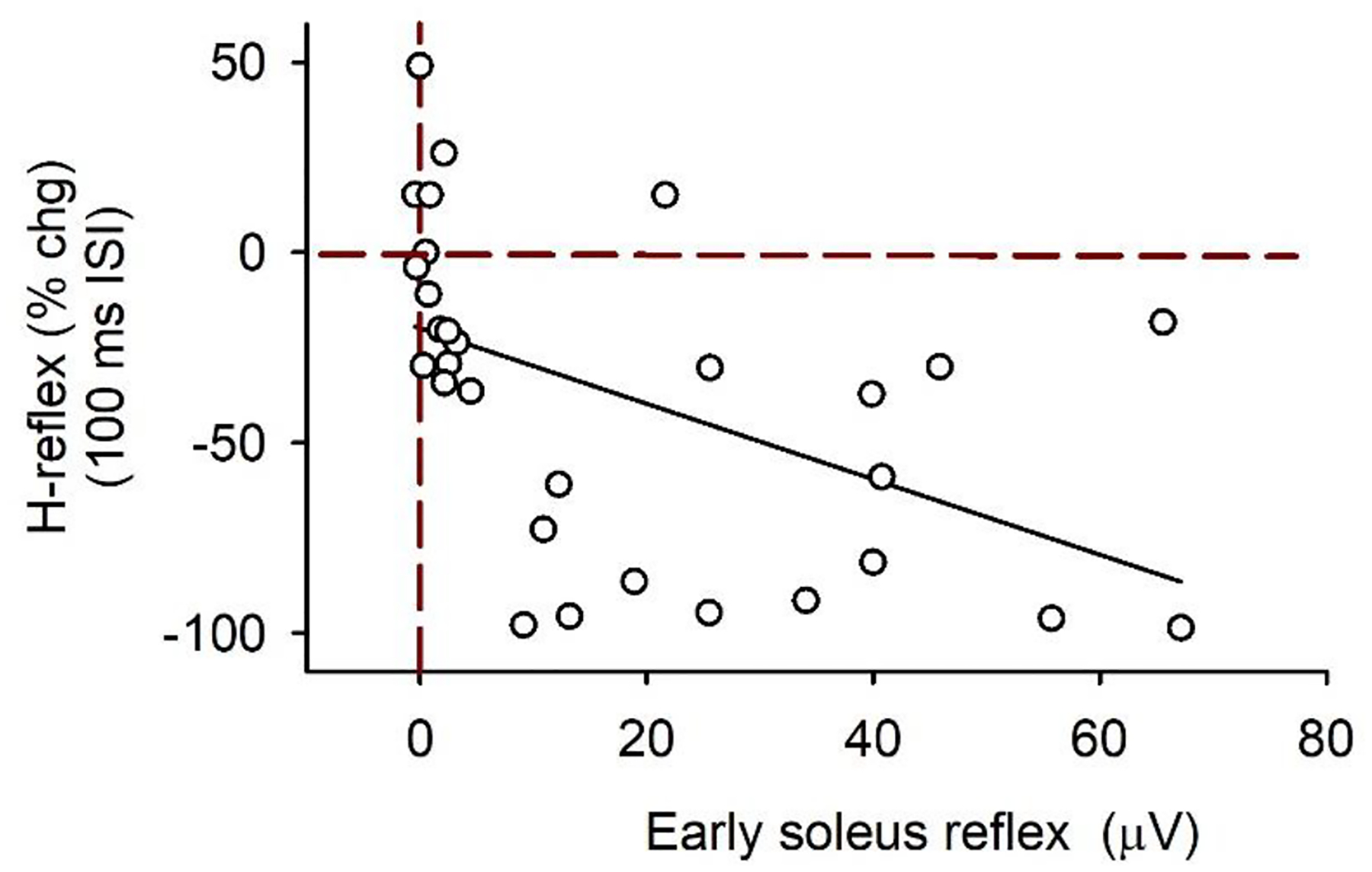
% change (chg) H-reflex at the 100 ms ISI plotted against the early soleus reflex (mean rectified EMG averaged over the 30–50 ms window after the CPN stimulation applied alone – the mean rectified pre-stimulus EMG) for both the 1.0 and 1.5 × MT CPN conditioning stimulation (3 pulses at 200 Hz) for participants with and without an early soleus reflex (n = 18). Data fitted to a linear line (slope = 1.0 % chg/mV). There was a significant correlation between the % chg H-reflex and the early soleus reflex (r = −0.50, P = 0.005, Pearson Product-Moment Correlation). There are 30 data points: typically 2 values from each participant (one from the 1.0 and 1.5 × MT trials each). In some recordings (n = 6), the TA M-wave obscured the early soleus reflex and this data was not used. Red vertical dashed line marks no change in the soleus EMG from the CPN stimulation above background. Red horizontal dashed line marks 0 % change in the conditioned soleus H-reflex at the 100 ms ISI.
Long lasting soleus H-reflex inhibition consistent with post-activation depression
Given that conditioning of the flexor CPN may produce spikes in the extensor soleus Ia afferents and post-activation depression of the H-reflex, we examined this further by exploring longer intervals between the CPN conditioning stimulation and the test H-reflex, where suppression should still occur if similar to rate-dependent depression (RDD) that is mediated by post-activation depression (Nielsen et al., 1993; Hultborn et al., 1996). Thus, we examined H-reflex inhibition at intervals between 500 to 2500 ms from the higher intensity CPN conditioning stimulation in the same 13 participants that had the earlier suppression of the H-reflex presented above. As shown for two participants, the soleus H-reflex measured at rest was maximally suppressed at the 500 ms ISI by a 1.5 × MT conditioning CPN stimulation (3 pulses at 200 Hz) and continued to be suppressed out to the 2500 ms ISI (Figs. 9Ai and Bi), which is similar to the suppression of monosynaptic EPSPs tested at the 1500 ms in mice when followed by a PAD-evoked spike and associated Ia-EPSP (Fig. 3E). Across the group, the H-reflex was modulated at these very long ISIs, being significantly suppressed at all ISIs from 500 to 2500 ms (* Fig. 9Ci, see legend for statistics). By itself, the 1.5 × MT CPN stimulation increased the firing rate of the soleus motor units for at least 500 ms in some participants (PSF Fig. 9Bii) and for a briefer period in others (PSF Fig. 9Aii). Across the group, both the PSF (Fig. 9Cii) and the rectified surface EMG (Fig. 9Ciii) returned to pre-stimulus levels by 500 ms while the H-reflex continued to be suppressed (see statistics in legend). In contrast, the 1.0 × MT CPN stimulation intensity did not suppress the soleus H-reflex at these long ISIs as plotted for the 500 ms ISI (Fig. 9Di), consistent with it having a weaker or absent early soleus reflex (Fig. 9Dii) that we take as evidence for PAD-evoked spikes. Thus, the suppressed H-reflex at these long ISIs was not likely related to any direct postsynaptic inhibition of the soleus motoneuron at the time the H-reflex was evoked but rather, may have been produced by the earlier soleus reflex activation described above. In support of this, H-reflex suppression was significantly correlated to the early increase in PSF when data for the 1.0 and 1.5 × MT CPN stimulation intensities were plotted together (Fig. 9Diii) or when compared to the amplitude of the early EMG response to CPN stimulation (from 30 to 50 ms, r = −0.74, P <0.001, Pearson’s Product, not shown).
Figure 9: CPN stimulation: late H-reflex suppression.
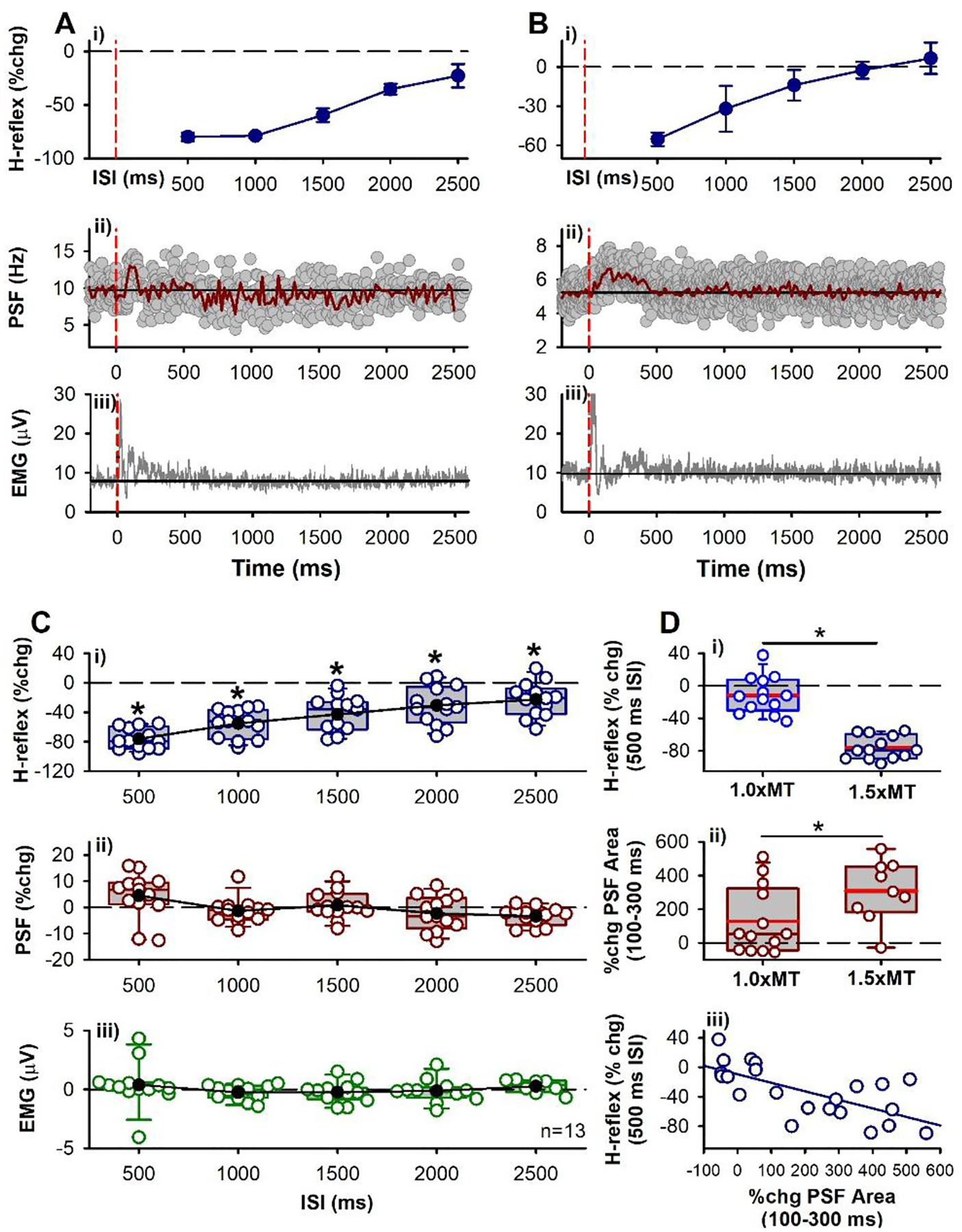
A-B) Representative data from two participants showing i) % change in H-reflex measured at rest following 1.5 × MT stimulation of CPN (3 pulses at 200 Hz), ii) PSF (37 sweeps in A, 98 sweeps in B) and iii) rectified soleus EMG (40 sweeps in A, 30 sweeps in B) following CPN stimulation alone during weak contraction in ii and iii. Vertical dashed red lines indicate onset of CPN conditioning stimulation. Error bars ± SD. C) Mean (black circles) and individual data points (open circles) from the 13 participants tested. There was an effect of ISI on the % change in the soleus H-reflex (i) (F[12,5] = 55.697, P < 0.001, one-way RM ANOVA) with all ISI’s significantly different from 0% change (P < 0.001, Tukey Test, marked by *). There was an effect of ISI on the % change PSF (ii) (F[12,5] = 4.393, P = 0.002) but not EMG (F[12,5] = 0.716, P = 0.614, one-way RM ANOVAs). The PSF and EMG data points occurred in the time bin that matched when the H-reflex was evoked at the various ISIs. Error bars ± SD. D) Box plot (similar to Figs. 3C&G) of: (i) % change soleus H-reflex at the 500 ms ISI for the 1.0 and 1.5 × MT CPN conditioning stimulation, significantly different (P = 0.040, Student’s t-test); (ii), % change in area of the PSF measured 100–300 ms after the 1.0 and 1.5 × MT CPN stimulation, significantly different (P < 0.001, Student’s t-test), the early PSF for 1.5 × MT in 4 participants was not usable due to stimulation artifact; (iii) % change H-reflex at 500 ms ISI plotted against % change PSF area (100 – 300 ms) with fitted linear line (blue, r = −0.74, P < 0.001, Pearson Product-Moment Correlation).
Post-activation depression of the soleus H-reflex during RDD
We next investigated whether the early reflex activation in the soleus muscle by the antagonist CPN stimulation causes a similar profile of long-lasting H-reflex inhibition to that expected from RDD, which has previously been demonstrated to be due to post-activation depression (Hultborn et al., 1996), thus providing further evidence that PAD-evoked spikes in the soleus Ia afferents produces a long-lasting post-activation depression of the soleus H-reflex. That is, in 7 of the 13 participants from above, we examined if the profile of long-lasting suppression of the soleus H-reflex from antagonist CPN conditioning (CPN→TN) was similar to the profile of H-reflex suppression evoked by repetitive TN stimulation during RDD (TN→TN), the latter used to quantify the profile of post-activation depression out to 2500 ms. Like with the CPN conditioning, repetitive stimulation of the TN measured at rest inhibited the H-reflex at all intervals tested out to 2500 ms, with the greatest suppression at the 500 ms ISI (compare Figs. 10Ai & Aii). The suppression of successive H-reflexes at the different TN stimulation frequencies typically plateaued after the 3rd H-reflex (H3, Fig. 10B right) and was more consistent following the CPN conditioning (Fig. 10B left). Overall, when averaging all the H-reflexes in a stimulation run for a given ISI across the group, the mean profile of H-reflex inhibition at the different ISIs was not different between antagonist CPN (open circles) or homonymous TN (closed circles) conditioning stimulation trials (Fig. 10C, see legend for statistics), with both forms of inhibition lasting out to the tested 2500 ms ISI. This suggested that the mechanism of H-reflex suppression was similar between the two stimulation protocols and likely due to post-activation depression, as defined in the Introduction, with similar mechanisms described previously for rate dependent depression (RDD).
Figure 10: Post-activation depression of the soleus H-reflex.
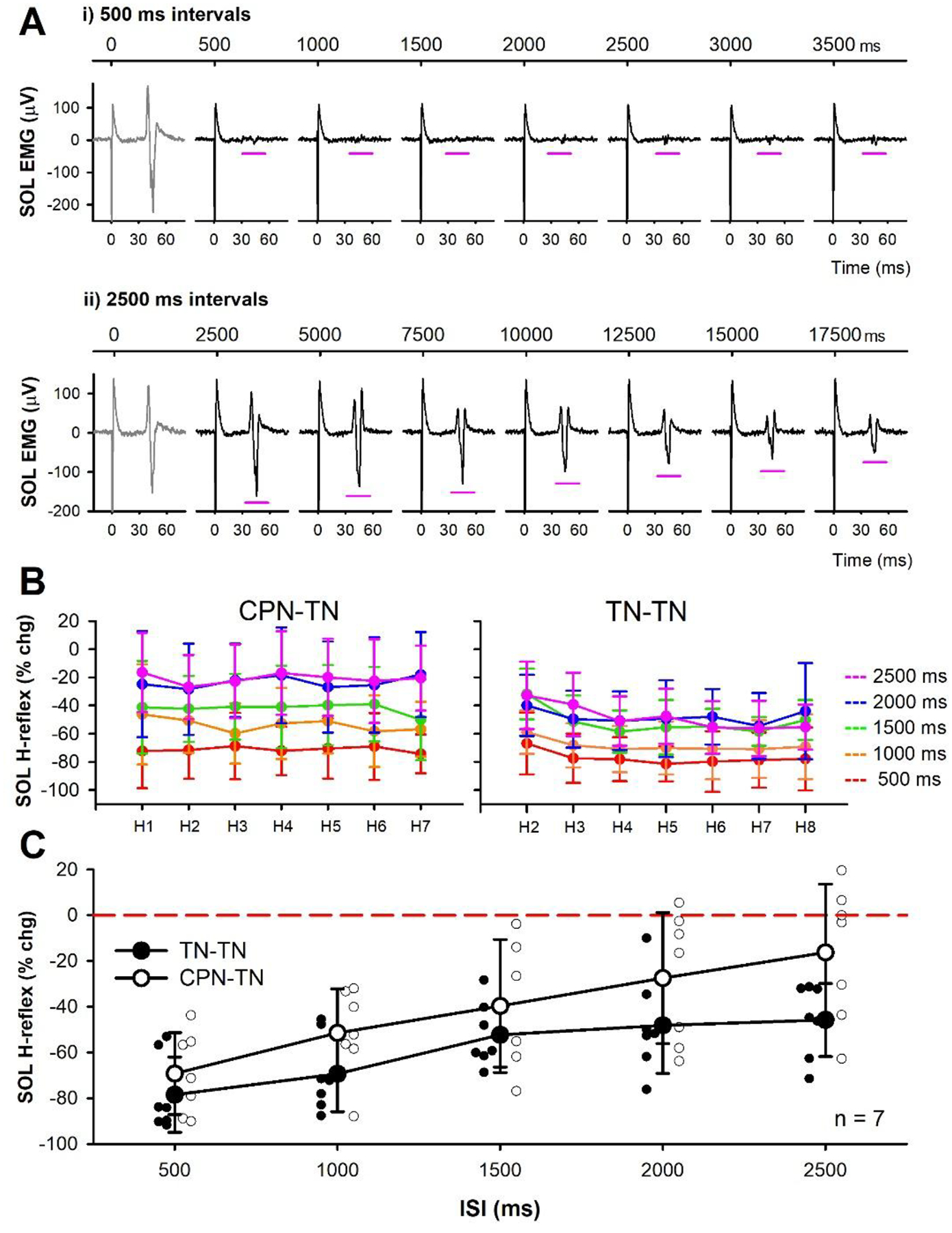
A) Example rate-dependent depression (RDD) of unrectified soleus EMG evoked by repetitive TN stimulation every 500 ms (i) and 2500 ms (ii) in one participant measured at rest. Continuous time displayed in top trace to indicate when TN stimulation was applied (marked by large stimulation artifact) and expanded time scale in bottom to display the H-reflex occurring in the time window marked by the pink horizontal lines (time set to 0 ms when TN stimulation was applied). B) Change in H-reflexes as a % of the mean test (unconditioned) H-reflex (CPN-TN, left) or as a % of H1 (TN-TN) for each ISI averaged across the 7 participants. C) The mean % change of all conditioned H-reflexes (CPN-TN: H1 to H7, TN-TN: H2 to H8) in a given ISI trial averaged across the group for both the CPN-TN stimulation (large white circles) and the TN-TN stimulation (RDD, large black circles), not significantly different (F[1,4] = 1.860, P = 0.152, two-way RM ANOVA). Small white and black circles represent individual participant data for CPN-TN and TN-TN trials, respectively. Error bars ± SD.
DISCUSSION
Inhibitory control mechanisms within the spinal cord are needed to regulate the large influx of sensory information from the periphery that would otherwise exceed the computing capacity of the central nervous system (Rudomin & Schmidt, 1999). One way to reduce the flow of sensory information to spinal targets is through presynaptic inhibition of afferent terminals. To regulate the transmission of proprioceptive information, the depolarization of primary (Ia) afferents, or PAD, from the activation of GABAA receptors was historically thought to inactivate sodium channels and/or shunt current at the afferent terminal and reduce neurotransmitter release to spinal motoneurons as one form of presynaptic inhibition (Willis, 2006). To demonstrate this, antagonist afferents were used to activate PAD in Ia afferents via GABAergic interneurons and shown to be accompanied by a parallel suppression of monosynaptic EPSPs in the motoneuron, though direct evidence for presynaptic inhibition via activation of terminal GABAA receptors and PAD is lacking. For example, numerous studies have failed to find GABAA receptors on Ia afferent terminals (Alvarez et al., 1996; Fink et al., 2014; Lucas-Osma et al., 2018; Hari et al., 2022). Instead these receptors are located more dorsally near the nodes of Ranvier near branch points where their activation facilitates afferent conduction and monosynaptic reflexes (Hari et al., 2022; Metz et al., 2022). We demonstrate here that the more likely mechanisms that produce the H-reflex suppression by antagonist afferents is both a direct long-lasting inhibition of the test motoneurons and/or an indirect post-activation depression of the Ia afferents mediating the H-reflex, two mechanisms that have been suggested previously but not tested explicitly (Frank, 1959; Eccles et al., 1961a; Hultborn et al., 1987a; Fink et al., 2014; Hari et al., 2022).
Postsynaptic inhibition of motoneurons from antagonist afferents
We demonstrate here that the extensor H-reflex is inhibited for up to a half second following vibration of the flexor tendon, similar to previous findings in cats (Curtis, 1998), which is too long to be explained by presynaptic inhibition from phasic PAD that only lasts for 100–200 ms, contrary to previous conclusions (Morin et al., 1984; Hultborn et al., 1987a; Hultborn et al., 1987b; Iles & Roberts, 1987; Roby-Brami & Bussel, 1990; Rossi et al., 1999). In humans, H-reflex suppression at ISIs of 40 to 500 ms was considered to be mediated primarily by PAD-induced presynaptic inhibition because it was thought that direct effects on the motoneuron from antagonist afferent conditioning did not last beyond the 40 ms ISI (Hultborn et al., 1987a; Hultborn et al., 1987b). However, as shown here with 3 pulses of TA tendon vibration and by others with a single pulse of CPN stimulation (Yavuz et al., 2018), inhibition of soleus motoneurons can last up to 100 – 300 ms following the conditioning stimulation when measured with the PSF. On average, the profile of the PSF was suppressed after the tendon vibration out to ~ 150 ms (Fig. 2), and this motoneuron inhibition likely contributes to the suppression of H-reflexes during this period. In cats and rodents, similar long-duration IPSPs and reduced neuronal time constants (τ, indicating postsynaptic shunting) have been induced in motoneurons from heteronymous afferent conditioning that also produced long-duration EPSP suppression (McCrea et al., 1990; Hari et al., 2022). The H-reflex suppression at the longer latencies is likely mediated by post activation depression as discussed below.
The prolonged postsynaptic inhibition of the soleus motoneurons may have resulted from multiple summated IPSPs of short duration that were triggered by multiple afferent inputs (Desmedt & Godaux, 1978), or from a single longer-duration IPSP. One source of prolonged postsynaptic inhibition could arise from the terminal projections of the GABAaxo interneurons themselves that also synapse onto motoneurons (Pierce & Mendell, 1993) and activate both postsynaptic GABAA and GABAB receptors (Stuart & Redman, 1992). The duration of this postsynaptic inhibition may be similar to the long duration (~ 200 ms) of PAD that these GABAaxo interneurons produce on the Ia afferent (Hari et al., 2022; Lin et al., 2022). Regardless of the source, when using a weak conditioning stimulation to suppress H-reflexes, like brief tendon vibration, sensitive methods are needed to measure if there are any direct actions on the motoneuron and for how long. The PSF provides a better representation of the IPSP evoked in the soleus motoneuron compared to the PSTH or surface EMG, the latter which better represent the occurrence of motoneuron discharge and are subject to synchronization artifact (Türker & Powers, 2005; Yavuz et al., 2015). In addition, we tested the excitability of the soleus motoneurons while they were tonically active during a weak voluntary contraction, so that any subtle inhibition from the tendon vibration or low intensity CPN stimulation could be revealed, in contrast to a resting motoneuron. A hyperpolarization of the distal dendrites by a low-intensity conditioning stimulation, which might not be detectible from intracellular somatic recordings in a resting motoneuron (Frank, 1959; McCrea et al., 1990), may produce a greater influence on the firing rate response of the cell (Hari et al., 2022). Thus, it is important to record single motor unit activity in a preactivated motoneuron to examine if the conditioning stimulation has any direct inhibitory effects on the motoneuron and the duration of its effect on the suppression of the H-reflex. However, the amount of inhibition may be underestimated in a preactivated motoneuron compared to at rest due to shunting of the postsynaptic inhibitory currents by voltage-gated channels in the depolarized state (Brownstone et al., 1994). Alternatively, the amount of inhibition may also be overestimated in a preactivated motoneuron compared to rest as the membrane potential is further from the reversal potential of chloride (Murray et al., 2011). Although still present, the estimated size and profile of inhibition measured from the PSF during a small contraction may be altered in the resting motoneuron when the H-reflexes were evoked, even though different levels of depolarization tend to have small effects on the measured IPSP (Turker & Powers, 1999).
Post-activation depression by antagonist afferents.
Our finding that direct electrical stimulation to the flexor CPN, and sometimes even flexor tendon vibration, causes early extensor soleus reflexes led us to examine whether these unusual, excitatory reciprocal reflexes may contribute to subsequent post-activation depression of the soleus H-reflex. Consistent with previous findings, we found that H-reflexes are suppressed by the antagonist CPN conditioning (Mizuno et al., 1971; El-Tohamy & Sedgwick, 1983; Capaday et al., 1995). However, we demonstrate that this inhibition only occurred when short-latency reflexes to the soleus muscle are evoked by the conditioning electrical stimulation to the flexor CPN. The inhibition of the extensor H-reflex by electrical CPN stimulation occurred even though there was little evidence of postsynaptic inhibition of the soleus motoneurons compared to conditioning with tendon vibration. We propose here that the H-reflex suppression following this early reflex response is likely mediated by post-activation depression of the soleus H-reflex induced by the antagonist CPN afferents activating PAD-evoked spikes in the soleus Ia afferents. In rodents we show that during a large and fast rising PAD, the membrane potential of the afferent can reach the sodium spiking threshold and produce action potentials that travel orthodromically down the Ia afferent to evoke monosynaptic EPSPs (early reflexes) in the motoneuron [see also (Frank, 1959; Duchen, 1986; Lucas-Osma et al., 2018)]. These monosynaptic EPSPs are likely produced by PAD-evoked spikes because they: 1) are tightly linked in time with a monosynaptic latency [see also (Duchen, 1986)], 2) repeat with a characteristic period of about the duration of the afferent AHP with a refractory period of 7–10 ms (140–100 Hz) in both rodent VR and human EMG recordings, and 3) are reduced by specifically hyperpolarizing the Ia afferent by blocking tonic PAD with L655708.
The PAD-evoked spike and the associated EPSP and early soleus reflex likely cause post-activation depression that can have multiple underlying mechanisms, as detailed in the Introduction. These include the depletion of transmitter packaging and release from the Ia afferent, the collision of GABAA receptor mediated dorsal root reflexes and the test Ia afferent volley [lasting up to 18 ms, see Fig. 11 in (Eccles et al., 1961a)], and the indirect activation of GABAB receptors on the afferent terminals triggered by the PAD-evoked spike to ultimately produce GABAB mediated presynaptic inhibition (Fig. 11). Likely all these mechanisms are involved in the early inhibition of the extensor soleus H-reflex by flexor conditioning, but only the GABAB action can account for the very long (>1 s) inhibition seen in our study and others (Eccles et al., 1961a; Curtis & Lacey, 1994, 1998) given that the recovery time constant from maximal transmitter depletion is around 300–400 ms (Neher & Sakaba, 2001). This raises the question of how are the GABAB receptors on soleus afferent terminals activated by the flexor nerve stimulation? We do not favor the simple idea that flexor nerve stimulation directly activates the trisynaptic circuit that drives the GABAergic neurons that innervate the soleus afferent terminals and associated GABAB receptors for two reasons. First, the inhibition of the soleus H-reflex, or related mice EPSPs, only occurs when it is linked to PAD-evoked spikes and associated PAD-evoked Ia-EPSPs or early soleus reflexes and thus, must somehow be indirectly linked to the GABAA receptors that mediate PAD, as previously argued from the actions of GABAA receptor blockers on this inhibition (Stuart & Redman, 1992). Second, the profile of inhibition from conditioning is similar in timing to RDD caused by repeated H-reflex activation, which has been shown to be mostly confined to inhibition mediated by the activation of the same afferents that are stimulated to evoke the H-reflex (thus also called homosynaptic depression) (Hultborn et al., 1996). This RDD is likely mediated by a restricted trisynaptic circuit, where the extensor afferent activates private GABAergic neurons that only activate extensor afferent terminals (Fig. 11). Thus, we favor the more complex scenario where flexor nerve stimulation evokes a widespread PAD and PAD-evoked spikes in extensor soleus afferents, which then activate a restricted trisynaptic circuit with GABAegic neurons that activate GABAB receptors on the extensor afferent terminal (Fig. 11). This raises interesting questions about whether there are indeed subpopulations of different GABAergic neurons that innervate extensor vs flexor afferent terminals, which needs to be investigated in the future (Curtis & Eccles, 1960b; Hultborn et al., 1996).
Figure 11. Schematic of PAD pathways mediating post-activation depression in the spinal cord.
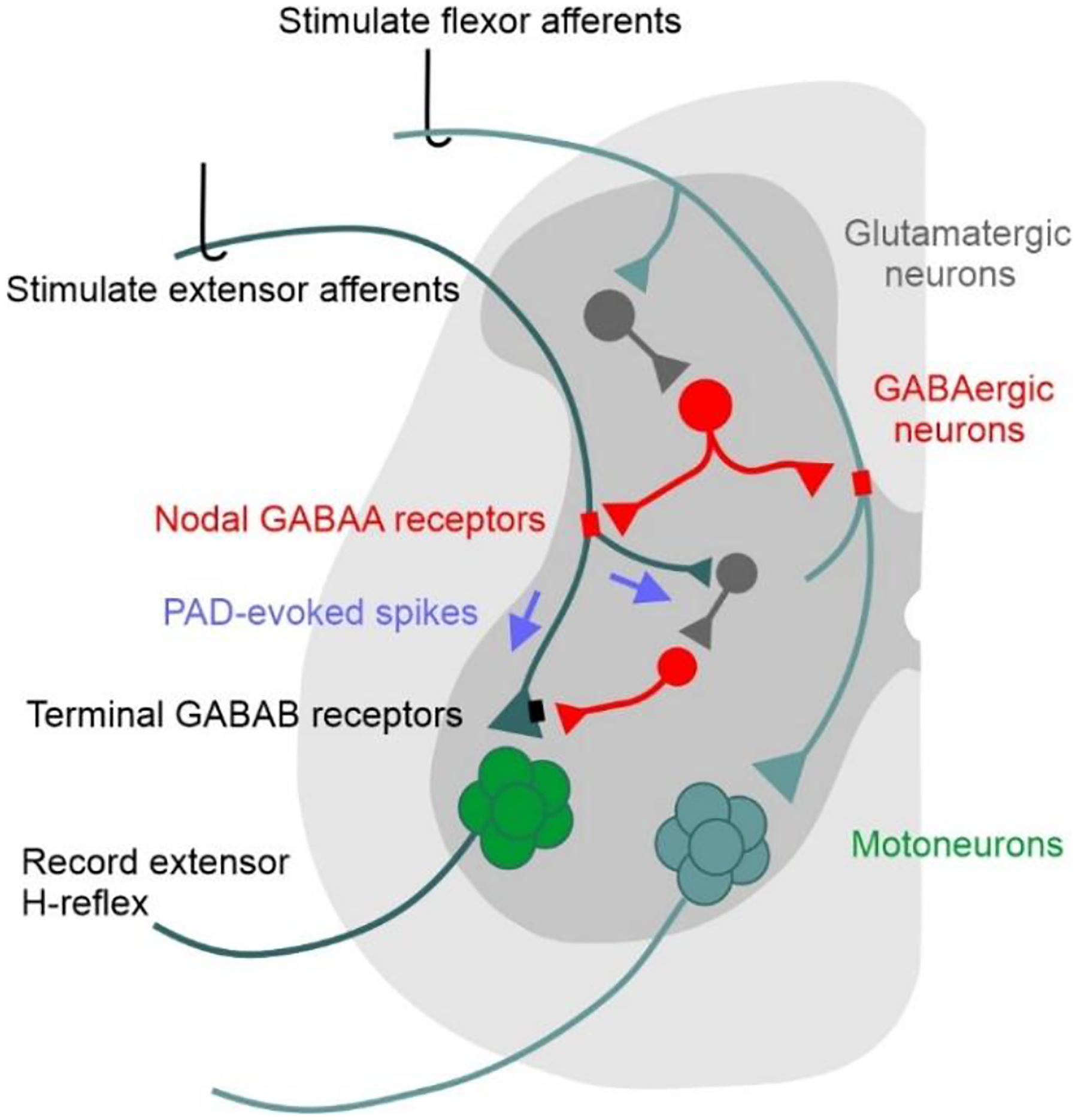
Activation of flexor afferents (cyan) activates a tri-synaptic dorsal PAD pathway (glutamatergic interneuron dark grey, GABAaxo neuron red) with axo-axonic projections to an extensor afferent (dark green), activating nodal GABAA receptors (red) and PAD in the extensor afferent. If the resulting PAD in the extensor afferent reaches sodium spiking threshold, PAD-evoked spikes (purple arrows) travel orthodromically to: 1) depolarize extensor afferent terminals, evoking an EPSP in the extensor motoneurons and a subsequent MSR/H-reflex; 2) activate a trisynaptic circuit containing GABAergic neurons that activate GABAB receptors (black) on the extensor afferent terminal. Thus, the PAD-evoked spike in the extensor Ia afferent can produce early and late post-activation depression of subsequently activated MSRs/H-reflexes by: 1) decreased afferent excitability from collision of orthodromic action potentials with antidromic PAD-evoked spikes; 2) transmitter depletion following the PAD-evoked spike entering the extensor Ia afferent; and/or 3) inhibition of the afferent terminals by GABAB receptors indirectly activated by the PAD-evoked spike (GABAB mediated presynaptic inhibition).
For the flexor CPN to evoke PAD in extensor afferents, they must activate a circuit that that has GABAergic interneurons with axoaxonic projections (GABAaxo interneurons) onto extensor afferents, as just mentioned (Fig. 11). This arrangement is broadly consistent with our understanding of the widespread nature of PAD. That is, it has been known for many years in both animals (Eccles et al., 1961b; Willis, 1999) and humans (Shefner et al., 1992) that stimulation of afferents from one nerve can produce PAD and related PAD-evoked spikes (DRRs) in afferents from neighbouring nerves, since GABAaxo interneurons are innervated by first order neurons [e.g., V3 interneurons (Lin et al., 2022)] with projections across many segments within the spinal cord (Jankowska et al., 1981c; Lidierth, 2006; Willis, 2006; Lucas-Osma et al., 2018). Furthermore, strong synchronous flexor nerve stimulation produces a larger PAD in extensor afferents compared to in flexor afferents (Eccles et al., 1961a; Rudomin & Schmidt, 1999) and is thus most likely to produce PAD-evoked spikes and associated post-activation depression. In contrast, asynchronous afferent activation from flexor muscle stretch appears not to produce PAD-evoked spikes or post-activation depression in extensor afferents (Hultborn et al., 1996), as is the case with weak dorsal root stimulation in mice (Fig. 3). Likewise, weak CPN stimulation or flexor vibration in humans likely produce a smaller amount of PAD-evoked spikes and early soleus reflex activity [see also (El-Tohamy & Sedgwick, 1983)], but may have been large enough to mask some of the early post-synaptic inhibition from these conditioning stimuli, especially following the low intensity TA vibration where the net inhibition of the PSF and EMG only occurred after 80 ms. Here the new addition to this older concept is that PAD is likely mediated by GABAA receptors at nodes, rather than at ventral terminals (Hari et al., 2022), though this does not change the known distributions of PAD (Fig. 11). Thus, the amplitude or presence of the early reflex response in the soleus muscle evoked from the antagonist CPN stimulation can be used as a gross indicator of PAD-evoked spikes in the soleus Ia afferents. In addition to producing heteronymous polysynaptic reflexes, these Ia spikes likely produce post-activation depression in the Ia afferent terminals to mediate the suppression of the conditioned H-reflexes, as we have discussed above. Indeed, the amplitude of the early soleus reflex EMG is correlated with the amount of H-reflex suppression at the 100 ms ISI (Fig 8), an interval previously attributed to PAD-induced presynaptic inhibition of the Ia afferent terminal (Mizuno et al., 1971; Burke et al., 1992; Iles & Pisini, 1992; Capaday et al., 1995; Iles, 1996; Knikou & Mummidisetty, 2014; Howells et al., 2020). Moreover, when an early soleus reflex is not produced following low-intensity CPN stimulation, the conditioned H-reflexes are instead facilitated, likely due to a facilitation of spikes through the soleus Ia branch points by nodal PAD (Hari et al., 2022; Metz et al., 2022). Nodal depolarization and facilitation of Ia afferent conductance likely also occurs when H-reflexes were suppressed following the early soleus reflex and may account for the decrease in H-reflex suppression near the 30 to 60 ms ISIs during the rising phase of PAD that divides the D1 and D2 phases of inhibition (Mizuno et al., 1971; El-Tohamy & Sedgwick, 1983; Berardelli et al., 1987; Faist et al., 1996). However, this facilitation is probably masked by the stronger post-activation depression of the Ia afferents from the PAD-evoked Ia spike(s).
Further evidence to support the conclusion that the profile of H-reflex inhibition from antagonist CPN conditioning is mediated by post-activation depression of the Ia afferent terminals is provided by its resemblance to the seconds long profile of rate dependent depression (RDD) from repeated TN stimulation that directly activates spikes in the soleus Ia afferents. Similar to RDD at long repetition intervals, H-reflexes are also suppressed out to 2500 ms after a strong conditioning CPN stimulation, well after any direct effect on the soleus motoneurons from the CPN stimulation had subsided. This is consistent with the long-duration suppression of the Ia-EPSP in mice when a PAD-evoked Ia spike occurred 1500 ms earlier, even though the motoneuron had long returned to its resting membrane potential [Fig. 3, see also (Duchen, 1986)]. Suppression of the soleus H-reflex at these long intervals after CPN conditioning occurs only when there is an early reflex activation of the soleus motoneurons, as measured by both the PSF and rectified EMG, with the PSF not affected by potential cross talk from the TA H-reflex that sometimes temporally overlaps with the early soleus reflex. Alternatively, if this early-latency soleus reflex was solely mediated by a polysynaptic pathway from the CPN that bypassed the soleus Ia afferents, the suppression of the H-reflex would likely not have lasted out to 2500 ms as we observe. This would also apply to any direct monosynaptic activation of soleus motoneurons by heteronymous Ia afferents from the peroneus muscle that also may have been activated during the CPN stimulation (Meunier et al., 1993). Future studies using microneurography or near-nerve recordings to measure PAD-evoked in humans, like in (Shefner et al., 1992), could provide more insight into the mechanism of post-activation depression. Likewise, it is important to examine if suppressing GABAA receptors in human participants, as with the α5 GABAA receptor neutral antagonist S44819 used in clinical stroke trials (Darmani et al., 2016; Chabriat et al., 2020), could reduce PAD evoked spikes and post-activation depression of H-reflexes as occurs in rodents (Hernandez-Reyes et al., 2019).
GABAB receptor activation.
Activation of GABAB receptors on Ia afferent terminals by the flexor CPN stimulation may also be involved in the suppression of the H-reflex and post-activation depression as we discuss above, though we detail the mechanisms further here. Activation of GABAB receptors produces presynaptic inhibition of Ia afferents and reflex transmission to motoneurons (Curtis & Lacey, 1994; Fink, 2013; Hari et al., 2022), likely by inhibiting voltage-dependent calcium channels on the presynaptic boutons to reduce the calcium-dependent exocytosis of neurotransmitter following the arrival of an action potential at the afferent terminal (Curtis & Lacey, 1994; Curtis et al., 1997; Howell & Pugh, 2016) or by inducing hyperpolarization of the afferent by opening G protein-gated, inwardly rectifying K+ (GIRK) channels (Shaye et al., 2021). In contrast to GABAA receptors, GABAB receptors are densely located on Ia afferent terminals (Hari et al., 2022) and when activated by a brief train of conditioning afferent stimulation, they can suppress monosynaptic EPSPs for up to 800 ms (Curtis & Lacey, 1994, 1998) or more (Salio et al., 2017; Hari et al., 2022). This GABAB mediated presynaptic inhibition is blocked by selective GABAB receptor antagonists (Curtis & Lacey, 1994; Fink, 2013; Hari et al., 2022), with evidence for a spontaneous amount of steady GABAB receptor mediated presynaptic inhibition at rest (Hari et al., 2022), demonstrating how long it can last. Thus, GABAB receptor activation on Ia afferent terminals could account for some of the long-lasting inhibition of the soleus H-reflex from the CPN conditioning stimulation or TA tendon vibration, requiring further study with specific antagonists to elucidate (Curtis et al., 1997). As mentioned above, we argue that these receptors are activated secondarily to PAD, with PAD-evoked spikes activating circuits that drive GABAergic innervation of extensor afferent terminals, as detailed in Figure 11.
Alternative mechanisms: Passive PAD current to Ia terminals.
Although there are sparse GABAA receptors at the afferent terminal, PAD from more proximal nodes may passively enter the afferent terminal to reduce the size of subsequent action potentials activated by the TN stimulation. However, because of the long distance (~ 800 μm) of the last node from the Ia afferent terminal (Hari et al., 2022) and the short space constant of the Ia axon (90 μm), the amount of depolarization at the Ia afferent terminal is small (Lucas-Osma et al., 2018) and only reduces the size of the action potential by 1% even when nearby. This reduction is not likely to have a noticeable effect on neurotransmitter release and suppression of H-reflexes as calculated from computer simulations of the Ia afferent (Hari et al., 2022).
Polysynaptic H-reflex pathway.
Excitatory interneurons in the fast, polysynaptic H-reflex pathway (Jankowska et al., 1981a; Burke et al., 1984) may also be suppressed by conditioning and lead to a decrease in the H-reflex that is unrelated to post-activation depression of the Ia afferent. Likewise, conditioning may also facilitate the activation of inhibitory [e.g., Ib] interneurons activated by the fast TN pathway but at later latencies. Although these effects are short-lasting (< 1s), further experiments are needed to demonstrate that flexor afferent conditioning decreases the probability of motor unit discharge within the first 0.5 ms of the H-reflex, when polysynaptic components of the H-reflex are unlikely to contribute.
Conclusion and clinical implications
We have provided several lines of evidence that H-reflex suppression from antagonist afferents is produced, in part, by direct effects on the motoneuron and from post-activation depression of Ia afferent transmission, the latter mediated by PAD-evoked Ia spikes triggered from the conditioning afferent input. These findings bring into question the interpretation of many human experiments claiming that H-reflex suppression by antagonist/heteronymous afferents are produced by PAD-mediated presynaptic inhibition of the Ia afferent terminal [reviewed in (Stein, 1995; Misiaszek, 2003; Hultborn, 2006; Willis, 2006)]. By extension, the idea that presynaptic inhibition of Ia afferents from PAD is reduced following nervous system injury or disease also needs to be reexamined, with examples including spinal cord injury (Ashby & Verrier, 1975; Mailis & Ashby, 1990; Roby-Brami & Bussel, 1990; Azouvi et al., 1993; Calancie et al., 1993; Faist et al., 1994; Aymard et al., 2000; Knikou & Mummidisetty, 2014; Caron et al., 2020), cerebral palsy (Mizuno et al., 1971; Achache et al., 2010), brain injury (Koelman et al., 1993; Faist et al., 1994), stroke (Milanov, 1992; Koelman et al., 1993) and multiple sclerosis (Azouvi et al., 1993; Koelman et al., 1993; Nielsen et al., 1995). Because the amount of H-reflex suppression has been incorrectly equated to the amplitude of PAD, changes in the activation of GABAA-receptor mediated PAD and its role in the regulation of afferent conduction in these various disorders needs to be reexamined. This is important because drug therapies that enhance GABAA receptor activation, such as the benzodiazepine diazepam (Valium), have been used to treat spasticity in some of these conditions (Simon & Yelnik, 2010; Chang et al., 2013). The greater use of GABAB antagonists, such as baclofen, to treat spasticity aligns better with our new understanding of the role of GABAB in presynaptic inhibition and post-activation depression (Li et al., 2004). Interestingly, diazepam increases PAD measured in the dorsal part of the afferent and also reduces the monosynaptic reflex (Stratten & Barnes, 1971). Thus, drug therapies that enhance the activation of GABAA receptors could actually increase afferent transmission while paradoxically increasing post-activation depression through the triggering of PAD-evoked Ia spikes or by directly facilitating inhibitory GABAA receptors on the motoneuron (Cartlidge et al., 1974; Simon & Yelnik, 2010; Chang et al., 2013). In light of our new findings, we need to investigate how PAD evoked in dorsal parts of the Ia afferent is involved in the abnormal control of afferent conduction and transmission to spinal neurons following injury or disease of the central nervous system in order to develop better methods to improve spared sensorimotor function and treat spasticity.
Key Points Summary.
Suppression of extensor H-reflexes by flexor afferent conditioning was thought to be mediated by GABAA receptor-mediated primary afferent depolarization (PAD) shunting action potentials in the Ia afferent terminal.
In line with recent findings that PAD has a facilitatory role in Ia afferent conduction, we show here that when large enough, PAD can evoke orthodromic spikes that travel to the Ia afferent terminal to evoke excitatory postsynaptic potentials in the motoneuron.
These PAD-evoked spikes also produce post-activation depression of Ia afferent terminals and may mediate the short and long-lasting suppression of extensor H-reflexes in response to flexor afferent conditioning.
Our findings highlight that we must reexamine how changes in the activation of GABAergic interneurons and PAD following nervous system injury or disease affects the regulation of Ia afferent transmission to spinal neurons and ultimately motor dysfunction in these disorders.
Acknowledgments
We thank Ms. Jennifer Duchcherer and Leo Sanelli for technical assistance.
Funding
This human work was supported by a National Science and Engineering Grant 05205 to M.A.G. and studentship funding to K.M. from the Neuroscience and Mental Health Institute and Faculty of Medicine and Dentistry at the University of Alberta. The animal work was supported by the Canadian Institutes of Health Research (CIHR MOP 14697 to D.J.B.) and the US National Institutes of Health (NIH, R01NS47567 to D.J.B).
Biography

Krista Metz received her PhD in Neuroscience from the University of Alberta, Edmonton, in 2022. She received her Bachelor’s degree in Kinesiology in 2016 from the University of Regina, Saskatchewan. Her PhD work focused on sensorimotor circuits in the human spinal cord and how changes to these circuits after injury to the central nervous system could be involved in sensorimotor impairment and spasticity. Currently, Krista works as a neurophysiologist for the Saskatchewan Health Authority at the Royal University Hospital in Saskatoon, Saskatchewan and continues research related to sensorimotor systems in the human spinal cord.
Footnotes
Preprint link: https://www.biorxiv.org/content/10.1101/2021.04.20.440509v1
Competing Interests
The authors have no competing interests to declare.
Data Availability
The human data is published in the Open Data Commons for Spinal Cord Injury http://dx.doi.org/10.34945/F5M88Z and the animal data is available upon request.
References
- Achache V, Roche N, Lamy JC, Boakye M, Lackmy A, Gastal A, Quentin V & Katz R. (2010). Transmission within several spinal pathways in adults with cerebral palsy. Brain 133, 1470–1483. [DOI] [PubMed] [Google Scholar]
- Afsharipour B, Manzur N, Duchcherer J, Fenrich KF, Thompson CK, Negro F, Quinlan KA, Bennett DJ & Gorassini MA. (2020). Estimation of self-sustained activity produced by persistent inward currents using firing rate profiles of multiple motor units in humans. J Neurophysiol 124, 63–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez FJ, Taylor-Blake B, Fyffe RE, De Blas AL & Light AR. (1996). Distribution of immunoreactivity for the beta 2 and beta 3 subunits of the GABAA receptor in the mammalian spinal cord. J Comp Neurol 365, 392–412. [DOI] [PubMed] [Google Scholar]
- Ashby P & Verrier M. (1975). Neurophysiological changes following spinal cord lesions in man. Can J Neurol Sci 2, 91–100. [DOI] [PubMed] [Google Scholar]
- Aymard C, Katz R, Lafitte C, Lo E, Pénicaud A, Pradat-Diehl P & Raoul S. (2000). Presynaptic inhibition and homosynaptic depression: a comparison between lower and upper limbs in normal human subjects and patients with hemiplegia. Brain 123 (Pt 8), 1688–1702. [DOI] [PubMed] [Google Scholar]
- Azouvi P, Roby-Brami A, Biraben A, Thiebaut JB, Thurel C & Bussel B. (1993). Effect of intrathecal baclofen on the monosynaptic reflex in humans: evidence for a postsynaptic action. J Neurol Neurosurg Psychiatry 56, 515–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldissera F, Hultborn H & Illert M. (1981). Integration in spinal systems. In Handbook of physiology sect 1 the nervous system, vol 2 motor control, ed. Brooks VB, pp. 509–595. American Physiology Society, Bethesda. [Google Scholar]
- Barron DH & Matthews BH. (1938). The interpretation of potential changes in the spinal cord. J Physiol 92, 276–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardelli A, Day BL, Marsden CD & Rothwell JC. (1987). Evidence favouring presynaptic inhibition between antagonist muscle afferents in the human forearm. J Physiol 391, 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulenguez P, Liabeuf S, Bos R, Bras H, Jean-Xavier C, Brocard C, Stil A, Darbon P, Cattaert D, Delpire E, Marsala M & Vinay L. (2010). Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat Med 16, 302–307. [DOI] [PubMed] [Google Scholar]
- Brownstone RM, Gossard JP & Hultborn H. (1994). Voltage-dependent excitation of motoneurones from spinal locomotor centres in the cat. Exp Brain Res 102, 34–44. [DOI] [PubMed] [Google Scholar]
- Burke D, Gandevia SC & McKeon B. (1984). Monosynaptic and oligosynaptic contributions to human ankle jerk and H-reflex. J Neurophysiol 52, 435–448. [DOI] [PubMed] [Google Scholar]
- Burke D, Gracies JM, Meunier S & Pierrot-Deseilligny E. (1992). Changes in presynaptic inhibition of afferents to propriospinal-like neurones in man during voluntary contractions. J Physiol 449, 673–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calancie B, Broton JG, Klose KJ, Traad M, Difini J & Ayyar DR. (1993). Evidence that alterations in presynaptic inhibition contribute to segmental hypo- and hyperexcitability after spinal cord injury in man. Electroencephalogr Clin Neurophysiol 89, 177–186. [DOI] [PubMed] [Google Scholar]
- Capaday C, Lavoie BA & Comeau F. (1995). Differential effects of a flexor nerve input on the human soleus H-reflex during standing versus walking. Can J Physiol Pharmacol 73, 436–449. [DOI] [PubMed] [Google Scholar]
- Caron G, Bilchak JN & Cote MP. (2020). Direct evidence for decreased presynaptic inhibition evoked by PBSt group I muscle afferents after chronic SCI and recovery with step-training in rats. J Physiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartlidge NE, Hudgson P & Weightman D. (1974). A comparison of baclofen and diazepam in the treatment of spasticity. J Neurol Sci 23, 17–24. [DOI] [PubMed] [Google Scholar]
- Chabriat H, Bassetti CL, Marx U, Picarel-Blanchot F, Sors A, Gruget C, Saba B, Wattez M, Audoli ML & Hermann DM. (2020). Randomized Efficacy and Safety Trial with Oral S 44819 after Recent ischemic cerebral Event (RESTORE BRAIN study): a placebo controlled phase II study. Trials 21, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang E, Ghosh N, Yanni D, Lee S, Alexandru D & Mozaffar T. (2013). A Review of Spasticity Treatments: Pharmacological and Interventional Approaches. Crit Rev Phys Rehabil Med 25, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crone C, Hultborn H, Mazières L, Morin C, Nielsen J & Pierrot-Deseilligny E. (1990). Sensitivity of monosynaptic test reflexes to facilitation and inhibition as a function of the test reflex size: a study in man and the cat. Exp Brain Res 81, 35–45. [DOI] [PubMed] [Google Scholar]
- Curtis DR. (1998). Two types of inhibition in the spinal cord. Oxford University Press, New York. [Google Scholar]
- Curtis DR & Eccles JC. (1960a). Synaptic action during and after repetitive stimulation. J Physiol 150, 374–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis DR & Eccles JC. (1960b). Synaptic action during and after repetitive stimulation. The Journal of physiology 150, 374–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis DR, Gynther BD, Lacey G & Beattie DT. (1997). Baclofen: reduction of presynaptic calcium influx in the cat spinal cord in vivo. Exp Brain Res 113, 520–533. [DOI] [PubMed] [Google Scholar]
- Curtis DR & Lacey G. (1994). GABA-B receptor-mediated spinal inhibition. Neuroreport 5, 540–542. [DOI] [PubMed] [Google Scholar]
- Curtis DR & Lacey G. (1998). Prolonged GABA(B) receptor-mediated synaptic inhibition in the cat spinal cord: an in vivo study. Exp Brain Res 121, 319–333. [DOI] [PubMed] [Google Scholar]
- Darmani G, Zipser CM, Böhmer GM, Deschet K, Muller-Dahlhaus F, Belardinelli P, Schwab M & Ziemann U. (2016). Effects of the Selective alpha5-GABAAR Antagonist S44819 on Excitability in the Human Brain: A TMS-EMG and TMS-EEG Phase I Study. J Neurosci 36, 12312–12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmedt JE & Godaux E. (1978). Mechanism of the vibration paradox: excitatory and inhibitory effects of tendon vibration on single soleus muscle motor units in man. J Physiol 285, 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. (1986). Excitation of mouse motoneurones by GABA-mediated primary afferent depolarization. Brain Res 379, 182–187. [DOI] [PubMed] [Google Scholar]
- Eccles JC, Eccles RM & Magni F. (1961a). Central inhibitory action attributable to presynaptic depolarization produced by muscle afferent volleys. J Physiol 159, 147–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Kozak W & Magni F. (1961b). Dorsal root reflexes of muscle group I afferent fibres. J Physiol 159, 128–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Schmidt R & Willis WD. (1963). Pharmacological Studies on Presynaptic Inhibition. J Physiol 168, 500–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguibar JR, Quevedo J & Rudomin P. (1997). Selective cortical and segmental control of primary afferent depolarization of single muscle afferents in the cat spinal cord. Experimental brain research 113, 411–430. [DOI] [PubMed] [Google Scholar]
- El-Tohamy A & Sedgwick EM. (1983). Spinal inhibition in man: depression of the soleus H reflex by stimulation of the nerve to the antagonist muscle. J Physiol 337, 497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faist M, Dietz V & Pierrot-Deseilligny E. (1996). Modulation, probably presynaptic in origin, of monosynaptic Ia excitation during human gait. Exp Brain Res 109, 441–449. [DOI] [PubMed] [Google Scholar]
- Faist M, Mazevet D, Dietz V & Pierrot-Deseilligny E. (1994). A quantitative assessment of presynaptic inhibition of Ia afferents in spastics. Differences in hemiplegics and paraplegics. Brain 117 (Pt 6), 1449–1455. [DOI] [PubMed] [Google Scholar]
- Fedirchuk B, Wenner P, Whelan PJ, Ho S, Tabak J & O’Donovan MJ. (1999). Spontaneous network activity transiently depresses synaptic transmission in the embryonic chick spinal cord. J Neurosci 19, 2102–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink A (2013). Exploring a behavioural role for presynaptic inhibition in spinal sensory-motor synapses. In Columbia University, pp. 1–293. Columbia University. [Google Scholar]
- Fink AJ, Croce KR, Huang ZJ, Abbott LF, Jessell TM & Azim E. (2014). Presynaptic inhibition of spinal sensory feedback ensures smooth movement. Nature 509, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank K (1959). Basic mechanisms of synaptic transmission in the central nervous system. Inst Radio Eng Trans Med Electron ME-6, 85–88. [Google Scholar]
- Hari K, Lucas-Osma AM, Metz K, Lin S, Pardell N, Roszko DA, Black S, Minarik A, Singla R, Stephens MJ, Pearce RA, Fouad K, Jones KE, Gorassini MA, Fenrich KK, Li Y & Bennett DJ. (2022). GABA facilitates spike propagation through branch points of sensory axons in the spinal cord. Nat Neurosci 25, 1288–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Reyes JE, Salinas-Abarca AB, Vidal-Cantu GC, Raya-Tafolla G, Elias-Vinas D, Granados-Soto V & Delgado-Lezama R. (2019). alpha5GABAA receptors play a pronociceptive role and avoid the rate-dependent depression of the Hoffmann reflex in diabetic neuropathic pain and reduce primary afferent excitability. Pain 160, 1448–1458. [DOI] [PubMed] [Google Scholar]
- Howell RD & Pugh JR. (2016). Biphasic modulation of parallel fibre synaptic transmission by co-activation of presynaptic GABAA and GABAB receptors in mice. J Physiol 594, 3651–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howells J, Sangari S, Matamala JM, Kiernan MC, Marchand-Pauvert V & Burke D. (2020). Interrogating interneurone function using threshold tracking of the H reflex in healthy subjects and patients with motor neurone disease. Clin Neurophysiol 131, 1986–1996. [DOI] [PubMed] [Google Scholar]
- Hughes DI, Mackie M, Nagy GG, Riddell JS, Maxwell DJ, Szabo G, Erdelyi F, Veress G, Szucs P, Antal M & Todd AJ. (2005). P boutons in lamina IX of the rodent spinal cord express high levels of glutamic acid decarboxylase-65 and originate from cells in deep medial dorsal horn. Proc Natl Acad Sci U S A 102, 9038–9043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultborn H (2006). Spinal reflexes, mechanisms and concepts: from Eccles to Lundberg and beyond. Prog Neurobiol 78, 215–232. [DOI] [PubMed] [Google Scholar]
- Hultborn H, Illert M, Nielsen J, Paul A, Ballegaard M & Wiese H. (1996). On the mechanism of the post-activation depression of the H-reflex in human subjects. Experimental brain research 108, 450–462. [DOI] [PubMed] [Google Scholar]
- Hultborn H, Meunier S, Morin C & Pierrot-Deseilligny E. (1987a). Assessing changes in presynaptic inhibition of I a fibres: a study in man and the cat. J Physiol 389, 729–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultborn H, Meunier S, Pierrot-Deseilligny E & Shindo M. (1987b). Changes in presynaptic inhibition of Ia fibres at the onset of voluntary contraction in man. J Physiol 389, 757–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iles JF. (1996). Evidence for cutaneous and corticospinal modulation of presynaptic inhibition of Ia afferents from the human lower limb. J Physiol 491 (Pt 1), 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iles JF & Pisini JV. (1992). Cortical modulation of transmission in spinal reflex pathways of man. J Physiol 455, 425–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iles JF & Roberts RC. (1987). Inhibition of monosynaptic reflexes in the human lower limb. J Physiol 385, 69–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowska E, McCrea D & Mackel R. (1981a). Oligosynaptic excitation of motoneurones by impulses in group Ia muscle spindle afferents in the cat. J Physiol 316, 411–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowska E, McCrea D, Rudomin P & Sykova E. (1981b). Observations on neuronal pathways subserving primary afferent depolarization. Journal of neurophysiology 46, 506–516. [DOI] [PubMed] [Google Scholar]
- Jankowska E, McCrea D, Rudomin P & Sykova E. (1981c). Observations on neuronal pathways subserving primary afferent depolarization. J Neurophysiol 46, 506–516. [DOI] [PubMed] [Google Scholar]
- Knikou M & Mummidisetty CK. (2014). Locomotor training improves premotoneuronal control after chronic spinal cord injury. J Neurophysiol 111, 2264–2275. [DOI] [PubMed] [Google Scholar]
- Koelman JH, Bour LJ, Hilgevoord AA, van Bruggen GJ & Ongerboer de Visser BW. (1993). Soleus H-reflex tests and clinical signs of the upper motor neuron syndrome. J Neurol Neurosurg Psychiatry 56, 776–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppanen L & Stys PK. (1997). Ion transport and membrane potential in CNS myelinated axons. II. Effects of metabolic inhibition. J Neurophysiol 78, 2095–2107. [DOI] [PubMed] [Google Scholar]
- Li Y, Li X, Harvey PJ & Bennett DJ. (2004). Effects of baclofen on spinal reflexes and persistent inward currents in motoneurons of chronic spinal rats with spasticity. J Neurophysiol 92, 2694–2703. [DOI] [PubMed] [Google Scholar]
- Lidierth M (2006). Local and diffuse mechanisms of primary afferent depolarization and presynaptic inhibition in the rat spinal cord. J Physiol 576, 309–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Hari K, Lucas-Osma AM, Black S, Khatmi A, Fouad K, Gorassini MA, Li Y, Fenrich KF & Bennett DJ. (2022). Locomotor-related propriospinal V3 neurons produce primary afferent depolarization and moduate sensory transmission to motoneurons. bioRxiv, doi: 10.1101/2022.1107.1104.498712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Li Y, Lucas-Osma AM, Hari K, Stephens MJ, Singla R, Heckman CJ, Zhang Y, Fouad K, Fenrich KK & Bennett DJ. (2019). Locomotor-related V3 interneurons initiate and coordinate muscles spasms after spinal cord injury. J Neurophysiol 121, 1352–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas-Osma AM, Li Y, Lin S, Black S, Singla R, Fouad K, Fenrich KK & Bennett DJ. (2018). Extrasynaptic alpha5GABAA receptors on proprioceptive afferents produce a tonic depolarization that modulates sodium channel function in the rat spinal cord. J Neurophysiol 120, 2953–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher HR, Ruenzel P, Fetz E & Henneman E. (1979). Postsynatpic population potentials recorded from ventral roots perfused with isotonic sucrose: connections of groups Ia and II spindle afferent fibers with large populations of motoneurons. J Neurophysiol 42, 1146–1164. [DOI] [PubMed] [Google Scholar]
- Mailis A & Ashby P. (1990). Alterations in group Ia projections to motoneurons following spinal lesions in humans. J Neurophysiol 64, 637–647. [DOI] [PubMed] [Google Scholar]
- Matthews PBC. (1996). Relationship of firing intervals of human motor units to the trajectory of post-spike after-hyperpolarization and synaptic noise. The Journal of physiology 492 (Pt 2), 597–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrea DA, Shefchyk SJ & Carlen PL. (1990). Large reductions in composite monosynaptic EPSP amplitude following conditioning stimulation are not accounted for by increased postsynaptic conductances in motoneurons. Neurosci Lett 109, 117–122. [DOI] [PubMed] [Google Scholar]
- Metz K, Concha-Matos I, Afsharipour B, Thompson CK, Negro F, Quinlan KA, Bennett DJ & Gorassini MA. (2022). Facilitation of Ia afferent transmission to motoneurons during cortical or sensory evoked primary afferent depolarization (PAD) in humans. Journal of Physiology In revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier S, Pierrot-Deseilligny E & Simonetta M. (1993). Pattern of monosynaptic heteronymous Ia connections in the human lower limb. Exp Brain Res 96, 534–544. [DOI] [PubMed] [Google Scholar]
- Milanov I (1992). A comparative study of methods for estimation of presynaptic inhibition. J Neurol 239, 287–292. [DOI] [PubMed] [Google Scholar]
- Misiaszek JE. (2003). The H-reflex as a tool in neurophysiology: its limitations and uses in understanding nervous system function. Muscle Nerve 28, 144–160. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Tanaka R & Yanagisawa N. (1971). Reciprocal group I inhibition on triceps surae motoneurons in man. J Neurophysiol 34, 1010–1017. [DOI] [PubMed] [Google Scholar]
- Morin C, Pierrot-Deseilligny E & Hultborn H. (1984). Evidence for presynaptic inhibition of muscle spindle Ia afferents in man. Neurosci Lett 44, 137–142. [DOI] [PubMed] [Google Scholar]
- Murray KC, Stephens MJ, Rank M, D’Amico J, Gorassini MA & Bennett DJ. (2011). Polysynaptic excitatory postsynaptic potentials that trigger spasms after spinal cord injury in rats are inhibited by 5-HT1B and 5-HT1F receptors. J Neurophysiol 106, 925–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima K, Rothwell JC, Day BL, Thompson PD & Marsden CD. (1990). Cutaneous effects on presynaptic inhibition of flexor Ia afferents in the human forearm. J Physiol 426, 369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negro F, Muceli S, Castronovo AM, Holobar A & Farina D. (2016). Multi-channel intramuscular and surface EMG decomposition by convolutive blind source separation. J Neural Eng 13, 026027. [DOI] [PubMed] [Google Scholar]
- Neher E & Sakaba T. (2001). Estimating transmitter release rates from postsynaptic current fluctuations. J Neurosci 21, 9638–9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J, Petersen N, Ballegaard M, Biering-Sørensen F & Kiehn O. (1993). H-reflexes are less depressed following muscle stretch in spastic spinal cord injured patients than in healthy subjects. Exp Brain Res 97, 173–176. [DOI] [PubMed] [Google Scholar]
- Nielsen J, Petersen N & Crone C. (1995). Changes in transmission across synapses of Ia afferents in spastic patients. Brain 118 (Pt 4), 995–1004. [DOI] [PubMed] [Google Scholar]
- Norton JA, Bennett DJ, Knash ME, Murray KC & Gorassini MA. (2008). Changes in sensory-evoked synaptic activation of motoneurons after spinal cord injury in man. Brain 131, 1478–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JP & Mendell LM. (1993). Quantitative ultrastructure of Ia boutons in the ventral horn: scaling and positional relationships. J Neurosci 13, 4748–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers RK & Binder MD. (2001). Input-output functions of mammalian motoneurons. Rev Physiol Biochem Pharmacol 143, 137–263. [DOI] [PubMed] [Google Scholar]
- Roby-Brami A & Bussel B. (1990). Effects of flexor reflex afferent stimulation on the soleus H reflex in patients with a complete spinal cord lesion: evidence for presynaptic inhibition of Ia transmission. Exp Brain Res 81, 593–601. [DOI] [PubMed] [Google Scholar]
- Rossi A, Decchi B & Ginanneschi F. (1999). Presynaptic excitability changes of group Ia fibres to muscle nociceptive stimulation in humans. Brain Res 818, 12–22. [DOI] [PubMed] [Google Scholar]
- Rudomin P (1990). Presynaptic inhibition of muscle spindle and tendon organ afferents in the mammalian spinal cord. Trends Neurosci 13, 499–505. [DOI] [PubMed] [Google Scholar]
- Rudomin P & Schmidt RF. (1999). Presynaptic inhibition in the vertebrate spinal cord revisited. Exp Brain Res 129, 1–37. [DOI] [PubMed] [Google Scholar]
- Salio C, Merighi A & Bardoni R. (2017). GABAB receptors-mediated tonic inhibition of glutamate release from Abeta fibers in rat laminae III/IV of the spinal cord dorsal horn. Mol Pain 13, 1744806917710041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler-Ivens S & Shields RK. (2000). Low frequency depression of H-reflexes in humans with acute and chronic spinal-cord injury. Exp Brain Res 133, 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaye H, Stauch B, Gati C & Cherezov V. (2021). Molecular mechanisms of metabotropic GABA(B) receptor function. Sci Adv 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shefner JM, Buchthal F & Krarup C. (1992). Recurrent potentials in human peripheral sensory nerve: possible evidence of primary afferent depolarization of the spinal cord. Muscle Nerve 15, 1354–1363. [DOI] [PubMed] [Google Scholar]
- Sherrington CS. (1908). On reciprocal innervation of antagonist muscles. Twelfth Note - Proprioceptive reflexes. Proc R Soc Lond B Biol Sci 80, 552–564. [Google Scholar]
- Simon O & Yelnik AP. (2010). Managing spasticity with drugs. Eur J Phys Rehabil Med 46, 401–410. [PubMed] [Google Scholar]
- Stein RB. (1995). Presynaptic inhibition in humans. Prog Neurobiol 47, 533–544. [DOI] [PubMed] [Google Scholar]
- Stratten WP & Barnes CD. (1971). Diazepam and presynaptic inhibition. Neuropharmacology 10, 685–696. [DOI] [PubMed] [Google Scholar]
- Stuart GJ & Redman SJ. (1992). The role of GABAA and GABAB receptors in presynaptic inhibition of Ia EPSPs in cat spinal motoneurones. J Physiol 447, 675–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turker KS & Powers RK. (1999). Effects of large excitatory and inhibitory inputs on motoneuron discharge rate and probability. J Neurophysiol 82, 829–840. [DOI] [PubMed] [Google Scholar]
- Türker KS & Powers RK. (2005). Black box revisited: a technique for estimating postsynaptic potentials in neurons. Trends Neurosci 28, 379–386. [DOI] [PubMed] [Google Scholar]
- Ueno M, Nakamura Y, Li J, Gu Z, Niehaus J, Maezawa M, Crone SA, Goulding M, Baccei ML & Yoshida Y. (2018). Corticospinal Circuits from the Sensory and Motor Cortices Differentially Regulate Skilled Movements through Distinct Spinal Interneurons. Cell Rep 23, 1286–1300 e1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis WD. (2006). John Eccles’ studies of spinal cord presynaptic inhibition. Prog Neurobiol 78, 189–214. [DOI] [PubMed] [Google Scholar]
- Willis WD Jr. (1999). Dorsal root potentials and dorsal root reflexes: a double-edged sword. Exp Brain Res 124, 395–421. [DOI] [PubMed] [Google Scholar]
- Yavuz US, Negro F, Diedrichs R & Farina D. (2018). Reciprocal inhibition between motor neurons of the tibialis anterior and triceps surae in humans. J Neurophysiol 119, 1699–1706. [DOI] [PubMed] [Google Scholar]
- Yavuz US, Negro F, Sebik O, Holobar A, Frommel C, Turker KS & Farina D. (2015). Estimating reflex responses in large populations of motor units by decomposition of the high-density surface electromyogram. J Physiol 593, 4305–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Vierock J, Yizhar O, Fenno LE, Tsunoda S, Kianianmomeni A, Prigge M, Berndt A, Cushman J, Polle J, Magnuson J, Hegemann P & Deisseroth K. (2011). The microbial opsin family of optogenetic tools. Cell 147, 1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The human data is published in the Open Data Commons for Spinal Cord Injury http://dx.doi.org/10.34945/F5M88Z and the animal data is available upon request.