

Abstract
Amorphous pharmaceuticals often possess a wide range of molecular conformations and bonding arrangements. The x-ray pair distribution function (PDF) method is a powerful technique for the characterization of variations in both intra-molecular and inter-molecular packing arrangements. Here, the x-ray PDF of amorphous Indomethacin is shown to be particularly sensitive to the preferred orientations of the chlorobenzyl ring found in isomers in the crystalline state. In some cases, the chlorobenzyl ring has no preferred torsional angle in the amorphous form, while in others evidence of distinct isomer orientations are observed. Amorphous samples with no preferred torsion angles of the chlorobenzyl ring are found to favor enhanced intermolecular hydrogen bonding, and this is reflected in the intensity of the first sharp diffraction peak. These significant variations in structure rule out amorphous Indomethacin as a possible standard for x-ray PDF measurements. At high humidity, time resolved PDF’s for >40 h reveal water molecules forming hydrogen bonds with Indomethacin molecules. A simple linear hydrogen bond model indicates that water molecules in the wet amorphous form have similar hydrogen bond strengths to those found between Indomethacin dimers or chains in the dry amorphous form.
Keywords: Structure, X-ray powder diffraction, Pair distribution function, NMR spectroscopy, Glass
Introduction
Amorphous Indomethacin is often regarded as a model compound, due to the wealth of information about the formation and characterization of this more soluble form.1–3 The intra-molecular and inter-molecular structural changes in amorphous pharmaceuticals associated with manufacturing, storage and exposure to high humidity can significantly affect their stability, solubility and bioavailability,4–6 yet are generally less well characterized. Indeed, the bioavailability of Indomethacin has been investigated in rabbits and it has been shown that the glassy form exhibits a 20–30% increase over the crystalline form.7 For metastable glassy and amorphous materials, the method of preparation and thermal history can significantly affect the structure, dissolution and crystallization behavior.8,9 Kamwar et al10 previously compared six amorphous Indomethacin samples prepared using different techniques, which exhibited a wide range of x-ray diffraction patterns in the region of the first sharp diffraction peak together with differing stabilities. Samples milled from the γ form were the least stable and the most stable form was bulk melt quenched glass. Generally, cryo-milled amorphous Indomethacin samples have a Tg a few degrees lower than quenched glassy samples and the much larger surface area leads to crystallization rates that are an order of magnitude faster.11 Temperature and humidity effects on the physical properties of amorphous molecules have been studied for decades,12,13 however details of the kinetics associated with structural rearrangements during hydration are less common. Amorphous Indomethacin can easily absorb small quantities of water, which acts as a plasticizer and lowers the glass transition temperature.1 The presence of water is known to increase molecular mobility, lower relaxation times and increases crystallization rates.14
High energy x-ray diffraction studies of (low-Z) amorphous pharmaceuticals is a relatively new application, that builds on the long history of Pair Distribution Function (PDF) analysis in glass diffraction,15 and enables more accurate and higher resolution investigations of organic materials.16 Here we present in-situ high energy x-ray measurements on both dry Indomethacin, and the early stages of humidity induced effects on amorphous Indomethacin. The characterization of dry amorphous Indomethacin is supported by solid state NMR (ssNMR), differential scattering calorimetry (DSC) and Raman spectroscopy measurements. While all of these techniques provide valuable insights into different aspects of the molecular structure and properties, x-ray PDF is shown to give a broad overview of the material structure, against which molecular models can be readily tested. Motivation for this study originated at the first Spring Pharmaceutical Synchrotron X-ray Powder Diffraction Workshop at Purdue University in May 2018. Amorphous Indomethacin was identified for use as a potential standard for total scattering (PDF) studies of amorphous pharmaceuticals at synchrotron beamlines across the globe. In this context we compare the total x-ray structure factors and corresponding pair distribution functions of several amorphous Indomethacin samples and the influence of water uptake on amorphous structure. The results highlight the issues associated with the characterization of metastable organic amorphous forms, and are discussed with respect to other techniques applied. In this way, we assess the usefulness of the x-ray PDF technique in exploring both the local and intermediate range ordering of molecules in different conformations.
Experimental
Materials and Sample Preparation for X-ray, NMR, DSC, and Raman
A summary of the samples studied in this work is shown in table 1. Crystalline γ-Indomethacin (>98% from Tokyo Chemical Industry) was used as received without further purification. Chlorobenzyl-d4-Indomethacin (98% from Cambridge Isotope Laboratories) was used as received in the crystalline solid form (α-polymorph) without further purification. Nuclear magnetic resonance (NMR) experiments were performed on the crystalline material as received. Subsequently, the crystalline samples were melted at 170 °C (sufficiently above the 161°C melting temperature (Tm), as previously reported,17,18 and measured in this work as 160.6 °C by differential scanning calorimetry (DSC)), and quenched in liquid nitrogen to form amorphous Indomethacin. The resulting amorphous sample was then loaded into a zirconia rotor for NMR data collection. In addition, small amounts of the sample were also used to collect Raman spectra and DSC thermograms as described in the supplementary information, along with descriptions of the recrystallization procedures for the other polymorphs.
Table 1.
Samples Prepared and Characterized in this Study.
| Sample | Laboratory | Preparation | Measurements | Storage & Transport | Number & phase from XRD |
|---|---|---|---|---|---|
|
| |||||
| I | Purdue University, USA | Melt quenched γ-form in liquid nitrogen. Cryoground. | 6-ID-D, APS | Liquid Nitrogen | 2 × Amorphous |
| II | Arizona State University, USA | Melt quenched γ-form in liquid nitrogen. Powder. | 6-ID-D, APS, NMR, Raman, DSC | Ice pack | 3 × Amorphous 7 × Crystalline |
| III | Arizona State University, USA | Melt quenched d4-chlorobenzoyl γ-form in liquid nitrogen. Powder. | 6-ID-D, APS, NMR, Raman, DSC | Ice pack | 1 × Amorphous |
| IV | Zurich, Switzerland | Melt quenched γ-form in liquid nitrogen. Powder. | I15–1 (XPDF), Diamond Light Source, UK. | Ice pack | 1 × Amorphous |
| V | Purdue University, USA | Melt quenched γ-form in liquid nitrogen. Cryoground. | 6-ID-D, APS | Liquid Nitrogen | 4 × Amorphous |
| VI | Arizona State University, USA | Melt quenched α-form in liquid nitrogen. Powder. | 6-ID-D, APS, NMR, Raman, DSC | Ice pack | 2 × Mixed Amorphous & small crystals 5 × Crystalline |
| VII | Arizona State University, USA | Melt quenched δ-form in liquid nitrogen. Powder. | 6-ID-D, APS, NMR, Raman, DSC | Ice pack | 3 × Crystalline 1 × Mixed Amorphous & small crystals |
NMR Experiments
Magic angle spinning (MAS) NMR data for chlorobenzyl-d4-Indomethacin was collected on a Bruker 400 MHz Avance III NMR spectrometer equipped with a 2.5 mm TriGamma MAS probe. 1H–13C Cross-polarization (CP)-MAS spectra were collected at a spinning frequency of 20 kHz, a 2 ms contact pulse, 20 ms acquisition time, 90 kHz 1H TPPM decoupling, and a 10 second relaxation delay. 2H MAS spectra was collected with 5 kHz MAS and a 2 μs 45 deg excitation pulse and dwell time of 1.25 μs. For non-deuterated Indomethacin stock, MAS NMR spectra were collected on a Varian 400 MHz NMR spectrometer using a 1.6 mm triple resonance Fast-MAS probe tuned to 1H and 13C frequencies of 100 MHz and 80 MHz, respectively. 1H–13C Cross-polarization (CP)-MAS spectra were collected at a spinning frequency of 20 kHz, a 1.5 ms contact pulse, 40 ms acquisition time, 80 kHz 1H TPPM decoupling, and a 20 second relaxation delay. All NMR data was processed using Bruker Topspin software.
High-energy x-ray Diffraction Experiments
Cryoground, fluffy amorphous Indomethacin samples were loaded cold, into horizontally mounted 1.5 mm diameter polyimide tubes and a portion of the tube removed, to allow the high energy x-ray beam to pass straight through the powdered sample and expose the sample to varying RH conditions within the chamber. High-energy x-ray measurements were performed on beamline 6-ID-D at the Advanced Photon Source (APS) at Argonne National Laboratory. The setup and correction procedures have been previously described in detail.19 Several independent experiments outlined in table 1 were carried out using a monochromatic x-ray beam E = 100 keV (λ=0.124 Å) collimated to a square 0.5 mm cross section. Two experiments on “dry” amorphous Indomethacin were performed at 7% RH using a Varex (CT4343) area detector (denoted samples I and V) and a time-resolved humidity experiment on amorphous Indomethacin sample I was performed at 80% RH using a Pilatus 2 M CdTe detector over a period of 40 h. In both cases NIST CeO2 powder was used for sample-detector distance calibration, which was set to 360 mm and 285 mm respectively in order to balance resolution and Q-range. The humidity chamber (Electro-Tech Systems, Inc. model 5503) was at a temperature of 22 °C throughout the experiment and diffraction measurements were taken every 100 s and summed every 10 runs. Supporting high energy x-ray experiments were carried out on amorphous Indomethacin samples at ambient humidity (samples II, IV at the APS) and sample IV at beamline I15–1 (XPDF) on the Diamond Light Source. In addition, sample III corresponding to the melt quenched d4-chlorobenzoyl γ-form used in the ssNMR measurements was also characterized with x-rays at the APS.
The total x-ray scattering data were analyzed as described previously using Fit2D and PDFgetX2.20 In brief, geometrical, polarization, background and attenuation corrections were applied to all data sets. For the Varex detector dark current corrections were applied. Additional masking was needed for the Pilatus 2 M detector due to the dead zones between detector elements and residual trapped excited states.19 The total x-ray structure factor S(Q) was subsequently extracted, where
And the associated differential x-ray pair distribution function (PDF)
Results & Discussion
The Indomethacin molecule is comprised of a largely hydrophobic indole and chlorobenzyl groups, and several hydrophilic groups: namely an amide, methoxyl and carboxylic acid that can act as donor or acceptor sites.21 γ Indomethacin is the stable crystalline form which exists only as the Z isomer22 and α is a denser metastable crystalline form comprising of three different isomers Z, E and α3.23 The structure of δ-Indomethacin has recently be found to consist of a dimer of the Z and E isomers,24 connected by double hydrogen bonding through their carboxylic acid groups. The three isomers, shown in Fig. 1, exhibit a large displacement of the chlorobenzyl group.
Figure 1.
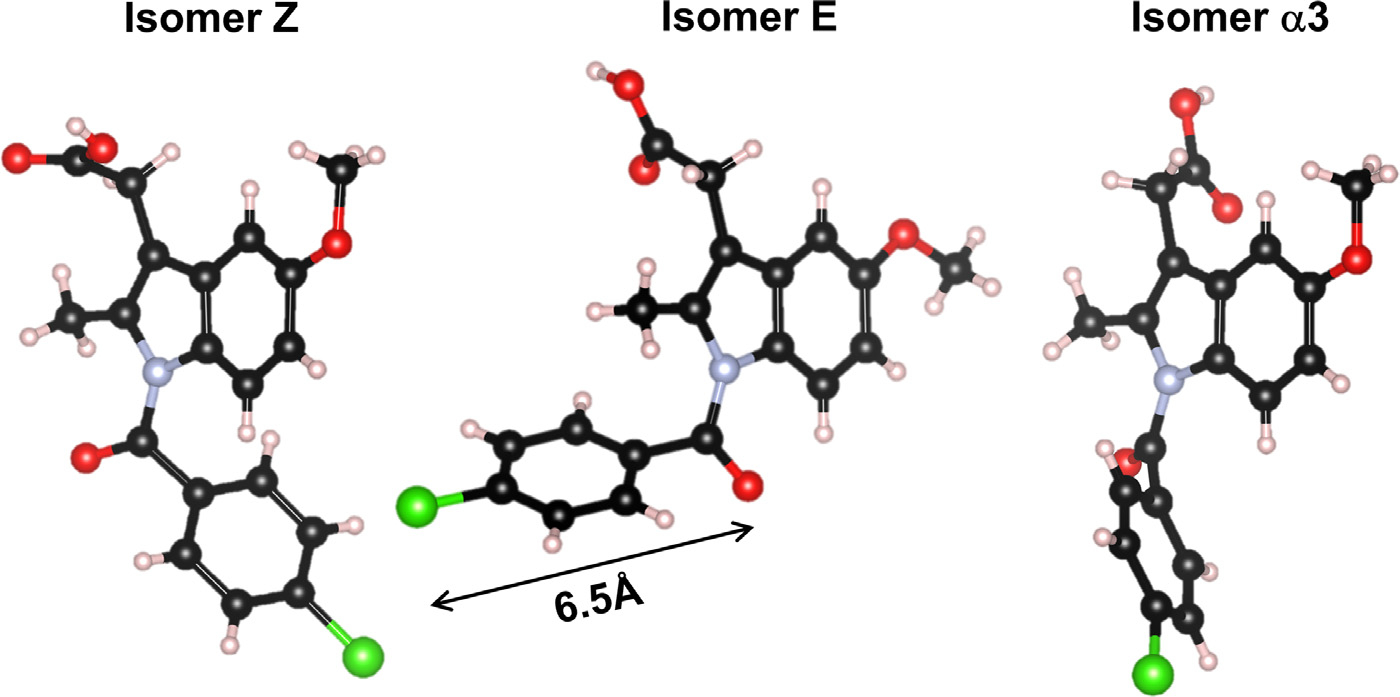
Three isomers for Indomethacin present in the α and γ forms.
NMR, dsc and Raman for Dry Amorphous Indomethacin
Samples made at ASU for NMR, DSC, and Raman characterization were made without humidity control. On the days that samples were made or handled, the relative humidity ranged from 11 to 21%. While this was marginally higher than the 7% RH dry condition for the x-ray experiments, it is far below the 43% RH limit known to result in crystallization of the γ form at temperatures below or near Tg.5 The 13C CP MAS spectra of the natural abundance crystalline γ- and α-Indomethacin samples are shown in Fig. 2 (a and d, respectively) along with the amorphous samples made from melt quenching each form (b and c). The chlorobenzyl-d4-Indomethacin was received as the α polymorph, and the 13C CP MAS spectra of the crystalline and melt quench amorphous forms for the same are given in Fig. 3a. The full resonance assignments have previously been reported for both polymorphs18,25 as well as the amorphous γ melt quench. Our results for both polymorphs and their respective amorphous forms, along with the crystalline chlorobenzyl-d4-α-Indomethacin sample agree with these studies,18,25 with the expected lower peak intensities in the range of 128–134 ppm for the chlorobenzyl ring due to small 1H–13C CP contribution in the >98% 2H sample. These samples also exhibit the characteristic transition temperatures and enthalpy changes (figure S1) that have been previously demonstrated for both polymorphs and Indomethacin glasses.5,26–28
Figure 2.
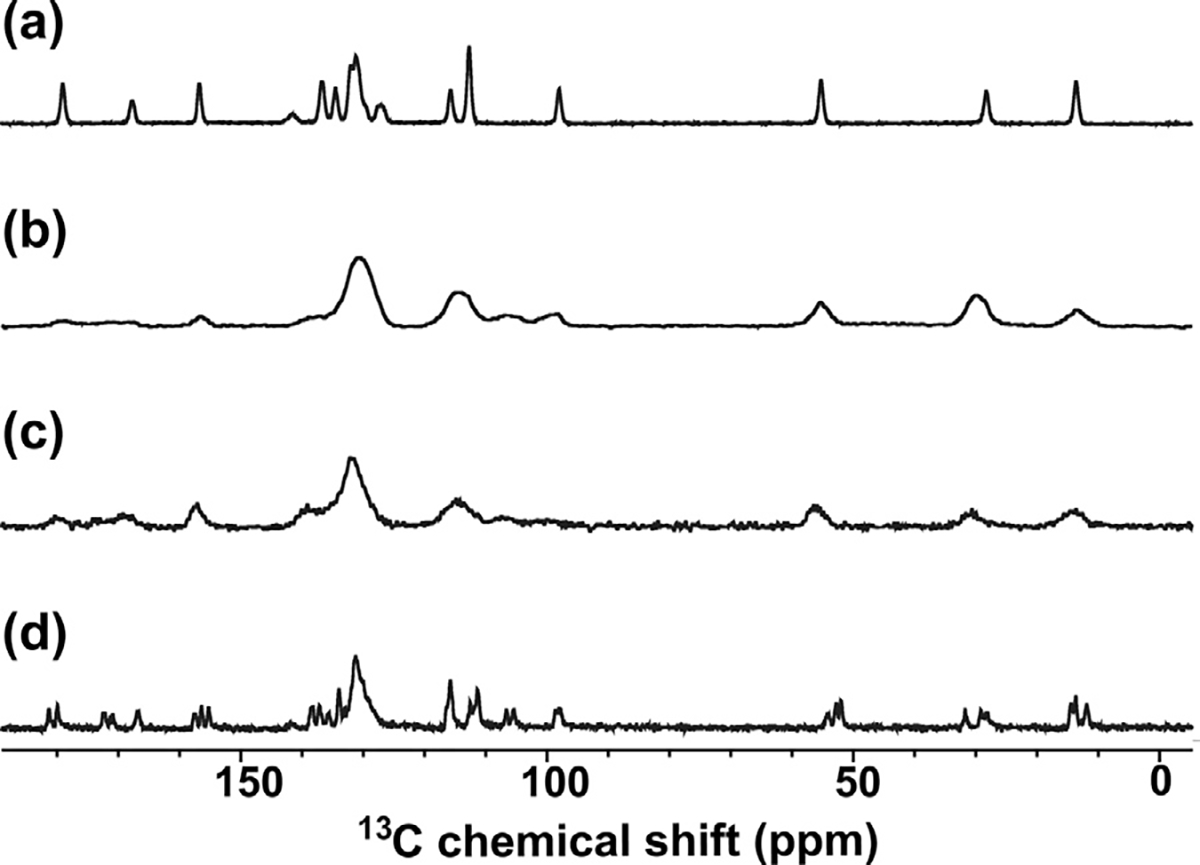
Solid state 1H→13C CP MAS (ωr = 20 kHz) NMR spectra of natural abundance Indomethacin polymorphs and their melt-quenched amorphous solids. (a) Crystalline γ-Indomethacin, (b) melt-quenched amorphous Indomethacin from crystalline γ-Indomethacin precursor; (c) melt-quenched amorphous Indomethacin from crystalline α-Indomethacin precursor, and (d) crystalline α-Indomethacin.
Figure 3.
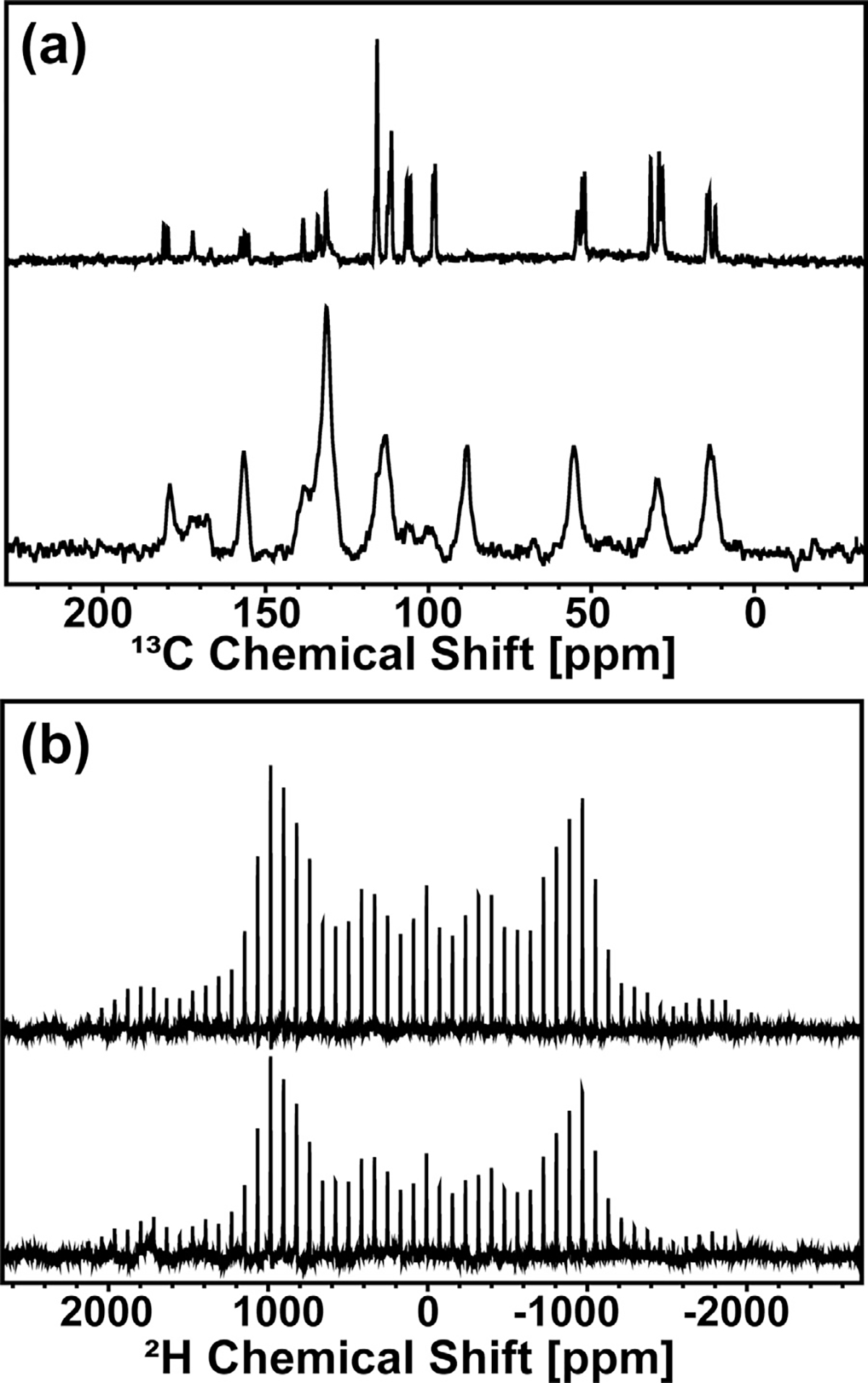
Solid state MAS NMR spectra of crystalline α polymorph (top) and melt quenched (bottom) chlorobenzyl-d4-Indomethacin (CIL, 98%). (a) 1H–13C CP MAS (ωr = 20 kHz) NMR spectra and (b) 2H MAS (ωr = 5 kHz) NMR spectra.
Unsurprisingly, melt-quenched samples prepared with the same thermal protocols have similar ssNMR spectra with closely matched peak integral values. However, the 13C spectrum for the amorphous chlorobenzyl-d4-α-Indomethacin sample (Fig. 3a; bottom) has a peak at 88.6 ppm corresponding to the carbon-4 peak of δ-indomethacin.39 This peak corresponds to a <0.5% impurity, estimated by comparing the peak enthalpy of the very small peak at 127°C for the 2H sample in the DSC thermogram to the melting peak enthalpy of the predominant α-polymorph of the same sample (see figure S1A). We have included the thermogram of the natural abundance δ-indomethacin for comparison; the 2H sample has slightly lower Tg and both melting transitions as is common for deuterated molecules compared with their proton-containing counterparts. The δ-phase does not recrystallize out of the melt-quenched amorphous solid from either α- or γ-Indomethacin, see figure S1B. Since δ-indomethacin is a dimer of the Z and E isomer configurations present in the α-crystalline phase; the spinning conditions for CP-MAS of 20 kHz can heat the rotor up to 35°C, allowing for the nucleation of these dimers within the amorphous solid. This results in the greater resolution of peaks in the amorphous 2H CPMAS compared with the amorphous α- or γ-phase samples, along with the peak shift of carbon-4 to 88.6 ppm, although the peaks are still broad compared with the crystalline samples due to their mostly amorphous nature. The larger rotor size used for the 2H samples due to the different probe needed for 2H NMR allowed for greater heating when spinning at the same 20 kHz speed, and thus this nucleation was only observed for amorphous 2H IMC and not for the natural abundance samples. We do not observe a significant nucleation of δ-phase in the 2H MAS data, because of the much lower spinning speed used for these measurements.
The 2H MAS NMR of 2H-chlorobenzyl-d4-Indomethacin allowed for measurements to observe the structural and dynamic processes of the chlorobenzyl ring in both crystalline and amorphous solid states by obtaining information about the 2H isotropic chemical shift.29,30 The chlorobenzyl ring in Indomethacin is shown to have significant configurational changes between the Z, E, and α3 isomers present in the α-polymorph. Fig. 3b gives the 2H MAS spectra of the α polymorph crystalline chlorobenzyl-d4-Indomethacin and the corresponding melt quenched glass. Both spectra exhibit a single manifold of spinning side-bands, with slight asymmetry in intensity due to the differences in the chlorobenzyl ring configuration relative to the adjacent carboxamide anisotropies. The two spectra are very similar, which gives further support to the observation that the same three isomers are present in the amorphous Indomethacin sample as are observed in the metastable α polymorph. The most obvious difference is that there are lower intensities for the spinning sidebands at the highest absolute ppm values for the amorphous sample, which is a well-known property of MAS NMR, where spinning side bands are sharper and more numerous for crystalline samples which have MAS rates lower than the chemical anisotropy of the molecule. There are only a few notable differences in relative peak intensities between the two samples: at −100 and −400 ppm the crystalline sample has higher intensity, and at 100 and 400 ppm the crystalline sample has lower intensity. Ring configuration changes result in major manifold changes; these very subtle differences are consistent with the presence of different amounts of δ-phase dimerization in the crystalline and amorphous samples.
The Raman spectrum of the crystalline 2H indomethacin sample matches that of the α polymorph, and confirms the low concentration of δ-phase (and absence of γ-phase) impurities (fig. S2). The Raman spectrum of natural abundance amorphous indomethacin (fig. S3) show three small peaks between 1550 and 1710 cm−1 for amorphous Indomethacin, which have been assigned to C = O stretch and hydrogen bonded carboxylic acid moieties in Indomethacin.26,31,32 A more detailed discussion of the DSC and Raman data, as well as powder x-ray diffraction data (fig. S4), is given in the supporting information.
High Energy X-ray Diffraction of Dry Amorphous Indomethacin
In all cases, only the starting γ-phase formed melt quenched x-ray amorphous samples. Melt quenched α- and δ-phases formed crystals or mixed amorphous and crystalline forms. The corrected and normalized x-ray intensities for dry amorphous Indomethacin are shown in Fig. 4(a). Also shown is the hydrated minus dry amorphous forms normalized to the quantum molecular form factor for water33 assuming the difference is due to water sorption. The maximum change in magnitude of the measured x-ray intensities between dry and wet Indomethacin occurs at the first peak position and is ~5%. This is correlated with a ~2% mass increase over a few days reported by others.1,34 Raman spectra have shown that the presence of water can be detected in amorphous Indomethacin after 10–20 min exposure to high humidity, but crystallization can occur anywhere between <1 day to 3 weeks.1,10 Fig. 4(b) shows the total atomic x-ray structure factors for amorphous Indomethacin multiplied by Q to highlight the weaker oscillations at high-Q.
Figure 4.
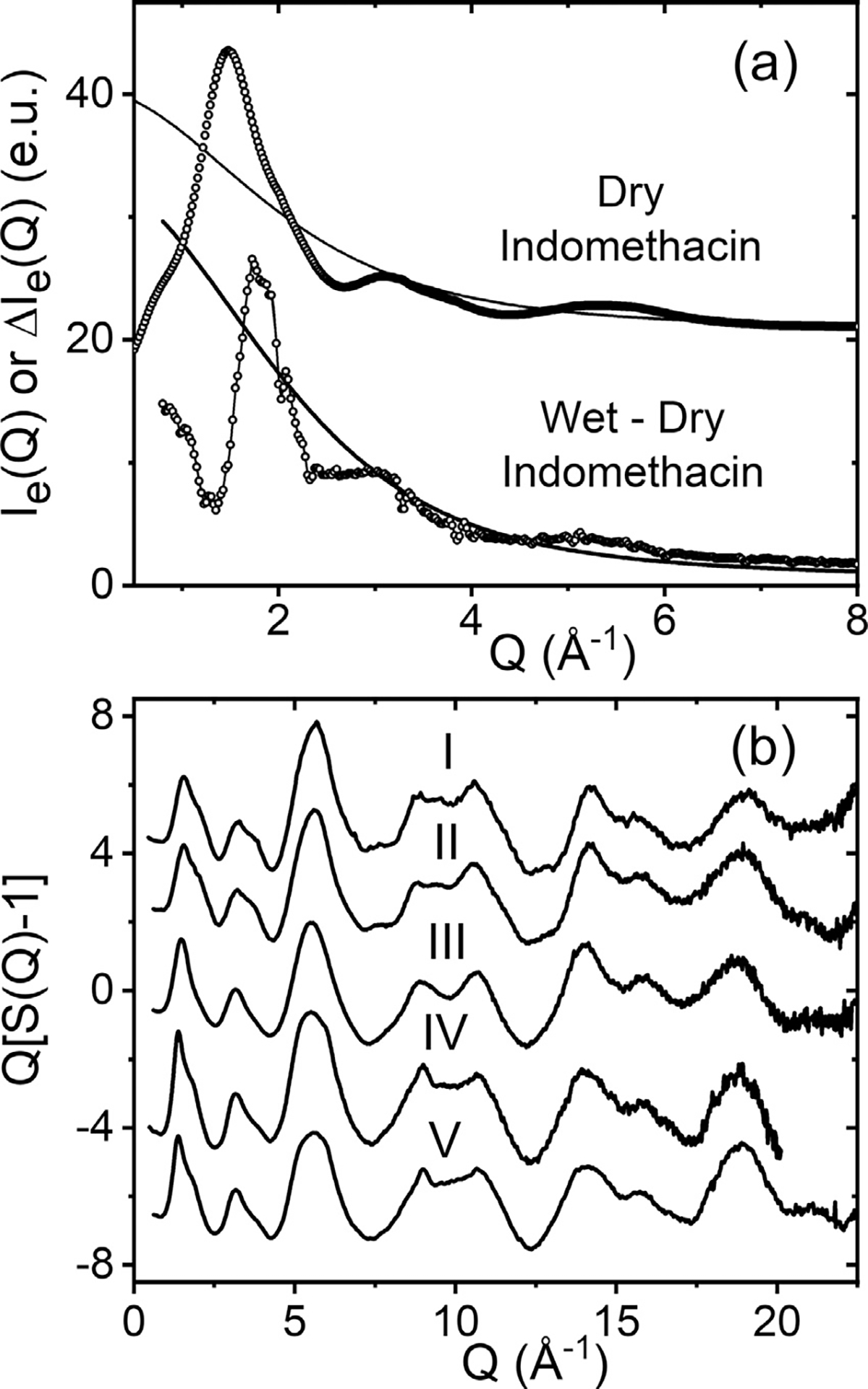
The measured x-ray intensities of amorphous Indomethacin. (a) dry Indomethacin (circles) normalized to the x-ray form factor (solid blue line) offset by +20. Wet minus dry amorphous Indomethacin, after >40 h exposure to 80% RH (circles), normalized to the molecular form factor of water (blue solid line, see text) (b) five independent measurements of the total x-ray structure factor for amorphous Indomethacin (see table 1).
The differential x-ray pair distribution functions for three dry amorphous Indomethacin samples are shown in Fig. 5. Early x-ray studies on amorphous Indomethacin using a laboratory x-ray diffractometer compared amorphous to γ and α Indomethacin but had a limited Q-range.3 More recently synchrotron based high-energy x-ray diffraction measurements have been performed over a wide Q-range to extract accurate pair distribution functions.2,35 Billinge et al.35 compared melt-quenched nano-crystalline Indomethacin to crystalline γ and α Indomethacin and found that it did not have the same local structure. Shi et al.2 studied amorphous films of Indomethacin and observed similar intramolecular components. Both of these PDF’s are generally similar to the three measured in this study, but with some variations in the 3–7 Å region.
Figure 5.
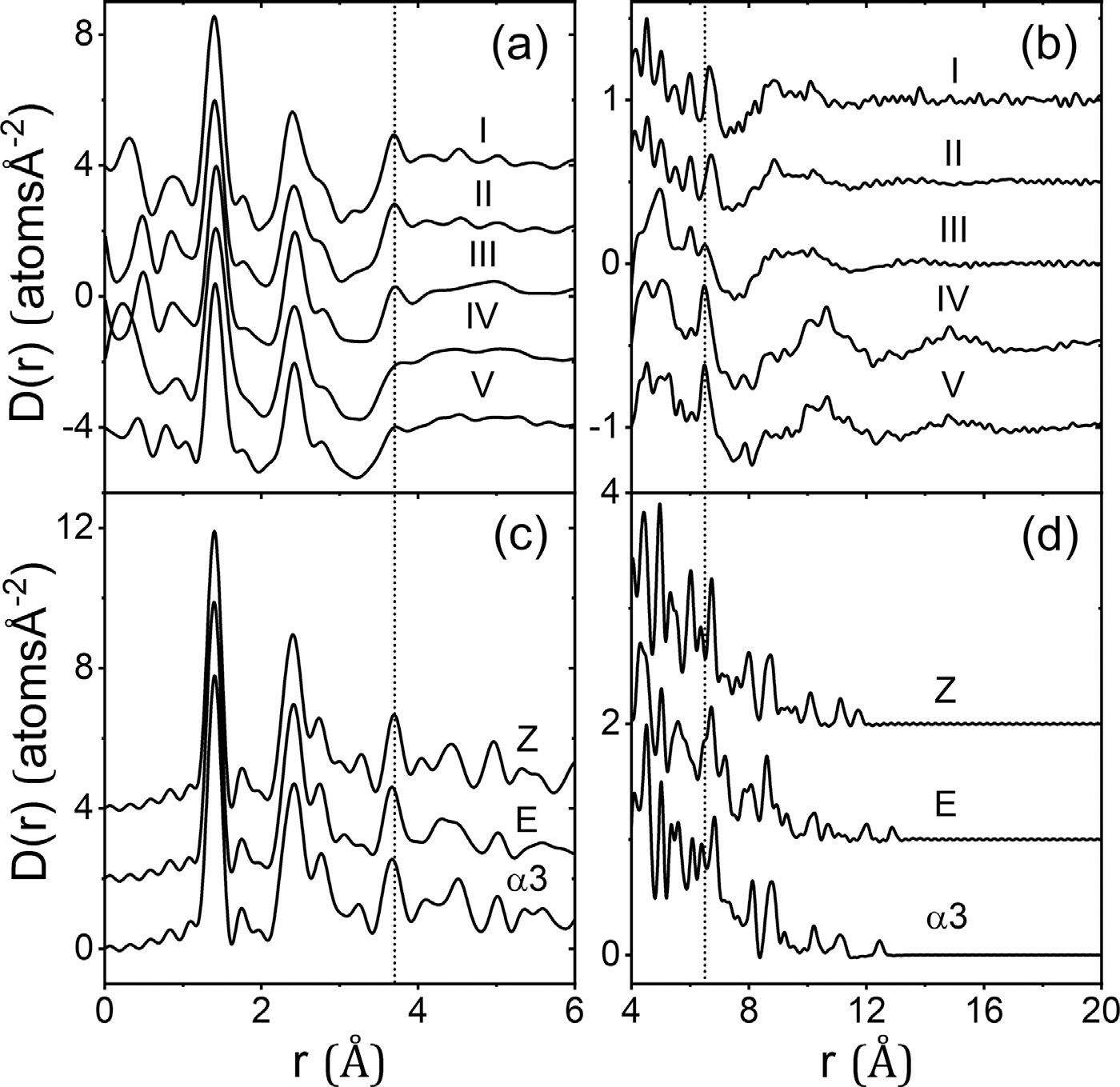
Five independently measured differential atomic pair-distribution functions D(r) for dry amorphous Indomethacin (a) and (b), compared to those calculated for the Z, E and α3 isomers (c) and (d), offset for clarity.
The x-ray PDF’s were compared to models of the three different isomers using the xINTERPDF program36 that calculates, fits and subtracts the PDF of a single molecule to separate the intra-molecular and inter-molecular contributions37. Samples IV and V show an absence of peaks at 3.7 Å, 4.4 Å and 5.0 Å compared to all three isomers. Samples I, II and III and the x-ray PDF’s of Billinge et al35 and Shi et al.2 all show a sharp peak at 3.7 Å. In the three isomers these peaks can be largely attributed to the geometries of the hindered interactions of the chlorobenzyl ring with respect to the amide ring. This suggests a near free rotation of the chlorobenzyl ring in samples IV and V, but a preferred geometry of the chlorobenzyl ring, more consistent with the known isomer orientations in samples I, II and the ssNMR findings. The 6.5 Å peak in the isomers can be mainly attributed to interactions between the strongly scattering Chlorine atom and the amide group, although overlapping correlations from hindered chlorobenzyl ring orientations may also contribute. We note that Raman spectroscopy performed on amorphous forms fabricated by different methods shows significant ring deformation,10 which may partly explain slight changes in shape and position. Taken together with the x-ray PDF data for samples I and II, the 2H MAS NMR data suggests that the variable rotation of the chlorobenzyl group is similar between the α polymorph and amorphous Indomethacin. In addition, there is more significant variation in the carboxylic acid driven dimerization or more complex hydrogen bonding chain patterns found in amorphous Indomethacin, especially in the presence of water during the glass formation or aging, the latter process leading to eventual crystallization of the γ polymorph at low temperatures and high%RH.
Carboxylic acid dimers are dominant intermolecular hydrogen bonded interactions in γ and α crystalline forms. The intermolecular hydrogen-bonded O–O distances in γ and α Indomethacin occur at 2.67 Å and 2.59–2.74 Å respectively.23 Molecular dynamics simulations of amorphous Indomethacin predict more complex patterns of hydrogen bonding chains.21,38 Interestingly, in Fig. 5 samples I, II and III, show a more ordered intramolecular structure in the region of 4<r (Å)<6 but no inter-molecular ordering in the region r>10 Å, whereas the opposite is observed for samples IV and V. The degree of intermolecular ordering is also reflected in the intensity of the first sharp diffraction peak (FSDP) in S(Q) which is ~20% less for samples I, II and III, compared to samples IV and V. In addition, the position of the FSDP shifts from Q = 1.36 Å−1 (V) to 1.53 Å−1 for sample I. Taking the real space changes together with those in the FSDP suggests a competition between preferred intra-molecular conformations of the molecule, and the degree of inter-molecular hydrogen bonding in the amorphous form. In particular, for forms with a variable rotation of the chlorobenzyl group, it appears the more flexible molecule can adopt orientations that favor inter-molecular hydrogen bonding. Furthermore, we note that similarly structured amorphous x-ray PDFs were obtained from samples prepared at different institutions (I & II versus IV & V), using slightly different procedures and stored under different conditions. Since Tg is significantly below room temperature, the increased ordering maybe due to sorbed water which enables amorphous indomethacin to crystallize “well below Tg over relatively short time scales”.1
High Energy X-ray Diffraction from Amorphous Indomethacin at High Humidity
At low water contents (<43% RH) or temperatures below or near Tg, amorphous Indomethacin crystallizes into the γ form.1,39 At high water contents or higher temperatures the α form crystallizes out.17,39 Crystallization of amorphous Indomethacin in aqueous solution as a function of pH has identified three more polymorphs ε, ζ and η but their crystal structures have not yet been solved,40 whereas the δ form crystallizes from methanol solution.24 The change in the differential electron pair-distribution function at 80%RH as a function of time is shown in Fig. 6. Here we show the electron form of the PDF (rather than the atomic form) to take advantage of the better statistics at high-Q. A main feature is the growth of a peak at 4.4 Å which plateaus at a time of >10 h of exposure, describing the formation of the metastable partially hydrated amorphous form. The rate of peak growth can be fit with a Fickian diffusion-type exponential decay curve, akin to that previously used to model water sorption in amorphous Indomethacin,1,34 see Fig. 6 insert.
Figure 6.
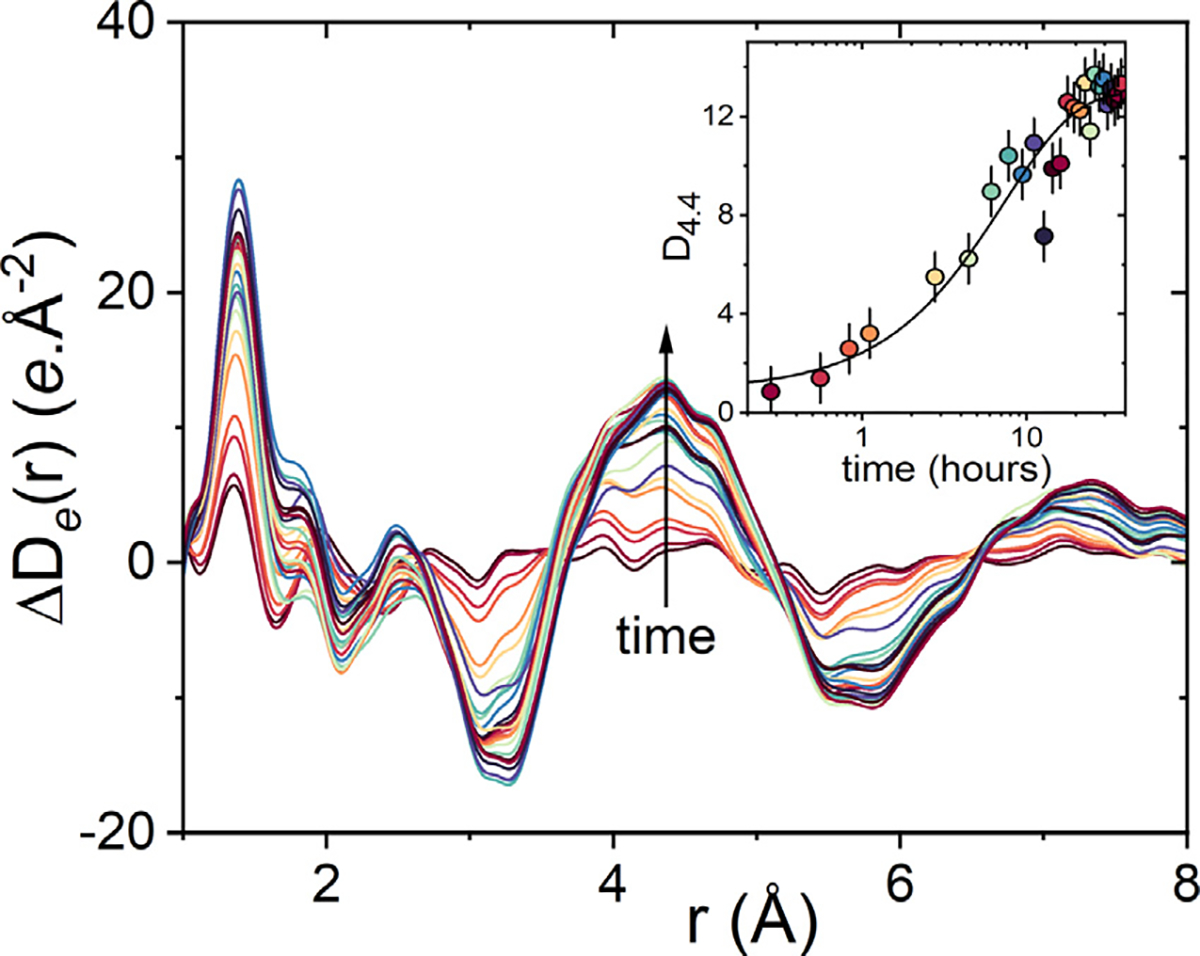
Changes in the measured differential electronic pair-distribution function ΔDe(r) exposed to 80% RH at room temperature relative to dry amorphous Indomethacin at time=0. The insert shows the changes in height of the peak at r = 4.4 Å as a function of time with the uptake of water.
Molecular dynamics simulations of hydrated amorphous Indomethacin show that most hydrogen bond donors occur at the carboxylic acid site, and that water molecules act as hydrogen donors with all oxygens in the molecule, but primarily at the amide and carboxylic groups.21 The water molecules are predicted form slightly stronger (i.e. ~3% shorter) hydrogen bonds with Indomethacin molecules than those found in Indomethacin dimers. To test this, a simple model was created with a water molecule in a linear hydrogen bond with the carboxylic acid site as shown in Fig. 7. The hydrogen bond distance was varied and ΔDe(r) calculated corresponding to only the interactions between the water molecule and an Indomethacin Z isomer. Since x-rays scatter more strongly from the oxygen and carbon atoms compared to hydrogen, the shortest distances in the PDF can be attributed to the water-carboxylic acid distances (centered on ~4.4 Å), followed by water-indole group correlations at ~7.4 Å. Based on this simple linear hydrogen bonding model an OH–O distance of ~2.8 Å was found, which is slightly longer than the ~2.7 Å distance observed in γ or α Indomethacin, but shorter than the first shell of bulk water at 2.9 Å 33 In an amorphous material with water molecules in many different hydrogen bonded orientations with Indomethacin the second peak at ~7.4 Å will become smeared out, and the 4.4 A peak would become the dominant feature in the x-ray PDF.
Figure 7.
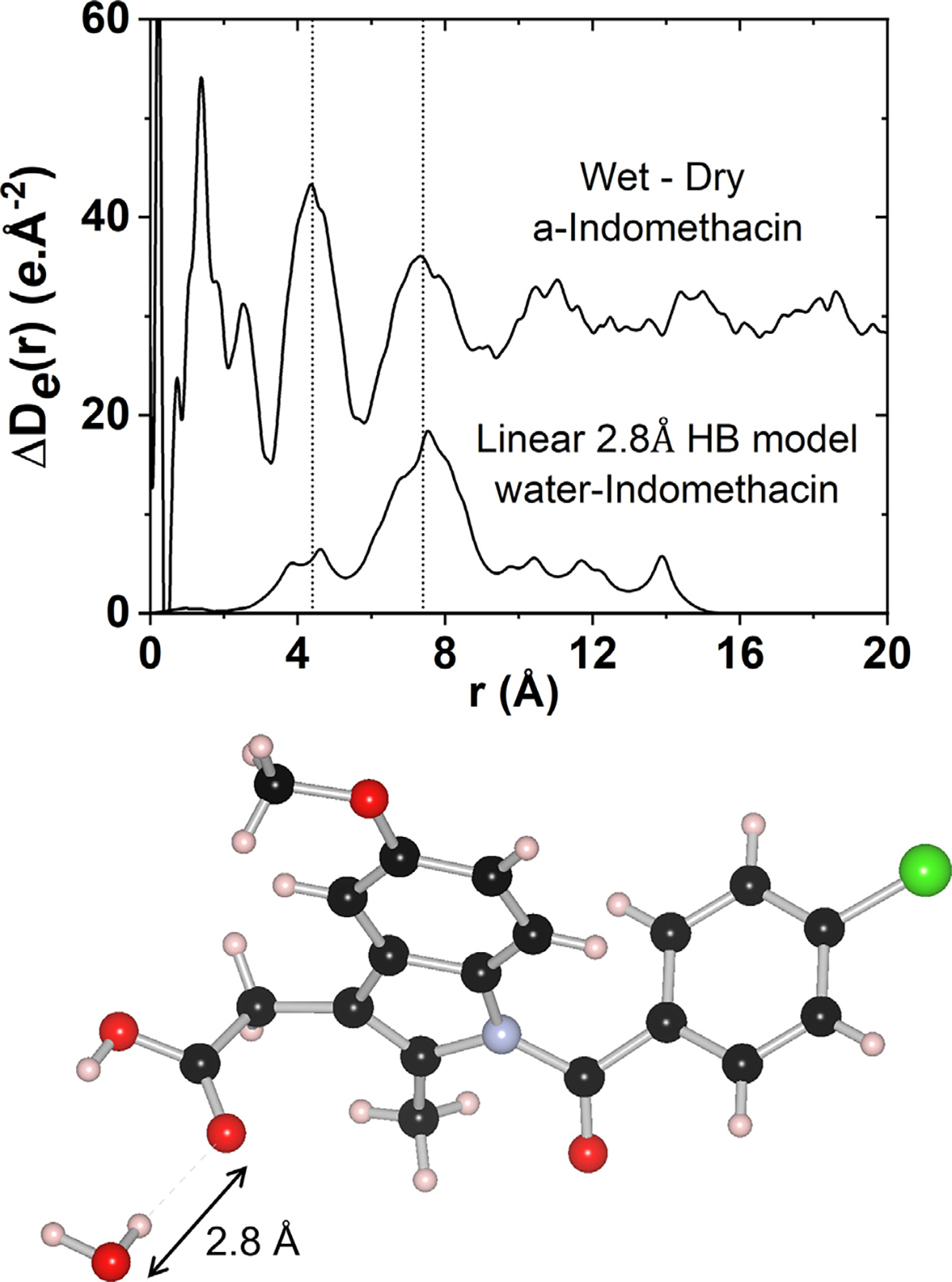
The measured differential electronic pair-distribution function ΔDe(r) of cryoground amorphous Indomethacin after exposure to 80% RH at room temperature for ~40 h, compared to a linear 2.8 Å inter-molecular hydrogen bond model between a water molecule and an Indomethacin Z isomer molecule.
Conclusions
Pharmaceuticals in the amorphous state are known to exhibit enhanced dissolution & bioavailabilities compared to their crystalline counterparts. However, the meta-stable nature of amorphous materials, storage conditions and elevated humidity conditions can result in the spontaneous conversion back to a stable crystalline form.1 This study confirms that not only the fabrication method can have an influence on the amorphous structure, but also the storage conditions. While some samples measured show a largely unhindered rotation of the chlorobenzyl ring in the dry amorphous form, others show preferred torsional angles consistent with those observed in the known isomers. High energy x-ray PDF measurements show that the hindered intra-molecular conformations associated with the preferred orientation of the chlorobenzyl ring occurs at the expense of intermolecular hydrogen bonding interactions. At high humidity, water molecules form hydrogen bonds with Indomethacin molecules leading to partial hydration of the amorphous form which stabilizes over the period of many (>10) hours. A simple linear hydrogen bond PDF model between a water molecule and an Indomethacin Z isomer, suggest the intermolecular hydrogen bond strengths are comparable to those found between Indomethacin dimers. The variability in structure of the Indomethacin molecule in the amorphous form makes it problematic to adopt as a standard for x-ray Pair Distribution Function measurements of glassy pharmaceuticals. A simpler molecule with a more rigid structure is recommended.
Supplementary Material
Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R44GM117701. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This research used resources of the Advanced Photon Source, a U.S. DOE Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357. The researchers and PI at ASU (JLY) would also like to acknowledge support from Arizona State University and the US National Science Foundation (NSF CHE 1856506 and NSF DMR BMAT 1809645).
Footnotes
Declaration of Interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary Materials
Supplementary material associated with this article can be found in the online version at doi:10.1016/j.xphs.2021.12.003.
References
- 1.Andronis V, Yoshioka M, Zografi G. Effects of sorbed water on the crystallization of indomethacin from the amorphous state. J Pharm Sci. 1997;86(3):346–351. [DOI] [PubMed] [Google Scholar]
- 2.Shi C, Teerakapibal R, Yu L, et al. Pair distribution functions of amorphous organic thin films from synchrotron X-ray scattering in transmission mode. IUCrJ. 2017;4(Pt 5):555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bates S, Zografi G, Engers D, et al. Analysis of amorphous and nanocrystalline solids from their X-ray diffraction patterns. Pharm Res. 2006;23(10):2333–2349. [DOI] [PubMed] [Google Scholar]
- 4.Yu L Amorphous pharmaceutical solids: preparation, characterization and stabilization. Adv Drug Deliv Rev. 2001;48(1):27–42. [DOI] [PubMed] [Google Scholar]
- 5.Andronis V, Zografi G. Crystal nucleation and growth of indomethacin polymorphs from the amorphous state. J Non Cryst Solids. 2000;271:236–248. [Google Scholar]
- 6.Khankari RK, Grant DJW. Pharmaceutical hydrates. Thermochim Acta. 1995;248:61–79. [Google Scholar]
- 7.Fukuoka E, Makita M, Yamamura S. Glassy state of pharmaceuticals. II. Bioinequivalence of glassy and crystalline indomethacin. Chem Pharm Bull. 1987;35(7):2943–2948. [DOI] [PubMed] [Google Scholar]
- 8.Greco K, Bogner R. Crystallization of amorphous indomethacin during dissolution: effect of processing and annealing. Mol Pharm. 2010;7(5):1406–1418. [DOI] [PubMed] [Google Scholar]
- 9.Chen S, Sheikh AY, Ho R. Evaluation of effects of pharmaceutical processing on structural disorders of active pharmaceutical ingredient crystals using nanoindentation and high-resolution total scattering pair distribution function analysis. J Pharm Sci. 2014;103(12):3879–3890. [DOI] [PubMed] [Google Scholar]
- 10.Karmwar P, Graeser K, Gordon KC, et al. Investigation of properties and recrystallisation behaviour of amorphous indomethacin samples prepared by different methods. Int J Pharm. 2011;417(1–2):94–100. [DOI] [PubMed] [Google Scholar]
- 11.Crowley KJ, Zografi G. Cryogenic grinding of indomethacin polymorphs and solvates: assessment of amorphous phase formation and amorphous phase physical stability. J Pharm Sci. 2002;91(2):492–507. [DOI] [PubMed] [Google Scholar]
- 12.Byrn SR ZG, Chen XS. Solid State Properties of Pharmaceutical Material. Hoboken: John Wiley & Sons, Inc; 2017. [Google Scholar]
- 13.Healy AM, Worku ZA, Kumar D, et al. Pharmaceutical solvates, hydrates and amorphous forms: a special emphasis on cocrystals. Adv Drug Deliv Rev. 2017;117:25–46. [DOI] [PubMed] [Google Scholar]
- 14.Hancock BC, Zografi G. Characteristics and significance of the amorphous state in pharmaceutical systems. J Pharm Sci. 1997;86(1):1–12. [DOI] [PubMed] [Google Scholar]
- 15.Benmore CJ. A review of high-energy x-ray diffraction from glasses and liquids. ISRN Materials Science. 2012;2012. [Google Scholar]
- 16.Benmore CJ, Weber JKR, Tailor AN, et al. Structural characterization and aging of glassy pharmaceuticals made using acoustic levitation. J Pharm Sci. 2013;102 (4):1290–1300. [DOI] [PubMed] [Google Scholar]
- 17.Yoshioka M, Hancock BC, Zografi G. Crystallization of indomethacin from the amorphous state below and above its glass transition temperature. J Pharm Sci. 1994;83(12):1700–1705. [DOI] [PubMed] [Google Scholar]
- 18.Masuda K, Tabata S, Sakata Y, et al. Comparison of molecular mobility in the glassy state between amorphous indomethacin and salicin based on spin-lattice relaxation times. Pharm. Res.. 2005;22(5):797–805. [DOI] [PubMed] [Google Scholar]
- 19.Skinner LB, Benmore CJ, Parise JB. Area detector corrections for high quality synchrotron X-ray structure factor measurements. Nucl Instrum Methods Phys Res Section A. 2012;662(1):61–70. [Google Scholar]
- 20.Qiu X, Thompson JW, Billinge SJL. PDFgetX2: a GUI-driven program to obtain pair distribution function from X-ray powder diffraction data. J Appl Crystallogr. 2004;37:678. [Google Scholar]
- 21.Xiang T-X, Anderson BD. Molecular dynamics simulation of amorphous indomethacin. Mol. Pharm.. 2013;10(1):102–114. [DOI] [PubMed] [Google Scholar]
- 22.Kistenmacher TJ, Marsh RE. Crystal and molecular structure of an antiinflammatory agent, indomethacin, 1-(p-chlorobenzoyl)-5-methoxy-2-methylindole-3-acetic acid. J. Am. Chem. Soc.. 1972;94(4):1340–1345. [DOI] [PubMed] [Google Scholar]
- 23.Chen X, Morris KR, Griesser UJ, et al. Reactivity differences of indomethacin solid forms with ammonia gas. J Am Chem Soc. 2002;124(50):15012–15019. [DOI] [PubMed] [Google Scholar]
- 24.Andrusenko I, Hamilton V, Lanza AE, et al. Structure determination, thermal stability and dissolution rate of d-indomethacin. Int J Pharm. 2021;608: 121067. [DOI] [PubMed] [Google Scholar]
- 25.Lu X, Xu W, Hanada M, et al. Solid-state NMR analysis of crystalline and amorphous Indomethacin: an experimental protocol for full resonance assignments. J Pharm Biomed Anal. 2019;165:47–55. [DOI] [PubMed] [Google Scholar]
- 26.Atef E, Chauhan H, Prasad D, et al. Quantifying solid-state mixtures of crystalline indomethacin by raman spectroscopy comparison with thermal analysis. ISRN Chromatography. 2012;2012: 892806. [Google Scholar]
- 27.Tita B, Marian E, Rusu G, et al. Effects of experimental conditions on the thermal behaviour of some non-steroidal anti-inflammatory drugs. Rev Chim. 2013:64. [Google Scholar]
- 28.Ben Osman Y, Liavitskaya T, Vyazovkin S. Polyvinylpyrrolidone affects thermal stability of drugs in solid dispersions. Int J Pharm. 2018;551(1–2):111–120. [DOI] [PubMed] [Google Scholar]
- 29.Hauch A, Bildsoe H, Jakobsen HJ, et al. 2H chemical shift anisotropies from high-field 2H MAS NMR spectroscopy. J Magn Reson. 2003;165(2):282–292. [DOI] [PubMed] [Google Scholar]
- 30.Kristensen JH, Hoatson GL, Vold RL. Investigation of multiaxial molecular dynamics by 2H MAS NMR spectroscopy. Solid State Nucl Magn Reson. 1998;13(1–2):1–37. [DOI] [PubMed] [Google Scholar]
- 31.Strachan CJ, Rades T, Gordon KC, et al. Raman spectroscopy for quantitative analysis of pharmaceutical solids. J Pharm Pharmacol. 2007;59(2):179–192. [DOI] [PubMed] [Google Scholar]
- 32.Heinz A, Savolainen M, Rades T, et al. Quantifying ternary mixtures of different solid-state forms of indomethacin by Raman and near-infrared spectroscopy. Eur J Pharm Sci. 2007;32(3):182–192. [DOI] [PubMed] [Google Scholar]
- 33.Skinner LB, Huang C, Schlesinger D, et al. Benchmark oxygen-oxygen pair-distribution function of ambient water from x-ray diffraction measurements with a wide Q-range. J Chem Phys. 2013;138:(7) 074506. [DOI] [PubMed] [Google Scholar]
- 34.Dawson KJ, Kearns KL, Ediger MD, et al. Highly stable indomethacin glasses resist uptake of water vapor. J Phys Chem B. 2009;113(8):2422–2427. [DOI] [PubMed] [Google Scholar]
- 35.Billinge S, Dykhne T, Juhas P, et al. Characterisation of amorphous and nanocrystalline molecular materials by total scattering. CrystEngComm. 2010;12:1366–1368. [Google Scholar]
- 36.Shi C xINTERPDF: a graphical user interface for analyzing intermolecular pair distribution functions of organic compounds from X-ray total scattering data. J Appl Crystallogr. 2018;51(5):1498–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mou Q, Benmore CJ, Yarger JL. X-ray Intermolecular Structure Factor (XISF): separation of intra- and intermolecular interactions from total X-ray scattering data. J Appl Crystallogr. 2015;48(3):950–952. [Google Scholar]
- 38.Gerges J, Affouard F. Insight from molecular dynamics simulations on the crystallization tendency of indomethacin polymorphs in the undercooled liquid state. J Pharm Sci. 2020;109(2):1086–1095. [DOI] [PubMed] [Google Scholar]
- 39.Otsuka M, Tanabe H. Stability test for amorphous materials in humidity controlled 96-well plates by near-infrared spectroscopy. Drug Dev Ind Pharm. 2012;38 (3):380–385. [DOI] [PubMed] [Google Scholar]
- 40.Surwase SA, Boetker JP, Saville D, et al. Indomethacin: new polymorphs of an old drug. Mol. Pharm.. 2013;10(12):4472–4480. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.