

Abstract
Receptor-mediated drug delivery presents an opportunity to enhance therapeutic efficiency by accumulating drug within the tissue of interest and reducing undesired, off-target effects. In cancer, receptor overexpression is a platform for binding and inhibiting pathways that shape biodistribution, toxicity, cell binding and uptake, and therapeutic function. This review will identify tumor-targeted drug delivery vehicles and receptors that show promise for clinical translation based on quantitative in vitro and in vivo data. The authors describe the rationale to engineer a targeted drug delivery vehicle based on the ligand, chemical conjugation method, and type of drug delivery vehicle. Recent advances in multivalent targeting and ligand organization on tumor accumulation are discussed. Revolutionizing receptor-mediated drug delivery may be leveraged in the therapeutic delivery of chemotherapy, gene editing tools, and epigenetic drugs.
Keywords: cancer, receptor targeting, targeted drug delivery, therapeutic, tumor accumulation
Graphical Abstract:
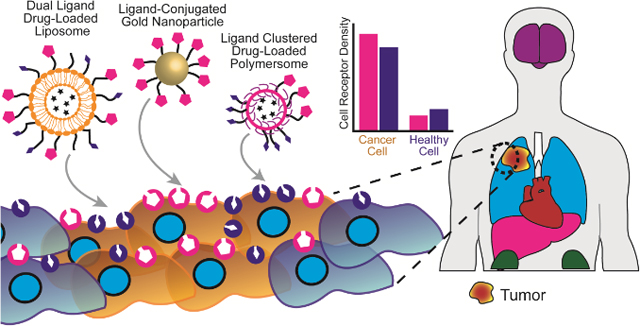
1. Introduction
Receptor-mediated targeting is a well-established method to enhance drug delivery vehicle (DDV) accumulation within specific tissues.[1] Much emphasis, over the past few decades, has focused on the therapeutic potential of treating both solid tumors and circulating tumor cells (CTCs).[2] Targeted DDVs demonstrated enhanced tumor internalization, diagnostic imaging, and prolonged survival in vivo relative to non-targeted DDVs.[3] Essentially, through ligand-based labeling approaches, DDVs can be ideally “addressed” to their desired destination with a high degree of precision. Thus, by taking advantage of receptor overexpression, unique binding motifs, and receptor patterning on the cell surface, DDVs can be functionalized with ligands that preferentially bind to receptors on the desired cells.[4] This occurs by way of “active” targeting in which ligand conjugated particles are bound and internalized by cancer cells at an accelerated rate due to the ligand–receptor interactions at the cell surface. By utilizing pathologically relevant receptor targets, DDVs administered systemically have a higher affinity for diseased tissue and a low affinity for surrounding healthy tissue, relative to non-targeted DDVs. In this way, receptor-mediated drug delivery alters the biodistribution of the drug by increasing accumulation in the site of interest. This is clinically important as reaching a therapeutic dosage locally and reducing systemic toxicity can be life-threatening or life-saving events for cancer patients.
The design of targeted DDVs can be optimized by altering the size, shape, material, ligand, and ligand orientation. For systemic delivery, spherical DDVs less than 200 nm in diameter are standard, which is balanced by the trade off between surface area and volume ratio. DDVs are prepared from biomaterials based on the size, hydrophobicity, and ideal release of the drug. There are a diverse range of ligand candidates for DDV functionalization, including monoclonal antibodies (mAbs), peptides, oligosaccharides, small molecules, and aptamers.[5] These ligands can be tethered to vehicles or conjugated directly to drugs by covalent (typically thiol or amide-based reactions, or “click” chemistry) or noncovalent attachment; the type of reaction is often based on the material of the DDV.[6] Multi-targeted and patterned DDVs improved cancer cell binding and the overall therapeutic benefit in vivo. The ideal DDV platform may be rationally designed by evaluating the drug, release mechanism, mode of delivery into the body, and target cells.
Utilizing these approaches, targeted DDVs were successfully translated from the research setting into clinical use, or are currently in preclinical trials.[7] Liposomal based DDVs were approved for use in cancer therapy, treating fungal diseases, analgesics, photodynamic therapy, and viral vaccines.[8] Specifically, FDA approved liposomal drug formulations, such as Mepact, Maribo, and Epaxal were proven to be effective for the treatment of nonmetastatic osteosarcoma, acute lymphoblastic leukemia, and hepatitis A, respectively.[9] Many targeted DDVs in clinical use are utilized for the treatment of multiple cancer types, as there is often broad overlap in malignant receptor overexpression across various carcinogenic tissues (Figure 1). The widely successful drug Pembrolizumab (Keytruda), a humanized IgG4 isotype mAb that targets the programmed cell death (PD-1) receptor of lymphocytes, is recommended by the FDA for the treatment of gastric cancer, melanoma, non-small cell lung cancer, squamous cell carcinoma of the head and neck, and urothelial carcinoma.[10] Receptor-targeted mAb-drug conjugates demonstrated promise to enhance chemotherapeutic efficacy.[11] Receptor-targeted DDVs have great promise as cancer therapeutics but, at the same time, exhibited limitations in their overall impact.
Figure 1.
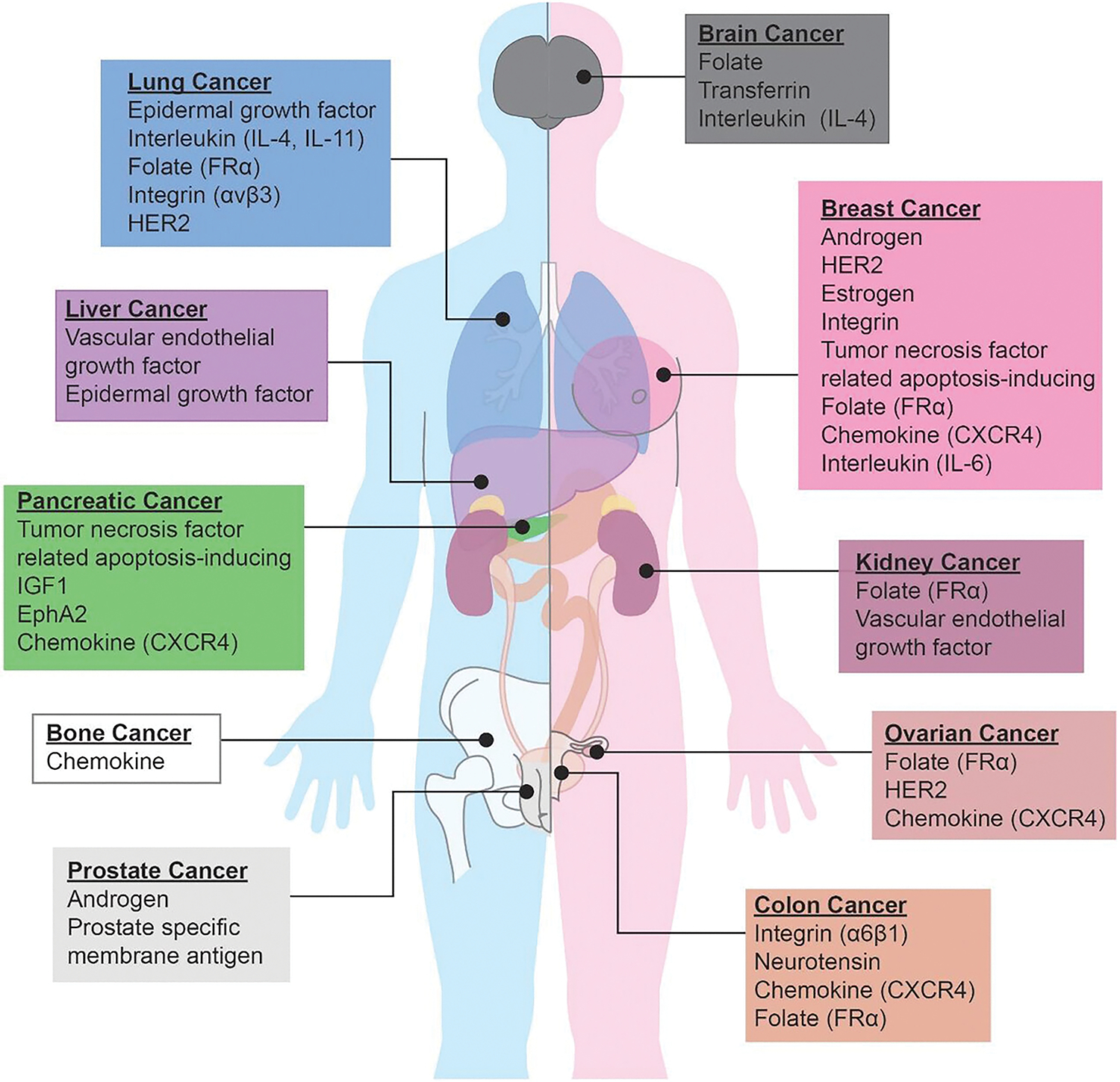
Common receptor overexpression associated with each different types of cancer.
Physiological challenges remain that limit widespread adoption of targeted DDVs and clinical translation. These challenges primarily pertain to immunogenic response, off-target toxicity, and nanocarrier instability in vivo. From a pharmaceutical stand point, the synthesis of targeted DDVs can be logistically challenging to scale up to industrial production levels, as well as expensive in terms of materials. Despite these present challenges, the development of clinically translatable targeted DDVs remains a substantial sector of research, as the curative potential is significant. Due to their high degree of customizability in both physiochemical composition and structure, as well as disease-specific ligand moiety, targeted DDVs have extended the ability to circumvent classical problems of chemotherapy treatment, such as multiple drug resistance and toxicity to healthy tissues.[12] DDVs alone can effect, or impede, the occurrence of physiological events via receptor-mediated pathways, such as hindering cancer proliferation, angiogenesis, and metastasis.[13]
In this review, we analyze the therapeutic effectiveness of previously developed receptor-mediated DDV platforms to demonstrate the rational design used to develop targeted DDVs. We then report on some of the most promising receptors or membrane-bound molecules used to recognize different cancers and suggest how these targeting approaches might be improved. We highlight the use of specific DDVs and their unique characteristics, including ligand surface patterning and ligand density optimization. Future directions toward developing novel DDVs to improve the clinical prognosis of cancer are discussed.
2. DDV Selection
DDVs are developed specifically for their ability to aid in the delivery of the cargo (Figure 2). Lipid-based DDVs recapitulate the structure of the native cell membrane, are relatively inert, and can easily be customized for a diverse range of applications. Polymeric vehicles are often selected for their capacity to control the release of cargo. Despite their differences, the size and shape of nanocarriers are critical for cellular uptake and biodistribution, while composition impacts biocompatibility, drug loading, and clearance (Table 1).
Figure 2.
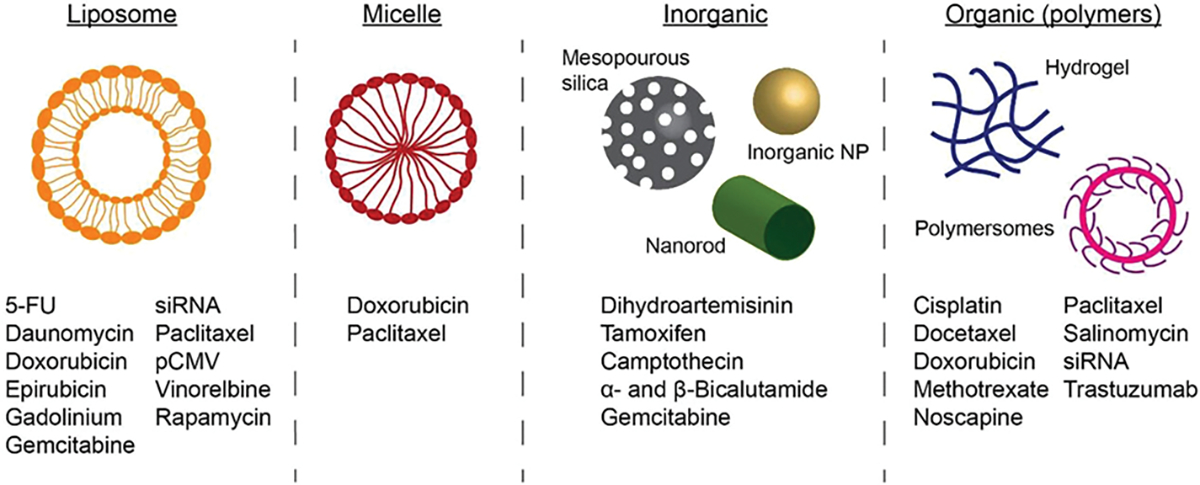
Drug loading in liposome, micelles, inorganic, and organic DDV. Refer to Table 2 for specific citations.
Table 1.
Positives and negatives of the different types of DDVs
| DDV | Positives | Negatives |
|---|---|---|
|
| ||
| Synthetic Liposomes/Micelles | Biocompatible | Smallest size 10nm |
| Biodegradable | Short drug release (days) | |
| Tailored chemical functionality | Limited shelf life | |
| Variable drug release kinetics | ||
| Polar/Nonpolar drug loading | ||
| Tunable mechanical properties | ||
| Self-assembly | ||
| Long-circulating | ||
|
|
||
| Bio-derived Liposomes/Micelles | Increased circulation | Limited cell membrane sources |
| Decreased immune response | Complex isolation | |
| Self-assembly | Quality control | |
|
|
||
| Inorganic Nanoparticles | Range of particle diameters | Generation of reactive oxygen species |
| Photothermal therapy | Apoptosis & necrosis | |
| Imaging contrast agent | Mitochondrial toxicity | |
| Custom shapes | Nonbiodegradable | |
| Hydrophobic, electrostatic, and chemical functionalization | ||
| Long shelf life | ||
|
|
||
| Organic Nanoparticles | High water content | Possible inflammatory response |
| “Click” chemistry, hydrophobic, electrostatic, or covalent attachment | Inactivation of encapsulated proteins | |
| Tailored drug release (days to years) | Accumulation of byproducts | |
| Long shelf life | ||
| pH, temp, re-dox, light responsive | ||
| Synthesis via self assembly, emulsions, precipitation, or other methods. | ||
2.1. Lipid-Based DDVs
Liposomes, micelles, lipid nano-emulsions (LNEs), and solid lipid nanoparticles (SLNs) are comprised predominantly of lipids, which self-assemble based on hydrophobic fatty acid tails and hydrophilic, electrostatically charged head groups. Lipids are the main component of the cell membrane, and are considered to be biocompatible and biodegradable. Lipid headgroups can easily be customized to alter their physiochemical properties or to provide chemical functionality. In addition, the hydrophobicity of the fatty acid chain (saturation and length) can alter particle stability and therefore drug retention and release.
Liposomes may be easily loaded with polar and nonpolar drugs, because of their lipid bilayer and aqueous core. This makes liposomes an ideal choice for chemotherapeutics, such as doxorubicin, and nucleic acids. Liposomes are spherical vesicles that range in size from 20 to 200 nm in diameter and may have one or more concentric lipid bilayers.[14] They are primarily composed of lipids with diacyl chains. Targeted liposomes are typically less than 200 nm in diameter and unilamellar (having one bilayer) as this permits systemic circulation and increases the drug payload.[7b] Liposomes can be functionalized by conjugating targeting ligands to chemically reactive lipids before or after DDV synthesis. Further, the mechanical properties of liposomes significantly affect their in vitro and in vivo behavior. It was recently shown that neutral, zwitterionic liposomes encapsulating calcium-alginate hydrogels with different cross-linking densities gave rise to liposomes with varying Young’s moduli. The in vitro cellular uptake of liposomes in both cancerous and noncancerous cells, as well as their in vivo tumor accumulation in an orthotopic breast cancer model, inversely correlated with the Young’s modulus of the liposomal formulation. This was determined to be due to differences in the endocytic pathway of liposomes with different moduli. Low-modulus liposomes were able to quickly and efficiently fuse with the cell membrane while high-modulus liposomes were endocytosed by the more energy consuming clathrin-mediated pathway.[15]
Micelles are composed of amphiphilic macromolecule copolymers, in which the hydrophilic head group faces outward and the single acyl chain comprises the inner core.[16] The micelle core is hydrophobic, making it well suited to accommodate moderately hydrophobic drug molecules, such as paclitaxel. Micelles are smaller than liposomes, typically 10–100 nm in diameter.[17] Targeted micelles were developed for delivering hydrophobic drugs to physiologically inaccessible tissues, such as the brain.[18] Additionally, micelle formulations were optimized for long circulation and targeted delivery in vivo.[16,17,19] Currently, there are a number of polymeric micelle formulations for chemotherapeutic delivery in clinical trials.[20]
Lipid nano-emulsions (LNEs) are oil-in-water emulsions, typically composed of the same lipids as liposomes and micelles, though their structural organization differs. LNEs have a single lipid layer shell that envelops a hydrophobic, lipid core and are approximately 20–200 nm in diameter.[21] The lipid core may be solid or liquid at physiological temperature. LNEs are better suited to encapsulate nonpolar drugs such as poly-methoxyflavones (PMFs).[22] LNEs are used to target cancer cells by modification of the lipid head groups with receptor-specific ligands.
Solid lipid nanoparticles (SLNs) are particles composed of lipids that have a low melting temperature. They are ideal carriers for hydrophobic drugs and nucleic acids. SLNs enhanced uptake and therapeutic delivery within the liver.[23] Phase I clinical trials conducted by Alnylam’s drug ALN-VSPO2 demonstrated their ability to deliver siRNA to the liver and provide a therapeutic benefit.[23] Patients with advanced solid liver tumors and metastatic lesions received a total of 140 doses of lipid nanoparticle encapsulating small interfering RNA (siRNA) to inhibit the expression of vascular endothelial growth factor (VEGF). Inhibition of disease progression with minimal side effects was achieved in 54.9% of patients, in which disease stability was noted for at least 2 months after treatment.[24] Thus, ALN-VSPO2 was touted as a well-tolerated therapeutic with antitumor activity.
In addition to synthetic lipid DDVs, DDVs may be derived from cell membranes.[25] These DDVs utilize cell membrane lipids, proteins, and sugars to impart stealth or specific cell properties (e.g., leukocyte). It was shown that leukocyte mimetic vehicles effectively targeted sites of inflammation caused by disease or cancer.[26] Red blood cell membranes were used to cloak encapsulated drug and increase circulation time.[27] Though immune responses are often minimized when utilizing biologically derived mimetic vehicles, there are other challenges which have limited their success and hindered clinical translation. Limited cell membrane sources, complexity of isolation processes, as well as issues concerning quality control and safety, have impeded their implementation as DDVs in the clinic.[28]
2.3. Inorganic Nanoparticle Based Systems
DDVs comprised of inorganic materials range in size (0.8–200 nm) and shape (nanospheres, nanoshells, and nanorods).[29] In some instances, inorganic DDVs exhibit multiple functions, such as exothermic reactors and contrasts agents that are useful in medicine. For example, gold nanoparticles (AuNPs), also referred to as gold nanoconjugates, that can be synthesized in a range of shapes and sizes,[30] are easily modified on the surface with ligands to target cells or drug,[31] and are useful in photothermal therapy[32] and imaging.[31b,33] Due to their advantageous chemical properties, they are ideal DDVs for chemotherapeutics.[33a,34]
AuNPs were developed for dual delivery of doxorubicin and bleomycin in vitro.[35] Antibodies against pancreatic cancer specific receptor overexpression, such as cetuximab, panitumumab, or trastuzumab, can be conjugated to AuNPs to stop proliferation of cancer cells or to identify tumors.[32c] Functionalization of AuNPs with specific ligands can be achieved via electrostatic and and chemical reactions. Due to their adsorption and scattering of electromagnetic radiation, AuNPs can be utilized for photothermal therapy (nanophotothermolysis) or radio frequency ablation, treatments typically administered when surgical resection is unlikely to be successful, has failed, or there are no clinical alternatives.[36] Targeted AuNPs have also been utilized for in vivo imaging, as they have favorable tissue penetration and optical contrast properties, compared to conventional dyes.[37] In vivo, PEGylated AuNPs can circulate for a half-life of up to 48 h.[38] However, the adoption of AuNPs for drug delivery systems is limited by toxic side effects in vivo, such as the generation of reactive oxygen species (ROS), apoptosis, necrosis, and mitochondrial toxicity.[39]
Similarly, iron oxide nanoparticles (IONPs) are used in a range of applications. IONPs were first developed in the late 1980s as contrast agents for MRI.[40] However, more recently, a series of methods for modifying the IONP surface with polymers was reported. Jain et al. developed a two-step conjugation protocol using oleic acid and a PEG oxide block copolymer (PEG-polypropylene oxide) to improve biocompatibility and enable drug and ligand conjugation.[41] Apart from IONPs, silicon oxide nanoparticles (SONPs) represent another potential DDV in which the oxide groups can readily be functionalized for targeted delivery.[29] SONPs are known to have good biocompatibility, low cost, and manufacturing controllability, in addition to being generally recognized as safe by the FDA, thereby improving their translation in the clinic.[42] To date, SONPs to target cancer cells are synthesized by conjugating a range of small molecules, peptides, or full proteins.[34c,43]
2.4. Organic Nanoparticle-Based Systems (Polymers)
Organic platforms have been extensively studied since their development in the 1960s.[44] More recently, there has been a large effort to develop and optimize drug-carrying hydrogel nanoparticles for cancer treatment. Hydrogels are polymer networks capable of absorbing large concentrations of water or biological fluid.[45] Hydrogel-based drug delivery nanoparticles are commonly composed of poly-lactic-co-glycolic (PLGA), alginate, chitosan, or hyaluronic acid. Surface functionalization for targeted delivery of these materials is typically achieved via “click” chemistry, electrostatic adsorption, or covalent attachment.[46]
PLGA is a well-established FDA-approved polymer known for its biodegradability. The ratio of lactic to glycolic acid is tuned to control the degradation of the DDV and the release of drug. PLGA-based systems were developed for prolonged delivery of estradiol, rifampicin, piroxicam, indomethacin, ibuprofen, lidocaine, cyclosporine A, and paclitaxel, to name a few.[47] PLGA NPs allow sustained release of drug payloads significantly longer than their lipid-based drug delivery counterparts, on the order of weeks compared to days.[48]
Alginate, an anionic polysaccharide derived from seaweed, undergoes coacervation with calcium ions to form a gel. When calcium ions are released, the hydrogel falls apart and drug is released.[49] Doxorubicin-loaded alginate DDVs with surface functionalization showed promise in their targeted delivery to liver cancer.[50] Alginate-based particles have shown versatility in a diverse range of therapeutic applications, such as oral drug administration,[51] controlled gastrointestinal release for optimal drug absorption,[52] and as drug excipients.[53]
Chitosan, a hydrophilic linear polysaccharide, is processed from the exoskeletons of crustaceans. Chitosan is widely utilized due to its biocompatibility, biodegradability, and low toxicity.[54] Drugs may be loaded into chitosan hydrogels via permeation, entrapment, or covalent bonding, which provides unique drug release profiles compared to lipid-based particles. Hybrid formulations incorporating both alginate and chitosan have demonstrated unique benefits over other encapsulation systems, such as: extended shelf life, enhanced entrapment efficiency, and sustained linear release of peptide in vitro.[55] Hybrid alginate-chitosan formulations were developed for the prolonged delivery of doxorubicin and paclitaxel.[56]
Polymeric vehicles are the DDV of choice for controlling drug release over long periods of time. Consideration of degradation products must be taken as PLGA degrades into acidic byproducts which can lower the pH, resulting in inflammation or loss of activity of therapeutic proteins. These factors impede the translation of polymer-based drug delivery into the clinic.
2.5. Smart Nanoparticles
“Smart” or stimuli-responsive nanoparticles are able to sense and react to a given environment.[57] These polymer-based DDVs are designed to be responsive to changes in pH, temperature, re-dox reaction, or light.[58] Thus, “smart” particles are capable of releasing drug payloads by external or physiological cues based on their engineered sensitivity.[59] These parameters can be incorporated into particle design, varying the particle permeability (or swelling) at different pH values or temperatures, for instance.[60] Typically, these particles are composed of polypeptides or block copolymer chains that self-assemble in aqueous solution. Smart nanoparticles were developed for the delivery of doxorubicin, paclitaxel, and cisplatin.[61] However, a number of challenges hinder the clinical translation of stimuli-responsive polymers, most important of which are the potential toxicity and accumulation within the body.[62]
Selection of the most appropriate material for a given drug delivery application is a critical design decision. When formulating nanoparticles, the ability to encapsulate efficiently, release in the desired location, and achieve the desired function are important. However, the synthesis of the desired DDV can also be critical to both the therapeutic outcome and ability to translate to the clinic. Costly methods that have low drug encapsulation or many processing steps can limit the potential of a DDV candidate. In addition, the choice of DDV material can be dependent on the target location, route of administration, and desired timeline of delivery. Typically, targeted DDVs are taken up by the cells of interest quickly to avoid uptake by the reticuloendothelial system (RES). Thus, all targeted DDVs, despite their benefits and limitations, are dependent on having strong cell adhesion with receptors on the cancer cell.
3. Common Tumor Targets for Receptor-Mediated Drug Delivery
Recognition of the target cancer cell by a cell surface receptor is a desirable method to deliver a therapeutic drug selectively to cancer cells (Table 2). Receptor-mediated DDVs increase accumulation within tumors and reduce tumor growth relative to non-targeted controls. Targeting limits the off-target cytotoxicity of chemotherapeutics by reducing drug concentrations in healthy tissue, while increasing concentrations in the tumor compared to the free drug (Figure 3A). Further, due to the selective receptor-mediated targeting of DDVs, a lower concentration (IC50) of the chemotherapeutic agent is required to treat tumors (Table 3). The receptors which have demonstrated promise as therapeutic targets in a wide range of cancer types are epidermal growth factor receptor (EGFR), interleukin, folate, and integrin αvβ3. These are highly expressed in lung, breast, colon, ovarian, and brain cancers and serve as physiological targets for therapeutic delivery.
Table 2.
Receptors commonly overexpressed in different types of cancer and previously DDVs developed to exploit this to targeted delivery (blue text, “smart” pH-responsive DDVs)
| Receptor | Ligand Target | Cancer | DDV | Drug cargo |
|---|---|---|---|---|
|
| ||||
| Transferrin | TfR ligand (7pep) | Breast | Micelle | Doxorubicin [170] |
| Transferrin | Breast | Iron oxide | Dihydroartemisinin [173] | |
| Niosome | Doxorubicin[171] | |||
| Liposome | Doxorubicin[172] | |||
| Glioma | Micelle | Paclitaxel[18] | ||
| Transferrin + TRAIL | Colon | HSA | Doxorubicin[194] | |
| Transferrin + Folate | Glioma | Liposome | Doxorubicin[191a] | |
| T7 peptide + TAT | Glioma | Liposome | Doxorubicin[195] | |
| TfR mAb | Glioma | Liposome | Daunomycin [2c] | |
|
| ||||
| Folate | Folic acid | Lung | Liposome | Doxorubicin[12] |
| Cervical | Organic | Methotrexate[99] | ||
| Organic | Docetaxel[100b] | |||
| Folate | Cervical | Micelle | Doxorubicin [96] | |
| Micelle | Paclitaxel[97] | |||
| Liposome | Doxorubicin[95a] | |||
| Conjugate | Doxorubicin[98b] | |||
| Breast | Micelle | Paclitaxel[97] | ||
| Liquid crystal | Docetaxel & Cisplatin[100a] | |||
| Organic | Doxorubicin[98a] | |||
| Carcinoma | Micelle | Fluorophore[185] | ||
| Folate + RGD | Carcinoma | Graphene oxide | Thermal[32a] | |
| Folate + Asp8 | Breast Metastasis | Liposome | Doxorubicin[191d] | |
| Folate + Transferrin | Glioma | Liposome | Doxorubicin[191a] | |
|
| ||||
| αvβ3 integrin | RGD | Endothelial | Micelle | Doxorubicin[107] |
| Glioma | Micelle | Paclitaxel[108] | ||
| Lung | Organic | Paclitaxel[106] | ||
| Melanoma | Conjugate | Doxorubicin[3] | ||
| Breast | Silica | Camptothecin[43] | ||
| RGD + pHA | Glioma | Liposome | Doxorubicin[191b] | |
| RGD + Estrone | Breast | Organic | Paclitaxel[193] | |
| RGD + YPSMA-1 mAb | Prostate | Micelle | Paclitaxel [190] | |
| RGD + Folate | Carcinoma | Graphene oxide | Thermal[32a] | |
|
| ||||
| PSMA | A10 PSMA Apt | Prostate | PLGA | Cisplatin[158] |
| PLGA | Docetaxel[217] | |||
| PLGA | shRNA[2b] | |||
| YPSMA-1 mAb + RGD | Prostate | Micelle | Paclitaxel [190] | |
| anti-PSMA + anti-CD14 mAb | Prostate | PAMAM | Fluorophore[114d] | |
|
| ||||
| HER2 | Trastuzumab | Breast | DMAEMA/HEMA | Docetaxel[32b] |
| Conjugate | Maytansinoid[11a] | |||
| Liposome | paclitaxel & rapamycin[180] | |||
| gelatin + HSA | Trastuzumab[177] | |||
| HSA | Doxorubicin[178] | |||
| PEI/PLGA | Paclitaxel[179] | |||
| anti-HER2 scFv | Breast | PLGA | Docetaxel[182] | |
| neu peptide (FCDGFYACYADV) | Breast | Liposome | Doxorubicin[181] | |
| KCCYSL (P6.1 peptide) | Breast | Liposome | Gadolinium[175a] | |
|
| ||||
| Estrogen | Estrone | Breast | Liposome | Doxorubicin [146a-d] |
| Gelatin | Noscapine[147] | |||
| Chitosan | Paclitaxel [148] | |||
| Estrone + RGD | Breast | Organic | Paclitaxel[193] | |
| 17β-Estradiol | Breast | Carbon nanotube | Doxorubicin[146e] | |
| Liposome | pCMV-beta-gal[144] | |||
| Tamoxifen | Breast | Gold | Tamoxifen [145] | |
|
| ||||
| CXCR4 | LFC131 peptide | Lung | Chitosan | Docetaxel[218] |
| Breast | PAMAM | Doxorubicin[125a] | ||
| anti-CXCR4 mAb | Breast | Liposome | Doxorubicin[125b] | |
| Peptide R | Lung | Liposome | Doxorubicin[124] | |
|
| ||||
| ICAM1 | anti-ICAM1 mAb | Breast | Iron oxide | N/A[152b] |
| Liposome | Lcn2 siRNA[153] | |||
| LFA-1 | Cervical | Urethane | Paclitaxel[152a] | |
|
| ||||
| Androgen | Testosterone | Prostate | Liposome | 5-FU[166] |
| α- & β-Bicalutamide | Prostate | Gold | α- & β-Bicalutamide[167] | |
|
| ||||
| CD | CD14) anti-CD14 mAb + anti-PSMA | Prostate | PAMAM | Fluorophore[114d] |
| CD22) anti-CD22 mAb | Lymphoma | Organic | Cyclophosphamide & Bortezomib [114c] | |
| CD44) Hyaluronic acid | Breast | Micelle | Paclitaxel[121] | |
| Organic | Doxorubicin[114a] | |||
| Melanoma | Liposome | Doxorubicin[117] | ||
| CD133. Aptamer | Bone | Organic | Salinomycin[123] | |
|
| ||||
| EGFR | anti-EGFR | Breast | Liposome | Doxorubicin, epirubicin, & vinorelbine[65a] |
| Lung | Liposome | Gemcitabine [68] | ||
| EGF | Oral | Inorganic | Thermal[32c] | |
| Cetuximab | Pancreatic | Inorganic | Gemcitabine[67] | |
|
| ||||
| IL | IL4) AP1 peptide | Colon | Liposome | Doxorubicin[82] |
| Glioma | Organic | Doxorubicin[76b] | ||
| IL4) Pep-1 | Lung | Liposome | Doxorubicin[79b] | |
| IL13) IL13 | Glioma | Organic | Docetaxel[4] | |
|
| ||||
| TNF | TRAIL + Transferrin | Colon | HSA | Doxorubicin[194] |
|
| ||||
| Glycyrrhetinic | glycyrrhetinic acid | Liver | Organic | Doxorubicin[50] |
|
| ||||
| VEGF | anti-VEGF mAb | Pancreatic | Conjugate | Gemcitabine[219] |
| AR7 + T7 peptide | Glioma | Liposome | Doxorubicin & Vincristine[191c] | |
Figure 3.
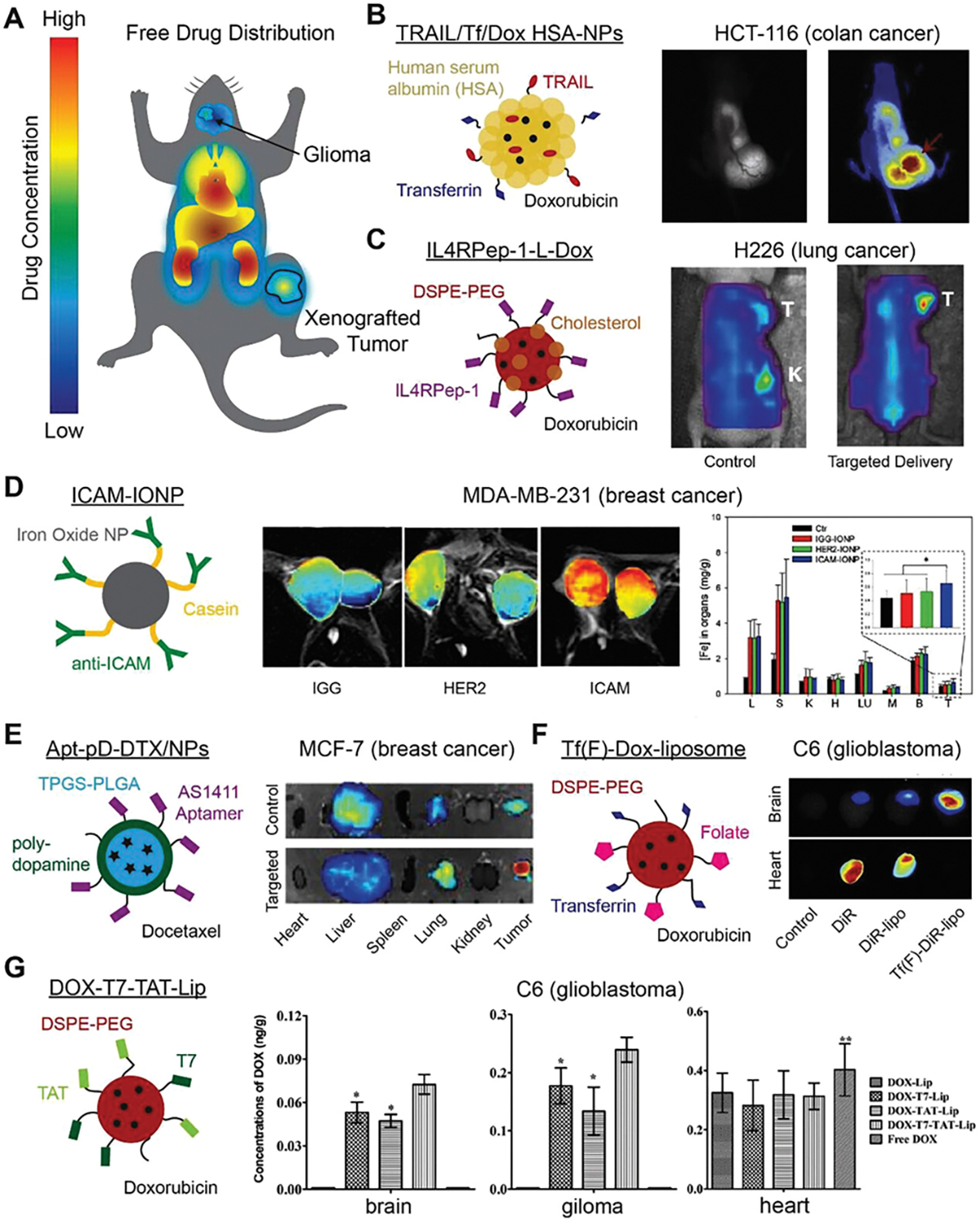
Representative state-of-the-art examples of biodistribution for targeted DDVs in the literature. A) Typical biodistribution profile of free drug has limited drug concentration in tumors and high concentrations in the heart, liver, and kidneys. B) Dual targeting polymeric NP increases doxorubicin concentrations in xenografted colon cancer tumors. Reproduced with permission.[194] Copyright 2012, Elsevier. C) Doxorubicin loaded IL4RPep-1 conjugated liposomes are delivered to the tumor in higher contentions than unconjugated controls. Reproduced with permission.[79b] Copyright 2015, Elsevier. D) Anti-ICAM1 conjugated NPs are taken up by TNBC more than unconjugated iron oxide NPs. Reproduced with permission.[152b] Copyright 2014, The Authors. E) Nucleolin targeting AS1411-conjugated polydopamine coated DDVs to target breast cancer reduce docetaxel concentrations in the liver, while increasing the amount of drug delivered to the tumor. Reproduced under the terms of the CC BY-NC license.[220] Copyright 2016, Ivyspring International Publisher. F) By targeting the transferrin and folate receptors, dual targeting liposomes traverse the blood–brain barrier and are selectivity taken up in the tumor. Reproduced with permission.[191a] Copyright 2013, Elsevier. G) TAT and T7 conjugated liposomes increase drug concentrations in a glioma, while remaining relatively low in the brain and unchanged in the heart. Reproduced with permission.[195] Copyright 2014, Elsevier.
Table 3.
Receptor-mediated DDVs increase the drug potency (IC50) of chemotherapeutic: Doxorubicin (Dox), Salinomycin (Sal), Maytansinoid (May), Vinorelbine (Vin), α- & β-Bicalutamide (α- β-Bic), Noscapine (Nos).
| Receptor | Ligand Target | DDV | Drug cargo | IC50 | ||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Targeted | Control | Drug | Units | Fold difference | ||||
|
| ||||||||
| Transferrin | Transferrin | Liposome | Dox[172] | 17.69 | >>100 | 59.08 | 3.34 | |
|
| ||||||||
| Folate | Folate | Micelle | Dox[96] | 0.068 | 0.43 | 0.047 | μg/mL | 0.69 |
| Conjugate | Dox[98b] | 5.6 | 9.8 | >>10 | μM | 1.79 | ||
| Organic | Dox[98a] | 3.25 | 95.97 | >>100 | μM | 30.77 | ||
|
| ||||||||
| αvβ3 integrin | RGD | Micelle | Pac[108] | 27.54 | 69.18 | 61.66 | nM | 2.24 |
| RGD + Estrone | Organic | Pac[193] | 14.4 | 38.5 | nM | 2.67 | ||
| RGD + YPSMA1 | Micelle | Pac[190] | 55.05 | 70.56 | 66.28 | ng/mL | 1.20 | |
|
| ||||||||
| PSMA | A10 PSMA Apt | Organic | Cis[158] | 0.03 | 0.13 | 2.4 | μM | 80 |
|
| ||||||||
| HER2 | Trastuzumab | Organic | Doc[32b] | 0.26 | 2.6 | μM | 10 | |
| Conjugate | May[11a] | 0.24 | 1 | nM | 4.17 | |||
|
| ||||||||
| Estrogen | Estrone | Liposome | Dox[146b] | 10 | 16 | 35 | μg/mL | 3.50 |
| Liposome | Dox[146d] | 0.8 | 3.3 | 0.15 | μM | 0.19 | ||
| Gelatin | Nos[147] | 21.2 | 32.1 | 43.3 | μmol/L | 2.04 | ||
| Organic | Pac[148] | 0.15 | 0.75 | 1.26 | μg/mL | 8.40 | ||
| 17β-Estradiol | Organic | Dox[146e] | 0.9 | 1.5 | 2.3 | μg/mL | 2.56 | |
| Organic | Dox[125a] | 210.4 | 1422 | 28.5 | μg/mL | 0.14 | ||
|
| ||||||||
| CXCR4 | anti-CXCR4 | Liposome | Dox[125b] | 7.2 | 50 | 3.6 | μg/mL | 0.50 |
| Peptide R | Liposome | Dox[124] | 0.2 | 0.46 | 0.31 | μg/mL | 1.55 | |
|
| ||||||||
| ICAM1 | LFA-1 | Organic | Pac[152a] | 1 | 37.5 | 63 | μM | 63 |
|
| ||||||||
| Androgen | α- & β-Bicalutamide | Inorganic | α- β- Bic[167] |
0.0016 | 33 | μM | 20625 | |
|
| ||||||||
| CD | CD44) Hyaluronic acid | Micelle | Pac[121] | 0.93 | 1.63 | μg/mL | 1.75 | |
| CD133) Apt | Organic | Sal[123] | 2.18 | 10.72 | 5.07 | μg/mL | 2.33 | |
|
| ||||||||
| EGFR | anti-EGFR | Liposome | Dox, | 1.1 | 0.8 | μg/mL | 0.73 | |
| Vin[65b] | 0.3 | 12 | 0.3 | μg/mL | 1 | |||
|
| ||||||||
| IL | IL4) AP1 Pep | Organic | Dox[76b] | 48.68 | 114.8 | 194.3 | ng/mL | 3.99 |
|
| ||||||||
| Glycyrrhetinic | glycyrrhetinic acid | Organic | Dox[50a] | 70.3 | 95.3 | ng/mL | 1.36 | |
| Organic | Dox[50b] | 79 | 47 | ng/mL | 0.59 | |||
3.1. Epidermal Growth Factor Receptor
EGFR is a 170 kDa transmembrane glycoprotein responsible for binding to EGF ligands and subsequent initiation of intracellular signaling pathways which regulate pathways responsible for cell proliferation, apoptosis, and migration.[63] Overexpression of EGFR is observed in aggressive head and neck, colon, renal, glioma, breast, nonsmall cell lung, and ovarian tumors.[64] Competitive binding of mAbs against EGFR demonstrated decreased cell proliferation by effectively blocking ligand dependent signaling of EGFR.[64] In vitro studies by Mamot et al. tested the therapeutic efficacy of methotrexate (MTX)-encapsulating EGFR-mAb targeted liposomes in MDA-MB-468 human breast cancer, U-87 human glioblastoma, and A-431 epidermoid cancer cell lines.[65] EGFR mAb–targeted delivery of MTX induced more than 20-fold higher cytotoxicity than non-targeted MTX-liposomes in MDA-MB-468 cells.
Acharya et al. formulated rapamycin (Rapa) encapsulating PLGA functionalized with mAbs to target EGFR on breast cancer MCF-7 cells.[66] Analysis using flow cytometry revealed a 13-fold increase of cell internalization of EGFR mAb labeled nanoparticles versus unconjugated nanoparticles, and 50 times higher internalization than that of free Rapa.[66] A 25% decrease in MCF-7 viability was noted when treated with EGFR-Rapa-NPs compared to treatment with unconjugated Rapa-NPs; a 10% decrease was observed compared to treatment with free Rapa.[66] Thus, targeted DDVs increased drug internalization and cancer cell death relative to non-targeted DDVs and free drug.
Work done by Patra et al. sought to deliver the poorly bioavailable and highly toxic chemotherapeutic gemcitabine (Gem) to pancreatic adenocarcinoma models in vitro and in vivo.[67] AuNPs conjugated with anti-EGFR mAb C225 and gemcitabine (Au-C225-Gem) were tested against mAb drug conjugate C225-Gem and nonspecifically labeled Au-IgG-Gem. In mice bearing pancreatic AsPC-1/luciferase tumors, treatment with Au-C225-Gem resulted in a significant decrease in tumor volume (≈80%) compared to treatment with free drug. This reduction in tumor volume occurred at lower dosage levels of Gem (2 mg kg−1).[67] Thus, when drug is successfully targeted in a receptor-mediated fashion, lower levels of drug can be used to achieve the same antitumor effect.
EGFR mAb functionalized, pH-sensitive immunoliposomes (PSLs) were synthesized by Kim et al. for delivery of Gem to nonsmall cell lung cancer (NSCLC) in vivo.[68] After a series of eight injections, mice treated with EGFR-targeted PSL-Gem DDVs had significantly greater reduction in NSCLC A549 tumor volume (two to threefold) than mice treated with nonfunctionalized PSL-Gem DDVs or free Gem. Immunohistological analysis of proliferating cell nuclear antigen (PCNA) revealed a significant increase in tumor inhibition and decrease in apoptotic index, amongst mice treated with EGFR-targeted PSL-Gem relative to nonfunctionalized PSL-Gem or free Gem.
Recently, the administration of EGFR-targeted DDVs were limited by inherent or acquired resistance to anti-EGFR therapies in color rectal,[69] lung,[70] and head and neck cancers.[71] These resistances arise through tumor-specific mutations to genes encoding EGFR receptors, resulting in an inability to inhibit the activation and subsequent downstream signaling pathways associated with oncogenesis.[71]
3.2. Interleukin Receptors
Due to their overexpression in cancerous tissues, interleukin receptors are ideal targets for developing novel DDVs. Among these interleukin receptors, IL-3, -4, -6, -11, -13, and -16 were examined as targets for receptor-mediated DDVs to improve the prognosis of cancer. The ligand–receptor complex undergoes receptor-mediated cell internalization to allow the cargo to be selectively delivered to the target cells. In this manner, the interleukin receptors in healthy cells function to regulate cell growth and proliferation, angiogenesis, and the immune system response. However, in tumor cells, overexpression of interleukin receptors can contribute to the malignant progression and apoptosis resistance of various cancer types. Therefore, not only do interleukin receptors make attractive targets for drug delivery, but they themselves can be targeted by pharmaceutical antagonists or agonists.
The IL-6 receptor, which is upregulated in ovarian cancer cells (RMG-1 and OVISE) with poor prognosis, was targeted with tocilizumab, an anti-IL-6 mAb.[72] In vitro, treatment with tocilizumab inhibited cancer invasion and proliferation,[72] making it a desirable candidate for therapeutic development. Conversely, RNA/DNA aptamers for the IL-6 receptor were used to deliver 5-fluorouracil (5-FU) for targeted chemotherapy.[73] Unlike the pharmaceutical antagonist/agonist, these IL-6 aptamers did not interfere with IL-6 signaling pathways, but were still internalized by receptor-mediated endocytosis along with their cargo molecules.[73,74] However, while the aptamer-mediated delivery of 5-FU decreased the proliferation of BaF3 hIL-6R cells by ≈25%, the 5-FU modified aptamer had decreased affinity (Kd = 151 ± 3 nm) in comparison with the unmodified aptamer (Kd = 21 ± 3 nm).[73] Consequently, such a system may be improved upon by conjugating the aptamer to a drug loaded nanocarrier to function as a targeted DDV for myeloma or hepatocellular carcinoma.[74,75]
The IL-4 receptor is known to be overexpressed in glioblastomas,[76] pancreatic,[77] ovarian,[78] lung,[79] renal,[80] breast,[81] and colorectal cancers.[82] IL-4 causes the direct inhibition of cancer in vitro.[77–79] Initial strategies to suppress tumor growth by targeting the IL-4 receptor sought to develop chimeric proteins composed of a circularly permuted IL-4 and a mutated form of a bacterial toxin (Pseudomonas exotoxin).[77,78,80,81b,83]. Wu et al. developed AP peptide (CRKRLDRN)-conjugated pH-responsive doxorubicin filled micelles to target the IL-4 receptor.[81a] Here, the combination of IL-4 receptor targeting and pH-mediated drug release resulted in a 40.4% or 13.4% reduction of breast cancer tumor (MDA-MB-231 xenograft) weight compared to treatment with free drug or non-targeted micelles, respectively.[81a] The AP peptide could also be used to target IL-4 receptors overexpressed on glioma,[76] colorectal cancer,[82] or lung cancer cells.[79b] Consequently, Yang et al. (2012) synthesized AP1 (CRKRLDRNC)-conjugated liposomes while Sun et al. synthesized similarly conjugated AP1 polymeric nanoparticles to target doxorubicin to glioblastomas.[76] Both Yang et al. and Chi et al. conjugated AP1 to liposomes for delivering doxorubicin to murine colorectal cancer IL-4 receptor positive cells (CT26-IL4R) and human squamous non-small cell lung cancer (H226), respectively.[79b,82] In these in vivo studies, tumor uptake of liposomes was enhanced with the use of the peptide specific ligand to IL-4R (Figure 3C) and tumor volume was significantly decreased in IL-4R peptide conjugated liposomes as compared to unconjugated liposomes.[79b,82]
Another viable interleukin targeting candidate is the IL-3 receptor, which is overexpressed on chronic myelogenous leukemia (CML) and acute myeloid leukemia blast.[84] Bellavia et al. engineered human embryonic kidney (HEK293T) cells to produce exosomes expressing IL-3 and lysosome-associated membrane protein 2 (LAMP2) fusion protein (IL3-Lamp2b).[84b] The engineered exosomes were loaded with Imatinib (IM) and able to target and reduce the size of IM-sensitive LAMA84 tumor xenografts in vivo.[84b] Further, these exosomes reduced the tumor volume of IM-resistant K562 xenografts by delivering functional siRNA against the breakpoint cluster region-Abelson (Bcr-Abl) oncoprotein,[84b,85] thereby overcoming pharmacological resistance common in CML.[86]
The IL-13 receptor represents a viable target for head and neck cancers. Madhankumar et al. synthesized IL-13-targeted DDVs for the delivery of doxorubicin to glioblastoma multiforme tissue in vitro and to glioma tumors in vivo.[87] In a glioma mouse model, subjects treated with IL-13 labeled liposomes grew tumors more than five times smaller than that of mice treated with non-targeted liposomes. Wang et al. improved upon this targeting scheme by substituting the complete IL-13 protein for a more stable and easily synthesized 32-amino acid peptide (IP) with high affinity for the IL-13 receptor.[88]. IP conjugated mesoporous silica nanoparticles loaded with doxorubicin were internalized by human glioma U251 cells, but not human astrocyte cells in vitro. Shortly following this study, a smaller nine-amino acid peptide (CGEMGWVRC; Pep-1) with high affinity for the alpha-2 subunit on the IL-13 receptor was isolated from a phage display library.[89] Wang et al. then demonstrated that a Pep-1 conjugated polymeric nanoparticle had improved accumulation and penetration into C6 tumor spheroids in vitro and significantly increased delivery to the brain and glioma region in vivo.[90]
IL-16-targeted approaches for drug delivery have been adopted for the delivery of docetaxel to glioblastomas in vivo. Gao et al. reported that the addition of a secondary receptor with the IL-16 targeting peptide improved the prognosis of glioblastoma in rodents.[91] Targeting neurovascular cells with the arginine-glycine-aspartic acid (RGD) integrin-binding motif and glioma C6 cells with the IL-16 peptide, resulted in a twofold increase in survivability compared to a 1.5-fold increase for each single targeting approach, relative to saline controls.
Lastly, human trials for IL-11 receptor-mediated drug targeting were investigated for the treatment of prostate, lung, and bone cancers.[92] Targeting the IL-11 receptor showed a significant reduction in the size of tumors in rodents and, in nonhuman primates and humans, had a favorable pharmacokinetics profile and tumor specific internalization.[92c] However, phase I clinical trials are necessary to examine the efficacy of IL-11 targeting in the treatment of cancer.
Interleukin receptor-mediated drug delivery is limited by the redundancy of shared activity, meaning that the inhibition of one type of interleukin can be compensated for by signaling from other interleukins.[93] The inhibition of interleukins can also lead to an impaired host immune response due to the imbalance of immune-regulatory cytokines.[93]
3.3. Folate Receptor
The folate receptor (FR), also known as folic acid or vitamin B9 receptor, is a cysteine-rich cell-surface glycoprotein that has three characterized subtypes: FRα, FRβ, and FRγ.[94] In healthy tissues, the folate receptor functions to bind and internalize folic acid (FA), which is necessary for the synthesis of nucleic acids and DNA repair.[94] More specifically, FRα is known to be overexpressed in multiple cancer types, including: breast, lung, colon, mesothelioma, ovarian, and renal carcinomas.[94] Early in vitro models by Lee et al. and Leamon et al. indicated the promise of folic acid–functionalized particles or drug-conjugates in directing chemotherapeutics to various tumor cell lines.[95] Further work was done to exploit FR-mediated drug delivery of adriamycin,[96] paclitaxel,[97] doxorubicin,[98] methotrexate,[99] docetaxel, and cisplatin.[100] Lee et al. formulated folate conjugated, self-organizing doxorubicin-loaded nanoparticles, which had significant anticancer efficacy in vivo. Tumor volume was reduced by 50% when treated with folate functionalized nanoparticles compared to free doxorubicin, over the course of 18 days in a tumor xenograft model.[101] Improved nuclear delivery of doxorubicin was achieved via folate-functionalized liposomes (FTLs).[102]
Delivery of folate-targeted DDVs encapsulating docetaxel (DC) and cisplatin (CIS) (FA-LCN-DC) were formulated by Thapa et al. for in vivo targeting of MDA-MB-231 human breast cancer tumor bearing mice.[100a] These nanoparticles induced a significant reduction of 42.8%, 23.7%, and 11.9% in tumor volume and increased survivability compared to free docetaxel, cisplatin, or the dual cocktail, respectively (Figure 4, yellow).[100a] Intracellular apoptotic markers, such as caspase-3 and poly ADP ribose polymerase, were also elevated in mice treated with FA-LCN-DC, indicating upregulation of cell-mediated death. Alternatively, proliferation and angiogenesis markers, Ki-67 and CD31, respectively, were significantly lower in FA-LCN-DC treated mice.
Figure 4.
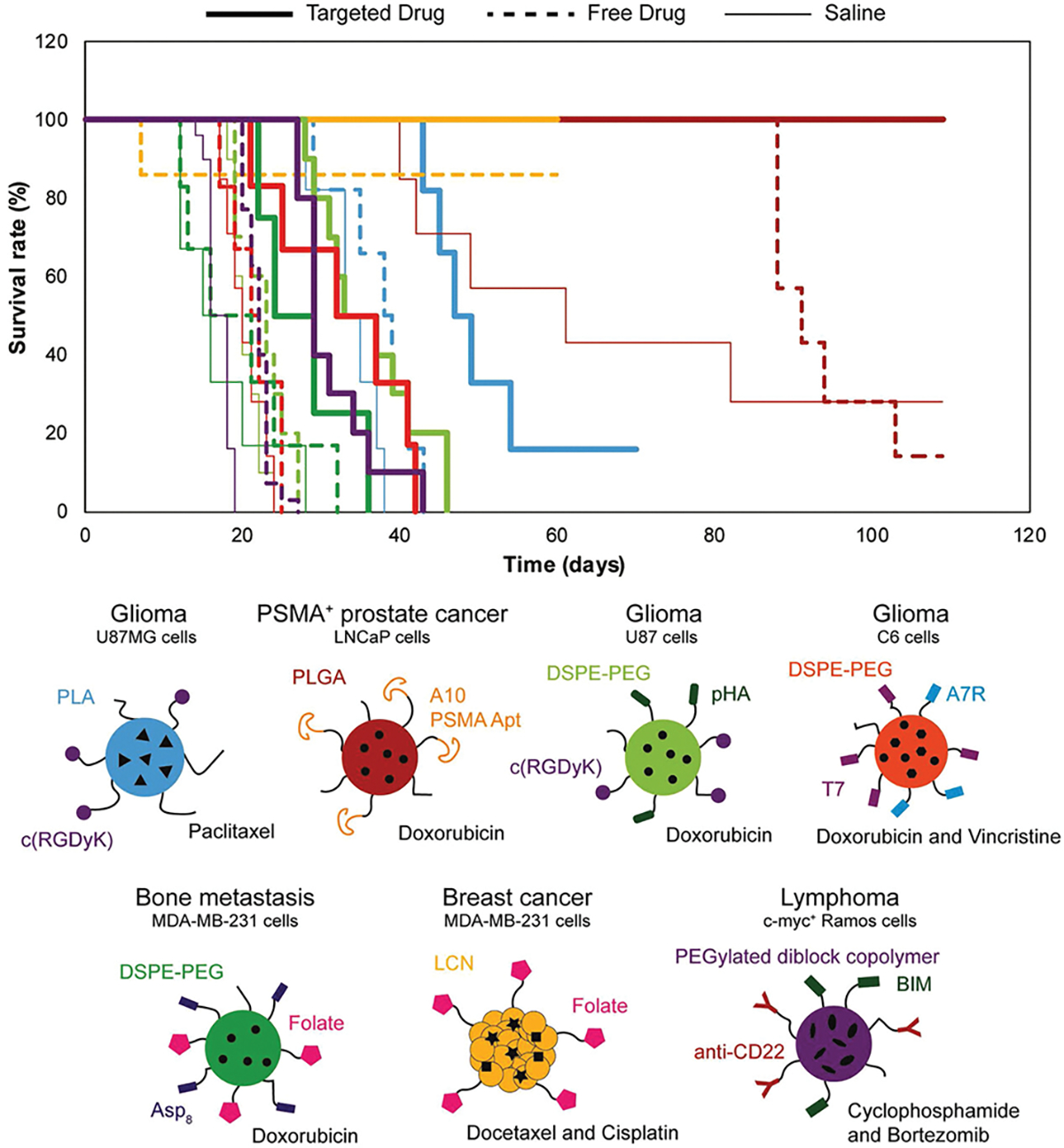
Kaplan–Meier survivability analysis of recently published DDVs shows that receptor-mediated drug delivery improves in vivo survivability compared to saline and free drug: blue,[108] red,[5a] light green,[191b] orange,[191c] green,[191d] yellow,[100a] purple,[114c]
Further in vivo work by Kukowska et al. with folate conjugated fifth generation polyamidoamine (PAMAM) dendritic polymers coupled to methotrexate (G5-FI-FA-MTX) demonstrated enhanced antitumor efficacy.[99] Severe combined immunodeficient female mice (SCID) bearing human KB tumor xenografts were treated with G5-FI-FA-MTX and various levels of free MTX. The survival rate of mice treated with G5-FA-MTX was approximately 30 days longer than those treated with free MTX.
Although many papers utilize folate receptors as a target, the use of folate clinically is limited due to expression on many normal tissues. This reduces the ability to selectively target in humans and results in less of an advantage relative to non-targeting DDV than observed in animal studies. A significant challenge in the field of targeted drug delivery is the design of animal studies that mimic the therapeutic benefit and toxicity profile in humans.
3.4. Integrin Receptor αvβ3
Integrins are a family of proteins that mediate cell attachment through binding to extracellular matrix (ECM) proteins to anchor cells within tissue.[103] Further, integrin receptors play a pivotal role in regulating cell functions responsible for the progression and metastasis of solid tumors.[104] The RGD motif, a cell binding peptide sequence identified from fibronectin, was discovered over 30 years ago and remains a highly explored therapeutic target.[105] Exploitation of this RGD recognition system for cell targeting and internalization is widely implemented in targeted delivery of paclitaxel,[106] doxorubicin,[3,107] and camptothecin.[43] Synthetic constructs of the RGD domain have enabled therapeutic and diagnostic targeting to cancer cells.
Arap et al. identified two synthetic integrin peptide ligands CDCRGDCFC (RGD-4C) and CNGRC from phage homing studies in tumors. Functionalized nanoparticles and drug-ligand conjugates for the delivery of doxorubicin were evaluated in mice bearing MDA-MB-435 tumors.[3] In vivo data demonstrated a reduction of tumor mass by 50% on average, as well as improved survival of mice treated with RGD-4C targeted DDVs compared to those treated with free doxorubicin.
RGD combined with residues d-tyrosine and lysine in a cyclic conformation c(RGDyK) was conjugated to paclitaxel loaded micelles for delivery to glioblastomas in vivo.[108] A glioblastoma U87MG mouse model, positive for αvβ3, was utilized for testing efficacy of c(RGDyK)-targeted, paclitaxel encapsulating micelles. Results demonstrated that these c(RGDyK)-targeted micelles accumulated within the tumor tissue, resulting in the greatest inhibition (≈50%) of tumor growth and survival compared to nonfunctionalized micelles encapsulating paclitaxel (Figure 4, blue).
Babu et al. developed PLGA-chitosan-based nanoparticles functionalized with RGD for the delivery of paclitaxel or cisplatin in cell lines which are known to overexpress the integrin αvβ3 receptor: A549, H1299, and H1975 lung cancer cells.[106] RGD-targeted DDVs were internalized significantly more than non-targeted particles or free paclitaxel in A549 and H1975 cells. Western blot analysis performed on samples with and without preincubation of excess RGD peptide, revealed inhibition of apoptotic integrin αvβ3 signaling (cleaved apoptotic protein, C-PARP, and caspase 9 levels) to be specifically modulated by RGD binding. Integrin-targeted therapeutics have demonstrated efficacy in the treatment of lung cancer and breast cancer in in vitro and in vivo models. A limitation to integrin-targeted therapeutics is the severity of side effects caused by integrin antagonists, such as the development of progressive multifocal leukoencephalopathy.[109]
4. Receptors Targets for Inhibiting Metastasis
Therapeutics designed to address the prevention and progression of metastatic cancer are in demand. Metastatic cancers account for 90% of all cancer-related deaths and are a leading cause of death worldwide.[110] Survival at 5 years post diagnosis is 23%, representing a 76% decrease in survival compared to patients with nonmetastatic breast cancer.[13] The “metastatic cascade” of cancer involves the following steps: 1) local invasion of surrounding tissues, 2) migration into nearby blood vessels and subsequent systemic circulation, 3) invasion into other organ systems, and 4) development of micro, and eventually macro, metastases.[111] Receptors, as well as signaling ligands, involved in processes of cell migration, attachment, and proliferation represent favorable targets for therapeutics.
4.1. Cluster of Differentiation (CD) Receptors
The family of cluster of differentiation (CD) receptors is a diverse set of immunoregulatory cell surface markers commonly found on cancer stem cells (CSCs). CSCs are a subpopulation of cancer cells which initiate tumor development and possess unique characteristics of self-renewal, quiescence, and overexpression of adenosine triphosphate-binding cassette (ABC) transporters. These features often lead to unfavorable cancer outcomes, such as drug resistance, tumor reoccurrence, and metastasis.[112] The CSC population within solid tumors varies widely (0.03–42%) depending on cancer type.[113]
CD receptors, 14, 22, 36, 44, and 133 have been noted as promising delivery targets to inhibit tumor metastasis and improve survivability (Figure 4, purple).[114] Inhibition of metastasis is achieved through a signaling blockade of CD receptors. CD36, also referred to as platelet glycol protein 4, is a fatty acid receptor which is noted to be “essential” in signaling metastatic progression.[115] A study by Pascual et al. demonstrated that by blocking these receptors with anti-CD36 neutralizing antibodies, lymph node metastasis was decreased by 80–90% and full remission was achieved in 15% of mice in an oral cancer model.[115] Similarly, models of CD44 blockade demonstrated inhibition of metastasis in vivo,[116] while targeting by Eliaz et al. indicated a marked increase in cancer cell–specific cytotoxicity from CD44 receptor-mediated drug delivery.[117] Hylauronic acid (HA)-conjugated liposomes encapsulating doxorubicin, targeting the CD44 receptor, were tested at various concentrations of doxorubicin (0.01, 0.1, 1, 10 μg mL−1) in high and low CD44 expressing B16F10 murine melanoma cells and CV-1 kidney cells, respectively. Resulting data indicated a 4.4-fold increase in potency of HA-targeted DDVs compared to administration of free drug, in B16F10 cells.
CD44-targeted mAb-conjugated particles were targeted to hepatocellular carcinoma (HCC) cells for the delivery of doxorubicin.[118] Clinically, the long-term survival of HCC is notably low, due in part to late diagnosis and accelerated metastasis.[119] Further approaches to target CD44 were implemented by Wei et al. in drug resistant MDA-MB-231/F breast cancer tumor cells, human pancreatic cancer cell line MIA PaCa-2, and MCF-7 human breast cancer cell spheroids.[120] In drug resistant cells, cholesterol-HA nanogels (CHA-nanogels) encapsulating etoposide or curcumin had 4.2 and 2.1-fold enhancement in cytotoxicity over free drug, respectively. Comparatively, CHA-nanogel formulations with salinomycin (SAL) and curcumin (CUR) demonstrated a 3.4 and 7.8-fold enhancement in cytotoxicity compared to their respective HA-drug conjugates, HA-SAL and HA-CUR.
Work done by Hu et al. demonstrated the in vitro efficacy of doxorubicin and/or cyclopamine encapsulated HA-cystamine-PLGA on high and low CD44 expressing breast cancer cell lines MCF-7 and MDA-MB-231, respectively.[114a] In vitro data noted that treatment with combined doxorubicin and cyclopamine encapsulating, HA-targeted nanoparticles resulted in nearly the complete elimination of CSC populations compared to treatment with free doxorubicin or cyclopamine alone. An MCF-7 human breast cancer xenograft model was adapted by Zhong et al. for the evaluation of CD44-targeted paclitaxel loaded micelles.[121] Biodistribution, in terms of the ratio of uptake in tumor-to-normal tissue, of HA-functionalized particles were significantly higher than that of free Taxol. A significant inhibition of tumor progression was noted in vivo after 30 days of treatment with HA-targeted DDVs compared to the control or free Taxol.
CD133-targeted, aptamer conjugated nanoparticles encapsulating salinomycin, demonstrated antitumor efficacy in mice bearing drug resistant Saos-2 osteosarcoma xenograft tumors. The RNA aptamer A15 was chosen for its ability to selectively bind to CD133 and mediate internalization after binding.[122] PEGylated PLGA nanoparticles loaded with salinomycin and functionalized with aptamer A15 (Ap-SAL-NP) significantly reduced tumor weight, by ≈15% and ≈50%, compared to nonfunctionalized SAL-NP or free SAL, respectively.[123]
4.2. Chemokine Receptors
Chemokine receptors are promising therapeutic targets as they are frequently overexpressed on cancer tumors and regulate immune cell processes, angiogenesis, and embryogenesis.[124] The chemokine receptor type 4 (CXCR4) is a G protein coupled receptor (GPCR) with seven trans-membrane domains and binds to the ligand CXCL12. CXCR4 is noted to play a critical role in breast cancer metastasis to the lung, bones, and distant lymph nodes by mobilizing cancer cells along chemokine gradients.[125] Further, CXCR4 surface density and organization was demonstrated to play a role in cellular adhesion and subsequent metastatic progression.[126]
Guo et al. demonstrated the importance of receptor surface density on the efficacy of CXCR4-targeted therapeutics. Metastatic breast cancer cell lines HCC1500 (highly invasive) and MDA-MB-175VII (noninvasive) expressed a 10 and 2.5-fold increase in CXCR4 mRNA expression, respectively, compared to noncancerous MCF10A epithelial cells. Both metastatic cell lines also exhibited a 20-fold increase in surface expression of CXCR4 over MCF10A cells. Based upon these trends of heightened CXCR4 expression, liposomes encapsulating doxorubicin and functionalized with the CXCR4 mAb were synthesized and evaluated for effectiveness in binding and subsequent cellular uptake, release of doxorubicin, and impact on cell viability. Liposomes with CXCR4 mAb surface conjugation of 1720 molecules per μm2 demonstrated significantly increased uptake in MDA-MB-175VII and HCC1500 cells, compared to the treatment with IgG control liposomes. HCC1500 and MDA-MB-175VII cell viability was reduced to 11% and 53%, respectively, when treated with 50 μg mL−1 doxorubicin loaded aCXCR4-Dox-LPs, representing a significant decrease compared to cells treated with non-targeting liposomes.
Further work from Guo et al. demonstrated the synergistic effects and anticancer efficacy of using CXCR4 targeting liposomes for the delivery of siRNA in triple negative breast cancer cell (TNBC) lines MDA-MB-436 and MDA-MB-231 and metastatic breast cancer cell lines MDA-MB-175VII and HCC1500.[13] pH-responsive liposomes conjugated with CXCR4 mAbs were prepared for gene delivery. Lipocalin-2 (Lcn2) was demonstrated to induce the epithelial to mesenchymal transition (EMT) and to be upregulated in a wide range of cancers: breast, ovarian, thyroid, pancreatic, and colon.[127] Accordingly, knockdown of Lcn2 resulted in decreased cancer migration and invasion in vitro, as well as reduced tumor growth and metastasis in vivo.[128] Both MDA-MB-436 and MDA-MB-231 exhibited overexpression of surface CXCR4 and significantly high levels of Lcn2, 2.8-fold higher, respectively, than in MCF10A cells. Delivery of siRNA via aCXCR4-Lcn2-pH to metastatic breast cancer cells resulted in a significant knockdown of gene expression, up to 84% in MDA-MB-231 cells followed by 78% in MDA-MB-436 cells, outperforming both the commercially available siRNA transfection reagent Lcn2-LIPO and non-pH-sensitive aCXCR4-Lcn2-LPs. Cell migration was also significantly halted. MDA-MB-436 and MDA-MB-231 cells treated with aCXCR4-Lcn2-pH exhibited a reduction in migration of 88% and 92%, respectively, compared to non-treated cells.
Chittasupho et al. adapted a similar approach, utilizing the CXCR4/CXCL12 axis for targeting of human breast cells with doxorubicin loaded ligand conjugated dendrimers.[125a] Low molecular weight linear peptide analogues of FC131 ((cyclo)(D-Tyr-Arg-Arg-L-3- (2-naphthyl)alanine-Gly)), a potent CXCR4 antagonist,[129] were synthesized and conjugated to the surface of polyamidoamine (PAMAM) dendritic polymer (LLCF131-D4). Doxorubicin was loaded into particles (LLCF131-DOX-D4) and tested in BT-549-Luc and T47D cells at concentrations of 0.25–1 mg mL−1 for 2 h of treatment. A significant reduction in the migration index and viability of cells treated with 0.5 mg mL−1 LLCF131-DOX-D4 was observed compared to controls.
CXCR4 is a regulator of proliferation and angiogenesis of both healthy and cancerous cells. The inhibition of chemokine signaling can negatively impact the function of normal immune and epithelial cells.[130] Allosteric effects caused by molecules that act as both CXCR activators and inhibitors may initiate cross talk within the CXCL-CXCR signaling axis. Taken together, these effects may cause unanticipated effects which can limit therapeutic efficacy.[130] Detailed research to elucidate mechanisms of CXCL-CXCR cross talk is necessary to aid in the development of safe and effective chemokine inhibitors for therapeutic use.[130]
4.3. Estrogen Receptor
Estrogen receptors (ERs) are a type of nuclear receptor which bind to hormone estrogen (estradiol) that act as transcription factors to regulate cell proliferation.[131] The ER plays an important role in the development and maintenance of sexual and reproductive function, as well as initiating and regulating morphogenesis of lung, mammary gland, and prostate development.[132] Further, as the ER is overexpressed in 50–80% of all breast cancers,[133] it represents an ideal target for receptor-mediated drug delivery. Some of the earliest examples of targeted delivery mediated by the ER was to conjugate the β-Estradiol, or estrogen analogs (tamoxifen), to the following cytotoxic molecules: porphyrins,[134] enediynes,[135] Taxol,[136] mitomycin C,[137] nitrosourea,[138] geldanamycin,[139] mustine,[140] ellipticine,[141] chlorambucil,[142] and 2-crotonyloxymethyl-(4R,5R,6R)-4,5,6-trihydroxy-2-cyclohexenone.[143] However, the ER-binding affinities of these conjugates was compromised due to the modification of the hormone. For example, conjugation of estradiol or tamoxifen to porphyrins resulted in a lower binding affinity to the ER (half maximal effective concentration, EC50, of 274 and 2.2 nm for the estradiol and tamoxifen porphyrin conjugates vs. 1 and 0.0075 nm for free estradiol and tamoxifen, respectively).[134a,134b] Yet despite this, researchers demonstrated that the ER-porphyrin conjugates were selectively taken up by ER-positive cells and exposure to red light induced cell death in vitro.[134b,134c] Reddy et al. reported on the development of a β-Estradiol conjugated stealth liposomes to deliver anticancer genes to breast cancer cells.[144] ER-targeting liposomes containing the β-Gal gene were incubated with ER-positive (MCF-7), ER-negative (MDA-MB-231), and control (human cervical carcinoma, Chinese hamster ovary, and murine sarcoma L-27) cells. Only MCF-7 cells were observed to have β-Gal activity, thereby demonstrating the high specificity for ER-positive cell internalization. The incorporation of the apoptosis inducing p53 gene within these β-Estradiol conjugated liposomes resulted in induced cell death in a target-specific manner in vitro. Similarly, the conjugation of an estrogen analog, tamoxifen, to AuNPs resulted in a 2.7-fold increase in potency toward MCF-7 cells in vitro relative to treatment with free tamoxifen.[145] Researchers have synthesized ER targeting estrone-conjugated liposomes and single walled carbon nanotubes to deliver doxorubicin,[146] gelatin NPs to deliver noscapine,[147] and chitosan NPs to deliver paclitaxel for breast cancer.[148] Rai et al. reported that these targeted liposomes had more than tenfold higher accumulation in the breast and uterus and lower concentrations in the heart compared with both free doxorubicin and non-conjugated liposomes.[146a] However, this liposome formulation also resulted in significant accumulation in the liver and spleen.
To overcome this limitation, Paliwal et al. synthesized ER-targeted, stealth liposomes.[[146b] These ER-targeted stealth liposomes were taken up significantly less by the reticuloendothelial system and resulted in a significant reduction in tumor volume compared to both free doxorubicin and non-targeting, stealth liposomes.[146b,146c] Lastly, an additional modification of this formulation to be pH-sensitive and only release the chemotherapeutic in the presence of the acidic tumor microenvironment, resulted in a further reduction in tumor volume, while maintaining a desirable biodistribution profile.[146d] Similarly, Yang et al. synthesized ER-targeted, pH-sensitive, polymeric DDVs that significantly reduced the tumor volume compared to non-targeting DDVs.[148]
4.4. Intracellular Adhesion Molecule-1 (ICAM-1)
ICAM-1 is a transmembrane glycoprotein found on endothelial and immune cells that is involved in immune cell recruitment to sites of inflammation.[149] Upon stimulation with inflammatory cytokines such as tumor necrosis factor alpha (TNF-α), interleukin 1β (IL1-β), and lipopolysaccharide (LPS), endothelial cells upregulate ICAM-1 on the cell surface to aide in the firm adhesion and extravasation of leukocytes through the inflamed endothelium into diseased tissue by transcellular and paracellular pathways.[150] Certain nanomaterials and cells that bind ICAM-1 cause it to form clusters in the cell membrane, inducing an intracellular signaling cascade that disrupts vascular junction and enhances endothelial permeability on inflamed endothelium.[25b,151] Many types of tumors exist in a state of chronic inflammation, resulting in the overexpression of ICAM-1 on tumor-associated vasculature. Additionally, some types of cancer constitutively overexpress ICAM-1 compared to the normal tissue from which they are derived.[152] Guo et al. has shown that ICAM-1 is significantly overexpressed on TNBC tissue and cell lines using histology and flow cytometry, respectively. Conjugation of ICAM-1 mAbs to casein-coated IONPs enhanced their TNBC cell uptake (2.4-fold to fourfold) and TNBC tumor targeting (Figure 3D) compared to IgG conjugated IONPS, which correlated well to the ICAM-1 expression on each cell line. ICAM-1-targeted IONP TNBC uptake was more pronounced than the enhanced uptake of HER2-targeted IONPs in a HER2-overexpressing breast cancer cell line.[152b]
The therapeutic potential of ICAM-1 was demonstrated in a later study through the synthesis of ICAM-1-targeted immunoliposomes for the treatment of TNBC. Guo et al. synthesized Ph-sensitive liposomes conjugated with ICAM-1 mAb and demonstrated that these liposomes achieved 3.5-fold higher cellular uptake in the TNBC cell line MDA-MB-231 over untargeted IgG immunoliposomes. Incubating these ICAM-1-targeted liposomes encapsulating anti-angiogenic Lcn2 siRNA achieved efficient knockdown of VEGF production in TNBC cells, which in turn led to reduced migration and proliferation of HUVEC and HMVEC cells cultured in TNBC conditioned media.[153]
A study performed by Fakhari et al. in which PLGA nanoparticles were synthesized with varying surface densities of cLABL peptide, a high affinity cyclic peptide derived from the native ICAM-1 ligand lymphocyte function-associated antigen 1 (LFA-1). DDV uptake was evaluated on cells from a lung cancer cell line A549 after stimulation with TNF-α for 24 h to increase ICAM-1 expression. The results of this study indicated that optimal binding occurred not at the highest possible surface densities, but rather intermediate to lower peptide densities (12 000–24 000 peptides μm−2), indicating that there exists an optimal peptide ligand surface density. Multivalent interactions with target receptors increased ligand binding.[154]
While receptor overexpression is a common consideration for the design of targeted DDVs, the precise relationship between the cell receptor surface density and DDV ligand density on enhancing cellular uptake is poorly understood. A recent study by Guo et al. showed that the difference in adhesion force between atomic force microscopy (AFM) tips conjugated with ICAM-1 mAb on live TNBC and nonneoplastic breast cells is a better predictor of a receptor’s potential as a therapeutic target for DDVs than PCR or flow cytometric analysis of the target receptor expression. The receptor expression determined by PCR and flow cytometric analysis indicated that ICAM-1 expression was 13.9 and 44.6 times higher on the TNBC cell line MDA-MB-231 than the nonneoplastic breast cell line MCF10A, respectively. This receptor expression contrasts with the relative adhesion force of the two cell lines, which indicated that MDA-MD-231 cells resulted in a 1.7-fold higher adhesion force over MCF10A cells. When an orthotopic TNBC tumor model was used to evaluate the effective targeting of ICAM-1-targeted liposomes, it was found that 48 h after injection ICAM-1-targeted liposomes achieved 1.6-fold higher tumor accumulation than control IgG liposomes, which corresponded closely to the relative adhesion force. This relationship was validated by correlating the relative binding force of ICAM-1 and EGFR on MDA-MB-231 and MCF10A cells to in vivo accumulation of liposomes targeted to each receptor. It was found that there exists a linear relationship between the relative binding force of mAb functionalized AFM tips with their specific receptor and the relative accumulation of the corresponding liposome in vivo.[155] Targeting ICAM-1 can be an effective strategy not only to enhance cellular internalization in cancer cells that overexpress the receptor, but also to enhance the accumulation of targeted DDVs in the tumor.
Park et al. synthesized a cross-linked urethane acrylate nonionomer (UAN) nanoparticle conjugated with the ICAM-1 binding domain of LFA-1 (Id-UANs). The UAN nanoparticles contained a hydrophobic core region and displayed nickel-nitriloacetic acid (NTA) functionalized PEG on their surface, which enabled efficient binding of the His tagged I domain of LFA-1 to the surface of the nanoparticle. LFA-1 was engineered to be stable in its high affinity conformation for both human and murine ICAM-1 in Escherichia coli. The Id-UANS accumulated in tissue that received a local LPS injection threefold greater than non-targeted controls. In an in vivo tumor model, mice were subcutaneously injected with the cervical cancer cell line HeLa and human embryonic kidney cell line 293T, which are ICAM-1 positive and ICAM-1 negative tumor phenotypes, respectively. Mice were intravenously injected with Id-UANs and NTA-UANs, resulting in a 5.1-and 2.1-fold increase in accumulation of Id-UANs over NTA-UANs in the respective tumors. Ex vivo flow cytometric analysis showed greater internalization of Id-UANS in tumor-associated cells than in other organs.[152a]
5. Receptor Targets for Drug Resistant Tumors
Resistance to cancer treatment accounts for 90% of treatment failure, resulting in poor prognosis in patients with metastatic cancer.[156] Intrinsic dysfunctions in intracellular apoptosis signaling are typically associated with the development of multiple drug resistance (MDR).[156] The overexpression of adenosine triphosphate-binding cassette (ABC) transporters was implicated in the pathogenesis of MDR.[157] ABC transporters efflux toxic drugs released in the cytoplasm, rendering them ineffective cytotoxic agents.[156] Thus, when first line chemotherapeutics lose their effectiveness, there are few treatment options remaining. As MDR is a persistent problem in the clinic, recent research has focused on the development of receptor-targeted therapeutics that can bypass the molecular mechanisms of MDR and effectively treat resistant cancers. Targeted drug delivery offers a promising alternative to classic delivery techniques and a potential avenue for the evasion of drug resistance mechanisms.
5.1. Prostate-Specific Membrane Antigens and Androgen Receptors
Prostate-specific membrane antigen (PSMA) is widely used as a receptor target against prostate cancer due to its overexpression in many prostate cancer cells.[158] Prostate cancer accounts for 19% of new cancer cases annually, making it the highest percentage of new cases reported amongst men.[159] A range of PSMA targeting agents, such as aptamers,[159] antibodies,[114d] and peptides,[160] were developed and conjugated to drug molecules and carriers to improve their targeting specificity to prostate cancer cells.[161] For instance, Lupold et al. developed two RNA aptamers (A9 and A10) that bind to PSMA positive prostate cancer cells.[162] Amongst these, the A10 aptamer was conjugated to polymeric nanoparticles and micelles for the delivery of traditional chemotherapeutics and siRNA for MDR prostate cancer cells. Farokhzad et al. synthesized A10 conjugated PLGA DDVs to deliver docetaxel to LNCaP prostate cancer cells in vitro and in vivo.[5a] In vivo, 100% of the mice survived when treated with the PSMA-targeted DDVs compared to 14% of the mice treated with free docetaxel (Figure 4, red).
The androgen receptor (AR) is located on the nuclear membrane, also widely referred to as nuclear receptor subfamily 3, group C, member 4 (NR3C4); it primarily binds testosterone and dihydrotestosterone. The AR is regarded as a “master regulator of genes” in castrate-resistant prostate cancer (CRPC) and acts as a transcription factor to regulate cancer genes for tumor growth.[163] In fact, AR signaling was demonstrated to be directly involved in prostate cancer progression. Binding of intracellular androgens to the ligand binding domain, promotes nuclear localization and subsequent expression of target genes.[164] Unlike many cancer-related receptor targets, androgen signaling occurs intracellularly at the cytosolic and nuclear levels, making it a more challenging target to effectively impact. Yet, their conjugates (steroidal ligands) can readily diffuse though the cell membrane and result in either cytosolic or nuclear delivery of drug conjugates or carriers.[165] Utilizing this approach, Mishra et al. synthesized AR-targeted DDVs by conjugating testosterone to 5-FU bearing liposomes.[166] These liposomes had high specificity to AR expressing tissues with a sustained release profile of 24 h. As an alternative to traditional chemotherapeutics, Yang et al. encapsulated ARHP8 siRNA in A10-conjugated PLGA DDVs.[163] Silencing the AR resulted in rapid prostate-specific antigen decline and tumor regression in 2 weeks in vivo, in both castration-resistant (22RV1) and androgen-responsive (LAPC-4 and LNCaP) prostate cancer cells.
Mutations in the AR binding domains can lead to castration- and chemotherapy-resistant prostate cancer cells, and is a major contributing factor to high morbidity of prostate cancer.[167] Dreaden demonstrated that anti-androgen-conjugated AuNPs lead to an increased pharmacological potency when treating treatment-resistant prostate cancers.[167] Success in targeted delivery to androgen receptors and PSMA is limited by resistance to AR-targeted therapies.[168] As previously mentioned, AR can be a physiologically challenging target, as it is a nuclear membrane receptor. Thus, targeting requires intracellular delivery of inhibitory ligand and cargo, presenting unique demands for material and vehicle design.
5.2. Transferrin Receptors
The transferrin receptor (Tfr) is a membrane-associated glyco-protein responsible for mediating iron acquisition via binding of the iron-bound transferrin plasma protein and subsequent receptor-mediated endocytosis.[169] Cellular uptake and storage of iron are critical to functions of iron proteins involved in oxygen transfer, metabolic processes, and DNA synthesis.[169] Tfr also represents a viable target for bypassing MDR. In a study conducted by Gao et al., a biodegradable pH-sensitive micellar doxorubicin DDV was devised with the surface conjugation of a modified seven peptide transferrin receptor ligand (7pep; amino-acid sequence: HAIYPRH).[170] Doxorubicin encapsulating micelles composed of 7pep conjugated 1,2-distearoyl-sn-glycero-3-phosphoethanolamine and polyethylene glycol-2000 (DSPE-PEG2000) were used in nude mice bearing drug-resistant MCF-7 xenografts (MCF-7/Adr) which overexpress Tfr.[170] Micelles were evaluated for internalization and therapeutic effect at formulations of varying 7pep modification (10%, 30%, 50%, 60% 7pep). Micelles with 60% Pep7 modification demonstrated statistically higher rates of internalization and were endocytosed via caveolae-mediated mechanisms. Compared to control micelles, 60% Pep7 modified micelles were internalized by tumor tissue 2.1-fold greater in vivo and significantly reduced MCF-7 cell viability in vitro.[170] Unfortunately, off target accumulation (heart, lung, and kidney) was found to be elevated in 7peptargeted micelles as compared to non-peptide conjugated HD micelles.
Ferris et al. synthesized Tfr-targeting, mesoporous silica nanoparticles (MSN) for the delivery of camptothecin (CPT) to determine uptake in pancreatic and pre-metastatic breast cancer cell lines, PANC-1 and BT-549, respectively. Analysis of cell cytotoxicity demonstrated that cell death was induced at a lower level of CPT in Tfr-targeted DDVs compared to non-targeted MSNs in PANC-1 cells.[43]
Tavano et al. developed transferrin (Tf) conjugated niosomes for the delivery of doxorubicin to MCF-7 and MDA-MB-231 breast cancer cells.[171] Niosomes composed of modified pluronic L64 surfactant (L64-ox) and cholesterol (chol), were synthesized with and without surface conjugation of Tf. L64-ox/Chol-D-Tf niosomes induced significantly higher cell death, as is noted in the reduction of cell viability at 48–96 h post treatment compared to cells treated with free doxorubicin.
Kobayashi developed a doxorubicin encapsulating DDV functionalized with a transferrin peptide to target SBC-3 and SBC-3/ADM small cell lung cancer cells.[172] As overexpression of P-glycoprotein (Pgp) is known to be implicated in the development of MDR, due to its role in mediating drug efflux,[172] DDVs that bypass this route of delivery may overcome MDR. Formulations of hydrogenated egg phosphatidylcholine (HEPC), egg phosphatidylcholine (EPC), 1,2-Dipalmitoyl-sn-glycero-3-phosphatidylethanolamine-N-[3-(2-pyridyldithio) propionate] (PDP-PE), and cholesterol (CHOL) were functionalized with ≈20–40 Tf molecules per liposome. These particles demonstrated a significant benefit over non-targeted DDVs in terms of degree of internalization, doxorubicin release within cancerous cells, and enhanced cytotoxicity.
Liu et al. developed targeted DDVs using dihydroartemisinin (DHA) and functionalized with Tf for internalization via the Tfr.[173] DHA is studied as a viable alternative for chemotherapy. The nanodrug (DHA-GO-Tf) was designed to induce cytotoxicity upon internalization via the generation of ROS induced by DHA, enhanced by the degradation of Tf during lysosomal endocytosis. Murine mammary cancer cells (EMT6) were utilized for in vitro testing of DHA-GO-Tf in which the DDVs demonstrated increased cellular uptake and a resulting significant decrease in cell viability over non-targeted DDVs. Ultimately DHA-GO-Tf killed up to 3× the cells as GO-DHA or GO-Tf DDV formulations.[173] An EMT6 xenograft model was adapted to evaluate the anticancer efficacy of DHA-GO-Tf DDVs in vivo. Xenograft bearing mice treated with intravenous injection of DHA-GO-Tf exhibited approximately ≈85% smaller tumor volumes than mice treated with DHA alone after 30 days. Survival studies within the same time frame revealed a 100% survival rate for EMT6 bearing mice treated with DHA-GO-Tf, compared to 50% survival when treated with DHA alone. These studies demonstrated the influence of synergistic treatment regimens and importance of target-based therapeutics.
Tfr-targeted drug delivery may enhance delivery of chemotherapeutics to surgically inaccessible tumors, however, limitations in clinical translation remain. The largest limitation is the high expression of Tfr in noncancerous cells, such as endocrine pancreas, liver, and brain endothelial cells.[174] This leads to internalization of Tf labeled DDVs into healthy cells and subsequent reduction in targeted antitumor efficacy.
5.3. HER2
The human epidermal growth factor receptor-2 (HER2) is a transmembrane glyco-protein of 185 kDa, with an intracellular tyrosine kinase domain.[175a] HER2 is known to be overexpressed in gastric (6–35%), lung (8–25%), breast and ovarian (25–30%) carcinomas.[175] High expression levels of HER2 are correlated with poor prognosis.[176] Immunotherapy-based tumor targeting with the anti-HER2 humanized mAb trastuzumab (Herceptin) has demonstrated therapeutic promise.[175a] Further, HER2-targeting may evade mechanisms of chemotherapy resistance.
A study by Wartlick et al. examined the effects of human serum albumin (HSA) nanoparticles modified with Herceptin on malignant breast carcinoma cell lines BT474 and SK-BR-3 (HER2 positive). Herceptin labeled NPs were internalized up to threefold higher in SK-BR-3 cells, compared to unlabeled HSA NPs. The formulation devised by Wartlick et al. was then further adapted to incorporate the loading of doxorubicin.[177] Herceptin-conjugated, doxorubicin-carrying HSA NPs developed by Anhorn et al. induced a greater than two and fivefold decrease in SK-BR-3 cell viability compared to cells treated with control Dox-NP-IgG and or NP-Herceptin formulations, respectively.[178]
Similarly, polyetheylenimine (PEI) and PLGA Herceptin bearing NPs (HER-PPNs) were developed by Yu et al. for delivery to HER2-overexpressing human breast cancer cell lines BT474 and SK-BR-3, as well as a HER2-negative MCF-7 cell line.[179] Herceptin was conjugated to paclitaxel encapsulating PEI/PLGA particles (PPNs) via electrostatic attachment (eHER-PPN) or chemical conjugation (cHER-PPN). eHER-PPNs were formulated at differing ratios of PPN to Herceptin and evaluated for HER2 binding efficiency; the optimal formulation was found to be 1:1. Cellular uptake and paclitaxel release were significantly greater in eHER-PPNs compared to cHER-PPNs in BT474 cells. Cell viability was significantly decreased in targeted eHER-PPNs compared to cHER-PPNs in vitro.[179] Further, BT474 cells treated with eHER-PPNs exhibited greater in vitro cell toxicity compared to those treated with a physical mixture of Herceptin and PPNs (pHER-NNPs) over the course of 72 h. This result suggests the increase in cytotoxicity of eHER-PPNs can be attributed specifically to HER2 receptor-mediated internalization of paclitaxel loaded particles, and is not a result of PPN and Herceptin cytotoxic superposition. Further support of this finding can be noted in the significant decrease in cell viability induced by eHER-PPNs versus blank PPNs (bPPNs) in cells treated with 100 and 200 μg mL−1 of nanoparticles.
Eloy et al. investigated the dual delivery of paclitaxel (PTX) and Rapa via anti-HER2 labeled immunoliposomes (ILs).[180] Particles were composed of soy phosphatidylcholine (SPC), DSPE-PEG2000, and cholesterol (Chol), and loaded with chemotherapeutics in the following ratio: SPC:Chol:DSPE-PEG2000:Rapa:PTX (10:2:0.5:1:0.33). The incorporation of SPC and Chol in the formulation of lipid-based vehicles was attributed to their biocompatibility and similarity to cell membrane composition, both of which allow for the particles to be efficiently internalized.[180] In vitro, triple negative 4T1 and HER2 positive SK-BR-3 breast cancer cells were utilized for in vitro and in vivo studies. Analysis by flow cytometry demonstrated a twofold higher uptake of ILs by SK-BR-3 cells compared to unlabeled liposomes. In vivo HER2-targeted ILs demonstrated significant cytotoxicity in tumor bearing mice. SK-BR-3 tumors were significantly reduced by over 50% throughout the course of 20 days of treatment, compared to liposomes lacking anti-HER2 functionalization or free PTX/Rapa.
Zahmatkeshan et al. examined the effect of varying ligand densities on delivery vehicles anticancer efficacy in human breast carcinoma cells lines SK-BR-3 and MDA-MB-231. Liposomes decorated with different concentrations of Anti-HER2/Neu (AHNP) peptide with a three glycine amino acid spacer (sequence: FCDG-FYACYADV) were tested in an in vivo mouse model for impact on tumor uptake, cell cytotoxicity, tumor growth, and tissue biodistribution.[181] Peptides were conjugated at levels of 25, 50, 100, and 200 molecules per vehicle and incubated with HER2-overexpressing cell lines. Tumor volume was significantly reduced in vehicles bearing 25 and 50 ligands per vehicle and completely eradicated in vehicles with 100 and 200 ligands, as compared to the negative control and Doxil mimic.[181] Survival outcomes for mice treated with AHNP liposomes of 100 and 200 peptides conjugated per vehicle were both sustained at a prodigious 100% over the course of 100 days post tumor inoculation.[181] Liposomes with 25 and 50 peptides per vehicle, however, demonstrated much lower rates of survival during the same time period.
Ringhieri et al. investigated the effects of conjugating the peptide KCCYSL (peptide P6.1) in various multimeric forms (monomer, dimer, and tetramer) to target HER2 overexpressing cells BT-474 and low HER2 expressing MDA-MB-231 cells. Using solid-phase synthesis and click chemistry procedures, Ringhieri et al. were able to maintain control over ligand orientation and density during attachment. Liposomes functionalized with P6.1 tetramers (tetrameric Lipo-T6) bound to BT-474 and MDA-MB-231 cells at nearly double the rate of nonfunctionalized liposomes (Lipo-NT), monomer (monomeric Lipo-M6), and dimer (dimeric Lipo-D6) functionalized liposomes at equal dosing concentrations (0.005, 0.01, 0.015, and 0.02 mm nanoparticles).[175a] Internalization of Lipo-T6 and anti-HER2-FITC labeled mAb (Herceptin) at 0.004 mm was found to be nearly equal after 60 and 90 min of incubation, demonstrating that the KCCYSL peptide is an equally effective targeting moiety and a potential therapeutic for patients who have grown resistant to herceptin treatment.[175a]
Le et al. evaluated the efficacy of docetaxel (Doc) loaded PLGA-poly(lactic acid) (PLA) nanoparticles functionalized with the single chain anti-HER2 mAb (scFv).[182] To test their NP formulations (scFv-Doc-NP), 3D tumor spheriods of BT-474 and HCT-116 cells were utilized, as spheroids may be more physiologically relevant models in vitro compared to 2D cultures which lack the spatial organization and diffusion limitations innate to 3D culture. Cellular uptake and central-tumor necrosis development was studied in mature BT-474 and HCT-116 spheroids. BT-474 tumor spheroids exhibited enhanced uptake in samples treated with 100 μg of scFv-Doc-NPs compared to the minimally expressing HER-2 HCT-116 spheroids. Spheriods were treated with 100 μg of Doc-loaded NPs for 5 days and evaluated for size and necrotic core development. Treatment with scFv-Doc-NPs resulted in greater tumor reduction as well as development of a larger necrotic center compared to treatment with nonfunctionalized Doc-NPs in BT-474 spheroids. The opposite effect was observed in HCT-116 spheriods, suggesting that the effect of mAb functionalization is highly dependent on the density of target receptor present on a tissue’s surface.
You et al. formulated HER2-targeted NPs capable of pH-sensitive doxorubicin release and thermal ablation of HER2 overexpressing (SK-BR-3) and minimally expressing (MCF-7) breast cancer cells.[32b] AuNPs were embedded in pH-responsive dithiolated dimethylaminoethyl methacrylate (dT-DMAEMA; 30 dT-DMAEMA/70 HEMA, mol/mol) and functionalized with Herceptin (≈117 molecules per nanoparticle) and PEG (HPG-Dox-30D70H). Flow cytometry analysis revealed a 5.4-fold increase in HPG-Dox-30D70H binding of SK-BR-3 cells relative to MCF-7 cells. Further, SK-BR-3 cells treated with HPG-Dox-30D70H demonstrated a significant reduction in viability when treated with laser irradiation, compared to cells treated with Dox-30D70H formulation with or without laser treatment. As anticipated, no significant impact on cell viability was noted in MCF-7. These “multifunctional” drug nanocarriers represent a site-specific and synergistic method to effectively target HER2 positive breast cancer.
Because HER2 lacks a natural ligand, the identification and synthesis of a high-affinity ligand with in vivo stability, is a significant challenge.[183] As with other receptor-targeted drugs, resistance to anti-HER2 therapy is a limiting factor of clinical progress.[184] SK-BR-3 overexpression of HER2 is approximately 200-fold higher than the overexpression of other cancer receptors, which is not necessarily representative of HER2 expressing patient tumors.[152b]
6. DDV Optimization
A number of techniques were developed to optimize target efficacy, improve ligand binding, and enhance cellular uptake of targeted drug delivery systems. Surface patterning of ligands on delivery vehicles in spatially distinct orientations has shown to enhance cell uptake and reduce nonspecific uptake in healthy tissues.[185] Spacing, density, and ligand orientation are arguably the most influential factors when it comes to functionalizing the surface of a DDV. Further, modeling and simulation-based approaches for vehicle design have proven to be useful and cost-effective tools for predicting optimal vehicle design and potential success in vivo.
6.1. Ligand Surface Patterning
The addition of ligands in precise patterns, density dependent arrays or clusters are viable techniques for enhancing vehicle binding and internalization to specific cells (Figure 5A). It was demonstrated that optimizing ligand density can have a dramatic effect on cellular uptake.[186] Moradi et al. synthesized folate-conjugated ovalbumin (OVA) coated polystyrene nanoparticles of varying folate density, targeted to bind folate receptors on Calu-3 cells (human airway endothelial cells). Here, higher folate densities on OVA nanoparticles (13.2 mol folate: 1 mol OVA) resulted in enhanced cellular internalization compared to OVA nanoparticles with less folate bound (1.5 mol folate: mol OVA). Ligand density may also affect the pathway by which a nanoparticle is internalized. OVA nanoparticles with higher ligand densities are internalized via caveolae-mediated endocytosis, while particles with lower ligand concentration are typically endocytosed via clathrin-mediated processes (Figure 5B).[187]
Figure 5.
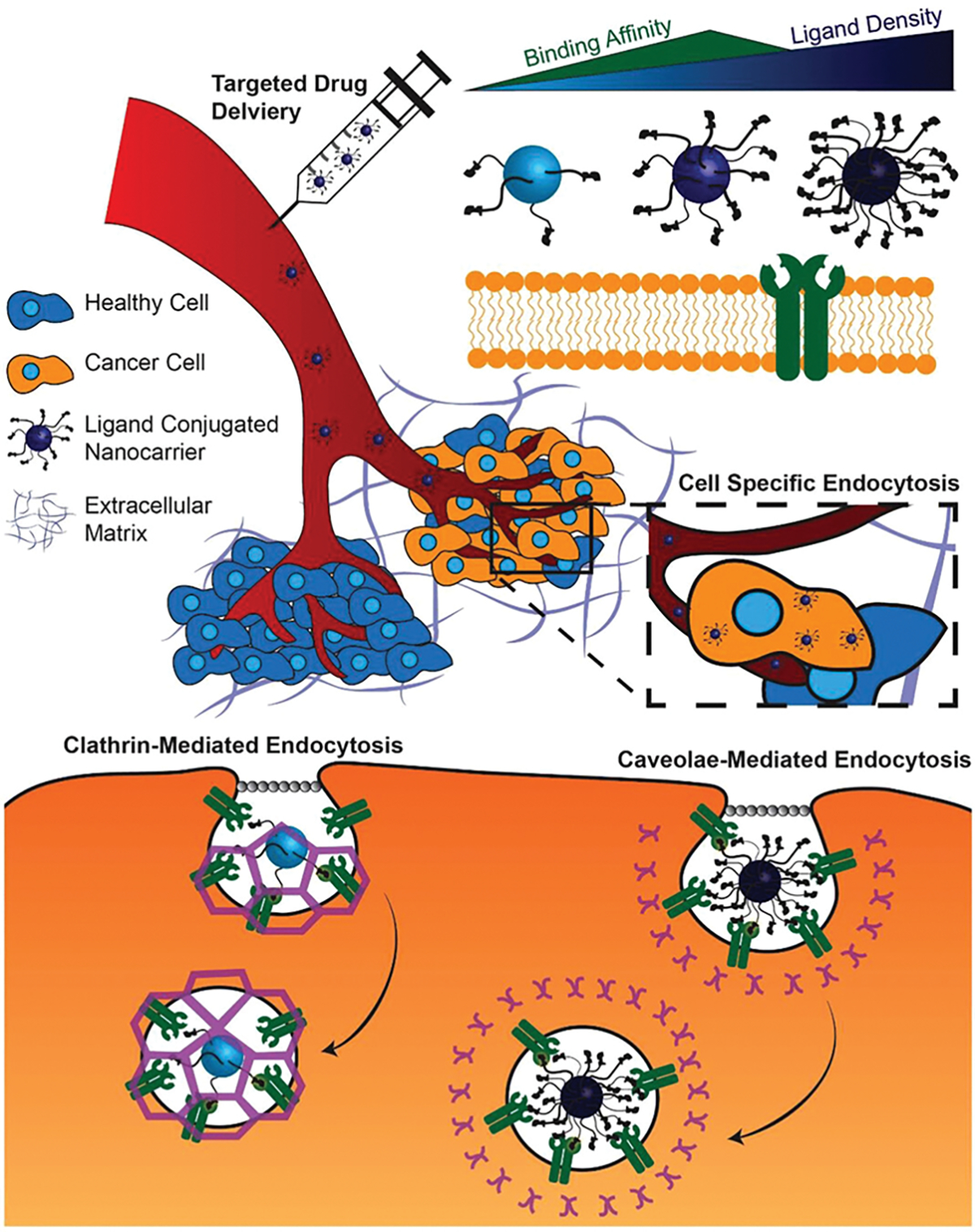
Receptor-mediated DDV can be optimized to improve receptor binding and cellular endocytosis by controlling the ligand density. DDVs with low (light blue) or high (dark blue) ligand density have lower receptor binding affinity compared to those with moderate (blue) ligand density. Further, low ligand density DDVs are typically internalized via clathrin-mediated endocytosis, while those with high ligand conjugation are endocytosed via caveolae-mediated endocytosis.
Other studies have demonstrated that ligand clustering can play an important role in influencing the binding affinity for a target receptor.[185] Poon et al. utilized self-assembling linear dendritic polymers (LDPs) to control the conjugation of folate in “patchy clusters” at varying distances apart, ultimately improving the nanoparticle’s binding affinity.[185] In vitro, formulations of micelles with varying percentages of functionalized LDP (1–16 molecules of folate per dendrimer) were evaluated for binding efficiency and cellular uptake in folate receptor overexpressing KB cancer cells (HeLa). Patchy micelles formulated with 20% folate conjugated LDP and 60% nonfunctionalized LDP exhibited the highest internalization in vitro. The 20%F-60%mix formulation also had the highest binding affinity of the eight formulations tested, as noted by the relatively high dissociation constant and relatively low EC50 values. In vivo, mice bearing KB tumors treated with 20%F-60%mix micelles had significantly higher accumulation within tumors relative to other micelle formulations tested. Through precise control over ligand spacing and density, patchy, ligand clustered micelles represent a novel DDV for surface patterning to enhance targeting efficacy. Adapting this technology for the delivery of therapeutic cargo may have a positive impact on drug efficacy, as well as minimize off-target cytotoxicity.
Liu et al. demonstrated that the peptide surface density on a liposome can be optimized to alter gene regulation of neoplastic cells and to reduce cell migration in vitro and metastasis in vivo.[188] The peptide DV1 binds with high affinity to chemokine receptor CXCR4, which is overexpressed on TNBC tissue and cell lines. This was confirmed by observing the exchange between DV1 and fluorescent CXCR4 specific mAbs on CXCR4 overexpressing cells. The potential of this peptide to form multivalent interactions with CXCR4 receptors clustered in lipid rafts was assessed by coating AFM tips with DV1 or CXCR4 mAbs and evaluating the relative binding force of each tip between neoplastic TNBC cells and nonneoplastic breast cells with low CXCR4 expression. While no significant difference in adhesion force was observed for the CXCR4 mAb tip, the DV1 tip bound 120% stronger to TNBC cell lines MDA-MB-231 and MDA-MB-436 compared to the nonneoplastic cell line MCF10A, indicating the ability of DV1 to form specific, multivalent interactions compared to its mAb counterpart. Liposomes were functionalized at different surface densities of DV1 ranging from 9000 peptides μm−2 to 74 000 peptides μm−2 using copper-free click chemistry. It was found that the liposome formulation with an intermediate surface density of 24 000 peptides μm−2 (24k) resulted in the greatest cell uptake in TNBC cells and were able to inhibit TNBC cell migration by 84% compared to 83% by CXCR4 mAb, while the 74 000 peptides μm−2 liposome only inhibited metastasis by 48%. The 24k formulation also demonstrated efficacy in vivo, where it prevented metastasis from a tail vein injection of MDA-MB-231-Luc cells for 31 days without any toxicity while LY2510924, a CXCR4 antagonists in clinical trials, did not show inhibition of metastasis. Weekly injections of the 24k formulation also prevented spontaneous metastasis from a primary tumor model for over 27 days. The mechanism by which this liposomal formulation acts is by downregulating p-115 RhoGEF and p85-PI3K, both proteins that are implicated in metastasis, in a DV1 density-dependent manner, where the 24k formulation results in the greatest downregulation of these proteins.[188]
6.2. Dual Target Ligand Patterning and Asymmetric Patterning
Another form of surface modification to enhance cellular internalization is by conjugating multiple targeted ligands to one vehicle, either homogeneously mixed or asymmetrically (Figure 6).[189] By exploiting multiple receptor-mediated binding interactions, dual functionalized drug carriers are more effective than their single ligand-conjugated counterparts. Dual-targeted nanoparticles functionalized with folate and RGD, resulted in a synergistic increase in cellular uptake and tumor accumulation in vivo.[32a] A dual ligand approach was also utilized in the treatment of prostate cancer. Guo et al. conjugated cRGDyK and antiprostate specific membrane antigen (anti-PSMA) mAb ligands to pH-responsive liposomes encapsulating paclitaxel.[190] These vehicles were internalized more frequently than control particles, and subsequently induced greater paclitaxel toxicity within 22Rv1 cells (prostate carcinoma cell line).
Figure 6.
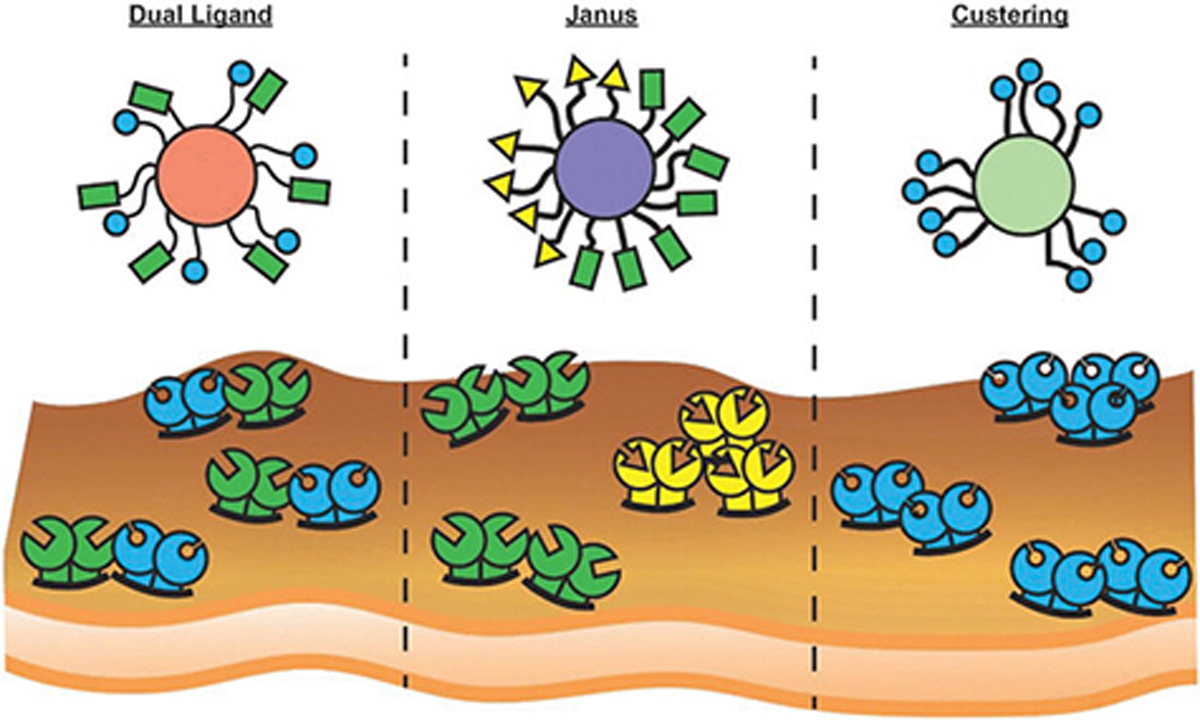
Dual, asymmetric, and clustered ligand conjugation systems can be tailored to exploit to increase receptor-mediated endocytosis for cells expressing different colocalized receptors (green and blue), distinct clusters of multiple receptors (green and yellow), or clustering of the same receptors (blue), respectively.
Dual targeted liposomes, such as those developed by Gao et al., Belhadj et al., Zhang et al., and Ke et al., were shown to improve in vivo survivability compared to free drug or single targeted liposomes (Figure 4, light green, orange, and green).[191] For example, Gao et al. functionalized liposomes with folate and Tf ligands for delivery of doxorubicin to gliomas.[191a] Folate and Tf were chosen for their ability to target tumors and penetrate the blood brain barrier (BBB), respectively. These dual targeting liposomes enabled significantly higher accumulation of doxorubicin compared to nonfunctionalized liposomes (Figure 3F). C6 glioma-bearing rats were evaluated for liposomal accumulation in tumors and off target organs, body-weight, and long-term survival in vivo. Dual targeting liposome treated rats weighed more and lived up to 6 days longer than rats treated with free doxorubicin. Tissue specific uptake was also noted, as functionalized dual targeting liposomes were preferentially internalized by the brain compared to the heart at 2 and 24 h after IV injection. Dual targeting liposomes exhibited limited toxicity in the heart, whereas free doxorubicin initiated inflammatory responses in heart tissue.
Kelly et al. synthesized a series of synthetic urea-based ligands with affinity for HSA and PSMA for targeted radiotherapy (TRT) of prostate cancer.[192] Side effects can become quite severe for patients receiving multiple rounds of α-emitters or β-emitters, thus the need for improved targeting technologies for these therapeutics is of clinical importance. As previously mentioned, PSMA is overexpressed in certain types of prostate cancer and a commonly utilized target for drug delivery. Human serum albumin (HSA) is highly abundant within the blood and has a long physiological half-life. Thus a ligand which possesses moderate affinity to HSA and high affinity to PSMA, may prolong vehicle circulation and enhance tumor targeting, respectively. A series of eight radiopharmaceutical candidates labeled with radioactive Iodine (131I) were screened for in vivo efficacy. In mice bearing human prostate cancer LNCaP xenografts, the dual targeting, RPS-027 formulation demonstrated the most promising biodistribution. Internalization of RPS-027 by tumor tissue compared to kidneys was fourfold to 20-fold higher relative to other RPS targeting formulations tested. The synthetic dual-targeting peptide ligands developed by Kelly et al. hold promise for advancing radiotherapy tissue specificity, as well as limiting off target side effects.
A hydrogel-based delivery system functionalized with estrone and RGD peptide ligands (Et-peptide-Taxol hydrogel) was devised for paclitaxel delivery to breast carcinoma via EGFR and αvβ3 integrin receptor-mediated endocytosis.[193] At concentrations of 50 and 100 nm, Et-peptide-Taxol hydrogel DDVs developed by Shu et al. induced greater cell toxicity in ER-positive MCF-7 human breast cancer cells than Taxol alone. In vivo, the Et-peptide-Taxol hydrogel was found to selectively accumulate in tumor tissue and bladder in MCF7 tumor bearing mice. Full renal clearance was achieved by 48 h, at which time Et-peptide-Taxol hydrogel particles were still highly retained within tumor tissue.[193]
Bae et al. utilized dual-ligand targeting in order to formulate chemotherapeutic encapsulating DDVs for targeting multiple tumor types using a single vehicle. HSA nanoparticles were functionalized with Tf and tumor necrosis factor related apoptosis-inducing ligand (TRAIL) for targeted delivery of doxorubicin to HCT 116, MCF-7, and CAPAN-1, human colon, breast, and pancreatic cancer cells, respectively. TRAIL/Tf/Dox HAS-NPs demonstrated enhanced accumulation within HCT116 tumor xenografts in mice over 32 h post systemic injection (Figure 3B). In vitro, TRAIL/Dox HSA-NPs induced up to a 92% reduction in cell viability demonstrating their ability to synergistically induce cell cytotoxicity in CAPAN-1 cells.[194]
Zong et al. developed liposomes dual-functionalized with T7 and TAT peptide ligands to target Tfr as well as enhance penetration of gliomas. TAT, the trans-activating peptide of the human immunodeficiency virus type 1, also known as a cell penetrating peptide (CPP), is crucial to viral replication due to its receptor-independent mechanism of internalization.[195] TAT is not limited by kinetics of receptor saturation and is able to transport cargo with high efficiency.[195] Functionalized DSPE-PEG liposomes encapsulating doxorubicin were tested in vitro for impact on cellular uptake and viability in C6 (rat brain glioma cells) and bEnd.3 (murine brain cells) cocultures. In vitro studies, demonstrated significantly higher uptake of T7-TAT labeled liposomes over liposomes labeled with only TAT or T7 alone in cells cultured independently and in the BBB model coculture. C6 tumor spheroids were utilized to provide a physiologically relevant in vitro model, in which TAT-T7-Lip were internalized significantly more and penetrated to greater tumor depths (up to 200 μm) compared to non-targeted liposomes. In vivo studies of doxorubicin plasma concentration over time as well as doxorubicin release within the brain, glioma, and heart were also conducted. TAT-T7-Lip was taken up significantly more in the brain and glioma, but significantly less in heart tissue, compared with free doxorubicin, Dox-TAT-Lip, or Dox-T7-Lip (Figure 3G).[195]
Optimizing ligand density and patterning of DDVs has shown great promise in in vivo studies. However, engineering multi-targeted DDVs is greatly hindered by the time and cost of synthesizing and testing of various formulations, as there is often no other method than trial-and-error. Further efforts to develop and validate high throughput screening models or simulations for predicting ligand–receptor binding interactions could provide a more cost-effective avenue for screening potential ligand densities and/or surface patterns.
6.3. Modeling-Based Approaches for Vehicle Targeting Optimization
More recently, computer-based modeling has emerged as a powerful tool for simulating processes in vitro and in vivo to cost-effectively screen and optimize systems. A dissipative particle dynamic simulation (DPDS) model of dual-ligand targeting was investigated by Xia et al. This predicted the binding affinity of random surface distributions of ligands.[196] Within the model, parameters of particle engulfment degree and length mismatch between ligands were studied in randomly patterned and Janus nanoparticles. The surface of a Janus nanoparticle possesses two or more distinct physical properties, allowing for different surface chemistries to occur on the same particle. This technique allows for precise spatial control over ligand conjugation to achieve asymmetrical functionalization. Typically, and as was the case with particles modeled by Xia et al., Janus particles are partitioned into two respective hemispheres for asymmetrical ligand conjugation. Simulation derived data revealed that dual ligand surface patterning increased engulfment degree of randomly patterned particles regardless of ligand length. The controlled arrangement of dual ligands on Janus particles exhibited a clear benefit over randomly patterned particles, as they were capable of achieving complete engulfment within 40% less time (reduction of 34.8 μs). Length mismatches between ligands were demonstrated to make the engulfment of nanoparticles energetically less favorable due to hydrophobic or electrostatic interactions. This phenomenon was observed as binding of particles with varying ligand lengths had comparatively low generation of free energy compared to particles with uniform ligand length.
Studies by Liu et al. developed a computational method based on Metropolis Monte Carlo and the weighed histogram analysis method (WHAM) to evaluate binding free energy landscapes of mAb functionalized spherical DDVs to the surface of endothelial cells.[197] Their investigation into the DDV targeting of lung endothelial cells in vivo and DDV-endothelial cell interaction using AFM rendered a highly accurate and experimentally validated computational model. Their model accurately predicted the critical threshold of DDV mAb density for competent binding to ICAM-1 glycoproteins on lung endothelial cells, providing a tool for predicting optimal DDV ligand density. Adhering to optimal density of ligands on functionalized DDVs ensures effective binding to the surface target, as well as avoidance of ligand-initiated immune response.[197] Adapting this computational model for the prediction of other tissue specific DDV-receptor interactions could be an excellent tool for the rational design and evaluation of potential in vivo effectiveness of new targeted DDVs.
Further Monte Carlo algorithm–based simulations were utilized in systematic studies to predict ideal design parameters of spherical vehicles.[198] These modeling based approaches to determine optimal ligand density and spacing in pattern formation have served as excellent predictive tools for success in vitro and in vivo.[199] Adaptation of Monte Carlo and DPDS-based models for targeted drug delivery highlights the impact of surface patterning on receptor-mediated cell binding, uptake efficiency, and off target uptake. These models provide a customizable and cost-effective technology for high throughput screening of nanoparticle optimization.
6.4. Ligand Modifications for Enhanced Uptake
DDV surfaces are engineered for long-circulation, penetration, and targeting capabilities. The addition of polyethylene glycol (PEG) to coat nanoparticles has improved stealth capabilities of DDVs. A successful example being the clinically translated Doxil (PEGylated formulation of doxorubicin). Nearly two decades ago, PEG coated nanoparticles were identified as a new technology for evading phagocytic uptake, increasing circulation half-life and decreasing protein adsorption.[200] Particles coated in PEG were coined “stealth” particles for their ability to circulate within the body undetected by the immune system for longer periods than non-PEG coated NPs. PEG can also play an important role in serving as an anchoring point for various ligand conjugation chemistries. However, humans can produce antibodies against PEGylated molecules or DDVs, which limits their use.
Elias et al. investigated the impact of the ligand density on a particle’s surface, receptor density on tissue of interest, and nanoparticle size (hydrodynamic diameter) on cell targeting efficacy.[186] In vitro studies focused on determining an optimum ligand density for the targeting of superparamagnetic iron oxide (SPIO) particles used to enhance contrast resolution for magnetic resonance (MR) imaging. High levels of SPIO are required to reach detectable MR contrast, though few studies had examined the effect of varying ligand density on the targeting efficiency of these particles. SPIO particles were functionalized with varying densities of HER2 affibody or folate ligands (6.4–35.8 affibodies per molecule SPIO) and evaluated for cellular internalization and subsequent T2 relaxation time. The effect of tissue receptor density appeared to be minimal as optimal ligand densities of 11.5 and 23 (ligands per SPIO) were observed in both cell lines, despite differences in receptor expression levels. Particles functionalized with 23 molecules affibody per SPIO particle demonstrated the highest uptake efficiency in HER2 overexpressing T6–17 cells. Folic acid conjugated particles (FA)-SPIO and HER2/neu homology derived small HER2-receptor binding peptide (AHNP) conjugated particles exhibited optimal T2 relaxation time and significant improvement in MR contrast at distinctly different levels of ligand conjugation: 133 and 20.6 molecules ligand per SPIO particle, respectively. Nanoparticle hydrodynamic diameter was also evaluated by comparing the uptake of 26 and 50 nm SPIOs with varying densities of HER2 conjugated. No significant difference in internalization was found between the differently sized particles when corresponding ligand densities were tested. This study demonstrated the importance of the ligand surface density on DDVs, regardless of vehicle diameter.
6.5. Ligand Conjugation Techniques for Enhanced Delivery
The use of platforms for ligand attachment has enhanced precision and diversity of ligand conjugation techniques. Click chemistry, a type of bioconjugation, has proven to be a useful tool in ligand attachment to nanoparticles.[201] The term “click chemistry” was coined by Dr. Sharpless in 1998, referring to the development of “powerful, selective, and modular ‘blocks’ that work reliably in both small- and large-scale applications.” Click chemistry reactions “must be modular, wide in scope, give very high yields, generate only inoffensive byproducts.”[202] This class of reactions permit the conjugation of specific biocompatible ligands, used for molecular probing or surface modification, rapidly and with high selectivity.[202] This technology is utilized for a diverse array of applications, including DNA sequencing, chemical synthesis, in situ probing and biosensing, and profiling enzymatic activity.[203] Bioconjugation via click chemistry is a useful tool for stereo specifically adjoining ligands to nanocarriers, also known as vehicle surface functionalization. Copper (I) catalyzed azide–alkyne (CuCAA) reactions are utilized for surface functionalization of nanoparticles.[204]
Bio-orthogonal copper-free click chemistry has demonstrated efficacy as an in vivo probe and as a platform for chemotherapeutic functionalization.[205] Photo-reactive click chemistry methods, such as radical mediated thiolene click chemistry, are useful in conjugating targeting ligands to nanocarriers.[206] A variety of easy to use click chemistry conjugation kits are now commercially available, such as the “click iT” series by ThermoFisher for the detection of cell apoptosis, proliferation, or RNA production via the conjugation of fluorescent probes. These kits are popular methods for quick, reliable, and reproducible bioconjugation.
Ligand conjugation through the use of sugars is another way in which DDVs are functionalized. Sugar-conjugation of triazole ligands to platinum and palladium compounds was performed by Yano et al. and evaluated for antitumor efficacy.[207] Further, the conjugation of ligands to the surface of DDVs can also be achieved through using simple chemical reactions. Carbodiimide (EDC) and N-hydroxysuccinimide (NHS) cross-link chemistry are utilized to tether peptide ligands to vehicles through the synthesis of a stable amide bond but often do not permit control of molecule orientation on the DDV surface.[208]
Another contemporary method of bioconjugation utilizes noncovalent attachment of histidine-modified peptides to lipid-based particles mediated by metal chelation-ligand reactions.[209] Interactions between nitrilotriacetic acid (NTA)-nickel and poly(histidine) are used to conjugate peptides to liposomes and for chromatographic protein purification; these interactions endure in serum and are safe in vivo.[209,210]
Residue-specific protein modifications have recently emerged as another highly specific bio-orthogonal method of ligand-conjugation for live cell imaging and therapeutic functionalization.[211] For example, mAb conjugation to cytotoxic therapeutics can be achieved by reacting the thiol side chain of genetically engineered antibodies with modified cysteine residues with maleimides.[211] This type of reaction quickly and efficiently yields stereospecific bioconjugated products.
Further conjugation methods utilizing thiol interactions include the site-specific addition and subsequent coupling of sulphuryl-modified molecules.[212] The disulfide linker is redox-responsive. When exposed to the reductive intracellular environment, the disulfide bond is cleaved by interaction with glutathione (thiol-disulfide exchange), releasing therapeutic pay load.[212] Glutathione-responsive DDVs have demonstrated anticancer efficacy in in vitro and in vivo applications.[213]
7. Conclusions
Herein, we have discussed the rationale design of parameters for receptor-mediated, targeted DDVs, as well as commonly exploited receptor targets for the treatment of a diverse array of cancers. Receptors implicated in pathways dealing with metastasis and MDR were also evaluated for their therapeutic potential to halt or evade these processes. We have highlighted the ability of targeted therapeutics to overcome challenges associated with classic chemotherapeutic administration, such as nonspecific cytotoxicity, rapid clearance by the renal system, and resistance. As further research uncovers new biomarkers to more accurately classify cancer by phenotype and progression, receptor-mediated drug delivery may be engineered to generate next-generation therapeutics.
As the treatment of cancer remains a prominent and challenging clinical problem, the innovative design of new therapeutics is of paramount importance. Exploiting physiological biomarkers and receptor colocalization to enhance therapeutic efficacy is a novel strategy to combat cancer more effectively. However, to investigate the necessary number of permutations to fully characterize and identify the optimal cancer targeting DDV with multiple binding ligands, variable ligand clustering, and different conjugation densities, high-throughput screening (HTS) approaches must be developed. The pharmaceutical industry employs a screening approach using microarrays,[214] and complex microfluidic devices,[215] to identify lead drug candidates, whilst minimizing the high cost of in vivo experiments. Therefore, to lower cost and time required to characterize novel multifaceted DDVs, research may be implemented to perform HTS during the development phase to find the most efficient DDVs. There has been notable work to address this need, such as HTS of barcoded nanoparticles in vivo.[216] However, there still exists a large gap in methodology and technology necessary to accurately adapt HTS approaches to the design of DDVs. Targeted drug delivery has shown promise in the treatment of diverse cancer types, demonstrating its versatility, customizability, and competency as a therapeutic platform and necessitates continued innovation and study.
Acknowledgements
D.T.A. acknowledges the support from the National Cancer Institute (NCI, 7DP2CA174495).
Biography

Debra Auguste is a professor of chemical engineering at Northeastern University. Prior to that, she was employed at the City College of New York and Harvard University. Her interests include targeted drug delivery, drug and gene delivery, and stimuli-sensitive materials. She received her S.B. from Massachusetts Institute of Technology in 1999 and her Ph.D. from Princeton University in 2005, both in chemical engineering. She was trained as a post-doctoral associate at the Massachusetts Institute of Technology.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Lee RJ, Low PS, J. Biol. Chem. 1994, 269, 3198. [PubMed] [Google Scholar]
- [2].(a) Wang M, Thanou M, Pharmacol Res. 2010, 62, 90; [DOI] [PubMed] [Google Scholar]; b) Yang J, Xie SX, Huang Y, Ling M, Liu J, Ran Y, Wang Y, Thrasher JB, Berkland C, Li B, Nanomedicine 2012, 7, 1297; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Huwyler J, Wu D, Pardridge WM, Proc. Natl. Acad. Sci. 1996, 93, 14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Arap W, Pasqualini R, Ruoslahti E, Science 1998, 279, 377. [DOI] [PubMed] [Google Scholar]
- [4].Gao H, Yang Z, Zhang S, Cao S, Shen S, Pang Z, Jiang X, Sci. Rep. 2013, 3, 2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].(a) Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW, Richie JP, Langer R, Proc. Natl. Acad. Sci. 2006, 103, 6315; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yang ZZ, Li JQ, Wang ZZ, Dong DW, Qi XR, Biomaterials 2014, 35, 5226; [DOI] [PubMed] [Google Scholar]; c) Benito JM, Gómez-García M, Ortiz Mellet C, Baussanne I, Defaye J, García Fernández JM, J. Am. Chem. Soc. 2004, 126, 10355; [DOI] [PubMed] [Google Scholar]; d) Nakagawa O, Ming X, Huang L, Juliano RL, J. Am. Chem. Soc. 2010, 132, 8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].(a) Said Hassane F, Frisch B, Schuber F, Bioconjug. Chem. 2006, 17, 849; [DOI] [PubMed] [Google Scholar]; (b) Friedman AD, Claypool SE, Liu R, Curr. Pharm. Des. 2013, 19, 6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].(a) Anselmo AC, Mitragotri S, Bioeng. Transl. Med. 2016, 1, 10; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Allen TM, Cullis PR, Adv. Drug Deliv. Rev. 2013, 65, 36; [DOI] [PubMed] [Google Scholar]; c) Merino M, Zalba S, Garrido MJ, J. Controlled Release 2018, 275, 162. [DOI] [PubMed] [Google Scholar]
- [8].(a) Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC, Clin. Pharmacol. Ther. 2008, 83, 761; [DOI] [PubMed] [Google Scholar]; b) Ozturk-Atar K, Eroglu H, Calis S, Drug Target J 2017, 10.1080/1061186X.2017.14010761. [DOI] [PubMed] [Google Scholar]
- [9].Bulbake U, Doppalapudi S, Kommineni N, Khan W, Pharmaceutics 2017, 9, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A, Nature 2014, 515, 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].(a) Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, Blattler WA, Lambert JM, Chari RV, Lutz RJ, Wong WL, Jacobson FS, Koeppen H, Schwall RH, Kenkare-Mitra SR, Spencer SD, Sliwkowski MX, Cancer Res.. 2008, 68, 9280; [DOI] [PubMed] [Google Scholar]; b) Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, Lu Y, Meng YG, Ng C, Yang J, Lee CC, Duenas E, Gorrell J, Katta V, Kim A, McDorman K, Flagella K, Venook R, Ross S, Spencer SD, Lee Wong W, Lowman HB, Vandlen R, Sliwkowski MX, Scheller RH, Polakis P, Mallet W, Nat. Biotechnol. 2008, 26, 925; [DOI] [PubMed] [Google Scholar]; c) Duncan R, Anticancer Drugs 1992, 3, 175. [DOI] [PubMed] [Google Scholar]
- [12].Goren D, Horowitz AT, Tzemach D, Tarshish M, Zalipsky S, Gabizon A, Clin. Cancer Res. 2000, 6, 1949. [PubMed] [Google Scholar]
- [13].Guo P, You JO, Yang J, Jia D, Moses MA, Auguste DT, Mol. Pharm. 2014, 11, 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) C. R. Dass, T. L. Walker, M. A. Burton, E. E. Decruz, J. Pharm. Pharmacol. 1997, 49, 972; [DOI] [PubMed] [Google Scholar]; b) Yuan F, Leunig M, Huang SK, Berk DA, Papahadjopoulos D, Jain RK, Cancer Res. 1994, 54, 3352; [PubMed] [Google Scholar]; c) Poste G, Papahadjopoulos D, Proc. Natl. Acad. Sci. 1976, 73, 1603; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hope M, Bally M, Webb G, Cullis P, Biochimica et Biophysica Acta (BBA)-Biomembranes 1985, 812, 55. [DOI] [PubMed] [Google Scholar]
- [15].Guo P, Liu D, Subramanyam K, Wang B, Yang J, Huang J, Auguste DT, Moses MA, Nat. Commun. 2018, 9, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Deshmukh AS, Chauhan PN, Noolvi MN, Chaturvedi K, Ganguly K, Shukla SS, Nadagouda MN, Aminabhavi TM, Int. J. Pharm. 2017, 532, 249. [DOI] [PubMed] [Google Scholar]
- [17].Sutton D, Nasongkla N, Blanco E, Gao J, Pharm. Res. 2007, 24, 1029. [DOI] [PubMed] [Google Scholar]
- [18].Zhang P, Hu L, Yin Q, Zhang Z, Feng L, Li Y, Control J. Release 2012, 159, 429. [DOI] [PubMed] [Google Scholar]
- [19].Kwon GS, Kataoka K, Adv. Drug. Deliv. Rev. 1995, 16, 295. [Google Scholar]
- [20].(a) Oerlemans C, Bult W, Bos M, Storm G, Nijsen JF, Hennink WE, Pharm. Res. 2010, 27, 2569; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Varela-Moreira A, Shi Y, Fens MHAM, Lammers T, Hennink WE, Schiffelers RM, Materials Chemistry Frontiers 2017, 1, 1485. [Google Scholar]
- [21].Shah P, Bhalodia D, Shelat P, Systematic Reviews in Pharmacy 2010, 1, 24. [Google Scholar]
- [22].Li Y, Zheng J, Xiao H, McClements DJ, Food Hydrocoll 2012, 27, 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kaczmarek JC, Kowalski PS, Anderson DG, Genome Med. 2017, 9, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cervantes A, Alsina M, Tabernero J, Infante JR, LoRusso P, Shapiro G, Paz-Ares LG, Falzone R, Hill J, Cehelsky J, White A, Toudjarska I, Bumcrot D, Meyers R, Hinkle G, Svrzikapa N, Sah DW, Vaishnaw A, Gollob J, Burris HA, J. Clin. Oncol. 2011, 29, 3025. [Google Scholar]
- [25].(a) Mitchell MJ, King MR, Expert Opin Drug Deliv. 2015, 12, 375; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Parodi A, Quattrocchi N, van de Ven AL, Chiappini C, Evangelopoulos M, Martinez JO, Brown BS, Khaled SZ, Yazdi IK, Enzo MV, Isenhart L, Ferrari M, Tasciotti E, Nat. Nanotechnol. 2013, 8, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].(a) Huang Y, Gao X, Chen J, Acta Pharmaceutica Sinica B 2018, 8, 4; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dong X, Chu D, Wang Z, Theranostics 2017, 7, 751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Muzykantov VR, Expert Opin Drug Deliv. 2010, 7, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li R, He Y, Zhang S, Qin J, Wang J, Acta Pharmaceutica Sinica B 2018, 8, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Xu ZP, Zeng QH, Lu GQ, Yu AB, Chem. Eng. Sci. 2006, 61, 1027. [Google Scholar]
- [30].Jain PK, Lee K, El-Sayed IH, El-Sayed MA, J. Phys. Chem. B 2006, 110, 7238. [DOI] [PubMed] [Google Scholar]
- [31].(a) Choi CJ, Alabi CA, Webster P, Davis ME, Proc. Natl. Acad. Sci. 2010, 107, 1235; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Brown SD, Nativo P, Smith J-A, Stirling D, Edwards PR, Venugopal B, Flint DJ, Plumb JA, Graham D, Wheate NJ, J. Am. Chem. Soc. 2010, 132, 4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].(a) Jang C, Lee JH, Sahu A, Tae G, Nanoscale 2015, 7, 18584; [DOI] [PubMed] [Google Scholar]; b) You JO, Guo P, Auguste DT, Angew. Chem. Int. Ed. 2013, 52, 4141; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Huang X, Jain PK, El-Sayed IH, El-Sayed MA, Photochem. Photobiol. 2006, 82, 412. [DOI] [PubMed] [Google Scholar]
- [33].(a) Wang F, Wang Y-C, Dou S, Xiong M-H, Sun T-M, Wang J, ACS Nano 2011, 5, 3679; [DOI] [PubMed] [Google Scholar]; b) Popovtzer R, Agrawal A, Kotov NA, Popovtzer A, Balter J, Carey TE, Kopelman R, Nano Lett. 2008, 8, 4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].(a) Patra CR, Bhattacharya R, Mukhopadhyay D, Mukherjee P, Adv. Drug Deliv. Rev. 2010, 62, 346; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chithrani BD, Ghazani AA, Chan WC, Nano Lett. 2006, 6, 662; [DOI] [PubMed] [Google Scholar]; c) Liong M, Lu J, Kovochich M, Xia T, Ruehm SG, Nel AE, Tamanoi F, Zink JI, ACS Nano 2008, 2, 889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Farooq MU, Novosad V, Rozhkova EA, Wali H, Ali A, Fateh AA, Neogi PB, Neogi A, Wang Z, Sci. Rep. 2018, 8, 2907. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [36].El-Sayed IH, Huang X, El-Sayed MA, Nano Lett. 2005, 5, 829. [DOI] [PubMed] [Google Scholar]
- [37].(a) Copland JA, Eghtedari M, Popov VL, Kotov N, Mamedova N, Motamedi M, Oraevsky AA, Mol. Imaging Biol. 2004, 6, 341; [DOI] [PubMed] [Google Scholar]; b) Jain PK, Lee KS, El-Sayed IH, El-Sayed MA, J. Phys. Chem. B 2006, 110, 7238. [DOI] [PubMed] [Google Scholar]
- [38].Hoshyar N, Gray S, Han H, Bao G, Nanomedicine 2016, 11, 673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jain S, Hirst DG, O’Sullivan JM, Br. J. Radiol. 2012, 85, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Patil US, Adireddy S, Jaiswal A, Mandava S, Lee BR, Chrisey DB, Int. J. Mol. Sci. 2015, 16, 24417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jain TK, Morales MA, Sahoo SK, Leslie-Pelecky DL, Labhasetwar V, Mol. Pharm. 2005, 2, 194. [DOI] [PubMed] [Google Scholar]
- [42].(a) Santra S, Zhang P, Wang K, Tapec R, Tan W, Anal. Chem. 2001, 73, 4988; [DOI] [PubMed] [Google Scholar]; b) Peng F, Su Y, Wei X, Lu Y, Zhou Y, Zhong Y, Lee ST, He Y, Angew. Chem. Int. Ed. 2013, 52, 1457; [DOI] [PubMed] [Google Scholar]; c) Song Y, Du D, Li L, Xu J, Dutta P, Lin Y, ACS Appl. Mater. Interfaces 2017, 9, 20410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ferris DP, Lu J, Gothard C, Yanes R, Thomas CR, Olsen JC, Stoddart JF, Tamanoi F, Zink JI, Small 2011, 7, 1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wichterle O, Ĺım D, Nature 1960, 185, 117. [Google Scholar]
- [45].(a) Peppas NA, Khare AR, Adv. Drug. Deliv. Rev. 1993, 11, 1; [Google Scholar]; b) Hamidi M, Azadi A, Rafiei P, Adv. Drug Deliv. Rev. 2008, 60, 1638. [DOI] [PubMed] [Google Scholar]
- [46].(a) Jiang Y, Chen J, Deng C, Suuronen EJ, Zhong Z, Biomaterials 2014, 35, 4969; [DOI] [PubMed] [Google Scholar]; b) Hu X, Tan H, Wang X, Chen P, Colloids Surf. A 2016, 489, 297. [Google Scholar]
- [47].Xu Y, Kim CS, Saylor DM, Koo D, J. Biomed. Mater. Res. B Appl. Biomater. 2017, 105, 1692. [DOI] [PubMed] [Google Scholar]
- [48].Lu L, Peter SJ, Lyman MD, Lai H-L, Leite SM, Tamada JA, Uyama S, Vacanti JP, Robert L, Mikos AG, Biomaterials 2000, 21, 1837. [DOI] [PubMed] [Google Scholar]
- [49].Lee KY, Mooney DJ, Prog. Polym. Sci. 2012, 37, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].(a) Zhang C, Wang W, Liu T, Wu Y, Guo H, Wang P, Tian Q, Wang Y, Yuan Z, Biomaterials 2012, 33, 2187; [DOI] [PubMed] [Google Scholar]; b) Tian Q, Zhang CN, Wang XH, Wang W, Huang W, Cha RT, Wang CH, Yuan Z, Liu M, Wan HY, Tang H, Biomaterials 2010, 31, 4748. [DOI] [PubMed] [Google Scholar]
- [51].Acartürk F, Takka S, J. Microencapsul. 1999, 16, 291. [DOI] [PubMed] [Google Scholar]
- [52].Whitehead L, Collett JH, Fell JT, Int. J. Pharm. 2000, 210, 45. [DOI] [PubMed] [Google Scholar]
- [53].Wong T, J. Pharm. Pharmacol. 2011, 63, 1497. [DOI] [PubMed] [Google Scholar]
- [54].Bhattarai N, Gunn J, Zhang M, Adv. Drug Deliv. Rev. 2010, 62, 83. [DOI] [PubMed] [Google Scholar]
- [55].Haidar ZS, Hamdy RC, Tabrizian M, Biomaterials 2008, 29, 1207. [DOI] [PubMed] [Google Scholar]
- [56].(a) Katuwavila NP, Perera ADLC, Samarakoon SR, Soysa P, Karunaratne V, Amaratunga GAJ, Karunaratne DN, Nanomat J. 2016, 2016, 1; [Google Scholar]; b) Wang F, Yang S, Yuan J, Gao Q, Huang C, J. Biomater. Appl. 2016, 31, 3. [DOI] [PubMed] [Google Scholar]
- [57].Yi H, Liu P, Sheng N, Gong P, Ma Y, Cai L, Nanoscale 2016, 8, 5985. [DOI] [PubMed] [Google Scholar]
- [58].Du J, O’Reilly RK, Soft Matter 2009, 5, 3544. [Google Scholar]
- [59].(a) Chen L, Jiang T, Cai C, Wang L, Lin J, Cao X, Adv. Healthc. Mater. 2014, 3, 1508; [DOI] [PubMed] [Google Scholar]; b) Bae Y, Nishiyama N, Fukushima S, Koyama H, Yasuhiro M, Kataoka K, Bioconj. Chem. 2005, 16, 122. [DOI] [PubMed] [Google Scholar]
- [60].Du J, Armes SP, J. Am. Chem. Soc. 2005, 127, 12800. [DOI] [PubMed] [Google Scholar]
- [61].Karimi M, Ghasemi A, Sahandi Zangabad P, Rahighi R, Moosavi Basri SM, Mirshekari H, Amiri M, Shafaei Pishabad Z, Aslani A, Bozorgomid M, Ghosh D, Beyzavi A, Vaseghi A, Aref AR, Haghani L, Bahrami S, Hamblin MR, Chem. Soc. Rev. 2016, 45, 1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hoffman AS, Adv. Drug. Deliv. Rev. 2013, 65, 10. [DOI] [PubMed] [Google Scholar]
- [63].Master AM, Sen Gupta A, Nanomedicine 2012, 7, 1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Herbst RS, Shin DM, Cancer 2002, 94, 1593. [DOI] [PubMed] [Google Scholar]
- [65].(a) Mamot C, Drummond DC, Noble CO, Kallab V, Guo Z, Hong K, Kirpotin DB, Park JW, Cancer Res.. 2005, 65, 11631; [DOI] [PubMed] [Google Scholar]; b) Mamot C, Drummond DC, Greiser U, Hong K, Kirpotin DB, Marks JD, Park JW, Cancer Res. 2003, 63, 3154. [PubMed] [Google Scholar]
- [66].Acharya S, Dilnawaz F, Sahoo SK, Biomaterials 2009, 30, 5737. [DOI] [PubMed] [Google Scholar]
- [67].Patra C, Bhattacharya R, Wang E, Katarya A, Lau JS, Dutta S, Muders M, Wang S, Buhrow SA, Safgren SL, Yaszemski MJ, Reid JM, Ames MM, Mukherjee P, Mukhopadhyay D, Cancer Res. 2008, 68, 1970. [DOI] [PubMed] [Google Scholar]
- [68].Kim I-Y, Kang Y-S, Lee D, Park H-J, Choi E-K, Oh Y-K, Son H-J, Kim J-S, J. Control. Release 2009, 140, 55. [DOI] [PubMed] [Google Scholar]
- [69].Zhao B, Wang L, Qiu H, Zhang M, Sun L, Peng P, Yu Q, Yuan X, Oncotarget 2017, 8, 3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Morgillo F, Corte C, Fasano M, Ciardiello F, ESMO Open 2016, 1, e000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Wykosky J, Fenton T, Furnari F, Cavenee WK, Chin. J. Cancer 2011, 30, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Isobe A, Sawada K, Kinose Y, Ohyagi-Hara C, Nakatsuka E, Makino H, Ogura T, Mizuno T, Suzuki N, Morii E, Nakamura K, Sawada I, Toda A, Hashimoto K, Mabuchi S, Ohta T, Morishige K-I, Kurachi H, Kimura T, PLoS One 2015, 10, e0118080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kruspe S, Hahn U, Angew. Chem. Int. Ed. 2014, 53, 10541. [DOI] [PubMed] [Google Scholar]
- [74].Hahn U, Int. J. Mol. Sci. 2017, 18, 2641.29211023 [Google Scholar]
- [75].Meyer C, Eydeler K, Magbanua E, Zivkovic T, Piganeau N, Lorenzen I, Grötzinger J, Mayer G, Rose-John S, Hahn U, RNA Biol. 2014, 9, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].(a) Yang F-Y, Wong T-T, Teng M-C, Liu R-S, Lu M, Liang H-F, Wei M-C, J. Control. Release 2012, 160, 652; [DOI] [PubMed] [Google Scholar]; b) Sun Z, Yan X, Liu Y, Huang L, Kong C, Qu X, Wang M, Gao R, Qin H, Oncotarget 2017, 8, 58823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Kawakami K, Kawakami M, Husain SR, Puri RK, Cancer Res. 2002, 62, 3575. [PubMed] [Google Scholar]
- [78].Kioi M, Takahashi S, Kawakami M, Kawakami K, Kreitman RJ, Puri RK, Cancer Res. 2005, 65, 8388. [DOI] [PubMed] [Google Scholar]
- [79].(a) Kawakami M, Kawakami K, Stepensky VA, Maki RA, Robin H, Muller W, Husain SR, Puri RK, Clin. Cancer Res. 2002, 8, 3503; [PubMed] [Google Scholar]; b) Chi L, Na M-H, Jung H-K, Vadevoo S, Kim C-W, Padmanaban G, Park T-I, Park J-Y, Hwang I, Park K, Liang F, Lu M, Park J, Kim I-S, Lee B-H, Control J. Release 2015, 209, 327. [DOI] [PubMed] [Google Scholar]
- [80].Puri RK, Leland P, Obiri NI, Husain SR, Mule J, Pastan I, Kreitman RJ, Cell. Immunol. 1996, 171, 80. [DOI] [PubMed] [Google Scholar]
- [81].(a) Wu X, Kim J, Koo H, Bae S, Shin H, Kim M, Lee B-H, Park R-W, Kim I-S, Choi K, Kwon I, Kim K, Lee D, Bioconjug. Chem. 2010, 21, 208; [DOI] [PubMed] [Google Scholar]; b) Leland P, Taguchi J, Husain SR, Kreitman RJ, Pastan I, Puri RK, Mol. Med. 2000, 6, 165. [PMC free article] [PubMed] [Google Scholar]
- [82].Yang C-Y, Liu H-W, Tsai Y-C, Tseng J-Y, Liang S-C, Chen C-Y, Lian W-N, Wei M-C, Lu M, Lu R-H, Lin C-H, Jiang J-K, Cancer Biol. Ther. 2015, 16, 1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Husain SR, Kreitman RJ, Pastan I, Puri RK, Nat. Med. 1999, 5, 817. [DOI] [PubMed] [Google Scholar]
- [84].(a) Testa U, Riccioni R, Militi S, Coccia E, Stellacci E, Samoggia P, Latagliata R, Mariani G, Rossini A, Battistini A, Lo-Coco F, Peschle C, Blood 2002, 100, 2980; [DOI] [PubMed] [Google Scholar]; b) Bellavia D, Raimondo S, Calabrese G, Forte S, Cristaldi M, Patinella A, Memeo L, Manno M, Raccosta S, Diana P, Cirrincione G, Giavaresi G, Monteleone F, Fontana S, Leo G, Alessandro R, Theranostics 2017, 7, 1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Dubé I, Dixon J, Beckett T, Grossman A, Weinstein M, Benn P, McKeithan T, Norman C, Pinkerton P, Genes Chromosomes Cancer 1989, 1, 106. [DOI] [PubMed] [Google Scholar]
- [86].Pinilla-Ibarz J, Sweet K, Emole J, Fradley M, Anticancer Res. 2015, 35, 6355. [PubMed] [Google Scholar]
- [87].Madhankumar AB, Slagle-Webb B, Mintz A, Sheehan JM, Connor JR, Mol. Cancer Ther. 2006, 5, 3162. [DOI] [PubMed] [Google Scholar]
- [88].Wang Y, Shi W, Song W, Wang L, Liu X, Chen J, Huang R, J. Mater. Chem. 2012, 22, 14608. [Google Scholar]
- [89].Pandya H, Gibo DM, Garg S, Kridel S, Debinski W, Neuro-oncol. 2012, 14, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Wang B, Lv L, Wang Z, Zhao Y, Wu L, Fang X, Xu Q, Xin H, Biomaterials 2014, 35, 5897. [DOI] [PubMed] [Google Scholar]
- [91].Gao H, Yang Z, Cao S, Xiong Y, Zhang S, Pang Z, Jiang X, Biomaterials 2014, 35, 2374. [DOI] [PubMed] [Google Scholar]
- [92].(a) Cardó-Vila M, Marchiò S, Sato M, Staquicini FI, Smith TL, Bronk JK, Yin G, Zurita AJ, Sun M, Behrens C, Sidman RL, Lee JJ, Hong WK, Wistuba II, Arap W, Pasqualini R, Am. J. Pathol. 2016, 186, 2162; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lewis VO, Devarajan E, Cardò-Vila M, Thomas DG, Kleinerman ES, Marchiò S, Sidman RL, Pasqualini R, Arap W, Proc. Natl. Acad. Sci. 2017, 114, 8065; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Pasqualini R, Millikan RE, Christianson DR, Cardó-Vila M, Driessen WHP, Giordano RJ, Hajitou A, Hoang AG, Wen S, Barnhart KF, Baze WB, Marcott VD, Hawke DH, Do KA, Navone NM, Efstathiou E, Troncoso P, Lobb RR, Logothetis CJ, Arap W, Cancer 2015, 121, 2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Rider P, Carmi Y, Cohen I, Int. J. Cell Biol. 2016, 2016, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Chen C, Ke J, Zhou XE, Yi W, Brunzelle JS, Li J, Yong EL, Xu HE, Melcher K, Nature 2013, 500, 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].(a) Lee RJ, Low PS, Biochim. Biophys. Acta, Biomembr. 1995, 1233, 134; [DOI] [PubMed] [Google Scholar]; b) Leamon CP, Reddy JA, Adv. Drug. Deliv. Rev. 2004, 56, 1127. [DOI] [PubMed] [Google Scholar]
- [96].Bae Y, Jang W-D, Nishiyama N, Fukushima S, Kataoka K, Mol. Biosyst. 2005, 1, 242. [DOI] [PubMed] [Google Scholar]
- [97].Park E, Kim S, Lee S, Lee Y, Control J. Release 2005, 109, 158. [DOI] [PubMed] [Google Scholar]
- [98].(a) Zhang Z, Lee S, Feng S-S, Biomaterials 2007, 28, 1889; [DOI] [PubMed] [Google Scholar]; b) Yoo H, Park T, Control J. Release 2004, 100, 247; [DOI] [PubMed] [Google Scholar]; c) Yoo HS, Park TG, Control Release J 2004, 100, 247. [DOI] [PubMed] [Google Scholar]
- [99].Kukowska-Latallo JF, Candido KA, Cao Z, Nigavekar SS, Majoros IJ, Thomas TP, Balogh LP, Khan MK, Baker JR, Cancer Res. 2005, 65, 5317. [DOI] [PubMed] [Google Scholar]
- [100].(a) Thapa RK, Choi JY, Gupta B, Ramasamy T, Poudel BK, Ku SK, Youn YS, Choi HG, Yong CS, Kim JO, Biomater. Sci. 2016, 4, 1340; [DOI] [PubMed] [Google Scholar]; b) Werner ME, Copp JA, Karve S, Cummings ND, Sukumar R, Li C, Napier ME, Chen RC, Cox AD, Wang AZ, ACS Nano 2011, 5, 8990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Lee KD, Choi SH, Kim DH, Lee HY, Choi KC, Arch. Pharm. Res. 2014, 37, 1546. [DOI] [PubMed] [Google Scholar]
- [102].Goren D, Horowitz AT, Tzemach D, Tarshish M, Zalipsky S, Gabizon A, Clin. Cancer Res. 2000, 6, 1949. [PubMed] [Google Scholar]
- [103].Damsky CH, Werb Z, Curr. Opin. Cell Biol. 1992, 4, 772. [DOI] [PubMed] [Google Scholar]
- [104].Desgrosellier JS, Cheresh DA, Nat. Rev. Cancer 2010, 10, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Ruoslahti E, Pierschbacher MD, Science 1987, 238, 491. [DOI] [PubMed] [Google Scholar]
- [106].Babu A, Amreddy N, Muralidharan R, Pathuri G, Gali H, Chen A, Zhao YD, Munshi A, Ramesh R, Sci. Rep. 2017, 7, 14674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Nasongkla N, Shuai X, Ai H, Weinberg BD, Pink J, Boothman DA, Gao J, Angew. Chem. Int. Ed. 2004, 43, 6323. [DOI] [PubMed] [Google Scholar]
- [108].Zhan C, Gu B, Xie C, Li J, Liu Y, Lu W, Controlled Release J 2010, 143, 136. [DOI] [PubMed] [Google Scholar]
- [109].Cox D, Brennan M, Moran N, Nat. Rev. Drug Discovery 2010, 9, 804. [DOI] [PubMed] [Google Scholar]
- [110].Zeeshan R, Mutahir Z, Bosn. J. Basic Med. Sci. 2017, 17, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Ganapathy V, Moghe PV, Roth CM, J. Control. Release 2015, 219, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Zhou B-BSB, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks PB, Nat. Rev. Drug Discovery 2009, 8, 806. [DOI] [PubMed] [Google Scholar]
- [113].Visvader JE, Lindeman GJ, Nat. Rev. Cancer 2008, 8, 755. [DOI] [PubMed] [Google Scholar]
- [114].(a) Hu K, Zhou H, Liu Y, Liu Z, Liu J, Tang J, Li J, Zhang J, Sheng W, Zhao Y, Wu Y, Chen C, Nanoscale 2015, 7, 8607; [DOI] [PubMed] [Google Scholar]; b) Vinogradov S, Wei X, Nanomedicine 2012, 7, 597; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Berguig GY, Convertine AJ, Frayo S, Kern HB, Procko E, Roy D, Srinivasan S, Margineantu DH, Booth G, Palanca-Wessels MC, Baker D, Hockenbery D, Press OW, Stayton PS, Mol. Ther. 2015, 23, 907; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Thomas TP, Patri AK, Myc A, Myaing M, Ye J, Norris TB, Baker JR, Biomacromolecules 2004, 5, 2269. [DOI] [PubMed] [Google Scholar]
- [115].Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, Berenguer A, Prats N, Toll A, Hueto JA, Bescos C, Di Croce L, Benitah SA, Nature 2017, 541, 41. [DOI] [PubMed] [Google Scholar]
- [116].(a) Sy MS, Guo YJ, Stamenkovic I, J. Exp. Med. 1992, 176, 623; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zawadzki V, Perschl A, Rösel M, Hekele A, Zöller M, Int. J. Cancer 1998, 75, 919. [DOI] [PubMed] [Google Scholar]
- [117].Eliaz RE, Szoka FC, Cancer Res. 2001, 61, 2592. [PubMed] [Google Scholar]
- [118].Wang L, Su W, Liu Z, Zhou M, Chen S, Chen Y, Lu D, Liu Y, Fan Y, Zheng Y, Han Z, Kong D, Wu JC, Xiang R, Li Z, Biomaterials 2012, 33, 5107. [DOI] [PubMed] [Google Scholar]
- [119].Tagaram HS, DiVittore NA, Barth BM, Kaiser JM, Avella D, Kimchi ET, Jiang Y, Isom HC, Kester M, Staveley-O’Carroll KF, Gut 2011, 60, 695. [DOI] [PubMed] [Google Scholar]
- [120].Wei X, Senanayake TH, Warren G, Vinogradov SV, Bioconjug. Chem. 2013, 24, 658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Zhong Y, Goltsche K, Cheng L, Xie F, Meng F, Deng C, Zhong Z, Haag R, Biomaterials 2016, 84, 250. [DOI] [PubMed] [Google Scholar]
- [122].Barbas AS, Mi J, Clary BM, White RR, Future Oncology 2010, 6, 1117. [DOI] [PubMed] [Google Scholar]
- [123].Yu Z, Ni M, Xiong M, Zhang X, Cai G, Chen H, Zeng Q, Int. J. Nanomed. 2015, 10, 2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Ierano C, Portella L, Lusa S, Salzano G, D’Alterio C, Napolitano M, Buoncervello M, Macchia D, Spada M, Barbieri A, Luciano A, Barone MV, Gabriele L, Caraglia M, Arra C, De Rosa G, Scala S, Nanoscale 2016, 8, 7562. [DOI] [PubMed] [Google Scholar]
- [125].(a) Chittasupho C, Anuchapreeda S, Sarisuta N, Eur. J. Pharm. Biopharm. 2017, 119, 310; [DOI] [PubMed] [Google Scholar]; b) Guo P, You JO, Yang J, Moses MA, Auguste DT, Biomaterials 2012, 33, 8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Wang B, Guo P, Auguste DT, Biomaterials 2015, 57, 161. [DOI] [PubMed] [Google Scholar]
- [127].Yang J, Moses MA, Cell Cycle 2009, 8, 2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Leng X, Ding T, Lin H, Wang Y, Hu L, Hu J, Feig B, Zhang W, Pusztai L, Symmans FW, Wu Y, Arlinghaus RB, Cancer Res. 2009, 69, 8579. [DOI] [PubMed] [Google Scholar]
- [129].Tamamura H, Tsutsumi H, Masuno H, Fujii N, Curr. Med. Chem. 2007, 14, 93. [DOI] [PubMed] [Google Scholar]
- [130].Xue LJ, Mao XB, Ren LL, Chu XY, Cancer Med. 2017, 6, 1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Lee HR, Kim TH, Choi KC, Lab Anim. Res. 2012, 28, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Strom A, Treuter E, Warner M, Gustafsson JA, Physiol. Rev. 2007, 87, 905. [DOI] [PubMed] [Google Scholar]
- [133].Osborne CK, Breast Cancer Res. Treat. 1998, 51, 227. [DOI] [PubMed] [Google Scholar]
- [134].(a) James DA, Swamy N, Paz N, Hanson RN, Ray R, Bioorg. Med. Chem. Lett. 1999, 9, 2379; [DOI] [PubMed] [Google Scholar]; b) Gacio A, Fernandez-Marcos C, Swamy N, Dunn D, Ray R, J. Cell. Biochem. 2006, 99, 665; [DOI] [PubMed] [Google Scholar]; c) Swamy N, Purohit A, Fernandez-Gacio A, Jones GB, Ray R, J. Cell. Biochem. 2006, 99, 966. [DOI] [PubMed] [Google Scholar]
- [135].McElvany KD, Carlson KE, Katzenellenbogen JA, Welch MJ, J. Steroid Biochem. 1983, 18, 635. [DOI] [PubMed] [Google Scholar]
- [136].Holmes FA, Walters RS, Theriault RL, Forman AD, Newton LK, Raber MN, Buzdar AU, Frye DK, Hortobagyi GN, Natl J. Cancer Inst. 1991, 83, 1797. [DOI] [PubMed] [Google Scholar]
- [137].Ishiki N, Onishi H, Machida Y, Int. J. Pharm. 2004, 279, 81. [DOI] [PubMed] [Google Scholar]
- [138].Lam HY, Ng PK, Goldenberg GJ, Wong CM, Cancer Treat Rep. 1987, 71, 901. [PubMed] [Google Scholar]
- [139].Kuduk SD, Zheng FF, Sepp-Lorenzino L, Rosen N, Danishefsky SJ, Bioorg. Med. Chem. Lett. 1999, 9, 1233. [DOI] [PubMed] [Google Scholar]
- [140].von Schoultz E, Lundgren E, Henriksson R, Anticancer Res.. 1990, 10, 693. [PubMed] [Google Scholar]
- [141].Devraj R, Barrett JF, Fernandez JA, Katzenellenbogen JA, Cushman M, J. Med. Chem. 1996, 39, 3367. [DOI] [PubMed] [Google Scholar]
- [142].Gupta A, Saha P, Descôteaux C, Leblanc V, Asselin É, Bérubé G, Bioorg. Med. Chem. Lett. 2010, 20, 1614. [DOI] [PubMed] [Google Scholar]
- [143].Oaksmith J, Ganem B, Tetrahedron Lett. 2009, 50, 3497. [Google Scholar]
- [144].Reddy BS, Banerjee R, Angew. Chem. Int. Ed. 2005, 44, 6723. [DOI] [PubMed] [Google Scholar]
- [145].Dreaden EC, Mwakwari SC, Sodji QH, Oyelere AK, El-Sayed MA, Bioconjug. Chem. 2009, 20, 2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].(a) Rai S, Paliwal R, Vaidya B, Khatri K, Goyal AK, Gupta PN, Vyas SP, J. Drug Target. 2008, 16, 455; [DOI] [PubMed] [Google Scholar]; b) Paliwal SR, Paliwal R, Mishra N, Mehta A, Vyas SP, Curr. Cancer Drug Targets 2010, 10, 343; [DOI] [PubMed] [Google Scholar]; c) Rai S, Paliwal R, Vyas SP, J. Biomed. Nanotechnol. 2011, 7, 121; [DOI] [PubMed] [Google Scholar]; d) Paliwal SR, Paliwal R, Pal HC, Saxena AK, Sharma PR, Gupta PN, Agrawal GP, Vyas SP, Mol. Pharm. 2012, 9, 176; [DOI] [PubMed] [Google Scholar]; e) Das M, Singh R, Datir SR, Jain S, Mol. Pharm. 2013, 10, 3404. [DOI] [PubMed] [Google Scholar]
- [147].Madan J, Gundala SR, Kasetti Y, Bharatam PV, Aneja R, Katyal A, Jain UK, Anticancer Drugs 2014, 25, 704. [DOI] [PubMed] [Google Scholar]
- [148].Yang H, Tang C, Yin C, Acta Biomater. 2018, 10.1016/j.actbio.2018.04.020. [DOI] [PubMed] [Google Scholar]
- [149].Rothlein R, Dustin ML, Marlin SD, Springer TA, Immunol J. 1986, 137, 1270. [PubMed] [Google Scholar]
- [150].(a) Myers CL, Wertheimer SJ, Schembri-King J, Parks T, Wallace RW, American Journal of Physiology-Cell Physiology 1992, 263, C767; [DOI] [PubMed] [Google Scholar]; (b) Millan J, Hewlett L, Glyn M, Toomre D, Clark P, Ridley AJ, Nat. Cell Biol. 2006, 8, 113. [DOI] [PubMed] [Google Scholar]
- [151].Sumagin R, Lomakina E, Sarelius IH, Am. J. Physio. Heart Circ. Physiol. 2008, 295, H969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].(a) Park S, Kang S, Chen X, Kim EJ, Kim J, Kim N, Kim J, Jin MM, Biomaterials 2013, 34, 598; [DOI] [PubMed] [Google Scholar]; (b) Guo P, Huang J, Wang L, Jia D, Yang J, Dillon DA, Zurakowski D, Mao H, Moses MA, Auguste DT, Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 14710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Guo P, Yang J, Jia D, Moses MA, Auguste DT, Theranostics 2016, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Fakhari A, Baoum A, Siahaan TJ, Le KB, Berkland C, J. Pharm. Sci. 2011, 100, 1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Guo P, Wang B, Liu D, Yang J, Subramanyam K, McCarthy CR, Hebert J, Moses MA, Auguste DT, Nano. Lett. 2018, 18, 2254. [DOI] [PubMed] [Google Scholar]
- [156].Longley DB, Johnston PG, Pathol J. 2005, 205, 275. [DOI] [PubMed] [Google Scholar]
- [157].Montanari F, Ecker GF, Adv. Drug. Deliv. Rev. 2015, 86, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ, Proc. Natl. Acad. Sci. 2008, 105, 17356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Siegel RL, Miller KD, Jemal A, CA Cancer J Clin. 2017, 67, 7. [DOI] [PubMed] [Google Scholar]
- [160].Jin W, Qin B, Chen Z, Liu H, Barve A, Cheng K, Int. J. Pharm. 2016, 513, 138. [DOI] [PubMed] [Google Scholar]
- [161].Barve A, Jin W, Cheng K, Controlled Release J 2014, 187, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Lupold SE, Hicke BJ, Lin Y, Coffey DS, Cancer Res. 2002, 62, 4029. [PubMed] [Google Scholar]
- [163].Yang J, Xie S-X, Huang Y, Ling M, Liu J, Ran Y, Wang Y, Thrasher BJ, Berkland C, Li B, Nanomedicine 2012, 7, 1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Dalal K, Che M, Que NS, Sharma A, Yang R, Lallous N, Borgmann H, Ozistanbullu D, Tse R, Ban F, Li H, Tam KJ, Roshan-Moniri M, LeBlanc E, Gleave ME, Gewirth DT, Dehm SM, Cherkasov A, Rennie PS, Mol. Cancer Ther. 2017, 16, 2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].(a) Lallous N, Dalal K, Cherkasov A, Rennie PS, Int. J. Mol. Sci. 2013, 14, 12496; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Green SM, Mostaghel EA, Nelson PS, Mol. Cell. Endocrinol. 2012, 360, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Mishra PK, Gulbake A, Jain A, Vyas SP, Jain SK, Drug Deliv. 2009, 16, 437. [DOI] [PubMed] [Google Scholar]
- [167].Dreaden EC, Gryder BE, Austin LA, Defo BA, Hayden SC, Pi M, Quarles DL, Oyelere AK, El-Sayed MA, Bioconjug. Chem. 2012, 23, 1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Kolinsky M, de Bono JS, Eur. Urol. 2016, 69, 841. [DOI] [PubMed] [Google Scholar]
- [169].Ponka P, Lok CN, Int. J. Biochem. Cell Biol 1999, 31, 1111. [DOI] [PubMed] [Google Scholar]
- [170].Gao W, Ye G, Duan X, Yang X, Yang VC, Int. J. Nanomedicine 2017, 12, 1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Tavano L, Muzzalupo R, Mauro L, Pellegrino M, Ando S, Picci N, Langmuir 2013, 29, 12638. [DOI] [PubMed] [Google Scholar]
- [172].Kobayashi T, Ishida T, Okada Y, Ise S, Harashima H, Kiwada H, Int. J. Pharm. 2007, 329, 94. [DOI] [PubMed] [Google Scholar]
- [173].Liu L, Wei Y, Zhai S, Chen Q, Xing D, Biomaterials 2015, 62, 35. [DOI] [PubMed] [Google Scholar]
- [174].Daniels TR, Delgado T, Helguera G, Penichet ML, Clin. Immunol. 2006, 121, 159. [DOI] [PubMed] [Google Scholar]
- [175].(a) Ringhieri P, Mannucci S, Conti G, Nicolato E, Fracasso G, Marzola P, Morelli G, Accardo A, Int. J. Nanomedicine 2017, 12, 501; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hirsh FR, Franklin WA, Veve R, Varella-Garcia M, Bunn PA, Semin. Oncol. 2002, 29, 51. [DOI] [PubMed] [Google Scholar]
- [176].Press MF, Cordon-Cardo C, Slamon DJ, Oncogene 1990, 5, 953. [PubMed] [Google Scholar]
- [177].Wartlick H, Michaelis K, Balthasar S, Strebhardt K, Kreuter J, Langer K, J. Drug Target 2004, 12, 461. [DOI] [PubMed] [Google Scholar]
- [178].Anhorn MG, Wagner S, Kreuter JR, Langer K, von Briesen H, Bioconjug. Chem. 2008, 19, 2321. [DOI] [PubMed] [Google Scholar]
- [179].Yu K, Zhao J, Zhang Z, Gao Y, Zhou Y, Teng L, Li Y, Int. J. Pharm. 2016, 497, 78. [DOI] [PubMed] [Google Scholar]
- [180].Eloy JO, Petrilli R, Chesca DL, Saggioro FP, Lee RJ, Marchetti JM, Eur. J. Pharm. Biopharm. 2017, 115, 159. [DOI] [PubMed] [Google Scholar]
- [181].Zahmatkeshan M, Gheybi F, Rezayat SM, Jaafari MR, Eur. J. Pharm. Sci. 2016, 86, 125. [DOI] [PubMed] [Google Scholar]
- [182].Le TTD, Pham TH, Nguyen TN, Ngo THG, Hoang TMN, Le QH, Adv. Nat. Sci.: Nanosci. Nanotechnol. 2016, 7, 025004. [Google Scholar]
- [183].Tai W, Mahato R, Cheng K, Controlled Release J 2010, 146, 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Diver EJ, Foster R, Rueda BR, Growdon WB, Oncologist 2015, 20, 1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Poon Z, Chen S, Engler AC, Lee HI, Atas E, von Maltzahn G, Bhatia SN, Hammond PT, Angew. Chem. Int. Ed. 2010, 49, 7266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Elias DR, Poloukhtine A, Popik V, Tsourkas A, Nanomedicine: Nanotechnology, Biology, and Medicine 2013, 9, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Moradi E, Vllasaliu D, Garnett M, Falcone F, Stolnik S, RSC Advances 2012, 2, 3025. [Google Scholar]
- [188].Liu D, Guo P, McCarthy C, Wang B, Tao Y, August DT, Nat. Commun. 2018, 9, 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Xie H, She ZG, Wang S, Sharma G, Smith JW, Langmuir 2012, 28, 4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Gao Y, Zhang C, Zhou Y, Li J, Zhao L, Li Y, Liu Y, Li X, Pharm. Res. 2015, 32, 2649. [DOI] [PubMed] [Google Scholar]
- [191].(a) Gao JQ, Lv Q, Li LM, Tang XJ, Li FZ, Hu YL, Han M, Biomaterials 2013, 34, 5628; [DOI] [PubMed] [Google Scholar]; b) Belhadj Z, Zhan C, Ying M, Wei X, Xie C, Yan Z, Lu W, Oncotarget 2017, 8, 66889; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang Y, Zhai M, Chen Z, Han X, Yu F, Li Z, Xie X, Han C, Yu L, Yang Y, Mei X, Drug Deliv.. 2017, 24, 1045; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ke X, Lin W, Li X, Wang H, Xiao X, Guo Z, Drug Deliv.. 2017, 24, 1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Kelly JM, Amor-Coarasa A, Nikolopoulou A, Wustemann T, Barelli P, Kim D, Williams C Jr., Zheng X, Bi C, Hu B, Warren JD, Hage DS, DiMagno SG, Babich JW, J. Nucl. Med. 2017, 58, 1442. [DOI] [PubMed] [Google Scholar]
- [193].Shu C, Li R, Yin Y, Yin D, Gu Y, Ding L, Zhong W, Chem. Commun. 2014, 50, 15423. [DOI] [PubMed] [Google Scholar]
- [194].Bae S, Ma K, Kim TH, Lee ES, Oh KT, Park ES, Lee KC, Youn YS, Biomaterials 2012, 33, 1536. [DOI] [PubMed] [Google Scholar]
- [195].Zong T, Mei L, Gao H, Shi K, Chen J, Wang Y, Zhang Q, Yang Y, He Q, J. Pharm. Sci. 2014, 103, 3891. [DOI] [PubMed] [Google Scholar]
- [196].Xia QS, Ding HM, Ma YQ, Nanoscale 2017, 9, 8982. [DOI] [PubMed] [Google Scholar]
- [197].Liu J, Weller GE, Zern B, Ayyaswamy PS, Eckmann DM, Muzykantov VR, Radhakrishnan R, Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 16530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Wang S, Dormidontova EE, Biomacromolecules 2010, 11, 1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].(a) Gopalakrishnan M, Forsten-Williams K, Nugent MA, Täuber UC, Biophys. J. 2005, 89, 3686; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Caré BR, Soula HA, BMC Syst. Biol. 2011, 5, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Gref R, Domb A, Quellec P, Blünk T, Muller R, Verbavatz J, Langer R, Adv. Drug Deliv. Rev. 1995, 16, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].von Maltzahn G, Ren Y, Park JH, Min DH, Kotamraju VR, Jayakumar J, Fogal V, Sailor MJ, Ruoslahti E, Bhatia SN, Bioconjug. Chem. 2008, 19, 1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Kolb HC, Finn MG, Sharpless KB, Angew. Chem. Int. Ed. 2001, 40, 2004. [DOI] [PubMed] [Google Scholar]
- [203].(a) Seo TS, Li Z, Ruparel H, Ju J, J. Org. Chem. 2003, 68, 609; [DOI] [PubMed] [Google Scholar]; b) Speers AE, Cravatt BF, Chem. Biol. 2004, 11, 535. [DOI] [PubMed] [Google Scholar]
- [204].(a) Elias DR, Cheng Z, Tsourkas A, Small 2010, 6, 2460; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lutz JF, Zarafshani Z, Adv. Drug Deliv. Rev. 2008, 60, 958; [DOI] [PubMed] [Google Scholar]; c) Yu H, Nie Y, Dohmen C, Li Y, Wagner E, Biomacromolecules 2011, 12, 2039. [DOI] [PubMed] [Google Scholar]
- [205].(a) Pathak RK, McNitt CD, Popik VV, Dhar S, Chemistry 2014, 20, 6861; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chang PV, Prescher JA, Sletten EM, Baskin JM, Miller IA, Agard NJ, Lo A, Bertozzi CR, Proc. Natl. Acad. Sci. 2010, 107, 1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Hoyle CE, Bowman CN, Angew. Chem. Int. Ed. 2010, 49, 1540. [DOI] [PubMed] [Google Scholar]
- [207].Yano S, Ohi H, Ashizaki M, Obata M, Mikata Y, Tanaka R, Nishioka T, Kinoshita I, Sugai Y, Okura I, Ogura S. i., Czaplewska JA, Gottschaldt M, Schubert US, Funabiki T, Morimoto K, Nakai M, Chem. Biodiversity 2012, 9, 1903. [DOI] [PubMed] [Google Scholar]
- [208].Sehgal D, Vijay IK, Anal. Biochem. 1994, 218, 87. [DOI] [PubMed] [Google Scholar]
- [209].Platt V, Huang Z, Cao L, Tiffany M, Riviere K, Szoka FC Jr., Bioconjug. Chem. 2010, 21, 892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Watson DS,Platt VM,Cao L, Venditto VJ, Szoka FC Jr., Clinical and Vaccine Immunology 2011, 18, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Krall N, da Cruz FP, Boutureira O, Bernardes GJ, Nat. Chem. 2016, 8, 103. [DOI] [PubMed] [Google Scholar]
- [212].Kramer K, Shields NJ, Poppe V, Young SL, Walker GF, Mol. Ther. 2017, 25, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z, J. Controlled Release 2011, 152, 2. [DOI] [PubMed] [Google Scholar]
- [214].Szymanski P, Markowicz M, Mikiciuk-Olasik E, Int. J. Mol. Sci. 2012, 13, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Chi CW, Ahmed AR, Dereli-Korkut Z, Wang S, Bioanalysis 2016, 8, 921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Dahlman JE, Kauffman KJ, Xing Y, Shaw TE, Mir FF, Dlott CC, Langer R, Anderson DG, Wang ET, Proc. Natl. Acad. Sci. 2017, 114, 2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Cheng J, Teply BA, Sherifi I, Sung J, Luther G, Gu FX, Levy-Nissenbaum E, Radovic-Moreno AF, Langer R, Farokhzad OC, Biomaterials 2007, 28, 869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Wang RT, Zhi XY, Yao SY, Zhang Y, Colloids Surf. B 2015, 133, 43. [DOI] [PubMed] [Google Scholar]
- [219].Bruns CJ, Shrader M, Harbison MT, Portera C, Solorzano CC, Jauch KW, Hicklin DJ, Radinsky R, Ellis LM, Int. J. Cancer 2002, 102, 101. [DOI] [PubMed] [Google Scholar]
- [220].Tao W, Zeng X, Wu J, Zhu X, Yu X, Zhang X, Zhang J, Liu G, Mei L, Theranostics 2016, 6, 470. [DOI] [PMC free article] [PubMed] [Google Scholar]